

Abstract
Objective
Multiple sclerosis (MS) is a chronic neuroinflammatory and neurodegenerative disease of unknown etiology. Although the prevalent view regards a CD4+‐lymphocyte autoimmune reaction against myelin at the root of the disease, recent studies propose autoimmunity as a secondary reaction to idiopathic brain damage. To gain knowledge about this possibility we investigated the presence of axonal and myelinic morphological alterations, which could implicate imbalance of axon‐myelin units as primary event in MS pathogenesis.
Methods
Using high resolution imaging histological brain specimens from patients with MS and non‐neurological/non‐MS controls, we explored molecular changes underpinning imbalanced interaction between axon and myelin in normal appearing white matter (NAWM), a region characterized by normal myelination and absent inflammatory activity.
Results
In MS brains, we detected blister‐like swellings formed by myelin detachment from axons, which were substantially less frequently retrieved in non‐neurological/non‐MS controls. Swellings in MS NAWM presented altered glutamate receptor expression, myelin associated glycoprotein (MAG) distribution, and lipid biochemical composition of myelin sheaths. Changes in tethering protein expression, widening of nodes of Ranvier and altered distribution of sodium channels in nodal regions of otherwise normally myelinated axons were also present in MS NAWM. Finally, we demonstrate a significant increase, compared with controls, in citrullinated proteins in myelin of MS cases, pointing toward biochemical modifications that may amplify the immunogenicity of MS myelin.
Interpretation
Collectively, the impaired interaction of myelin and axons potentially leads to myelin disintegration. Conceptually, the ensuing release of (post‐translationally modified) myelin antigens may elicit a subsequent immune attack in MS. ANN NEUROL 2021;89:711–725
Multiple sclerosis (MS) is a chronic inflammatory central nervous system (CNS) disease with axonal demyelination as the main pathological hallmark. 1 According to a standard etiological model, MS pathology is initiated by peripherally activated CD4+ T‐lymphocytes infiltrating the CNS. 2 , 3 , 4 However, this “outside‐in” theory is challenged by recent studies, arguing that MS pathological features cannot be solely explained by a primary autoimmune myelin attack. 3 For instance, oligodendrocyte apoptosis and widening of myelin lamellae often appear spatially segregated from high inflammatory activity regions in MS brains. 5 , 6 , 7 Furthermore, inner‐sheath myelin associated glycoprotein (MAG) loss temporally precedes that of other structural molecules in a subset of newly forming lesions. 8 , 9 Finally, clinical studies repeatedly underlined that standard immunosuppressant therapy, effective in suppressing relapsing disease, is largely ineffective for progressive MS forms. 3 , 10
Altogether, these findings are consistent with an “inside‐out” paradigm that posits that MS autoimmunity occurs subsequently to primary CNS cytodegeneration. 3 , 11 Nevertheless, the underlying degenerative processes are poorly understood.
We asked whether disturbance of the recently documented synaptic‐type communication between axon and myelin (the axo‐myelinic synapse [AMS]) might implicate a novel proximal cause of autoimmunity eliciting MS injury. 12 , 13 The AMS exploits glutamate release for axon‐myelin communication supporting action potential propagation. 13 Dysfunction of this signaling arrangement culminates in altered stimulation of myelinic glutamate receptors (eg, NMDARs). 14 Chronic over/under stimulation of Ca2+‐permeable NMDARs might induce aberrant myelinic Ca2+ accumulation, potentially eliciting a cascade of pathophysiological events, including activation of proteases, lipases, and peptidyl arginine deiminases (PADs). 15 The PADs constitute a family of enzymes that catalyze conversion of protein arginine residues into citrulline. Citrullination is a common protein post‐translational modification in tissues affected by (auto)immune mediated inflammation. 16 Whether this results from or precedes autoimmune inflammation is not completely understood. In a cuprizone (CPZ)‐induced mouse demyelination model, myelin basic protein (MBP) citrullination precedes and triggers autoimmune inflammatory demyelination. 12 In MS brains, citrullinated MBP is elevated in white matter lesions (WMLs), 17 , 18 and citrullination has been shown to potentiate the pathogenic activity of immunodominant myelin antigen myelin oligodendrocyte glycoprotein (MOG) in MS marmoset models. 11 Last, MS‐specific accumulation of extracellular leptomeningeal myelin proteins (such as proteolipid protein [PLP] and MBP 19 ) indicates an intense clearing up mechanism for myelin debris.
Although these findings suggest that myelin citrullination recapitulates early MS degeneration, the sequence of events potentially leading to myelin Ca2+ accumulation, biochemical modification, and myelin disintegration in MS remains unknown. Here, we tested the hypothesis that MS normal‐appearing white matter (NAWM) contains subtle structural abnormalities, which may precede myelin disintegration. We report the presence of blister‐like myelin swellings formed by myelin detachment from axons in NAWM and defective expression of glutamate receptors and tethering/adhesion molecules in blisters and other morphological alterations. Myelin blisters are much less prevalent in healthy brains or brains from Alzheimer's disease (AD) or encephalitis cases.
Materials and Methods
Patient Material
Four percent formalin‐fixated control, MS disease, and AD cases brain blocks were obtained from the Netherlands Brain Bank (NBB) and the Normal Aging Brain Collection Amsterdam. Per patient, 2 blocks containing the anterior part of the corpus callosum (CC), and the subcortical/periventricular white matter (sWM, extracallosal) were selected. The block surface of MS cases contained 94.1 ± 1.7% of NAWM, 3.2 ± 1.2% of WML and 3.9 ± 1.6% of diffusely abnormal white matter (WM). Of the lesions, 18.7% were active, 18.7% were chronically active, and 62.5% were inactive. To analyze the data, only the NAWM not falling in the vicinity of a lesion was taken into account (>0.5–1mm from lesion borders). To compare MS NAWM with inflammatory control WM, we obtained 3 additional aged‐matched paraffin embedded specimens (from the NBB) from encephalitis cases.
For the study of meningeal citrulline, we obtained 4 additional paraffin‐embedded MS specimens (from the NBB) and 6 cases (3 MS and 3 controls) from the UK MS Tissue Bank at Imperial College, London, UK (www.imperial.ac.uk/medicine/multiple-sclerosis-and-parkinsons-tissue-bank; Table). All procedures complied with local ethical and legal guidelines. Informed consent for brain autopsy, use of brain tissue, and clinical information for scientific research was given by either the donor or the next of kin.
TABLE 1.
Demographics of the Cases Used for the Whole Study
| Sample ID | Sex (n.s.) | Age (n.s.) | Onset (yr) first episode | Disease duration (yr) | PMD (min) (n.s.) | MS type | Cause of death | Used for |
|---|---|---|---|---|---|---|---|---|
| 1 | F | 73 | 43 | 30 | 430 | PPMS | Euthanasia | Figures 1–5 |
| 2 | F | 59 | 38 | 22 | 530 | SPMS | Euthanasia | Figures 1–5 |
| 3 | F | 77 | 50 | 27 | 430 | SPMS | Lung failure | Figures 1–5 |
| 4 | M | 66 | 39 | 27 | 510 | SPMS | Pneumonia | Figures 1–5 |
| 5 | M | 65 | 29 | 36 | 594 | SPMS | Euthanasia | Figures 3H–K to 5 |
| 6 | M | 70 | 32 | 38 | 570 | SPMS | Euthanasia | Figures 3H–K to 5 |
| 7 | F | 83 | 49 | 34 | 370 | PPMS | Ovary carcinoma | Figures 3H–K to 5 |
| 8 | M | 72 | 29 | 43 | 445 | SPMS | General malaise | Figures 3H–K to 5 |
| 9 | F | 77 | 38 | 39 | 585 | SPMS | Aspiration pneumonia | Figures 4 and 5 |
| 10 | F | 59 | 20 | 39 | 1,260 | SPMS | Unknown | Figure 5 |
| 11 | F | 35 | 30 | 5 | 540 | SPMS | Unknown | Figure 5 |
| 12 | M | 51 | 33 | 18 | 1,020 | SPMS | Respiratory failure | Figure 5 |
| 13 | F | 35 | 25 | 10 | 620 | SPMS | Euthanasia | Figure 5 |
| 14 | F | 61 | 59 | 2 | 600 | SPMS | Euthanasia | Figure 5 |
| 15 | M | 70 | 24 | 46 | 415 | SPMS | Heart failure | Figure 5 |
| 16 | F | 61 | 59 | 2 | 270 | SPMS | Euthanasia | LFB example Figure 1 |
| 17 | M | 67 | 20 | 47 | 360 | SPMS | Cardiac failure | LFB example Figure 1 |
| 18 | F | 59 | – | – | 490 | CTRL | Euthanasia | Figures 1–5 |
| 19 | F | 63 | – | – | 490 | CTRL | Euthanasia | Figures 1–5 |
| 20 | M | 68 | – | – | 520 | CTRL | Euthanasia | Figures 1–5 |
| 21 | F | 69 | – | – | 780 | CTRL | Pulmonary embolism | Figures 1–5 |
| 22 | F | 72 | – | – | 445 | CTRL | Hart failure | Figures 1–5 |
| 23 | M | 35 | – | – | 1,320 | CTRL | Tongue carcinoma | Figure 5 |
| 24 | F | 60 | – | – | 780 | CTRL | Ovarian carcinoma | Figure 5 |
| 25 | M | 68 | – | – | 1,800 | CTRL | Heart failure | Figure 5 |
| 26 | M | 62 | – | – | 395 | CTRL | Lung carcinoma | Figure 5 |
| 27 | M | 70 | – | – | 450 | CTRL | Pancreas carcinoma | Figure 5 |
| 28 | F | 71 | – | – | 615 | CTRL | Pneumonia | Figure 5 |
| 29 | M | 90 | 82 | 8 | 360 | AD | Heart failure | Figure 1 |
| 30 | M | 82 | 75 | 7 | 555 | AD | COPD exacerbation | Figure 1 |
| 31 | F | 86 | 84 | 2 | 510 | AD | Unknown | Figure 1 |
| 32 | M | 67 | Unknown | Unknown | 710 | AD | Heart failure | Figure 1 |
| 33 | F | 53 | Unknown | Unknown | 330 | Encephalitis | Cachexia | Figure 1 |
| 34 | F | 66 | Unknown | Unknown | 360 | Encephalitis | Sepsis | Figure 1 |
| 35 | M | 60 | Unknown | Unknown | 300 | Encephalitis | Cachexia | Figure 1 |
COPD = chronic obstructive pulmonary disease; MS = multiple sclerosis; n.s. = not significant; PMD = post‐mortem delay; PPMS = primary progressive MS; SPMS = secondary progressive MS.
For the experiments from Figures 1–5c a maximum of 9 MS cases, and 5 non‐neurological controls were used. For the analysis in Figure 1E, F, 4 matched Alzheimer's disease (AD) cases were also included (CTRL vs MS vs AD; age in years: 66.20 ± 2.31 vs 71.33 ± 2.43 vs 81.25 ± 5.02; Welch's analysis of variance (ANOVA) test; W(2.00,7.01) = 3.66; p = 0.081; gender F:M: 4:1 vs 5:4 vs 1:3; chi squared test; chi squared (1) = 0.461; p = 0.497; post‐mortem delay in minutes: 545 ± 59.96 vs 496 ± 26.76 vs 533 ± 72.04; Kruskal‐Wallis test; K = 0.162; p = 0.930). In addition 3 encephalitis cases have been used for the analysis in Figure 1F (CTRL vs MS vs encephalitis [ENC]; age in years ENC: 59.67 ± 3.76; Welch's ANOVA test; W(2.00,4.743) = 1.384; p = 0.260). In Figure 5e, 7 controls and 8 MS cases were included (CTRL vs MS; age in years: 61.29 ± 4.66 vs 55.88 ± 5.32; Mann Whitney test; U = 18.50; p = 0.293; gender F:M: 3:4 vs 6:2; Fisher's exact test; p = 0.315; post‐mortem delay in minutes: 835.70 ± 199.90 vs 676.90 ± 106.60; Mann Whitney test; U = 25; p = 0.779).
Immunohistochemistry Staining
For experiments with fixed material, blocks were cryoprotected (10% sucrose in phosphate buffer [PBS]), frozen, and subsequently sliced (10μm) using a cryostat (Leica Biosystem, Germany). For experiments with paraffin material, blocks were sliced (10μm) using a microtome (Leica Biosystem, Germany) followed by deparaffination in 100% xylene (3 × 10 minutes) and ethanol (2 × 5 minutes 100% and 5 minutes 96%, Sigma‐Aldrich, St. Louis, MO). Antigen retrieval (tris‐EDTA, pH9), endogenous peroxidase blockade (1% H2O2), and blocking nonspecific binding (3% normal donkey/goat serum, 20 minutes) were performed. After overnight incubation with primary antibodies (Table S1) in block buffer and biotinylated antibody incubation (1:200, 2 hours; Jackson Immunoresearch, UK), sections were incubated with either the avidin‐biotin complex (1:400, 15 minutes, ABC; Thermo Fisher, Waltham, MA) for tertiary binding to tyramide (Alexa 488 or 594, 1:100, Thermo Fisher), or EnVision HRP kit (Agilent Technologies, Santa Clara, CA) for DAB (DAKO kit, Denmark), stay yellow (SY; 1:50–10 minutes; Abcam, UK), and Vector SG (VSG; 3 drops kit in 2.5ml PBS for 10 minutes; Vector Laboratories, UK) staining. An Empress alkaline phosphatase (AP) kit was used for liquid permanent red (LPR; 1:100–10 minutes, Agilent Technologies). Furthermore, non‐biotinylated secondary antibodies coupled with Alexa 488/546/594/647(1:200; Thermo Fisher) were used for some triple stainings. Slides stained with fluorophores were mounted with glass coverslips using Mowiol (Sigma‐Aldrich) plus anti‐fading agent DABCO, whereas for the other slides Entellan (Sigma‐Aldrich) was used.
Luxol Fast Blue Staining
Deparaffinized sections (see above) were incubated in a 0.1% Luxol Fast Blue (LFB; Gurr, UK) solution in a stove (58°C) overnight. Washing steps were performed consecutively in 96% alcohol (2–3 seconds) and milli‐Q (MQ) water (3 seconds). Immediately thereafter, differentiation steps were performed in 0.05% Lithium carbonate solution (5 seconds; Merck Millipore, Germany) and 70% ethanol (5–7 seconds) until the grey matter was colorless and white matter remained blue. The sections were then rinsed in MQ water and dehydrated in a series of ethanol 96% (3–5 minutes), 100% (2 × 5 minutes), and xylene (3 × 5 minutes) and mounted with Entellan and a coverslip.
Microscopy
Slide Scanner
PLP/major histocompatibility complex II (MHC‐II)/LFB‐stained whole slices were acquired using an MCID slide scanner (20×, Leica Biosystem, Germany) and a DM5000 photomicroscope (Leica Microsystem, Germany).
Fluorescent Microscopy
Z‐stack images of tethering protein expression were acquired using an inverted fluorescent microscope (Axio Observer, Zeiss, Germany). Samples were set under the microscope and 40x/ N.A. 1.30 x, y, z images of 10 regions of interest (ROIs; x*y = 273.14*203.26μm) per slice were acquired (Z‐axis step width = 0.74μm; exposure time = 1,000ms) using SlideBookReader 6.0 (Intelligent Imaging System, Huntsville, AL).
Confocal Microscopy
Z‐stacks (1024*1024 pixels, N.A. 1.40) were deconvoluted and used to represent graphically the structure of interest, using an SP8 Leica confocal (Leica Biosystem, Germany). In some cases, signal brightness was increased for clarity. Colocalization analysis was performed acquiring 2‐dimensional pictures of NMDAR and PLP staining (1024*1024 pixels, scanning frequency = 600 Hz, 6 lines of accumulation).
Nuance Spectral Camera
Images were acquired from chromogen stained slides using a multispectral imaging system (Nuance version 3.0.2; Perkin Elmer, Hopkinton, MA). Cubic autoexposure (binning 1 × 1) and reference acquisition preceded cube acquisition of the image. References for the different chromogens were then acquired from single staining, saved, and used to unmix the channels from double/triple staining.
Coherent Anti‐Stokes Raman Scattering
Two hundred micrometer thick brain sections were preserved with 0.003% sodium azide in PBS. The coherent anti‐stokes Raman scattering (CARS) microscope consisted of a pulsed laser (5 ps pulse width) operating at 1,032nm providing the Stokes beam. The pump beam was generated by an optical parametric oscillator (OPO; Levante Emerald, Germany; pumped by the doubled 1,032nm), tunable between 750 and 900nm. Combining the 2 beams allows excitation of vibrational modes over a wide range of wavelengths, including the CH stretch at ≈ 2,850cm−1 and OH (water) at ≈3,000–3,500cm−1. The 512 × 512 pixel NAWM images were acquired using an inverted microscope (N.A. objective 1.20) and LabView (National Instruments, Austin, TX).
Image Acquisition and Quantification
Analysis was performed using Fiji ImageJ, QuPath 0.2.2, and MATLAB (for CARS analysis; Mathworks, Natick, MA). Overall, images passed background subtraction, median filtering, and auto‐threshold (kept constant for all the analysis). NAWM was defined as a region where the percentage of PLP immunoreactivity in MS cases was similar to that of the control subjects and where MHC‐II immunoreactivity was not found (including in form microglia clusters).
Analysis of swellings to quantify the presence of swelling formations was executed acquiring 4/5 images per case/region (DM5000/Nuance spectral camera, 63x objective/ N.A. 1.40). Swellings were selected based on the following criteria: (1) presence of visible spherical shape of myelin extending in the z‐axis, and clear presence of axon staining (SMI312) inside the swelling, and (2) clear presence of myelinated axons at the edge of the swelling. Swelling count was followed by swelling type classification (expressed as percentage of total swelling numbers) discriminating among: (1) blisters, representing local myelin detachment from its axon, (2) blebs formed by swelling of the axon, and (3) swellings putatively containing axon degeneration as assessed by SMI312 staining fragmentation (Fig 1E). To determine MAG, NR subunit, tethering protein expression, and CARS analysis in individual swellings, the swelling subtypes were pooled. MAG expression in swellings and peri‐swelling regions was expressed as the percentage of MAG staining present in these areas. Tethering protein analysis was performed by acquiring 10 ROIs (see above) per case. Z‐stack images were then filtered, and a 3D object recognition plug‐in was run to measure the objects in the x, y, and z dimensions (volume). The volume occupied by tethering/periaxonal protein expression was examined to circumvent the difficulties in analyzing the particle density for proteins, like MAG, which are compactly expressed along myelin sheaths. NMDAR colocalization analysis was performed on x and y pictures of 4 ROIs per case. The pictures were used to identify swellings, and the average amount/percentage of NMDAR signal colocalized with PLP. Both channels were processed (PLP channel: subtract background −50, filter ‐median 0.5, auto threshold‐Li‐; NR3a/NR2c channel: subtraction of median intensity 36 from original picture already spatially filtered with median 0.5, further subtraction of 5, auto threshold –triangle‐). For colocalization analysis, JACoP‐ImageJ plugin and custom‐made JAVA script were used. Analysis of NF155/Caspr for the nodal length was performed acquiring 4 images per case (final magnification 63x/ N.A. 1.40). Measurement of the distance between NF155/Caspr immunoreactivity and axonal width was made by zooming in the image (2–4 times). The same parameters were used to locate the swelling formations along axons with respect to the Caspr expression and to analyze voltage‐gated sodium channels (Nav) data. For CARS analysis, equal squares in myelinated fibers (up to 3) or swelling (up to 3) were drawn using a custom‐made MATLAB script (available upon request). Intensity analysis was performed inside the squares only. Analysis of NAWM and meningeal citrulline was performed acquiring three/four 10 times images per section per case (10 times final magnification/ N.A. 0.40). The same analysis was performed to estimate the amount of citrulline inside the swellings (63 times final magnification).
FIGURE 1.
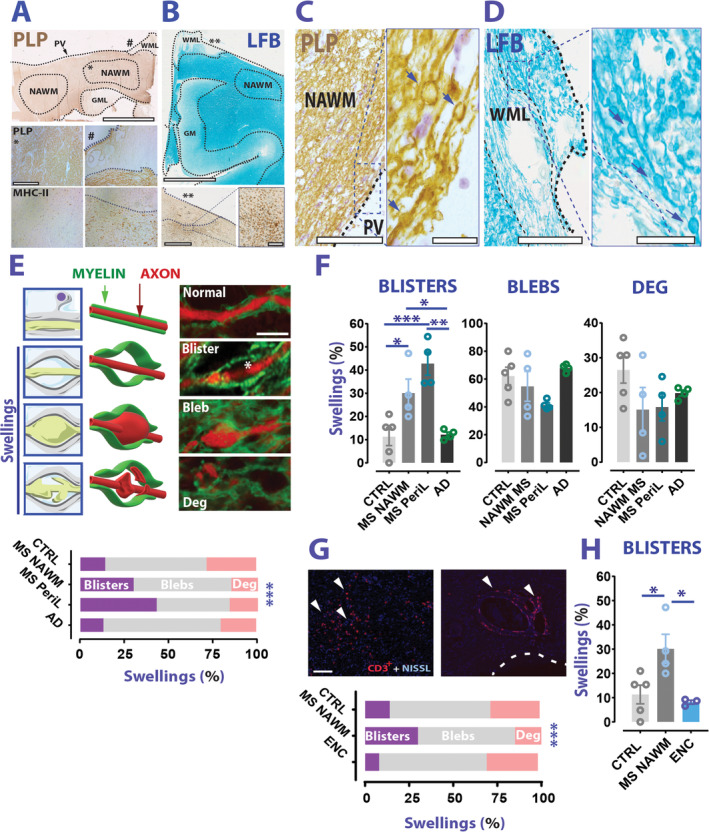
Blistering of AMS presents more frequently in MS than non‐MS WM. (A) Top panel: Example of PLP staining on MS brain sections (brown). The dashed lines indicate a boundary for NAWM, or a WML, or a GML. Bottom panel: Photomicroscope acquisitions of PLP and MHC‐II from the same case show NAWM (*) and a chronic WML (#). Thin‐dotted lines show the lesion borders while thick‐dotted lines mark the periventricular region (PV). (B) Top panel: LFB staining from an MS case. Bottom panel and inset: MHC‐II staining performed on the same case highlights a chronic active lesion (**). (C, D) Examples of PLP and LFB staining images of swelling formations (arrows) retrieved in NAWM (left) and lesion/perilesion (right) of MS cases. (E) Top panel: Cartoons (left), 3D drawings (middle), and examples (right) of the different types of swelling analyzed. Bottom graph: Distribution of swelling types among CTRL, MS, and AD (chi squared test; chi squared (6) = 108.6; p < 0.0001). (F) Blister percentage is significantly increased in MS than CTRLs and AD (Blisters: 1‐way ANOVA; F(3,13) = 12.17; p = 0.0004; Sidak's multiple comparison test; p = 0.031 MS NAWM vs CTRLs; p = 0.0009 MS perilesion vs CTRLs; p = 0.042 MS NAWM vs AD; p = 0.002 MS perilesion vs AD; blebs: Brown‐Forsythe ANOVA; F(3.0,5.563) = 3.156; p = 0.114 CTRLs vs MS vs AD; degenerative: 1‐way ANOVA; F(3,13) = 1.675; p = 0.221 CTRLs vs MS vs CTRLs). (G) Top panel: Examples of inflammatory regions in the WM of an encephalitis case. Arrowheads indicate CD3+ cells. Dotted line indicates the ventricular region. Bottom graph: distribution of swelling types between CTRLs, MS NAWM, and encephalitis (ENC) WM (chi squared test; chi squared (6) = 108.6; p < 0.0001). (H) Blister percentage is significantly higher in MS NAWM than EN WM (Blisters: 1‐way ANOVA; F (2,9)=6.672; p = 0.0167; Sidak's multiple comparison test; p = 0.032 MS NAWM vs ENC). Scale bars: 5 mm in A (top panel) and B; 500μm in A (bottom panel); 1mm in B bottom panel (inset scale bar is 200μm); 200μm in C and D (inset is 25μm); 2.5μm in E, 100μm in G. Data in F, H are reported as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001. AD = Alzheimer's disease; AMS = axo‐myelinic synapse; ANOVA = analysis of variance; CTRL = control; GM = grey matter; GML = grey matter lesion; LFB = Luxol Fast Blue; MHC II = major histocompatibility complex II; MS = multiple sclerosis; NAWM = normal appearing white matter; PLP = proteolipid protein; WML = white matter lesion.
Exclusion Criteria
Two patients from the whole sample were excluded from the study (including matching analysis) because of misdiagnosis and technical issues with the material. Photographs showing extended portions of folded material or excessive background staining were excluded from final analysis.
Statistical Analysis
Analysis was performed using MATLAB, and Prism version 7.0 (GraphPad Software, San Diego, CA). Parametric/nonparametric t test was used to compare results from MS cases with controls. Chi squared and Fisher's test was used to analyze swelling and Nav distributions. Analysis of variance (ANOVA)s/mixed models (followed by post hoc test) were used to compare the percentage of swellings between controls and patients, and MAG staining in swellings and myelinated fibers, and preceded by Shapiro–Wilk normality test/KS test.
Results
Blistering of Axo‐Myelinic Units in MS NAWM
The analysis of representative portions of MS NAWM (see Fig 1A–C) and control (CTRL) WM for histological abnormalities, retrieved both from the CC and the sWM, revealed typical morphological abnormalities similar to previously described myelin/axonal swellings. 20 These swellings were observed in the NAWM and in the peri‐lesion WM in MS cases, as well as in control material and in a cohort of AD cases. Closer examination of the axon‐myelin interaction inside these swellings revealed presence of distinct morphological patterns (Fig 1E). In most cases, we observed typical axonal enlargement inside swellings (axonal swellings/blebs), with a portion of them showing neurofilament abnormalities, suggesting axon degeneration. Furthermore, in many other cases, we observed blister‐like structures where myelin detaches from a largely intact axon cylinder (myelin swellings/blisters). In a total of 1,119 analyzed swellings (615 in MS NAWM/peri‐lesion, 211 in CTRL WM, and 293 in AD WM) myelin blisters appeared significantly more frequent in MS than CTRLs and AD cases (Fig 1E, F).
To exclude a possible role of inflammation in the increased percentage of myelin blisters in MS NAWM, we additionally analyzed 493 swellings from encephalitis cases. Also in this case, the percentage of myelin blisters was significantly less prevalent than in MS NAWM (Fig 1G, H).
Altogether, these results show that myelin blisters are morphological alterations more frequently present in MS than non‐MS cases and that they are found in the absence of overt inflammation.
Molecular Fingerprints of MS Swellings
We characterized the myelin/axonal swellings by studying their location, myelin lipid biochemistry, NMDAR expression, and MAG content, which together form the complex machinery that the AMU exploits for functioning.
Paranodal contactin associated protein (CASPR) staining revealed a strikingly different swelling distribution in myelinated axonal tracts in MS NAWM versus CTRLs. While in control cases most swellings localized in proximity to or included the node of Ranvier, MS swellings localized equally in the nodal/paranodal and internodal regions of the axon (Fig 2A).
FIGURE 2.
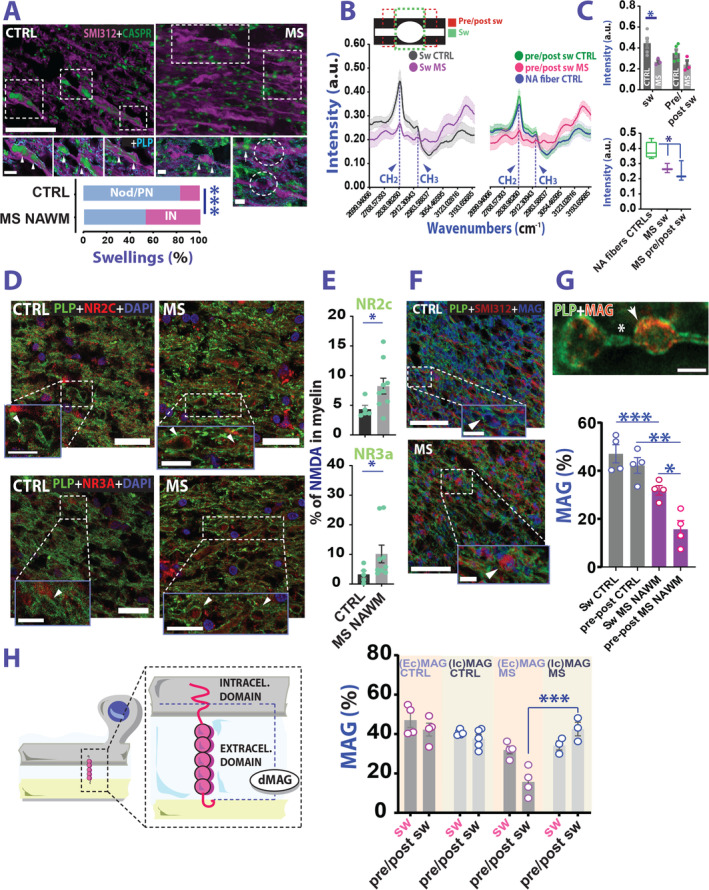
Biochemical and structural fingerprints of MS swellings. (A) Example of CASPR staining to locate the swellings along axonal tract. Graph reports a differential distribution of swelling in our dataset (Nodal‐paranodal/internodal: 83.1%/16.9% and 53%/47% CTRL vs MS; Fisher exact test; p = 0.0001). (B) Graphical representation of CARS analysis (top) and intensity peak graphs of the analyzed regions (bottom). (C) Top graph: Analysis of CH2 peak in swellings (sw) and peri‐swelling region (pre/post sw) in MS NAWM and CTRLs (CH2: RM ANOVA; myelin location: F(1,6) = 8.743; p = 0.025; group effect: F(1,6) = 6.937; p = 0.039; interaction myelin location x group: F(1,6) = 2.659; p = 0.154; Sidak's multiple comparison test; t(12.00) = 3.041; p = 0.020; swelling CTRL vs swelling MS; p = 0.161 pre/post swelling CTRL vs pre/post swelling MS). Bottom graph: Comparison between CH2 intensity peak in swellings and peri‐swellings versus normally myelinated fibers from controls (Welch's t test; t(4.713) = 3.485; p = 0.019; normal appearing CTRL fibers vs MS swellings; t(3.877) = 2.944; p = 0.044; normal appearing CTRL fibers vs MS pre/post swelling). (D) Example of NR2c (top panel) and NR3a (bottom panel) NMDA receptor subunit expression in the swelling formations. (E) Both NR2c and NR3a NMDAR subunits are overexpressed in the MS NAWM swelling formations, compared with controls (CTRL vs MS NR2c CC: Mann–Whitney test; U = 7; p = 0.042; NR3a CC: Mann–Whitney test; U = 7; p = 0.04). (F) Example of MAG staining inside the swellings. (G) While MAG is expressed in MS NAWM swellings (arrow top panel), it is absent in the peri‐swelling region (*). MAG analysis inside the swellings and peri‐swelling regions (bottom panel) reveals a difference between MS and CTRLs (CTRL vs MS NAWM; RM ANOVA: interaction group × location, F(1,6) = 8.877, p = 0.025; Sidak's multiple comparison test: swelling: p = 0.012; pre/post swelling: p = 0.0002). (H) Left panel: Graphic representation of MAG hydrolysis. Right graph: Analysis showing that in the peri‐swelling region intracellular MAG accumulates in MS (REML mixed effect model: interaction location × group × epitope, F(1,12) = 13.83, p = 0.003; swelling intra‐ vs extracellular MAG in MS: Tukey's multiple comparison test: p = 0.999; extra vs intracellular MAG pre/post swelling MS: p < 0.0001). Scale bars: 10 μm in A (inset scale is 5 μm), 25 μm in D (inset scale is 10 μm) and 50 μm in F (inset scale is 10μm). In G scale bar is 5μm. Data in C (top graph), E, and G, and H are reported as mean ± SEM, wheras in C bottom graph is mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001. ANOVA = analysis of variance; CARS = coherent anti‐stokes Raman scattering; CASPR = contactin associated protein; CTRL = control; GM = grey matter; GML = grey matter lesion; MAG = myelin associated glycoprotein; MS = multiple sclerosis; NAWM = normal appearing white matter; NMDA = N‐methyl‐D‐aspartate; NMDAR = N‐methyl‐D‐aspartate receptor; PLP = proteolipid protein; REML = restricted maximum likelihood; WML = white matter lesion.
Using CARS spectroscopy analysis over small myelin fractions, we observed a significant decrease in myelin lipid carbon‐hydrogen CH2 bonds in MS swellings compared with CTRLs, suggesting a reduction in the long‐chain fatty acid myelin composition (Fig 2B, C).
CNS myelin is known to express functional NMDARs that likely support important physiological functions. 13 , 14 Despite being characterized by a greater variability in the expression level, corpus callosum NMDAR analysis in the myelin of swellings showed an upregulation of the NR2c and NR3a subunits in MS versus control subjects (Fig 2D, E).
MS myelin/axonal swellings were found associated with aberrant MAG expression both inside the swellings and in the peri‐swelling region compared with CTRLs (Fig 2F, G). Although less abundant than in control material, most of the MS MAG staining was observed inside the swelling compared with flanking normally myelinated parts of the fiber (peri‐swelling), hinting at a compensatory mechanism for protecting the AMS at the level of its morphological alterations. Previous studies reported that in patients with MS, MAG is degraded much faster into a less functional truncated form (dMAG) than in healthy individuals. Hence, we studied whether the reduced expression of MAG in our samples might be due to dMAG formation (Fig 2H). 21 To this end, we stained our material with antibodies selective for the amino acid sequence 531 to 581, an MAG epitope localized at the cytoplasmic tail right before MAG truncation site. 22 In this way, we could discriminate the expression level of intracellular versus extracellular MAG. Overall, our analysis indicated that in the MS swelling region no difference could be detected between staining with either extra‐ or intracellular MAG antibodies. However, in the peri‐swelling region, despite a dramatic reduction of extracellular MAG, staining with intracellular MAG reported comparable results with control and MS swelling (see Fig 2). Together, these data suggest contribution of MAG breakdown/truncation to swelling formation in MS.
Axon‐Myelin Adhesion and Nodal Regions are Affected in MS NAWM
We tested whether changes in seemingly normally myelinated tissue portions of WM of patients with MS might represent unstable axon‐glia interaction. We investigated the expression of a family of tethering/periaxonal proteins relevant to action potential propagation. 23 In MS NAWM versus controls, we observed a heterogeneous expression level change of these molecules (Fig 3A–C) both in the CC and sWM. Although we spotted selective density reduction of contactin‐1 and MAG in the CC and sWM, respectively (Fig 3C ), the expression levels of juxtaparanodal contactin‐2 as well as paranodal neurofascin‐155 (NF155) and CASPR (Fig 3D) in the same regions were increased. These alterations hint at structural abnormalities at Ranvier's nodes. In line with this possibility, myelinated axons in MS NAWM exhibited significantly longer nodal regions compared to control axons (Fig 3E–G) as assessed through positive NF155 and CASPR staining. These results suggest that subtle nodal pathology occurs in NAWM regions of patients with MS. In addition, we spotted a frequent overlap of nodal sodium channels (Nav) and CASPR paranodal staining in MS material (Fig 3H, I). This pattern was often accompanied by nodal Nav negativity (Fig 3H–K), especially in cases of elongated nodal distance (>3μm; Fig 3K) suggesting that in MS NAWM either a possible paranodal intrusion of the node or an Nav displacement to paranodal axolemma occurs, likely affecting action potential propagation along the axon.
FIGURE 3.
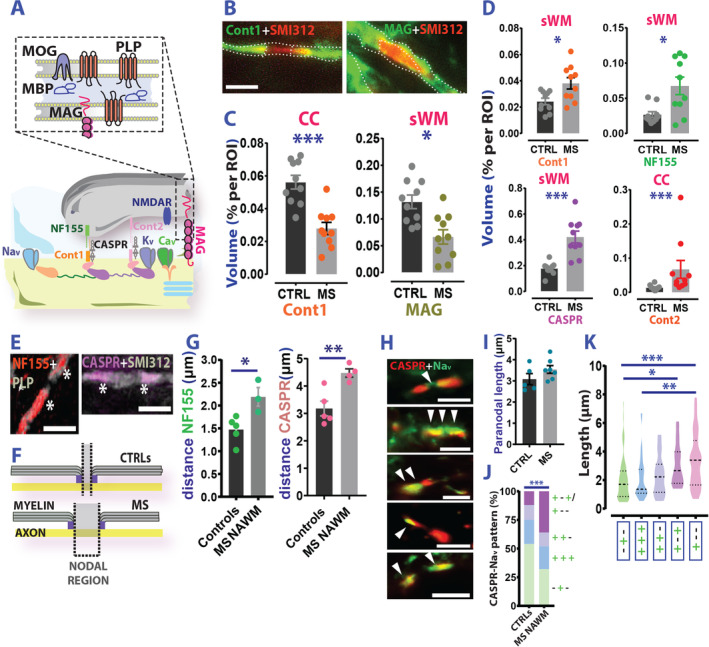
(Inter)nodal pathology in MS NAWM. (A) Schematic representation of the main structural/tethering proteins expressed in the AMS. (B) Photomicroscope fluorescent images of Cont‐1 (left) and MAG (right) in the axonal tract. (C) Region of interest (ROI) analysis (expressed as percentage of volume) of cont‐1 and MAG reactivity in CTRLs versus patients with MS CC and sWM, respectively (CTRL vs MS NAWM Cont 1: Welch's t test; t(17.73) = 5.038; p < 0.0001; MAG: Welch's t test; t(17.94) = 3.55; p = 0.023) (D) Analysis (CTRL vs MS NAWM) of the expression level of the main tethering proteins across different ROIs of MS (n = 4) and controls (n = 5) reports an increase rather than a decrease of Cont‐1 (sWM: Welch's t test; t(13.25) = 2.769; p = 0.016), NF155 (sWM: Mann–Whitney test; U = 23; p = 0.043), CASPR (sWM: Welch's t test; t(10.90) = 5.038; p = 0.0004) Cont 2 (CC: Mann–Whitney test; U = 7; p = 0.0005). (E) Left panel: Example of NF155 (left) and CASPR (right) staining. (F) Graphic representation of the elongated length of the Ranvier's node in MS. (G) Analysis of distance between adjacent NF155 (left graph) and CASPR (right graph) molecules in MS and CTRLs (CTRL vs MS NAWM NF155: Welch's t test; t(3.610) = 2.970; p = 0.047; CASPR: Welch's t test; t(6.119) = 4.199; p = 0.005). (H) Examples of Nav expression in MS NAWM/CTRL WM nodes. From top to bottom: nodal Nav flanking paranodal CASPR; bilateral Nav, and unilateral Nav displacement to the paranode; and 2 cases showing Nav paranodal invasion patterns with evident nodal Nav negativity. (I) Analysis of paranodal length in MS and controls (Welch's t test; t(7.618) = 1,427; p = 0.193; CTRL vs MS NAWM). (J) Distribution of the Nav/CASPR expression patterns in MS NAWM and control axons (chi squared test; chi squared (3)=23.04; p < 0.0001; CTRL vs MS). (K) Nav redistribution in the paranodal region with reduced expression in the nodal region is a specific feature of axons with augmented nodal length (Kruskal‐Wallis test; KW = 24.66; p < 0.0001; Dunn's multiple comparison test; p = 0.023; −+− vs +−+; p = 0.054; +++ vs +−−; p = 0.0003; −+− vs +−−) Scale bars: 5μm in B and 10μm in D. In H are from top to bottom: 5, 10, 5, 10, and 10μm. Data in C and F are reported as mean ± SEM, whereas in J thick dotted lines represent mean and thin dotted lines the SD. *p < 0.05; **p < 0.01; ***p < 0.001. AMS = axo‐myelinic synapse; CASPR = contactin associated protein; CC = corpus callosum; CTRL = control; MAG = myelin associated glycoprotein; MBP = myelin basic protein; MOG = myelin oligodendrocyte glycoprotein; MS = multiple sclerosis; NAWM = normal appearing white matter; PLP = proteolipid protein; ROI = region of interest; sWM = subcortical/periventricular white matter; WML = white matter lesion.
Dysregulation of NMDA Receptors in MS NAWM Myelin
Nodal aberrations and AMS instability may alter axo‐myelinic glutamate signaling. 13 Therefore, we studied the expression of NMDARs in normal axons of patients with MS. We focused on the glutamate‐sensitive NMDAR subunit NR2c, and the glycine‐sensitive NR1 and NR3a subunits. 24 In line with studies in mice, 14 immunohistochemistry experiments confirmed expression of these subunits in human myelin (Fig 4A–D). Normal axon analysis revealed that both the percentage of myelin occupied by NMDAR NR3a subunits, as well as the amount of NR3a subunit expression within myelin were enhanced in MS NAWM compared with control WM. This effect was particularly significant in the CC (Fig 4E–G). Interestingly, NR2c expression remained unaffected. Hence, contrary to observations in swellings, here, we detect enhanced expression only of glycine‐sensitive NMDAR subunits.
FIGURE 4.
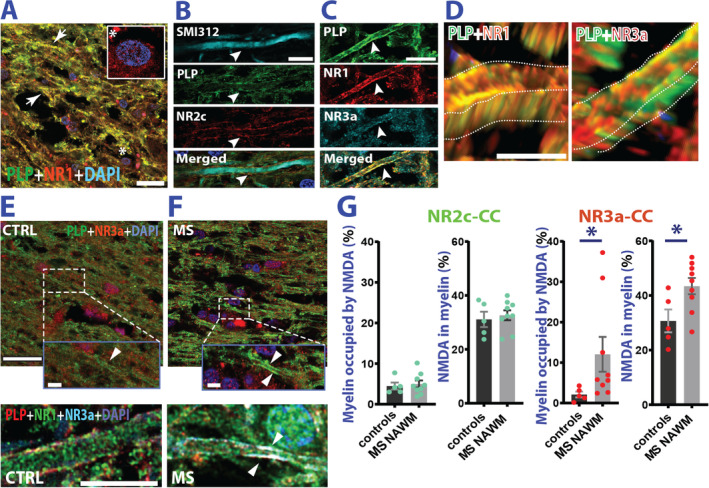
Myelin NMDA receptors are dysregulated in MS NAWM. (A) Confocal example of double staining for PLP and NMDAR NR1 obligatory subunit. NR1 is present in myelin (arrows), and cell bodies (DAPI staining *) (B) SMI‐312 (axonal neurofilament), PLP (myelin), and NMDA receptor subunit NR2c triple staining shows preferential distribution of NMDARs in the periaxonal space (arrowhead). Blue color:DAPI staining. (C) Triple staining for NMDAR NR1 and NR3a subunits shows colocalization with the myelin (PLP). (D) NR3a seem mostly expressed in the inner myelin sheath (left panel), whereas NR1 seems ubiquitously expressed in the entire myelin. (E, F) Top panels: Examples of PLP and NR3a subunit co‐expression in the corpus callosum of CTRLs and MS cases. Bottom panels: Magnification of axonal processes reporting co‐expression of PLP, NR1, and NR3a subunits in CTRL (E) and MS cases (F). (G) Analysis of myelin occupied by either NR2c or NR3a in MS NAWM corpus callosum. While expression of NR2c is unaffected (CTRL vs MS NAWM myelin occupied by NR2c: Welch's t test; t(10.70) = 0.413; p = 0.688; NR2c in myelin: Welch's t test; t(7.362) = 0.451; p = 0.665) NR3a is overexpressed in MS compared with controls (CTRL vs MS NAWM myelin occupied by NR3a: Mann–Whitney test; U = 4; p = 0.012; NR3a in myelin: Welch's t test; t(8.060) = 2.577; p = 0.038). Scale bars are 20μm in A; 10μm in B and C and 2.5μm in D. E and F top panel scale bar is 25μm (inset scale bar is 5μm). Bottom panel for E and F scale bar is 10μm Data are reported as mean ± SEM. *p < 0.05. CTRL = control; MS = multiple sclerosis; NAWM = normal appearing white matter; NMDA = N‐methyl‐D‐aspartate; NMDAR = N‐methyl‐D‐aspartate receptor; PLP = proteolipid protein.
Biochemical Changes in MS NAWM May Precede Immune Attack
A frequently observed post‐translational modification in tissues affected by (auto)immune‐mediated inflammation that enhances antigenicity of proteins is the enzymatic substitution of positively charged arginine residues for neutrally charged citrulline (citrullination). In MS NAWM, we observed enrichment of citrullinated proteins compared with control WM (Fig 5A–C). Citrullination tends to accumulate in myelin (see Fig 5B) but could also be detected in blood vessels and, in particular, in leptomeningeal/perivascular spaces of MS cases (Fig 5D, E), which show increased citrulline accumulation compared with controls (Fig 5F). We did not find augmented myelin protein citrullination in MS swellings compared with CTRLs (Fig 5G, H). Hence, it is possible that myelin protein citrullination precedes swelling formation, serving, together with structural and lipid alterations, as prodromal event upstream of an autoimmune attack.
FIGURE 5.
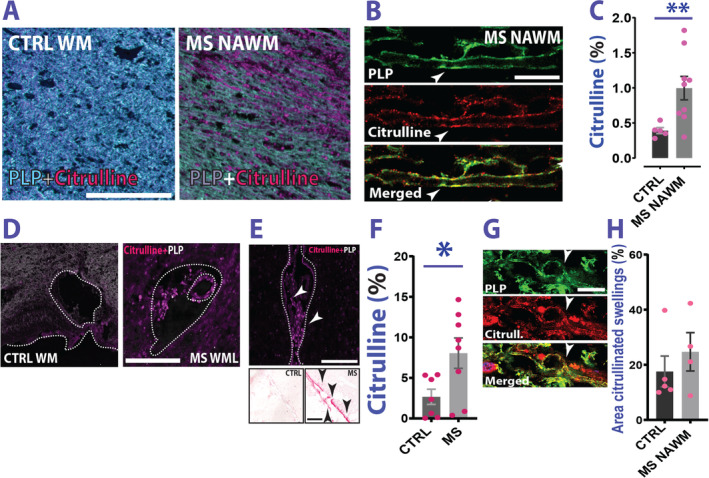
Citrulline content in MS. (A) Spectral nuance camera examples of citrulline content in CTRL WM and MS NAWM. (B) Citrulline staining co‐localizes with PLP staining (arrowheads). (C) Analysis of the area fraction occupied by citrullinated proteins in MS NAWM versus CTRLs reveals higher content in MS cases (Welch's t test; t(9.004) = 3.517; p = 0.006; CTRL vs MS NAWM). (D) MS WM blood vessel presents with citrulline content. (E) Meninges in patients with MS also contain citrullinated proteins. Top panel: Image of the leptomeninges of an MS case in a region adjacent to a cortical lesion area. Arrowheads indicate citrullinated proteins accumulating in the meningeal space. Bottom panel: Representative image of a staining performed in a cohort of MS and CTRL cases showing citrulline accumulation (red) in the meninges (arrowheads). (F) Analysis of area (%) occupied by citrulline in the meninges in MS versus controls (Welch's t test; t(10.77) = 2.496; p = 0.030 CTRL meninges/cortex vs MS meninges/cortex). (G) Example of citrullinated swellings identified by PLP staining. (H) Analysis of citrullinated area in swellings of MS versus controls does not show a significant difference (Mann–Whitney test; U = 7; p = 0.556; CTRL vs MS NAWM). Scale bars are 200μm in A; 10μm in B, 50μm in D, 200μm in E, and 10μm in G. Data are reported as mean ± SEM. *p < 0.05; **p < 0.01. CTRL = control; MS = multiple sclerosis; NAWM = normal appearing white matter; PLP = proteolipid protein; WM = white matter.
Discussion
Results from genomewide association studies added to beneficial effects of monoclonal antibodies and the presence of cerebrospinal fluid (CSF) oligoclonal immunoglobulin G (IgG) support a central pathogenic role of the immune system in MS. 25 However, whether autoimmunity is a primary or secondary pathological event is debated.
This study documents axon‐myelin unit changes in MS NAWM not detectably affected by inflammation. We posit that the increased blistering of myelin by focal detachment from its axons potentially contributes to early MS pathological manifestations, initiating myelin degradation and eliciting a subsequent immune reaction against myelin debris. Myelin blisters share similarities with reported intramyelinic edema. 26 Although the exact relationship between the latter and myelin blisters is still unknown, it seems that edema is a much larger morphological alteration, which results from tissue water inclusion between lamellae.
In addition, we describe: (1) another swelling class characterized by axonal enlargements (blebs), and (2) a condition where the SMI312 staining was fragmented. Although the latter category might indicate axonal degeneration, it is not clear whether it represents a separate class, a blister/bleb degeneration stage, or a simple abnormal neurofilament distribution inside the swellings.
In MS samples, these changes correlate with structural/biochemical alterations in the swellings hinting at imbalanced axon‐myelin communication through the recently described glutamate‐mediated form of axon‐myelinic communication. 13
Although a primary role of the immune system is difficult to exclude, the subtle changes we observed in areas characterized by absence of overt inflammation seem consistent with an inside‐out paradigm of MS. According to this hypothesis, CNS events, histologically presenting as blisters, initiate myelin injury and immunogenic debris release against which autoreactive T/B‐cells in lymphoid organs react. 3 Conceptually, the ensuing CNS‐targeted autoimmune process leads to inflammation through essentially similar pathogenic mechanisms as in the outside‐in MS paradigm. Proper AMS functioning requires an ~ 20 nm distance between the inner myelin lamella and the axon, 13 a condition which is clearly disturbed in the blister‐like swellings. Results of this miscommunication can be the triggering of aberrant myelinic Ca2+ signaling leading to myelin degradation. Consistently, we spotted an augmented myelin blister presence in perilesional WM regions, supporting its putative role in lesion formation.
An important factor in AMS destabilization is glutamate, which signals through NMDAR, triggering myelinic Ca2+ accumulation. 13 Here, we show that MS NAWM presents altered myelinic expression of NR2c and NR3a NMDAR subunits. 24 Interestingly, whereas the glycine‐sensitive NR3a subunit is considered neuroprotective, 27 the glutamate‐sensitive NR2c mediates neurodegeneration. 28 Therefore, whereas NR3a upregulation in normal axons putatively protects myelin against Ca2+ overload in face of a degenerative insult, a sudden NR2c increase rather promotes blisters/blebs formation.
Such uncontrolled myelinic Ca2+ entry might instigate a cascade of myelin destabilizing mechanisms, such as phospholipase A2‐IVa translocation from oligodendrocytes to myelin, 29 and myelin lipid ratio changes, 30 provoking myelinic insulation failure. This condition, together with our previous findings in MS NAWM, 31 might explain the lipid biochemical alterations in MS swellings (see Fig 2B). Interestingly, the same lipid changes are absent in CTRL swellings, suggesting the existence of distinct biochemical mechanisms governing the formation of MS and non‐MS swellings.
On the other hand, Ca2+ can also initiate a proteolytic cascade leading to activation of the calpain‐cathepsin axis, 32 and the hydrolysis of MAG. 22 MAG is a protein complex important for axon‐glia maintenance and a marker of normal periaxonal spacing. 33 In MS, the conversion rate of MAG to truncated dMAG is faster than in controls. 21 Regardless of the debated mechanism responsible for MAG hydrolysis, 34 dMAG is deprived of its membrane‐anchoring domain, which impairs its functionality. In line with this notion, we spotted a reduction of extracellular MAG reactivity in MS peri‐swelling regions, a condition likely involved in swelling formation, which seems compensated by increased intra‐swelling MAG expression to restore axo‐glial maintenance. Although studies in MAG−/− mice reported normal myelination, 35 ultrastructural investigations showed subtle changes in this model, including dysfunctional myelin outfolds, 36 dying‐back oligodendropathy, 37 and nodal region formation delays. 38 These changes support a primary role of MAG in AMS destabilization in absence of frank demyelination, a condition which aligns with MAG reduction in MS swelling formations when compared with controls.
Notably, the reported nodal formation delays 38 also support a direct effect of altered MAG functionality in the Ranvier's node physiology, contributing to abnormal ion influx in the axonal space. This concept is reinforced by altered expression levels of MS NAWM paranodal tethering proteins, crucial to maintain nodal regions. 23 Interestingly, we observed an increase in tethering protein volume compared with controls with exception of contactin‐1 in the CC. This enhancement might reveal a compensatory mechanism, which occurs to circumvent contactin‐1 reduction in the CC, a region associated with early MS lesion formation, 39 in an attempt to preserve axonal physiology/myelin stability. Therefore, overexpression of other paranodal tethering molecules (eg, contactin‐2) in the CC, and contactin‐1 in sWM, might prevent the aberrant axonal physiology caused by contactin‐1 reduction. Interestingly, a recent study reported that simultaneous depletion of both contactin‐1/NF155/CASPR complex and MAG deeply alters axon myelination, 40 alternatively explaining why in our samples a reduction of either contactin‐1 or MAG is accompanied by unaltered expression (or even an overexpression) of the other.
Furthermore, the overall tethering protein overexpression in MS NAWM might also indirectly explain why MS swellings expand throughout the axonal tract more often than CTRL swellings (see Fig 2A). In this case, a tighter paranodal junction in MS might have prevented the formation of swellings in proximity of the node. This condition, together with dMAG formation and lipid insulation failure, may have promoted a proliferation of internodal swellings in MS.
Finally, a general increase in tethering protein volume may also be due to paranodal length alterations, the presence of swellings, and/or internodal distance reduction in MS NAWM. Although conceivable, we believe that these possibilities do not influence our results. Paranodal length was not different in MS versus controls (see Fig 3I) and distribution analysis along axonal tract showed that about 50% of the swellings were located far from nodal/paranodal regions (see Fig 2A). Moreover, contactin‐1 expression density analysis retrieved the same bidirectional alteration spotted in CC and sWM volume analysis (data not shown) suggesting that, if a shorter internodal length is ubiquitously present in MS NAWM compared with controls, this is a less relevant parameter to explain our results.
In line with studies in MS inflammatory lesions, 41 here, we also report aberrant nodal elongation in MS axons distant from inflammatory activity. This effect may hold consequences for axonal physiology, 23 causing myelin retraction‐dependent potassium channel (Kv) redistribution and/or altered Nav turnover/distribution. 23 , 41 Although both possibilities are plausible, the latter seems more directly supported by our results indicating Nav paranodal displacement in MS NAWM axons. This pattern is more frequent in conditions of elongated nodal regions supporting altered action potential propagation due to excessive current outflow in the paranodal region. 23 MS patient brain imaging reported greater extra/intracellular Na+ concentration in the NAWM than in WM of healthy volunteers. 42 Hence, nodal region enlargement/paranodal Nav displacement in MS might be responsible for higher Na+ intake into the axonal space. This effect might increase axonal Ca2+ influx either inverting the axonal sodium‐calcium exchanger (NCX) operation or triggering a reversal of the glutamate/glycine transporter into efflux mode, 43 or eliciting Ca2+ efflux from mitochondria. 44 Furthermore, other aberrant conditions like Ca2+‐permeable axonal nanoruptures 45 should be also taken into account.
Predictably, the observed structural/morphological alterations correlate with changes in the MS NAWM biochemistry. Studies in CPZ mouse models showed MBP citrullination prior to demyelination 12 and investigations in marmoset models and patients with MS showed enhanced myelin protein citrullination. 17 , 18 Here, we document that MS NAWM is highly citrullinated. This process suggests myelin modifications in regions minimally influenced by immune attack, where the release of highly immunogenic myelin fragments may provoke a subsequent immune response against myelin debris. This possibility may explain the presence of citrulline content in MS blood vessels and leptomeninges.
Interestingly, our data do not show a direct link between myelin citrullination and MS swelling formation (see Fig 5H). This suggests that myelin protein citrullination may happen when the glutamate‐mediated communication still operates normally. Notably, citrullinated MBP less efficiently compacts myelin, and increases susceptibility to digestion by proteases, leading to increased risk of myelin exposure to proteolytic attack. 46
In conclusion, we propose the contribution of previously overlooked aberrant molecular processes in axon‐myelin units to the initiation of the complex MS pathological process.
The morphological reflection of these processes may be the local myelin detachment from axons, forming the characteristic blisters (see Fig 6 for a proposed model). Conceptually, the disturbed communication within axon‐myelin units triggers the progressive axon and myelin degeneration, leading among others to the enhanced release of potentially immunogenic debris. Studies in a translationally highly relevant MS animal model, marmoset experimental autoimmune encephalitis (EAE), revealed that autoimmune reactions against MS myelin can elicit MS‐like pathology in the animal's CNS. In the cascade of pathogenic events, the myelin constituent myelin oligodendrocyte glycoprotein has a core pathogenic role, which is strongly enhanced by protein citrullination. 47 , 48 This sequence of events aligns with Wilkin's primary lesion theory, which conceptualizes autoimmunity as a genetically predisposed immune system hyperreaction against antigens released from an idiopathic primary lesion in the target organ. 49
FIGURE 6.

Proposed sequence of events in MS. Graphic representation of the hypothesized cascade of mechanisms involved in AMS imbalance in MS. Red pathways 1a (as indicated by arrows) shows the possible instigator in AMS imbalance. Elongation of the Ranvier's node might induce a re‐distribution of Nav channels (eg, the persistent firing channels Nav1.6 41 ). High axonal Na+ entrance might promote an NCX 43 inversion increasing the intracellular Ca2+ entrance (2, blue pathway), resulting in a higher probability of glutamate release from axon to myelin. At the same time, myelin lipid aberrations (red pathway, 1b) might reduce the axonal shielding from external ions, favoring Ca2+ entrance in the axon (where Cav are expressed 13 ). This effect might also add to that shown in 1a. Higher glutamate release triggers Ca2+ increase in the myelin, activating PAD enzymes to citrullinate myelin proteins 12 (black pathway, 4). Release of citrullinated proteins might instigate an immune response against the debris. Ca2+ entrance might also activate the calpain‐cathepsin axis, 32 detrimental for lysosomes, which ultimately would promote dMAG formation 22 (violet pathway, 5). This effect instigates myelin detachment from its axon. Finally, Ca2+ increase in the myelin can also favor the translocation of PLA‐Iva enzymes, 29 which affects the lysosomal functionality and lead to lipid biochemical changes. The latter alteration may consequently make the lipid membrane more permeable to external ions (including Ca2+, brown pathway, 6). Detachment of myelin causes the retrieved blisters (7), causing AMS instability and hampering the lactate‐dependent signal to axonal mitochondria, 13 instigating virtual hypoxia, axonal swelling (8), and, ultimately due to insufficient axonal trophic support, axonal degeneration (9). AMS = axo‐myelinic synapse; dMAG = degraded myelin associated glycoprotein; MAG = myelin associated glycoprotein; MS = multiple sclerosis; PAD = peptidyl arginine deiminase.
Author Contributions
A.L., G.J.S., B.t'H., and J.J.G. contributed to conception and design of the study. A.L., L.P., I.F., H.O., and G.J.S. contributed to acquisition and analysis of the data. A.L., B.t'H., A.M.vD., P.K.S., G.J.S., and J.J.G. contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest
The authors declared no conflict of interest.
Supporting information
TABLE S1 List of the Principal Antibodies Used for the Whole Study.
Acknowledgment
This study was supported by Stichting MS Research/Stichting Klimmen tegen MS (MoveS) (pilot project number 16‐954a/b MS), Ammodo KNAW award (2017) awarded to Prof. Dr. Jeroen J. G. Geurts, and Monique Blom‐de Wagt grant Stichting MS Research (18‐997 MS) awarded to Dr. Antonio Luchicchi. We also thank Stichting Sandy MoveS for additional support. We thank Dr. Maarten E. Witte and Dr. Laura E. Jonkman for providing brain blocks where meninges where highly preserved and Alzheimer's disease blocks, respectively, and Mr. John Bol, Mrs. Angela Ingrassia, Mr. Dennis Karabag, Mrs. Kimberley Catsman, and Mr. Jos Nijhof for their excellent technical assistance.
References
- 1. Ringold S, Lynm C, Glass RM. JAMA patient page. Multiple sclerosis. JAMA 2005;293:514. [DOI] [PubMed] [Google Scholar]
- 2. Chitnis T. The role of CD4 T cells in the pathogenesis of multiple sclerosis. Int Rev Neurobiol 2007;79:43–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stys PK, Zamponi GW, van Minnen J, Geurts JJ. Will the real multiple sclerosis please stand up? Nat Rev Neurosci 2012;13:507–514. [DOI] [PubMed] [Google Scholar]
- 4. Lassmann H, Ransohoff RM. The CD4‐Th1 model for multiple sclerosis: a critical [correction of crucial] re‐appraisal. Trends Immunol 2004;25:132–137. [DOI] [PubMed] [Google Scholar]
- 5. Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol 2004;55:458–468. [DOI] [PubMed] [Google Scholar]
- 6. Henderson AP, Barnett MH, Parratt JD, Prineas JW. Multiple sclerosis: distribution of inflammatory cells in newly forming lesions. Ann Neurol 2009;66:739–753. [DOI] [PubMed] [Google Scholar]
- 7. Rodriguez M, Scheithauer B. Ultrastructure of multiple sclerosis. Ultrastruct Pathol 1994;18:3–13. [DOI] [PubMed] [Google Scholar]
- 8. Aboul‐Enein F, Rauschka H, Kornek B, et al. Preferential loss of myelin‐associated glycoprotein reflects hypoxia‐like white matter damage in stroke and inflammatory brain diseases. J Neuropathol Exp Neurol 2003;62:25–33. [DOI] [PubMed] [Google Scholar]
- 9. Lucchinetti C, Bruck W, Parisi J, et al. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 2000;47:707–717. [DOI] [PubMed] [Google Scholar]
- 10. Filippi M, Rovaris M, Rice GP, et al. The effect of cladribine on T(1) 'black hole' changes in progressive MS. J Neurol Sci 2000;176:42–44. [DOI] [PubMed] [Google Scholar]
- 11. Hart ´t, BA HRQ, Laman JD. Multiple sclerosis ‐ a response‐to‐damage model. Trends Mol Med 2009;15:235–244. [DOI] [PubMed] [Google Scholar]
- 12. Caprariello AV, Rogers JA, Morgan ML, et al. Biochemically altered myelin triggers autoimmune demyelination. Proc Natl Acad Sci U S A 2018;115:5528–5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Micu I, Plemel JR, Caprariello AV, et al. Axo‐myelinic neurotransmission: a novel mode of cell signalling in the central nervous system. Nat Rev Neurosci 2018;19:49–58. [DOI] [PubMed] [Google Scholar]
- 14. Micu I, Jiang Q, Coderre E, et al. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature 2006;439:988–992. [DOI] [PubMed] [Google Scholar]
- 15. Wood DD, Ackerley CA, Brand B, et al. Myelin localization of peptidylarginine deiminases 2 and 4: comparison of PAD2 and PAD4 activities. Lab Invest 2008;88:354–364. [DOI] [PubMed] [Google Scholar]
- 16. Gyorgy B, Toth E, Tarcsa E, et al. Citrullination: a posttranslational modification in health and disease. Int J Biochem Cell Biol 2006;38:1662–1677. [DOI] [PubMed] [Google Scholar]
- 17. Moscarello MA, Wood DD, Ackerley C, Boulias C. Myelin in multiple sclerosis is developmentally immature. J Clin Invest 1994;94:146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moscarello MA, Lei H, Mastronardi FG, et al. Inhibition of peptidyl‐arginine deiminases reverses protein‐hypercitrullination and disease in mouse models of multiple sclerosis. Dis Model Mech 2013;6:467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kooi EJ, van Horssen J, Witte ME, et al. Abundant extracellular myelin in the meninges of patients with multiple sclerosis. Neuropathol Appl Neurobiol 2009;35:283–295. [DOI] [PubMed] [Google Scholar]
- 20. Trapp BD, Peterson J, Ransohoff RM, et al. Axonal transection in the lesions of multiple sclerosis. N Engl J Med 1998;338:278–285. [DOI] [PubMed] [Google Scholar]
- 21. Moller JR. Rapid conversion of myelin‐associated glycoprotein to a soluble derivative in primates. Brain Res 1996;741:27–31. [DOI] [PubMed] [Google Scholar]
- 22. Stebbins JW, Jaffe H, Fales HM, Moller JR. Determination of a native proteolytic site in myelin‐associated glycoprotein. Biochemistry 1997;36:2221–2226. [DOI] [PubMed] [Google Scholar]
- 23. Arancibia‐Carcamo IL, Attwell D. The node of Ranvier in CNS pathology. Acta Neuropathol 2014;128:161–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pina‐Crespo JC, Talantova M, Micu I, et al. Excitatory glycine responses of CNS myelin mediated by NR1/NR3 "NMDA" receptor subunits. J Neurosci 2010;30:11501–11505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Filippi M, Bar‐Or A, Piehl F, et al. Multiple sclerosis. Nat Rev Dis Primers 2018;4:43. [DOI] [PubMed] [Google Scholar]
- 26. Hoegg‐Beiler MB, Sirisi S, Orozco IJ, et al. Disrupting MLC1 and GlialCAM and ClC‐2 interactions in leukodystrophy entails glial chloride channel dysfunction. Nat Commun 2014;5:3475. [DOI] [PubMed] [Google Scholar]
- 27. Nakanishi N, Tu S, Shin Y, et al. Neuroprotection by the NR3A subunit of the NMDA receptor. J Neurosci 2009;29:5260–5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kadotani H, Namura S, Katsuura G, et al. Attenuation of focal cerebral infarct in mice lacking NMDA receptor subunit NR2C. Neuroreport 1998;9:471–475. [DOI] [PubMed] [Google Scholar]
- 29. Gijon MA, Spencer DM, Kaiser AL, Leslie CC. Role of phosphorylation sites and the C2 domain in regulation of cytosolic phospholipase A2. J Cell Biol 1999;145:1219–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thelen AM, Zoncu R. Emerging roles for the lysosome in lipid metabolism. Trends Cell Biol 2017;27:833–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Poon KWC, Brideau C, Klaver R, et al. Lipid biochemical changes detected in normal appearing white matter of chronic multiple sclerosis by spectral coherent Raman imaging. Chem Sci 2018;9:1586–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Llorens F, Thune K, Sikorska B, et al. Altered ca(2+) homeostasis induces Calpain‐Cathepsin axis activation in sporadic Creutzfeldt‐Jakob disease. Acta Neuropathol Commun 2017;5:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Filbin MT. Myelin‐associated glycoprotein: a role in myelination and in the inhibition of axonal regeneration? Curr Opin Neurobiol 1995;5:588–595. [DOI] [PubMed] [Google Scholar]
- 34. Paivalainen S, Suokas M, Lahti O, Heape AM. Degraded myelin‐associated glycoprotein (dMAG) formation from pure human brain myelin‐associated glycoprotein (MAG) is not mediated by calpain or cathepsin L‐like activities. J Neurochem 2003;84:533–545. [DOI] [PubMed] [Google Scholar]
- 35. Li C, Tropak MB, Gerlai R, et al. Myelination in the absence of myelin‐associated glycoprotein. Nature 1994;369:747–750. [DOI] [PubMed] [Google Scholar]
- 36. Pernet V, Joly S, Christ F, et al. Nogo‐a and myelin‐associated glycoprotein differently regulate oligodendrocyte maturation and myelin formation. J Neurosci 2008;28:7435–7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lassmann H, Bartsch U, Montag D, Schachner M. Dying‐back oligodendrogliopathy: a late sequel of myelin‐associated glycoprotein deficiency. Glia 1997;19:104–110. [PubMed] [Google Scholar]
- 38. Marcus J, Dupree JL, Popko B. Myelin‐associated glycoprotein and myelin galactolipids stabilize developing axo‐glial interactions. J Cell Biol 2002;156:567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Evangelou N, Konz D, Esiri MM, et al. Regional axonal loss in the corpus callosum correlates with cerebral white matter lesion volume and distribution in multiple sclerosis. Brain 2000;123:1845–1849. [DOI] [PubMed] [Google Scholar]
- 40. Djannatian M, Timmler S, Arends M, et al. Two adhesive systems cooperatively regulate axon ensheathment and myelin growth in the CNS. Nat Commun 2019;10:4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Howell OW, Palser A, Polito A, et al. Disruption of neurofascin localization reveals early changes preceding demyelination and remyelination in multiple sclerosis. Brain 2006;129:3173–3185. [DOI] [PubMed] [Google Scholar]
- 42. Inglese M, Madelin G, Oesingmann N, et al. Brain tissue sodium concentration in multiple sclerosis: a sodium imaging study at 3 tesla. Brain 2010;133:847–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stys PK. General mechanisms of axonal damage and its prevention. J Neurol Sci 2005;233:3–13. [DOI] [PubMed] [Google Scholar]
- 44. Palty R, Hershfinkel M, Sekler I. Molecular identity and functional properties of the mitochondrial Na+/Ca2+ exchanger. J Biol Chem 2012;287:31650–31657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Witte ME, Schumacher AM, Mahler CF, et al. Calcium influx through plasma‐membrane Nanoruptures drives axon degeneration in a model of multiple sclerosis. Neuron. 2019;101:615–24 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pritzker LB, Joshi S, Gowan JJ, et al. Deimination of myelin basic protein. 1. Effect of deimination of arginyl residues of myelin basic protein on its structure and susceptibility to digestion by cathepsin D. Biochemistry 2000;39:5374–5381. [DOI] [PubMed] [Google Scholar]
- 47. Araman C, van Gent ME, Meeuwenoord NJ, et al. Amyloid‐like behavior of site‐specifically Citrullinated myelin oligodendrocyte protein (MOG) peptide fragments inside EBV‐infected B‐cells influences their cytotoxicity and autoimmunogenicity. Biochemistry 2019;58:763–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hart ´t, BA KYS, Morandi E, et al. EBV infection and multiple sclerosis: lessons from a marmoset model. Trends Mol Med 2016;22:1012–1024. [DOI] [PubMed] [Google Scholar]
- 49. Wilkin TJ. The primary lesion theory of autoimmunity: a speculative hypothesis. Autoimmunity 1990;7:225–235. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 List of the Principal Antibodies Used for the Whole Study.