

Abstract
The behavior of the redox‐active aminotroponiminate (ATI) ligand in the coordination sphere of bismuth has been investigated in neutral and cationic compounds, [Bi(ATI)3] and [Bi(ATI)2Ln][A] (L=neutral ligand; n=0, 1; A=counteranion). Their coordination chemistry in solution and in the solid state has been analyzed through (variable‐temperature) NMR spectroscopy, line‐shape analysis, and single‐crystal X‐ray diffraction analyses, and their Lewis acidity has been evaluated by using the Gutmann–Beckett method (and modifications thereof). Cyclic voltammetry, in combination with DFT calculations, indicates that switching between ligand‐ and metal‐centered redox events is possible by altering the charge of the compounds from 0 in neutral species to +1 in cationic compounds. This adds important facets to the rich redox chemistry of ATIs and to the redox chemistry of bismuth compounds, which is, so far, largely unexplored.
Keywords: aminotroponiminates, bismuth, cationic species, redox chemistry, redox-active ligands
Central control: A series of bismuth aminotroponiminate cations are discussed and differences between neutral and cationic compounds in terms of coordination chemistry, Lewis acidity, and redox behavior are pinpointed. Specifically, cationization allows switching from ligand‐centered redox events to metal‐centered redox events, facilitating rare examples of quasi‐reversible electron transfer at bismuth.

Introduction
In recent years, redox‐active ligands have been established as a versatile and valuable tool to diversify and control the properties and reactivity of coordination entities. [1] In compounds of first‐row transition metals, for instance, redox‐active ligands have been used to stabilize unusual oxidation states of the central atom [2] or to foster reactions that involve two‐electron processes rather than single‐electron transfer. [3] In compounds of p‐block elements, redox‐active ligands have been used to enable facile and reversible electron transfer and to open up reaction pathways for controlled radical reactions.[ 1c , 1g , 4 ]
It has been demonstrated that redox‐active ligands may also be directly involved in selective bond‐forming events, such as the dimerization of radical species, which result from a high spin density at specific sites of the redox‐active entity. [5] However, strategies to control such reactivity remain rare. Aluminum complex A has been reported to undergo dimerization through C−C bond formation to give A 2 (Scheme 1 a, left), whereas the closely related species B is isolable in the monomeric form (Scheme 1 a, right). [4a] This has been ascribed to intramolecular ferromagnetic coupling between the radical ligands in B, which leads to a reduced effective spin density in these compounds. We have demonstrated that rhodium compounds of aminotroponiminates (ATIs) are susceptible to reversible (ligand‐centered) electron transfer (Scheme 1 b (left), C→D), whereas the corresponding alkali‐metal complexes readily undergo reductively induced dimerization (Scheme 1 b (right), C→0.5 E). [6] Thus, modifying the metal bound to a redox‐active ligand can have a dramatic influence on ligand‐centered redox events and reactivity.
Scheme 1.

Controlling the properties of open‐shell main‐group compounds: a) through magnetic coupling (dimerization vs. isolable species); b) through the choice of the central atom (reversible electron transfer vs. dimerization). X=Cl, O3SCF3; R=2,6‐iPr2‐C6H3.
Although the chemistry of redox‐active ligands is well developed for central atoms across large parts of the periodic table, [1] the redox chemistry of heavier Group 15 elements, and the heaviest congener bismuth, in particular, has been focused on metal(oid)‐centered radical species and redox‐shuttling.[ 5 , 7 ] Remarkable advances in the field include the incorporation of BiV/BiIII and proposed BiIII/BiI redox couples in catalytic cycles, [8] the first examples of persistent and isolable bismuth radical complexes, [9] the generation of an organometallic biradical, [10] unusual reactivity patterns (e.g., towards P4 and S8), [11] and catalytic applications in cycloisomerization and dehydrocoupling reactions, [12] olefin polymerization, [13] and photochemistry. [12c] These findings were largely based on strategies in which the radical center (if present) was stabilized through bulky ligands or through reversible radical recombination reactions. In contrast, the use of (potentially) redox‐active ligands in the field of bismuth chemistry is only little explored. Examples include bismuth(I) and bismuth(III) complexes with ferrocenyl groups in the ligand backbone (F, G), [14] as well as bismuth(III) pyridine dipyrrolide complexes H (Scheme 2). [15]
Scheme 2.

Bismuth complexes with redox‐active ligands (F, G) and potentially redox‐active ligands (H). R/R′=tBu/tBu, Ph/mesityl; Dtp=3,5‐tBu2‐C6H3.
Herein, we report on the synthesis, isolation, characterization, and redox properties of the first series of cationic bismuth ATI complexes.
Results and Discussion
Synthesis and structure
Starting from a reported sodium ATI compound [Na(ATIPh/iPr)(thf)], with its unsymmetric Ph/iPr substitution pattern, neutral and cationic bismuth ATIs 1 and 2‐X were synthesized in straightforward salt elimination protocols (Scheme 3; X=OTf, SbF6, BArF; OTf=O3SCF3; BArF=B(3,5‐(CF3)2C6H3)4). Following the same synthetic approach, cationic bismuth ATIs 3‐X and 4‐X based on ATIiPr/iPr, which has been reported in the literature, and the new ATIPhSMe/iPr ligand were obtained (Scheme 3; for details on the synthesis and characterization of [Na(ATIPhSMe/iPr)(thf)] and related species, see the Experimental Section and the Supporting Information). The products were isolated as red to orange solids.
Scheme 3.

Synthesis of neutral and cationic bismuth ATI complexes.
NMR spectroscopic analysis in solution revealed the expected signal patterns for bismuth complexes with one set of magnetically equivalent ATI ligands in all cases. In the case of neutral 1, a second set of resonances with minor intensities of 6 % was detected. This was ascribed to fac/mer isomerism in this M(AB)3‐type compound;[ 14b , 16 ] the main isomer is the fac species, according to DFT calculations (ΔG (fac‐1→mer‐1)= +1.9 kcal mol−1) and structural analysis of 1 in the solid state (see below). In all compounds with the [ATIPh/iPr]− ligand, the resonances for all protons of the phenyl groups were significantly broadened at room temperature. Variable‐temperature (VT) 1H NMR spectroscopy revealed coalescence temperatures of 40 °C for neutral 1 and 15–25 °C for cationic 2‐X and five well‐resolved signals for the phenyl groups in the low‐temperature scenario. This indicates a hindered rotation of the phenyl groups around the NATI−CPh bond. Line‐shape analyses and Eyring plots revealed activation parameters of ΔG ≠(298 K)=14.9 kcal mol−1 for neutral 1 and ΔG ≠(298 K)=13.6–13.9 kcal mol−1 for cationic 2‐X (for details, see the Supporting Information). Thus, the ATI ligands in neutral and cationic complexes experience similar steric clash in the coordination sphere of bismuth, although a significantly less crowded coordination environment and a more easily accessible metal center is found for the cationic species (see below). This was ascribed to the coordination geometry of the cationic species being dictated by orbital interactions rather than Coulomb interactions.
Compounds 1, 2‐X, 3‐X, and 4‐BArF were characterized by single‐crystal X‐ray diffraction analyses. Compound 1 crystallized in the monoclinic space group P21/n with Z=4 and shows a fac configuration (Figure 1 a). The central atom is found in a distorted octahedral coordination geometry: angles of ligands in cis orientation range from 65 to 118°. Strong deviations from ideal symmetry are due to the small bite angle of the ATI ligand (chelating: N‐Bi‐N, 65–67°). In addition, the HOMO‐3 shows significant contributions from an s‐type bismuth atomic orbital, which is polarized towards the empty space created by deviations from an octahedral symmetry, as determined by DFT calculations (Figure 1 b; for details, see the Supporting Information). This may be associated with a stereochemically active lone pair at bismuth. Due to its fac configuration, compound 1 bears three pairs of trans‐oriented NPh and NiPr groups. Delocalization of electron density in the ATI ligands prohibits the unequivocal assignment of amido and imino groups. Nevertheless, it is apparent that the C−NiPr bonds (1.31–1.33 Å) are, on average, slightly shorter than the C−NPh bonds (1.34–1.35 Å). In agreement with this observation, the Bi−NiPr bonds (2.53–2.58 Å) are significantly longer than the Bi−NPh bonds (2.35–2.38 Å). This is further supported by the Wiberg bond indices (WBIs), which are considerably smaller for the Bi−NiPr bonds (WBI=0.22–0.23) than for the Bi‐NPh bonds (WBI=0.32–0.33). This is in contrast to the only other homoleptic bismuth ATI complex [Bi(ATIFc/iPr)3], which crystallizes in a mer configuration, leading to an even more pronounced variation in Bi−N bond lengths (2.33–2.61 Å). [14b]
Figure 1.

a) Molecular structure of [Bi(ATIPh/iPr)3] (1) in the solid state. Displacement ellipsoids are shown at the 50 % probability level. Carbon atoms of Ph and iPr groups are shown in the wireframe model and hydrogen atoms are omitted for clarity. Bi1−N1 2.552(4), Bi1−N2 2.384(4), Bi1−N3 2.531(4), Bi1−N4 2.354(4), Bi1−N5 2.581(4), Bi1−N6 2.368(4), N1−C1 1.313(6), N2−C2 1.349(6), N3−C17 1.326(6), N4−C18 1.354(6), N5−C33 1.319(7), N6−C34 1.342(6) Å; N1‐Bi1‐N6 155.87(14), N2‐Bi1‐N3 158.65(13), N4‐Bi1‐N5 154.62(14), N1‐Bi1‐N2 64.89(12), N3‐Bi1‐N4 67.16(13), N5‐Bi1‐N6 64.89(13), N1‐Bi1‐N5 117.64(13)°. b) HOMO‐3 of compound 1 (isovalue=0.05), as determined by DFT calculations. This molecular orbital (MO) shows contributions by an s‐type bismuth atomic orbital that is polarized towards the hemisphere in which the NiPr groups are localized and may be associated with a stereochemically active lone pair (for details, see the Supporting Information).
The molecular structures of the cationic bismuth ATIs 2‐X, 3‐X, and 4‐BArF are shown in Figure 2. Selected crystallographic and structural information are summarized in Table 1. The coordination geometries of the cationic species are bisphenoidal (2‐BArF, 3‐OTf, 4‐BArF) or square pyramidal (2‐OTf, 2‐SbF6, 3‐SbF6; τ 5=0.18–0.28), if only the ATI and thf ligands are considered. If additional weak bonding interactions with counteranions or the thioether group in 4‐BArF are also taken into account, the coordination geometries are best described as square pyramidal (2‐BArF; τ 5=0.22), distorted octahedral (3‐OTf, 3‐SbF6), and irregular (2‐OTf, 2‐SbF6, 4‐BArF). The Bi−N bond lengths of the cationic species (2.21–2.38 Å) are, on average, significantly shorter than those in neutral 1 (2.35–2.58 Å), although the same or even higher coordination numbers are reached in the cationic species. This is due to the interactions between the bismuth atoms of the cationic species and their counteranions or neutral donor groups (thf or thioether) being weak compared with the Bi−NATI interactions in neutral 1. In all cationic species, the Bi−N bonds that face another Bi−N bond in the trans position (2.31–2.38 Å) are significantly longer than those that have a Bi−X bond or a free coordination site in the trans position (2.21–2.25 Å; X=OTHF, OOTf, Sthioether, FBArF), that is, there is a considerable thermodynamic trans effect in these compounds.[ 14b , 17 ] Notably, this is also observed in compounds 3‐X, in which the substituents at nitrogen are identical and any impact of the substitution pattern on Bi−N bond lengths is thus ruled out. The Bi−OTHF bond lengths in the cationic bismuth ATIs (2.81–2.94 Å) are within the broad range of Bi−OTHF bond lengths reported for other cationic bismuth amides (e.g., 2.57–3.19 Å in [Bi(NiPr2)2(thf)3][BArF]). [18] As a trend, a stronger electron‐donating ability of the ATI ligand ([ATIiPr/iPr]− in 3‐SbF6 versus [ATIPh/iPr]− in 2‐SbF6) [19] and the counteranion (OTf− in 2‐OTf vs. [SbF6]− in 2‐SbF6) increase the Bi−OTHF bond lengths (Table 1). The Bi⋅⋅⋅Sthioether interactions in 4‐BArF are very weak (if they exist) based on distance criteria (Bi⋅⋅⋅S 3.87 Å; identical to the sum of the van der Waals radii), which is ascribed to geometrical constraints in the ligand framework. While bonding interactions between the bismuth atom and the counteranion are significant in the case of the triflate species (Bi⋅⋅⋅OOTf 2.87–3.22 Å; 10–20 % below the sum of the van der Waals radii), they are less pronounced for the hexafluoroantimonates (Bi⋅⋅⋅F 3.08–3.14 Å; 11–13 % below the sum of the van der Waals radii), [20] and only minor in the BArF species (Bi⋅⋅⋅F 3.29–3.47 Å; 2–7 % below the sum of the van der Waals radii). The cationic species generally form typical mononuclear complexes in the solid state. As an exception to this, compound 3‐OTf crystallizes as a coordination polymer due to a bridging coordination mode of the triflate anion (for details, see the Supporting Information). Surprisingly, the cationic species 2‐BArF, which forms only very weak interactions with the borate counteranion, crystallizes from solutions of difluorobenzene without any thf ligands bound to the bismuth atom. This shows that the thf adduct (as obtained from the isolation of analytically pure bulk material) may release its neutral ligand in solutions of moderately polar and weakly donating solvents.
Figure 2.
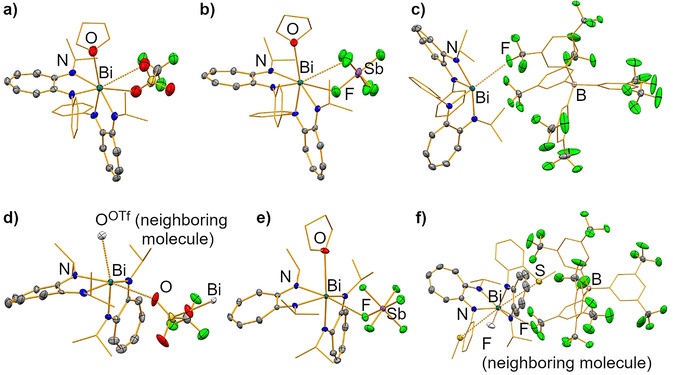
Molecular structures of compounds 2‐X (a–c), 3‐X (d, e), and 4‐BArF (f) in the solid state. Displacement ellipsoids are shown at the 50 % probability level. Carbon atoms, except for those of the ATI backbone, are shown as wireframe. Hydrogen atoms and lattice‐bound solvent molecules are omitted for clarity. Only one of the six crystallographically independent, but chemically identical, formula units is shown in d). Atoms that exceed one formula unit are shown as colorless ellipsoids (d, f). For details, see the Supporting Information.
Table 1.
Selected crystallographic information and structural parameters of compounds 1, 2‐X, 3‐X, and 4‐BArF. For details, see the Supporting Information.
|
|
1 |
2‐OTf |
2‐SbF6 |
2‐BArF |
3‐OTf[a] |
3‐SbF6 |
4‐BArF |
|
|---|---|---|---|---|---|---|---|---|
|
crystal system |
monoclinic |
monoclinic |
monoclinic |
triclinic |
monoclinic |
monoclinic |
orthorhombic |
|
|
space group |
P21/n |
P21/n |
P21/n |
P
|
P21 |
Cc |
C2221 |
|
|
Z |
4 |
4 |
4 |
2 |
2 |
4 |
4 |
|
|
R/R′ |
Ph/iPr |
Ph/iPr |
Ph/iPr |
Ph/iPr |
iPr/iPr |
iPr/iPr |
PhSMe/iPr |
|
|
coordination number with (without) weak interactions[b] |
6 |
7 (5) |
7 (5) |
5 (4) |
6 (4) |
6 (5) |
8 (4) |
|
|
Bi−OTHF [Å] |
– |
2.943(11) |
2.809(5) |
– |
– |
2.888(4) |
2×Bi⋅⋅⋅SMeR; 3.8693(15); 3.8693(15)[c] |
|
|
Bi⋅⋅⋅anion; [Å] |
– |
2×Bi⋅⋅⋅O; 3.120(14); 3.215(13) |
2×Bi⋅⋅⋅F; 3.084(7); 3.437(7) |
1×Bi⋅⋅⋅F; 3.2908(19) |
2×Bi⋅⋅⋅O; 2.873(6); 2.940(6) |
1×Bi⋅⋅⋅F; 3.139(3) |
2×Bi⋅⋅⋅F; 3.470(6); 3.470(6) |
|
|
Bi−N (other N atom in trans position) [Å] |
shortest Bi−N: 2.354(4); longest Bi−N: 2.581(4) |
2.360(4); 2.383(4) |
2.311(5); 2.370(5) |
2.327(2); 2.3389(19) |
2.336(7); 2.341(8) |
2.339(3); 2.376(3) |
2.338(5); 2.338(5) |
|
|
Bi−N (X or free coordination site in trans position)[d] [Å] |
2.241(4); 2.250(4) |
2.221(4); 2.249(4) |
2.2068(19); 2.210(2); |
2.195(10); 2.226(6) |
2.226(3); 2.243(3) |
2.219(5); 2.219(5) |
[a] Data for one of the six crystallographically independent, but chemically identical, formula units are presented (for details, see the Supporting Information). [b] For the determination of the coordination number in compounds 2‐X, 3‐X, and 4‐X, Bi⋅⋅⋅OOTf, Bi⋅⋅⋅F, and Bi⋅⋅⋅Sthioether, interactions were considered to be weak. [c] No thf ligand is present in 4‐BArF and the interatomic distance between the Bi atom and S atoms of the thioether functional groups are listed instead. [d] X=OTHF, OOTf, Sthioether, F , F .
Lewis acidity
The Lewis acidity of molecular cationic bismuth compounds can be a crucial factor for the realization of unusual phenomena and reactivity patterns in coordination chemistry, [21] group‐transfer reactions, [18] CH activation, [22] small‐molecule activation, [23] and catalytic applications.[ 13b , 24 ] It has recently been quantified by using the Gutmann–Beckett (GB) method, that is, through adduct formation of the bismuth species with OPEt3. [25] We have recently reported that the use of EPMe3 (E=S, Se) instead of OPEt3 can be exploited for the assessment of the soft character of a Lewis acid, which is demonstrated to be especially pronounced for bismuth cations. [26] Because the Lewis acidity of cationic bismuth compounds based on chelating N,N‐donor ligands has not been quantified, to date, selected bismuth ATI complexes were investigated with the original and modified GB methods (Table 2).
Table 2.
31P NMR chemical shifts and acceptor numbers (ANs), according to the original GB method (OPEt3 as a Lewis base) and modifications thereof (SPMe3, SePMe3 as Lewis bases).[a]
|
Compound |
OPEt3 |
SPMe3 |
SePMe3 |
|||
|---|---|---|---|---|---|---|
|
|
δ 31P [ppm] |
AN |
δ 31P [ppm] |
AN |
δ 31P [ppm] |
AN |
|
1 |
50.3 |
21 |
|
|
|
|
|
2‐OTf |
50.4 |
21 |
|
|
|
|
|
2‐SbF6 |
53.4 |
27 |
|
|
|
|
|
2‐BArF |
52.4 |
25 |
|
|
|
|
|
3‐OTf |
51.1 |
22 |
|
|
|
|
|
4‐BArF |
54.0 |
29 |
30.9 |
11 |
8.9 |
6 |
[a] One equivalent of OPEt3, SPMe3, or SePMe3 was added to solutions of the respective bismuth compound in dichloromethane. For details, see the Experimental Section.
By using one equivalent of OPEt3 as a donor in dichloromethane, all bismuth ATIs showed relatively low acceptor numbers of AN(OPEt3)=21–29. As a trend, bismuth ATI cations without significant bonding interactions to the counteranion showed slightly larger ANs of 25–29 than those with Bi⋅⋅⋅OTf interactions or neutral 1 [27] (AN(OPEt3)=21–22).
These ANs are larger than that of B(NMe2)3 (AN(OPEt3)=9) and similar to that of B(OMe)3 (AN(OPEt3)=23). [28] Compound 4‐BArF was additionally investigated with the modified GB method, but also showed low ANs if the soft donors EPMe3 were applied (AN(SPMe3)=11; AN(SePMe3)=6). Thus, the ATI ligand (an example of a bidentate monoanionic N,N‐donor ligand) quenches large parts of the Lewis acidity of the bismuth center towards external Lewis bases, even in cationic species. This raises the question of whether the reduction of cationic bismuth ATIs would predominantly be a ligand‐ or a metal‐centered event (or an intermediate scenario; see below).
Redox properties
The redox‐active nature of ATI ligands has recently been established. Specifically, it has been demonstrated that alkali‐metal ATI complexes undergo reductively induced dimerization reactions, whereas rhodium ATI species are susceptible to reversible, ligand‐centered electron transfer (Scheme 1 b). [6] To shed some light on the redox properties of the bismuth complexes presented herein, they were analyzed by cyclic voltammetry (all potentials referenced vs. ferrocene/ferrocenium; for details, see the Experimental Section and the Supporting Information). An irreversible redox event in the range of 0.27–0.39 V was observed for all compounds (see the Supporting Information). Under reducing conditions, neutral bismuth ATI complex 1 showed a reduction wave at −2.26 V, with the corresponding oxidation wave appearing at −1.20 V (Figure 3 a and Table 3, entry 1). This indicates that a chemical reaction takes place after electron transfer. The ratio, I pc/I pa, of these redox waves is close to one and the shape of the cyclic voltammogram does not change significantly upon increasing the number of cycles, which indicates the reversibility of the ECEC sequence. This behavior (as determined from cyclic voltammetry) is strongly reminiscent of the situation found for the corresponding sodium compound, [Na(ATIPh/iPr)(thf)], which undergoes a highly selective, chemically reversible, reductively induced dimerization with the formation of a C−C bond between two ATI moieties (cf. C→E in Scheme 1 b). [6b] In contrast, no indications for such redox behavior were obtained in the cyclic voltammetry analyses of the cationic bismuth ATI species 2‐X, 3‐X, and 4‐BArF. While complexes with [OTf]− or [SbF6]− counteranions show partially reversible redox events, compounds 2‐BArF and 4‐BArF show quasi‐reversible redox events at strongly negative potentials of −2.61 and −2.57 V, respectively (shown for 2‐BArF in Figure 3 b and Table 3, entries 2–7). While the redox potentials of cationic bismuth ATIs with [ATIPh/iPr]− and [ATIPhSMe/iPr]− ligands range from −2.57 to −2.64 V, those for complexes with the [ATIiPr/iPr]− ligand were found from −2.71 to −2.80 V. This corresponds to a cathodic shift of up to 230 mV and reflects the more electron‐donating nature of the [ATIiPr/iPr]− ligand, which has previously been discussed in the context of catalytic applications. [19]
Figure 3.
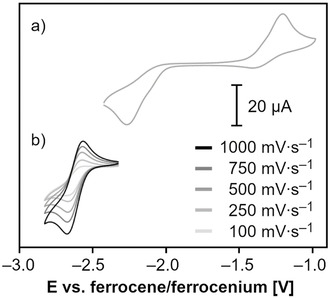
Cyclic voltammograms of 1 (a) and [Bi(ATIPh/iPr)2][BArF] (2‐BArF) (b) in THF/0.1 m [N(nBu)4][PF6] at a temperature of 23 °C and scan rates in the range of 100–1000 mV s−1.
Table 3.
Redox properties of bismuth ATI complexes under reducing conditions, as deduced from cyclic voltammetry in THF/0.1 m [N(nBu)4][PF6] at 23 °C and scan rates of 50–5000 mV s−1 (for details, see the Supporting Information).
|
Entry |
Compound |
E 1/2 or E pc/E pa [V][a] |
Classification[b] |
Reversibility[c] |
|---|---|---|---|---|
|
1 |
1 |
−2.26/−1.20[d] |
ECEC |
qr |
|
2 |
2‐OTf |
−2.63 |
EE |
pr |
|
3 |
2‐SbF6 |
−2.64 |
EE |
pr |
|
4 |
2‐BArF |
−2.61 |
EE |
qr |
|
5 |
3‐OTf |
−2.71 |
EE |
pr |
|
6 |
3‐SbF6 |
−2.80 |
EE |
pr |
|
7 |
4‐BArF |
−2.57 |
EE |
qr |
[a] Potentials are referenced versus the ferrocene/ferrocenium couple; entry 1: E pc/E pa; entries 2–7: E 1/2. [b] E=electron transfer; C=chemical reaction. [c] qr=quasi‐reversible; pr=partially reversible. [d] There is an additional redox event at −2.66 V (for details, see the Supporting Information).
Overall, these findings demonstrate that the redox properties of bismuth ATI compounds can be controlled by the choice of the charge of the complex. While the redox behavior of the neutral species resembles that of alkali‐metal ATI complexes, the cationic species show a redox behavior reminiscent of the corresponding rhodium compound. DFT calculations were performed to rationalize these differences (for details, see the Experimental Section and the Supporting Information). Frontier orbital analysis of 1 and 2‐BArF revealed that the LUMO is exclusively ligand‐centered in the neutral compound, but has significant contributions from a bismuth p‐type atomic orbital in the cationic species (Figure 4 a,b). These findings are in agreement with an analysis of the spin‐density distribution in the reduced species Na‐1‐rad and 2‐rad. Compound Na‐1‐rad shows a negligible spin density of 0.6 % at the bismuth atom, and an overwhelming spin density of 97 % at one ATI ligand, which interacts with the sodium countercation (with spin densities of up to 27 % at individual atoms of this ligand; Figure 4 c). In contrast, compound 2‐rad may be described as a bismuth‐centered radical (72 % spin density) supported by two redox‐active ATI ligands, each of which has a spin density of 14 % (with up to 3 % spin density at individual atoms of these ligands; Figure 4 d). [29]
Figure 4.

Structures obtained from geometry optimizations by DFT calculations. LUMOs of 1 (a) and 2‐BArF (b; the counteranion is included in the calculation, but omitted in the figure for clarity) at isovalues of 0.03. Spin densities of [Na(thf)2][Bi(ATIPh/iPr)3] (Na‐1‐rad) (c) and [Bi(ATIPh/iPr)2] (2‐rad) (d) at isovalues of 0.002.
Conclusion
We have prepared the first examples of cationic bismuth ATIs, [Bi(ATI)2Ln][A] (L=neutral ligand; n=0–1; A=counteranion). Depending on the choice of the counteranion, contact ion pairs, solvent‐separated ion pairs, or “naked” bismuth ATI cations with weak Bi⋅⋅⋅[F] interactions were obtained ([F]=fluorine atom of BArF). According to VT‐NMR spectroscopy studies and line‐shape analyses, the ATI ligands in neutral and cationic species experience similar steric clash, which was assigned to a predominantly orbital‐controlled coordination chemistry in the cationic compounds. The Lewis acidity of bismuth ATI cations towards hard and soft donors was relatively weak, according to the GB method (and its modifications), which was ascribed to the bidentate nature of the monoanionic ATI ligand. In turn, the empty p orbital of the bismuth atom in cationic species was accessible under reducing conditions and allowed for quasi‐reversible electron transfer at bismuth (according to cyclic voltammetry and DFT calculations). In contrast, investigations into the redox chemistry of the neutral bismuth ATI complex [Bi(ATIiPr/iPr)3] indicated ligand‐centered redox events. Thus, we demonstrated that the choice of the charge in bismuth ATI complexes allowed switching from ligand‐centered redox events (as previously reported for alkali‐metal complexes) to metal‐centered, quasi‐reversible redox events. The latter was analogous to Rh ATI compounds in terms of reversibility, but are distinct in terms of the localization of spin density in the reduced species (metal‐centered in [Bi(ATI)2]., but ligand‐centered in [Rh(ATI)(cod)].−; cod=1,5‐cyclooctadienyl). These findings open up perspectives for switchable redox‐catalysis based on the heavy main‐group element bismuth.
Experimental Section
General
All air‐ and moisture‐sensitive manipulations were carried out by using standard vacuum‐line Schlenk or glove box techniques in an atmosphere of purified argon. Solvents were degassed and purified, according to standard laboratory procedures. NMR spectra were recorded on Bruker instruments operating at 200, 400, or 500 MHz with respect to 1H. 19F and 31P NMR spectra were recorded with proton decoupling. All chemical shifts (δ) are reported in ppm. 1H and 13C NMR chemical shifts are reported relative to SiMe4 by using the residual 1H and 13C chemical shifts of the solvent as a secondary standard. 19F and 31P NMR chemical shifts are reported relative to CFCl3 and 85 % aqueous H3PO4, respectively, as external standards. Unless stated otherwise, NMR spectra were recorded at 23 °C. Elemental analyses were performed on a Leco or a Carlo Erba instrument. For MS analyses, an Exactive plus instrument (Thermo Scientific) was used. Cyclic voltammograms were recorded by using a Gamry Instruments Reference 600 potentiostat at 23 °C in THF containing 0.1 m [N(nBu)4][PF6], unless otherwise noted. A standard three‐electrode cell configuration was employed by using a platinum disk working electrode, a platinum wire counter electrode, and a silver wire separated by a Vycor tip as the reference electrode. Formal redox potentials are referenced to the ferrocene/ferrocenium redox couple. Single crystals suitable for X‐ray diffraction were coated with perfluorinated polyether oil in a glove box, transferred to a nylon loop, and then transferred to the goniometer of a diffractometer equipped with a molybdenum (λ=0.71073 Å) or copper (λ=1.5418 Å) X‐ray tube. The structures were solved using the intrinsic phasing method, completed by Fourier synthesis and refined by full‐matrix least‐squares procedures. Deposition numbers 2044644, 2044645, 2044646, 2044647, 2044648, 2044649, 2044650, 2044875, and 2044876 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Computational details
DFT calculations were performed with the Gaussian program by using the 6‐31G(d,p) (H, B, C, N, O, F), [30] 6‐311G(d,p) (Na), [31] and LaNL2DZ (Bi) [32] basis sets and the (U)B3LYP functional. [33] The D3 version of Grimme's dispersion model with the original D3 damping function was applied. [34] Frequency analyses of the reported structures showed no imaginary frequencies. Thermodynamic parameters were calculated at a temperature of 298.15 K and a pressure of 1.00 atm. Further details are given in the Supporting Information. Cartesian coordinates of optimized structures are provided in .xyz format.
General procedure for the original and modified GB methods
Equimolar amounts of the potential Lewis acid (usually 10 mg of the bismuth complex) and the Lewis base (OPEt3, SPMe3, or SePMe3) were dissolved in CD2Cl2 (0.5 mL) and 1H and 31P NMR spectra were recorded.
Compound 1
NaN(SiMe3)2 (42 mg, 0.23 mmol) and BiCl3 (24 mg, 76 μmol) were subsequently added to a solution of N‐iPr‐2‐phenylaminotroponimine (54 mg, 0.23 mmol) in THF (2 mL). All volatile compounds were removed from the red reaction mixture under reduced pressure. The residue was suspended in toluene (3×1.5 mL) and the resulting suspension was filtered. All volatile compounds were removed from the deep‐red filtrate under reduced pressure to give a deep‐red solid, which was dried in vacuo (58 mg, 63 μmol, 83 %). M.p. 198 °C (dec); 1H NMR (500 MHz, −40 °C, [D8]toluene): δ=1.23 (d, 3 J(H,H)=5.5 Hz, 9 H; Me1), 1.32 (d, 3 J(H,H)=5.8 Hz, 9 H; Me2), 3.96–4.03 (br s, 3 H; CHMe2), 4.80 (d, 3 J(H,H)=8.2 Hz, 3 H; o‐Ph1), 5.83 (d, 3 J(H,H)=11.2 Hz, 3 H; 3‐H), 6.00 (br dd, 3 J(H,H)=8.2, 8.9 Hz, 3 H; 5‐H), 6.43 (d, 3 J(H,H)=11.8 Hz, 3 H; 7‐H), 6.44 (br dd, 3 J(H,H)=9.8, 11.2 Hz, 3 H; 4‐H), 6.77–6.80 (m, 3 H; p‐Ph), 6.87 (br dd, 3 J(H,H)=8.2, 11.8 Hz, 3 H; 6‐H), 6.97 (d, 3 J(H,H)=8.2 Hz, 3 H; o‐Ph2), 6.98–7.02 ppm (m, 6 H; m‐Ph1, 2); 13C NMR (126 MHz, −40 °C, [D8]toluene): δ=23.9 (s, Me2), 24.1 (s, Me1), 49.9 (s, CHMe2), 116.8 (s, 7‐C), 117.4 (s, 5‐C), 117.4 (s, 3‐C), 124.7 (s, p‐Ph), 127.7 (s, o‐Ph2), 128.5 (s, m‐Ph1/m‐Ph2), 129.2 (s, m‐Ph2/m‐Ph1), 133.3 (s, 6‐C), 133.6 (s, 4‐C), 151.4 (s, ipso‐Ph), 163.5 (s, 1‐C), 168.0 ppm (s, 2‐C); elemental analysis calcd (%) for C48H51N6Bi (920.95 g mol−1): C 62.60, H 5.58, N 9.13; found: C 62.79, H 5.41, N 9.09.
Compound 2‐OTf
A solution of Na(ATIiPr/Ph)(thf)0.84 (50 mg, 156 μmol) in THF (1 mL) was added dropwise to a suspension of sodium triflate (13.5 mg, 78 μmol) and BiCl3 (25 mg, 78 μmol) under stirring. The red suspension was filtered and layered with pentane (0.5 mL). After 24 h, the red crystalline precipitate was isolated by filtration, washed with pentane (3×3 mL), and dried in vacuo. Crystals suitable for single‐crystal X‐ray analysis were obtained by layering a solution of 2‐OTf (20 mg) in THF (0.5 mL) with pentane (0.1 mL). Yield: 33 mg, 37 μmol (with n=0.75 equiv of THF), 47 %; 1H NMR (500 MHz, −30 °C, [D5]pyridine): δ=0.70 (d, 3 J(H,H)=5.9 Hz, 6 H; CHMe), 1.35 (d, 3 J(H,H)=6.0 Hz, 6 H; CHMe), 1.48–1.50 (m, n×4 H; β‐THF), 3.26–3.20 (br m, 2 H; CHMe2), 3.57–3.59 (m, n×4 H; α‐THF), 6.30 (d, 3 J(H,H)=11.0 Hz, 2 H; 3‐H), 6.35 (d, 3 J(H,H)=7.8 Hz, 2 H; o‐Ph, partially overlapped by 3‐H), 6.37 (d, 3 J(H,H)=11.7 Hz, 2 H; 7‐H), 6.73 (dd, 3 J(H,H)=8.7, 9.8 Hz, 2 H; 5‐H), 7.13 (d, 3 J(H,H)=7.6 Hz, 2 H; o‐Ph), 7.18 (m, 2 H; 6‐H, partially overlapped by [D5]pyridine), 7.22 (m, 2 H; m‐Ph, partially overlapped by [D5]pyridine), 7.27 (dd, 3 J(H,H)=6.5, 8.3 Hz, 2 H; p‐Ph), 7.45 (m, 2 H; m‐Ph, partially overlapped by 4‐H), 7.47 ppm (m, 2 H; 4‐H); 13C NMR (126 MHz, −30 °C, [D5]pyridine): δ=21.0 (s, CHMe), 26.2 (s, β‐THF), 26.7 (s, CHMe), 50.0 (s, CHMe2), 68.3 (s, α‐THF), 120.6 (s, 3‐C), 121.7 (s, 7‐C), 124.5 (s, 5‐C), 127.8 (s, m‐Ph), 128.1 (s, o‐Ph), 129.4 (s, o‐Ph), 130.7 (s, p‐Ph), 130.7 (s, p‐Ph), 148.1 (s, ipso‐Ph), 165.1 (s, 1‐C), 170.3 ppm (s, 2‐C); 19F NMR (470 MHz, [D5]pyridine): δ=−76.07 ppm; elemental analysis calcd (%) for C33H34N4BiF3SO3 ⋅(C4H8O) (904.80 g mol−1): C 49.12, H 4.68, N 6.19, S 3.54; found: C 49.03, H 4.58, N 6.41, S 3.32.
Compound 2‐SbF6
A solution of Na(ATIiPr/Ph) (25 mg, 96 μmol) in THF (0.5 mL) was added dropwise to a suspension of silver hexafluoroantimonate(V) (17 mg, 48 μmol) and BiCl3 (15 mg, 48 μmol) under stirring. After 10 min, the red suspension was filtered, and the volume of the filtrate was reduced to half. After 24 h, the red crystalline precipitate was isolated by filtration, washed with pentane (3×1 mL), and dried in vacuo (38 mg, 39 μmol (with n=0.75 equiv of THF), 81 %). 1H NMR (500 MHz, −35 °C, [D5]pyridine): δ=0.59 (d, 3 J(H,H)=5.9 Hz, 6 H; CHMe), 1.25 (d, 3 J(H,H)=5.8 Hz, 6 H; CHMe), 1.46–1.49 (m, n×4 H; β‐THF), 3.19–3.23 (br m, 2 H; CHMe2), 3.56–3.59 (m, n×4 H; α‐THF), 6.33 (d, 3 J(H,H)=7.8 Hz, 2 H; o‐Ph, partially overlapped by 4‐H), 6.34 (d, 3 J(H,H)=11.1 Hz, 2 H; 3‐H), 6.37 (d, 3 J(H,H)=12.1 Hz, 2 H; 7‐H), 6.78 (dd, 3 J(H,H)=8.8, 9.7 Hz, 2 H; 5‐H), 7.01 (d, 3 J(H,H)=8.0 Hz, 2 H; o‐Ph), 7.22–7.25 (m, 2 H; 6‐H, partially overlapped by m‐Ph), 7.23–7.26 (m, 2 H; m‐Ph, partially overlapped by 6‐H), 7.28 (dd, 3 J(H,H)=7.4, 8.3 Hz, 2 H; p‐Ph), 7.45 (dd, 3 J(H,H)=7.2, 8.1 Hz, 2 H; m‐Ph), 7.51 ppm (dd, 3 J(H,H)=9.4, 11.1 Hz, 2 H; 4‐H); 13C NMR (126 MHz, −35 °C, [D5]pyridine): δ=20.9 (s, CHMe), 26.2 (s, β‐THF), 26.8 (s, CHMe), 49.9 (s, CHMe2), 68.3 (s, α‐THF), 120.8 (s, 3‐C), 124.5 (s, 7‐C), 125.0 (s, 5‐C), 127.9 (s, m‐Ph), 129.3 (s, o‐Ph), 129.5 (s, o‐Ph), 130.7 (s, p‐Ph), 130.8 (s, p‐Ph), 147.8 (s, ipso‐Ph), 165.0 (s, 1‐C), 170.2 ppm (s, 2‐C); elemental analysis calcd (%) for C32H34N4BiF6Sb⋅(C4H8O) (991.49 g mol−1): C 43.61, H 4.27, N 5.65; found: C 43.87, H 4.24, N 5.65.
Compound 2‐BArF
Na(ATIiPr/Ph) (200 mg, 602 μmol) was added dropwise to a solution of sodium tetrakis[3,5‐bis(trifluoromethyl)phenyl]borate (267 mg, 301 μmol) and BiCl3 (95 mg, 301 μmol) in THF (4 mL) under stirring. After 1 h, the red suspension was filtered, and all volatile compounds were removed under reduced pressure. The crude product was washed with hexane (2×2 mL) and dried in vacuo (416 mg, 257 μmol (with n=1 equiv of THF), 85 %). 1H NMR (500 MHz, −40 °C, [D8]THF): δ=1.10 (d, 3 J(H,H)=6.0 Hz, 6 H; CHMe), 1.48 (d, 3 J(H,H)=6.3 Hz, 6 H; CHMe), 1.77–1.79 (m, n×4 H; β‐THF), 3.39–3.44 (br m, 2 H; CHMe2), 3.61–3.63 (m, n×4 H; α‐THF), 6.14 (d, 3 J(H,H)=11.1 Hz, 2 H; 3‐H), 6.46 (d, 3 J(H,H)=7.8 Hz, 2 H; o‐Ph1), 6.48 (d, 3 J(H,H)=11.8 Hz, 2 H; 7‐H), 6.67 (dd, 3 J(H,H)=8.6, 9.8 Hz, 2 H; 5‐H), 7.15 (dd, 3 J(H,H)=9.8, 11.1 Hz, 2 H; 4‐H), 7.18 (d, 3 J(H,H)=7.8 Hz, 2 H; o‐Ph2), 7.28 (ddd, 3 J(H,H)=7.6, 7.6 Hz, 4 J(H,H)=1.7 Hz, 2 H; m‐Ph1), 7.35 (br dd, 3 J(H,H)=7.3, 7.3 Hz, 2 H; p‐Ph), 7.45 (m, 3 J(H,H)=8.6, 11.8 Hz, 4 J(H,H)=1.5 Hz, 2 H; 6‐H), 7.59 (ddd, 3 J(H,H)=7.3, 7.8 Hz, 4 J(H,H)=1.7 Hz, 2 H; m‐Ph2), 7.76 (br s, 4 H; 4‐C6H3(CF3)2), 7.86 (br s, 8 H; 2,6‐C6H3(CF3)2) ppm; 13C NMR (126 MHz, −40 °C, [D8]THF): δ=21.1 (s, Me1), 26.2 (s, β‐THF), 26.5 (s, Me2), 50.2 (s, CHMe2), 68.0 (s, α‐THF), 118.1 (s, 4‐C6H3(CF3)2), 120.8 (s, 3‐C), 124.4 (s, 5‐C), 125.0 (s, 7‐C), 125.2 (quart, 1 J(C,F)=271.9 Hz, CF3), 127.9 (s, p‐Ph), 128.6 (s, o‐Ph2), 129.5 (s, o‐Ph1), 129.7 (br quart, 2 J(C,F)=34.5 Hz, 3,5‐C6H3(CF3)2), 130.7 (s, m‐Ph1, 2), 135.2 (br s, 4‐C, 2,6‐C6H3(CF3)2 (overlapping)), 135.4 (s, 6‐C), 147.8 (s, ipso‐Ph), 162.7 (quart, 1 J(B,C)=49.9 Hz, 1‐C6H3(CF3)2), 165.0 (s, 1‐C), 170.2 ppm (s, 2‐C); elemental analysis calcd (%) for C64H46N4BF24Bi⋅(C4H8O) (1618.95 g mol−1): C 50.45, H 3.36, N 3.46; found: C 50.73, H 3.20, N 3.48.
Compound 3‐OTf
A solution of Na(ATIiPr/iPr) (200 mg, 808 μmol) in THF (1.5 mL) was added dropwise to a suspension of sodium triflate (70 mg, 404 μmol) and BiCl3 (127 mg, 404 μmol) under stirring. The red suspension was filtered and all volatile compounds of the filtrate were removed under reduced pressure to give an orange solid, which was washed with pentane (3×3 mL) and dried in vacuo. Crystals suitable for single‐crystal X‐ray analysis were obtained by layering a solution of 3‐OTf (20 mg) in THF (0.5 mL) with pentane (0.1 mL). Yield: 162 mg, 210 μmol, 52 %; 1H NMR (500 MHz, [D5]pyridine): δ=1.37 (d, 3 J(H,H)=6.7 Hz, 24 H; CHMe), 4.73–4.81 (sept, 3 J(H,H)=6.7 Hz, 4 H; CHMe2), 6.71 (t, 3 J(H,H)=9.1 Hz, 2 H; 5‐H), 6.92 (d, 3 J(H,H)=11.5 Hz, 4 H; 3‐H, 7‐H), 7.48 ppm (dd, 3 J(H,H)=11.8, 9.1 Hz, 4 H; 4‐H, 7‐H); 13C NMR (126 MHz, [D5]pyridine): δ=22.6 (s, CHMe), 52.3 (s, CHMe2), 120.86 (s, 3‐C, 7‐C), 120.87 (s, 3‐C, 7‐C), 123.5 (s, 5‐C), 135.0 (s, 4‐C, 6‐C), 168.2 ppm (s, 1‐C, 2‐C); 19F NMR (470 MHz, [D5]pyridine): δ=−76.16 ppm; elemental analysis calcd (%) for C27H38N4BiF3SO3 (764.66 g mol−1): C 42.41, H 5.01, N 7.33, S 4.19; found: C 42.61, H 4.98, N 7.49, S 3.96.
Compound 3‐SbF6
A solution of Na(ATIiPr/iPr) (25 mg, 101 μmol) in THF (1 mL) was added dropwise to a suspension of silver hexafluoroantimonate(V) (17 mg, 51 μmol) and BiCl3 (16 mg, 51 μmol) in a plastic vial under stirring. The red suspension was filtered and hexane was added (5 mL). The brown precipitate was isolated by filtration, washed with hexane (3×3 mL), and dried in vacuo. Drying for prolonged periods of time reproducibly led to the start of decomposition of the compound. Therefore, the amount of THF and residual hexane had to be checked individually for every batch. Typically, n equivalents of THF and m equivalents of hexane were detected, with n=0.07–1.0 and m=0.0–0.5. Yield: 31 mg, 33 μmol (with n=0.9 equiv of THF and 0.25 equiv of hexane), 65 %; 1H NMR (400 MHz, CD2Cl2): δ=1.48 (d, 3 J(H,H)=6.7 Hz, 24 H; CHMe), 1.80–1.83 (m, n×4 H; β‐THF), 3.66–3.70 (m, n×4 H; α‐THF), 4.77–4.87 (sept, 3 J(H,H)=6.7 Hz, 4 H; CHMe2), 6.74 (t, 3 J(H,H)=9.2 Hz, 2 H; 5‐H), 6.88 (d, 3 J(H,H)=11.5 Hz, 4 H; 3‐H, 7‐H), 7.42 ppm (dd, 3 J(H,H)=11.8, 9.2 Hz, 4 H; 4‐H, 7‐H); 13C NMR (101 MHz, CD2Cl2): δ=23.0 (s, CHMe), 26.2 (s, β‐THF), 52.4 (s, CHMe2), 68.3 (s, α‐THF), 121.6 (s, 3‐C, 7‐C), 125.2 (s, 5‐C), 135.1 (s, 4‐C, 6‐C), 167.4 ppm (s, 1‐C, 2‐C); elemental analysis calcd (%) for C26H38N4BiF6Sb⋅(C4H8O)0.07 (856.40 g mol−1): C 36.86, H 4.54, N 6.54; found: C 36.46, H 4.68, N 6.35. Fast decomposition of 3‐SbF6 was observed in THF at ambient temperature, which was accompanied by the precipitation of a dark solid. According to 1H NMR spectroscopy, 23 % of the free ligand were detected after about 30 min. After 5 d in solution, the complex was fully decomposed and only the free ligand was detected.
Compound 4‐BArF
Na(ATIiPr/PhSMe)(thf)0.5(hex)0.5 (100 mg, 259 μmol) was added dropwise to a solution of BiCl3 (41 mg, 130 μmol) in THF (4 mL) and sodium tetrakis[3,5‐bis(trifluoromethyl)phenyl]borate (115 mg, 130 μmol) was added under stirring. After 20 min, the red suspension was filtered, and all volatile compounds were removed under reduced pressure. The crude product was washed with hexane (4×2 mL) and dried in vacuo (190 mg, 113 μmol (with n=0.5 equiv of THF), 87 %); 1H NMR (500 MHz, −40 °C, CD2Cl2): δ=1.10 (d, 3 J(H,H)=6.0 Hz, 6 H; CHMe), 1.33 (d, 3 J(H,H)=6.3 Hz, 6 H; CHMe), 2.44 (s, 6 H; Me), 3.26–3.34 (br m, 2 H; CHMe2), 5.33 (s, n×2 H; CH2Cl2), 6.17 (d, 3 J(H,H)=10.9 Hz, 2 H; 3‐H), 6.47 (d, 3 J(H,H)=11.9 Hz, 2 H; 7‐H), 6.50 (d, 3 J(H,H)=7.9 Hz, 2 H; 6‐Harom), 6.72 (dd, 3 J(H,H)=8.7, 9.7 Hz, 2 H; 5‐H), 7.07 (dd, 3 J(H,H)=9.4, 11.4 Hz, 2 H; 4‐H), 7.22 (dd, 3 J(H,H)=6.7, 8.3 Hz, 2 H; 5‐Harom), 7.33 (d, 3 J(H,H)=7.9 Hz, 2 H; 3‐Harom), 7.41 (br dd, 3 J(H,H)=6.5, 8.2 Hz, 2 H; 4‐Harom), 7.48 (br dd, 3 J(H,H)=9.3, 11.2 Hz, 4 J(H,H)=1.5 Hz, 2 H; 6‐H), 7.55 (br s, 4 H; 4‐C6H3(CF3)2), 7.72 (br s, 8 H; 2,6‐C6H3(CF3)2) ppm; 13C NMR (126 MHz, −40 °C, CD2Cl2): δ=13.6 (s, Me), 20.8 (s, Me1), 25.2 (s, Me2), 49.7 (s, CHMe2), 117.4 (s, 4‐C6H3(CF3)2), 117.9 (s, 3‐C), 124.3 (s, 3‐Carom), 124.4 (quart, 1 J(C,F)=273.0 Hz, CF3), 124.5 (s, 7‐C), 125.5 (s, 5‐C), 125.6 (s, 5‐Carom), 128.5 (quart, 2 J(C,F)=50.0 Hz, 3,5‐C6H3(CF3)2 overlapping), 128.6 (s, 6‐Carom), 128.9 (s, 4‐Carom), 134.6 (s, 2,6‐C6H3(CF3)2), 135.1 (s, 4‐C), 135.5 (s, 6‐C), 137.0 (s, 2‐Carom), 141.0 (s, ipso‐Carom), 161.6 (quart, 1 J BC=50.0 Hz, 1‐C6H3(CF3)2), 163.5 (s, 1‐C), 168.3 ppm (s, 2‐C); 19F NMR (470 MHz, −40 °C, CD2Cl2): δ=−62.55 ppm; elemental analysis calcd (%) for C66H50N4S2BF24Bi⋅(CH2Cl2)0.5 (1681.49 g mol−1; obtained by recrystallization from CH2Cl2/pentane): C 47.50, H 3.06, N 3.33; found: C 47.44, H 3.03, N 3.18.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Funding through the Fonds der Chemischen Industrie and the Deutsche Forschungsgemeinschaft is gratefully acknowledged. C.L. thanks Prof. Holger Braunschweig for continued support. Open access funding enabled and organized by Projekt DEAL.
A. Hanft, K. Radacki, C. Lichtenberg, Chem. Eur. J. 2021, 27, 6230.
Dedicated to Professor Wolfgang Kaim on the occasion of his 70th birthday
References
- 1.
- 1a. Luca O. R., Crabtree R. H., Chem. Soc. Rev. 2013, 42, 1440–1459; [DOI] [PubMed] [Google Scholar]
- 1b.”Redox Non-innocent Ligands: Reactivity and Catalysis“: de Bruin B., Gualco P., Paul N. D., in Ligand Design in Metal Chemistry: Reactivity and Catalysis, Wiley, Chichester, 2016, pp. 176—204; [Google Scholar]
- 1c. Berben L. A., Chem. Eur. J. 2015, 21, 2734–2742; [DOI] [PubMed] [Google Scholar]
- 1d. Wei J., Diaconescu P. L., Acc. Chem. Res. 2019, 52, 415–424; [DOI] [PubMed] [Google Scholar]
- 1e. Lyaskovskyy V., de Bruin B., ACS Catal. 2012, 2, 270–279; [Google Scholar]
- 1f. van der Vlugt J. I., Chem. Eur. J. 2019, 25, 2651–2662; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1g. Hanft A., Lichtenberg C., Eur. J. Inorg. Chem. 2018, 3361–3373; [Google Scholar]
- 1h. Kaim W., Inorg. Chem. 2011, 50, 9752–9765; [DOI] [PubMed] [Google Scholar]
- 1i. Kaim W., Eur. J. Inorg. Chem. 2012, 343–348. [Google Scholar]
- 2.
- 2a. Vogt M., de Bruin B., Berke H., Trincado M., Grützmacher H., Chem. Sci. 2011, 2, 723–727; [Google Scholar]
- 2b. Lichtenberg C., Viciu L., Adelhardt M., Sutter J., Meyer K., de Bruin B., Grützmacher H., Angew. Chem. Int. Ed. 2015, 54, 5766–5771; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5858–5863; [Google Scholar]
- 2c. Lichtenberg C., Viciu L., Vogt M., Rodríguez-Lugo R. E., Adelhardt M., Sutter J., Khusniyarov M. M., Meyer K., de Bruin B., Bill E., Grützmacher H., Chem. Commun. 2015, 51, 13890–13893; [DOI] [PubMed] [Google Scholar]
- 2d. Lichtenberg C., Adelhardt M., Gianetti T. L., Meyer K., de Bruin B., Grützmacher H., ACS Catal. 2015, 5, 6230–6240. [Google Scholar]
- 3.
- 3a. Chirik P. J., Wieghardt K., Science 2010, 327, 794–795; [DOI] [PubMed] [Google Scholar]
- 3b. Bouwkamp M. W., Bowman A. C., Lobkovsky E., Chirik P. J., J. Am. Chem. Soc. 2006, 128, 13340–13341. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Myers T. W., Kazem N., Stoll S., Britt R. D., Shanmugam M., Berben L. A., J. Am. Chem. Soc. 2011, 133, 8662–8672; [DOI] [PubMed] [Google Scholar]
- 4b. Magdzinski E., Gobbo P., Workentin M. S., Ragogna P. J., Inorg. Chem. 2013, 52, 11311–11319. [DOI] [PubMed] [Google Scholar]
- 5. Lichtenberg C., Chem. Eur. J. 2020, 26, 9674–9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Lichtenberg C., Organometallics 2016, 35, 894–902; [Google Scholar]
- 6b. Lichtenberg C., Krummenacher I., Chem. Commun. 2016, 52, 10044–10047; [DOI] [PubMed] [Google Scholar]
- 6c. Hanft A., Krummenacher I., Lichtenberg C., Chem. Eur. J. 2019, 25, 11883–11891. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Helling C., Schulz S., Eur. J. Inorg. Chem. 2020, 3209–3221; [Google Scholar]
- 7b.C. Lichtenberg, Radical Compounds of Antimony and Bismuth in Encyclopedia of Inorganic and Bioinorganic Chemistry, Wiley, 2020.
- 8.
- 8a. Planas O., Wang F., Leutzsch M., Cornella J., Science 2020, 367, 313–317; [DOI] [PubMed] [Google Scholar]
- 8b. Wang F., Planas O., Cornella J., J. Am. Chem. Soc. 2019, 141, 4235–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Ishida S., Hirakawa F., Furukawa K., Yoza K., Iwamoto T., Angew. Chem. Int. Ed. 2014, 53, 11172–11176; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11354–11358; [Google Scholar]
- 9b. Schwamm R. J., Harmer J. R., Lein M., Fitchett C. M., Granville S., Coles M. P., Angew. Chem. Int. Ed. 2015, 54, 10630–10633; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10776–10779; [Google Scholar]
- 9c. Ganesamoorthy C., Helling C., Wölper C., Frank W., Bill E., Cutsail G. E., Schulz S., Nat. Commun. 2018, 9, 87; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9d. Lichtenberg C., Angew. Chem. Int. Ed. 2016, 55, 484–486; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 494–496. [Google Scholar]
- 10.
- 10a. Mukhopadhyay D. P., Schleier D., Wirsing S., Ramler J., Kaiser D., Reusch E., Hemberger P., Preitschopf T., Krummenacher I., Engels B., Fischer I., Lichtenberg C., Chem. Sci. 2020, 11, 7562–7568; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b.for the investigation of a dinuclear bismuth biradical see: Bresien J., Hinz A., Schulz A., Villinger A., Dalton Trans. 2018, 47, 4433–4436. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Schwamm R. J., Lein M., Coles M. P., Fitchett C. M., Angew. Chem. Int. Ed. 2016, 55, 14798–14801; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15018–15021; [Google Scholar]
- 11b. Schwamm R. J., Lein M., Coles M. P., Fitchett C. M., J. Am. Chem. Soc. 2017, 139, 16490–16493. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Ramler J., Krummenacher I., Lichtenberg C., Angew. Chem. Int. Ed. 2019, 58, 12924–12929; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 13056–13062; [Google Scholar]
- 12b. Schwamm R. J., Lein M., Coles M. P., Fitchett C. M., Chem. Commun. 2018, 54, 916–919; [DOI] [PubMed] [Google Scholar]
- 12c. Ramler J., Krummenacher I., Lichtenberg C., Chem. Eur. J. 2020, 26, 14551–14555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Yamago S., Kayahara E., Kotani M., Ray B., Kwak Y., Goto A., Fukuda T., Angew. Chem. Int. Ed. 2007, 46, 1304–1306; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 1326–1328; [Google Scholar]
- 13b. Lichtenberg C., Pan F., Spaniol T. P., Englert U., Okuda J., Angew. Chem. Int. Ed. 2012, 51, 13011–13015; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 13186–13190. [Google Scholar]
- 14.
- 14a. Sakagami M., Sasamori T., Sakai H., Furukawa Y., Tokitoh N., Chem. Asian J. 2013, 8, 690–693; [DOI] [PubMed] [Google Scholar]
- 14b. Hanft A., Lichtenberg C., Dalton Trans. 2018, 47, 10578–10589. [DOI] [PubMed] [Google Scholar]
- 15. Turner Z. R., Inorg. Chem. 2019, 58, 14212–14227. [DOI] [PubMed] [Google Scholar]
- 16. Raston C. L., Skelton B. W., Tolhurst V.-A., White A. H., J. Chem. Soc. Dalton Trans. 2000, 1279–1285. [Google Scholar]
- 17. Balasubramaniam S., Kumar S., Andrews A. P., Jemmis E. D., Venugopal A., Eur. J. Inorg. Chem. 2020, 2530–2536. [Google Scholar]
- 18. Dengel H., Lichtenberg C., Chem. Eur. J. 2016, 22, 18465–18475. [DOI] [PubMed] [Google Scholar]
- 19. Hanft A., Jürgensen M., Bertermann R., Lichtenberg C., ChemCatChem 2018, 10, 4018–4027. [Google Scholar]
- 20.An additional weak Bi⋅⋅⋅F contact with an interatomic distance of 3.44 Å is also present in 2-SbF6.
- 21.
- 21a. Tschersich C., Hoof S., Frank N., Herwig C., Limberg C., Inorg. Chem. 2016, 55, 1837–1842; [DOI] [PubMed] [Google Scholar]
- 21b. Chitnis S. S., Burford N., Decken A., Ferguson M. J., Inorg. Chem. 2013, 52, 7242–7248; [DOI] [PubMed] [Google Scholar]
- 21c. Ramler J., Radacki K., Abbenseth J., Lichtenberg C., Dalton Trans. 2020, 49, 9024–9034. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Ritschel B., Poater J., Dengel H., Bickelhaupt F. M., Lichtenberg C., Angew. Chem. Int. Ed. 2018, 57, 3825–3829; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3887–3891; [Google Scholar]
- 22b. Ritschel B., Lichtenberg C., Synlett 2018, 29, 2213–2217. [Google Scholar]
- 23. Ramler J., Poater J., Hirsch F., Ritschel B., Fischer I., Bickelhaupt F. M., Lichtenberg C., Chem. Sci. 2019, 10, 4169–4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.
- 24a. Balasubramaniam S., Kumar S., Andrews A. P., Varghese B., Jemmis E. D., Venugopal A., Eur. J. Inorg. Chem. 2019, 3265–3269; [Google Scholar]
- 24b. Kannan R., Balasubramaniam S., Kumar S., Chambenahalli R., Jemmis E. D., Venugopal A., Chem. Eur. J. 2020, 26, 12717–12721. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Johnson J. A., Venugopal A., J. Chem. Sci. 2019, 131, 114; [Google Scholar]
- 25b. Kannan R., Kumar S., Andrews A. P., Jemmis E. D., Venugopal A., Inorg. Chem. 2017, 56, 9391–9395; [DOI] [PubMed] [Google Scholar]
- 25c. Ramler J., Hofmann K., Lichtenberg C., Inorg. Chem. 2020, 59, 3367–3376. [DOI] [PubMed] [Google Scholar]
- 26. Ramler J., Lichtenberg C., Chem. Eur. J. 2020, 26, 10250–10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coordination of OPEt3 or EPMe3 (E = S, Se) to compound 1 would either result in dissociation of one NiPr group (turning one ATI ligand into a monodentate ligand and keeping a coordination number of six for bismuth) or it would lead to a coordination number of seven at bismuth. For examples of neutral molecular bismuth compounds with a coordination number of seven, see:
- 27a. Murphy J. N., Kerton F. M., Dawe L. N., J. Chem. Crystallogr. 2014, 44, 108–114; [Google Scholar]
- 27b. Bowmaker G. A., Hannaway F. M. M., Junk P. C., Lee A. M., Skelton B. W., White A. H., Aust. J. Chem. 1998, 51, 331–336. [Google Scholar]
- 28. Beckett M. A., Strickland G. C., Holland J. R., Sukumar Varma K., Polymer 1996, 37, 4629–4631. [Google Scholar]
- 29.Although compound 2-rad has two NiPr groups in the apical positions of the bisphenoidal coordination geometry, an isomer with one NiPr and one NPh group in the apical positions (2-rad-isom) was found to be only slightly higher in energy (ΔH=+3.0 kcal mol−1; ΔG=+3.6 kcal mol−1) and showed a very similar spin density distribution (for details, see the Supporting Information).
- 30. Hariharan P. C., Pople J. A., Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar]
- 31.
- 31a. Krishnan R., Binkley J. S., Seeger R., Pople J. A., J. Chem. Phys. 1980, 72, 650–654; [Google Scholar]
- 31b. McLean A. D., Chandler G. S., J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar]
- 32.
- 32a. Dunning T. H., Hay P. J., in Modern Theoretical Chemistry, Vol. 3 (Ed.: Schaefer H. F.), Springer, Boston, 1977, pp. 1–28; [Google Scholar]
- 32b. Wadt W. R., Hay P. J., J. Chem. Phys. 1985, 82, 284–298. [Google Scholar]
- 33. Becke A. D., J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- 34. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary