

Abstract
Nucleosidic and oligonucleotidic diarylethenes (DAEs) are an emerging class of photochromes with high application potential. However, their further development is hampered by the poor understanding of how the chemical structure modulates the photochromic properties. Here we synthesized 26 systematically varied deoxyuridine‐ and deoxycytidine‐derived DAEs and analyzed reaction quantum yields, composition of the photostationary states, thermal and photochemical stability, and reversibility. This analysis identified two high‐performance photoswitches with near‐quantitative, fully reversible back‐and‐forth switching and no detectable thermal or photochemical deterioration. When incorporated into an oligonucleotide with the sequence of a promotor, the nucleotides maintained their photochromism and allowed the modulation of the transcription activity of T7 RNA polymerase with an up to 2.4‐fold turn‐off factor, demonstrating the potential for optochemical control of biological processes.
Keywords: diarylethenes, in vitro transcription, nucleic acids, photochromism, photoresponsive DNA
From a library of 26 deoxyuridine‐ and deoxycytidine‐derived diarylethenes we identified two high‐performance photoswitches with near‐quantitative, fully reversible back‐and‐forth switching, high reaction quantum yields and no detectable thermal or photochemical deterioration. Incorporated into an oligonucleotide, these nucleotides allowed the optochemical modulation of the activity of T7 RNA polymerase.

Introduction
The development of tools for the selective manipulation and control of biomolecules is critical for the progress in many areas of the life sciences. Among those, photochromic molecules are of major interest, as they use light as modulation channel.[ 1 , 2 , 3 , 4 , 5 ] The non‐invasive nature of light and the high spatio‐temporal resolution offer key advantages over electrical or chemical stimuli.[ 6 , 7 ] However, biomolecules, and in particular nucleic acids, usually do not possess photochromic properties. Therefore, nucleic acids need to be modified with suitable photoactive moieties in order to gain remote control over their structure and function. A covalent linkage to photoswitchable entities is a common approach to make biomolecules sensitive towards reversible photochemical stimuli. Azobenzenes[ 8 , 9 , 10 , 11 , 12 , 13 , 14 ] and diarylethenes (DAEs)[ 15 , 16 , 17 , 18 , 19 ] have most commonly been used for this purpose, while spiropyrans,[ 20 , 21 , 22 , 23 ] fulgides,[ 24 , 25 ] and hemithioindigo‐based switches [26] were applied less frequently.
The core structure of the classical DAE photoswitches shown in Figure 1 A evolved over the years from the stilbenes. It is characterized by a conjugated hexatriene system and was originally developed as an optical information storage medium in materials sciences.[ 27 , 28 , 29 , 30 ] Recently, such systems have also been used in biological applications, such as photopharmacology [31] and single molecule localization microscopy. [32] DAEs undergo a reversible photochemical electrocyclic ring closure, which enlarges the conjugated π electron system, rendering the compound colored in the closed form and colorless in the open.
Figure 1.

DAE photoswitches. A) Classical design and isomerization reaction. The reactive α carbon atoms are marked. B) Deazapurine nucleoside‐based photoswitches.[ 33 , 34 ] C) Pyrimidine nucleoside‐based photoswitches. [35]
The photophysical and photochemical properties of DAE photoswitches can be modulated and optimized by variation of the substitution pattern (R).[ 36 , 37 , 38 ]
In 2010, our group reported the first DAE‐based photochromic nucleosides in which a purine nucleobase (of a nucleoside) represented one of the two aryl systems of the diarylethene (Figure 1 B). Importantly and different from most other approaches towards photoswitchable nucleic acids, the nucleobase formed an active constituent of the photoswitch (rather than an appendage), changing bonding and hybridization upon switching.[ 33 , 34 ] This new class of purine nucleoside DAEs, and especially the deoxyadenosine‐derived ones, turned out to have favorable switching characteristics with respect to their photostationary states (PSS), reversibility and fatigue resistance. [39]
In 2013 our group developed a 2nd type of DAE‐based photochromic nucleosides, which incorporated two rare features: a six‐membered heterocyclic pyrimidine ring acted as one of the aryls, and only one rather than the common two alkyl group was present at the reactive α carbon atoms (Figure 1 C). Such a structural setup of a DAE was unprecedented at the time. To our surprise, these pyrimidine‐based DAEs showed significant photochromism, high reversibility and thermal stability of the closed‐ring isomer. However, a major drawback of this system was that UV‐irradiation led to a PSSUV that contained ≤50 % of the closed‐ring form under the conditions used at that time, thereby limiting the use of these compounds in applications where a higher dynamic range of response is required. [35] Additionally, only a few examples were synthesized (phenyl‐, naphthyl‐ and methyl‐substituted at the 5‐position of the thiophene, as shown in Figure 1 C), and a comprehensive photophysical characterization was lacking. Therefore, the previously gathered data are not sufficient to understand and generalize the properties of these pyrimidine nucleoside‐based DAEs. In order to address these shortcomings, we undertook a systematic investigation of structure–activity relationships by chemical variation. Our primary goal was to understand which structural features render a candidate a good photochrome, including the following key properties: quantitative switching in both directions, high quantum yields for cyclization and cycloreversion, intensive color (i.e., high ϵ max, vis) of the closed‐ring form, thermal stability of the closed isomer, reversible switching and fatigue resistance. The most promising pyrimidine nucleoside DAEs should then be incorporated into oligonucleotides and their properties compared to the previous generation.[ 40 , 41 ] As the control of biological structure and function by light would benefit tremendously from near‐quantitative on/off switching, we aimed at generating high‐performance photochromic DAE oligonucleotides for future applications in synthetic biology and nanotechnology.
Results and Discussion
In general, the absorption of chromophores can be modulated either by extending the conjugated π‐system, or by incorporating auxochromic groups, i.e., electron‐withdrawing or electron‐donating groups.[ 42 , 43 ] Both approaches have been applied extensively to (non‐nucleosidic) DAEs.[ 27 , 44 ] Based on literature precedents and synthetic feasibility, we selected 13 substituents for incorporation into 2′‐deoxyuridine (dU) and 2′‐deoxycytidine (dC) DAE photoswitches (Scheme 1 A).
Scheme 1.

Design and synthesis of dU‐and dC‐based DAE switches. A) Arrangement of the substituents on the thiophene moiety from electron donating (EDG, red) to electron withdrawing (EWG, blue). B) General synthetic approach involving a Suzuki coupling. C) Synthesis of the boronic acid pinacolate esters with residues that are inert against n‐BuLi (Top: Non‐sensitive R) and residues that require borylation via Miyaura borylation (Bottom: sensitive R). a) Ar‐X, Na2CO3 (aq.), Pd(dppf)Cl2, 80 °C, THF; b) n‐BuLi, B(OBu)3, −78 °C, THF; c) dibromocyclopentene, Na2CO3 (aq.), Pd(dppf)Cl2, 80 °C, THF; d) n‐BuLi, BPinOiPr, −78 °C, THF; e) B2Pin2, KOAc, Pd(dppf)Cl2, 120 °C, dioxane; f) dibromocyclopentene, Na2CO3 (aq.), Pd(dppf)Cl2, 80 °C, THF; g) B2Pin2, KOAc, Pd(dppf)Cl2, 120 °C, dioxane.
Synthesis of the Nucleosidic DAEs
Position 5 of the thiophene ring was chosen as the primary site of structural variation, since the nucleobase, including its ability to base‐pair, should be kept as “natural” as possible (as demonstrated previously). [33] Specifically, we introduced the electron‐donating conjugated benzodithiole (Bdt), N‐N‐dimethylamino‐naphthalene (NpNMe2) and N‐N‐dimethylaniline (PhNMe2) groups. We varied the length of the conjugated system from methyl (Me) via phenyl (Ph) and 2‐naphthyl (Np) to anthracenyl (An) and introduced electron‐withdrawing 4‐pyridyl (4Py), 2‐pyridyl (2Py), bis(trifluormethyl)‐phenyl (Ph(CF3)2), tert‐butylester‐phenyl (PhtBu), p‐nitrophenyl (PhNO2) and cyano (CN) groups. This panel allows a comprehensive investigation of π‐system lengths and auxochromic effects on various photochemical and photophysical parameters. The general synthetic approach is based on our earlier work. [33] First, the modified boronic acid pinacolate esters of the cyclopentene‐bridged thiophene 4R had to be synthesized. These compounds were coupled to 5‐iodo‐2′‐deoxyuridine and 5‐iodo‐2′‐deoxycytidine, respectively, in a Suzuki cross‐coupling reaction (Scheme 1 B). Two main routes were established for the synthesis of the 13 boronic acid pinacolate esters (Scheme 1 C):
For residues that can withstand treatment with n‐BuLi, the synthetic procedure described by Singer et al. [33] was used, in which the thiophene derivatives 2R were furnished by Suzuki cross‐coupling of (4‐bromo‐5‐methylthiophen‐2‐yl) boronic acid 1 with the corresponding aryl halides (Ar‐X). Then, in a one‐pot reaction, 2R was activated (as boronic acid) in situ, and the cyclopentene bridge was attached in a second Suzuki reaction with 1,2‐dibromocyclopentene to give 3R. In the last step, 3R was converted to the boronic acid pinacolate ester 4R.
For more sensitive residues R (CN, PhNO2, Ph(CF3)2, PhtBu and Bdt), pinacolate ester 4R had to be synthesized via a different route, namely by Miyaura borylation and subsequent Suzuki cross‐coupling. [45] For synthetic details and standard analytical characterization, see Supporting Information.
Photochromism
A first assessment of the photochromism of the synthesized compounds was performed by UV/Vis spectroscopy of each nucleosidic DAE upon UV light irradiation (310–340 nm, Figure 2, Figure S1,S2). In both the dUR and the dCR series, all compounds except the anthracene derivative showed photochromism, i.e., the formation of a new absorption band in the visible wavelength range (460–510 nm) upon irradiation with UV‐light, indicative of the electrocyclic ring closure reaction. For nearly all compounds, this irradiation resulted in a PSSUV (for exceptions see below). In both classes of switches (dUR and dCR) there was a clear influence of the substituent on the absorption spectrum. Variation of R allowed to shift the closed‐ring absorption maximum over a range of 85 nm (for dU switches) and 70 nm (for dC switches) (Table 1).
Figure 2.

Absorption spectra, reversibility and thermal stability of dU‐based (A, C, E) and dC‐based (B, D, F) photoswitches. This selection includes switches with electron donating (A, B), electron neutral (C, D) and electron withdrawing (E, F) substituents. The absorption spectra of the open (black line) and calculated closed (red line) isomer and the PSSUV (dashed blue line) are shown. In each panel the reversibility (top right) and the thermal stability (low right) are shown. The yellow area indicates the spectral separation of dU . For obtaining the PSSUV absorption spectra, the samples were irradiated with their optimal wavelength (see footnotes of Table 1) at 50–70 μM in water/ethanol mixtures.
Table 1.
Photophysical properties of the dU and dC photoswitches.
|
|

Residues that enlarge the conjugated system, for example, from methyl over phenyl to naphthyl, shifted the closed‐ring absorption maximum to longer wavelengths (Figure S1,S2). Hammett parameter correlations indicated a relationship between the electronic character of the substituent at the thiophene moiety and the newly formed absorption maximum (Figure 3 B). (Modified) Hammett parameters σ′ were obtained from 13C shifts of the carbon in para position to the substituents (Figure 3 A). [46] On this scale, a value of σ′>1 implies an electron‐withdrawing character of the substituent, while σ′<1 indicates an electron‐donating character. Auxochromes like PhNMe2, PhtBu (Figure 2 E/F, Figure 3 B), or PhNO2 caused a shift of the absorption maximum to higher wavelength due to their −M or +M (mesomeric) effects, while more electron‐neutral substituents like Ph showed a hypsochromic shift of the absorption maximum (Figure 2 C/D). Similar tendencies have been observed earlier for symmetrical dithienylethenes.[ 37 , 38 ] Only dUAn and dCAn did not show any photochromicity, while dUBdt showed a biphasic behavior where first a broad new band with a maximum at 550 nm appeared, which then gradually disappeared in favor of another intensive band at 525 nm, suggesting rapid decomposition of the initially formed closed‐ring form (Figure S1).
Figure 3.
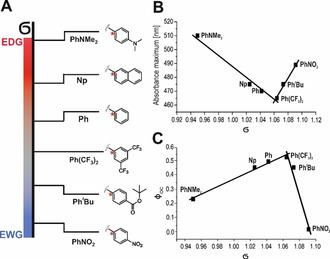
Dependence of selected photophysical properties on the electronic character of the substituents of dU switches. A) Schematic arrangement of the electron withdrawing (EWG) and electron donating (EDG) substituents on the thiophene moiety according to their 13C‐NMR‐derived Hammett parameters (σ′) for all derivatives that can be analyzed by this formalism. The carbon atom C4 that was used for the calculation of the Hammett parameters is marked with a star. B) Hammett correlation of σ′ with the absorption maximum in the visible wavelength range of the closed‐ring form. C) Hammett correlation of σ′ with the ring‐closing quantum yields (ΦOC).
In direct comparison with the dU switch carrying the same substituent R, the absorption maximum of the dC switch is bathochromically shifted, for example, 475 nm for dUNp vs. 494 nm for dCNp (see Figure 2 A/B, Table 1). This bathochromic shift is likely caused by the electronic differences between the two nucleobases. The carbonyl function in uridine exerts a −I (inductive) and −M effect, whereas the amino group of cytidine has a −I and +M effect. Both groups act as auxochromes, however the amino group of cytidine seems to have a stronger influence on the absorption spectrum. An exception from this rule is the Ph(CF3)2 substituent where λ max, vis of the dC switch is hypsochromically shifted, relative to the dU switch.
Ring Opening and Reversibility
Upon irradiation with Vis light matching the absorption maximum of the closed‐ring form, complete discoloration was observed for 8 of the 12 photochromic dU switches and for 3 of the 12 dC switches, indicating that for these compounds, the PSSvis was close to 100 % open‐ring isomer (Figure 2 A/C/D/E, Figure S3,S4). These compounds could be switched back and forth by alternating UV and Vis light irradiation over several cycles, essentially reaching the same absorption maxima and minima after each cycle. This behavior indicates the absence of significant irreversible side reactions. The remaining compounds exhibited different patterns of irreversibility and fatigue (Figure 2 B/F, Figure S3,S4). These patterns were particularly pronounced for the electron‐donating substituents Bdt, NpNMe2, PhNMe2 (Figure S3,S4). In general, the members of the dC series exhibited decreased reversibility and increased fatigue, compared to the dU series (Figure 2 A/B, E/F). The introduction of electron‐withdrawing trifluormethyl substituents at aromatic rings, which was reported to significantly improve the fatigue resistance of dithienylethenes, [47] surprisingly reduced the reversibility in our nucleosidic DAEs (Figure S5, compare dCPh and dC ).
Thermal Stability
DAEs are known to undergo not only photochemical, but also thermal reactions. In particular, the closed‐ring form may revert via a thermal pathway back to the open‐ring isomer. This is an undesired property for some applications while it is beneficial for others. We investigated the thermal stability of all photochromic nucleosides by monitoring the absorbance at the closed‐ring maximum over one hour at 25 °C, 50 °C and 70 °C (Figure S6,S7). Most dU switches showed <5 % decrease in absorbance over 1 h even at 70 °C (Figure 2 A/C/E), while many dC switches deteriorated significantly at 50 °C (Figure 2 B/D/F). The thermal stability of DAEs is known to correlate with the aromatic stabilization energy of the heterocycles. The superiority of uridine switches in terms of thermal stability is based on their lower aromatic stabilization energy. [48]
Switching Yields—Composition of the PSSUV
After this first qualitative assessment of the photochromic properties we selected those compounds that were sufficiently thermostable and fatigue‐resistant for a quantitative characterization. For 11 dU and 10 dC switches, the composition of the PSSUV induced by UV irradiation could be determined by HPLC (Table 1, Figure S8,S9). The dU switches generally showed higher switching yields than the dC switches (see dotted blue spectra in Figure 2). The percentage of closed‐ring form in the PSSUV varied in the dU series from 14 % for dU to 93 % for the dU . Substituents without a heteroatom (dUMe, dUPh, dUNp) and thus without significant electron‐donating or ‐withdrawing effects generally showed switching yields between 50–78 % (Table 1). Our best photochromic nucleosides (dU , dU2Py, dU4Py) showed an almost quantitative conversion to the closed form (Table 1), which is comparable to many non‐nucleosidic DAEs. [44] Based on the known composition of the PSSUV, we calculated the spectra of the pure closed‐ring isomers. The extinction coefficients ϵ at the absorption maximum of closed‐ring form of the dU series were generally higher than those of the dC series (Figure 2, Table 1). As expected, DAE switches without aromatic substituents generally had the lowest ϵ values, followed by one‐ring aromatic and heteroaromatic substituents, and the highest values were determined for larger conjugated and push‐pull‐systems. For our two best compounds (in terms of PSSUV composition), dU and dU2Py, the strong absorption band in the UV‐region was hypsochromically shifted upon irradiation with UV light (by 29 nm and 19 nm, respectively), causing an increased spectral separation between the open and closed isomers in the region around 340 nm (see yellow area in Figure 2 E). This results in a favorable situation for the cyclization reaction, since in this case the open isomer has a higher extinction coefficient and therefore absorbs stronger than the closed one, driving ring closure more strongly than ring opening and significantly effecting the composition of the PSSUV. Such spectral shifts were less pronounced in the dC series, explaining their lower switching efficiency (Figure 2 B/D/F). Besides the spectral separation, which is reflected in the ratio of the extinction coefficients of the two isomers, the ratio of the quantum yields of ring opening and ring closing plays a significant role for the composition of the PSSUV as illustrated by Equation 1, which holds true in the absence of side reactions:
| (1) |
Quantum yields
Quantum yields were determined using a modified quantum yield determination setup by Megerle et al. [49] In order to determine reaction quantum yields, the excited state of the photochromic nucleosides should not enter additional irreversible reaction pathways. Therefore, quantum yields were determined for 16 selected compounds (Table 1). UV‐induced cyclization quantum yields were generally high for both the dU and dC series, ranging from 0.23 (dU ) to 0.65 (dU2Py) with the exception of dU (φ OC=0.02). These values agree well with those for classical DAEs.[ 30 , 37 ] Additionally, we observed a correlation between the Hammett parameter (σ′) and φ OC (Figure 3 C). Particularly high cyclization quantum yields were found for switches with electron‐neutral substituents, while strong auxochromes such as PhNMe2 or PhNO2 led to a much lower quantum yield φ OC. For most compounds, UV irradiation stimulated both cyclization and ring opening. The ratio of cyclization and cycloreversion quantum yields measured upon UV irradiation was found to differ significantly between the dU and the dC series. While it is usually >1 for the dU switches, it is smaller for the dC switches. This shows that for uridine‐based photochromes the cyclization is preferred over the cycloreversion, which is not the case for the cytidines, furthermore explaining their different PSSUV compositions. Those uridine switches that do not have a phenyl ring at the periphery of the thiophene seem to be an exception in this respect, since their quantum yields of the ring opening reaction are significantly increased (dUMe φ CO=0.38, dUCN φ CO=0.78). The photochromic nucleosides dUPh, dUNp, dU and dU2Py showed a clear preference for the cyclization reaction with ratios between 1.35 and 2.14. Thus, together with a large spectral separation in the UV range, a high ratio of cyclization and cycloreversion quantum yields is responsible for the almost quantitative switching of dU2Py and dU , which makes them our best‐performing candidates for the incorporation into oligonucleotides. An enormous difference in comparison to classical DAEs is apparent for the cycloreversion quantum yields φ CO when irradiated at the absorption band in the visible wavelength range (470 nm–505 nm). There, the quantum yields of the nucleoside‐based DAEs are more than one order of magnitude larger than those of non‐nucleosidic DAEs (which are typically only a few percent). [27] A similar tendency has already been observed by Buckup et al. by transient absorption experiments of some dU switches. [50] This overall high quantum yield for the ring opening reaction is especially interesting for applications where a fast back‐and‐forth switching of the two isomers is desired. Considering the most important properties for applications in biology (near‐quantitative light‐induced switching in both directions, reversibility, thermal stability) combined, our investigation revealed two dU switches that were excellent with respect to all three properties, namely dU2Py and dU , while dUPh, dUMe, dU4Py and dU had a slightly lower overall performance. dU2Py and dU not only rival many non‐nucleosidic DAEs, [27] but beat most azobenzene‐based photochromic nucleosides.[ 14 , 31 , 51 ] The most important difference to the literature‐known azobenzene C‐nucleosides [12] and C5‐azobenzene‐substituted dU switches [52] is the quantitative switching upon Vis irradiation (ring opening for DAE, cis → trans for azobenzenes), which allows for a higher dynamic range. Our high performance dU switches also show an increased thermal stability compared to ortho methylated azobenzene switches, for example, 4‐carboxy‐2′,6′‐dimethylazobenzene (2′,6′‐Me‐Azo), which were incorporated into DNA via the phosphate‐backbone. The thermal isomerization half‐life of dU2Py at 70 °C is more than 3 times higher (123 h) than that of 2′,6′‐Me‐Azo (36 h) which was determined at only 60 °C. [53] The 2 excellent and the 4 good photochromic dU derivatives all have visible absorption maxima of the closed‐ring form ≤475 nm, indicating that our strategies for bathochromic shifting (extension of the π‐system, addition of auxochromic groups) achieved the desired goal only at the cost of a reduced overall performance. The members of the dC series typically perform worse than the dU derivatives in all three criteria. dC2Py is the only promising candidate, if experiments at or slightly above room temperature are planned.
Photochromic Oligonucleotides
For use in automated solid‐phase synthesis, phosphoramidites containing the dU2Py and dU structures had to be synthesized. Starting from commercial 5‐iodo‐2′‐deoxyuridine, the 5′‐hydroxyl group was protected as the dimethoxytrityl ether, followed by a Suzuki cross‐coupling reaction with the corresponding boronic acid pinacolate ester 4R and reaction with 2‐cyanoethyl N,N‐diisopropylchlorophosphoramidite (Figure 4 A), generating phosphoramidites 10R. Incorporation of 10R into 19mer oligonucleotides was accomplished with high incorporation efficiency indistinguishable from commercial standard phosphoramidites (Figure S10). After HPLC purification, the identity of the modified oligonucleotides was confirmed by mass spectrometry (Figure 4 B). Both oligonucleotides showed photochromism, and a 6 nm bathochromic shift of λ max,vis of the photoswitchable DNA compared to the free nucleoside (Figure 5). The percentage of closed‐ring form in the PSSUV after UV irradiation was only slightly decreased (by 5–6 %), compared to the nucleoside (Figure S11). However, the photochromic oligonucleotides showed strongly reduced quantum yields (4‐fold for dU2Py and 2‐fold for dU ). These effects were primarily due to the different steric and electronic environment of a nucleoside DAE inside an oligonucleotide, while the effects of solvents and buffers [54] were less important (Figure S12,13). Since the ratio of ring closure and ring opening quantum yield upon UV irradiation in the oligonucleotide was still >1, only minor changes in the PSSUV composition were observed. Due to the reduced quantum yields, slightly longer irradiation times were necessary to reach the PSSUV, compared to the nucleosides. Both modified oligonucleotides showed excellent thermal stability of their closed‐ring isomer at 25 °C and 37 °C and high reversibility (Figure 5). Upon hybridization with a fully complementary 19mer strand, the duplexes retained their photochromicity. Duplex formation, however, influenced the composition of the PSSUV differently for the two modifications (Figure 4 C). In the dU ‐containing duplex, the percentage of closed‐ring isomer was reduced by 17 %, compared to the single‐strand. No significant decrease in the quantum yields of the ring opening reactions (upon UV and Vis irradiation) was detected. However, the quantum yield for ring closure was reduced by a factor of 2.5, explaining the altered PSSUV composition. For the dU2Py‐containing strand, duplex formation led to an PSSUV after UV‐irradiation containing only 20 % closed‐ring isomer, which could be explained by the strong decrease of the ring closing quantum yield (0.17 (ss)→0.02 (ds)) and the concurrent increase of the ring opening quantum yield (0.12 (ss)→0.21 (ds), Figure 4 C). Compared to Azo‐ and DM‐Azo‐derivatized oligonucleotides,[ 14 , 31 , 51 ] our photochromic T7‐PhtBu oligonucleotide has a higher switching efficiency upon UV irradiation (88 % vs. 77 % in the single strand and 70 % vs. 30 % in the duplex).
Figure 4.

Design, synthesis and properties of photochromic oligonucleotides. A) General synthetic approach. a) DMT‐Cl, DMAP, rt, pyridine; b) 4R, Cs2CO3 (aq.), Pd(dppf)Cl2, 120 °C, acetonitrile/water (2/1); c) CEP‐Cl, N(Et)(iPr)2, rt, DCM; d) complementary strand, 95 °C, buffer. B) Synthesized photochromic DNA strands with determined masses. C) Photophysical properties and melting temperatures of the synthesized DNA strands.
Figure 5.

Absorption spectra, reversibility and thermal stability of T7‐2Py‐(ss) (A) and T7‐PhtBu‐(ss) (B). The absorption spectra of the open (black line) and calculated closed (red line) isomer and the PSSUV (dashed blue line) are shown. The inset shows the zoomed‐in range between 300 and 750 nm and the spectral separation is highlighted in yellow. In each panel, the reversibility (top right) and the thermal stability (low right) are shown. For obtaining the PSSUV absorption spectra, the samples were irradiated with 340 nm at 35–40 μM in TRIS‐buffer (40 mM, pH 7) with 22 mM MgCl2.
Upon Vis irradiation, our oligonucleotides exhibit quantitative switching, while this cannot be achieved for the azobenzenes due to a spectral overlap of the two isomers. In addition, our photochromic oligonucleotides show an increased thermal stability (half‐lives at 37 °C: 88.5 h for T7‐PhtBu (ss) and 67 h for T7‐2Py (ss) vs. 36.5 h for S‐Azo‐DM in the single strand).
To analyze the influence of the bulky DAE substitution on DNA duplex stability, melting curves were recorded. In the open form, the incorporation of dU2Py did not alter the melting temperature, whereas dU induced a destabilization of the duplex (ΔT=−3.7 °C). This could be due to the steric demand of the tert‐butyl ester group. The cyclization of the DAE unit caused a further destabilization of the duplex by approximately 1 °C for both compounds (Figure S14).
Photocontrol of Transcription In Vitro
The sequence of the 19mer photochromic oligonucleotides was chosen to represent the non‐templating strand of a promoter for the DNA‐dependent RNA polymerase of bacteriophage T7, an enzyme commonly used for RNA synthesis by in vitro transcription. After combination with a templating strand, a photochromic promoter is formed. Our goal was to test whether an “optochemical” control of enzymatic RNA synthesis is possible with dU2Py‐ or dU ‐modified promoters. There are—to our knowledge—only very few reports on transcription control by incorporation of a single reversible photoswitch. Asanuma and co‐workers inserted a threoninol‐linked azobenzene at various positions of both templating and non‐templating strand of a T7 promotor, and monitored the enzymatic synthesis before and after UV irradiation of the DNA duplex. [31] At different positions, transcription was suppressed in the trans‐form but proceeded faster in the cis‐form. The activation factors for single modifications were mostly between 1.5‐ and 2.3‐fold, and the activated form was at least 30 % less active than the unmodified promoter. Cahová and Jäschke observed that replacement of dT at position −2 by dUPh inhibited transcription activity by ≈50 %, while UV irradiation restored activity almost quantitatively. [35] Based on the crystal structure of the T7 polymerase [55] we decided to incorporate the photochromic nucleosides dU2Py and dU at position −8 of the non‐template strand, one of the most responsive positions for photoregulation observed in the azobenzene study. [31] The modified non‐template strand was hybridized with the template strand consisting of the complementary promotor sequence linked to the sequence encoding the malachite green aptamer (MGA). [56] Transcribed MGA forms a fluorescent complex with its target malachite green (MG), so that progress of the transcription can be monitored continuously in real time by fluorescence (Figure 6 A).[ 57 , 58 , 59 ] After a short initiation phase, a linear increase in fluorescence was observed, from which we calculated the initial transcription rates. The photoregulation efficiency (α) of the transcription was defined as the ratio of initial transcription rates (k obs) of irradiated and non‐irradiated template. In the open‐ring form, both modified promoters showed almost the same activity as the unmodified T7 promotor (relative activity β of 96 % and 92 %, respectively, Figure 6 B), indicating that modifications at this position do not perturb the interaction with the enzyme. However, in the PSSUV after UV irradiation, a strong decrease of the transcription rate up to 2.39 fold for the dU and 1.66 fold for dU2Py was observed. These results demonstrate that photoregulation of transcription is possible using our optimized nucleosidic DAEs and suggest that reversible photoswitching in situ can be achieved after further optimization.
Figure 6.

Photocontrol of transcription A) Design of the in vitro transcription assay. The DAE‐modified strand (red) is hybridized to a complementary strand (blue) that is linked to the sequence of the malachite green aptamer (MGA). Transcription by T7 RNA polymerase generates the aptamer which binds to its ligand malachite green (MG), causing a fluorescence increase. B) Time course of the in vitro transcription visualizing the increase of fluorescence intensity. The photoregulation efficiency was defined as α and the relative activity compared to the positive control was defined as β. C) Measured rate constants for transcriptions.
Conclusion
In this work we provided the first systematic structure–activity study of pyrimidine nucleoside‐based DAE photoswitches. A collection of 26 different dU‐ and dC‐based DAEs was synthesized of which 24 showed the typical photoinduced electrocyclic ring closure and opening. While we could shift the visible absorption maximum of the closed‐ring form considerably into the bathochromic direction by addition of auxochromes or extension of the π‐system, none of these red‐shifted compounds had the desired combination of properties required for biological applications (near‐quantitative switching in both directions, full reversibility, thermal stability). We could establish a correlation between the electronic character of the substituents reflected in the Hammett parameter and the absorption maximum of the closed‐ring isomer. Our winning DAEs all had rather simple, small, and electronically mostly neutral substituents, such as methyl, pyridyl, or (substituted) phenyl, indicating that the options for tuning the absorption spectrum of the closed‐ring form via thiophene modification are more limited than we thought. Our investigation revealed further that it is much easier to generate high‐performance dU‐switches than dC switches.
A remarkable feature of the nucleoside‐based DAEs reported here is their exceptionally high cycloreversion quantum yield, which is of great interest, e.g., for applications as a short term information storage medium.[ 40 , 60 , 61 ] Based on our analysis of 26 nucleosidic DAEs, two dU switches (dU , dU2Py) were identified as promising candidates for the incorporation into the T7 promoter to control the activity of its polymerase. The photochromism of the monomers was maintained in the environment of the oligonucleotide, and for dU even in the duplex. The transcription rate of the polymerase was modulated with an up to 2.4‐fold turn on/off‐factor with a single modification site. Future work will focus on increasing the turn‐off factor of the transcription rate by modifying the template strand that is directly recognized by the polymerase. This system offers great potential for the reversible optochemical control over genes of interest.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors thank Dr. R. Wirth and D. Englert for discussion of the manuscript. We acknowledge H. Rudy, D. Wolf and T. Timmermann for technical assistance and K. Haubrich and V. Haller for their assistance in the early stage of the experiments (all Heidelberg University). We also acknowledge H. Derondeau (LMU Munich) for her help in calculating the quantum yields. Open access funding enabled and organized by Projekt DEAL.
T. Kolmar, S. M. Büllmann, C. Sarter, K. Höfer, A. Jäschke, Angew. Chem. Int. Ed. 2021, 60, 8164.
References
- 1. Brieke C., Rohrbach F., Gottschalk A., Mayer G., Heckel A., Angew. Chem. Int. Ed. 2012, 51, 8446–8476; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 8572–8604. [Google Scholar]
- 2. Gautier A., Gauron C., Volovitch M., Bensimon D., Jullien L., Vriz S., Nat. Chem. Biol. 2014, 10, 533–541. [DOI] [PubMed] [Google Scholar]
- 3. Jäschke A., FEBS Lett. 2012, 586, 2106–2111. [DOI] [PubMed] [Google Scholar]
- 4. Szymański W., Beierle J. M., Kistemaker H. A. V., Velema W. A., Feringa B. L., Chem. Rev. 2013, 113, 6114–6178. [DOI] [PubMed] [Google Scholar]
- 5. Bath J., Turberfield A. J., Nat. Nanotechnol. 2007, 2, 275–284. [DOI] [PubMed] [Google Scholar]
- 6. Minkin V. I. in Molecular Switches (Eds.: Feringa B. L., Browne W. R.), Wiley-VCH, Weinheim, 2011, pp. 37–74. [Google Scholar]
- 7. Loudwig S., Bayley H. in Dynamic Studies in Biology: Phototriggers, Photoswitches and Caged Biomolecules (Eds.: Goeldner M., Givens R.), Wiley-VCH, Weinheim, 2005, pp. 253–340. [Google Scholar]
- 8. Asanuma H., Ito T., Yoshida T., Liang X., Komiyama M., Angew. Chem. Int. Ed. 1999, 38, 2393–2395; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1999, 111, 2547–2549. [Google Scholar]
- 9. Broichhagen J., Schönberger M., Cork S. C., Frank J. A., Marchetti P., Bugliani M., Shapiro A. M. J., Trapp S., Rutter G. A., Hodson D. J., Trauner D., Nat. Commun. 2014, 5, 5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Damijonaitis A., Broichhagen J., Urushima T., Hüll K., Nagpal J., Laprell L., Schönberger M., Woodmansee D. H., Rafiq A., Sumser M. P., Kummer W., Gottschalk A., Trauner D., ACS Chem. Neurosci. 2015, 6, 701–707. [DOI] [PubMed] [Google Scholar]
- 11. Frank J. A., Moroni M., Moshourab R., Sumser M., Lewin G. R., Trauner D., Nat. Commun. 2015, 6, 7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goldau T., Murayama K., Brieke C., Steinwand S., Mondal P., Biswas M., Burghardt I., Wachtveitl J., Asanuma H., Heckel A., Chem. Eur. J. 2015, 21, 2845–2854. [DOI] [PubMed] [Google Scholar]
- 13. Horstmann B., Korbus M., Friedmann T., Wolff C., Thiele C. M., Meyer-Almes F. J., Bioorg. Chem. 2014, 57, 155–161. [DOI] [PubMed] [Google Scholar]
- 14. Kamiya Y., Takagi T., Ooi H., Ito H., Liang X., Asanuma H., ACS Synth. Biol. 2015, 4, 365–370. [DOI] [PubMed] [Google Scholar]
- 15. Barrois S., Wagenknecht H. A., Beilstein J. Org. Chem. 2012, 8, 905–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen X., Wehle S., Kuzmanovic N., Merget B., Holzgrabe U., König B., Sotriffer C. A., Decker M., ACS Chem. Neurosci. 2014, 5, 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fujimoto K., Kajino M., Sakaguchi I., Inouye M., Chem. Eur. J. 2012, 18, 9834–9840. [DOI] [PubMed] [Google Scholar]
- 18. Fujimoto K., Maruyama T., Okada Y., Itou T., Inouye M., Tetrahedron 2013, 69, 6170–6175. [Google Scholar]
- 19. Göstl R., Hecht S., Chem. Eur. J. 2015, 21, 4422–4427. [DOI] [PubMed] [Google Scholar]
- 20. Lion-Dagan M., Willner I., J. Photochem. Photobiol. A 1997, 108, 247–252. [DOI] [PubMed] [Google Scholar]
- 21. Özçoban C., Halbritter T., Steinwand S., Herzig L. M., Kohl-Landgraf J., Askari N., Groher F., Fürtig B., Richter C., Schwalbe H., Suess B., Wachtveitl J., Heckel A., Org. Lett. 2015, 17, 1517–1520. [DOI] [PubMed] [Google Scholar]
- 22. Sakata T., Yan Y., Marriott G., Proc. Natl. Acad. Sci. USA 2005, 102, 4759–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsai Y. H., Essig S., James J. R., Lang K., Chin J. W., Nat. Chem. 2015, 7, 554–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Willner I., Rubin S., Wonner J., Effenberger F., Bäuerle P., J. Am. Chem. Soc. 1992, 114, 3150–3151. [Google Scholar]
- 25. Yokoyama Y., Chem. Rev. 2000, 100, 1717–1739. [DOI] [PubMed] [Google Scholar]
- 26. Wiedbrauk S., Dube H., Tetrahedron Lett. 2015, 56, 4266–4274. [Google Scholar]
- 27. Irie M., Chem. Rev. 2000, 100, 1685–1716. [DOI] [PubMed] [Google Scholar]
- 28. Irie M., Lifka T., Kobatake S., Kato N., J. Am. Chem. Soc. 2000, 122, 4871–4876. [Google Scholar]
- 29. Irie M., Lifka T., Uchida K., Kobatake S., Shindo Y., Chem. Commun. 1999, 747–748. [Google Scholar]
- 30. Irie M., Mohri M., J. Org. Chem. 1988, 53, 803–808. [Google Scholar]
- 31. Liu M., Asanuma H., Komiyama M., J. Am. Chem. Soc. 2006, 128, 1009–1015. [DOI] [PubMed] [Google Scholar]
- 32. Roubinet B., Weber M., Shojaei H., Bates M., Bossi M. L., Belov V. N., Irie M., Hell S. W., J. Am. Chem. Soc. 2017, 139, 6611–6620. [DOI] [PubMed] [Google Scholar]
- 33. Singer M., Jäschke A., J. Am. Chem. Soc. 2010, 132, 8372–8377. [DOI] [PubMed] [Google Scholar]
- 34. Singer M., Nierth A., Jäschke A., Eur. J. Org. Chem. 2013, 2766–2769. [Google Scholar]
- 35. Cahová H., Jäschke A., Angew. Chem. Int. Ed. 2013, 52, 3186–3190; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3268–3272. [Google Scholar]
- 36. Irie M., Photochem. Photobiol. Sci. 2010, 9, 1535–1542. [DOI] [PubMed] [Google Scholar]
- 37. Irie M., Sakemura K., Okinaka M., Uchida K., J. Org. Chem. 1995, 60, 8305–8309. [Google Scholar]
- 38. Gilat S. L., Kawai S. H., Lehn J.-M., Chem. Eur. J. 1995, 1, 275–284. [Google Scholar]
- 39. Sarter C., Heimes M., Jäschke A., Beilstein J. Org. Chem. 2016, 12, 1103–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kellis D. L., Sarter C., Cannon B. L., Davis P. H., Graugnard E., Lee J., Pensack R. D., Kolmar T., Jäschke A., Yurke B., Knowlton W. B., ACS Nano 2019, 13, 2986–2994. [DOI] [PubMed] [Google Scholar]
- 41. Sarter C., Dey S., Jäschke A., ACS Omega 2019, 4, 12125–12129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wizinger R., Chimia 1961, 15, 89–105. [Google Scholar]
- 43. Zollinger H., Color Chemistry—Syntheses, Properties, and Applications of Organic Dyes and Pigments, Wiley-VCH, Weinheim, 2003, pp. 15–20. [Google Scholar]
- 44. Irie M., Fukaminato T., Matsuda K., Kobatake S., Chem. Rev. 2014, 114, 12174–12277. [DOI] [PubMed] [Google Scholar]
- 45. Ishiyama T., Murata M., Miyaura N., J. Org. Chem. 1995, 60, 7508–7510. [Google Scholar]
- 46. Gillanders F., Giordano L., Díaz S. A., Jovin T. M., Jares-Erijman E. A., Photochem. Photobiol. Sci. 2014, 13, 603–612. [DOI] [PubMed] [Google Scholar]
- 47. Herder M., Schmidt B. M., Grubert L., Pätzel M., Schwarz J., Hecht S., J. Am. Chem. Soc. 2015, 137, 2738–2747. [DOI] [PubMed] [Google Scholar]
- 48. Sun G., Nicklaus M. C., Theor. Chem. Acc. 2007, 117, 323–332. [Google Scholar]
- 49. Megerle U., Lechner R., König B., Riedle E., Photochem. Photobiol. Sci. 2010, 9, 1400–1406. [DOI] [PubMed] [Google Scholar]
- 50. Buckup T., Sarter C., Volpp H. R., Jäschke A., Motzkus M., J. Phys. Chem. Lett. 2015, 6, 4717–4721. [DOI] [PubMed] [Google Scholar]
- 51. Liang X., Wakuda R., Fujioka K., Asanuma H., FEBS J. 2010, 277, 1551–1561. [DOI] [PubMed] [Google Scholar]
- 52. Mori S., Morihiro K., Obika S., Molecules 2014, 19, 5109–5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nishioka H., Liang X., Asanuma H., Chem. Eur. J. 2010, 16, 2054–2062. [DOI] [PubMed] [Google Scholar]
- 54. Ishibashi Y., Umesato T., Fujiwara M., Une K., Yoneda Y., Sotome H., Katayama T., Kobatake S., Asahi T., Irie M., Miyasaka H., J. Phys. Chem. C 2016, 120, 1170–1177. [Google Scholar]
- 55. Cheetham G. M., Jeruzalmi D., Steitz T. A., Nature 1999, 399, 80–83. [DOI] [PubMed] [Google Scholar]
- 56. Babendure J. R., Adams S. R., Tsien R. Y., J. Am. Chem. Soc. 2003, 125, 14716–14717. [DOI] [PubMed] [Google Scholar]
- 57. Höfer K., Langejürgen L. V., Jäschke A., J. Am. Chem. Soc. 2013, 135, 13692–13694. [DOI] [PubMed] [Google Scholar]
- 58. Ilgu M., Ray J., Bendickson L., Wang T., Geraskin I. M., Kraus G. A., Nilsen-Hamilton M., Methods 2016, 98, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Furukawa K., Abe H., Abe N., Harada M., Tsuneda S., Ito Y., Bioorg. Med. Chem. Lett. 2008, 18, 4562–4565. [DOI] [PubMed] [Google Scholar]
- 60. Ward C. L., Elles C. G., J. Phys. Chem. Lett. 2012, 3, 2995–3000. [DOI] [PubMed] [Google Scholar]
- 61. Ward C. L., Elles C. G., J. Phys. Chem. A 2014, 118, 10011–10019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary