

Abstract
Introduction
We investigated how components of immunity relate to biomarkers of Alzheimer's disease (AD) in plasma and explored the influence of AD genetic risk factors in the population‐based Rotterdam Study.
Methods
In 7397 persons, we calculated the granulocyte‐to‐lymphocyte ratio (GLR), platelet‐to‐lymphocyte ratio (PLR), and systemic immune‐inflammation index (SII). In 3615 of these persons, plasma amyloid‐beta (Aβ)42 and Aβ40 were measured. Next, we constructed an overall genetic risk score (GRS) based on genome‐wide significant variants, both including and excluding APOE ε4.
Results
All innate immunity phenotypes were related to higher Aβ, most strongly with a doubling in GLR leading to a 1.9% higher Aβ42 (95% confidence interval [95% CI] 0.4 to 3.3%) and 3.2% higher Aβ40 (95% CI 2.0 to 4.3%). Higher AD GRS including APOE ε4 was associated with higher immunity markers.
Discussion
Higher levels of immunity markers were associated with higher Aβ in plasma. Participants with a higher genetic predisposition to AD had higher immunity markers, where these effects were mainly driven by APOE ε4.
Keywords: amyloid, cohort study, dementia, immunity, population‐based
1. INTRODUCTION
Alzheimer's disease (AD) is characterized pathologically by accumulation of amyloid beta (Aβ) as amyloid plaques and phosphorylated tau as neurofibrillary tangles. 1 This accumulation gave rise to the amyloid hypothesis, posing that Aβ activates a cascade of pathologic changes. 2 As an extension to the amyloid hypothesis, the antimicrobial protection model was recently proposed suggesting that Aβ oligomerization is not intrinsically pathological, but emerges as an innate immune response. 3 Genetic variants identified through genome‐wide association studies (GWAS) support this role for immunity. 4 It has also been found that higher activation of the innate immune system and lower activation of the adaptive immune system leads to higher dementia risk. 5 Yet, the link between immunity and AD‐related brain pathology is largely unknown.
In the early 1990s, apolipoprotein E (apoE) was found to be a component of amyloid plaques. 6 , 7 However, how apoE's function as a lipid‐carrier is in itself related to AD, either in relation to Aβ or other factors, is not entirely clear. Lipids serve more than pure nutritional purposes; they also play essential roles in immune regulation. 8 Innate immunity in turn affects Aβ and tau pathology buildup. Insoluble tau isolated from postmortem AD brain is shown to be taken up by microglia in vitro and in vivo. 9 This taking in of tau may participate in tau spreading by microglia subsequently releasing some form of tau. 10
To study further how AD relates to the immune response, we may use serum levels of various blood cells which reflect a systemic immune response 11 and relate them with biomarkers of AD pathology and progression. Serum levels of granulocytes and platelets are known biomarkers of the innate immunity, whereas lymphocyte levels may yield information on the adaptive immunity. 12 Combining these measurements into ratios is thought to reflect the relative balance between innate and adaptive immunity even better, obtaining the granulocyte‐to‐lymphocyte ratio (GLR), platelet‐to‐lymphocyte ratio (PLR), and systemic immune‐inflammation index (SII) as phenotypical markers of immunity. 13 Whereas for the diagnosis of AD, biomarkers are grouped into those of β amyloid deposition, pathologic tau, and neurodegeneration (ATN) according to the recent classification by Jack et al., 14 yielding plasma Aβ, total tau, and neurofilament light chain (NfL) as biomarkers of AD‐related brain pathology, respectively.
In an earlier study investigating the role of various biological pathways based on a genetic risk score, we found an important role for the immune response pathway in early AD pathology. 15 Relating phenotypical immunity and AD‐related markers to each other and additionally to genetic predisposition to AD may help to understand the role of immunity biomarkers in AD pathology and their relation with AD genetic risk, and ultimately identify therapeutic strategies targeting upstream events of altered immune response in amyloidosis and neurodegeneration. We previously found that higher levels of the GLR, PLR, and SII over time reflect higher innate immunity and increased dementia risk. 5 Since we also previously found that low Aβ42 and high NfL plasma levels were associated with risk of AD, 16 we hypothesized that higher innate immunity affects the serum levels of Aβ42 to be lower and NfL to be higher. We tested this hypothesis by investigating whether serum GLR, PLR, and SII were associated with Aβ, total tau, and NfL and how genetic predisposition to AD affected these markers.
2. METHODS
2.1. Study population
The present study is embedded within the Rotterdam Study, a prospective population‐based cohort study in Rotterdam, the Netherlands. The Rotterdam Study started in 1989 with 7983 persons (response rate: 78%) aged ≥55 years and residing in the district of Ommoord, a suburb of Rotterdam. This first subcohort (RS‐I) was extended with a second subcohort (RS‐II) in 2000, consisting of 3011 persons (response rate: 67%), and with a third subcohort (RS‐III) in 2006, composed of 3932 persons aged ≥45 years (response rate: 65%). The design of the Rotterdam Study has been described in detail previously. 17 In brief, participants were examined at study entry and follow‐up visits every 3 to 5 years. They were interviewed at home by a trained research nurse, followed by two visits at the research facility for additional interviewing and laboratory assessments. The Rotterdam Study has been approved by the Medical Ethics Committee of the Erasmus Medical Center and by the board of the Netherlands Ministry of Health, Welfare, and Sports. Written informed consent was obtained from all participants.
RESEARCH IN CONTEXT
Systematic review: Alzheimer's disease (AD) is characterized pathologically by accumulation of amyloid plaques and tau tangles. Genetic variants identified through genome‐wide association studies suggest a role for immunity. However, the link between immunity and biomarkers of neurodegeneration is largely unknown. We investigated how biomarkers of immunity relate to established AD biomarkers, amyloid beta (Aβ), total tau, and neurofilament light chain (NfL), in plasma. We also explored the influence of genetic risk factors of AD in these associations.
Interpretation: In this study, we found that higher levels of serum innate immunity markers were associated with lower Aβ42/40 ratio, lower total tau and higher NfL in serum. A higher genetic predisposition to AD was significantly associated with higher serum immunity markers and with lower Aβ42 and Aβ42/40 ratio, where these genetic effects were mainly driven by the APOE ε4 variant.
Future directions: Knowledge of the potential pro‐inflammatory role of APOE ε4 should encourage future studies to find ways to scale down innate immune responses in APOE ε4 carriers to limit AD‐related brain pathology to prevent AD.
Laboratory tests for granulocytes, platelets, and lymphocytes were introduced from 2002 onwards, corresponding with the following assessment rounds in the Rotterdam Study (baseline for this study): fourth round of RS‐I (RS‐I‐4), second round of RS‐II (RS‐II‐2), and first round of RS‐III (RS‐III‐1). Plasma biomarkers of AD‐related brain pathology were only assessed in RS‐I‐4 and RS‐II‐2. Blood was drawn from 11,496 participants for genotyping. Of these participants, 7413 underwent granulocyte, platelet, and lymphocyte assessments, while 3627 participants underwent plasma Aβ, total tau, and NfL assessment. We excluded participants with missing APOE genotype and participants with incomplete assessments of serum markers, leaving 7397 participants for analysis with complete blood assessment for immunity markers and 3615 participants with complete plasma Aβ, total tau, and NfL assessment (Figure 1).
FIGURE 1.
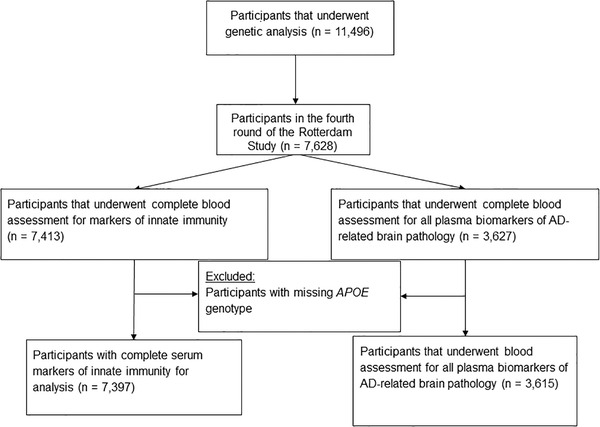
Flow diagram
2.2. Genotyping
DNA genotyping was performed at the internal genotyping facility of the Erasmus Medical Center, Rotterdam. All samples were genotyped with the 550, 550, duo or 610 K Illumina arrays. Genotyping quality control criteria include call rate <95%, Hardy‐Weinberg equilibrium of P < 1.0 × 10−6, and minor allele frequency <1%. Moreover, study samples with excess autosomal heterozygosity, call rate <97.5%, ethnic outliers, and duplicate or family relationships were excluded during quality control analysis. Genetic variants were imputed from the Haplotype Reference Consortium reference panel (version 1.0), 18 using the Michigan imputation server. 19 The server uses SHAPEIT2 (v2.r790) 20 to phase the genotype data and performs imputation with Minimac 3 software. 21 For this study, we used genetic variants that had imputation quality (R2) >0.5. APOE genotype was determined separately by PCR on coded DNA samples in the baseline cohort and with a bi‐allelic Tacqman assay in the extensions of the Rotterdam Study. 22
2.3. Assessment of blood cell counts and their derived ratios
Full blood count measurements were performed using the COULTER® Ac·T diff2™ Hematology Analyzer (Beckman Coulter, San Diego, California, USA) directly after the blood sample was drawn. Laboratory measurements included absolute granulocyte, platelet, and lymphocyte counts in 109/L. The GLR and PLR were calculated as the ratio of granulocyte count to lymphocyte count, and as the ratio of platelet count to lymphocyte count, respectively. The SII was defined as platelet count times the GLR. 13 We use the GLR as a proxy measure for the commonly used neutrophil‐to‐lymphocyte ratio, as granulocytes are the most abundant subtype of neutrophils.
2.4. Assessment of plasma Aβ, total tau, and NfL
Blood was sampled in EDTA‐treated containers and centrifuged. Subsequently plasma was aliquoted and frozen at −80⁰C according to standard procedures. Measurements were done in two separate batches. The first batch included in total 2000 samples, obtained from a random selection of 1000 participants from sub‐cohort RS‐I‐4 and 1000 from RS‐II‐2. The second batch included in total 3094 samples from the remaining participants.
All measurements were performed at Quanterix (Lexington, MA, USA) on a single molecule array (Simoa) HD‐1 analyzer platform. 23 Samples were tested in duplicate. Two quality control (QC) samples were run on each plate for each analyte. NfL was measured with the NF‐light advantage kit. 24 The Simoa Human Neurology 3‐Plex A assay (N3PA) was used for measuring the concentration of total tau, Aβ42, and Aβ40. When duplicates or single measurements were missing (the majority of missing samples were due to system failures [n = 279], and few because of insufficient volume [n = 47]) or in the case the concentration coefficient of variation (CV) exceeded 20% (14 to 87 samples) or control samples were out of range (none), participant data were excluded from the analyses. 16
2.5. Genetic risk scores
We computed genetic risk scores by selecting late‐onset AD‐associated single‐nucleotide polymorphisms (SNPs) reaching genome‐wide significance (P < 5.0 × 10−8). Among common variants, we considered only variants identified by the International Genomics of Alzheimer's Project (IGAP) meta‐analyses. 4 In addition, we included four rare variants which can be classified under immune response based on their functional role, 25 , 26 leading to a total of 28 independent genome‐wide significant AD‐associated variants. A weighted genetic risk score was constructed using the effect sizes (log of odds ratio) of the genome‐wide significant variants from the IGAP meta‐analysis as weights and their respective allele dosages from imputed genotype data of our study cohorts. A genetic risk score was constructed as the sum of the products of SNP dosages and their corresponding weights. We constructed genetic risk scores in three ways: (1) combining all 28 selected variants, (2) excluding the APOE ε4 variant to identify the joint independent effect of all other genome‐wide significant SNPs, and (3) clustering the variants into the immune response pathway. We classified AD‐associated SNPs into immune system pathways based on information on current investigations and reviews. 4 , 27 , 28 , 29 Of the 28 SNPs, 9 could be clustered into the immune response pathway‐based genetic risk score (Table S1). All genetic risk scores were standardized to allow direct comparison of results.
2.6. Covariates
Potential confounding factors were chosen on the basis of previous literature. 13 , 30 All covariates were measured at the same rounds as the assessments of AD‐related brain pathology and immunity markers. Smoking habits were categorized as current versus former and never smoking. Body mass index (BMI) was calculated as weight in kilograms per height in meters squared. Blood pressure was measured twice at the right brachial artery with the participant in sitting position, from which the mean was used. Diabetes mellitus was defined as use of antidiabetic medication, fasting serum glucose level ≥7.1 mmol/L (≥127.9 mg/dL), or random non‐fasting serum glucose level ≥11.1 mmol/L (≥200.0 mg/dL). 31 Additional information from the serum samples was collected on high sensitivity C‐reactive protein (hs‐CRP) and creatinine in a subsample (N = 1342).
2.7. Statistical analysis
As the GLR, PLR, and SII had skewed distributions, analysis was based on the natural logarithmic (Ln) transformation of their values. For the same reason, all plasma biomarkers of AD‐related brain pathology, except Aβ40 which had a normal distribution, were Ln transformed. We first determined the association between the GLR, PLR, and SII with plasma biomarkers of AD‐related brain pathology using linear regression. For this analysis, we conducted two different transformations. First, because we transformed both exposure and outcome (logYi = α + βlogXi + ε), we reported mean differences in percentages with 95% confidence intervals (CIs) obtained by exponentiating Ln(2) times the estimated coefficients from the linear regression model to facilitate interpretation of results; these mean differences correspond to the percentage change in plasma Aβ, total tau, or NfL for a doubling of the GLR, PLR, and SII. For this analysis, Aβ40 was also Ln transformed. Second, we standardized all blood markers to allow direct comparison of the results. We adjusted all models for age, sex, and study cohort (model I). In addition, the following covariates were added to a second model (model II): smoking, diabetes, BMI, systolic blood pressure, and APOE ε4 carriership. Since plasma Aβ, total tau, and NfL were measured in two batches, we additionally corrected for batch effects in models. We performed additional adjustment for platelet count when analyzing GLR in model II. We assessed effect measure modification by APOE genotype by stratifying for participants having the ε2/ε2 or ε2/ε3, ε3/ε3, and ε3/ε4 or ε4/ε4 genotype. We formally tested interaction between APOE genotype and ratios of blood cell counts on the multiplicative scale by adding interaction terms to model II. In addition, we assessed effect measure modification by activity of the innate immune system by stratifying for median granulocyte count (as a pure innate immunity marker rather than studying the effect of the balance between innate and adaptive immunity) when assessing the associations between genetic risk scores and plasma biomarkers of AD‐related brain pathology as outcome. We formally tested interaction between genetic risk scores and granulocyte counts on the multiplicative scale by adding interaction terms to the model. As a sensitivity analysis, we also adjusted for creatinine and hs‐CRP in a subsample of participants in which these markers were measured. Because hs‐CRP is also an important inflammatory biomarker, we also assessed the association between Ln(hs‐CRP) as exposure and the standardized plasma biomarkers of AD‐related brain pathology as outcome using linear regression. We adjusted all models for age, sex, cohort, smoking, diabetes, BMI, systolic blood pressure, APOE ε4 carriership, batch effects, platelet count, and creatinine.
We then determined associations between all three genetic risk scores per standard deviation (SD) increase as exposure with the standardized immunity markers (Ln[GLR], Ln[PLR] and Ln[SII]) and with the standardized plasma biomarkers of AD‐related brain pathology (Ln[Aβ42, Aβ40], Ln[Aβ42/40 ratio], Ln[total tau] and Ln[NfL]) as separate outcomes, using linear regression. These analyses were all adjusted for age, sex, and study cohort.
Furthermore, we investigated the association between APOE‐allele carriership as exposure with serum markers of immunity and plasma biomarkers of AD‐related brain pathology as outcomes. For this analysis, we defined APOE‐allele carriership as either carrying ε2 (ε2/ε2 or ε2/ε3 genotype), being homozygous for ε3 (ε3/ε3 genotype) or carrying ε4 (ε3/ε4 or ε4/ε4 genotype). People with the ε2/ε4 genotype were excluded from the analyses and ε3 homozygosity was considered the reference group. Analyses were adjusted for age, sex, and study cohort. We calculated mean levels of hs‐CRP and creatinine levels within the different APOE genotypes.
Missing covariate data (maximum 0.7%) were imputed using 5‐fold Multiple Imputation by Chained Equations based on determinant, outcome, and included covariates. All analyses were performed using RStudio version 1.0.153 (R version 3.6.1, RStudio, Inc., Boston, MA). We corrected for multiple testing using Bonferroni adjustment for all analyses using the total population (ie, not stratified or no subgroup analyses): for the associations between serum markers of immunity and plasma biomarkers of AD‐related brain pathology, results are considered statistically significant if the P‐value is below .05/(6×5) = .002; for the associations of genetic risk scores reflecting AD including and excluding APOE ε4 and immune response with plasma biomarkers of AD‐related brain pathology and serum markers of immunity, results were considered statistically significant if the P‐value is below .05/(9×5) = .001; and for the associations of APOE genotypes with plasma biomarkers of AD‐related brain pathology and serum markers of immunity, results are considered statistically significant if the P‐value is below .05/16 = .003. For these analyses, a suggestive association was considered at the level of alpha = 0.05. All other analyses were considered statistically significant at the level of alpha = 0.05.
3. RESULTS
Characteristics of study participants are displayed in Table 1. The mean age of participants with serum markers of innate immunity was 66.1 (±10.4) years of which 4208 (57%) were women, and the mean age of the sample undergoing serum AD marker measurements was 71.9 (±7.3) years of which 2059 (57%) were women.
TABLE 1.
Characteristics of study population
| Characteristic | Sample with serum markers of immunity (N = 7397) | Sample with plasma biomarkers of AD‐related brain pathology (N = 3615) |
|---|---|---|
| Women | 4208 (56.9%) | 2059 (56.8%) |
| Age (years) | 66.1 ± 10.4 | 71.9 ± 7.3 |
| Study cohort | ||
| Cohort 1 | 2656 (35.9%) | 2159 (59.7%) |
| Cohort 2 | 1704 (23.0%) | 1456 (40.3%) |
| Cohort 3 | 3037 (41.1%) | – |
| Current smokers | 1427 (19.4%) | 536 (14.9%) |
| Diabetes mellitus type 2 | 433 (5.9%) | 214 (5.9%) |
| Body mass index (kg/m2) | 27.6 ± 4.3 | 27.6 ± 4.1 |
| Systolic blood pressure (mmHg) | 142.8 ± 21.9 | 149.2 ± 21.0 |
| APOE genotype | ||
| ε2/ε2 | 46 (0.6%) | 31 (0.9%) |
| ε2/ε3 | 952 (12.9%) | 490 (13.6%) |
| ε2/ε4 | 202 (2.7%) | 103 (2.8%) |
| ε3/ε3 | 4293 (58.0%) | 2119 (58.6%) |
| ε3/ε4 | 1741 (23.5%) | 810 (22.4%) |
| ε4/ε4 | 163 (2.2%) | 62 (1.7%) |
| Granulocyte count, × 103/μL | 4.0 ± 1.4 | 4.0 ± 1.3 |
| Platelet count, × 103/μL | 268.2 ± 67.2 | 256.4 ± 64.4 |
| Lymphocyte count, × 103/μL | 2.3 ± 1.2 | 2.2 ± 1.3 |
| Granulocyte‐to‐lymphocyte ratio | 1.9 ± 0.9 | 2.0 ± 0.9 |
| Platelet‐to‐lymphocyte ratio | 128.2 ± 47.3 | 130.0 ± 49.8 |
| Systemic immune‐inflammation index | 517.3 ± 280.1 | 522.0 ± 290.9 |
| Amyloid‐beta 42 (pg/mL) | – | 10.6 ± 3.0 |
| Amyloid‐beta 40 (pg/mL) | – | 265.6 ± 54.6 |
| Amyloid‐beta 42/40 ratio | – | 0.04 ± 0.009 |
| Total tau (pg/mL) | – | 2.6 ± 2.5 |
| Neurofilament light chain (pg/mL) | – | 15.7 ± 11.6 |
N = number of participants included in study. Data presented as mean (standard deviation) for continuous variables and number (percentages) for categorical variables. Data here are unimputed. Number of missing values for the immunity cohort: 39 (0.5%) for smoking, 19 (0.3%) for diabetes, 20 (0.3%) for BMI, 37 (0.5%) for systolic blood pressure, 0 for APOE carriership. Number of missing values for the sample undergoing serum AD measurements are: 25 (0.7%) for smoking, 8 (0.2%) for diabetes, 0 for BMI, 12 (0.3%) for systolic blood pressure and 0 for APOE genotype.
We found that a doubling of SII was associated with a 1.2% higher serum Aβ42, albeit not statistically significant (95% confidence interval [95% CI] −0.005% to 2.3%, P = .051) and a 1.8% higher serum Aβ40 (95% CI 0.9 to 2.7%, P = < .001). Estimates were even higher for a doubling of GLR, leading to a 1.9% higher serum Aβ42 (95% CI 0.4 to 3.3%, P = .010) and 3.2% higher serum Aβ40 (95% CI 2.0 to 4.3%, P = < .001). The Aβ42/40 ratio was lower with a doubling in GLR only (−1.2%, 95% CI −2.3 to −0.1%, P = .028). Only a doubling in PLR was significantly associated with lower total tau (−3.6%, 95% CI −5.5% to −1.7%, P = < .001). NfL was higher with all innate immunity phenotypes, most strongly with a doubling in GLR (4.7%, 95% CI 2.5 to 7.1%, P = < .001) (Table 2 and Table S2). Stratification did not reveal statistically significant differences across strata of APOE carriership, with the effects in the APOE ε3/ε4 or ε4/ε4 stratum being only non‐significantly stronger than in the other strata, especially for NfL (Figure 2 and Table S2). In addition, the associations between all genetic risk scores with lower Aβ42/40 ratio became lower in participants with granulocyte counts higher than the median (mean difference = −0.18 [95% CI −0.23 to −0.13, P = < .001] for the overall AD genetic risk score, mean difference = −0.06 [95% CI −0.11 to −0.02, P = .007] for the overall AD genetic risk score excluding APOE, and mean difference = −0.05 [95% CI −0.10 to −0.004, P = .035] for the immune response pathway‐based genetic risk score), yet the interaction remained non‐significant (Table S3). When additionally adjusting for hs‐CRP and creatinine (in a subsample, N = 1341), the associations between GLR and SII with higher Aβ42 and NfL disappeared, the associations between immunity markers with higher Aβ40 were borderline significant, and the associations with lower Aβ42/40 ratio and lower total tau persisted (Table S4). Higher hs‐CRP was significantly associated with a higher Aβ40 and higher total tau (Table S5).
TABLE 2.
Associations between serum markers of immunity and plasma biomarkers of AD‐related brain pathology
| Percentage change in Aβ, total tau or NFL, 95% CI | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Per doubling in serum markers of immunity | Aβ42 | P | Aβ40 | P | Aβ42/40 ratio | P | Tau | P | NFL | P |
| Model I | ||||||||||
| GLR a | 1.6 (0.2; 3.1) | 0.025 | 3.2 (2.1; 4.3) | <0.001 | −1.5 (−2.6; −0.4) | 0.008 | 0.1 (−1.8; 2.0) | <0.001 | 4.9 (2.5; 7.2) | <.001 |
| PLR | −0.7 (‐2.2; 0.8) | 0.351 | −0.2 (−1.4; 0.9) | 0.688 | −0.5 (−1.6; 0.7) | <0.001 | −4.4 (−6.3; −2.5) | <0.001 | 2.9 (0.5; 5.3) | .018 |
| SII | 1.0 (−0.1; 2.2) | 0.078 | 1.8 (0.9; 2.7) | <0.001 | −0.8 (−1.7; 0.1) | 0.093 | −0.4 (−1.9; 1.1) | 0.590 | 3.3 (1.5; 5.2) | <.001 |
| Model II | ||||||||||
| GLR a | 1.9 (0.4; 3.3) | 0.010 | 3.2 (2.0; 4.3) | <0.001 | −1.2 (−2.3; −0.1) | 0.028 | −0.1 (−1.9; 1.8) | 0.927 | 4.7 (2.5; 7.1) | <.001 |
| PLR | −0.8 (−2.2; 0.7) | 0.314 | −0.2 (−1.3; 1.0) | 0.790 | −0.6 (−1.8; 0.6) | 0.309 | −3.6 (−5.5; −1.7) | <0.001 | 1.5 (−0.8; 3.9) | .205 |
| SII | 1.2 (0.00; 2.3) | 0.051 | 1.8 (0.9; 2.7) | <0.001 | −0.6 (−1.5; 0.3) | 0.190 | −0.5 (−2.0; 1.0) | 0.539 | 2.9 (1.1; 4.8) | .002 |
Model I adjusted for age, sex, study cohort, and batch effects. Model II adjusted for age, sex, study cohort, smoking, diabetes, BMI, platelets, systolic blood pressure, APOE ε4, and batch effects. The GLR, PLR, and SII reflect the balance between innate and adaptive immunity, with higher markers indicating an imbalance towards higher innate immunity. Abbreviations: Aβ40, amyloid‐beta 40; Aβ42, amyloid‐beta 42; Aβ42/40 ratio, amyloid‐beta 42‐to‐40 ratio; CI, confidence interval; GLR, granulocyte‐to‐lymphocyte ratio; Ln, natural logarithmic transformation; NFL, neurofilament light chain; PLR, platelet‐to‐lymphocyte ratio; SII, systemic immune‐inflammation index; Tau, total tau.
Additional adjustment for platelet count. Results are considered statistically significant if the P‐value is below .05/(6×5) = .002.
FIGURE 2.
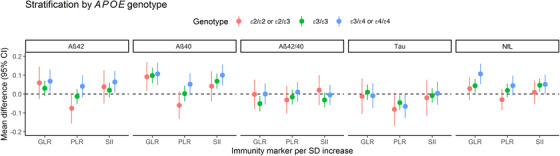
Associations between serum markers of immunity and plasma biomarkers of AD‐related brain pathology. Model adjusted for age, sex, study cohort, smoking, diabetes, BMI, platelets, systolic blood pressure, APOE ε4, and batch effects. Additional adjustment for platelet count when analyzing Ln(GLR). All phenotypical markers were Ln(transformed) except for Aβ40. The GLR, PLR, and SII reflect the balance between innate and adaptive immunity, with higher markers indicating an imbalance towards higher innate immunity. Abbreviations: Aβ40, amyloid‐beta 40; Aβ42, amyloid‐beta 42; Aβ42/40 ratio, amyloid‐beta 42‐to‐40 ratio; CI, confidence interval; ε2/ε2 or ε2/ε3, apolipoprotein Eε2/ε2 genotype; ε3/ε3, apolipoprotein Eε3/ε3 genotype; ε3/ε4 or ε4/ε4, apolipoprotein Eε3/ε4 or ε4/ε4 genotype; GLR, granulocyte‐to‐lymphocyte ratio; Ln, natural logarithmic transformation; NFL, neurofilament light chain; PLR, platelet‐to‐lymphocyte ratio; SD, standard deviation; SII, systemic immune‐inflammation index
Furthermore, we found that a standard deviation (SD) increase in the overall AD genetic risk score including APOE ε4 was associated with higher GLR (mean difference in Ln[GLR] [SD] = 0.024, 95% CI 0.002 to 0.046, P = .032) and significantly with lower Aβ42 and Aβ42/40 ratio (mean difference in Aβ42 [SD] = −0.129, 95% CI −0.162 to −0.095, P = < .001, and Ln[Aβ42/40] [SD] = −0.157, 95% CI −0.190 to −0.124, P = < .001, respectively). These associations were mainly driven by the APOE ε4 variant (Figure 3). The genetic risk score reflecting the immune response showed a suggestive association with lower Aβ42/40 ratio (mean difference = −0.038, 95% CI −0.070 to −0.006, P = .020), but not with the serum markers of innate immunity (Figure 3 and Table S6). We found that APOE ε2 carriers displayed lower serum markers of innate immunity levels compared to ε3/ε3, while these markers were elevated in APOE ε4 (Figure 4 and Table S7). Mean levels of hs‐CRP and creatinine levels within the different APOE genotypes are shown in Table S8.
FIGURE 3.
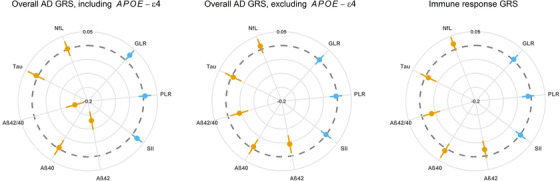
Associations of genetic risk scores reflecting AD including and excluding APOE ε4 and immune response with plasma biomarkers of AD‐related brain pathology and serum markers of immunity. Model adjusted for age, sex, and study cohort. GRS are per SD increase. All phenotypical markers were Ln(transformed) except for Aβ40. Gray dotted circle indicates the null with associations becoming increasingly negative towards the center of the circle. Orange indicates the plasma biomarkers of AD‐related brain pathology while blue indicates the immunity phenotypes. The GLR, PLR, and SII reflect the balance between innate and adaptive immunity, with higher markers indicating an imbalance towards higher innate immunity. Abbreviations: AD, Alzheimer's disease; Aβ40, amyloid‐beta 40; Aβ42, amyloid‐beta 42; Aβ42/40, amyloid‐beta 42‐to‐40 ratio; GLR, granulocyte‐to‐lymphocyte ratio; GRS, genetic risk score; Ln, natural logarithmic transformation; NFL, neurofilament light chain; PLR, platelet‐to‐lymphocyte ratio; SII, systemic immune‐inflammation index
FIGURE 4.
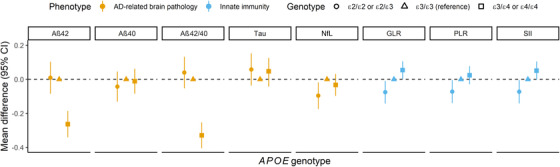
Associations of APOE genotypes with plasma biomarkers of AD‐related brain pathology and serum markers of immunity. Adjusted for age, sex, and study cohort. All phenotypical markers were Ln(transformed) except for Aβ40. The GLR, PLR, and SII reflect the balance between innate and adaptive immunity, with higher markers indicating an imbalance towards higher innate immunity. Abbreviations: AD, Alzheimer's disease; APOE, apolipoprotein E; Aβ40, amyloid‐beta 40; Aβ42, amyloid‐beta 42; Aβ42/40, amyloid‐beta 42‐to‐40 ratio; CI, confidence interval; ε2/ε2 or ε2/ε3, apolipoprotein Eε2/ε2 genotype; ε3/ε3, apolipoprotein Eε3/ε3 genotype; ε3/ε4 or ε4/ε4, apolipoprotein Eε3/ε4 or ε4/ε4 genotype; GLR, granulocyte‐to‐lymphocyte ratio; GRS, genetic risk score; Ln, natural log transformation; NFL, neurofilament light chain; SD, standard deviation; SII, systemic immune inflammation index
4. DISCUSSION
In this study, we found associations between higher levels of GLR, PLR, and SII reflecting higher innate immunity, with higher Aβ42 and 40, lower Aβ42/40 ratio, lower total tau, and higher NfL in plasma. In APOE ε4 carriers, these associations were even stronger. The overall genetic risk score including APOE ε4 was associated with higher GLR and SII and with lower Aβ42 and Aβ42/40 ratio. These effects were mainly driven by the APOE ε4 variant. The genetic risk score reflecting the immune response was associated with a lower Aβ42/40 ratio, but not with the serum immunity markers. Furthermore, we found that APOE ε2 carriers displayed lower serum markers of innate immunity compared to APOE ε3/ε3, while these markers were elevated in APOE ε4 carriers.
Interestingly, we found that higher innate immunity was associated with higher serum Aβ42, but with even higher Aβ40. The 40‐residue peptide represents the most abundant Aβ isoform in the brain, 32 while the 42‐residue shows a significant increase with certain forms of AD. Our findings are concordant with the notion that Aβ functions as an antimicrobial peptide (AMP), because the physiochemical and biological properties previously reported for Aβ are similar to those of AMPs. In addition, experiments have shown that Aβ is active against at least eight common and clinically relevant microorganisms. 33 Activity was isoform‐specific for six organisms with Aβ42 showing greater potency compared to Aβ40. 33 Taking all evidence together, we propose that APOE ε4 carriers display stronger innate immune responses, and thus produce more/excess Aβ in response to pathogens. Of the two isoforms, Aβ42 will aggregate in the brain (possibly due to its larger size) while Aβ40 will not aggregate (or to a lesser extent) and as a consequence will be higher in the serum. Further study is needed to confirm this hypothesis.
Furthermore, we found that both higher serum markers of innate immunity and the genetic risk score reflecting the immune response associate with the lower serum Aβ42/40 ratio. APOE ε4 carriership, which is the major genetic risk factor for AD, also displays a lower serum Aβ42/40 ratio as well as lower Aβ42. In line with this, a meta‐analysis of prospective cohort studies has shown that lower serum Aβ40, and even lower Aβ42 and consequently lower Aβ42/40 ratio, lead to higher AD risk. 34 Previous reports of associations of low plasma Aβ42/40 ratio with increased amyloid brain uptake, as measured by Pittsburgh compound B (PiB) positron emission tomography (PET) scan, support the notion that lower plasma Aβ reflects the aggregation of Aβ in the brain. 35 , 36 , 37 , 38 In this context, our finding that APOE ε4 carriers have higher activity of innate immunity compared to ε3/ε3, while we see the opposite in APOE ε2 carriers suggests that APOE ε4 might have an overactive innate immune response. Our results support experimental studies showing the capacity of APOE to modulate inflammation. Indeed, in healthy humans challenged with intravenous lipopolysacharide (LPS) infusion, ε4 carriers demonstrated significantly higher elevation of body temperature and plasma tumor necrosis factor levels than ε4 non‐carriers. 39 In this same study, when whole blood isolates from human subjects were stimulated ex‐vivo with Toll‐like receptor ligands, increased production of a wide panel of cytokines and chemokines was observed in blood from ε4+ donors compared with ε4−donors. A higher immune response associated with the ε4 allele is also observed in human APOE‐targeted replacement mice and in cultured microglia and/or macrophages upon LPS stimulation. 39 , 40 , 41 How APOE achieves this is not yet well understood. 8 Evidence shows that lipid rafts play an essential role in immune activation by serving as platforms for signaling complexes. 42 APOE ε4 is reported to be less effective than APOE ε3 in inducing cholesterol efflux from macrophages, 43 which leads to cholesterol accumulation on cell membranes. 39 , 43 This mechanism has been proposed to explain the higher immune reactivity associated with APOE ε4, 8 but additional studies are needed to explore this or other potential mechanisms further.
In addition to the role of immune response pathways in Aβ, our data show that higher innate immunity is related to lower plasma total tau. Others assessed the effect of microglial activation on tau pathogenesis, where they found that microglial activation is shown to precede tau pathology in a tauopathy mouse model (Yoshiyama et al. 2007) and administering an immunosuppressant drug FK506 from an early age drastically reduces tau pathology. 44 According to previous studies, higher plasma tau is associated with AD dementia, 16 , 45 although correlations were weak 45 and non‐linear (J‐shape), 16 making the role of immunity in tau elusive. Future studies are warranted with the same crude markers of the immune system in relation to phospho tau.
The associations between immunity markers with higher Aβ42 and NfL disappeared when additionally adjusting for hs‐CRP and creatinine. There could be two explanations: (1) This analysis was performed within a subsample of the total population, impeding comparison of the results in the total population and decreasing the power of the analysis, or (2) We found that higher hs‐CRP was also associated with higher NfL, albeit not statistically significant, suggesting that hs‐CRP could be acting as a mediator.
Our result that the immune GRS was not related to the immunity biomarker levels is surprising, especially since several immune genes are key players for innate immunity, including TREM2. 46 TREM2 overexpression is thought to enhance microglial phagocytotic capacity, but transcription analyses show mixed microglial activation patterns with suppression of certain disease‐associated microglia (DAM) genes, but further activation patterns of other DAM genes, 47 making the role of TREM2 in AD unclear. Further studies are needed to identify the precise gene signatures of microglia that mediate pathology‐ and neurodegeneration‐associated sterile inflammation.
Our study has several limitations. First, ours is a cross‐sectional study, limiting our ability to draw causal inferences. Longitudinal cohort studies are required to confirm our findings. Second, we were limited to studying crude markers of the immune system in this population‐based setting. These types of measures have been associated with other outcomes such as those related to cancer, pancreatitis, and many other responses. 48 These ratios could be proxies for different systemic inflammatory responses that affect the mobilization of bone marrow‐derived cells and the egress of leukocytes into tissue. 49 It could also be related to immunosenescence and this might explain the association with NfL because it also increases with age. 50 Third, we were similarly limited to crude markers of AD‐related brain pathology which are not diagnostic of AD, especially since the ATN‐classification includes AD biomarkers as measured by PET, structural MRI or CSF and we do not have phospho tau measured. However, plasma and CSF levels of these biomarkers are strongly correlated. 51 , 52 , 53 Also NfL is a global biomarker and not specific for AD. Fourth, we were unable to categorize the age groups into early and late onset AD, due to the low number of early onset AD patients within our population‐based cohort. It would be helpful to categorize the biomarkers according to early and late onset AD in future studies, because Aβ has a better correlation in early onset AD. Early onset AD differs from late onset AD not only with respect to genetic predisposition and pathology but also in relation to the clinical outcome and the natural course. Only 11% of early AD patients have familial mutations with APP, PSEN1 and PSEN2. For example, patients with early AD have a greater burden in the precuneus and parietal lobes and to a lesser extent in the frontal lobes. 54 Fifth, we did not offer replication data. We hope that our study provides a stimulus for other cohort studies to replicate our findings. Lastly, our study contains predominantly Caucasians, which limits its external generalizability. Further study in other ethnicities is needed to identify potential ethnic differences.
In summary, in this population‐based study we showed that higher innate immunity, as reflected by higher levels of serum GLR, PLR, and SII, were associated with higher plasma Aβ42, 40, and NfL, and with lower Aβ42/40 and total tau. Furthermore, APOE ε2 carriers displayed lower serum markers of innate immunity levels compared to ε3/ε3, while these markers were elevated in APOE ε4. Knowledge of the potential pro‐inflammatory role of APOE ε4 should encourage future studies to find ways to scale down innate immune responses in APOE ε4 carriers to limit AD‐related brain pathology in order to prevent AD.
CONFLICTS OF INTEREST
None.
AUTHOR CONTRIBUTIONS
| Participants | Affiliated institutions |
|---|---|
| Lana Fani | Epidemiology, Erasmus MC University Medical Center, Rotterdam, the Netherlands |
| Shahzad Ahmad | Epidemiology, Erasmus MC University Medical Center, Rotterdam, the Netherlands |
| Division of Systems Biomedicine and Pharmacology, Leiden Academic Centre for Drug Research, Leiden University, Leiden, the Netherlands. | |
| M. Kamran Ikram | Epidemiology, Erasmus MC University Medical Center, Rotterdam, the Netherlands |
| Neurology, Erasmus MC University Medical Center, Rotterdam, the Netherlands | |
| Mohsen Ghanbari | Epidemiology, Erasmus MC University Medical Center, Rotterdam, the Netherlands |
| M. Arfan Ikram | Epidemiology, Erasmus MC University Medical Center, Rotterdam, the Netherlands |
All authors have made a substantial intellectual contribution to the conception and design of the study (MKI, MAI, LF, SA), acquisition of data (MKI, LF, MG), analysis and interpretation of data (LF, MKI, MAI), drafting the manuscript (LF), or drafting a significant portion of the manuscript or figures (LF, MAI). All authors approved the final version of the manuscript for publication. MAI had full access to the data in the study and takes responsibility for data integrity and accuracy of data analysis.
Supporting information
Supplementary information
ACKNOWLEDGMENTS
We gratefully acknowledge the study participants of the Ommoord district and their general practitioners and pharmacists for their devotion in contributing to the Rotterdam Study. We also thank all staff who facilitated assessment of participants in the Rotterdam Study throughout the years.
This work was supported by the European Union's Horizon 2020 research and innovation program (grant number 667375; “CoSTREAM”); the Erasmus Medical Center and Erasmus University Rotterdam; the Netherlands Organization for Scientific Research (NWO; grant numbers 948‐00‐010, 918‐46‐615); the Netherlands Organization for Health Research and Development (ZonMw); the Research Institute for Diseases in the Elderly (RIDE); the Ministry of Education, Culture and Science; the Ministry of Health, Welfare and Sports; the European Commission (DG XII); and the Municipality of Rotterdam.
This study was partly funded by ZonMw Memorabel (grant number 73305095005) and Alzheimer Nederland through the Netherlands Consortium of Dementia Cohorts (NCDC) in the context of Deltaplan Dementie. Further funding was obtained from the Netherlands CardioVascular Research Initiative, the Dutch Heart Foundation (CVON 2018‐28 Heart Brain Connection Cross‐roads), Dutch Federation of University Medical Centres, the Netherlands Organisation for Health Research and Development, and the Royal Netherlands Academy of Sciences.
The funding source had no role in study design, collection, analysis, interpretation of data, writing of the report or decision to submit the article for publication.
Fani L, Ahmad S, Ikram MK, Ghanbari M, Ikram MA. Immunity and amyloid‐beta, total tau and neurofilament light chain: findings from a community‐based cohort study. Alzheimer's Dement. 2021;17:446–456. 10.1002/alz.12212
REFERENCES
- 1. Strittmatter W, Saunders AM, Schmechel D, et al. Apolipoprotein E: high‐avidity binding to beta‐amyloid and increased frequency of type 4 allele in late‐onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:1977‐1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hardy J. The discovery of Alzheimer‐causing mutations in the APP gene and the formulation of the “amyloid cascade hypothesis. FEBS J. 2017;284:1040‐1044. [DOI] [PubMed] [Google Scholar]
- 3. Moir RD, Lathe R, Tanzi RE. The antimicrobial protection hypothesis of Alzheimer's disease. Alzheimers Dement. 2018;14:1602‐1614. [DOI] [PubMed] [Google Scholar]
- 4. Kunkle BW, Grenier‐Boley B, Sims R, et al. Genetic meta‐analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51:414‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van der Willik KD, Fani L, Rizopoulos D, et al. Balance between innate versus adaptive immune system and the risk of dementia: a population‐based cohort study. J Neuroinflammation. 2019;16:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer's disease and kuru plaque amyloid in Creutzfeldt‐Jakob disease. Brain Res. 1991;541:163‐166. [DOI] [PubMed] [Google Scholar]
- 7. Wisniewski T, Frangione B. Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci Lett. 1992;135:235‐238. [DOI] [PubMed] [Google Scholar]
- 8. Shi Y, Holtzman DM. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol. 2018;18:759‐772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bolós M, Llorens‐Martín M, Jurado‐Arjona J, Hernández F, Rábano A, Avila J. Direct evidence of internalization of tau by microglia in vitro and in vivo. J Alzheimers Dis. 2016;50:77‐87. [DOI] [PubMed] [Google Scholar]
- 10. Asai H, Ikezu S, Tsunoda S, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18:1584‐1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mócsai A. Diverse novel functions of neutrophils in immunity, inflammation, and beyond. J Exp Med. 2013;210:1283‐1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bonilla FA, Oettgen HC. Adaptive immunity. J Allergy Clin Immunol. 2010;125:S33‐S40. [DOI] [PubMed] [Google Scholar]
- 13. Fest J, Ruiter R, Ikram MA, Voortman T, van Eijck CHJ, Stricker BH. Reference values for white blood‐cell‐based inflammatory markers in the Rotterdam Study: a population‐based prospective cohort study. Sci Rep. 2018;8:10566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jack CR Jr, Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ahmad S, Bannister C, van der Lee SJ, et al. Disentangling the biological pathways involved in early features of Alzheimer's disease in the Rotterdam Study. Alzheimers Dement. 2018;14:848‐857. [DOI] [PubMed] [Google Scholar]
- 16. de Wolf F, Ghanbari M, Licher S, et al. Plasma tau, neurofilament light chain and amyloid‐β levels and risk of dementia; a population‐based cohort study. Brain. 2020;143(4):1220‐1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ikram MA, Brusselle G, Ghanbari M, et al. Objectives, design and main findings until 2020 from the Rotterdam Study. Eur J Epidemiol. 2020;35:483‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McCarthy S, Das S, Kretzschmar W, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48:1279‐1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Das S, Forer L, Schönherr S, et al. Next‐generation genotype imputation service and methods. Nat Genet. 2016;48:1284‐1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Delaneau O, Marchini J. The 1000 Genomes Project Consortium. Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel. Nat Commun. 2014;5:3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome‐wide association studies through pre‐phasing. Nat Genet. 2012;44:955‐959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ikram MA, Brusselle GGO, Murad SD, et al. The Rotterdam Study: 2018 update on objectives, design and main results. Eur J Epidemiol. 2017;32:807‐850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rissin DM, Fournier DR, Piech T, et al. Simultaneous detection of single molecules and singulated ensembles of molecules enables immunoassays with broad dynamic range. Anal Chem. 2011;83:2279‐2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rohrer JD, Woollacott IO, Dick KM, et al. Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia. Neurology. 2016;87:1329‐1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013;368:107‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sims R, van der Lee SJ, Naj AC, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial‐mediated innate immunity in Alzheimer's disease. Nat Genet. 2017;49:1373‐1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dourlen P, Kilinc D, Malmanche N, Chapuis J, Lambert JC. The new genetic landscape of Alzheimer's disease: from amyloid cascade to genetically driven synaptic failure hypothesis?. Acta Neuropathol. 2019;138(2):221‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. International Genomics of Alzheimer's Disease Consortium (IGAP). Convergent genetic and expression data implicate immunity in Alzheimer's disease. Alzheimers Dement. 2015;11:658‐671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guerreiro R, Bras J, Hardy J. SnapShot: genetics of Alzheimer's disease. Cell. 2013;155:968‐e1. [DOI] [PubMed] [Google Scholar]
- 30. Toledo JB, Vanderstichele H, Figurski M, et al. Factors affecting Aβ plasma levels and their utility as biomarkers in ADNI. Acta Neuropathol. 2011;122:401‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Diabetes mellitus . Report of a WHO Study Group. World Health Organization technical report series. 1985;727:1‐113. [PubMed] [Google Scholar]
- 32. Mori H, Takio K, Ogawara M, Selkoe DJ. Mass spectrometry of purified amyloid beta protein in Alzheimer's disease. J Biol Chem. 1992;267:17082‐17086. [PubMed] [Google Scholar]
- 33. Soscia SJ, Kirby JE, Washicosky KJ, et al. The Alzheimer's disease‐associated amyloid beta‐protein is an antimicrobial peptide. PLoS One. 2010;5:e9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chouraki V, Beiser A, Younkin L, et al. Plasma amyloid‐β and risk of Alzheimer's disease in the Framingham Heart Study. Alzheimers Dement. 2015;11:249‐257.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Devanand DP, Schupf N, Stern Y, et al. Plasma Aβ and PET PiB binding are inversely related in mild cognitive impairment. Neurology. 2011;77:125‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rembach A, Watt AD, Wilson WJ, et al. Plasma amyloid‐β levels are significantly associated with a transition toward Alzheimer's disease as measured by cognitive decline and change in neocortical amyloid burden. J Alzheimers Dis. 2014;40:95‐104. [DOI] [PubMed] [Google Scholar]
- 37. Swaminathan S, Risacher SL, Yoder KK, et al. Association of plasma and cortical amyloid beta is modulated by APOE ε4 status. Alzheimers Dement. 2014;10:e9‐e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Risacher SL, Fandos N, Romero J, et al. Plasma amyloid beta levels are associated with cerebral amyloid and tau deposition. Alzheimers Dement (Amst). 2019;11:510‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gale SC, Gao L, Mikacenic C, et al. APOε4 is associated with enhanced in vivo innate immune responses in human subjects. J Allergy Clin Immunol. 2014;134:127‐134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vitek MP, Brown CM, Colton CA. APOE genotype‐specific differences in the innate immune response. Neurobiol Aging. 2009;30:1350‐1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhu Y, Nwabuisi‐Heath E, Dumanis SB, et al. APOE genotype alters glial activation and loss of synaptic markers in mice. Glia. 2012;60:559‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fessler MB, Parks JS. Intracellular lipid flux and membrane microdomains as organizing principles in inflammatory cell signaling. J Immunol. 2011;187:1529‐1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Okoro EU, Zhao Y, Guo Z, Zhou L, Lin X, Yang H. Apolipoprotein E4 is deficient in inducing macrophage ABCA1 expression and stimulating the Sp1 signaling pathway. PLoS One. 2012;7:e44430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yoshiyama Y, Higuchi M, Zhang B, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337‐351. [DOI] [PubMed] [Google Scholar]
- 45. Mattsson N, Zetterberg H, Janelidze S, et al. Plasma tau in Alzheimer disease. Neurology. 2016;87:1827‐1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Boutajangout A, Wisniewski T. The innate immune system in Alzheimer's disease. Int J Cell Biol. 2013;2013:576383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee CYD, Daggett A, Gu X, et al. Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer's disease models. Neuron. 2018;97:1032‐1048.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu H, Tabuchi T, Takemura A, et al. The granulocyte/lymphocyte ratio as an independent predictor of tumour growth, metastasis and progression: its clinical applications. Mol Med Rep. 2008;1:699‐704. [DOI] [PubMed] [Google Scholar]
- 49. Furze RC, Rankin SM. Neutrophil mobilization and clearance in the bone marrow. Immunology. 2008;125:281‐288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Martin C, Burdon PC, Bridger G, Gutierrez‐Ramos JC, Williams TJ, Rankin SM. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity. 2003;19:583‐593. [DOI] [PubMed] [Google Scholar]
- 51. Hanon O, Vidal JS, Lehmann S, et al. Plasma amyloid levels within the Alzheimer's process and correlations with central biomarkers. Alzheimers Dement. 2018;14:858‐868. [DOI] [PubMed] [Google Scholar]
- 52. Kovacs GG, Andreasson U, Liman V, et al. Plasma and cerebrospinal fluid tau and neurofilament concentrations in rapidly progressive neurological syndromes: a neuropathology‐based cohort. Eur J Neurol. 2017;24:1326‐e77. [DOI] [PubMed] [Google Scholar]
- 53. Mielke MM, Syrjanen JA, Blennow K, et al. Plasma and CSF neurofilament light: relation to longitudinal neuroimaging and cognitive measures. Neurology. 2019;93:e252‐e260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bekris LM, Yu CE, Bird TD, Tsuang DW. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol. 2010;23:213‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information