

Abstract
Previous studies showed that the FeII/α‐ketoglutarate dependent dioxygenase AsqJ induces a skeletal rearrangement in viridicatin biosynthesis in Aspergillus nidulans, generating a quinolone scaffold from benzo[1,4]diazepine‐2,5‐dione substrates. We report that AsqJ catalyzes an additional, entirely different reaction, simply by a change in substituent in the benzodiazepinedione substrate. This new mechanism is established by substrate screening, application of functional probes, and computational analysis. AsqJ excises H2CO from the heterocyclic ring structure of suitable benzo[1,4]diazepine‐2,5‐dione substrates to generate quinazolinones. This novel AsqJ catalysis pathway is governed by a single substituent within the complex substrate. This unique substrate‐directed reactivity of AsqJ enables the targeted biocatalytic generation of either quinolones or quinazolinones, two alkaloid frameworks of exceptional biomedical relevance.
Keywords: biocatalysis, biosynthesis, enzyme mechanism, FeII/α-ketoglutarate dependent dioxygenases, natural products
AsqJ catalyzes a complex rearrangement sequence in quinolone biosynthesis. We show the AsqJ biocatalytic potential to significantly exceed this natural function. An unprecedented reaction pathway leading to quinazolinones is uncovered, functionally and mechanistically characterized in detail, revealing a unique substrate‐dependent product selectivity in enzyme catalysis.
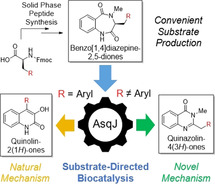
Introduction
Quinolones are one of the most important classes of antibiotics. Synthetic compounds, such as ciprofloxacin (1) or moxifloxacin (2, Figure 1 a), show broad spectrum activity against Gram‐negative and Gram‐positive bacteria by binding to bacterial topoisomerases and thereby interfering with DNA replication and hence cell division.[ 1 , 2 ] In recent years, naturally occurring constitutional isomers of such synthetic quinolin‐4(1H)‐ones have been discovered, the quinolin‐2(1H)‐ones. This includes the insecticidal and antibiotic yaequinolone B (3), [3] the cytotoxic aspoquinolone A (4), [4] and 4′‐methoxyviridicatin (5 a), which is assumed to be an intermediate in the biosynthesis of quinolones such as 4.[ 4 , 5 ] Because of the interesting structural and biological properties of natural quinolones, their biosynthesis has been studied intensively in recent years. [6] The dioxygenase AsqJ from Aspergillus nidulans is a key enzyme in the biosynthesis of 5 a.[ 5 , 7 ] Starting from a N 4‐methylated benzo[1,4]diazepin‐2,5‐dione 6 a, which is assembled by the non‐ribosomal peptide synthetase AsqK from a O‐methyl‐tyrosine and anthranilic acid building block, AsqJ induces an impressive reaction cascade (Figure 1 b). [7]
Figure 1.
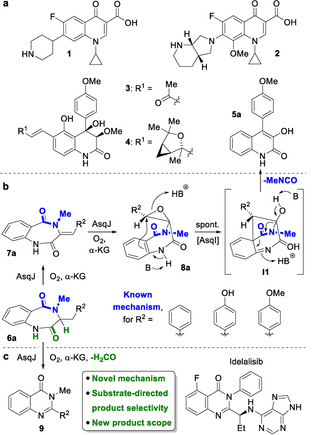
a) Structures of synthetic (1,2) and natural (3–5) quinolones. b) Mechanism of natural quinolone formation from 6 a to 5 a catalyzed by AsqJ. B=Basic residues in the AsqJ active site. [7] c) Unprecedented reactivity of AsqJ leading to the biomedically important quinazolinone scaffold 9.
Initial AsqJ‐catalyzed desaturation delivers 7 a, which is followed by further oxidation of the double bond to give epoxide 8 a (Figure 1 b, see Supporting Information, Figure S128, for detailed mechanism). This leads to a conformational ring‐flip that is favored by π‐stacking of the two aromatic rings and His134 within the catalytic pocket of AsqJ, forcing 8 a into a boat‐like conformation. Spontaneous fragmentation via intermediate I1 results in quinolone 5 a, overall leading to a remarkable ring contraction by excision of methylisocyanate. The final fragmentation can also be accelerated by the cyclopenase AsqI. [8]
Inspired by this fascinating mechanism of AsqJ together with the history and biomedical significance of the product class, we aimed to exploit this enzyme for biocatalytic generation of novel quinolones. In the course of this work, we discovered that the enzyme is very promiscuous as desired, but does not always catalyze quinolone formation; we found an unprecedented reaction pathway of AsqJ with certain classes of substituents to give an alternative heterocyclic product class: quinazolinones 9 (Figure 1 c). This structural framework is of biomedical importance, as exemplified by the anticancer drug idelalisib. Quinazolinone formation by AsqJ follows an enzymatic pathway that diverges from the known AsqJ mechanism, by redirecting the initial hydrogen abstraction by the iron‐oxo intermediate. We now report on the discovery of this unprecedented bimodal enzyme activity of AsqJ, its mechanistic elucidation and its application in biocatalytic quinazolinone synthesis.
Results and Discussion
Discovery of an Unprecedented AsqJ Reactivity
To validate the activity of AsqJ, we initially prepared a small set of substrates following the synthetic route published by Bräuer et al. [7] Besides the natural substrate of AsqJ 6 a, analogs 6 b and 6 c were prepared to test the current hypothesis[ 7 , 9 ] that a benzylic substituent R is required for enzymatic conversion. As expected, conversion of 6 a by AsqJ (see Supporting Information, chapter 3.1 for experimental details) led to the formation of the corresponding enzymatic dehydro‐reaction intermediate 7 a and the rearranged product 5 a (Figure 2). Compound 6 b lacking the aromatic O‐methyl group showed better conversion than the natural substrate 6 a while also leading to the anticipated products 7 b and 5 b. [5] The substrate analogs of 6 a and 6 b devoid of an N 4‐Me‐substitution did not show any conversion, as previously reported (data not shown, see SI, Figures S24, S25).[ 7 , 9 ] Substrate 6 c lacking a benzylic R group showed very efficient conversion to a single product (Figure 2). Unlike the conversion of 6 a and 6 b, this product had a reduced retention time when compared to its precursor and thus increased polarity. HPLC‐MS recorded a mass of m/z of 216.1, significantly higher than the expected rearrangement product (203.1 Da). This was in accordance with a potential formal loss of formaldehyde when compared to 6 c (246.1 Da) and thus suggested an exciting new reactivity of AsqJ.
Figure 2.
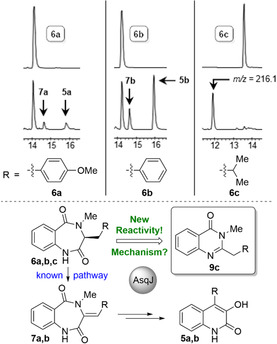
Initial AsqJ enzyme assays. Top: Enzymatic conversion of 6 a–c using recombinant AsqJ (retention times are given in minutes). Bottom: dual function of AsqJ leading to quinolones 5 a,b, or quinazolinone 9 c following a novel reaction mechanism.
To facilitate the identification of the novel product formed from 6 c, the enzymatic assay was scaled up from 100 μL to a total volume of 800 μL and the product purified by preparative HPLC for NMR analysis. The 1H‐NMR spectrum was closely related to that of the starting material 6 c. Surprisingly, the N 4‐methyl group was still present, as indicated by a singlet at 3.54 ppm (see SI, Figure S123). The 1H signal at the former C 3‐atom of the substrate vanished. Together with the high‐resolution mass data (m/z=217.1336 [M+H]+), this led to the assumption that AsqJ generates a quinazolin‐4(3H)‐one 9 c (calcd. for C13H17N2O+=217.1335 [M+H]+). To unambiguously corroborate this hypothesis, the proposed product 9 c was prepared by synthesis (see SI, Figure S2). Comparison of the analytical data of synthetic versus enzymatically produced 9 c clearly confirmed the proposed structure (see SI, Figure S126). We thus uncovered a novel reaction pathway catalyzed by AsqJ that gives access to a second class of heterocyclic products, quinazolinone alkaloids. This not only suggests an unprecedented additional reactivity of AsqJ, but is also of high significance for potential biotechnological applications. Quinazolinone scaffolds are found in many important biomedical agents, including the anticancer drugs idelalisib (cf. Figure 1; Zydelig, Gilead Sciences) gefitinib (Iressa, Astra Zeneca) and erlotinib (Tarceva, Roche) or the sedatives methaqualone and diproqualone. We thus set out to fully explore this exciting new reaction pathway of AsqJ.
Evaluating AsqJ Substrate and Product Scope
To explore the reactivity, specificity and mechanism of AsqJ, access to a broad range of substrates facilitating a systematic evaluation of substrate/product relationships (SPRs) was necessary. Given the unsatisfactory outcomes of the published synthetic sequence[ 7 , 10 ] in our hands (2–11 % yield for 6 a–6 c), we thus aimed to develop an efficient, flexible and reliable general synthetic alternative to N 4‐methylated benzo[1,4]diazepin‐2,5‐diones 6. We hence established a route taking advantage of Solid Phase Peptide Synthesis (SPPS) applying a polystyrene resin bearing 4‐hydroxybenzyl alcohol (Wang resin) combined with final cyclization and concomitant offloading from the resin (Scheme 1 a; see SI: chapter 4.1 for every substrate and step).[ 11 , 12 ]
Scheme 1.
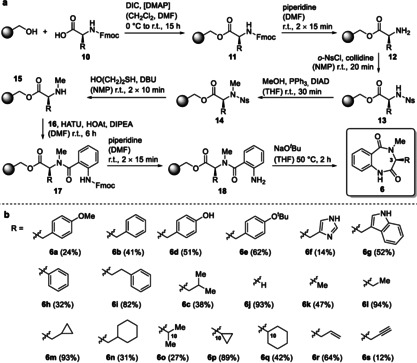
Synthesis of substrates to evaluate the reaction profile of AsqJ. a) SPPS‐based synthetic route to benzo[1,4]diazepine‐2,5‐diones 6. b) Synthesized substrates 6 a–s.
After loading the first amino acid 10 onto the resin using the standard Steglich esterification protocol with DIC and DMAP in DCM and DMF to 11, Fmoc deprotection with 20 % piperidine in DMF gave free amine 12. Introduction of the o‐nitrobenzylsulfonyl protection group (Ns) with NsCl and collidine in NMP led to sulfonamide 13. N‐Methylation proceeded under Mitsunobu conditions with PPh3 and DIAD in THF to the corresponding substituted sulfonamide 14. The Ns group was cleaved using mercaptoethanol with DBU in NMP to yield the N‐methylated amine 15. 2‐Fmoc‐aminobenzoic acid (16) was coupled to 15 using HATU, HOAt and DIPEA to give dipeptide 17. The Fmoc group was cleaved off as described above, resulting in the amine 18. In a final cyclization step using sodium tert‐butoxide as a base in THF, [12] the corresponding benzo[1,4]diazepine‐2,5‐dione 6 was generated. In case of substrates 6 d and 6 f, a final side chain deprotection with TFA in DCM was necessary. This synthetic approach proved to be highly streamlined also in terms of product purification, since only one final chromatographic step was needed. The reliability of this route was shown by the wide range of synthesized compounds, yielding a library of 19 congeners of the desired compound class, comprising aromatic and heteroaromatic (6 a, b, d–i) linear aliphatic (6 c, j–l, o), cyclic aliphatic (6 m, n, p, q), and linear alkene (6 r) and alkyne (6 s) substrates (Scheme 1 b).
With substrates 6 a–s in hands, an in‐depth evaluation of substrate specificity and product outcome of AsqJ was possible (Scheme 2 a). HPLC‐MS analyses of these reactions clearly established a strong substrate‐product relationship of AsqJ. All substrates with a benzylic residue at the C 3‐carbon (6 a, b, d, f, g) were converted to quinolin‐2(1H)‐ones 5 following the established enzymatic reaction pathway. Only 6 e carrying a sterically demanding O‐tert‐Bu protective group at the phenol showed no turnover by AsqJ. Furthermore, substrates with an aliphatic side chain with a tertiary carbon at position C 10, i.e., compounds 6 o–q, showed no conversion. Remarkably, all other tested substrates 6 c, h–n were shown to be converted to the new product class, quinazolin‐4(3H)‐ones 9.
Scheme 2.
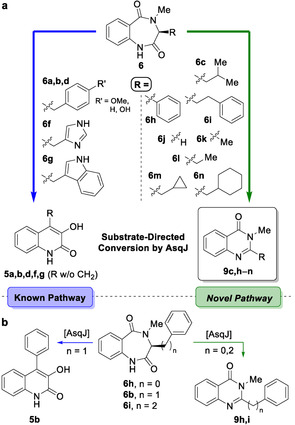
Evaluation of the substrate specificity of AsqJ. a) Substrate‐dependent product selectivity of AsqJ. b) Divergent enzymatic conversion of substrates 6 b, h, i, all bearing aromatic side chains.
In addition to the detection and characterization of these products by HPLC‐MS, the quinazolinones resulting from turnover of the methyl (6 k) and cyclohexyl (6 n) substituted substrates were also synthesized (see SI, Figure S2), leading to an unambiguous further corroboration of the structures of 9 k and 9 n, respectively. Taken together, the discovered novel transformation of AsqJ was thus firmly established as the major pathway across all tested substrates (Scheme 2 a).
Most surprisingly, substrates 6 h and 6 i, derived of phenylglycine and homophenylalanine, respectively, were likewise converted to the corresponding quinazolin‐4(3H)‐ones 9 h and 9 i, despite their aromatic substituent R (Scheme 2 b). AsqJ thus exclusively converts substrates with a methylene‐bridged aromatic substituent R following the naturally observed pathway to the quinolone structural motif, hence corroborating the published mechanistic preconditions for this rearrangement.[ 7 , 9 ] All other substrates are converted to the newly uncovered product class, raising questions on the molecular mechanism leading to these quinazolinones.
Elucidating the Novel Reaction Pathway
For the two accepted cyclic aliphatic substrates 6 m, n delivering quinazolinones 9 m, n, the respective dehydrogenated derivatives 7 m, n were also observed. In addition, the alkene and alkyne substrates 6 r and 6 s, respectively, exclusively delivered the corresponding desaturated intermediates 7 r and 7 s (see SI, Figures S39, S40, S44, S45), raising the question if these unsaturated enzymatic products are central intermediates in both the known and the novel enzymatic pathway. To test this possibility, the unsaturated analog 7 l was synthesized. This compound was the substance of choice, because its corresponding substrate precursor 6 l is efficiently converted to quinazolinone 9 l by AsqJ. Synthetic 7 l was obtained as a mixture of E/Z‐isomers (2:1, see SI, chapter 2.4) by NaOtBu‐induced elimination of tBuOH from threonine‐derived substrate 6 t. In contrast to 6 l, its unsaturated derivative 7 l was not converted by AsqJ (Scheme 3 a). It can therefore be precluded that the newly discovered enzymatic reaction pathway of AsqJ proceeds via an initial desaturation reaction, unlike the known natural reaction cascade.
Scheme 3.
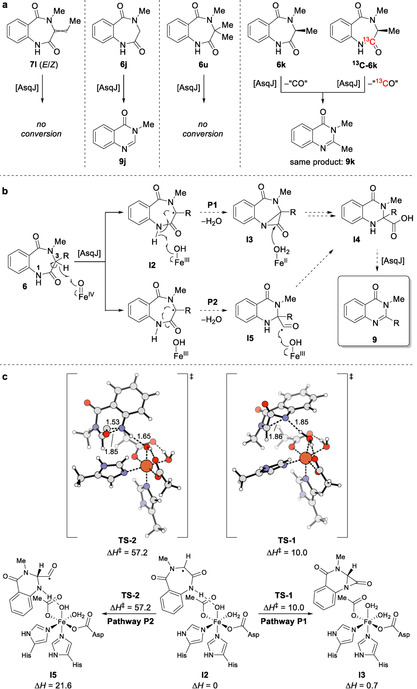
Pathway elucidation by in vitro analyses and theoretical investigations. a) Mechanistic probes and their conversion by AsqJ to obtain first mechanistic insights. b) Proposed enzymatic pathways P1 and P2 to the newly discovered product class. c) Calculated AsqJ model for P1 versus P2 at the uB3LYP‐D3(BJ)/def2‐TZVP//uB3LYP‐D3(BJ)/def2‐SVP level of theory. Pathway P1 is strongly preferred with a low barrier for α‐lactam formation.
Of particular interest was the turnover of glycine‐derived substrate 6 j, which likewise was converted by AsqJ following the quinazolin‐4(3H)‐one pathway to give 9 j, thus providing evidence that the new reaction proceeds without any mechanistic involvement of a side chain (Scheme 3 a). This indicated that the mechanism starts with radical formation at the C 3‐position. Indeed, evaluation of 6 u (synthesis shown in SI, chapter 2.1 and 2.3) with a dimethyl substitution at C 3 that precludes radical formation at this position as substrate for AsqJ did not show any conversion. This led to the assumption that initial radical formation at C 3 ultimately leads to excision of C 2=O from the ring structure during enzymatic conversion. To gain further evidence for this proposal, a 13C‐labelled derivative 13C‐6 k was synthesized starting from N‐Fmoc‐l‐alanine‐1‐13C (13C‐10 k) by SPPS according to Scheme 1 a and subjected to reaction with AsqJ. This experiment corroborated that exactly this carbon is excised to transform the 6.7‐ into the 6.6‐membered bicycle (Scheme 3 a).
Based on these combined results, the mechanism of the quinazolinone formation may proceed via one of the pathways shown in Scheme 3 b. The reaction sequence is assumed to begin with a hydrogen radical abstraction at C 3 of substrate 6 by the hypervalent iron(IV) oxo species in the active site of AsqJ, thus generating a carbon‐centered radical I2. From this intermediate, two different pathways (P1, P2) are conceivable. In pathway P1, a second radical hydrogen abstraction at the nitrogen followed by bond formation between the two resulting radical centers leads to formation of an α‐lactam intermediate I3. Hydrolytic cleavage of the amide bond delivers the enzyme‐bound carboxylic acid I4; subsequent deprotonation, addition of O2 to the AsqJ iron center, and elimination of CO2 yields the observed product 9 (cf. Figure S127 for mechanistic details). In pathway P2, the new N 1‐C 3‐bond is directly formed at the expense of the amide bond, resulting in formyl radical I5. Hydroxyl abstraction from the AsqJ iron center by the formyl radical unites the two proposed routes at carboxylic acid I4 (cf. Figure S127). Alternatively, pathway P2 may proceed directly from radical I2 to carboxylic acid I4 through concomitant amide bond homolysis and hydroxyl abstraction from the AsqJ iron center. Elimination of CO2, as described for P1 above, again yields the observed product 9.
To determine the feasibility of each proposed mechanistic route leading to 9, we undertook density functional theory calculations. Utilizing a previously‐validated representative system for AsqJ, [13] we constructed a model of the catalytic iron moiety and calculated the energy surface for R=H (6 j) at the uB3LYP‐D3(BJ)/def2‐TZVP//uB3LYP‐D3(BJ)/def2‐SVP level of theory (Scheme 3 c).[ 14 , 15 , 16 , 17 , 18 ] These calculations demonstrate that the formation of the formyl radical I5 is strongly disfavored, exhibiting an unsurmountable transition state barrier of 57.2 kcal mol−1 leading to a product >20 kcal mol−1 higher in energy than the initial radical I2. The P2 pathway—or variants proceeding directly to the carboxylic acid, which would require further bond reorganization—can thus not be operational. In contrast, formation of the α‐lactam I3 is only slightly endergonic (+0.7 kcal mol−1) with a transition state barrier of 10.0 kcal mol−1. The significant difference in energies required for these pathways provides convincing evidence that the reaction mechanism proceeds through the α‐lactam I3 and pathway P1. Hydrolytic cleavage of I3 by one of the proximal water molecules thereby leads to production of a carboxylic acid derivative I4 (see SI, Figure S127). [19] Subsequent elimination of carbon dioxide from this structure in analogy to a previously‐described non‐heme iron mechanism (with initial abstraction of β‐NH instead of β‐CH) would yield the observed product 9 j. [20] Most interestingly, re‐inspection of a previously unidentified second, more polar product of the transformation of 6 j with AsqJ revealed this compound to be carboxylic acid I4 j (see SI, Figure S35), hence experimentally validating the formation of this putative new pathway intermediate. Incubation of isolated I4 j with AsqJ did not result in turnover to 6 j or any other product (see Figure S49). It can therefore be assumed that I4 j cannot re‐enter the AsqJ active site if released before complete turnover to 6 j. Interestingly, the carboxylic acid intermediate I4 was exclusively detected for the glycine‐derived substrate 6 j (Scheme 3 b, R=H), suggesting an important role of the side‐chain substituent R in keeping all pathway intermediates efficiently bound to the enzyme active site during catalysis, hence facilitating full conversion to the respective quinazolinones 9. This function of R for efficient substrate binding as well as its role in the selection of previously known versus new reaction pathway are currently explored in our laboratories by further in‐depth structural and computational studies.
Conclusion
Enzymes catalyzing oxidative transformations play major roles in the biosynthesis of complex natural products. They are important in atom‐transfer reactions, such as halogenations or hydroxylations, contribute to shaping three‐dimensional structures, for example, by phenol‐oxidative cross‐linking, [21] and can even induce massive skeletal rearrangement reactions. The fungal dioxygenase AsqJ, known to catalyze formation of quinolone scaffolds 5 from N 4‐methylated benzo[1,4]diazepin‐2,5‐diones 6, was previously shown to catalyze a pathway leading to elimination of N‐methyl isocyanate following a remarkable rearrangement reaction (cf. Figure S128).[ 5 , 7 ] Our work shows that the biocatalytic potential of AsqJ can significantly exceed this natural biosynthetic function. Combining synthetic organic chemistry with experimental and computational mechanistic studies, we have uncovered a novel reaction pathway leading to quinazolin‐4(3H)‐one scaffolds 9. Remarkably, selection of the reaction pathway is achieved by a single, simple structural change in the substitution pattern of the benzo[1,4]diazepine‐2,5‐dione substrate. Substituents direct the site of initial radical formation in the corresponding substrates; different initial radicals undergo two entirely different reaction pathways. AsqJ thus represents a unique example of substrate‐dependent product selectivity in enzyme catalysis. While in Nature this dual function of AsqJ is suppressed by the selectivity of the non‐ribosomal peptide synthetase AsqK that exclusively provides the benzo[1,4]diazepin‐2,5‐dione 6 a as precursor, this remarkable enzymatic feature of AsqJ can now be fully exploited in vitro for the chemo‐enzymatic synthesis of biomedically valuable quinolone and quinazolinone alkaloids.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Prof. Michael Groll (TU München, Biochemistry) for providing the AsqJ expression plasmid enabling our studies, Dr. T. Lübken and his team (TU Dresden, Organic Chemistry I) for measuring NMR spectra and F. Drescher (TU Dresden, Bioanalytical Chemistry) for conducting HR‐MS measurements. This project was generously funded by the German Research Foundation (DFG, GU 1233/1‐1) and the US National Institute of General Medical Sciences, National Institutes of Health (GM‐124480). All calculations were performed on computing resources made available by the UCLA Institute of Digital Research and Education. Open access funding enabled and organized by Projekt DEAL.
M. Einsiedler, C. S. Jamieson, M. A. Maskeri, K. N. Houk, T. A. M. Gulder, Angew. Chem. Int. Ed. 2021, 60, 8297.
Dedicated to Professor Siegfried Hünig on the occasion of his 100th birthday
Contributor Information
Prof. Kendall N. Houk, Email: houk@chem.ucla.edu.
Prof. Tobias A. M. Gulder, Email: tobias.gulder@tu-dresden.de.
References
- 1. Pham T. D. M., Ziora Z. M., Blaskovich M. A. T., MedChemComm 2019, 10, 1719–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aldred K. J., Kerns R. J., Osheroff N., Biochemistry 2014, 53, 1565–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Uchida R., Imasato R., Tomoda H., Ōmura S., J. Antibiot. 2006, 59, 652–658. [DOI] [PubMed] [Google Scholar]
- 4. Scherlach K., Hertweck C., Org. Biomol. Chem. 2006, 4, 3517–3520. [DOI] [PubMed] [Google Scholar]
- 5. Ishikawa N., Tanaka H., Koyama F., Noguchi H., Wang C. C. C., Hotta K., Watanabe K., Angew. Chem. Int. Ed. 2014, 53, 12880–12884; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13094–13098. [Google Scholar]
- 6. Zou Y., Garcia-Borràs M., Tang M. C., Hirayama Y., Li D. H., Li L., Watanabe K., Houk K. N., Tang Y., Nat. Chem. Biol. 2017, 13, 325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bräuer A., Beck P., Hintermann L., Groll M., Angew. Chem. Int. Ed. 2016, 55, 422–426; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 432–436. [Google Scholar]
- 8. Kishimoto S., Hara K., Hashimoto H., Hirayama Y., Champagne P. A., Houk K. N., Tang Y., Watanabe K., Nat. Commun. 2018, 9, 2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mader S. L., Bräuer A., Groll M., Kaila V. R. I., Nat. Commun. 2018, 9, 1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Al-Said N. H., Monatsh. Chem. 2010, 141, 1249–1251. [Google Scholar]
- 11. Biron E., Chatterjee J., Kessler H., J. Pept. Sci. 2006, 12, 213–219. [DOI] [PubMed] [Google Scholar]
- 12. Mayer J. P., Zhang J., Bjergarde K., Lenz D. M., Gaudino J. J., Tetrahedron Lett. 1996, 37, 8081–8084. [Google Scholar]
- 13. Wojdyla Z., Borowski T., J. Biol. Inorg. Chem. 2018, 23, 795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Becke A. D., J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- 15. Grimme S., Ehrlich S., Goerigk L., J. Comput. Chem. 2011, 32, 1456–1465. [DOI] [PubMed] [Google Scholar]
- 16. Lee C., Yang W., Parr R. G., Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- 17. Stephens P. J., Devlin F. J., Chabalowski C. F., Frisch M. J., J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar]
- 18. Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- 19. Lengyel I., Sheehan J. C., Angew. Chem. Int. Ed. Engl. 1968, 7, 25–36; [Google Scholar]; Angew. Chem. 1968, 80, 27–37. [Google Scholar]
- 20. Huang J.-L., Tang Y., Yu C.-P., Sanyal D., Jia X., Liu X., Guo Y., Chang W.-C., Biochemistry 2018, 57, 1838–1841. [DOI] [PubMed] [Google Scholar]
- 21. Aldemir H., Richarz R., Gulder T. A. M., Angew. Chem. Int. Ed. 2014, 53, 8286–8293; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 8426–8433. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary