

Abstract
Based on the results from previous high‐pressure experiments on the gadolinite‐type mineral datolite, CaBSiO4(OH), the behavior of the isostructural borates β‐HfB2O5 and β‐ZrB2O5 have been studied by synchrotron‐based in situ high‐pressure single‐crystal X‐ray diffraction experiments. On compression to 120 GPa, both borate layer‐structures are preserved. Additionally, at ≈114 GPa, the formation of a second phase can be observed in both compounds. The new high‐pressure modification γ‐ZrB2O5 features a rearrangement of the corner‐sharing BO4 tetrahedra, while still maintaining the four‐ and eight‐membered rings. The new phase γ‐HfB2O5 contains ten‐membered rings including the rare structural motif of edge‐sharing BO4 tetrahedra with exceptionally short B−O and B⋅⋅⋅B distances. For both structures, unusually high coordination numbers are found for the transition metal cations, with ninefold coordinated Hf4+, and tenfold coordinated Zr4+, respectively. These findings remarkably show the potential of cold compression as a low‐energy pathway to discover metastable structures that exhibit new coordinations and structural motifs.
Keywords: borates, diamond anvil cell, gadolinite structure, high-pressure chemistry, synchrotron radiation
Push the boundaries: The behavior of β‐HfB2O5 and β‐ZrB2O5 under extreme pressure up to 120 GPa has been studied by synchrotron‐based in situ high‐pressure X‐ray diffraction in a diamond anvil cell. Both compounds remain stable to the highest applied pressure and for both experiments, a second additional phase, named γ‐HfB2O5 and γ‐ZrB2O5, respectively, was found at pressures above 114 GPa.
Introduction
Due to their exceptional physical and chemical properties, the minerals of the gadolinite supergroup [1] have been investigated for the past decades for potential use in the electrical engineering industry or as materials for radiation shields.[ 2 , 3 , 4 , 5 , 6 ] In petrology and geochemistry the gadolinite group minerals also serve as markers for geological reconstructions.[ 7 , 8 , 9 ]
The members of the gadolinite supergroup are represented by the general chemical formula A 2 MQ 2 T 2O8 φ 2 (A=Ca, RE, Pb, Mn, Bi; M=Fe, Vac., Mg, Mn, Zn, Cu, Al; Q=B, Be, Li; T=Si, P, As, B, Be, S; φ=O, OH, F) [10] and their structures feature eight‐ and four‐membered rings, made up of tetrahedra centered by the Q and T‐site atoms. Prominent representatives of the gadolinite group (space group P21/c) are minerals including datolite CaBSiO4(OH),[ 11 , 12 , 13 , 14 ] homilite Ca2B2FeSi2O8(OH)2, [15] hingganite‐(Y) Y2Be2Si2O8(OH)2, [16] and hingganite‐(Yb) Yb2Be2Si2O8(OH)2, [17] minasgeraisite Y2Be2CaSi2O8(OH)2, [18] and gadolinite‐(Y) Y2Be2FeSi2O8O2 itself.[ 19 , 20 ] In 2007 and 2008, researchers led by Huppertz found the “simplest” structural variants of all compounds of the gadolinite supergroup, namely β‐HfB2O5 and β‐ZrB2O5.[ 21 , 22 ] They represent the first ternary compounds in this structure family with Hf4+ or Zr4+ on the A site and boron on the Q and T sites, corresponding to “(Hf2/Zr2)B2B2O8O2”→HfB2O5/ZrB2O5.
Considering the importance of the gadolinite‐supergroup minerals there is relatively little information on their behavior under high‐pressure and/or high‐temperature conditions. To the best of our knowledge, there are only such studies concerning the borosilicate datolite, CaBSiO4(OH),[ 9 , 23 , 24 , 25 , 26 , 27 , 28 ] and, very recently, hingganite‐(Y). [29] While the crystal structure of hingganite‐(Y) is preserved up to pressures of 47 GPa, a displacive phase transition can be observed in datolite between 27 and 33 GPa. The application of such high pressures leads to the formation of solely fivefold coordinated Si atoms, resulting in the splitting of the eight‐membered rings into two five‐membered rings, separated by edge‐sharing SiO5 trigonal bipyramids. [28] The present study was in part motivated by a natural desire to synthesize a compound with boron in the coordination higher than four.
To date, only a few structural studies of borates under extreme pressure conditions using diamond anvil cells were performed. For FeBO3 and GdFe3(BO3)4, optical absorption spectra were recorded up to pressures of 82 GPa and 60 GPa, respectively. Additional electrical resistance measurements for FeBO3 were conducted up to 140 GPa. In both cases, electronic transitions were observed at 46 GPa for FeBO3 and at 26 and 43 GPa for GdFe3(BO3)4, but no additional structural information was given.[ 30 , 31 ] In addition to these iron borates, SrB4O7:Sm2+ was studied up to 130 GPa for its potential application as an optical sensor in the diamond anvil cell.[ 32 , 33 , 34 , 35 , 36 ]
To expand current information from surveys into this field and in the context of the investigations into datolite, the high‐pressure behavior of the isostructural borates β‐HfB2O5 and β‐ZrB2O5 were investigated to aspire to similar structural changes as that described for datolite. The results of these high‐pressure studies up to 120 GPa are presented in the following.
Experimental Section
Single crystals of β‐ZrB2O5 and β‐HfB2O5 were synthesized under high‐pressure and high‐temperature conditions in a Walker‐type multianvil apparatus according to the procedure described by Knyrim and Huppertz.[ 21 , 22 ] In situ high‐pressure single‐crystal X‐ray diffraction experiments (SCXRD) were performed at the experimental station P02.2 (Extreme Conditions Beamline) at the synchrotron Petra III (Hamburg, Germany). Preselected single crystals of β‐ZrB2O5 and β‐HfB2O5 were placed inside the sample chamber of a diamond anvil cell (DAC) along with a ruby sphere for pressure estimation. The DAC was loaded with neon as pressure‐transmitting medium. The samples of β‐ZrB2O5 and β‐HfB2O5 were gradually pressurized up to 120 GPa. Neon is known to be hydrostatic up to 15 GPa [37] therefore most of the experiment has been performed under quasihydrostatic conditions. SCXRD data were collected at each pressure step of ≈5–10 GPa. In total, 17 high‐pressure structural refinements have been performed for each crystal. The starting crystallographic parameters for β‐ZrB2O5 and β‐HfB2O5 were taken from the structural refinements reported by Knyrim and Huppertz.[ 21 , 22 ] More details on the synchrotron XRD measurements and the structure refinement is provided in the Supporting Information.
Results and Discussion
β‐Phases of ZrB2O5 and HfB2O5 at 120 GPa
The crystal structure of β‐ZrB2O5 and β‐HfB2O5, synthesized at 7.5 GPa in the multianvil press, is composed of eight‐ and four‐membered rings of BO4 tetrahedra that form layers in the bc plane. Between those layers, the Zr4+ and Hf4+ cations, respectively, are coordinated by eight oxygen atoms and form square‐antiprisms. Upon further compression up to 120 GPa, both compounds, β‐ZrB2O5 and β‐HfB2O5, preserve this structural arrangement (Figure 1). As expected, a shrinkage of the cell parameters during the compression process was observed. At 120 GPa, the highest pressure achieved, the unit cell volume decreases to 74.7 % of the ambient pressure volume in β‐ZrB2O5, and to 75.2 % in β‐HfB2O5. Also, the β angles increase for both compounds (Figure S1 in Supporting Information). A comparison of the unit cell parameters at atmospheric pressure and at nearly 120 GPa is given in Table 1. A graphical representation of the pressure dependence of the lattice parameters of β‐ZrB2O5 and β‐HfB2O5 is shown in Figure 2 and Figure 3, respectively. More detailed data for all pressure points is provided in the Supporting Information (Tables S1 and S2; for the normalized unit cell parameters see Figure S2).
Figure 1.
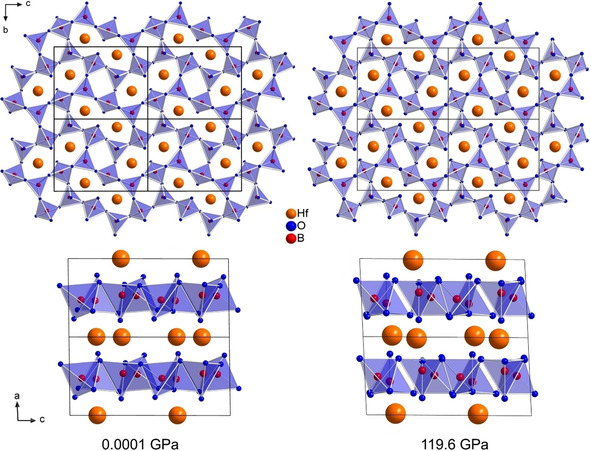
Comparison of β‐HfB2O5 at ambient pressure (left) and at nearly 120 GPa (right).
Table 1.
Comparison of the lattice parameters of β‐ZrB2O5 and β‐HfB2O5 at ambient pressure and at ≈120 GPa.
|
Compound |
β‐ZrB2O5 |
β‐HfB2O5 |
||
|
Pressure [GPa] |
0.0001 |
119.6 |
0.0001 |
119.6 |
|
Space group |
P21/c |
P21/c |
||
|
a [Å] |
4.4021(2) |
3.901(2) |
4.3843(3) |
3.8918(7) |
|
b [Å] |
6.9315(3) |
6.434(2) |
6.9048(6) |
6.4311(8) |
|
c [Å] |
8.9924(3) |
8.11(4) |
8.9727(6) |
8.17(2) |
|
β [°] |
90.93(3) |
92.95(8) |
90.76(1) |
92.90(4) |
|
V [Å3] |
272.1(2) |
203.3(10) |
271.6(1) |
204.2(4) |
Figure 2.
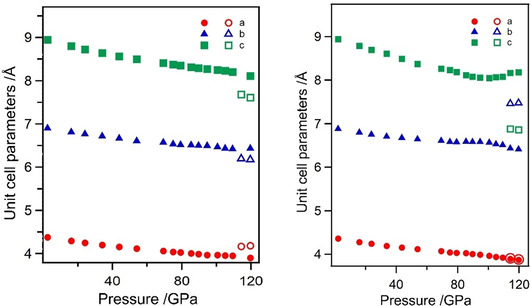
Course of the cell parameter of β‐ZrB2O5 (left) and β‐HfB2O5 (right) during the compression process. The outlined symbols indicate the simultaneous existence of the new phases γ‐ZrB2O5 (left) and γ‐HfB2O5 (right), respectively.
Figure 3.
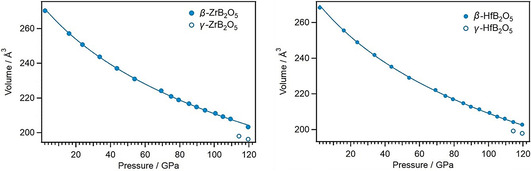
Reduction of the cell volume of β‐ZrB2O5 (left) and β‐HfB2O5 (right) with increasing pressure. The outlined symbols indicate the simultaneous existence of the new phases γ‐ZrB2O5 (left) and γ‐HfB2O5 (right), respectively.
At pressures above ≈114 GPa, the appearance of new reflections in the diffraction patterns indicated the formation of a second phase for both the zirconium and the hafnium borates (Figure 2 and 3). The structures of these new high‐pressure phases, designated as γ‐ZrB2O5 and γ‐HfB2O5, have been solved and refined. In the following, the structural transformations will be discussed, starting with the zirconium borate γ‐ZrB2O5.
Crystal structure of γ‐ZrB2O5 at 120 GPa
At approximately 114 GPa, a displacive phase transition from β‐ZrB2O5 to γ‐ZrB2O5 takes place. The wavelike arrangement of BO4 tetrahedra (marked red in Figure S3, top left) orientate themselves in opposite directions alongside the c‐axis, counteracting the applied pressure and leading to an abrupt decline in the cell parameter values for b and more pronounced for c. In the structure of the new phase γ‐ZrB2O5, the eight‐ and four‐membered rings from the β‐phase are preserved (Figure 4 and Figure S3). This alignment is responsible for the tilting of the layers. In the β‐phase, the layers run parallel to the c‐axis, and in the γ‐phase, they are inclined by ≈29° (Figure 4, right). In this new arrangement at 120 GPa, the BO4 polyhedra diverge significantly from the ideal tetrahedral arrangement, leading to B−O distances in the range of 1.333(7) to 1.440(6) Å, with an average value of ≈1.382 Å, but in about the same range of the bond lengths in the even more distorted β‐ZrB2O5 at 120 GPa, which lie in the range of 1.28(9)–1.72(2) Å (average=≈1.4 Å). As expected, these values are much shorter than in BO4 tetrahedra at ambient pressure (1.476 Å[ 38 , 39 ]). The O‐B‐O angles also lie in a wide range between 88.1(4) and 117.6(4)°. Here, the resulting mean value of 109.25° agrees well with the ideal tetrahedral angle. All bond lengths and angles for γ‐ZrB2O5 are listed in Tables 2 and 3, the values for β‐ZrB2O5 at 120 GPa can be found in Tables S3 and S4.
Figure 4.

Layered structure of γ‐ZrB2O5 still containing eight‐ and four‐membered rings depicted along [ 0 0] (left) along [0 0] (right).
Table 2.
Interatomic B−O distances [Å] for γ‐ZrB2O5 and γ‐HfB2O5 at 119.6 GPa (standard deviations in parentheses).
|
γ‐ZrB2O5 |
|
|
|
||
|---|---|---|---|---|---|
|
B1 |
‐O4 |
1.354(6) |
B2 |
‐O2 |
1.333(7) |
|
|
‐O5 |
1.363(6) |
|
‐O3 |
1.380(7) |
|
|
‐O1 |
1.371(6) |
|
‐O5 |
1.426(6) |
|
|
‐O2 |
1.382(6) |
|
‐O1 |
1.440(6) |
|
Ø |
|
1.368 |
Ø |
|
1.395 |
|
γ‐HfB2O5 |
|
|
|
||
|---|---|---|---|---|---|
|
B1 |
‐O5 |
1.26(3) |
B2 |
‐O1 |
1.38(2) |
|
|
‐O2 |
1.40(4) |
|
‐O4 |
1.39(6) |
|
|
‐O3 |
1.42(4) |
|
‐O1 |
1.43(4) |
|
|
‐O4 |
1.42(5) |
|
‐O3 |
1.48(3) |
|
Ø |
|
1.38 |
Ø |
|
1.42 |
Table 3.
Bond angles [°] for γ‐ZrB2O5 and γ‐HfB2O5 at 119.6 GPa (standard deviations in parentheses).
|
γ‐ZrB2O5 |
|
|
|
|---|---|---|---|
|
O5‐B1‐O2 |
101.1(3) |
O5‐B2‐O1 |
88.1(4) |
|
O4‐B1‐O1 |
104.8(4) |
O3‐B2‐O1 |
111.0(4) |
|
O4‐B1‐O5 |
106.4(4) |
O3‐B2‐O5 |
111.0(4) |
|
O5‐B1‐O1 |
112.3(4) |
O2‐B2‐O5 |
112.3(4) |
|
O4‐B1‐O2 |
115.2(5) |
O2‐B2‐O3 |
114.1(5) |
|
O1‐B1‐O2 |
116.9(4) |
O2‐B2‐O1 |
117.6(4) |
|
Ø |
109.5 |
Ø |
109.0 |
|
γ‐HfB2O5 |
|
|
|
|---|---|---|---|
|
O3‐B1‐O4 |
91(2) |
O4‐B2‐O3 |
100(2) |
|
O2‐B1‐O4 |
99(2) |
O1‐B2‐O1 |
101(2) |
|
O2‐B1‐O4 |
100(4) |
O1‐B2‐O3 |
103(3) |
|
O5‐B1‐O2 |
113(2) |
O1‐B2‐O3 |
113(2) |
|
O5‐B1‐O4 |
116(2) |
O1‐B2‐O4 |
117(4) |
|
O5‐B1‐O3 |
133(5) |
O4‐B2‐O1 |
123(2) |
|
Ø |
108.5 |
Ø |
109.5 |
The tilting of the layers is also responsible for the increased coordination number of ten for the zirconium cation (Figures S3, bottom and S4). Half of the O4 atoms in the β‐phase shift in the c‐direction and the other half in the opposite direction, leading simultaneously to a displacement of the oxygen atoms O3 and O5 for the newly formed polymorph γ‐ZrB2O5 (Figure S3, middle). These two atoms O3 and O5 are now in closer proximity to the Zr4+ cations and account for the enlarged coordination number. To the best of our knowledge, such a high coordination was never observed for Zr4+. The Zr−O distances at 120 GPa are around the same values in γ‐ZrB2O5 (1.958(4)–2.377(4) Å) and β‐ZrB2O5 (1.93(4)–2.10(5) Å) with slightly longer distances for the increased coordination number (Table 4).
Table 4.
Interatomic Hf/Zr−O distances [Å] for γ‐ZrB2O5 and γ‐HfB2O5 at 119.6 GPa (standard deviations in parentheses).
|
γ‐ZrB2O5 |
γ‐HfB2O5 |
||||
|---|---|---|---|---|---|
|
Zr |
‐O4 |
1.958(4) |
Hf |
‐O5 |
1.95(2) |
|
|
‐O5 |
2.018(4) |
|
‐O2 |
1.97(2) |
|
|
‐O4 |
2.028(4) |
|
‐O4 |
2.03(2) |
|
|
‐O3 |
2.029(4) |
|
‐O3 |
2.04(3) |
|
|
‐O1 |
2.110(4) |
|
‐O5 |
2.09(3) |
|
|
‐O2 |
2.136(3) |
|
‐O3 |
2.10(4) |
|
|
‐O1 |
2.175(4) |
|
‐O2 |
2.11(2) |
|
|
‐O5 |
2.240(3) |
|
‐O1 |
2.16(3) |
|
|
‐O3 |
2.243(4) |
|
‐O4 |
2.27(2) |
|
|
‐O3 |
2.377(4) |
|
|
|
|
Ø |
|
2.131 |
Ø |
|
2.08 |
Crystal structure of γ‐HfB2O5 at 120 GPa
In contrast to the phase transition from β‐ZrB2O5 to γ‐ZrB2O5 discussed before, the phase transition of β‐HfB2O5 at about 114 GPa is reconstructive and accompanied by a reorganization of the BO4 tetrahedra. As a consequence of the extreme pressure applied to β‐HfB2O5, the bonds between B2 and O2 atoms are broken apart, thus opening the four‐ and eight‐membered rings (Figure 5). New bonds between the B2 and O1 atoms of two different BO4 tetrahedra can now form, leading to edge‐sharing BO4 tetrahedra (Figure 6 a), a relatively rare structural motif in the structural chemistry of borates. In comparison with other borates containing these B2O6 groups, the B⋅⋅⋅B distance of 1.79(6) Å at 120 GPa in γ‐HfB2O5 is exceptionally short. In other borates containing this structural motif, the B⋅⋅⋅B distances typically range from 2.072 Å in Dy4B6O15 (Figure 6 b), [40] 2.04 Å in α‐Gd2B4O9, [41] and 2.088 Å in HP‐NiB2O4, [42] to 2.17 Å in HP‐CsB5O8, [43] and 2.21 Å in HP‐KB3O5 (Figure 6 c). [44] The slightly longer distances in the latter two compounds originate from the oxygen atoms forming the common edge, which are coordinated by a third boron atom and not a metal cation like the aforementioned compounds. In all of the compounds with edge‐sharing BO4 tetrahedra, the B−O bonds inside the B2O2 rings are longer than those outside the ring. This is not the case in the here presented γ‐HfB2O5, which is attributed to the high applied pressure of 120 GPa. To maximize the distance between the two boron cations located in the centers of the two edge‐sharing tetrahedra, the O‐B‐O angle inside of the B2O2 ring is usually very small (here: 101(2)°). In this case, the B‐O‐B angle constitutes the smallest angle inside the B2O2 ring, with only 79(2)°. Comparing this to all of the other known borates that contain edge‐sharing BO4 tetrahedra, only in HP‐MB3O5 (M=K, Rb, Tl)[ 44 , 45 , 46 ] and HP‐Cs1−x(H3O)xB3O5 (x=0.5–0.7) [47] the B‐O‐B angle is the smallest angle inside the B2O2 ring. Within these compounds, the oxygen atoms at the common edge are coordinated by a third boron atom and not by a metal cation. Additionally, the values of these two angles inside the B2O2 ring in γ‐HfB2O5 depart considerably from all those of known compounds, causing the greatest distortion of such a B2O6 group compared to all borate compounds containing edge‐sharing BO4 tetrahedra. Also, the ratio of edge‐sharing to corner sharing BO4 tetrahedra in γ‐HfB2O5 is strikingly high. In this phase, this ratio is 1:1, that is, there are equal amounts of edge‐sharing and corner‐sharing BO4 tetrahedra, which is the second largest ratio of this rare structural motif in all known borates and related compounds. Only HP‐MB2O4 (M=Ni, Co, Fe)[ 42 , 48 , 49 ] consists of solely edge‐sharing BO4 tetrahedra, and therefore contains more B2O6 groups. Considering just compounds where corner‐sharing BO4 tetrahedra are also present, γ‐HfB2O5 comprises the highest ratio of edge‐sharing to corner‐sharing tetrahedra.
Figure 5.

Layered structure of γ‐HfB2O5 with ten‐membered rings depicted along [ 0 0] (left) and along [0 0] (right). Corner‐sharing BO4 tetrahedra are colored in blue, edge‐sharing BO4 tetrahedra in red.
Figure 6.
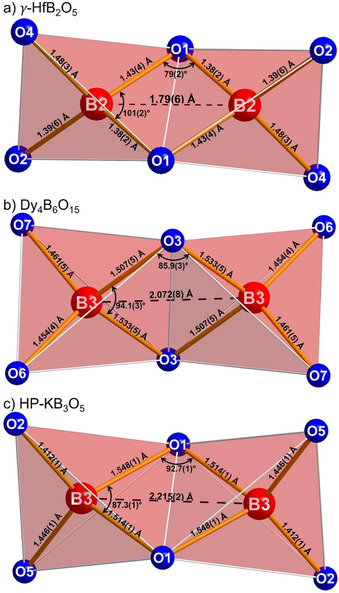
a) Edge‐sharing BO4 tetrahedra in γ‐HfB2O5 in comparison to b) Dy4B6O15 and c) HP‐KB3O5.
Examining all the B−O bonds in γ‐HfB2O5, a variation of bonding distances is found to be larger than in γ‐ZrB2O5. In γ‐HfB2O5, they vary within the range of 1.26(3) to 1.48(3) Å, with the longest distances, as expected, within the edge‐sharing tetrahedra. Similar variations of bond lengths occur in silicates at high pressure and mark the beginnings of a transition from SiO4 to SiO5 polyhedra, as for example in danburite, [50] titanite‐like silicate CaSi2O5, [51] and high‐pressure forms of enstatite. [52] In the present borate compound, this could designate the start of a possible transition to BO5 polyhedra. The mean value for all the B−O distances is 1.40 Å, which is in the same range as for β‐HfB2O5 at 120 GPa (B−O distances from 1.35(1)–1.45(2) Å; average=≈1.38 Å). The O‐B‐O angles in γ‐HfB2O5 vary from 91(2) to 133(5)° with both the smallest and the widest angle inside the corner‐sharing tetrahedra, featuring, therefore, the greatest distortion. All bond distances and angles of γ‐HfB2O5 are listed in the Table 2, the values for β‐HfB2O5 at 120 GPa can be found in Tables S3 and S4.
As in the zirconium compound, the coordination of the hafnium cation by oxygen atoms in γ‐HfB2O5 increases from an eightfold to a ninefold coordination. The Hf−O distances range from 1.95(2) to 2.27(2) Å (Table 4), which again is in the same range as the Hf−O distances in β‐HfB2O5 at 120 GPa (1.96(2)–2.086(7) Å). A comparison of the coordination polyhedra is illustrated in Figure S6.
Tables 5 and 6 present all the relevant data from the crystal structure refinement, as well as the atomic coordinates for both phases γ‐ZrB2O5 and γ‐HfB2O5. [53] Additionally, the charge distributions for both compounds were calculated according to the CHARDI concept (∑Q)[ 54 , 55 ] to further verify the determined structures. All formal charges are in good agreement with the formal valence state of the cations and anions (Table S5). Only the oxygen atom O5 in γ‐HfB2O5 features a lower charge (−1.64) than the expected −2. This can be attributed to the fact that the O5 atom is coordinated by two hafnium cations and only one boron cation. In contrast to O3, which has a similar environment, O5 is further away and therefore receives less attraction from the boron cation.
Table 5.
Crystal data and structure refinement of γ‐ZrB2O5 and γ‐HfB2O5 at 119.6 GPa.
|
Empirical formula |
γ‐ZrB2O5 |
γ‐HfB2O5 |
|
Molar mass [g mol−1] |
192.84 |
280.11 |
|
Crystal system |
monoclinic |
|
|
Space group |
P21/n (no. 14) |
P21/c (no. 14) |
|
T [K] |
285(2) |
|
|
Wavelength [Å] |
0.2901 |
|
|
a [Å] |
4.1859(7) |
3.8804(13) |
|
b [Å] |
6.1734(12) |
7.476(3) |
|
c [Å] |
7.6078(11) |
6.86(2) |
|
β [°] |
93.343(14) |
96.22(10) |
|
V [Å3] |
196.26(6) |
197.9(6) |
|
Z |
4 |
|
|
Calculated density [g cm−3] |
6.526 |
9.400 |
|
Max. θ [°] |
18.047 |
12.870 |
|
Index ranges |
−7≤h≤6, −10≤k≤9, −12≤l≤14 |
−6≤h≤6, −13≤k≤13, −5≤l≤4 |
|
Reflections collected |
1017 |
550 |
|
Independent reflections |
629 [Rint=0.0459, R sigma=0.0488] |
276 [Rint=0.0185, R sigma=0.0205] |
|
Data/ restraints/ parameters |
629/0/38 |
276/0/38 |
|
Goodness‐of‐fit on F 2 |
1.063 |
1.169 |
|
R1/wR2 indices [I≥2σ(I)] |
0.0490/0.1236 |
0.0565/0.1593 |
|
R1/wR2 indices (all data) |
0.0504/0.1264 |
0.0608/0.1659 |
|
Largest diff. peak/hole [e Å−3] |
3.54/−2.41 |
3.02/−2.99 |
Table 6.
Atomic coordinates, and equivalent isotropic displacement parameters U eq [Å2] (standard deviations in parentheses) for γ‐ZrB2O5 and γ‐HfB2O5 at 119.6 GPa. All atoms are located at the Wyckoff position 4e.
|
γ‐ZrB2O5 |
|
|
|
|
|---|---|---|---|---|
|
Atom |
x |
y |
z |
U eq |
|
Zr1 |
0.0168(3) |
0.1152(2) |
0.6763(6) |
0.0085(6) |
|
B1 |
0.511(5) |
0.221(4) |
0.41(2) |
0.0025(5) |
|
B2 |
0.443(9) |
0.074(5) |
1.13(2) |
0.0031(6) |
|
O1 |
0.794(6) |
0.101(3) |
0.16(1) |
0.0036(5) |
|
O2 |
0.311(6) |
0.880(2) |
0.14(1) |
0.0035(5) |
|
O3 |
0.277(6) |
0.204(4) |
0.1(1) |
0.0050(7) |
|
O4 |
0.278(7) |
0.149(4) |
0.31(1) |
0.0042(5) |
|
O5 |
0.747(5) |
0.080(3) |
0.458(9) |
0.0035(5) |
|
| ||||
|
γ‐HfB2O5 | ||||
|---|---|---|---|---|
|
Atom |
x |
y |
z |
U eq |
|
Hf1 |
0.4392(2) |
0.37445(7) |
0.3009(2) |
0.0126(9) |
|
B1 |
0.011(5) |
0.170(2) |
0.125(8) |
0.017(3) |
|
B2 |
0.135(4) |
0.077(2) |
0.441(6) |
0.010(2) |
|
O1 |
0.229(3) |
0.560(2) |
0.076(5) |
0.018(2) |
|
O2 |
0.092(3) |
0.811(2) |
0.261(5) |
0.018(2) |
|
O3 |
0.264(3) |
0.259(2) |
0.032(5) |
0.027(2) |
|
O4 |
0.268(3) |
0.089(2) |
0.258(5) |
0.034(2) |
|
O5 |
0.0755(3) |
0.071(2) |
0.063(5) |
0.015(2) |
Comparison to gadolinite‐type minerals datolite and hingganite‐(Y)
The observed high‐pressure pathways of β‐ZrB2O5 and β‐HfB2O5 differ from the behavior recently described for the isostructural borosilicate datolite, CaBSiO4(OH), that is built up of four‐ and eight‐membered rings of alternating SiO4 and BO4 tetrahedra. Datolite undergoes a displacive phase transition between 27 and 33 GPa with the formation of additional Si−O bonds across the eight‐membered rings (Figure S7, top left). In contrast, β‐ZrB2O5 and β‐HfB2O5 reveal striking sustainability up to ≈120 GPa, the highest pressure achieved in this study. The bulk moduli of β‐ZrB2O5 and β‐HfB2O5 were determined to 228(1) and 223(1) GPa, respectively. The boron‐oxygen distances at 120 GPa inside the eight‐membered rings are of 2.36 and 2.45 Å in β‐HfB2O5 and β‐ZrB2O5, respectively, and therefore too long to be considered a bonding distance (Figure S7, bottom left, and right). The new phases γ‐ZrB2O5 and γ‐HfB2O5 formed at ≈114 GPa do not feature an increased boron coordination number and possess structural motifs different from datolite‐II. Similar to β‐ZrB2O5 and β‐HfB2O5, another member of the gadolinite‐type minerals, hingganite‐(Y), Y2□Be2Si2O8(OH)2 persists its structure up to 47 GPa at least. [29] Gorelova et al. have explained the increased persistence of the initial structure of hingganite‐(Y) in comparison to datolite by the nature of the interlayer cation, that is, by its size and charge. Our observations are in line with this assumption. The evolution of MO8 (M=Ca2+, Y3+, Zr4+) polyhedral volumes in datolite, hingganite‐(Y) and β‐ZrB2O5 are compared in Figure S6 while their bulk moduli are given in Table S6. Due to the small size (0.83 Å) [56] and high charge of Zr4+, the ZrO8 polyhedron is twice as stiff as YO8 in hingganite‐(Y) and three times stiffer than CaO8 in datolite (Table S6). It is likely that the compressibility of the tetrahedra also contributes to the increased persistence of the initial crystal structure. The B1O4 and B2O4 units show the highest stiffness among TO4 tetrahedra in datolite, hingganite‐(Y), and β‐ZrB2O5 (Table S6). Accordingly, the BO4 tetrahedra in β‐ZrB2O5 undergo an insignificant geometrical distortion upon compression as is evident from the evolution of quadratic elongation and bond angle variance parameters (Figure S9). [57] Interestingly, the pronounced increase of these parameters at ≈120 GPa likely indicates the upcoming phase transition at higher pressures, in line with previous reports on the pressure‐induced evolution of structures based on tetrahedral layers and frameworks.[ 28 , 50 , 58 , 59 , 60 ] The high ratio of charge to cation size for both Zr4+ and B3+ results in the increased stiffness of ZrO8 and BO4 polyhedra and, thus, of the overall crystal structure. Indeed, the bulk modulus of β‐ZrB2O5 is considerably larger than those of datolite (106(4) GPa) and hingganite‐(Y) (124(1) GPa).
Conclusions
The presented study of the high‐pressure behavior of the gadolinite‐type borates β‐ZrB2O5 and β‐HfB2O5 up to 120 GPa offers new fundamental insights into the properties of borates at extreme conditions. Using synchrotron single‐crystal X‐ray diffraction in a diamond anvil cell it was shown that the structure of the two compounds was preserved up to the highest pressures achieved in this study. At pressures of about 114 GPa, a phase transition was observed in both β‐ZrB2O5 and β‐HfB2O5, which resulted in the synthesis of the previously unknown polymorphs γ‐ZrB2O5 and γ‐HfB2O5. The structure of γ‐ZrB2O5 features layers containing four‐ and eight‐membered rings, similar to those characteristic for β‐ZrB2O5, but the layers are tilted. The structure of the new hafnium borate polymorph, γ‐HfB2O5, features ten‐membered rings along with the relatively rare structural motif of edge‐sharing BO4 tetrahedra, mostly occurring in high‐pressure compounds. An extreme contraction of B−O distances and distortion of O‐B‐O angles are typical for the high‐pressure borates. In both structures, the coordination number of the cations increase in comparison to that (equal to eight) in their ambient pressure counterparts: In γ‐HfB2O5, Hf4+ is ninefold coordinated by oxygen, while in γ‐ZrB2O5, Zr4+ is tenfold coordinated. In the presented work, the high‐pressure behavior of the two borates β‐HfB2O5 and β‐ZrB2O5 was found to be different to that of the isostructural silicate datolite, CaBSiO4(OH), up to 120 GPa. Thus, subsequent experiments to at least 180 GPa are desirable to figure out if further pressure increase could promote turning boron's coordination to fivefold, analogous to that of silicon in datolite.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors acknowledge the Deutsches Elektronen‐Synchrotron (DESY, PETRA III) for provision of beamtime at the P02.2 beamline. N.D. and L.D. thank the Federal Ministry of Education and Research, Germany (BMBF, grant no. 05K19WC1) and the Deutsche Forschungsgemeinschaft (DFG projects DU 954‐11/1, DU 393‐9/2, and DU 393‐13/1) for financial support. N.D. thanks the Swedish Government Strategic Research Area in Materials Science on Functional Materials at Linköping University (Faculty Grant SFO‐Mat‐LiU No. 2009 00971).
A. Pakhomova, B. Fuchs, L. S. Dubrovinsky, N. Dubrovinskaia, H. Huppertz, Chem. Eur. J. 2021, 27, 6007.
Contributor Information
Dr. Anna Pakhomova, Email: anna.pakhomova@desy.de.
Birgit Fuchs, http://www‐c724.uibk.ac.at/aac/.
Prof. Dr. Hubert Huppertz, Email: hubert.huppertz@uibk.ac.at.
References
- 1. Hålenius U., Hatert F., Pasero M., Mills S. J., Mineral. Mag. 2016, 80, 1135–1144. [Google Scholar]
- 2. Konerskaya L. P., Orlova R. G., Bogdanis É. P., Konerskii V. D., Guseva N. P., Glass Ceram. 1988, 45, 199–201. [Google Scholar]
- 3. Farsiyants S. Y., Opaleichuk L. S., Romanova V. I., Glass Ceram. 1989, 46, 338–339. [Google Scholar]
- 4. Tarasevich B. P., Isaeva L. B., Kuznetsov E. V., Zhenzhurist I. A., Glass Ceram. 1990, 47, 175–178. [Google Scholar]
- 5. Mann K. S., Sidhu G. S., Ann. Nucl. Energy 2012, 40, 241–252. [Google Scholar]
- 6. Malczewski D., Dziurowicz M., Am. Mineral. 2015, 100, 1378–1385. [Google Scholar]
- 7. Zaccarini F., Morales-Ruano S., Scacchetti M., Garuti G., Heide K., Geochem. 2008, 68, 265–277. [Google Scholar]
- 8. Lyalina L. M., Selivanova E. A., Savchenko Y. E., Zozulya D. R., Kadyrova G. I., Geol. Ore Deposits 2014, 56, 675–684. [Google Scholar]
- 9. Goryainov S. V., Krylov A. S., Vtyurin A. N., Pan Y., J. Raman Spectrosc. 2015, 46, 177–181. [Google Scholar]
- 10. Bacik P., Miyawaki R., Atencio D., Camara F., Fridrichova J., Eur. J. Mineral. 2017, 29, 1067–1082. [Google Scholar]
- 11. Ito T., Mori H., Acta Crystallogr. 1953, 6, 24–32. [Google Scholar]
- 12. Pant A. K., Cruickshank D. W. J., Z. Kristallogr., Kristallgeom., Kristallphys., Kristallchem. 1967, 125, 286–297. [Google Scholar]
- 13. Franklin J., Foit F., Phillips M. W., Gibbs G. V., Am. Mineral. 1973, 58, 909–914. [Google Scholar]
- 14. Sugitani Y., Watanabe M., Nagashima K., Acta Crystallogr. B 1972, 28, 326–327. [Google Scholar]
- 15. Miyawaki R., Nakai I., Nagashima K., Acta Crystallogr. C 1985, 41, 13–15. [Google Scholar]
- 16. Demartin F., Minaglia A., Gramaccioli C. M., Can. Mineral. 2001, 39, 1105–1114. [Google Scholar]
- 17. Voloshin A. V., Pakhomovskii Y. A., Men'shikov Y. P., Povarennykh A. S., Matvienko E. N., Yakubovich O. V., Dokl. Akad. Nauk SSSR 1983, 270, 1188–1192. [Google Scholar]
- 18. Foord E. E., Gaines R. V., Crock J. G., William J., Simmons B., Barbosa C. P., Am. Mineral. 1986, 71, 603–607. [Google Scholar]
- 19. Pavlov P. V., Belov N. V., Kristallografiya 1959, 4, 324–340. [Google Scholar]
- 20. Miyawaki R., Nakai I., Nagashima K., Am. Mineral. 1984, 69, 948–953. [Google Scholar]
- 21. Knyrim J. S., Huppertz H., J. Solid State Chem. 2007, 180, 742–748. [Google Scholar]
- 22. Knyrim J. S., Huppertz H., Z. Naturforsch. B 2008, 63, 707–712. [Google Scholar]
- 23. Tarney J., Nicol A. W., Marriner G. F., Mineral. Mag. 1973, 39, 158–175. [Google Scholar]
- 24. Kimata M., Neues Jahrb. Mineral. Monatsh. 1978, 2, 58–70. [Google Scholar]
- 25. Perchiazzi N., Gualtieri A. F., Merlino S., Kampf A. R., Am. Mineral. 2004, 89, 767–776. [Google Scholar]
- 26. Rinaldi R., Gatta G. D., Angel R. J., Am. Mineral. 2010, 95, 1413–1421. [Google Scholar]
- 27. Krzhizhanovskaya M. G., Gorelova L. A., Bubnova R. S., Pekov I. V., Krivovichev S. V., Phys. Chem. Miner. 2018, 45, 463–473. [Google Scholar]
- 28. Gorelova L. A., Pakhomova A. S., Aprilis G., Dubrovinsky L. S., Krivovichev S. V., Inorg. Chem. Front. 2018, 5, 1653–1660. [Google Scholar]
- 29. Gorelova L. A., Pakhomova A. S., Krivovichev S. V., Kasatkin A. V., Dubrovinsky L. S., Phys. Chem. Miner. 2020, 47, 22. [Google Scholar]
- 30. Troyan I. A., Eremets M. I., Gavrilyuk A. G., Lyubutin I. S., Sarkisyan V. A., JETP Lett. 2003, 78, 13–16. [Google Scholar]
- 31. Gavriliuk A. G., Kharlamova S. A., Lyubutin I. S., Troyan I. A., Ovchinnikov S. G., Potseluiko A. M., Eremets M. I., Boehler R., JETP Lett. 2004, 80, 426–432. [Google Scholar]
- 32. Lacam A., Chateau C., J. Appl. Phys. 1989, 66, 366–372. [Google Scholar]
- 33. Datchi F., LeToullec R., Loubeyre P., J. Appl. Phys. 1997, 81, 3333–3339. [Google Scholar]
- 34. Goncharov A. F., Zaug J. M., Crowhurst J. C., Gregoryanz E., J. Appl. Phys. 2005, 97, 094917. [Google Scholar]
- 35. Jing Q., Wu Q., Liu L., Xu J.-a., Bi Y., Liu Y., Chen H., Liu S., Zhang Y., Xiong L., Li Y., Liu J., J. Appl. Phys. 2013, 113, 023507. [Google Scholar]
- 36. Romanenko A. V., Rashchenko S. V., Kurnosov A., Dubrovinsky L., Goryainov S. V., Likhacheva A. Y., Litasov K. D., J. Appl. Phys. 2018, 124, 165902. [Google Scholar]
- 37. Klotz S., Chervin J. C., Munsch P., Le Marchand G., J. Phys. D 2009, 42, 075413. [Google Scholar]
- 38. Zobetz E., Z. Kristallogr. 1982, 160, 81–92. [Google Scholar]
- 39. Zobetz E., Z. Kristallogr. 1990, 191, 45–57. [Google Scholar]
- 40. Huppertz H., von der Eltz B., J. Am. Chem. Soc. 2002, 124, 9376–9377. [DOI] [PubMed] [Google Scholar]
- 41. Emme H., Huppertz H., Chem. Eur. J. 2003, 9, 3623–3633. [DOI] [PubMed] [Google Scholar]
- 42. Knyrim J. S., Roessner F., Jakob S., Johrendt D., Kinski I., Glaum R., Huppertz H., Angew. Chem. Int. Ed. 2007, 46, 9097–9100; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 9256–9259. [Google Scholar]
- 43. Sohr G., Többens D. M., Schmedt auf der Günne J., Huppertz H., Chem. Eur. J. 2014, 20, 17059–17067. [DOI] [PubMed] [Google Scholar]
- 44. Neumair S. C., Vanicek S., Kaindl R., Többens D. M., Martineau C., Taulelle F., Senker J., Huppertz H., Eur. J. Inorg. Chem. 2011, 4147–4152. [Google Scholar]
- 45. Sohr G., Neumair S. C., Huppertz H., Z. Naturforsch. B 2012, 67, 1197–1204. [Google Scholar]
- 46. Sohr G., Perfler L., Huppertz H., Z. Naturforsch. B 2014, 69, 1260–1268. [Google Scholar]
- 47. Sohr G., Neumair S. C., Heymann G., Wurst K., Schmedt auf der Günne J., Huppertz H., Chem. Eur. J. 2014, 20, 4316–4323. [DOI] [PubMed] [Google Scholar]
- 48. Neumair S. C., Kaindl R., Huppertz H., Z. Naturforsch. B 2010, 65, 1311–1317. [Google Scholar]
- 49. Neumair S. C., Glaum R., Huppertz H., Z. Naturforsch. B 2009, 64, 883–890. [Google Scholar]
- 50. Pakhomova A., Bykova E., Bykov M., Glazyrin K., Gasharova B., Liermann H.-P., Mezouar M., Gorelova L., Krivovichev S., Dubrovinsky L., IUCrJ 2017, 4, 671–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Angel R. J., Ross N. L., Seifert F., Fliervoet T. F., Nature 1996, 384, 441–444. [Google Scholar]
- 52. Finkelstein G. J., Dera P. K., Duffy T. S., Phys. Earth Planet. Inter. 2015, 244, 78–86. [Google Scholar]
- 53. Deposition Numbers 2047708 (114.3 GPa), 2047709 (119.6 GPa) contain the supplementary crystallographic data for γ-ZrB2O5 and Deposition Numbers 2047710 (114.3 GPa), 2047711 (119.6 GPa) contain the supplementary crystallographic data for γ-HfB2O5. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
- 54. Hoppe R., Voigt S., Glaum H., Kissel J., Müller H. P., Bernet K., J. Less-Common Met. 1989, 156, 105–122. [Google Scholar]
- 55. Nespolo M., Guillot B., J. Appl. Crystallogr. 2016, 49, 317–321. [Google Scholar]
- 56. Shannon R., Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar]
- 57. Robinson K., Gibbs G. V., Ribbe P. H., Science 1971, 172, 567–570. [DOI] [PubMed] [Google Scholar]
- 58. Pakhomova A., Simonova D., Koemets I., Koemets E., Aprilis G., Bykov M., Gorelova L., Fedotenko T., Prakapenka V., Dubrovinsky L., Nat. Commun. 2020, 11, 2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pakhomova A., Aprilis G., Bykov M., Gorelova L., Krivovichev S. S., Belov M. P., Abrikosov I. A., Dubrovinsky L., Nat. Commun. 2019, 10, 2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gorelova L. A., Pakhomova A. S., Krivovichev S. V., Dubrovinsky L. S., Kasatkin A. V., Sci. Rep. 2019, 9, 12652. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary