

Summary
As a young bacteriologist just launching my career during the early days of the ‘microbial revolution’ in the 1980s, I was fortunate to participate in some early discoveries, and collaborate in the development of cross‐disciplinary methods now commonly referred to as "metagenomics". My early scientific career focused on applying phylogenetic and genomic approaches to characterize ‘wild’ bacteria, archaea and viruses in their natural habitats, with an emphasis on marine systems. These central interests have not changed very much for me over the past three decades, but knowledge, methodological advances and new theoretical perspectives about the microbial world certainly have. In this invited ‘How we did it’ perspective, I trace some of the trajectories of my lab's collective efforts over the years, including phylogenetic surveys of microbial assemblages in marine plankton and sediments, development of microbial community gene‐ and genome‐enabled surveys, and application of genome‐guided, cultivation‐independent functional characterization of novel enzymes, pathways and their relationships to in situ biogeochemistry. Throughout this short review, I attempt to acknowledge, all the mentors, students, postdocs and collaborators who enabled this research. Inevitably, a brief autobiographical review like this cannot be fully comprehensive, so sincere apologies to any of my great colleagues who are not explicitly mentioned herein. I salute you all as well!
Introduction
The past 30 years of microbial biology have witnessed remarkable and transformative advances across its many diverse sub‐disciplines. Perspectives that have particularly advanced and matured in the context of microbial biology in the past three decades include:
Evolutionary perspectives ‐ The flowering of molecular phylogenetics and later, phylogenomics, that facilitate quantitative inference of the evolutionary relationships among all life forms, heralding a new age of comparative genomic evolutionary biology.
Ecological perspectives ‐ Community structure and spatiotemporal variability of natural microbial communities can now be quantitatively assessed in relatively unbiased ways, ushering in a new era in microbial ecology and microbiome science.
Population biology perspectives ‐ Culture‐based and cultivation‐independent population genetics and genomics now provide new mechanistic insight into the origins of genomic variability, microheterogeneity, gene exchange and speciation in microbial populations.
Biogeochemical perspectives ‐ The recognition of new microorganisms, genes and biochemical pathways in native microbial communities has led to new understandings of how biogeochemical cycles have evolved and function in natural habitats.
Systems biology perspectives, from genomes to biomes ‐ That the whole (organelle, cell, multicellular organism, population, community and ecosystem) is more than the sum of its parts has long been appreciated. Recent developments now allow more fully exploring the interdependencies of complex, multispecies biological systems at a variety of organizational levels, which inform a new understanding of emergent ecosystem function.
Early days
During my thesis work at the Scripps Institution of Oceanography in the early 1980s, I became keenly aware of difficulties associated with bacterial taxonomic identification ‐ even for bacteria that we could cultivate. I was lucky to participate in undergraduate research with Paul Baumann at UC Davis, in studies of evolutionary relationships among marine bacteria. At that time, Paul was leveraging immunological approaches to measure protein similarity in bacteria, since sequencing techniques were not broadly available then (DeLong et al., 1984). So, I had some knowledge of the current state of bacterial taxonomic and evolutionary approaches when I started at Scripps. My thesis work focused on an impressive collection of deep‐sea pressure‐loving bacteria (then called barophiles, now piezophiles) that my advisor Art Yayanos had assembled over the years (Yayanos, 1995). Some of these bacterial isolates were recovered from depths as great as 10 000 m, requiring hydrostatic growth pressures of >300 atm to grow (Yayanos et al., 1981; Yayanos, 1986, 1995). We found that these piezophilic bacteria displayed a type of ‘homeoviscous adaptation’ in their membrane lipids, presumably counteracting the membrane‐solidifying effects of elevated hydrostatic pressures of the deep sea (DeLong and Yayanos, 1985). We also found that many piezophilic isolates biosynthesized large amounts of polyunsaturated fatty acids (PUFAs) and incorporated them into their membrane lipids, yet another adaptation to deep‐sea low‐temperature and high‐pressure (DeLong and Yayanos, 1986). That bacteria could synthesize PUFAs at all was somewhat controversial, since de novo PUFA biosynthesis was thought to be restricted to select eukaryote groups, in particular, microalgae at the time. It is now broadly recognized, however, that deep‐sea and other psychrophilic bacteria commonly synthesize and incorporate PUFAs in their membrane lipids, using a unique polyketide biosynthetic pathway (Metz et al., 2001; Allen and Bartlett, 2002; Allemann and Allen, 2020).
Despite some progress, when I had finished writing my thesis in 1986, I did not know the precise taxonomic identity of the deep‐sea isolates we would have been working with, which was enormously frustrating. A contemporary student might ask: ‘Why the taxonomic ignorance on organisms you were cultivating successfully in the lab?’ Simply put, standard bacterial chemotaxonomic identification techniques (typically performed at mesophilic temperatures in liquid media or on agar plates) did not perform well, and were not practically applicable, at 2°C and hundreds of atmospheres of hydrostatic pressure. At that time, 5S rRNA sequencing approaches were just becoming available, so Jody Deming and Rita Colwell teamed up with George Fox to sequence the 5S rRNAs of two barophilic bacteria and found that they belonged to the Vibrio‐Photobacterium clade (Deming et al., 1984). That was about all we knew at that time. When I finished my Ph.D. work in 1986 then, I was keenly aware of the challenges of bacterial identification, and I was anxious to explore new ways to identify and characterize naturally occurring microbes. After completing my thesis dissertation, I was very fortunate to land a postdoc in Norm Pace's group at Indiana University, Bloomington.
Cultivation‐independent phylogenetic surveys of natural marine microbial communities
The 1980s marked an authentic ‘paradigm shift’ in the true Kuhnian sense (Kuhn, 1962) for biology and microbiology writ large. During that time, Carl Woese and Norman Pace were championing new approaches that revealed for the very first time, the global evolutionary and ecological details of the natural microbial world. Thanks to Woese, the quantitative evolutionary relationships of ALL cellular life on Earth could be determined, leading him and his colleagues to define a whole new Domain of life, the Archaea (Woese and Fox, 1977; Woese, 1987; Woese et al., 1990; Pace et al., 2012). Soon thereafter, Pace realized that quantitative macromolecular phylogenetic techniques could provide a solution to a frustrating, long‐standing conundrum in microbial ecology, popularly termed ‘the great plate count anomaly’ (Staley and Konopka, 1985). The problem then (and still today) was that only a small fraction of naturally occurring microbial species could be readily isolated and studied in pure culture in the laboratory. And even if most bacteria could be cultivated in this way, this approach overlooks a significant amount of in situ community processes and ecological insights. With singular insights and extraordinary energy, Pace and colleagues ushered in rRNA‐based cultivation‐independent phylogenetic survey methods, first by directly sequencing 5S rRNAs (Stahl et al., 1984, 1985), and later via small and large subunit rRNA sequencing (Lane et al., 1985; Pace et al., 1985; Olsen et al., 1986; Pace et al., 1986). These early efforts fundamentally changed practice, knowledge and theory in microbial biology writ large (Pace, 1997; Hugenholtz et al., 1998; DeLong and Pace, 2001; Pace et al., 2012). Indeed, these new approaches paved the way for the development of comparative microbial genomics and metagenomics that were to blossom a decade later, thanks to improved DNA sequencing technologies (Pace, 2018).
The Pace lab was an exciting place to be in 1986. At that time, the central scientific focus of Norm's lab was not molecular microbial ecology. Rather, most Pace lab folks back then were studying catalytic RNAs (ribozymes). Pace's activities had been pivotal in studies of the ribozyme RNAase P, and in developing a theory about the early evolution of RNA catalysis (Gardiner and Pace, 1980; Marsh and Pace, 1985; Pace and Marsh, 1985; Reich et al., 1986; Waugh and Pace, 1986). In parallel, Norm's lab also advanced state‐of‐the‐art of rRNA sequencing and molecular phylogenetics, including the development of direct 16S rRNA sequencing, using a Sanger dideoxynucleotide termination approach, enabled by the use of reverse transcriptase (Lane et al., 1985; Field et al., 1988; Lane et al., 1988; Giovannoni et al., 1988b). The skillsets and perspectives of all the RNA chemists, geneticists, microbiologists and biochemists working in the Pace group back then provided a rich backdrop for Norm's other professional ‘hobby’ (besides caving!). That ‘hobby’ (Norm's term!) involved developing approaches in ‘molecular microbial ecology’, the nascent merger between molecular phylogenetics and microbial ecology. One of Norm's favourite sayings (among others) in those days was ‘Symbiosis gets more done’! So many visitors passed through Norm's lab for short stays to learn the Pace lab's new rRNA sequencing approaches, among them Colleen Cavanaugh, Dan Distel and Jed Fuhrman.
Several key papers had just been published in the mid‐1980s that introduced Norm's cultivation‐independent, phylogenetically oriented microbial survey approaches (Pace et al., 1985; Olsen et al., 1986; Pace et al., 1986; Pace, 1997). And these methods had recently been field‐tested via direct 5S rRNA sequencing, by Dave Stahl and colleague's characterization of hydrothermal vent tubeworm symbionts, and Yellowstone hotspring microbial communities (Stahl et al., 1984, 1985). That set the stage for the group of us that worked in what we jokingly called ‘the back lab’ (the Pace lab low rent district), trying to further develop and apply molecular ecology methods and approaches. Residents in the ‘back lab’ then included postdocs Steve Giovannoni, Sean Turner, myself and Tom Schmidt, and graduate students Gene Wickham, Sue Barns and Esther Angert, among others. Phil Hugenholtz joined the group sometime after I had left.
After joining Norm's lab, I quickly got started on two ongoing projects. One was aimed at phylogenetic identification of single microbial cells, using rRNA‐targeted oligodeoxynucleotide probes. Giovannoni et al. (1988a) had demonstrated this was feasible using domain‐specific radiolabeled oligodeoxynucleotide probes, coupled with microautoradiography. My task was to extend the approach using fluorescently labelled oligodeoxynucleotide probes and identify single cells using epifluorescence microscopy. At that time, this required synthesizing one's own fluorescent probes, purifying them and testing their efficacy. Using that approach with domain‐specific and species‐specific probes, we demonstrated that ‘phylogenetic stains’ (rRNA‐targeted, taxon‐specific fluorescently labelled oligodeoxynucleotide probes) were effective in identifying individual cell types using epifluorescence microscopy (Fig. 1; DeLong et al., 1989). We also demonstrated using quantitative confocal microscopy that the fluorescent signal strength from individual cells was proportional to their ribosomal RNA content and growth rate (Fig. 1), an important consideration for later improvements to the technique (Amann and Fuchs, 2008). While the ‘phylogenetic stains’ moniker did not quite stick, the general method certainly did, and the approach (with various improvements and modifications) has now found widespread use in microbial ecology and beyond.
Fig 1.

Phylogenetic stains. Fluorescent in situ hybridization (FISH) using rRNA‐targeted, fluor‐labelled oligonucleotide probes complementary to rRNA sequences to identify single microbial cells. DeLong et al. (1989).
A. Bacillus megaterium and Saccharomyces cerevisiae hybridized with a fluorescein labelled universal probe and a rhodamine labelled eukaryote probe simultaneously. Cells in the same field were visualized with phase contrast (left), and epifluorescence using fluorescein filters (centre), or rhodamine filters (right).
B. Bacillus megaterium and Saccharomyces cerevisiae hybridized with a fluorescein labelled eukaryote probe and a rhodamine labelled bacterial probe simultaneously, and visualized with phase contrast (left), or epifluorescence photographed using a double exposure of fluorescein and rhodamine filter sets (right).
C. Single‐cell fluorescence intensity of individual E. coli cells labelled with a fluorescein‐labelled universal probe as a function of growth (solid circles). RNA content (solid triangle) and RNA/DNA per cell (open triangle) versus growth rate are also shown. The open circles show the fluorescence intensity of cells hybridized with a fluorescein‐labelled, negative control probe that does not bind to rRNA. DeLong et al. (1989). [Color figure can be viewed at wileyonlinelibrary.com]
Another Pace lab ONR‐funded lab project at the time was aimed at phylogenetic identification of uncultivated oligotrophic marine picoplankton. Steve Giovannoni initially worked on this project in the Pace lab, before leaving for his new post at Oregon State University. There he applied the polymerase chain reaction (PCR) to amplify rRNA genes from Sargasso Sea picoplankton DNA, leading to the discovery of the SAR11 clade of Alphaproteobacteria (Giovannoni et al., 1990). Meanwhile back in the Pace lab, Tom Schmidt and I continued to test a more ‘classical’ approach that involved cloning and sequencing of large DNA fragments into bacteriophage lambda vectors (Fig. 2). This approach required large amounts of DNA so, at Dave Karl's invitation, we spent a good amount of time on the second Hawaii Ocean Time‐series cruise (https://hahana.soest.hawaii.edu/hot/) in December 1988, concentrating cells from >8000 L of seawater via tangential flow filtration (See Supplemental movie clip). The concentrated 560 mg (wet weight) of picoplankton biomass yielded ~2 mg of mixed‐population DNA (Schmidt et al., 1991). Subsequent restriction enzyme partial digestion and density gradient size fractionation yielded DNA fragments of 10–20 kbp that were ligated into the phage lambda cloning vector, yielding ~10 million recombinant clones. At the time, this type of molecular cloning was a very finicky process and it was largely a do‐it‐yourself venture. After several failed attempts, I was ecstatic when my first positive library results finally came in, as phage plaques on an E. coli lawn. Subsequent library screening with radiolabeled rRNA‐targeted probes identified the rRNA gene‐containing lambda clones, which were then selected for 16S rRNA gene sequencing by Tom Schmidt. We were surprised to see that the most abundant bacterial rRNAs from the North Pacific Subtropical Gyre (NPSG) (that now are called Pelagibacter and Prochlorococcus) were closely related to the most abundant groups found in the Sargasso Sea via the PCR approach (Fig. 2; Giovannoni et al., 1990; Schmidt et al., 1991).
Fig 2.

Bacterioplankton phylogenetic analyses via cloning and sequencing of large DNA fragments containing rRNA genes.
A. The methodological flow chart and results are shown.
B. Station ALOHA seawater (left) and the tangential flow retentate of bacterioplankton concentrated ~1000‐fold (right; See Supplementary movie, Suppl. 1).
C. Lambda clone plaque lifts of rRNA‐containing clones hybridized with radiolabeled bacterial (left) or eukaryote (right) rRNA probes.
D. Phylogenetic identity of cloned rRNAs from cyanobacteria.
E. Phylogenetic identity of cloned rRNAs from proteobacteria. [Color figure can be viewed at wileyonlinelibrary.com]
My postdoc in the Pace lab was transformative and set the trajectory of the rest of my professional career. It was clear that the quest to further characterize ‘wild’ microbial assemblages was worth pursuing since there was still so much left to learn. Phylogenetic characterization of natural assemblages obviously had great potential to advance our understanding of microbial ecology. Importantly, I also realized that it was now possible to go much farther than just rRNA‐based phylogenetic analyses. For what we had accomplished already in the late 1980s is now what is more commonly referred to as ‘metagenomics’. Indeed, with a nod to the potential of genomic analyses and ‘chromosome walking’ to characterize uncultivated microbes, we had noted then: ‘The approach used here, analysis of rRNA gene sequences retrieved from a shotgun library of naturally occurring picoplankton DNA, seems the most rigorous way to obtain all representatives of the population. The shotgun library, with 10‐ to 20‐kb inserts, is also a source of other sequences associated with the rRNA genes or other genes of interest.’ (Schmidt et al., 1991). This realization, that the dissection of genomes and functional gene repertoires of naturally occurring microbes was now possible using such approaches, strongly guided my future research efforts (for example, Stein et al., 1996). Somewhat later, the general approaches we had already been applying for a decade to marine microbial assemblages were also applied in soil, and the term ‘metagenome’ was coined (Schmidt et al., 1991; Handelsman et al., 1998).
Setting sail solo – marine snow, Antarctic Archaea and other adventures
After my postdoc in the Pace lab, my career from 1990 to 2004 included time as an Assistant Scientist at Woods Hole Oceanographic Institution, a professor at UC Santa Barbara, and a Senior Scientist at the Monterey Bay Aquarium Research Institute. These were exciting times in environmental microbiology and microbial ecology, during which many discoveries were enabled by technological improvements in PCR, gene cloning, DNA sequencing and stable isotope approaches. I was lucky over those years to work with some very bright, motivated students and postdocs (Fig. 3), whose efforts led to many new discoveries and methodological advances described below.
Fig 3.

DeLong lab group photos.
A. At UC Santa Barbara. Left to right, Ke Ying Wu, Christina Preston, Ramon Massana, Carol Kosman, Paul Fowler, Alison Murray, Trent Taylor, Henry Cittone (Not present for this photo are other critical UCSB team members, including Christa Schleper and Victoria Orphan).
B. At MBARI. Left to right, Christina Preston, Victoria Orphan, Ed DeLong, Marion Leclerc, Oded Béjà, Marcelino Suzuki (Not present for this photo but critical members of the MBARI team include Steve Hallam, Pete Girguis, Trent Taylor and Grieg Steward). [Color figure can be viewed at wileyonlinelibrary.com]
At Woods Hole, realizing that the culture‐independent survey approach provided new opportunities to revisit studies of niche partitioning and microbial biogeochemistry in natural habitats, I began a project investigating marine snow particles. That turned out to be a lot of fun, since collecting marine snow was facilitated by SCUBA diving, an early passion in my youth. After getting research diving certified, I was very lucky to be able to work with some of the very best blue‐water divers and field ecologists at that time: Dave Caron then at WHOI, and Alice Alldredge at UCSB. Their expert guidance fueled my in situ marine snow research. An early hypothesis then proposed that archaeal methanogenesis might be taking place in anoxic pockets of marine snow, which might explain the mysterious marine subsurface methane maximum. But when I set out to find putative methanogenic Archaea within marine snow particles collected in coastal environments (Fig. 2), I recovered just the opposite result! While Archaea in marine snow particles were not evident, high archaeal abundances in the controls, the free‐living coastal picoplankton (DeLong, 1992), were common! And it turned out those coastal Archaea were comprised of two new different types. One fell within the (then) Crenarchaeota (Marine Group I Archaea, now classified within the family Nitrosopumilaceae; (Konneke et al., 2005)), and one in the Euryarchaeota (Marine Group II Archaea, now classified within Poseidoniales; (Rinke et al., 2019)). And these new archaeal phylotypes it turned out were ubiquitous, from coastal seas, to frigid Antarctic waters (Fig. 4), to the deep ocean (DeLong, 1992; Fuhrman et al., 1992; DeLong et al., 1994; Massana et al., 1997; DeLong, 1998; Murray et al., 1998; Murray et al., 1999; Karner et al., 2001). Indeed, one specific crenarchaeal phylotype, now classified within the family Nitrosopumilaceae of archaeal ammonia‐oxidizers (Konneke et al., 2005), occurred as a symbiont in one specific coastal sponge species (Preston et al., 1996; Hallam et al., 2006a; Hallam et al., 2006b). The discovery of Marine Group I and Group II Archaea in marine habitats led to many follow‐on studies in my group at UCSB, which are described in greater detail elsewhere (DeLong, 2020). (The recognition that Marine Group I Archaea are chemoautotrophic ammonia‐oxidizers, is described in an earlier article in this series by Stahl (2020)).
Fig 4.

Antarctic Archaea.
A. The author drilling through sea ice, to sample the seawater below for Archaea.
B. Phylogenetic identification of PCR‐amplified archaeal rRNA genes from Antarctic surface waters (DeLong et al., 1994).
C. Time‐series analyses of archaeal relative abundances in Antarctic surface waters using radiolabeled rRNA‐targeted probes (Murray et al., 1998). [Color figure can be viewed at wileyonlinelibrary.com]
Although analyses of coastal marine snow particles did not provide evidence for archaeal methanogenesis, they did reveal extensive niche partitioning between free‐living versus particle attached marine bacteria. Specifically, the marine snow‐attached bacterial phylotypes were distinct, encompassing groups including Cytophaga, Colwellia, Flavobacteria and Planctomycetes relatives that encompass members well known for copiotrophic and/or surface‐associated lifestyles (Fig. 5; DeLong et al., 1993). Plastid rRNA genes were also amplified by the bacterial primers used and were not surprisingly also abundant on the phytodetrital marine snow. Bulk seawater surrounding the marine snow particles in contrast contained the ‘usual suspects’ ‐ including Pelagibacter relatives and Gammaproteobacteria (DeLong et al., 1993). Working in my lab at UCSB, Johannes Rath confirmed and extended these results in Adriatic marine snow samples in samples collected by Gerhard Herndl (Rath et al., 1998). Similar strategies have now been applied to marine coastal, lake water and deep‐sea particle‐associated microbial assemblages and continue to expand our knowledge of the nuances of niche partitioning and biogeochemistry on particulate organic matter (Crump et al., 1999; Simon et al., 2002; Grossart et al., 2005; Ganesh et al., 2014; Fontanez et al., 2015; Pelve et al., 2017; Boeuf et al., 2019).
Fig 5.

Marine snow sampling.
A. The author blue‐water SCUBA diving and collecting marine snow in syringes.
B. Phylogenetic analysis of free‐living versus marine snow‐attached bacterial populations (DeLong et al., 1993). [Color figure can be viewed at wileyonlinelibrary.com]
Genomic, biogeochemical and biochemical inference of uncultivated marine microbial physiologies and phenotypes
Soon after receiving tenure at UCSB in 1997, I was contacted by a former WHOI graduate student whose thesis committee I had served on, who was then a postdoctoral fellow at the MBARI ‐ Chris Scholin. Chris had asked if I would come to give a seminar and meet folks at MBARI, since they were looking to hire a new Scientist. So, I visited MBARI, one thing led to another, and I ended up spending a wonderful 7 years there with a remarkable group of scientists, engineers, marine technicians, graduate students and postdocs. (MBARI's mission includes the development and operation of in situ instrumentation, autonomous and remotely operated underwater vehicle systems, and research vessels, which continue to provide rich opportunities for its resident scientists, engineers, postdocs and students. Chris Scholin is now MBARI's President and CEO).
At MBARI, I soon got involved in a collaborative project with Peter Brewer, and John Hayes and Kai‐Uwe Hinrichs at WHOI, which aimed to characterize the microbial biogeochemistry at cold‐water methane seeps off the coast of northern California near Eel River. Geologists had known for some time that in situ methane consumption near methane seeps and in anoxic sediments tended to coincide with the disappearance of sulfate. One attractive hypothesis was that in anoxic habitats, anaerobic oxidation of methane (catalysed by postulated ‘reverse methanogens’) was coupled to sulfate reduction mediated by commonly known sulfate‐reducing bacteria. Kai‐Uwe Hinrichs and John Hayes at WHOI, and our team at MBARI, were able to provide the first coupled phylogenetic and compound‐specific stable isotopic evidence that revealed the likely organisms catalysing the oxidation of methane in anoxic marine sediments (Fig. 6). Hinrichs and Hayes data showed that archaeal‐specific lipids extracted from Eel River sediments were highly depleted in 13C (Hinrichs et al., 1999). The only natural carbon source that could account for such 13C depletion was methane itself, suggesting that indeed, Archaea were consuming methane. In the same Eel River sediments, my rRNA‐based phylogenetic data revealed the prevalence of several new Archaeal groups that were only distantly to other known methanogens (Fig. 6; Hinrichs et al., 1999). The most parsimonious explanation of these results was that some archaeal lineages in different clades had evolved to become specialized for either methane production, or for methane consumption. Our results also demonstrated the power of combining molecular isotopic and phylogenetic evidence to infer in situ biogeochemical processes. Follow on studies quickly appeared. Using FISH probes targeting one of the Eel River archaeal groups affiliated with the Methanosarcinales (BA‐2H11, Fig. 6) called ANME‐2 that we had found (Hinrichs et al., 1999; Orphan et al., 2001b), Boetius et al. demonstrated it was tightly associated with sulfate‐reducing bacteria in Hydrate Ridge sediments (Boetius et al., 2000; DeLong, 2000). This supported the notion that consortia of archaeal methane‐consumers working with bacterial sulfate‐reducers catalysed methane oxidation coupled to sulfate reduction in anoxic marine sediments.
Fig 6.

Anaerobic oxidation of methane by consortia of marine Archaea and sulfate‐reducing bacteria.
A. Chromatograms of archaeal lipids from Eel River Basin methane seeps, showing characteristic archaeal lipids that are extremely isotopically depleted in 13C, implying their carbon sources must also be very isotopically light (Hinrichs et al., 1999).
B. Phylogenetic tree of archaeal rRNA genes found in sediments at the Eel River Basin methane seeps. The Eel‐BA‐2H11 group was abundant (Hinrichs et al., 1999) and represents the ANME‐2 lineage (Orphan et al., 2001b). Both ANME‐1 and ANME‐2 are known to be archaea that catalyse the anaerobic oxidation of methane (Hinrichs et al., 1999; Orphan et al., 2000; Orphan et al., 2001a, 2002).
C. Epifluorescence photomicrograph of ANME‐2 methane‐oxidizing archaea (red) surrounded by sulfate‐reducing bacteria (green). (Orphan et al., 2002).
D. The use of FISH‐SIMS to identify archaeal–bacterial methane‐oxidizing consortia microscopically, and then map carbon isotope distributions through the aggregates (Orphan et al., 2001a). The more negative carbon isotopic values map to the location of the ANME‐2 Archaea. [Color figure can be viewed at wileyonlinelibrary.com]
Working in my lab at MBARI, Victoria Orphan soon made pivotal contributions to furthering our understanding of archaeal anaerobic oxidation of methane. Victoria had started in my group as an undergraduate student at UCSB and then continued on as a graduate student with me at MBARI (Fig. 3). Victoria's early thesis work had focused on microbes in hot oil reservoirs (Orphan et al., 2000), but after some cajoling, I managed to convince her that studies of methane seeps might be an interesting addition to her thesis. She quickly followed up on more detailed phylogenetic analyses of methane seep sediment bacterial and archaeal communities that further demonstrated that several distinct lineages of Archaea (ANME‐1 and ANME‐2) were unique to methane seep sites, while most of their putative sulfate‐reducing partners were more widespread (Orphan et al., 2001b). Working in my lab, and collaborating with Kevin McKeegan, Chris House and Kai‐Uwe Hinrichs, Victoria developed a new technique that allowed direct coupling of molecular phylogenetic and isotopic analyses (Orphan et al., 2001a). The method combined serial use of fluorescent in situ hybridization (FISH, aka ‘phylogenetic stains’; DeLong et al., 1989) with secondary ion mass spectrometry, applied to the same cells in a fixed position on a microscope slide. Resulting data from methane seep assemblages directly showed a dramatic 13C isotopic depletion in ANME‐2 archaeal cells, relative to other co‐occurring microbes in method seep sediments, confirming that methane was their primary carbon source (Fig. 6; Orphan et al., 2001a). This was the first application of ion microprobe mass spectrometry for interrogating active microbial cells and demonstrated the feasibility of determining the phylogenetic identity and activity of individual microbial cells. Since then, with nano‐SIMS technological improvements, it has begun to find more widespread application. Using the same approach, Victoria and colleagues showed that the other major archaeal group found at methane seeps, ANME‐1, was also capable of anaerobic methane consumption (Orphan et al., 2002). Steve Hallam and Pete Girguis, MBARI postdocs in my group, also contributed importantly to furthering our understanding of the biochemistry and physiology of the anaerobic oxidation of methane at cold seeps (Girguis et al., 2003; Hallam et al., 2003; Hallam et al., 2004).
Another major focus for us in those years was the development of environmental genomic approaches (a.k.a. eco‐genomics or metagenomics) to better dissect and infer the biochemical and physiological properties of yet‐uncultivated microbes. At USCB I began expanding on approaches for cloning large genomic DNA fragments directly from mixed microbial populations. Jeff Stein and I, working with Hiroaki Shizuya in Mel Simon's lab, applied high fidelity cloning vectors ‐ bacterial artificial chromosomes (BACs) and fosmids (Kim et al., 1996) ‐ to study marine Archaea (DeLong, 2020). We were able to successfully characterize the genes and genome fragments from planktonic Archaea and archaeal sponge symbionts (Stein et al., 1996; Schleper et al., 1997; Schleper et al., 1998; Béjà et al., 2002b). And beyond this, preliminary biochemical and phenotypic characterization of uncultivated Archaea was achieved by Christa Scheper in my lab at UCSB. She was able to clone, heterologously express and biochemically characterize a cold‐water archaeal DNA polymerase, and showed that it was thermolabile at temperatures above 40°C, unlike its most similar thermostable homologues from hyperthermophilic Archaea (Schleper et al., 1997). These early successes inspired us to continue developing chromosome walking and genome sequencing approaches for characterizing natural microbial communities, before high throughput, automated DNA sequencing approaches were broadly available.
In the late 1990s, Oded Béjà joined my lab as a Postdoc at MBARI, and we focused on developing BAC libraries for characterizing the genes and genomes of bacterioplankton populations in Monterey Bay. The strategy was at least in theory simple, and similar in approach to our earlier studies (Fig. 2; Schmidt et al., 1991; Stein et al., 1996; Schleper et al., 1998). This was a challenging project, since one had to preserve the large size and integrity of the DNA, while at the same time be able to digest and ligate the large fragments into the BAC vector and electroporate the recombinant DNA into E. coli. Oded's skilled hands were able to achieve what few others have done, before or since: namely, the construction of high‐quality BAC libraries (average insert sizes of 80 kb, maximal insert sizes >150 kb) from a natural microbial population (Béjà et al., 2000b). Rapid screening approaches we developed allowed identification of BAC clones that contained rRNA operons, allowing us to link phylogenetic identity (from rRNA phylogeny) to other functional genes in genomes of uncultivated microorganisms (Fig. 7; Béjà et al., 2000a; Béjà et al., 2000b). Application and extension of this general approach led to a number of surprising discoveries (Béjà et al., 2000a; Béjà et al., 2002a; Hallam et al., 2004; Sabehi et al., 2004; Frigaard et al., 2006).
Fig 7.
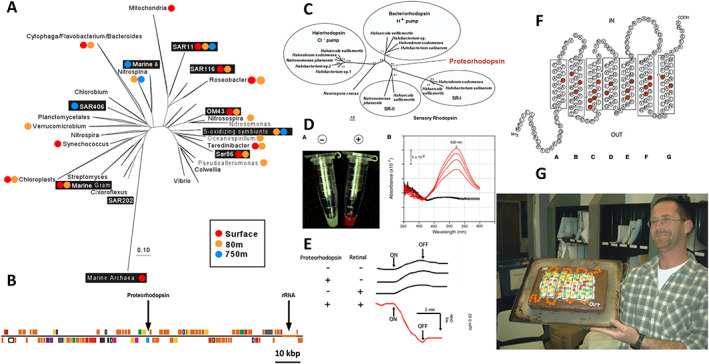
Bacterial rhodopsin ‐ proteorhodopsin is a light‐driven proton pump.
A. Phylogenetic tree of 16S rRNA genes, with black boxes representing bacterioplankton BAC clones from Monterey Bay. Coloured dots indicate depths from which each phylotype was recovered.
B. Diagram of a 120 kbp BAC clone from a SAR86 bacterium that encodes both the 16S rRNA as well as proteorhodopsin.
C. Phylogenetic relationships of opsin genes from extremely halophilic Archaea and the bacterial proteorhodopsin (Béjà et al., 2000a).
D. Escherichia coli cells expressing proteorhodopsin in the presence of exogenously added all‐trans retinal (red) versus negative control (white). To the right is a graph of the time series absorption spectra of proteorhodopsin‐containing E. coli membranes, showing the increase in absorbance as retinal binds over time. Black line, no proteorhodopsin negative control.
E. Results of light‐driven media acidification experiments, in the presence or absence of proteorhodopsin, retinal and light. Light‐driven proton pumping causes media acidification.
F. The inferred secondary structure of proteorhodopsin, with conserved retinal binding sites shown in red.
G. Oded Béjà, with his birthday cake showing the secondary structure of proteorhodopsin. The transmembrane residues are indicated with yellow and green M&M's®, and the conserved retinal binding residues with red M&M's. Baking credit, Christina Preston. Photo credit, Steve Haddock. [Color figure can be viewed at wileyonlinelibrary.com]
BAC libraries prepared from Monterey Bay bacterioplankton contained a variety of different phylogenetic groups that included Alphaproteobacteria, Gammaproteobacteria, Flavobacteria, Cyanobacteria, Gram‐positive bacteria and Archaea (Fig. 7; Béjà et al., 2000b). Sequencing large DNA fragments by shotgun cloning, sequencing and assembly was the state‐of‐the‐art back then, so progress was a bit slower compared with today. But the second BAC clone that we sequenced, from an uncultivated bacterium of the Gammaproteobacteria SAR86 clade, yielded a surprising result: About 50 kbp upstream of the SAR86 rRNA operon was a gene that had never been encountered before in bacteria (Fig. 7); (Béjà et al., 2000a)! It encoded a membrane‐associated photoprotein that shared the highest sequence similarity to the opsins found in extremely halophilic Archaea, and it contained all the conserved amino acid residues for binding the opsin chromophore trans retinal (Fig. 7; Béjà et al., 2000a). Likewise, its predicted tertiary structure mirrored that of other known haloarchaeal rhodopsins. In a nod to its phylogenetic origin, we called this new bacterial opsin ‘proteorhodopsin’. But did this newly discovered bacterial retinylidene protein gene truly encode a functional light‐driven ion pump (or possibly a sensory opsin), as had been previously described in extremely halophilic Archaea?
Previous success with heterologous expression of a DNA polymerase from uncultivated Archaea in E. coli encouraged us to try the same approach to study the biochemical properties of these novel bacterial proteorhodopsins. And fortuitously, Oded was ‘pre‐adapted’ for this project, since his thesis work had focused in part on the heterologous expression of membrane proteins in E. coli. After several attempts using engineered signal sequences upstream of the cloned rhodopsin, Oded found that the native SAR86 signal peptide actually worked the best for expressing the bacterial proteorhodopsin in E. coli membranes. When all‐trans retinal was added to the opsin‐expressing E. coli cells, they turned bright red (absorbance peak at 520 nm), a chromic shift that occurs when retinal binds to opsins. So, the cloned proteorhodopsin could at least properly bind to the chromophore retinal (Fig. 7; Béjà et al., 2000a). After this success, I encouraged Oded to try some of the classical, light‐activated proton pumping experiments that were originally performed with the archaeon Halobacterium halobium in early bacteriorhodopsin research (Oesterhelt and Stoeckenius, 1973) ‐ only this time using proteorhodopsin‐expressing E. coli cells. Luck was again on our side! As postulated, outward transport of protons was observed only in proteorhodopsin‐containing E. coli cells in the presence of retinal and light, and was completely abolished in the presence of protonophores (Fig. 7). Furthermore, in right‐side‐out membrane vesicles containing retinal‐bound proteorhodopsin, a light‐driven membrane electrical potential of 90 mV was generated (Béjà et al., 2000a). Together, these data showed that proteorhodopsin actively translocated protons in light and was capable of generating a physiologically relevant membrane potential (Béjà et al., 2000a). And since the photocycle of proteorhodopsin was ‘fast’, about 15 ms (characteristic of ion pumping rhodopsins), we could effectively rule out the possibility it was a sensory opsin. In total, the data suggested ‘a new type of phototrophy in the sea’, an hypothesis that has subsequently stood the test of time. In a follow‐up study (in collaboration with John and Elena Spudich and Marion Leclerc), we showed that membrane preparations from naturally occurring bacterioplankton exhibited the same photochemical reaction cycle as heterologously expressed proteorhodopsin, which was completely eliminated when the chromophore retinal was removed by bleaching with hydroxylamine (Béjà et al., 2001). We also were able to show that different proteorhodopsins from various depths or geographic regions had variable light absorption maxima, ranging from 490 to 527 nm (Béjà et al., 2001).
Laboratory and field studies of diverse bacterial (and other) opsins by many others soon followed our initial discovery of proteorhodopsin (DeLong and Béjà, 2010; Pinhassi et al., 2016). We had originally thought we were lucky in finding this seemingly rare bacterial opsin, since none had ever before been observed. As it turned out, however, bacterial opsins are quite common and occur in most bacterial clades found in ocean surface waters, and elsewhere ‐ they are both ubiquitous and abundant. Further opsin surprises came when I was examining large DNA fragment library sequences from the NPSG (DeLong, 2005) and noticed a proteorhodopsin gene directly adjacent to a Marine Group II archaeal (now Poseidoniales; (Rinke et al., 2019)) rRNA gene! Postdoc Niels‐Ulrik Frigaard at MIT followed up this observation with some nice analyses, showing that about 10% of Group II Archaea (e.g. Poseidoniales) in the photic zone contained a proteorhodopsin gene adjacent to their SSU rRNA (Frigaard et al., 2006).
In follow on work, a research scientist in my group at MIT, Asuncion (Chon) Martinez, showed that some large DNA fragment clones from NPSG surface waters when induced to high copy number in E. coli produced a reddish pigment (Fig. 8; see clone HF10_19P19) (Martinez et al., 2007). We were surprised to learn these clones encoded a genetically linked full suite of retinal biosynthesis genes adjacent to a proteorhodopsin gene that together produced a functional photosystem (without exogenous addition of retinal). In a nod to early classic experiments showing bacteriorhodopsin‐enabled light‐driven photophosphorylation in Halobacterium halobium (Danon and Stoeckenius, 1974; Hartmann and Oesterhelt, 1977), Chon followed up with similar experiments in recombinant E. coli. She performed knockout mutations demonstrating the function of each gene and showed that just these seven photosystem proteins were necessary and sufficient to enable light‐driven photophosphorylation in E. coli (Fig. 8; Martinez et al., 2007). In principle, just seven genes (or less) are required for converting a heterotroph into a phototroph! This observation helps to explain in part the frequent lateral transfer, retention and ubiquitous presence of these photoprotein genes in bacteria. And we now know that light‐driven ion transport in marine bacterial opsins is not just restricted to proton translocation. In a visit to my lab at MIT, Susumu Yoshizawa later showed that within the genome of a single flavobacterium, three different bacterial opsins are present (Fig. 8); (Yoshizawa et al., 2014). When expressed, each opsin exhibited light‐driven translocation of a different ion, either H+, Na+, or Cl− (Yoshizawa et al., 2014). Although the function of the flavobacterial inward translocating Cl− ion opsin pump is similar to that of halorhodopsins found in haloarchaea, its sequence and evolutionary origins are completely different. The bacterial Cl− translocating opsins have apparently have arisen via convergent evolution and are specifically related to Na+ ion transporting bacterial homologues (Yoshizawa et al., 2014).
Fig 8.

Proteorhodopsin photosystem photophosphorylation, and the different ‘flavours’ of bacterial ion‐pumping opsins.
A. Top, diagram showing the arrangement of proteorhodopsin photosystem genes in fosmid clones from Station ALOHA that heterologously express an active photosystem in E coli. crt, carotenoid biosynthetic genes; PR, proteorhodopsin; blh, β‐carotene dioxygenase; idi, isopentenyl diphosphate isomerase. Bottom, knockout mutants in proteorhodopsin photosystem genes and their phenotypes (Martinez et al., 2007).
B. Experimental demonstration that the proteorhodopsin photosystem is necessary and sufficient for light‐driven photophosphorylation in E. coli. Top, diagram showing processes and experimental parameters tested in E. coli cells carrying proteorhodopsin photosystem genes. CCCP, carbonylcyanide m‐chlorophenylhydrazone; DCCD, N,N‐dicyclohexylcarbodiimide. Middle, light‐driven media acidification under different experimental conditions. Bottom, light‐driven ATP synthesis (photophosphorylation) in E. coli cells containing proteorhodopsin photosystem genes (Martinez et al., 2007).
C. Cartoon showing light‐driven bacterial opsins that translocate either protons, sodium ions or chloride ions (Pinhassi et al., 2016). [Color figure can be viewed at wileyonlinelibrary.com]
In later work, Chon and Oscar Sosa (a grad student in my group at MIT) leveraged cultivation‐independent genetic and biochemical approaches to characterize phosphonate degradation pathways in marine microbial communities. It was known that in the marine environment, biogenic phosphonates might be important sources of the limiting nutrient phosphorus. Indeed, with colleagues Dave Karl and Dan Repeta at WHOI, we showed that bacterial biodegradation of methylphosphonate appeart to be the ultimate source of the aerobic, subsurface methane maxima found throughout the world's oceans (Karl et al., 2008; Martinez et al., 2013; Repeta et al., 2016; Sosa et al., 2017; Sosa et al., 2019). To further investigate the pathways of phosphonate degradation, Chon used large‐insert clone libraries from bacterioplankton, to complement E. coli deletion mutants in the C‐P lyase pathway that were unable to use phosphonates. She found a number of clones that produced enzymes enabling the degradation phosphonates, some known (like C‐P lyase), and some novel (Martinez et al., 2010). Two new genes Chon discovered in this way, phnY and phnZ, encoded the enzymes PhnY and PhnZ that could degrade phosphonates by a previously undescribed pathway (Martinez et al., 2010). In collaboration with David Zechel, it was shown that PhnY was a α‐ketoglutarate/Fe(II)‐dependent dioxygenase, and PhnZ was an Fe(II)‐dependent oxidative enzyme that utilized a new catalytic mechanism for the cleavage of the carbon‐phosphorus bond (McSorley et al., 2012). Once again, the expression of genes and characterization of proteins from uncultivated marine bacterioplankton had led to the discovery of a new metabolic pathway.
Ecosystems biology in the sea ‐ some first steps
When I moved from MBARI to MIT in 2004 (Fig. 9), the technology revolution and democratization of DNA sequencing was in full swing. It had only been a decade since the first full free‐living bacterial genome had been sequenced (Fleischmann et al., 1995), and it was also clear that these new developments would enable ‘systems biology’ approaches in naturally occurring microbial communities (DeLong, 2002a; DeLong, 2002b; DeLong, 2004; DeLong, 2005). We had proposed just such approaches, when David Karl, Jonathan Zehr, Penny Chisholm and I proposed the establishment of a new NSF Science and Technology Centre, called the ‘Centre for Microbial Oceanography: Research and Education’ (C‐MORE; https://hahana.soest.hawaii.edu/cmoreserver/). Indeed, when C‐MORE was successfully established and we started our first summer course in 2006, the state‐of‐the‐art at the time for automated DNA sequencing was fluor‐labelled dideoxynucleotide Sanger sequencing (DeLong and Karl, 2005). Over the course of the 10‐year program, we worked with students first using that technology, then through Roche 454 pyrosequencing technologies, and finally, with the massively parallel, bridge‐amplification‐based Solexa/Illumina sequencing technologies still in wide use today. It was exhilarating, but also dizzying, to try and keep up with the rapidly changing technologies from 2006 to 2016! Yet these new technological approaches made possible some of the microbial community systems biology efforts that we had proposed for C‐MORE.
Fig 9.

DeLong lab group photo at MIT.
Left to right: John Eppley, Asuncion (Chon) Martinez, Justin Buck, Ed DeLong, Rachel Barry, Yanmei Shi, Julie Maresca, Laure‐Anne Ventouras, Gene Tyson, Tsultrim Palden, Hiroyuki Kimura, Frank Stewart, Elizabeth Ottesen, Jay McCarren and Vinh Pham. (Not present for this photo are other critical members of the MIT team that include Jessica Bryant, Scott Gifford, Niels‐Ulrik Frigaard, Steve Hallam, Tracy Mincer, Oscar Sosa, Mike Valliere and Susumu Yoshizawa). [Color figure can be viewed at wileyonlinelibrary.com]
These technological developments helped us to finish the first full draft genome sequence of a member Marine Group I Archaea, which is now known to be affiliated with the ammonia‐oxidizing Archaea of the family Nitrosopumilaceae (Konneke et al., 2005). This first cold‐water archaeal genome sequence was the result of the work of many: Chris Preston whose thesis work focused on the discovery of the archaeal sponge symbiont Cenarchaeum symbiosum, Christa Schleper who constructed fosmid libraries enriched with the C. symbiosum genome, colleagues from the JGI who assisted with the sequencing of our fosmid clones, and Steve Hallam and Kostas Konstantinidis who led the genome annotation and analysis effort (Hallam, JGI). We found genes for metabolic pathways involved in urea transport and degradation, ammonia transport, and its oxidation by an archaeal ammonia monooxygenase complex, and CO2 fixation by a variant of the autotrophic hydroxypropionate/hydroxybutyrate cycle (Hallam et al., 2006a; Hallam et al., 2006b). The C. symbiosum genome sequence was also a useful reference to map metagenomic fragments of Marine Group I Archaea (e.g. Nitrosopumilaceae) in the environment (Hallam et al., 2006b). Today, many of the detailed nuances of Nitrosopumilaceae ecology and evolution continue to be explored, via comparative genome sequence analyses (Qin et al., 2020).
With the advent of ‘next generation’ genome sequencing, many new approaches in both gene‐centric and genome‐centric microbial community analyses could begin to be explored. Computational biologist John Eppley, first at MIT and then at UH, has been a key player enabling all of our 'omics analyses over many years. From the perspective of gene‐centric approaches, we became interested in the repertoire of genes and taxa that occupy the open ocean water column from surface waters to the abyss. Working with Dave Karl at Station ALOHA in the NPSG, initially using ‘old‐fashioned’ techniques (fosmid end Sanger sequencing), we found that genes and taxa were distinctly stratified in specific layers in the open ocean water column (Fig. 10); (DeLong et al., 2006). Some of those gene distributions made physiological and ecological sense (e.g. photolyases and chlorophyll biosynthesis genes peaked in the euphotic zone), while others were more unexpected (peaks of intracellular phage at 75 m depth and transposase genes at 4000 m depth) (DeLong et al., 2006), that now are more readily interpretable on the basis of new and more extensive data and analyses (Konstantinidis et al., 2009; Mende et al., 2017; Luo et al., 2020). In a later study based on time‐series metagenomic sequence data, we discovered interesting depth‐dependent trends in genomic GC content, genome size and predicted proteome nitrogen content across the entire microbial communities at Station ALOHA (Fig. 10; Mende et al., 2017). Graduate student Jess Bryant, postdoc Daniel Mende, and colleagues showed that across entire microbial communities at Station ALOHA, GC content, genome size and proteome nitrogen and carbon content were correlated with nutrient availability, in particular dissolved inorganic nitrogen (Fig. 10; Mende et al., 2017). These data support the hypotheses that nutrient limitation is a central driver in the evolution of core bacterial and archaeal genomic and proteomic properties. Further evidence now suggests that such resource‐driven natural selection has shaped genome and proteome composition, codon usage, and even the evolution of the genetic code itself, across all three domains of life (Shenhav and Zeevi, 2020).
Fig 10.

Microbial genomics in the open ocean's interior at Station ALOHA.
A. Dendrogram and Venn diagram showing the overlapping suites of metagenomic sequences shared between, or unique to, different depths in the water column (DeLong, 2005).
B. Heat map showing the relative abundance of specific microbial genes found in metagenomes throughout the water column at Station ALOHA (DeLong et al., 2006).
C. Left, dendrogram showing the overall similarity of metagenomes sampled at different times and depths. Right, heat map of various corresponding parameters sampled at different times and depths (Mende et al., 2017).
D. Top left, the average %GC of genes in metagenomes sampled at different depths and times at Station ALOHA. White triangles represent the average at each depth. Top right, average difference from the mean %GC of all genes, of different taxa at each depth. Bottom, N‐ARSC and C‐ARSC as a function of GC content, in protein‐coding genes recovered in the metagenomes sampled over space and time at Station ALOHA. N‐ARSC and C‐ARSC represent the number of nitrogen or carbon atoms (respectively) per amino‐acid‐residue side chain (Mende et al., 2017). [Color figure can be viewed at wileyonlinelibrary.com]
Technological advances also enabled the application of high‐throughput ‘RNA‐Seq’ approaches to natural microbial communities, in ‘metatranscriptomic’ approaches (Frias‐Lopez et al., 2008). Yanmei Shi, a grad student in my lab at MIT then, spearheaded this effort, and in collaboration with Penny Chisholm's group, we showed the efficacy of the approach for analyses of multispecies transcript abundances in sympatric bacterioplankton communities. Puzzling, however, was the presence of highly abundant RNAs that did not correspond to any known protein‐coding genes. Yanmei and Gene Tyson (then a postdoc in my group) subsequently showed that many of these abundant RNA species were in fact regulatory noncoding RNA molecules, including riboswitches (Shi et al., 2009). The inherent population variability even in a single sample also allowed alignment of RNA homologues and identification of secondary structure‐preserving compensatory base changes, that aided in their identification and structural identification. Although much of the field was focused on identifying noncoding RNA in genomic DNA, metatranscriptomes proved to be rich sources for discovering new regulatory noncoding RNAs (Shi et al., 2009; Weinberg et al., 2009).
The newfound ability to measure environmental parameters synoptically with massively parallel queries of metagenomes and metatranscriptomes (and to a lesser extent, metaproteomes and metabolomes) quickly enabled the advancement of new systems biology‐like approaches in the marine environment (DeLong, 2002a; DeLong, 2002b; DeLong, 2009). While at MIT, and later at the University of Hawaii (UH), we were fortunate to be able to team up with colleagues at MBARI to test some of these approaches, to examine the circadian rhythms of gene expression in planktonic microbial communities. After some initial ground‐truthing and methodological development (Stewart et al., 2010; Ottesen et al., 2011), we used the robotic platform called the Environmental Sample Processor (Fig. 11) to simultaneously drift with and sample sympatric microbial communities over time in coastal and open ocean habitats (Ottesen et al., 2013; Ottesen et al., 2014). This Lagrangian sampling strategy helps to avoid sampling heterogeneous populations that arrive from other water sources and currents, enabling fine‐scale sampling of the same population over time. One fascinating discovery from some of our first field experiments led by Liz Ottesen, then a postdoc in my group at MIT, was that regular, predictable oscillating diel cycles of gene transcripts in heterotrophic bacterial and archaeal populations (Fig. 11; Ottesen et al., 2013; Ottesen et al., 2014). This is postulated to be driven by diurnal fluctuations of external metabolites produced by phytoplankton. Liz further found that diel oscillations in different heterotrophic bacteria suggested population‐specific timing of transcript abundance maxima across a variety of metabolic gene suites. Postdoc Frank Aylward at the University of Hawaii (Fig. 12) later showed that the microbial community transcriptional oscillations of key metabolic pathways are conserved across the Pacific Ocean basin, from the coast to the open ocean (Fig. 11; Aylward et al., 2015). This is a compelling phenomenon to imagine: As the Earth turns, and sunlight streaks across the oceans, marine microbial communities orchestrate their diel transcriptional oscillations in a continuous symphonic wave across the globe! Following up on this work, Frank went on to demonstrate that in the NPSG, cyanobacterial viruses also exhibit diel cycle in transcription, presumably keeping time with the reproduction cycles of their hosts (Aylward et al., 2017; Chen and Zeng, 2020). And light and resource‐driven microbial circadian cycles are not limited to the oceanic microbiomes. The human gut microbiome also exhibits similar diurnal oscillations, in response to host circadian rhythms and feeding activities, that may provide feed backs to the metabolic homeostatisis of the whole human host‐microbiome system (Thaiss et al., 2014; Thaiss et al., 2015).
Fig 11.

Diel oscillations in the transcriptomes of naturally occurring bacterioplankton.
A. A cartoon of the Environmental Sample Processor (bottom) suspended from a float, in the configuration used for surface water Lagrangian sampling of bacterioplankton.
B. Timing of periodically expressed transcripts. Left, histograms showing the number of diel transcripts with peak expressions binned in 1‐h intervals. Right, Peak expression time of all transcripts in selected KEGG pathways (grey circles). Red circles denote significantly periodic diel transcripts (Ottesen et al., 2014).
C. Whole‐community transcriptional networks in coastal and open ocean marine microbial plankton are predominantly structured around the 24‐h metabolic cycles of the photoautotrophic members. Left, the overall number of transcripts in each phylogenetic group is shown in the bar charts (D, H). Middle, whole‐community transcriptome networks represent correlations across transcripts identified in California coast and North Pacific Subtropical Gyre planktonic microbial assemblages. Networks are coloured according to microbial group (A, E) and module (i.e. set of coexpressed transcripts; B; F). Right, trendlines representing the average 24‐h expression profile for the 10 largest transcriptional modules are shown (C, G) (Aylward et al., 2015). [Color figure can be viewed at wileyonlinelibrary.com]
Fig 12.

DeLong lab group photo at the University of Hawaii, Manoa. Left to right: Ed DeLong, Anna Romano, Paul Den Uyl, Elaine Luo, Eint Ki, Carla Gimple (front), Andy Burger (back), Byron Pedler‐Sherwood, Bethanie Edwards, Dan Olson, Fuyan Li, Daniel Mende, Elisha Wood‐Charlson and Frank Aylward. (Not present for this photo are other critical members of the UH team that include Dominique Boeuf, Jessica Bryant, Jonathan Cardwell, John Eppley, Andy Leu, Torben Nielsen, Oscar Sosa and Kirsten Poff.) [Color figure can be viewed at wileyonlinelibrary.com]
And of course, these approaches lend themselves well to comparative ecosystem analyses. In collaborative work with Osvaldo Ulloa, Don Canfield, Ricardo Letelier and others, we dove into microbial community omics of oxygen minimum zones. These oxygen‐depleted regions are critical to ocean systems, since in their extreme states they can create ‘dead zones’, as well as significant bioavailable nitrogen loss from the ocean (Ulloa et al., 2012). While at MIT, postdoc Frank Stewart led the collaborative charge for our group, along with Osvaldo Ulloa, Don Canfield and others. In the course of that work, Frank and colleagues showed that that microbial phylogenetic and protein‐coding functional diversity both declined steeply along with the transition from oxygen‐rich surface water to the permanent OMZs (Bryant et al., 2012). Metatranscriptome profiling further documented dominant nitrogen cycling pathways in OMZs off the Chilean coast, that included both oxidative (nitrification) and reductive (anammox, denitrification) components, as well as some specific sulfur cycling genes (Stewart et al., 2012a). With Don Canfield, Osvaldo Ulloa and collaborators, combined omics and process rate measurements indicated that both sulfate reduction and sulfide oxidation contributed to energy flux and elemental cycling in OMZs (Canfield et al., 2010). The challenges of incubation studies in the most extreme regions devoid of any oxygen also became very apparent in the course of these studies (Stewart et al., 2012b; Dalsgaard et al., 2014). Collectively, these and other studies indicated that extreme OMZs are essentially anoxic marine zones that correspond to an intermediate state poised between fully oxic systems versus fully sulfidic systems (Ulloa et al., 2012).
It has been exciting to see the accelerating developments in ocean ecosystems biology, as technology marches on and analytical approaches continue to mature (DeLong, 2002a; DeLong, 2002b; DeLong, 2009; Logares et al., 2020; Sunagawa et al., 2020). Simultaneous, cross‐taxon field surveys of metazoan, protozoan, fungal, bacterial, archaeal and viral dynamics are now becoming more possible. Leveraging such all‐taxon dynamic surveys across nested scales of space and time will undoubtedly provide more detailed four‐dimensional perspectives so necessary to understand the interplay of environmental, evolutionary and ecological forces that shape ecosystems. These are still early days, but the potential is great to further advance both reductionist‐level understanding as well as ecosystem‐level perspectives of how the Earth system functions.
Lessons learned
There are many! A few lessons that strike me as truly important are listed below:
Technology continually marches on
As we make new instruments and measure new things, new vistas continually open up, especially for microbiology. The evolution of stable isotopic techniques for qualitative and quantitative inference of in situ microbial physiologies is a one good example. Another, of course, is the ongoing march of the ‘omics’. Even DNA sequencing, which has seen such dramatic changes over the past two decades, remains in flux. The power of single‐molecule, long read sequencing, for example, has the potential to overcome some of the shortcomings of short read assembly approaches, allowing the observation of new biological phenomena in situ (Beaulaurier et al., 2020). New omic technologies, coupled with more quantitative environmental sampling and measurement techniques at nested scaled, will facilitate simultaneous observations of microbial community genome, transcriptome, proteome and metabolome dynamics, to improve theory and modelling of complex microbial community system dynamics. And of course, allied technological advancements ‐ in microscopy, analytical chemistry, single cell physiology, compuational biology, and beyond ‐ will propel new undertstandings that we cannot even imagine currently.
Expect the unexpected
Even well‐accepted paradigms are often based on limited data and observations. Novel scientific paradigms do not typically appear gradually, over long periods of iterative science, however. Instead, new ideas and paradigms in science tend to blossom quickly, as the right sets of scientific circumstances promote saltatory, punctuated advancement (Kuhn, 1962). Over the course of my own career old certainties like ‘PUFAs are not biosynthesized by bacteria’, ‘Archaea only inhabit extreme environments.’, ‘Nitrification is performed only by a few bacterial groups.’, ‘Non‐chlorophyll containing bacterioplankton are either strictly heterotrophic or chemolithotrophic’ have all fallen the wayside. It certainly is important to keep one's mind open as new observational opportunities become available, and light is shed in previously dark corners. This of course is not a new concept, and Pastuer said it so well: ‘Dans les champs de l'observation le hasard ne favorise que les esprits prepares.’ (I guess the Boy Scouts had this one correct too: ‘Be prepared!’). All to say, expect the unexpected!
Symbiosis gets more done
I respectfully borrow one of my favourite adages from Norm Pace (known to his disciples as a ‘Normism’). What is true of biological symbiotic associations is also true of human collaborations, even more so. One of the real joys of science is when complementary sets of different expertise combine in new ways to build bridges across previous disciplinary boundaries. That is often where the action is, for new and fundamental advancements in scientific knowledge. I feel very fortunate to have experienced a few such transdisciplinary leaps!
Passing the baton
Human knowledge is a cross‐generational form of memory, which allows us collectively to grow ideas beyond what any single person or group could ever conceive of in a single generation. We all truly stand on the shoulders of giants, as our collective societal memory, knowledge and accomplishments grow and accumulate across the centuries! This is important to realize, even in our own smaller generational passing of knowledge and practice, as trained apprentices become independent practitioners. It is not always easy, but it is certainly important, to strive to pass the baton on as gracefully as possible when students and postdocs carry forward into their own independent scientific careers. One does not want to quit the race of course, but relay races do not work very well if batons do not get passed on. It is always a conversation, but obviously an important one at transitional mentorship stages.
To conclude, I am continually struck by what a remarkable time it has been and continues to be, for contemporary microbial biologists studying the natural microbial world. A rising tide truly lifts all boats! Previously unseen vistas continue to emerge into view, challenging our imaginations and technical capabilities, and thrusting us past former, sometimes unnecessarily narrow conceptual boundaries. What a great time it continues to be for microbial biologists!
Supporting information
Supplement movie file. Concentrating bacterioplankton on HOT Cruise #2, 1988 (https://hahana.soest.hawaii.edu/hot/csreports/cs2.html)
Acknowledgements
I am grateful and indebted to all of my colleagues, past and present, including those mentioned here and those not. My work over the past 30 years has been generously supported by the Office of Naval Research, NOAA, EPA, the U.S. National Science Foundation, the Monterey Bay Aquarium Research Institute, the David and Lucile Packard Foundation, the U.S. Department of Energy, the Agouron Institute, the Gordon and Betty Moore Foundation, and the Simons Foundation. The author's lab is currently supported by the Simons Foundation award ID #721223, and the Gordon and Betty Moore Foundation award ID #6000.
References
- Allemann, M.N. , and Allen, E.E. (2020) Genetic regulation of the bacterial omega‐3 polyunsaturated fatty acid biosynthesis pathway. J Bacteriol 202: JB.00050–20. 10.1128/JB.00050-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, E.E. , and Bartlett, D.H. (2002) Structure and regulation of the omega‐3 polyunsaturated fatty acid synthase genes from the deep‐sea bacterium Photobacterium profundum strain SS9. Microbiology 148: 1903–1913. [DOI] [PubMed] [Google Scholar]
- Amann, R. , and Fuchs, B.M. (2008) Single‐cell identification in microbial communities by improved fluorescence in situ hybridization techniques. Nat Rev Microbiol 6: 339–348. [DOI] [PubMed] [Google Scholar]
- Aylward, F.O. , Boeuf, D. , Mende, D.R. , Wood‐Charlson, E.M. , Vislova, A. , Eppley, J.M. , et al. (2017) Diel cycling and long‐term persistence of viruses in the ocean's euphotic zone. Proc Natl Acad Sci U S A 114: 11446–11451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylward, F.O. , Eppley, J.M. , Smith, J.M. , Chavez, F.P. , Scholin, C.A. , and DeLong, E.F. (2015) Microbial community transcriptional networks are conserved in three domains at ocean basin scales. Proc Natl Acad Sci U S A 112: 5443–5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulaurier, J. , Luo, E. , Eppley, J.M. , Uyl, P.D. , Dai, X. , Burger, A. , et al. (2020) Assembly‐free single‐molecule sequencing recovers complete virus genomes from natural microbial communities. Genome Res 30: 437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béjà, O. , Aravind, L. , Koonin, E.V. , Suzuki, M.T. , Hadd, A. , Nguyen, L.P. , et al. (2000a) Bacterial rhodopsin: evidence for a new type of phototrophy in the sea. Science 289: 1902–1906. [DOI] [PubMed] [Google Scholar]
- Béjà, O. , Koonin, E.V. , Aravind, L. , Taylor, L.T. , Seitz, H. , Stein, J.L. , et al. (2002b) Comparative genomic analysis of archaeal genotypic variants in a single population and in two different oceanic provinces. Appl Environ Microbiol 68: 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béjà, O. , Spudich, E.N. , Spudich, J.L. , Leclerc, M. , and DeLong, E.F. (2001) Proteorhodopsin phototrophy in the ocean. Nature 411: 786–789. [DOI] [PubMed] [Google Scholar]
- Béjà, O. , Suzuki, M.T. , Heidelberg, J.F. , Nelson, W.C. , Preston, C.M. , Hamada, T. , et al. (2002a) Unsuspected diversity among marine aerobic anoxygenic phototrophs. Nature 415: 630–633. [DOI] [PubMed] [Google Scholar]
- Béjà, O. , Suzuki, M.T. , Koonin, E.V. , Aravind, L. , Hadd, A. , Nguyen, L.P. , et al. (2000b) Construction and analysis of bacterial artificial chromosome libraries from a marine microbial assemblage. Environ Microbiol 2: 516–529. [DOI] [PubMed] [Google Scholar]
- Boetius, A. , Ravenschlag, K. , Schubert, C.J. , Rickert, D. , Widdel, F. , Gieseke, A. , et al. (2000) A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 407: 623–626. [DOI] [PubMed] [Google Scholar]
- Boeuf, D. , Edwards, B.R. , Eppley, J.M. , Hu, S.K. , Poff, K.E. , Romano, A.E. , et al. (2019) Biological composition and microbial dynamics of sinking particulate organic matter at abyssal depths in the oligotrophic open ocean. Proc Natl Acad Sci U S A 116: 11824–11832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant, J.A. , Stewart, F.J. , Eppley, J.M. , and DeLong, E.F. (2012) Microbial community phylogenetic and trait diversity declines with depth in a marine oxygen minimum zone. Ecology 93: 1659–1673. [DOI] [PubMed] [Google Scholar]
- Canfield, D.E. , Stewart, F.J. , Thamdrup, B. , De Brabandere, L. , Dalsgaard, T. , Delong, E.F. , et al. (2010) A cryptic sulfur cycle in oxygen‐minimum‐zone waters off the Chilean coast. Science 330: 1375–1378. [DOI] [PubMed] [Google Scholar]
- Chen, Y. , and Zeng, Q. (2020) Temporal transcriptional patterns of cyanophage genes suggest synchronized infection of cyanobacteria in the oceans. Microbiome 8: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump, B.C. , Armbrust, E.V. , and Baross, J.A. (1999) Phylogenetic analysis of particle‐attached and free‐living bacterial communities in the Columbia river, its estuary, and the adjacent coastal ocean. Appl Environ Microbiol 65: 3192–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalsgaard, T. , Stewart, F.J. , Thamdrup, B. , De Brabandere, L. , Revsbech, N.P. , Ulloa, O. , et al. (2014) Oxygen at nanomolar levels reversibly suppresses process rates and gene expression in anammox and denitrification in the oxygen minimum zone off northern Chile. MBio 5: e01966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danon, A. , and Stoeckenius, W. (1974) Photophosphorylation in Halobacterium halobium. Proc Natl Acad Sci U S A 71: 1234–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong, E. (1998) Archael means and extremes. Science 280: 542–543. [DOI] [PubMed] [Google Scholar]
- DeLong, E.F. (1992) Archaea in coastal marine environments. Proc Natl Acad Sci U S A 89: 5685–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong, E.F. (2000) Resolving a methane mystery. Nature 407: 577–579. [DOI] [PubMed] [Google Scholar]
- DeLong, E.F. (2002a) Towards microbial systems science: integrating microbial perspective, from genomes to biomes. Environ Microbiol 4: 9–10. [DOI] [PubMed] [Google Scholar]
- DeLong, E.F. (2002b) Microbial population genomics and ecology. Curr Opin Microbiol 5: 520–524. [DOI] [PubMed] [Google Scholar]
- DeLong, E.F. (2004) Microbial population genomics and ecology: the road ahead. Environ Microbiol 6: 875–878. [DOI] [PubMed] [Google Scholar]
- DeLong, E.F. (2005) Microbial community genomics in the ocean. Nat Rev Microbiol 3: 459–469. [DOI] [PubMed] [Google Scholar]
- DeLong, E.F. (2009) The microbial ocean from genomes to biomes. Nature 459: 200–206. [DOI] [PubMed] [Google Scholar]
- DeLong, E.F. (2020) Exploring marine planktonic archaea: then and now. Front Microbiol 11: 616086. 10.3389/fmicb.2020.616086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong, E.F. , Baumann, L. , Bowditch, R.D. , and Baumann, P. (1984) Evolutionary relationships of superoxide dismutases and glutamine synthetases from marine species of Alteromonas. Arch Microbiol 138: 170–178. [Google Scholar]
- DeLong, E.F. , and Béjà, O. (2010) The light‐driven proton pump proteorhodopsin enhances bacterial survival during tough times. PLoS Biol 8: e1000359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong, E.F. , Franks, D.G. , and Alldredge, A.L. (1993) Phylogenetic diversity of aggregate‐attached versus free‐living marine bacterial assemblages. Limnol Oceanogr 38: 924–934. [Google Scholar]
- DeLong, E.F. , and Karl, D.M. (2005) Genomic perspectives in microbial oceanography. Nature 437: 336–342. [DOI] [PubMed] [Google Scholar]
- DeLong, E.F. , and Pace, N.R. (2001) Environmental diversity of bacteria and archaea. Syst Biol 50: 470–478. [PubMed] [Google Scholar]
- DeLong, E.F. , Preston, C.M. , Mincer, T. , Rich, V. , Hallam, S.J. , Frigaard, N.U. , et al. (2006) Community genomics among stratified microbial assemblages in the ocean's interior. Science 311: 496–503. [DOI] [PubMed] [Google Scholar]
- DeLong, E.F. , Wickham, G.S. , and Pace, N.R. (1989) Phylogenetic stains: ribosomal RNA‐based probes for the identification of single cells. Science 243: 1360–1363. [DOI] [PubMed] [Google Scholar]
- DeLong, E.F. , Wu, K.Y. , Prezelin, B.B. , and Jovine, R.V. (1994) High abundance of Archaea in Antarctic marine picoplankton. Nature 371: 695–697. [DOI] [PubMed] [Google Scholar]
- DeLong, E.F. , and Yayanos, A.A. (1985) Adaptation of the membrane lipids of a deep‐sea bacterium to changes in hydrostatic pressure. Science 228: 1101–1103. [DOI] [PubMed] [Google Scholar]
- DeLong, E.F. , and Yayanos, A.A. (1986) Biochemical function and ecological significance of novel bacterial lipids in deep‐sea procaryotes. Appl Environ Microbiol 51: 730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deming, J.W. , Hada, H. , Colwell, R.R. , Luehrsen, K.R. , and Fox, G.E. (1984) The ribonucleotide sequence of 5s rRNA from two strains of deep‐sea barophilic bacteria. J Gen Microbiol 130: 1911–1920. [DOI] [PubMed] [Google Scholar]
- Field, K.G. , Olsen, G.J. , Lane, D.J. , Giovannoni, S.J. , Ghiselin, M.T. , Raff, E.C. , et al. (1988) Molecular phylogeny of the animal kingdom. Science 239: 748–753. [DOI] [PubMed] [Google Scholar]
- Fleischmann, R.D. , Adams, M.D. , White, O. , Clayton, R.A. , Kirkness, E.F. , Kerlavage, A.R. , et al. (1995) Whole‐genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269: 496–512. [DOI] [PubMed] [Google Scholar]
- Fontanez, K.M. , Eppley, J.M. , Samo, T.J. , Karl, D.M. , and DeLong, E.F. (2015) Microbial community structure and function on sinking particles in the North Pacific Subtropical Gyre. Front Microbiol 6: 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frias‐Lopez, J. , Shi, Y. , Tyson, G.W. , Coleman, M.L. , Schuster, S.C. , Chisholm, S.W. , and Delong, E.F. (2008) Microbial community gene expression in ocean surface waters. Proc Natl Acad Sci U S A 105: 3805–3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigaard, N.U. , Martinez, A. , Mincer, T.J. , and DeLong, E.F. (2006) Proteorhodopsin lateral gene transfer between marine planktonic bacteria and archaea. Nature 439: 847–850. [DOI] [PubMed] [Google Scholar]
- Fuhrman, J.A. , McCallum, K. , and Davis, A.A. (1992) Novel major archaebacterial group from marine plankton. Nature 356: 148–149. [DOI] [PubMed] [Google Scholar]
- Ganesh, S. , Parris, D.J. , DeLong, E.F. , and Stewart, F.J. (2014) Metagenomic analysis of size‐fractionated picoplankton in a marine oxygen minimum zone. ISME J 8: 187–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner, K. , and Pace, N.R. (1980) RNase P of Bacillus subtilis has a RNA component. J Biol Chem 255: 7507–7509. [PubMed] [Google Scholar]
- Giovannoni, S.J. , Britschgi, T.B. , Moyer, C.L. , and Field, K.G. (1990) Genetic diversity in Sargasso Sea bacterioplankton. Nature 345: 60–63. [DOI] [PubMed] [Google Scholar]
- Giovannoni, S.J. , DeLong, E.F. , Olsen, G.J. , and Pace, N.R. (1988a) Phylogenetic group‐specific oligodeoxynucleotide probes for identification of single microbial cells. J Bacteriol 170: 720–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni, S.J. , Turner, S. , Olsen, G.J. , Barns, S. , Lane, D.J. , and Pace, N.R. (1988b) Evolutionary relationships among cyanobacteria and green chloroplasts. J Bacteriol 170: 3584–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girguis, P.R. , Orphan, V.J. , Hallam, S.J. , and DeLong, E.F. (2003) Growth and methane oxidation rates of anaerobic methanotrophic archaea in a continuous‐flow bioreactor. Appl Environ Microbiol 69: 5472–5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossart, H.P. , Levold, F. , Allgaier, M. , Simon, M. , and Brinkhoff, T. (2005) Marine diatom species harbour distinct bacterial communities. Environ Microbiol 7: 860–873. [DOI] [PubMed] [Google Scholar]
- Hallam, S.J. , Girguis, P.R. , Preston, C.M. , Richardson, P.M. , and DeLong, E.F. (2003) Identification of methyl coenzyme M reductase A (mcrA) genes associated with methane‐oxidizing archaea. Appl Environ Microbiol 69: 5483–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallam, S.J. , Konstantinidis, K.T. , Putnam, N. , Schleper, C. , Watanabe, Y. , Sugahara, J. , et al. (2006b) Genomic analysis of the uncultivated marine crenarchaeote Cenarchaeum symbiosum. Proc Natl Acad Sci U S A 103: 18296–18301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallam, S.J. , Mincer, T.J. , Schleper, C. , Preston, C.M. , Roberts, K. , Richardson, P.M. , and DeLong, E.F. (2006a) Pathways of carbon assimilation and ammonia oxidation suggested by environmental genomic analyses of marine Crenarchaeota. PLoS Biol 4: e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallam, S.J. , Putnam, N. , Preston, C.M. , Detter, J.C. , Rokhsar, D. , Richardson, P.M. , and DeLong, E.F. (2004) Reverse methanogenesis: testing the hypothesis with environmental genomics. Science 305: 1457–1462. [DOI] [PubMed] [Google Scholar]
- Handelsman, J. , Rondon, M.R. , Brady, S.F. , Clardy, J. , and Goodman, R.M. (1998) Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem Biol 5: R245–R249. [DOI] [PubMed] [Google Scholar]
- Hartmann, R. , and Oesterhelt, D. (1977) Bacteriorhodopsin‐mediated photophosphorylation in Halobacterium halobium. Eur J Biochem 77: 325–335. [DOI] [PubMed] [Google Scholar]
- Hinrichs, K.U. , Hayes, J.M. , Sylva, S.P. , Brewer, P.G. , and DeLong, E.F. (1999) Methane‐consuming archaebacteria in marine sediments. Nature 398: 802–805. [DOI] [PubMed] [Google Scholar]
- Hugenholtz, P. , Goebel, B.M. , and Pace, N.R. (1998) Impact of culture‐independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 180: 4765–4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karl, D.M. , Beversdorf, L. , Björkman, K.M. , Church, M.J. , Martinez, A. , and Delongm, E.F. (2008) Aerobic production of methane in the sea. Nat Geosci 1: 473–478. [Google Scholar]
- Karner, M.B. , DeLong, E.F. , and Karl, D.M. (2001) Archaeal dominance in the mesopelagic zone of the Pacific Ocean. Nature 409: 507–510. [DOI] [PubMed] [Google Scholar]
- Kim, U.J. , Birren, B.W. , Slepak, T. , Mancino, V. , Boysen, C. , Kang, H.L. , et al. (1996) Construction and characterization of a human bacterial artificial chromosome library. Genomics 34: 213–218. [DOI] [PubMed] [Google Scholar]
- Konneke, M. , Bernhard, A.E. , de la Torre, J.R. , Walker, C.B. , Waterbury, J.B. , and Stahl, D.A. (2005) Isolation of an autotrophic ammonia‐oxidizing marine archaeon. Nature 437: 543–546. [DOI] [PubMed] [Google Scholar]
- Konstantinidis, K.T. , Braff, J. , Karl, D.M. , and DeLong, E.F. (2009) Comparative metagenomic analysis of a microbial community residing at a depth of 4,000 meters at station ALOHA in the North Pacific subtropical gyre. Appl Environ Microbiol 75: 5345–5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn, T.S. (1962) The Structure of Scientific Revolutions, 1st ed. Chicago, IL: The University of Chicago Press. [Google Scholar]
- Lane, D.J. , Field, K.G. , Olsen, G.J. , and Pace, N.R. (1988) Reverse transcriptase sequencing of ribosomal RNA for phylogenetic analysis. Methods Enzymol 167: 138–144. [DOI] [PubMed] [Google Scholar]
- Lane, D.J. , Pace, B. , Olsen, G.J. , Stahl, D.A. , Sogin, M.L. , and Pace, N.R. (1985) Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci U S A 82: 6955–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logares, R. , Deutschmann, I.M. , Junger, P.C. , Giner, C.R. , Krabberod, A.K. , Schmidt, T.S.B. , et al. (2020) Disentangling the mechanisms shaping the surface ocean microbiota. Microbiome 8: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, E. , Eppley, J.M. , Romano, A.E. , Mende, D.R. , and DeLong, E.F. (2020) Double‐stranded DNA virioplankton dynamics and reproductive strategies in the oligotrophic open ocean water column. ISME J 14: 1304–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh, T.L. , and Pace, N.R. (1985) Ribonuclease P catalysis differs from ribosomal RNA self‐splicing. Science 229: 79–81. [DOI] [PubMed] [Google Scholar]
- Martinez, A. , Bradley, A.S. , Waldbauer, J.R. , Summons, R.E. , and DeLong, E.F. (2007) Proteorhodopsin photosystem gene expression enables photophosphorylation in a heterologous host. Proc Natl Acad Sci U S A 104: 5590–5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez, A. , Tyson, G.W. , and Delong, E.F. (2010) Widespread known and novel phosphonate utilization pathways in marine bacteria revealed by functional screening and metagenomic analyses. Environ Microbiol 12: 222–238. [DOI] [PubMed] [Google Scholar]
- Martinez, A. , Ventouras, L.A. , Wilson, S.T. , Karl, D.M. , and Delong, E.F. (2013) Metatranscriptomic and functional metagenomic analysis of methylphosphonate utilization by marine bacteria. Front Microbiol 4: 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massana, R. , Murray, A.E. , Preston, C.M. , and DeLong, E.F. (1997) Vertical distribution and phylogenetic characterization of marine planktonic Archaea in the Santa Barbara Channel. Appl Environ Microbiol 63: 50–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McSorley, F.R. , Wyatt, P.B. , Martinez, A. , DeLong, E.F. , Hove‐Jensen, B. , and Zechel, D.L. (2012) PhnY and PhnZ comprise a new oxidative pathway for enzymatic cleavage of a carbon‐phosphorus bond. J Am Chem Soc 134: 8364–8367. [DOI] [PubMed] [Google Scholar]
- Mende, D.R. , Bryant, J.A. , Aylward, F.O. , Eppley, J.M. , Nielsen, T. , Karl, D.M. , and DeLong, E.F. (2017) Environmental drivers of a microbial genomic transition zone in the ocean's interior. Nat Microbiol 2: 1367–1373. [DOI] [PubMed] [Google Scholar]
- Metz, J.G. , Roessler, P. , Facciotti, D. , Levering, C. , Dittrich, F. , Lassner, M. , et al. (2001) Production of polyunsaturated fatty acids by polyketide synthases in both prokaryotes and eukaryotes. Science 293: 290–293. [DOI] [PubMed] [Google Scholar]
- Murray, A.E. , Blakis, A. , Massana, R. , Strawzewski, S. , Passow, U. , Alldredge, A. , and DeLong, E.F. (1999) A time series assessment of planktonic archaeal variability in the Santa Barbara Channel. Aquatic Microb Ecol 20: 129–145. [Google Scholar]
- Murray, A.E. , Preston, C.M. , Massana, R. , Taylor, L.T. , Blakis, A. , Wu, K. , and DeLong, E.F. (1998) Seasonal and spatial variability of bacterial and archaeal assemblages in the coastal waters near Anvers Island, Antarctica. Appl Environ Microbiol 64: 2585–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oesterhelt, D. , and Stoeckenius, W. (1973) Functions of a new photoreceptor membrane. Proc Natl Acad Sci U S A 70: 2853–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen, G.J. , Lane, D.J. , Giovannoni, S.J. , Pace, N.R. , and Stahl, D.A. (1986) Microbial ecology and evolution: a ribosomal RNA approach. Annu Rev Microbiol 40: 337–365. [DOI] [PubMed] [Google Scholar]
- Orphan, V.J. , Hinrichs, K.U. , Ussler, W., 3rd , Paull, C.K. , Taylor, L.T. , Sylva, S.P. , et al. (2001b) Comparative analysis of methane‐oxidizing archaea and sulfate‐reducing bacteria in anoxic marine sediments. Appl Environ Microbiol 67: 1922–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orphan, V.J. , House, C.H. , Hinrichs, K.U. , McKeegan, K.D. , and DeLong, E.F. (2001a) Methane‐consuming archaea revealed by directly coupled isotopic and phylogenetic analysis. Science 293: 484–487. [DOI] [PubMed] [Google Scholar]
- Orphan, V.J. , House, C.H. , Hinrichs, K.U. , McKeegan, K.D. , and DeLong, E.F. (2002) Multiple archaeal groups mediate methane oxidation in anoxic cold seep sediments. Proc Natl Acad Sci U S A 99: 7663–7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orphan, V.J. , Taylor, L.T. , Hafenbradl, D. , and Delong, E.F. (2000) Culture‐dependent and culture‐independent characterization of microbial assemblages associated with high‐temperature petroleum reservoirs. Appl Environ Microbiol 66: 700–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottesen, E.A. , Marin, R., 3rd , Preston, C.M. , Young, C.R. , Ryan, J.P. , Scholin, C.A. , and DeLong, E.F. (2011) Metatranscriptomic analysis of autonomously collected and preserved marine bacterioplankton. ISME J 5: 1881–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottesen, E.A. , Young, C.R. , Eppley, J.M. , Ryan, J.P. , Chavez, F.P. , Scholin, C.A. , and DeLong, E.F. (2013) Pattern and synchrony of gene expression among sympatric marine microbial populations. Proc Natl Acad Sci U S A 110: E488–E497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottesen, E.A. , Young, C.R. , Gifford, S.M. , Eppley, J.M. , Marin, R., 3rd , Schuster, S.C. , et al. (2014) Ocean microbes. Multispecies diel transcriptional oscillations in open ocean heterotrophic bacterial assemblages. Science 345: 207–212. [DOI] [PubMed] [Google Scholar]
- Pace, N.R. (1997) A molecular view of microbial diversity and the biosphere. Science 276: 734–740. [DOI] [PubMed] [Google Scholar]
- Pace, N.R. (2018) The small things can matter. PLoS Biol 16: e3000009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace, N.R. , and Marsh, T.L. (1985) RNA catalysis and the origin of life. Orig Life Evol Biosph 16: 97–116. [DOI] [PubMed] [Google Scholar]
- Pace, N.R. , Sapp, J. , and Goldenfeld, N. (2012) Phylogeny and beyond: scientific, historical, and conceptual significance of the first tree of life. Proc Natl Acad Sci U S A 109: 1011–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace, N.R. , Stahl, D.A. , and Lane, D.J. (1985) Analyzing natural microbial populations by rRNA sequences. Am Soc Microbiol News 51: 4–12. [Google Scholar]
- Pace, N.R. , Stahl, D.A. , Lane, D.J. , and Olsen, G.J. (1986) The Analysis of Natural Microbial Populations by Ribosomal RNA Sequences. Boston, MA: Springer. [Google Scholar]
- Pelve, E.A. , Fontanez, K.M. , and DeLong, E.F. (2017) Bacterial succession on sinking particles in the Ocean's interior. Front Microbiol 8: 2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinhassi, J. , DeLong, E.F. , Béjà, O. , Gonzalez, J.M. , and Pedros‐Alio, C. (2016) Marine bacterial and archaeal ion‐pumping rhodopsins: genetic diversity, physiology, and ecology. Microbiol Mol Biol Rev 80: 929–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston, C.M. , Wu, K.Y. , Molinski, T.F. , and DeLong, E.F. (1996) A psychrophilic crenarchaeon inhabits a marine sponge: Cenarchaeum symbiosum gen. nov., sp. nov. Proc Natl Acad Sci U S A 93: 6241–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, W. , Zheng, Y. , Zhao, F. , Wang, Y. , Urakawa, H. , Martens‐Habbena, W. , et al. (2020) Alternative strategies of nutrient acquisition and energy conservation map to the biogeography of marine ammonia‐oxidizing archaea. ISME J 14: 2595–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath, J. , Wu, K.Y. , Herndl, G.J. , and DeLong, E.F. (1998) High phylogenetic diversity in a marine‐snow‐associated bacterial assemblage. Aquat Microb Ecol 14: 261–269. [Google Scholar]
- Reich, C. , Gardiner, K.J. , Olsen, G.J. , Pace, B. , Marsh, T.L. , and Pace, N.R. (1986) The RNA component of the Bacillus subtilis RNase P. sequence, activity, and partial secondary structure. J Biol Chem 261: 7888–7893. [PubMed] [Google Scholar]
- Repeta, D.J. , Ferrón, S. , Sosa, O.A. , Johnson, C.G. , Repeta, L.D. , Acker, M. , et al. (2016) Marine methane paradox explained by bacterial degradation of dissolved organic matter. Nat Geosci 9: 884–887. [Google Scholar]
- Rinke, C. , Rubino, F. , Messer, L.F. , Youssef, N. , Parks, D.H. , Chuvochina, M. , et al. (2019) A phylogenomic and ecological analysis of the globally abundant marine group II archaea (Ca. Poseidoniales ord. nov.). ISME J 13: 663–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabehi, G. , Béjà, O. , Suzuki, M.T. , Preston, C.M. , and DeLong, E.F. (2004) Different SAR86 subgroups harbour divergent proteorhodopsins. Environ Microbiol 6: 903–910. [DOI] [PubMed] [Google Scholar]
- Schleper, C. , DeLong, E.F. , Preston, C.M. , Feldman, R.A. , Wu, K.Y. , and Swanson, R.V. (1998) Genomic analysis reveals chromosomal variation in natural populations of the uncultured psychrophilic archaeon Cenarchaeum symbiosum . J Bacteriol 180: 5003–5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleper, C. , Swanson, R.V. , Mathur, E.J. , and DeLong, E.F. (1997) Characterization of a DNA polymerase from the uncultivated psychrophilic archaeon Cenarchaeum symbiosum . J Bacteriol 179: 7803–7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, T.M. , DeLong, E.F. , and Pace, N.R. (1991) Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing. J Bacteriol 173: 4371–4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenhav, L. , and Zeevi, D. (2020) Resource conservation manifests in the genetic code. Science 370: 683–687. [DOI] [PubMed] [Google Scholar]
- Shi, Y. , Tyson, G.W. , and DeLong, E.F. (2009) Metatranscriptomics reveals unique microbial small RNAs in the ocean's water column. Nature 459: 266–269. [DOI] [PubMed] [Google Scholar]
- Simon, M. , Grossart, H.P. , Schweitzer, B. , and Ploug, H. (2002) Microbial ecology of organic aggregates in aquatic ecosystems. Aquat Microb Ecol 28: 175–211. [Google Scholar]
- Sosa, O.A. , Repeta, D.J. , DeLong, E.F. , Ashkezari, M.D. , and Karl, D.M. (2019) Phosphate‐limited ocean regions select for bacterial populations enriched in the carbon‐phosphorus lyase pathway for phosphonate degradation. Environ Microbiol 21: 2402–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa, O.A. , Repeta, D.J. , Ferron, S. , Bryant, J.A. , Mende, D.R. , Karl, D.M. , and DeLong, E.F. (2017) Isolation and characterization of bacteria that degrade phosphonates in marine dissolved organic matter. Front Microbiol 8: 1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl, D.A. (2020) The path leading to the discovery of the ammoniaoxidizing archaea. Environ Microbiol 22: 4507–4519. [DOI] [PubMed] [Google Scholar]
- Stahl, D.A. , Lane, D.J. , Olsen, G.J. , and Pace, N.R. (1984) Analysis of hydrothermal vent‐associated symbionts by ribosomal RNA sequences. Science 224: 409–411. [DOI] [PubMed] [Google Scholar]
- Stahl, D.A. , Lane, D.J. , Olsen, G.J. , and Pace, N.R. (1985) Characterization of a Yellowstone hot spring microbial community by 5S rRNA sequences. Appl Environ Microbiol 49: 1379–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley, J.T. , and Konopka, A. (1985) Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annu Rev Microbiol 39: 321–346. [DOI] [PubMed] [Google Scholar]
- Stein, J.L. , Marsh, T.L. , Wu, K.Y. , Shizuya, H. , and DeLong, E.F. (1996) Characterization of uncultivated prokaryotes: isolation and analysis of a 40‐kilobase‐pair genome fragment from a planktonic marine archaeon. J Bacteriol 178: 591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, F.J. , Dalsgaard, T. , Young, C.R. , Thamdrup, B. , Revsbech, N.P. , Ulloa, O. , et al. (2012b) Experimental incubations elicit profound changes in community transcription in OMZ bacterioplankton. PLoS One 7: e37118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, F.J. , Ottesen, E.A. , and DeLong, E.F. (2010) Development and quantitative analyses of a universal rRNA‐subtraction protocol for microbial metatranscriptomics. ISME J 4: 896–907. [DOI] [PubMed] [Google Scholar]
- Stewart, F.J. , Ulloa, O. , and DeLong, E.F. (2012a) Microbial metatranscriptomics in a permanent marine oxygen minimum zone. Environ Microbiol 14: 23–40. [DOI] [PubMed] [Google Scholar]
- Sunagawa, S. , Acinas, S.G. , Bork, P. , Bowler, C. , Tara Oceans, C. , Eveillard, D. , et al. (2020) Tara Oceans: towards global ocean ecosystems biology. Nat Rev Microbiol 18: 428–445. [DOI] [PubMed] [Google Scholar]
- Thaiss, C.A. , Zeevi, D. , Levy, M. , Zilberman‐Schapira, G. , Suez, J. , and Tengeler, A.C. (2014) Transkingdom Control of Microbiota Diurnal Oscillations Promotes Metabolic Homeostasis. Cell 159(3): 514–529. 10.1016/j.cell.2014.09.048. [DOI] [PubMed] [Google Scholar]
- Thaiss, C.A. , Zeevi, D. , Levy, M. , Segal, E. , and Elinav, E. (2015) A day in the life of the meta‐organism: diurnal rhythms of the intestinal microbiome and its host. Gut Microbes 6(2): 137–142. 10.1080/19490976.2015.1016690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulloa, O. , Canfield, D.E. , DeLong, E.F. , Letelier, R.M. , and Stewart, F.J. (2012) Microbial oceanography of anoxic oxygen minimum zones. Proc Natl Acad Sci U S A 109: 15996–16003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waugh, D.S. , and Pace, N.R. (1986) Catalysis by RNA. Bioessays 4: 56–61. [DOI] [PubMed] [Google Scholar]
- Weinberg, Z. , Perreault, J. , Meyer, M.M. , and Breaker, R.R. (2009) Exceptional structured noncoding RNAs revealed by bacterial metagenome analysis. Nature 462: 656–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese, C.R. (1987) Bacterial evolution. Microbiol Rev 51: 221–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese, C.R. , and Fox, G.E. (1977) Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci U S A 74: 5088–5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese, C.R. , Kandler, O. , and Wheelis, M.L. (1990) Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A 87: 4576–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yayanos, A.A. (1986) Evolutional and ecological implications of the properties of deep‐sea barophilic bacteria. Proc Natl Acad Sci U S A 83: 9542–9546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yayanos, A.A. (1995) Microbiology to 10,500 meters in the deep sea. Annu Rev Microbiol 49: 777–805. [DOI] [PubMed] [Google Scholar]
- Yayanos, A.A. , Dietz, A.S. , and Van Boxtel, R. (1981) Obligately barophilic bacterium from the Mariana trench. Proc Natl Acad Sci U S A 78: 5212–5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa, S. , Kumagai, Y. , Kim, H. , Ogura, Y. , Hayashi, T. , Iwasaki, W. , et al. (2014) Functional characterization of flavobacteria rhodopsins reveals a unique class of light‐driven chloride pump in bacteria. Proc Natl Acad Sci U S A 111: 6732–6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement movie file. Concentrating bacterioplankton on HOT Cruise #2, 1988 (https://hahana.soest.hawaii.edu/hot/csreports/cs2.html)