

Summary
Prevascularized 3D microtissues have been shown to be an effective cell delivery vehicle for cardiac repair. To this end, our lab has explored the development of self-organizing, prevascularized human cardiac organoids by co-seeding human cardiomyocytes with cardiac fibroblasts, endothelial cells, and stromal cells into agarose microwells. We hypothesized that this prevascularization process is facilitated by the endogenous upregulation of hypoxia-inducible factor (HIF) pathway in the avascular 3D microtissues. In this study, we used Molidustat, a selective PHD (prolyl hydroxylase domain enzymes) inhibitor that stabilizes HIF-α, to treat human cardiac organoids, which resulted in 150 ± 61% improvement in endothelial expression (CD31) and 220 ± 20% improvement in the number of lumens per organoids. We hypothesized that the improved endothelial expression seen in Molidustat treated human cardiac organoids was dependent upon upregulation of VEGF, a well-known downstream target of HIF pathway. Through the use of immunofluorescent staining and ELISA assays, we determined that Molidustat treatment improved VEGF expression of non-endothelial cells and resulted in improved co-localization of supporting cell types and endothelial structures. We further demonstrated that Molidustat treated human cardiac organoids maintain cardiac functionality. Lastly, we showed that Molidustat treatment improves survival of cardiac organoids when exposed to both hypoxic and ischemic conditions in vitro. For the first time, we demonstrate that targeted HIF-α stabilization provides a robust strategy to improve endothelial expression and lumen formation in cardiac microtissues, which will provide a powerful framework for prevascularization of various microtissues in developing successful cell transplantation therapies.
Keywords: HIF, prevascularization, human cardiac organoid, human induced stem cell-derived cardiomyocyte, tissue engineering, HIF prolyl-hydroxylase inhibitor
1. Introduction
Human pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) hold remarkable promise to treat cardiovascular diseases, the leading cause of death worldwide (Barad, Schick, Zeevi-Levin, Itskovitz-Eldor, & Binah, 2014; Burridge et al., 2014; Hirt, Hansen, & Eschenhagen, 2014; Lian et al., 2012; Prowse et al., 2014; Vunjak-Novakovic et al., 2010). Current transplantation strategies have been mainly focused on direct injection of dissociated human pluripotent stem cell-derived cardiomyocytes into injured hearts (Chong et al., 2014; Laflamme et al., 2007; Liu et al., 2018; Shiba et al., 2012). However, this approach has been limited by low cell survival and poor engraftment after cell transplantation (Heusch, 2016; Laflamme et al., 2007; Laflamme & Murry, 2005; Robey, Saiget, Reinecke, & Murry, 2008; Shiba et al., 2016). To address these challenges, significant efforts have been devoted to developing prevascularized cardiac tissue patches to enhance anastomosis with host tissue and improve cardiac cell survival and engraftment in an ischemic environment (Kim et al., 2010; Tulloch et al., 2011). Current strategies for developing prevascularized cardiac tissues have been focused on the utilization of vascular cells (e.g., endothelial cells and stromal cells) to promote endothelial network formation within the constructs (Kim et al., 2010; Schaefer, Guzman, Riemenschneider, Kamp, & Tranquillo, 2018; Tulloch et al., 2011).
Despite these advances, the clinical translation of prevascularized cardiac patches has been limited in that they are susceptible to ischemic strain post-transplantation, resulting in necrotic regions at the center of the graft (Weinberger et al., 2016). As an alternative, microtissues have increasingly received attention, as their 3D structure improves cell retention and experiences less ischemic strain when compared to cardiac patches due to their smaller sizes (Gunter et al., 2016). In addition, microtissues can serve as “building blocks” for modular fabrication of large-scale tissue structures with significantly improved cell density and demonstrated perfusibility when compared to traditional fabrication strategies (De Moor et al., 2018; Skylar-Scott et al., 2019).
To this end, our lab has explored the development of prevascularized human cardiac organoids which leverages the self-organization of vascular cells in a manner that resembles the intramyocardial organization events in coronary vasculogenesis (Richards et al., 2017). We hypothesize that the prevascularization process is driven, at least in part, by the hypoxia gradient which develops in avascular 3D spherical microtissues (Cheema, Brown, Alp, & MacRobert, 2008; Fennema, Rivron, Rouwkema, van Blitterswijk, & de Boer, 2013; Jain, 2003; Moore, Moore, & McFetridge, 2013). The hypoxia-inducible factor (HIF) pathway is a well-documented, hypoxia sensitive regulatory cascade known for its role in directing neovascularization and anastomosis (Gonzalez-King et al., 2017; Ong & Hausenloy, 2012; Semenza, Nejfelt, Chi, & Antonarakis, 1991). Upregulation of HIF pathway requires a stable concentration of HIF-α, which then binds with HIF-β and forms the HIF transcription complex for downstream gene activation (Ladoux & Frelin, 1997). Under normoxic conditions, prolyl hydroxylase domain enzymes (PHDs) modify HIF-α via prolyl-hydroxylation, allowing for Von Hippel-Lindau (VHL) protein to target HIF-α for degradation (Ivan et al., 2001; Jaakkola et al., 2001). As oxygen levels decrease, PHDs undergo conformational change that inhibits their activity, leading to HIF-α accumulation required for downstream transcription activation (Speer et al., 2013). We hypothesized that through the stabilization of HIF-α with PHD inhibition, it may be possible to further enhance prevascularization within human cardiac organoids through the targeted activation of downstream pro-angiogenic pathways. While previous studies have explored HIF-α stabilization in the heart for the treatment of ischemic cardiomyopathy, to the best of our knowledge, no previous research has exploited the modulation of HIF pathway to promote endothelial network formation within engineered cardiac tissues. Further, the recent progress in the development of selective PHD inhibitors (e.g., Molidustat) to stabilize HIF-α has resulted in multiple Phase II/III clinical trials to treat anemia (Yeh et al., 2017). This supports a translational path to use PHD inhibitors to improve prevascularization within cardiac tissue engineering constructs. Compared to the use of proangiogenic growth factors (e.g., VEGF), small molecule based PHD inhibition provides a robust method to improve organoid prevascularization.
In this study, we show that treatment of human cardiac organoids with Molidustat significantly improves endothelial expression and lumen formation through the upregulation of pro-angiogenic factor VEGF. For the first time, we demonstrate HIF-α stabilization provides a robust strategy to improve endothelial expression and lumen formation in cardiac microtissues. This has laid down the foundation to develop human cardiac organoids with enhanced prevascularization.
2. Materials and Methods
2.1. Cell culture
Human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) (iCell Cardiomyocytes, Cellular Dynamics International-CDI, Madison, WI, USA) were cultured according to the manufacturer’s protocol. Briefly, hiPSC-derived cardiomyocytes were plated on 0.1% gelatin coated 6-well plates in iCell Cardiomyocyte Plating Medium (CDI) at a density of about 3 × 105 to 4.0 × 105 cells/well and incubated at 37 °C in 5% CO2 for 4 days. Two days after plating, the plating medium was removed and replaced with 4 mL of iCell Cardiomyocytes Maintenance Medium (CDI). After 4 days of monolayer pre-culture, cells were detached using trypLE Express (Gibco Life Technologies, Grand Island, NY) and prepared for organoid fabrication. Human cardiac ventricular fibroblasts (cFBs) (Lonza, Basel, Switzerland), cultured in FGM-2 media (Lonza), were used at passage 3–4 for organoid fabrication. Human umbilical vein endothelial cells (HUVECs) (Lonza), cultured in EGM-3 media (Lonza) were used at passage 2–3 for organoid fabrication. Human adipose-derived stem cells (hADSCs) (Lonza) were cultured in low glucose Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin, 1% glutamine and 1% antimycin (Gibco Life Technologies, Grand Island, NY). hADSCs were used at passage 3–4 for organoid fabrication. Culture media for cardiac organoids was comprised of a ratiometric combination of cell-specific media reflecting the cell ratio of the organoid. In organoid media, CDI hiPSC-CM Maintenance Media (supplied without glucose) was substituted with glucose-containing F12/DMEM media with 10% FBS, 1% glutamine, and 1% non-essential amino acids (Gibco).
2.2. Organoid fabrication and treatment
As described in our previous publication (Richards et al., 2017), the agarose hydrogel molds were prepared using 2% agarose (Sigma Aldrich, St. Louis, MO) and master micro-molds from Microtissues, Inc (Providence, RI) as negative replicates to create non-adhesive agarose hydrogels molds containing 35 microwells with hemispheric bottoms (800 μm diameter, 800 μm deep) to facilitate the formation of spherical microtissues. Working cell suspensions of each cell type were used at ~5.0 × 106 cells/mL to make organoid cell ratio mixtures and mixed with 1 volume media for a final concentration of either ~0.85 × 106 cells/mL or ~2.5 × 106 cells/mL. Cells were combined in an approximate ratio of 55% cardiomyocytes, 24% fibroblasts, 14% endothelial cells, and 7% adult stem cells. Approximately 75 μL of each cell suspension was pipetted into an agarose mold for each treatment group. When dispensed to their molds, the differing cell suspensions resulted in organoids of distinctly different and varying diameter. Day 0 (D0) of the experiment was marked after 4 days of organoid assembly. Pharmacological compounds and physiological stimuli were used to investigate the effects of HIF-α and downstream angiogenic upregulation. Molidustat/BAY 85–3924 (Selleckchem) was used to stabilize HIF-α via PHD inhibition at a concentration of 5 μM in organoid maintenance media for 10 days. Semaxanib/SU5416 (Selleckchem) was used to selectively inhibit VEGFR (Flk-1/KDR) at a concentration of 20 μM in organoid maintenance media for 10 days. Hypoxic culture conditions were achieved using a C-Chamber (BioSpherix, Parish, NY) and cultured at 12% O2 for 10 days. Supplemental VEGF culture conditions were achieved by adding soluble hVEGF (Lonza) at a concentration of 2.5 μg/mL for 10 days.
2.3. Immunofluorescent staining and imaging
Freshly collected organoids were flash frozen in Tissue-Tek OCT compound (Sakura, Torrance, CA). Embedded organoids were cryosectioned into 7 μm sections onto glass slides. Sections were incubated with primary antibody diluted in PBST (1:200) overnight at 4 °C or 2 hrs at room temperature: mouse anti-alpha sarcomeric actinin (αSA) (Abcam ab52917), rabbit anti-vimentin (Abcam ab92547), mouse anti-CD31 (BD Biosciences 550274), rabbit anti-von Willebrand factor (vWF) (Abcam ab6994), rabbit anti-vascular endothelial growth factor (VEGF) (Abcam ab52917), rabbit anti-HIF 1α (Abcam ab51608). Sections were incubated with complement secondary antibodies diluted in PBST for 2 hr at room temperature: goat anti-mouse Alexa Fluor 546 (Invitrogen A11030), goat anti-rabbit Alexa Fluor 647 (Invitrogen A21244). Nuclei were counterstained with DAPI (Molecular Probes/Invitrogen R37606) diluted in PBST for 15 min at room temperature. TCS SP5 AOBS laser scanning confocal microscope (Leica Microsystems, Inc., Exton, PA) was used for imaging, with the majority of images being composite stacks of 5–8 μm at 0.5 μm steps.
2.4. Immunfluorescent image analysis
Fluorescent protein expression was calculated using ImageJ software (National Institutes of Health) as the antibody-positive fluorescence area coverage: endothelial density was calculated as CD31- or vWF-positive covered area divided by organoid area, vimentin density was the ratio of vimentin-positive covered area divided by organoid area, and α-SA-positive density was the ratio of α-SA-positive covered area divided by organoid area. This was done by first measuring the area of the organoid cross-section using ImageJ. Next, the desired fluorescent channel was converted to 8-bit in order to calculate threshold intensity. The measured area of threshold intensity was then divided by the corresponding cross-sectional area of the corresponding organoid to determine percent area of expression. ImageJ was also utilized to quantify the number of lumens per organoid and lumen area as a percentage of organoid area. This was accomplished by manually identifying lumen structures (via CD-31 antigen-specific staining) in each organoid cross-section, measuring the area of each individual lumen, and dividing the sum of these lumen structures by the total area of the respective organoid. Lumens were defined as circular structures with a diameter of at least 3 μm and the majority of the structure’s circumference staining positive for endothelial-specific markers. For vascular morphogenic quantification, immunofluorescent CD31 images of sectioned organoids were analyzed by open source AngioTool software according to their online manual. Vascular morphogenesis characteristics including vessels area, vessels % area, total vessels length, average vessels length, and junctions density were automatically quantified. Representative sections from at least 5 organoids were analyzed for each condition. Output data from Angiotool was exported to GraphPad Prism for statistical analysis via Mann-Whitney nonparametric test. Co-localization of fluorescently labeled proteins was achieved via co-staining of compatible antibodies (mouse anti-CD31 and rabbit anti-vimentin). Using ImageJ, the images were split into two channels and compared using the Coloc 2 analysis tool. The R-value outputs were converted via Z-transform and averaged to acquire the results. Representative sections from at least 5 organoids were analyzed for each condition.
2.5. Contraction analysis of beating spheroids
Videos of beating spheroids/organoids from each group were recorded at each condition using a Carl Zeiss Axiovert A1 Inverted Microscope and Zen 2011 software (Zeiss, Göttingen, Germany). Tissue pacing was achieved via electrical stimulation (C-Pace unit, Ion Optix, Milton, MA) of agarose molds suspended in culture media. After 21 total days of culture, spheroids/organoids were stimulated (12V/5ms) at increasing frequency (0.5–1.5 Hz). The percentage of spheroids/organoids keeping pace with external stimulation was calculated. Additionally, threshold edge-detecting in ImageJ was used on high contrast spheroid picture series and graphed to realize beating profiles of fractional area change (i.e., contraction amplitude), from which contraction amplitude was calculated (n=15 spheroids/organoids). Contraction amplitudes were calculated as the percent change in fractional area change amplitude between contraction and relaxation.
2.6. Calcium transient imaging
Life Technologies’s Fluo-4 Direct Calcium Assay Kit (Life Technologies F10471) was used to label calcium in the whole organoids based on the manufacturer protocol. Briefly, organoids stained with a working solution of 1:1 calcium dye solution to media and incubated at 37 °C, 5% CO2, 20% O2 for 30 min. Carl Zeiss Axiovert A1 Inverted Microscope completed with GFP fluorescence imaging capacity (Zeiss) was used to collect the videos of the calcium transient of whole spheroids with a capture rate of 20 frames per second. Mean gray value of the whole organoid calcium transient (controlled for area for pre- and post- treatment) was measured using ImageJ software (National Institutes of Health) to construct calcium transient profiles. Normalized F/F0 was calculated as the peak calcium fluorescence (F) divided by the start fluorescence level (F0), then divided by the F/F0 of the baseline transient before treatment. Time to 50% calcium decay (sec) was calculated as the difference in time between the start of the calcium transient and the half-peak (F50) of the calcium decay.
2.7. VEGF protein expression via ELISA
To quantify VEGF secretion from individual organoid cell types, a Human VEGF Quantikine ELISA Kit (R&D Systems DVE00) was used. HUVECs, ADSCs, and cvFBs were plated in 24-well plates at 50,000 cells/well while iPSC-CMs were plated in a 96-well plate at 20,000 cells/well. The cells were incubated for 48 hours before being treated with 5 μM Molidustat dissolved in the respective cell culture media. After 24 hours of treatment, cell culture supernatant was collected and immediately transferred to a −80°C freezer before analysis. The supernatants were analyzed with the ELISA kit according to the manufacturer’s instructions. At least 3 replicates were analyzed for each condition. A microplate reader set to 450 nm with wavelength correction at 540 nm was used to read the optical density (O.D.) of each well. VEGF concentrations (pg/mL) were calculated using a standard curve. Molidustat-treated conditions were normalized to the average concentration of the control group to determine fold change.
2.8. In vitro ischemic treatment
Ischemic conditions were simulated in vitro through a combination of hypoxic culture conditions and ischemic media. Hypoxic conditions were produced through the use of a hypoxia chamber (ProOx Model 110, BioSpherix, Parish, NY) set to 1% O2. Ischemic media was produced through the dilution of organoid maintenance media (see above) with glucose-free/serum-free medium (Glucose Free DMEM, Gibco) at 90% total volume. Control and Molidustat treated organoids were cultured in either hypoxic (1% O2) or ischemic conditions (1% O2, 1/10 organoid maintenance media) for 24 hours or 72 hours. In the case of the 24 hours testing conditions, we quantified at least n=5 organoids.
2.9. Staining and quantification of nucleus-specific markers
Roche In Situ Cell Death Detection Kit (Sigma 11684795910) was used to visualize the viability of cells in frozen sections of cardiac organoids based on the Roche protocol (TUNEL Staining). TCS SP5 AOBS laser scanning confocal microscope (Leica Microsystems) was used for imaging. Quantification of viability was achieved using ImageJ particle analysis. Briefly, we quantified the overall number of nuclei represented per organoid through DAPI staining, followed by a quantification of the total number of TUNEL-positive nuclei for the corresponding organoid. We then used the complementary percentage of the ratio between TUNEL-positive nuclei to total nuclei to estimate the percentage of viable cells per organoid cross-section, defining the “Viability Index” for each organoid culture condition. To study the effect of culture conditions on cell viability, the changes of viability (Δ% Viability Index) in each culture condition compared to the ischemic condition were calculated for each organoid size group. A similar method of quantification was used for the staining and analysis of phospho-histone H3 (PHH3, Millipore 06–570). PHH3 was co-stained with DAPI, and the percentage of PHH3-positive nuclei per organoid cross-section was estimated using ImageJ. For each culturing condition and time-point during the 1–3 day time point, we quantified at least n=5 organoids.
2.10. Statistical analysis
Differences between experimental groups were analyzed on JMP Pro 12 Statistical software (SAS, Cary, NC) using Student’s t-test, matched pairs comparison, one-way ANOVA, and two-way ANOVA with Tukey’s post-hoc test, and p<0.05 was considered significantly difference for all statistical tests.
3. Results and Discussion
3.1. Molidustat treatment improved endothelial expression and lumen formation within human cardiac organoids
Our lab has explored the development of human cardiac organoids by co-seeding hiPSC-CMs with endothelial cells (HUVECs; human umbilical vein endothelial cells), stromal cells (hADSCs; human adipose-derived stem cells) and cardiac fibroblasts (hcvFBs; human cardiac ventricular fibroblasts) into non-adhesive agarose microwells (Richards et al., 2017). Similar to intramyocardial organization events in coronary vasculogenesis, these cells self-organize into prevascularized human cardiac organoids containing endothelial networks. To further enhance endothelial network formation and organization, we treated the organoids with Molidustat (5 μM), as it is a drug designed to selectively inhibit PHDs to stabilize HIF-α (Supplemental Fig 1). The therapeutic potential of Molidustat has been well documented and is currently in phase II clinical trial for treating anemia (Yeh et al., 2017). 5 μM was selected for treatment as this concentration showed minimal cell death compared to higher concentrations (50 and 250 μM) according to TUNEL staining (Supplemental Figs 2, 3). Additionally, cardiac organoids treated with Molidustat (5 μM) resulted in significantly improved expression of HIF-α after 10 days of culture as detected by immunofluorescant staining (Supplemental Fig 4).
When treated with Molidustat (5 μM), there was no discernable difference in bright-field observations (Fig 1A) or cardiomyocyte organization (Fig 1B). Cardiomyocyte structure for both control and Molidustat treated groups reflected results reported in our previous studies, with clear sarcomere networks organized at the periphery of the organoids. This indicates the Molidustat treatment does not significantly influence hiPSC-CM contractile structure, consistent with the previous studies that showed HIF-α stabilization does not negatively affect contractile structures of cardiomyocytes in vivo (Hyvarinen et al., 2010).
Figure 1. Human cardiac organoids treated with Molidustat result in improved endothelial expression and lumen formation.
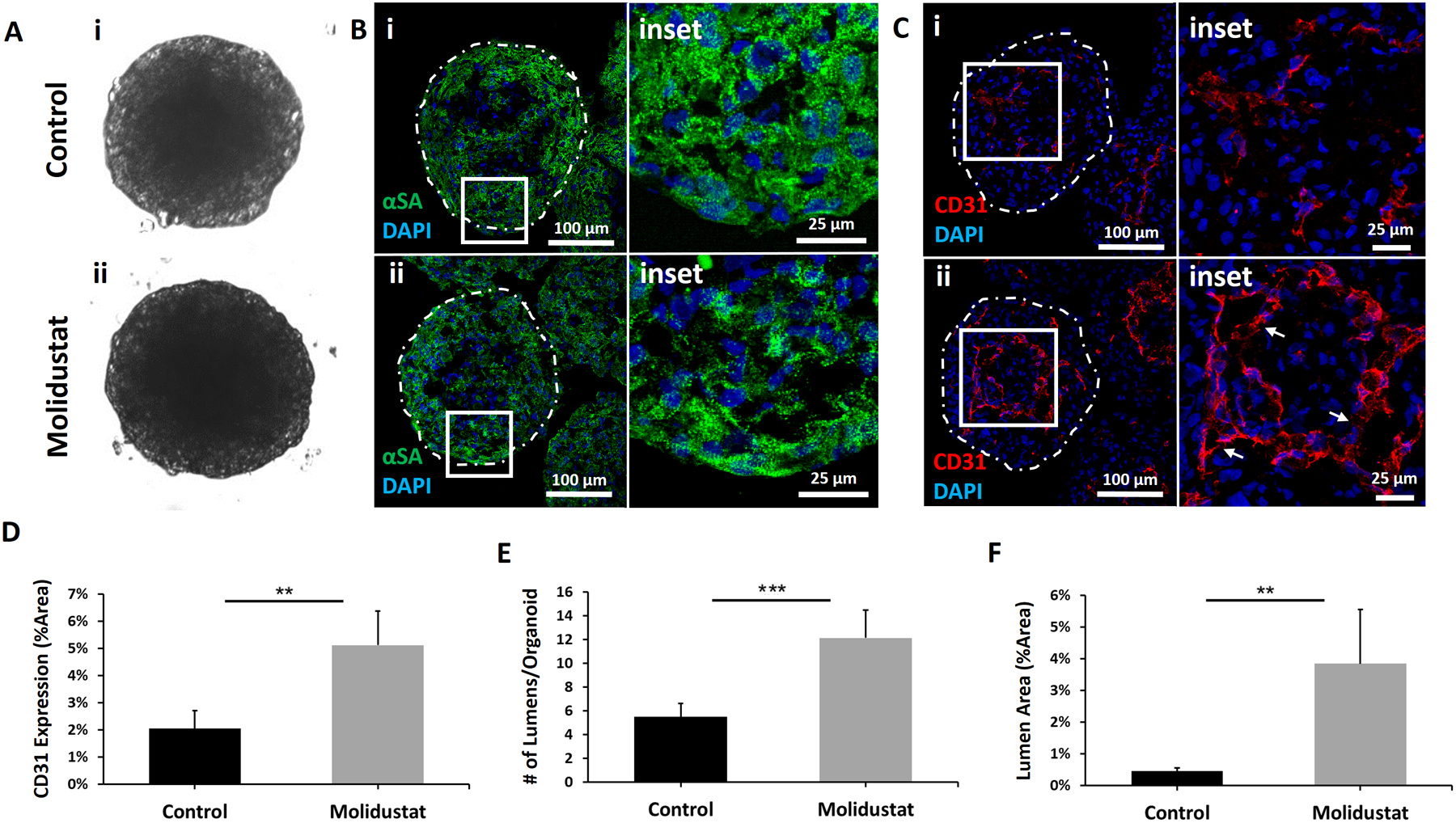
Representative images of Control and Molidustat treated organoids after 10 days of culture: (A) brightfield, (B) immunofluorescent staining of cardiac marker α-sarcomeric actinin (αSA) showing comparable development of sarcomeric structures, and (C) immunofluorescent staining of endothelial marker CD31 showing improved organization of endothelial cells in Molidustat treated organoids. The white arrows in the inset (ii) point to the lumen structures. Green- αSA Red- CD31, Blue-DAPI. (D) Percent area expression of CD31 comparing Control and Molidustat treated organoids. (E) Total number of lumens per cardiac organoid when comparing Control and Molidustat treated groups. (F) Total sum of lumen area as a percentage of cardiac organoid area when comparing Control and Molidustat treated groups. n=at least 5 biological replicates. Mann-Whitney nonparametric test and two tailed confidence interval. (**) represents p<0.005, (***) represents p<0.0005.
While endothelial marker CD31 staining revealed abundant lumen and cord-like structures in both control and Molidustat treated organoids with morphology similar to that of prevascularized cardiac patches reported in the literature (Kreutziger et al., 2011) (Fig 1C), there was a clear difference in quantifiable expression and organization of endothelial cells between control and Molidustat treated groups (Fig 1C–F). Endothelial marker CD31 staining revealed a 150 ± 61% increase in endothelial expression for the Molidustat treated group when compared to control (Fig 1D), a 220 ± 20% increase in number of lumens per organoid (Fig 1E) and an 840 ± 44% increase in the lumen area per organoid (Fig 1F). The significant increase in endothelial expression and lumen formation in the Molidustat treated organoids demonstrates the potential of modulating HIF pathway to promote prevascularization within cardiac microtissues. In order to assess whether the increased expression of endothelial markers was a result of increased cell proliferation in Molidustat treated organoids, both control and treated groups were stained for the mitosis-marker phospho-histone H3 (PHH3) (Supplemental Fig 5). After 4 days of pre-culture with Molidustat, organoids were harvested at Day 1 for proliferation analysis. These results indicate that at Day 1, there was no difference in proliferation with respect to control organoids. Interestingly, the Molidustat treated organoids showed a significant improvement in CD31 expression when compared to control, indicating the Molidustat treatment mainly improves endothelial cells spreading and self-organization instead of proliferation (Supplemental Fig 5c,d).
3.2. Cardiac organoid diameter influenced effects of Molidustat treatment
As our previous studies have shown, the hypoxia gradient in cardiac microtissues is affected by the sizes of the microtissues. Our results support that there is an increasing hypoxic tension which develops in the center of 3D spherical microtissues with the increasing diameter of 3D spherical microtissues (Coyle, Yao, Richards, & Mei, 2019; Richards et al., 2017). To this end, we investigated the impact of diameter on the development of endothelial networks in the cardiac organoids. The microtissues of different diameters were fabricated by varying initial cell seeding concentrations (Fig 2A–C). When compared to the control organoids, the effects of Molidustat treatment decreased with increasing size of the organoids (Fig 2D, Supplemental Fig 6), which supported our hypothesis that oxygen tension plays a role in the development of endothelial networks in cardiac organoids. While there was a significant increase in vWF expression found in the 275 μm diameter organoids and a trended increase in 325 μm diameter organoids, little difference in the vWF expression was found in 400 μm organoids. Notably, we observed improvement in endothelial organization between Molidustat treated 400 μm organoids and control (i.e., increased lumen structures).
Figure 2. Human cardiac organoid diameter influences results of Molidustat treatment on endothelial organization.

Human cardiac organoids with increased diameter result in varied expression of endothelial markers for (A) Control and (B) Molidustat treatment. Cardiac organoid diameter= (i) 275 μm, (ii) 325 μm, and (iii) 400 μm. Staining for cardiac-specific markers show consistent organization of hiPSC-CM development. Green- αSA Red- vWF, Blue-DAPI. (C) Diameter measurements show consistent size groupings of 275 μm, 325 μm, and 400 μm, with no significant difference in diameter between control and treatment groups. (D) Endothelial-specific marker vWF expression showed significant improvement for Molidustat treated organoids when compared to control for both 275 μm and 325 μm diameter organoids, but not between treatment groups of 400 μm diameter. (E) Total number of lumens per cardiac organoid when comparing Control and Molidustat treated groups of 400 μm diameter. (F) Total sum of lumen area as a percentage of cardiac organoid area when comparing Control and Molidustat treated groups of 400 μm diameter. n=at least 5 biological replicates. Mann-Whitney nonparametric test and two tailed confidence interval. (*) represents p<0.05, (**) represents p<0.005, (***) represents p<0.0005.
To further assess whether the Molidustat treatment can promote prevascularization for the 400 μm organoids, we examined the number of lumens per organoid (Fig 2E) and lumen area per organoid (Fig 2F). Interestingly, the improvements in number of lumens per organoid between control and Molidustat treated 400 μm diameter groups were found to be proportional when compared to the difference between 325 μm groups, with both being 220 ± ~20% improvement (Fig 1E and Fig 2E). Improvements in lumen area per organoid exhibit a similar trend between 325 μm and 400 μm diameter groups, with increases of 840 ± 44% (Fig 1F) and 660 ± 28% (Fig 2F) respectively. These results clearly demonstrate the Molidustat treatment is capable of promoting endothelial lumen formation even for the 400 μm organoids. In addition to using Molidustat to upregulate HIF pathway, we also exposed 325 μm cardiac organoids to the reduced oxygen (12% from 21%). However, the reduced oxygen did not elicit a significant improvement in vWF expression from control (Supplemental Fig 7). This clearly shows that targeted HIF-α stabilization provides a controlled and powerful method to promote prevascularization within cardiac microtissues regardless of endogenous oxygen tension.
3.3. Molidustat treated organoids maintain cardiac functionality
After confirming that Molidustat treated cardiac organoids showed the improved expression and lumen formation of endothelial cells, we examined functionality of the Molidustat treated organoids. To investigate the effects of Molidustat on the hiPSC-CMs functionality, we examined cardiac specific function of both hiPSC-CM spheroids (microtissues composed of 100% hiPSC-CMs) and cardiac organoids (microtissues composed of 55% hPSC-CMs and 45% supporting cells). Both hiPSC-CM spheroids and cardiac organoids contain equal numbers of cardiomyocytes per microtissue. Comparing these two microtissue compositions allows for the separation of the impact of Molidustat on hiPSC-CMs alone, while also taking into consideration potential downstream effects via paracrine signaling initiated by Molidustat to the supporting cells. To examine the electrical properties of the hiPSC-CM spheroids and organoids, we used an electrical stimulation apparatus to pace the control and Molidustat treated groups at physiologically relevant frequencies (Fig 3A). For frequencies of 0.5, 1.0, and 1.5Hz, near 100% pacing were maintained for both spheroids and organoids. This indicates the potential capacity of Molidustat treated organoids to be paced by physiological stimulation after transplantation into human patients. To examine the contractile properties, we compared the difference in area between relaxation and contraction for hiPSC-CM spheroids and cardiac organoids while being electrically paced (Fractional Area Change; FAC) (Fig 3B). Each testing condition started within the same statistical range for 0.5Hz, and they trended downward with each increase of pacing. Admittedly, there did appear to be a trended decrease in FAC change between Molidustat and control cardiac organoids at similar paces. This could be a result of a proportionally higher expression of endothelial and supporting cells relative to cardiomyocytes, as this trend was not observed in the hiPSC-CM spheroids.
Figure 3. Molidustat treated organoids maintain cardiac functionality.
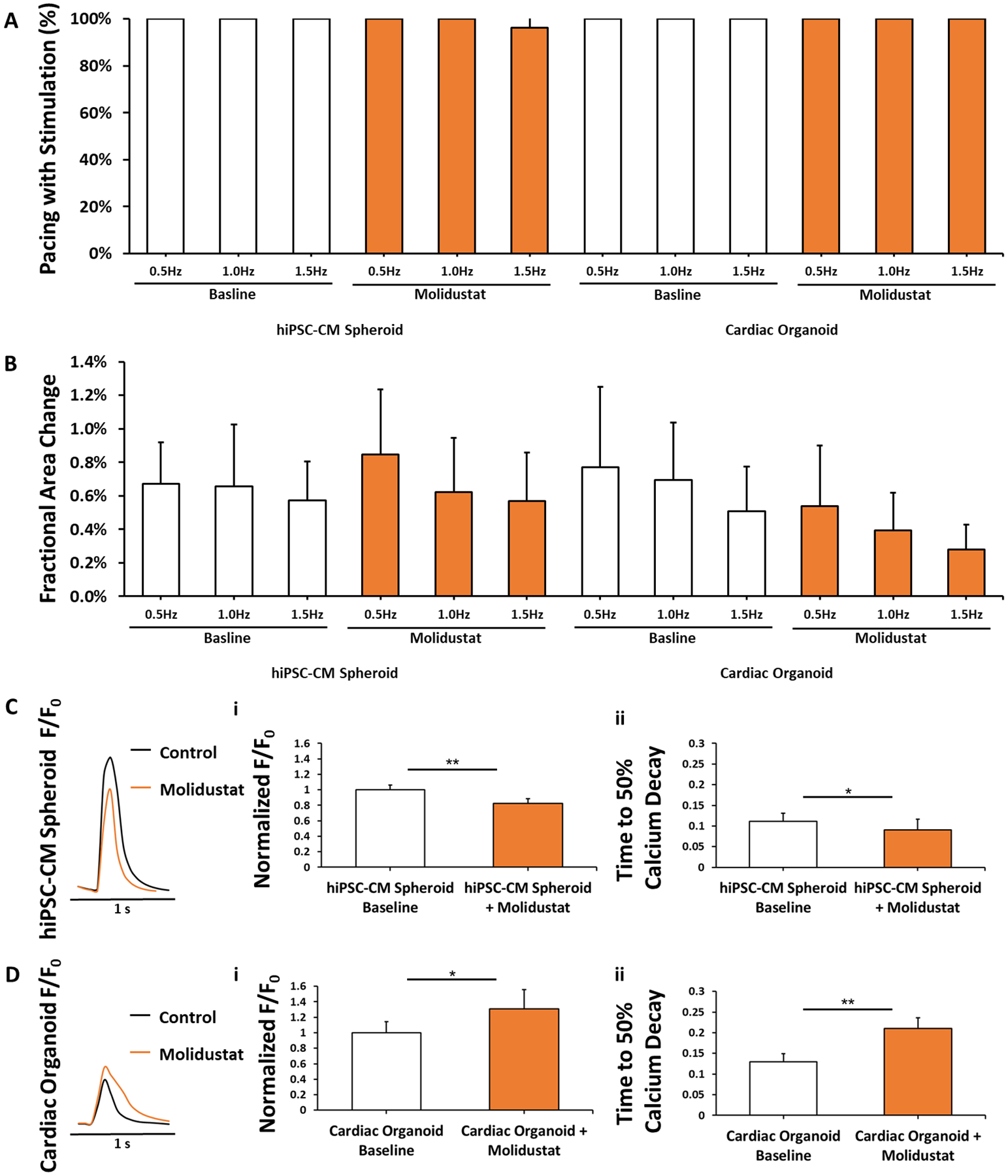
(A) Electrical pacing of hiPSC-CM Spheroids, hiPSC-CM Spheroids + Molidustat, Cardiac Organoids, and Cardiac Organoids + Molidustat at 0.5, 1.0, and 1.5Hz. n=15 biological replicates. (B) Fractional area change (FAC) of hiPSC-CMs, hiPSC-CMs + Molidustat, Cardiac Organoids, and Cardiac Organoids + Molidustat at 0.5, 1.0, and 1.5Hz. n=15 biological replicates. (C) Molidustat treated hiPSC-CM spheroids showed a decrease in both (i) peak calcium fluorescence and (ii) time to 50% calcium decay when compared to control. n=5 biological replicates. (D) Molidustat treated cardiac organoids showed a significant increase in both (i) peak calcium fluorescence and (ii) time to 50% calcium decay. n=5 biological replicates. Mann-Whitney nonparametric test and two tailed confidence interval. (*) represents p<0.05, (**) represents p<0.005.
We then investigated the effects of Molidustat on calcium handling properties of hiPSC-CMs due to their important role in cardiac contractions, as well as electrical signal generation and propagation. It is worth noting that peak calcium transient of both control and treated organoids were significantly lower than that of both control and treated hiPSC-CMs, which was attributed to the lower concentration of hiPSC-CMs in the organoids (~55%) than hiPSC-CM spheroids (100%). For hiPSC-CM spheroids (Fig 3C), the Molidustat treatment led to a decrease in both the peak calcium transient and the time to 50% calcium decay under unstimulated conditions. In contrast, Molidustat treated organoids showed a significant increase in the peak calcium transient when compared to control (Fig 3D), which was accompanied with a significant increase in the time to 50% calcium decay in treated organoids. While these results demonstrate that hiPSC-CMs in both Molidustat treated hiPSC-CM spheroids and organoids display functional calcium handling properties, their difference may be attributed to the effects of Molidustat treatment on the supporting cells (e.g., fibroblasts). For example, hypoxia is well known to promote cardiac fibroblast proliferation and ECM remodeling (Wang et al., 2013; Watson et al., 2014), which may in turn have an impact on calcium handling properties of hiPSC-CMs in the organoids. Notably, our results are in agreement with previous studies showing HIF stabilization in vivo did not cause cardiac function abnormality (Hyvarinen et al., 2010).
3.4. Molidustat treatment improves endothelial network formation via upregulation of VEGF
It has been well documented that HIF-α regulates neovascularization via upregulation of vascular endothelial growth factor (VEGF) (Forsythe et al., 1996; Rey & Semenza, 2010). As a result, we hypothesized that the improved endothelial expression seen in Molidustat treated human cardiac organoids was due, at least in part, to the increased expression of VEGF. To determine the effect of VEGF signaling on organoid development, we cultured our human cardiac organoids with Semaxanib, a competitive inhibitor for VEGF receptor (Flk-1/KDR), which has been shown to have anti-angiogenic properties (T. A. Fong et al., 1999; Hoff, Wolff, Bogaard, Waldrum, & Abbruzzese, 2006). We also chose to co-culture Molidustat and Semaxanib with organoids to assess whether VEGF inhibition could negate the improved endothelial expression and networking shown in Molidustat treated organoids. For both Semaxanib and Semaxanib + Molidustat treated organoids, there was a significant decrease in expression of endothelial marker CD31 (Fig 4A, C) relative to control and Molidustat groups. These findings show that the improved networking induced via Molidustat treatment could be negated by the inhibition of VEGF, demonstrating the mechanistic dependence on the VEGF signaling pathway. We also investigated the expression of VEGF via immunofluorescent staining (Fig 4B, D). For the Molidustat treated organoids, there was a significant increase in VEGF expression when normalized to control and compared to the other groups. While there was a trended decrease in the Semaxanib and Semaxanib + Molidustat groups when compared to control, these results were not statistically different. These data suggest that improved expression of endothelial markers in Molidustat treated organoids may be a result of improved expression of VEGF, and that inhibition of the VEGF pathway will significantly reduce endothelial cell expression and networking. Interestingly, when we added supplemental VEGF (2.5 μg/mL) to control organoids, we did not observe the enhanced networking seen in Molidustat treated groups (Supplemental Fig 8), which is consistent with results found in the literature for attempts to improve prevascularization with supplemental VEGF (Rivron et al., 2012). This suggests that targeting upstream angiogenic pathways in situ is more effective for endothelial cells than exogenous VEGF delivery (Rey & Semenza, 2010). In addition, these results demonstrate the advantages of using a small molecule (Molidustat) which targets the upstream proangiogenic pathway when compared to a large molecule (VEGF) which targets the downstream proangiogenic pathway in order to improve microtissue prevascularization.
Figure 4. Molidustat improves endothelial expression via upregulation of VEGF.
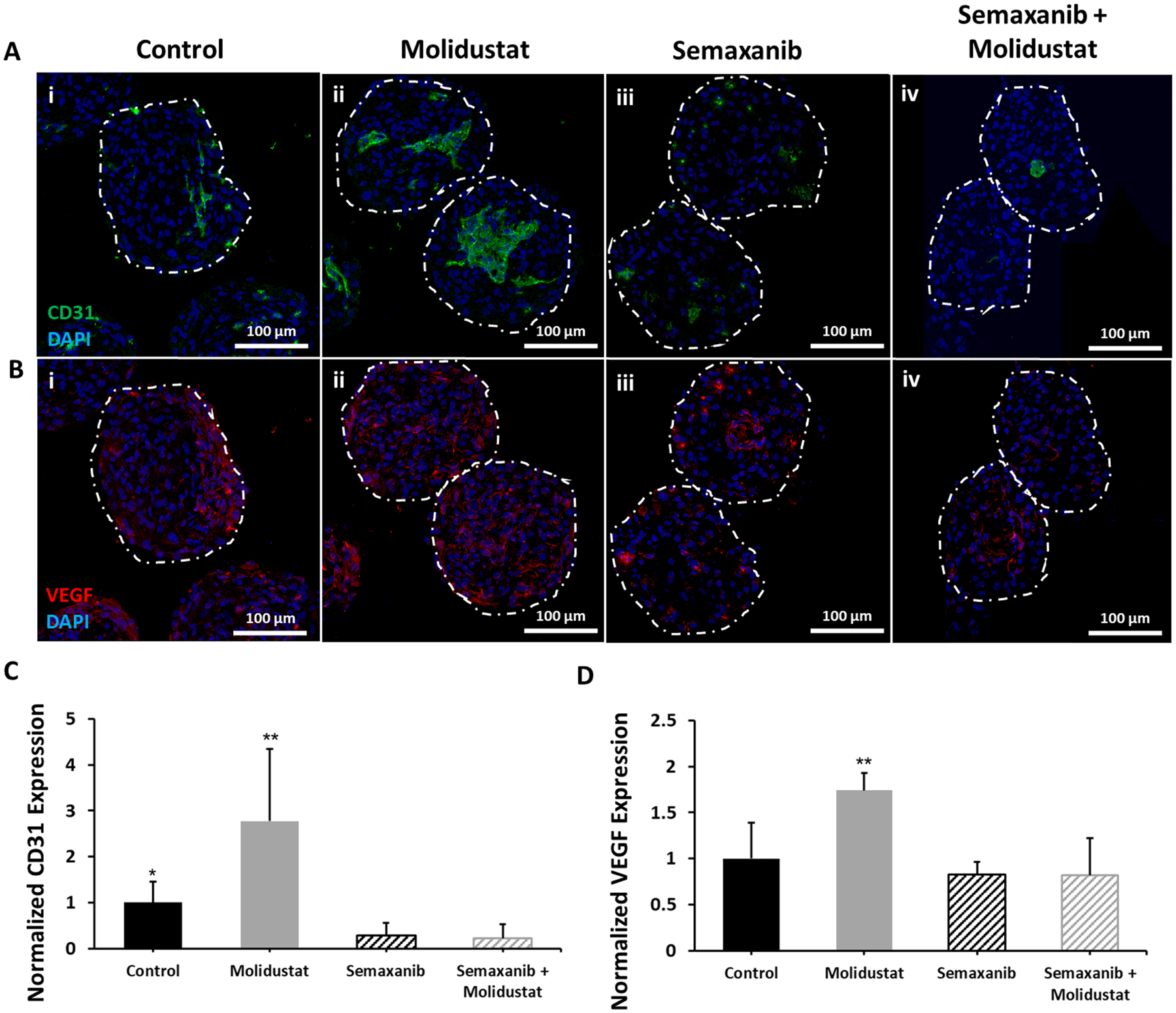
Representative images comparing (i) Control, (ii) Molidustat, (iii) Semaxanib, and (iv) Semaxanib + Molidustat treated organoids after 10 days of culture for (A) immunofluorescent staining of endothelial marker CD31 showing increased endothelial expression for Molidustat treated organoids and a decrease in endothelial expression for Semaxanib and Semaxanib + Molidustat treated organoids, and (B) immunofluorescent staining of VEGF showing increased expression for Molidustat treated organoids. Green- CD31, Red- VEGF, Blue- DAPI. Normalized expression of (C) CD31 and (D) VEGF as a function of organoid area relative to Control. n=5 biological replicates. Mann-Whitney nonparametric test and two tailed confidence interval. (*) represents p<0.05, (**) represents p<0.005.
In order to further validate the role of Molidustat in upregulating HIF-1α and VEGF, we conducted an ELISA study quantifying differences in protein expression between control and Molidustat treatment for each of the four cell types used in our cardiac organoids (Fig 5A). In this study, three out of the four cell types (hiPSC-CMs, cvFBs, and hADSCs) showed significant upregulation of VEGF secretion, with HUVECS showing virtually no secretion of VEGF in either case. Interestingly, previous studies indicate that autocrine signaling of VEGF plays a critical role in supporting endothelial cell viability and vascular integrity (G. H. Fong, 2009). Notably, there was a significant increase in co-localization of CD31 for both vimentin (a mesenchymal cell filament marker, Fig 5B, C) and NG2 (a cell-adhesion proteoglycan indicating pericyte activity, Fig 5D, E) for Molidustat treated groups when compared to control, indicating the upregulated VEGF secretion of vimentin+ and NG2+ cells (e.g., cardiac fibroblasts, hADSCs) plays a critical role in promoting endothelial network formation after Molidustat treatment. The pericyte activity of fibroblasts and mesenchymal stem cells described here has been well documented as being a critical component in the self-organization of endothelial structures in multi-cellular cardiac tissues. Here, we demonstrate that it is possible to enhance this support function through the activation of the HIF-1α pathway via Molidustat treatment (Tulloch et al., 2011). However, it cannot be ruled out that this increased co-localization of CD31 and vimentin may be a result of other mechanisms, such as endothelial to mesenchymal transitioning (EndoMT), as HIF- α signaling has been shown to regulate this transdifferentiation process (Piera-Velazquez & Jimenez, 2012; Zhu et al., 2006).
Figure 5. Molidustat upregulates VEGF secretion of non-endothelial cell types.
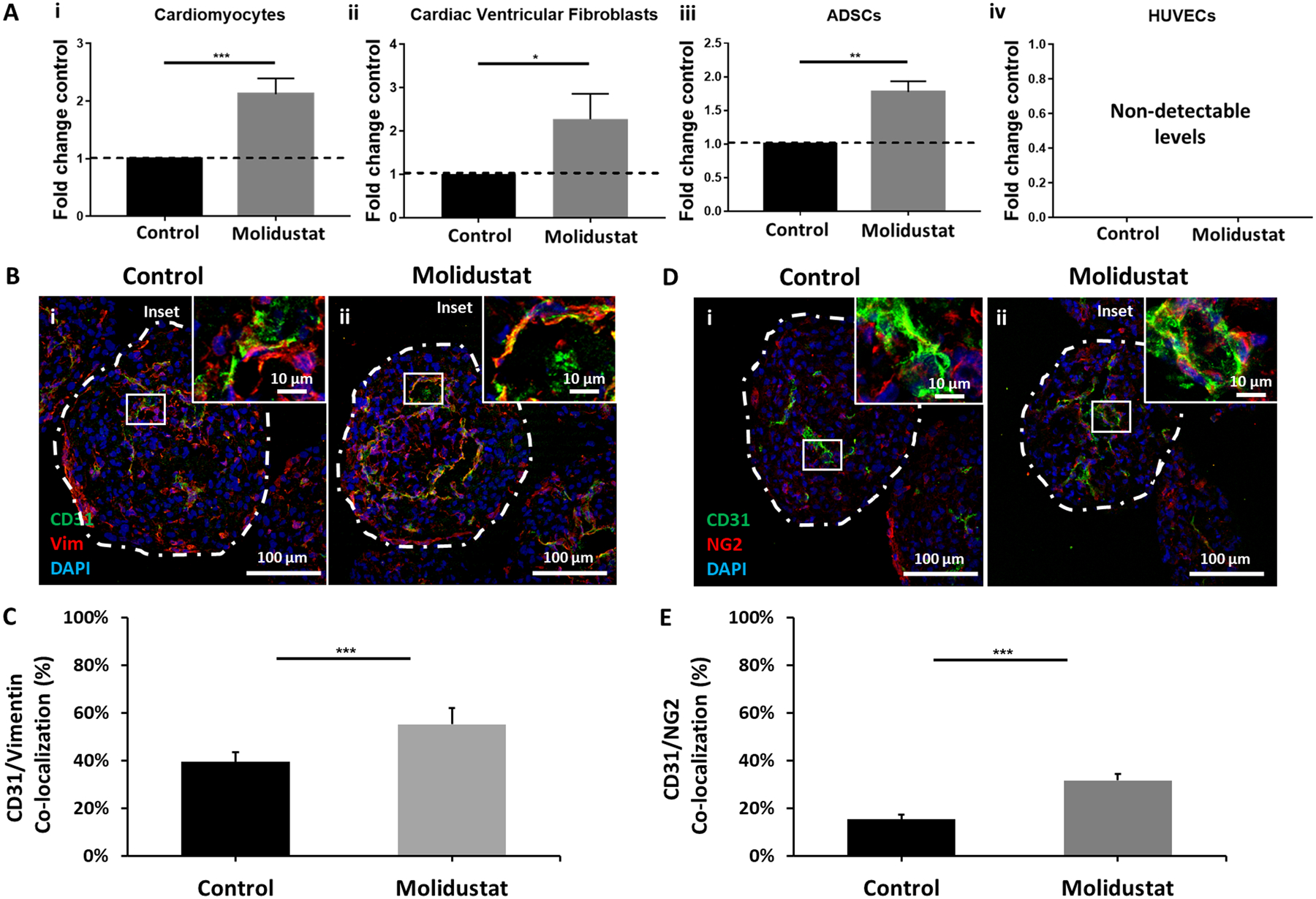
(A) ELISA results show significant increase of VEGF secretion in (i) hiPSC-CMs, (ii) cardiac ventricular fibroblasts, and (iii) adipose-derived stem cells (ADSCs); (iv) Endothelial cells (HUVECS) did not express detectable concentration of VEGF in either group. (B) Vimentin expression showed (C) improved co-localization with endothelial cell-specific marker CD31 for (ii) Molidustat treated organoids when compared to (i) Control. n=at least 5 biological replicates. (D) NG2 expression showed (E) improved co-localization with endothelial cell-specific marker CD31 for (ii) Molidustat treated organoids when compared to (i) Control. n=at least 5 biological replicates.Mann-Whitney nonparametric test and two tailed confidence interval. (*) represents p<0.05, (**) represents p<0.005, (***) represents p<0.0005.
3.5. Molidustat improves survival of cardiac organoids under in vitro models of hypoxic and ischemic conditions
Our previous results showed the presence of lumen-like vasculature within cardiac organoids can improve cell survival in hypoxic conditions (Richards et al., 2017). To assess whether Molidustat treatment can improve cardiac organoid survival, we cultured both control and Molidustat treated cardiac organoids in hypoxia (1% O2) and ischemia (1% O2; 1:10 media dilution) for 24 hours. There was a significant improvement in cardiac organoid survival in both conditions for the treated groups when compared to control (Fig 6). According to staining of structural proteins, there were non-significant changes in the expression of endothelial and cardiac markers after 24 hours of hypoxic conditioning (Supplemental Fig 9). Under 24 hours of ischemic conditions, vWF staining showed significant decreases for both control and Molidustat treated groups. However, the significantly improved expression of endothelial markers for Molidustat treated organoids when compared to control was observed across all culture conditions. Interestingly, the majority of the cells indicating apoptosis (TUNEL) co-localized to the endothelial specific regions of the cardiac organoids, suggesting that these cells are more vulnerable to hypoxic and ischemic strain relative to other regions of the organoids. (Supplemental Fig 9d,e) This data also suggest that the makeup of cells undergoing apoptosis is proportional across conditions and treatments, with there being an overall lower percentage of apoptotic nuclei for Molidustat treated groups, as seen in Fig 6. After 72 hours of culture, there was substantial improvement in the Molidustat treatment groups over control groups (Supplemental Fig 10). These data demonstrated that treatment with Molidustat not only improves endothelial networking, but also improves cardiac organoid survival in ischemic conditions post-transplantation. These results support previous studies that showed HIF-α stabilization provides cardioprotection for myocardial infarction injuries (Hyvarinen et al., 2010; Levent et al., 2020). Notably, upregulation of HIF-α is also shown to be responsible for a shift from aerobic to anaerobic metabolism under hypoxic conditions, so it is possible that this may play a role in cardiac organoid survival under the described treatment (Ong & Hausenloy, 2012).
Figure 6. Molidustat treatment improves survival of cardiac organoids when exposed to hypoxic and ischemic conditions.
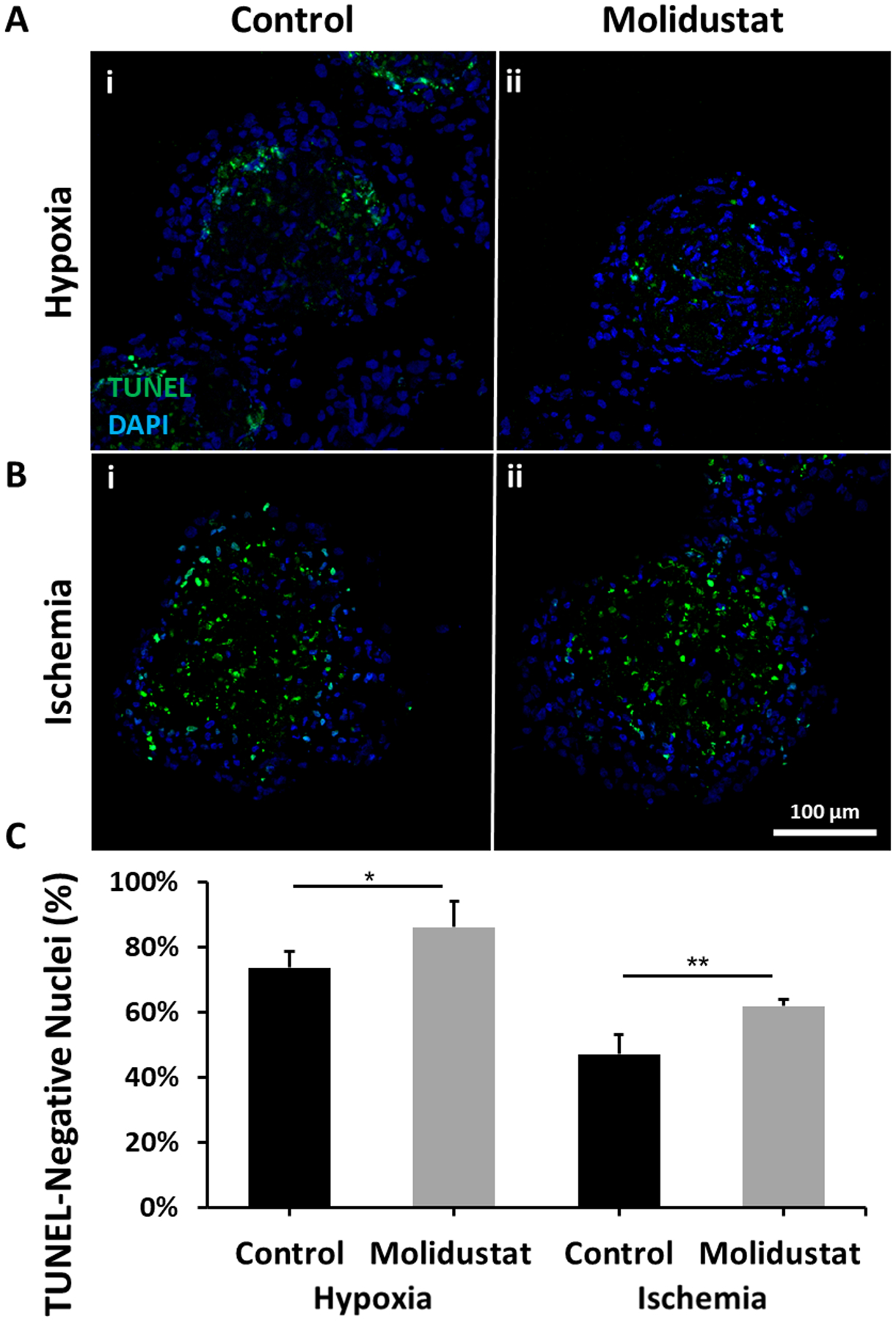
Viability assay shows that after 24 hours of exposure to (A) Hypoxia (1% O2) and (B) Ischemia (1% O2 and 1/10th media dilution), there is a (C) significant reduction in the number of TUNEL positive cells in (ii) Molidustat treated organoids when compared to (i) Control. n= at least 5 biological replicates. Mann-Whitney nonparametric test and two tailed confidence interval. (*) represents p<0.05, (**) represents p<0.005.
4. Conclusion
Here, we demonstrated that treatment of human cardiac organoids with Molidustat dramatically improved endothelial expression and lumen formation. We also show that hiPSC-CMs in the Molidustat treated organoids maintain cardiac structures and functions. The Molidustat improved endothelial network formation is, at least partially, attributed to upregulated VEGF secretion of vimentin+/NG2+ cells. Finally, we demonstrated the Molidustat treatment improved cell survival under hypoxic and ischemic conditions. For the first time, we demonstrate that HIF-α stabilization via Molidustat treatment provides a powerful and robust method to improve prevascularization within cardiac tissue engineering constructs. Compared to the use of proangiogenic growth factors (e.g., VEGF), small molecule PHD inhibitors provide a straightforward method to promote prevascularization within the cardiac microtissues. The approach outlined in this research provides a powerful framework for robust prevascularization of a wide variety of microtissues to further improve their translational capacity either as an injectable cell delivery vehicle or as an improved “building block” for the fabrication of highly vascularized tissue constructs for regenerative therapies.
Supplementary Material
Supplemental Figure 1. Mechanism of HIF-α under PHD inhibition and normoxic conditions. Under normoxic conditions, HIF-α is targeted by prolyl hydroxylase domain (PHD)-containing enzymes for hydroxylation. These hydroxyl groups recruit the von Hippel Lindau (VHL) ubiquitin E3 ligase, which results in ubiquitination and degradation of HIF-α. Under conditions where PHD is inhibited (e.g. hypoxia or PHD competitive inhibitors), allowing for HIF-α accumulation. At sufficiently high concentrations, HIF-α and HIF-β bind to form the HIF transcription factor. This transcription factor crosses the nuclear membrane and interacts with the transcriptional co-activator P300 to active the Hypoxia Response Element. Once activated, the HRE results in changes to angiogenesis, cell metabolism, and cell survival.
Supplemental Figure 2. Molidustat does not negatively impact viability of cardiac microtissues. (A, B) Viability assay shows that (B) Molidustat treatment concentration of 5 μM after 10 days of culture does not increase the number of TUNEL positive cells when compared to (B) Control in (i) hiPSC-CM spheroids or (ii) human cardiac organoids. (C) Quantification of number of nuclei that are not TUNEL positive. n=at least 5 biological replicates. Mann-Whitney nonparametric test and two tailed confidence interval.
Supplemental Figure 3. Higher concentrations of Molidustat show signs of microtissue toxicity. Brightfield microscopy of human cardiac organoids show that concentrations 50 μM and 250 μM show signs of microtissue toxicity and deterioration.
Supplemental Figure 4. Molidustat treatment results in improved expression of HIF-α. (A) Immunofluorescant staining for HIF-1α in Control(i) and Molidustat-treated (ii) cardiac organoids after 10 days of culture. Red- HIF-α, Blue- DAPI. (B) Quantified expression of HIF-α as a function of cardiac organoid area comparing Control and Molidustat-treated cardiac organoids after 10 days of culture. n=at least 4 biological replicates. Mann-Whitney nonparametric test and two tailed confidence interval. (*) represents p<0.05.
Supplemental Figure 5. Molidustat treatment does not result in increased cell proliferation. (A) Immunofluorescent staining for mitosis-marker PHH3 in Control (i) and Molidustat-treated (ii) organoids after 1 day of culture. Red- PHH3, Blue- DAPI. (B) Quantified expression of PHH3 as a percentage of nucleic co-staining identified via DAPI. n=at least 8 biological replicates (C) Immunofluorescent staining for CD31 in Control (i) and Molidustat-treated (ii) cardiac organoids after 1 day of culture. Green- CD31, Blue- DAPI. (D) Quantified expression of CD31 as a function of cardiac organoid area comparing Control and Molidustat-treated cardiac organoids after 1 day of culture. (E) Immunofluorescent co-staining for PHH3 and CD31 in Control (i) and Molidustat-treated (ii) cardiac organoids after 1 day of culture. Red- PHH3, Green- CD31, Blue- DAPI. (F) Quantified expression of PHH3+ nuclei co-localized to CD31+ regions as a percentage of the total number of PHH3+ nuclei of the cardiac organoids. n=at least 8 biological replicates. Mann-Whitney nonparametric test and two tailed confidence interval. (*) represents p<0.05.
Supplemental Figure 6. AngioTool analysis of endothelial network for human cardiac organoids with different sizes. AngioTool analysis of endothelial network for (A) 275 μm, (B) 325 μm, and (C) 400 μm diameter human cardiac organoids for the selected vasculature assessment measurements including vessel area (i), vessel percentage area (ii), total vessel length (iii), average vessel length (iv), and total number of junctions (v). n=at least 5 biological replicates. Mann-Whitney nonparametric test and two tailed confidence interval. (*) represents p<0.05, (**) represents p<0.005.
Supplemental Figure 7. Treatment of human cardiac organoids with reduced oxygen does not recapitulate results of Molidustat treatment. (A,D) Cardiac-specific staining for αSA shows that neither (ii) Molidustat treatment nor (iii) 12% Oxygen treatment negatively impacts cardiomyocyte organization when compared to (i) control. Quantification of ?SA shown as a function of area when normalized to Control. (B,E) Endothelial cell-specific staining for CD31 shows that there is a non-significant difference between (i) control and (iii) 12% Oxygen treatment, while there is a significant increase in CD31 expression for (ii) Molidustat treated organoids. Quantification of CD31 shown as a function of area when normalized to Control. (C, F) Viability assay shows that there is a non-significant difference in TUNEL-positive nuclei for (ii) Molidustat treated or (iii) 12% Oxygen treated human cardiac organoids when compared to (i) control. n=at least 5 biological replicates. Mann-Whitney nonparametric test and two tailed confidence interval. (**) represents p<0.005.
Supplemental Figure 8. Treatment of human cardiac organoids with supplemental VEGF does not recapitulate results of Molidustat treatment. (A) Endothelial cell-specific staining for CD31 shows that there is a non-significant difference between (i) control and (iii) Supplemental VEGF treatment (2.5 μg/mL), while there is a significant increase in CD31 expression for (ii) Molidustat treated organoids. (B) VEGF specific staining shows that there is a non-significant difference between (i) control and (iii) Supplemental VEGF treatment, while there is a significant increase in VEGF expression for (ii) Molidustat treated organoids. (C) Quantification of CD31 shown as a function of area. (D) Quantification of VEGF shown as a function of area. n= at least 5 biological replicates. Mann-Whitney nonparametric test and two tailed confidence interval. (*) represents p<0.05, (**) represents p<0.005.
Supplemental Figure 9. Molidustat treatment results in improved expression of endothelial markers under ischemic conditions. (A) Immunofluorescent staining of cardiac-specific (αSA) and endothelial specific (vWF) markers of control (i) and Molidustat treated (ii) cardiac organoids after 24 hours of Hypoxic and Ischemic conditions. Green- αSA, Red- vWF, Blue- DAPI. (B) Quantification of endothelial specific marker (vWF) as a function of area. (C) Quantification of cardiac specific marker (αSA) as a function of area. (D) Immunofluorescent co-staining of apoptotic nucleic marker (TUNEL) and vWF of control (i) and Molidustat treated (ii) cardiac organoids after 24 hours of Hypoxic and Ischemic conditions. Green- TUNEL, Red- vWF, Blue- DAPI. (E) Quantified expression of TUNEL+ nuclei co-localized to vWF+ regions as a percentage of the total number of TUNEL+ nuclei of the cardiac organoids for each of the testing conditions. n= at least 5 biological replicates. Mann-Whitney nonparametric test and two tailed confidence interval. (**) represents p<0.005.
Supplemental Figure 10. Molidustat treatment improves survival of cardiac organoids when exposed to hypoxic and ischemic conditions for 72 hours. Viability assay shows that after 72 hours of exposure to (A) Hypoxia (1% O2) and (B) Ischemia (1% O2 and 1/10th media dilution), there is a (C) qualitative improvement in the number of TUNEL negative nuclei in Molidustat treated organoids when compared to control. n= at least 2 biological replicates. Mann-Whitney nonparametric test and two tailed confidence interval.
Acknowledgements
The work is supported by the National Institutes of Health (1F31HL145979, 1R01HL133308, 8P20 GM103444, U54 GM104941), the startup funds from Clemson University, the National Science Foundation (NSF - EPS-0903795, NSF1655740), and the NIH Cardiovascular Training Grant (T32 HL007260), and US Department of Veterans Affairs Merit Review (I01 BX002327). This study used the services of the Morphology, Imaging, and Instrumentation Core, which is supported by NIH-NIGMS P30 GM103342 to the South Carolina COBRE for Developmentally Based Cardiovascular Diseases.
Footnotes
Conflicts of Interest Statement
The authors have no conflicts of interest to report.
References
- Barad L, Schick R, Zeevi-Levin N, Itskovitz-Eldor J, & Binah O (2014). Human embryonic stem cells vs human induced pluripotent stem cells for cardiac repair. Can J Cardiol, 30(11), 1279–1287. doi: 10.1016/j.cjca.2014.06.023 [DOI] [PubMed] [Google Scholar]
- Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, … Wu JC (2014). Chemically defined generation of human cardiomyocytes. Nat Methods, 11(8), 855–860. doi: 10.1038/nmeth.2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheema U, Brown RA, Alp B, & MacRobert AJ (2008). Spatially defined oxygen gradients and vascular endothelial growth factor expression in an engineered 3D cell model. Cell Mol Life Sci, 65(1), 177–186. doi: 10.1007/s00018-007-7356-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong JJ, Yang X, Don CW, Minami E, Liu YW, Weyers JJ, … Murry CE (2014). Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature, 510(7504), 273–277. doi: 10.1038/nature13233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle R, Yao J, Richards D, & Mei Y (2019). The Effects of Metabolic Substrate Availability on Human Adipose-Derived Stem Cell Spheroid Survival. Tissue Eng Part A, 25(7–8), 620–631. doi: 10.1089/ten.TEA.2018.0163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Moor L, Merovci I, Baetens S, Verstraeten J, Kowalska P, Krysko DV, … Declercq H (2018). High-throughput fabrication of vascularized spheroids for bioprinting. Biofabrication, 10(3), 035009. doi: 10.1088/1758-5090/aac7e6 [DOI] [PubMed] [Google Scholar]
- Fennema E, Rivron N, Rouwkema J, van Blitterswijk C, & de Boer J (2013). Spheroid culture as a tool for creating 3D complex tissues. Trends Biotechnol, 31(2), 108–115. doi: 10.1016/j.tibtech.2012.12.003 [DOI] [PubMed] [Google Scholar]
- Fong GH (2009). Regulation of angiogenesis by oxygen sensing mechanisms. J Mol Med (Berl), 87(6), 549–560. doi: 10.1007/s00109-009-0458-z [DOI] [PubMed] [Google Scholar]
- Fong TA, Shawver LK, Sun L, Tang C, App H, Powell TJ, … McMahon G (1999). SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res, 59(1), 99–106. [PubMed] [Google Scholar]
- Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, & Semenza GL (1996). Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol, 16(9), 4604–4613. doi: 10.1128/mcb.16.9.4604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-King H, Garcia NA, Ontoria-Oviedo I, Ciria M, Montero JA, & Sepulveda P (2017). Hypoxia Inducible Factor-1alpha Potentiates Jagged 1-Mediated Angiogenesis by Mesenchymal Stem Cell-Derived Exosomes. Stem Cells, 35(7), 1747–1759. doi: 10.1002/stem.2618 [DOI] [PubMed] [Google Scholar]
- Gunter J, Wolint P, Bopp A, Steiger J, Cambria E, Hoerstrup SP, & Emmert MY (2016). Microtissues in Cardiovascular Medicine: Regenerative Potential Based on a 3D Microenvironment. Stem Cells Int, 2016, 9098523. doi: 10.1155/2016/9098523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusch G (2016). Myocardial Ischemia: Lack of Coronary Blood Flow or Myocardial Oxygen Supply/Demand Imbalance? Circ Res, 119(2), 194–196. doi: 10.1161/CIRCRESAHA.116.308925 [DOI] [PubMed] [Google Scholar]
- Hirt MN, Hansen A, & Eschenhagen T (2014). Cardiac tissue engineering: state of the art. Circ Res, 114(2), 354–367. doi: 10.1161/CIRCRESAHA.114.300522 [DOI] [PubMed] [Google Scholar]
- Hoff PM, Wolff RA, Bogaard K, Waldrum S, & Abbruzzese JL (2006). A Phase I study of escalating doses of the tyrosine kinase inhibitor semaxanib (SU5416) in combination with irinotecan in patients with advanced colorectal carcinoma. Jpn J Clin Oncol, 36(2), 100–103. doi: 10.1093/jjco/hyi229 [DOI] [PubMed] [Google Scholar]
- Hyvarinen J, Hassinen IE, Sormunen R, Maki JM, Kivirikko KI, Koivunen P, & Myllyharju J (2010). Hearts of hypoxia-inducible factor prolyl 4-hydroxylase-2 hypomorphic mice show protection against acute ischemia-reperfusion injury. J Biol Chem, 285(18), 13646–13657. doi: 10.1074/jbc.M109.084855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, … Kaelin WG Jr. (2001). HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science, 292(5516), 464–468. doi: 10.1126/science.1059817 [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, … Ratcliffe PJ (2001). Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science, 292(5516), 468–472. doi: 10.1126/science.1059796 [DOI] [PubMed] [Google Scholar]
- Jain RK (2003). Molecular regulation of vessel maturation. Nat Med, 9(6), 685–693. doi: 10.1038/nm0603-685 [DOI] [PubMed] [Google Scholar]
- Kim C, Majdi M, Xia P, Wei KA, Talantova M, Spiering S, … Chen HS (2010). Non-cardiomyocytes influence the electrophysiological maturation of human embryonic stem cell-derived cardiomyocytes during differentiation. Stem Cells Dev, 19(6), 783–795. doi: 10.1089/scd.2009.0349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutziger KL, Muskheli V, Johnson P, Braun K, Wight TN, & Murry CE (2011). Developing vasculature and stroma in engineered human myocardium. Tissue Eng Part A, 17(9–10), 1219–1228. doi: 10.1089/ten.TEA.2010.0557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladoux A, & Frelin C (1997). Cardiac expressions of HIF-1 alpha and HLF/EPAS, two basic loop helix/PAS domain transcription factors involved in adaptative responses to hypoxic stresses. Biochem Biophys Res Commun, 240(3), 552–556. doi: 10.1006/bbrc.1997.7708 [DOI] [PubMed] [Google Scholar]
- Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, … Murry CE (2007). Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol, 25(9), 1015–1024. doi: 10.1038/nbt1327 [DOI] [PubMed] [Google Scholar]
- Laflamme MA, & Murry CE (2005). Regenerating the heart. Nat Biotechnol, 23(7), 845–856. doi: 10.1038/nbt1117 [DOI] [PubMed] [Google Scholar]
- Levent E, Noack C, Zelarayan LC, Katschinski DM, Zimmermann WH, & Tiburcy M (2020). Inhibition of Prolyl-Hydroxylase Domain Enzymes Protects From Reoxygenation Injury in Engineered Human Myocardium. Circulation, 142(17), 1694–1696. doi: 10.1161/CIRCULATIONAHA.119.044471 [DOI] [PubMed] [Google Scholar]
- Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, … Palecek SP (2012). Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci U S A, 109(27), E1848–1857. doi: 10.1073/pnas.1200250109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YW, Chen B, Yang X, Fugate JA, Kalucki FA, Futakuchi-Tsuchida A, … Murry CE (2018). Human embryonic stem cell-derived cardiomyocytes restore function in infarcted hearts of non-human primates. Nat Biotechnol, 36(7), 597–605. doi: 10.1038/nbt.4162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M, Moore R, & McFetridge PS (2013). Directed oxygen gradients initiate a robust early remodeling response in engineered vascular grafts. Tissue Eng Part A, 19(17–18), 2005–2013. doi: 10.1089/ten.TEA.2012.0592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SG, & Hausenloy DJ (2012). Hypoxia-inducible factor as a therapeutic target for cardioprotection. Pharmacol Ther, 136(1), 69–81. doi: 10.1016/j.pharmthera.2012.07.005 [DOI] [PubMed] [Google Scholar]
- Piera-Velazquez S, & Jimenez SA (2012). Molecular mechanisms of endothelial to mesenchymal cell transition (EndoMT) in experimentally induced fibrotic diseases. Fibrogenesis Tissue Repair, 5(Suppl 1), S7. doi: 10.1186/1755-1536-5-S1-S7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prowse AB, Timmins NE, Yau TM, Li RK, Weisel RD, Keller G, & Zandstra PW (2014). Transforming the promise of pluripotent stem cell-derived cardiomyocytes to a therapy: challenges and solutions for clinical trials. Can J Cardiol, 30(11), 1335–1349. doi: 10.1016/j.cjca.2014.08.005 [DOI] [PubMed] [Google Scholar]
- Rey S, & Semenza GL (2010). Hypoxia-inducible factor-1-dependent mechanisms of vascularization and vascular remodelling. Cardiovasc Res, 86(2), 236–242. doi: 10.1093/cvr/cvq045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards DJ, Coyle RC, Tan Y, Jia J, Wong K, Toomer K, … Mei Y (2017). Inspiration from heart development: Biomimetic development of functional human cardiac organoids. Biomaterials, 142, 112–123. doi: 10.1016/j.biomaterials.2017.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivron NC, Raiss CC, Liu J, Nandakumar A, Sticht C, Gretz N, … van Blitterswijk CA (2012). Sonic Hedgehog-activated engineered blood vessels enhance bone tissue formation. Proc Natl Acad Sci U S A, 109(12), 4413–4418. doi: 10.1073/pnas.1117627109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey TE, Saiget MK, Reinecke H, & Murry CE (2008). Systems approaches to preventing transplanted cell death in cardiac repair. J Mol Cell Cardiol, 45(4), 567–581. doi: 10.1016/j.yjmcc.2008.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer JA, Guzman PA, Riemenschneider SB, Kamp TJ, & Tranquillo RT (2018). A cardiac patch from aligned microvessel and cardiomyocyte patches. J Tissue Eng Regen Med, 12(2), 546–556. doi: 10.1002/term.2568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL, Nejfelt MK, Chi SM, & Antonarakis SE (1991). Hypoxia-inducible nuclear factors bind to an enhancer element located 3’ to the human erythropoietin gene. Proc Natl Acad Sci U S A, 88(13), 5680–5684. doi: 10.1073/pnas.88.13.5680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba Y, Fernandes S, Zhu WZ, Filice D, Muskheli V, Kim J, … Laflamme MA (2012). Human ES-cell-derived cardiomyocytes electrically couple and suppress arrhythmias in injured hearts. Nature, 489(7415), 322–325. doi: 10.1038/nature11317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba Y, Gomibuchi T, Seto T, Wada Y, Ichimura H, Tanaka Y, … Ikeda U (2016). Allogeneic transplantation of iPS cell-derived cardiomyocytes regenerates primate hearts. Nature, 538(7625), 388–391. doi: 10.1038/nature19815 [DOI] [PubMed] [Google Scholar]
- Skylar-Scott MA, Uzel SGM, Nam LL, Ahrens JH, Truby RL, Damaraju S, & Lewis JA (2019). Biomanufacturing of organ-specific tissues with high cellular density and embedded vascular channels. Sci Adv, 5(9), eaaw2459. doi: 10.1126/sciadv.aaw2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speer RE, Karuppagounder SS, Basso M, Sleiman SF, Kumar A, Brand D, … Ratan RR (2013). Hypoxia-inducible factor prolyl hydroxylases as targets for neuroprotection by “antioxidant” metal chelators: From ferroptosis to stroke. Free Radic Biol Med, 62, 26–36. doi: 10.1016/j.freeradbiomed.2013.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulloch NL, Muskheli V, Razumova MV, Korte FS, Regnier M, Hauch KD, … Murry CE (2011). Growth of engineered human myocardium with mechanical loading and vascular coculture. Circ Res, 109(1), 47–59. doi: 10.1161/CIRCRESAHA.110.237206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vunjak-Novakovic G, Tandon N, Godier A, Maidhof R, Marsano A, Martens TP, & Radisic M (2010). Challenges in cardiac tissue engineering. Tissue Eng Part B Rev, 16(2), 169–187. doi: 10.1089/ten.TEB.2009.0352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Chen A, Lieu DK, Karakikes I, Chen G, Keung W, … Li RA (2013). Effect of engineered anisotropy on the susceptibility of human pluripotent stem cell-derived ventricular cardiomyocytes to arrhythmias. Biomaterials, 34(35), 8878–8886. doi: 10.1016/j.biomaterials.2013.07.039 [DOI] [PubMed] [Google Scholar]
- Watson CJ, Collier P, Tea I, Neary R, Watson JA, Robinson C, … Baugh JA (2014). Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Hum Mol Genet, 23(8), 2176–2188. doi: 10.1093/hmg/ddt614 [DOI] [PubMed] [Google Scholar]
- Weinberger F, Breckwoldt K, Pecha S, Kelly A, Geertz B, Starbatty J, … Eschenhagen T (2016). Cardiac repair in guinea pigs with human engineered heart tissue from induced pluripotent stem cells. Sci Transl Med, 8(363), 363ra148. doi: 10.1126/scitranslmed.aaf8781 [DOI] [PubMed] [Google Scholar]
- Yeh TL, Leissing TM, Abboud MI, Thinnes CC, Atasoylu O, Holt-Martyn JP, … Schofield CJ (2017). Molecular and cellular mechanisms of HIF prolyl hydroxylase inhibitors in clinical trials. Chem Sci, 8(11), 7651–7668. doi: 10.1039/c7sc02103h [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P, Huang L, Ge X, Yan F, Wu R, & Ao Q (2006). Transdifferentiation of pulmonary arteriolar endothelial cells into smooth muscle-like cells regulated by myocardin involved in hypoxia-induced pulmonary vascular remodelling. Int J Exp Pathol, 87(6), 463–474. doi: 10.1111/j.1365-2613.2006.00503.x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Mechanism of HIF-α under PHD inhibition and normoxic conditions. Under normoxic conditions, HIF-α is targeted by prolyl hydroxylase domain (PHD)-containing enzymes for hydroxylation. These hydroxyl groups recruit the von Hippel Lindau (VHL) ubiquitin E3 ligase, which results in ubiquitination and degradation of HIF-α. Under conditions where PHD is inhibited (e.g. hypoxia or PHD competitive inhibitors), allowing for HIF-α accumulation. At sufficiently high concentrations, HIF-α and HIF-β bind to form the HIF transcription factor. This transcription factor crosses the nuclear membrane and interacts with the transcriptional co-activator P300 to active the Hypoxia Response Element. Once activated, the HRE results in changes to angiogenesis, cell metabolism, and cell survival.
Supplemental Figure 2. Molidustat does not negatively impact viability of cardiac microtissues. (A, B) Viability assay shows that (B) Molidustat treatment concentration of 5 μM after 10 days of culture does not increase the number of TUNEL positive cells when compared to (B) Control in (i) hiPSC-CM spheroids or (ii) human cardiac organoids. (C) Quantification of number of nuclei that are not TUNEL positive. n=at least 5 biological replicates. Mann-Whitney nonparametric test and two tailed confidence interval.
Supplemental Figure 3. Higher concentrations of Molidustat show signs of microtissue toxicity. Brightfield microscopy of human cardiac organoids show that concentrations 50 μM and 250 μM show signs of microtissue toxicity and deterioration.
Supplemental Figure 4. Molidustat treatment results in improved expression of HIF-α. (A) Immunofluorescant staining for HIF-1α in Control(i) and Molidustat-treated (ii) cardiac organoids after 10 days of culture. Red- HIF-α, Blue- DAPI. (B) Quantified expression of HIF-α as a function of cardiac organoid area comparing Control and Molidustat-treated cardiac organoids after 10 days of culture. n=at least 4 biological replicates. Mann-Whitney nonparametric test and two tailed confidence interval. (*) represents p<0.05.
Supplemental Figure 5. Molidustat treatment does not result in increased cell proliferation. (A) Immunofluorescent staining for mitosis-marker PHH3 in Control (i) and Molidustat-treated (ii) organoids after 1 day of culture. Red- PHH3, Blue- DAPI. (B) Quantified expression of PHH3 as a percentage of nucleic co-staining identified via DAPI. n=at least 8 biological replicates (C) Immunofluorescent staining for CD31 in Control (i) and Molidustat-treated (ii) cardiac organoids after 1 day of culture. Green- CD31, Blue- DAPI. (D) Quantified expression of CD31 as a function of cardiac organoid area comparing Control and Molidustat-treated cardiac organoids after 1 day of culture. (E) Immunofluorescent co-staining for PHH3 and CD31 in Control (i) and Molidustat-treated (ii) cardiac organoids after 1 day of culture. Red- PHH3, Green- CD31, Blue- DAPI. (F) Quantified expression of PHH3+ nuclei co-localized to CD31+ regions as a percentage of the total number of PHH3+ nuclei of the cardiac organoids. n=at least 8 biological replicates. Mann-Whitney nonparametric test and two tailed confidence interval. (*) represents p<0.05.
Supplemental Figure 6. AngioTool analysis of endothelial network for human cardiac organoids with different sizes. AngioTool analysis of endothelial network for (A) 275 μm, (B) 325 μm, and (C) 400 μm diameter human cardiac organoids for the selected vasculature assessment measurements including vessel area (i), vessel percentage area (ii), total vessel length (iii), average vessel length (iv), and total number of junctions (v). n=at least 5 biological replicates. Mann-Whitney nonparametric test and two tailed confidence interval. (*) represents p<0.05, (**) represents p<0.005.
Supplemental Figure 7. Treatment of human cardiac organoids with reduced oxygen does not recapitulate results of Molidustat treatment. (A,D) Cardiac-specific staining for αSA shows that neither (ii) Molidustat treatment nor (iii) 12% Oxygen treatment negatively impacts cardiomyocyte organization when compared to (i) control. Quantification of ?SA shown as a function of area when normalized to Control. (B,E) Endothelial cell-specific staining for CD31 shows that there is a non-significant difference between (i) control and (iii) 12% Oxygen treatment, while there is a significant increase in CD31 expression for (ii) Molidustat treated organoids. Quantification of CD31 shown as a function of area when normalized to Control. (C, F) Viability assay shows that there is a non-significant difference in TUNEL-positive nuclei for (ii) Molidustat treated or (iii) 12% Oxygen treated human cardiac organoids when compared to (i) control. n=at least 5 biological replicates. Mann-Whitney nonparametric test and two tailed confidence interval. (**) represents p<0.005.
Supplemental Figure 8. Treatment of human cardiac organoids with supplemental VEGF does not recapitulate results of Molidustat treatment. (A) Endothelial cell-specific staining for CD31 shows that there is a non-significant difference between (i) control and (iii) Supplemental VEGF treatment (2.5 μg/mL), while there is a significant increase in CD31 expression for (ii) Molidustat treated organoids. (B) VEGF specific staining shows that there is a non-significant difference between (i) control and (iii) Supplemental VEGF treatment, while there is a significant increase in VEGF expression for (ii) Molidustat treated organoids. (C) Quantification of CD31 shown as a function of area. (D) Quantification of VEGF shown as a function of area. n= at least 5 biological replicates. Mann-Whitney nonparametric test and two tailed confidence interval. (*) represents p<0.05, (**) represents p<0.005.
Supplemental Figure 9. Molidustat treatment results in improved expression of endothelial markers under ischemic conditions. (A) Immunofluorescent staining of cardiac-specific (αSA) and endothelial specific (vWF) markers of control (i) and Molidustat treated (ii) cardiac organoids after 24 hours of Hypoxic and Ischemic conditions. Green- αSA, Red- vWF, Blue- DAPI. (B) Quantification of endothelial specific marker (vWF) as a function of area. (C) Quantification of cardiac specific marker (αSA) as a function of area. (D) Immunofluorescent co-staining of apoptotic nucleic marker (TUNEL) and vWF of control (i) and Molidustat treated (ii) cardiac organoids after 24 hours of Hypoxic and Ischemic conditions. Green- TUNEL, Red- vWF, Blue- DAPI. (E) Quantified expression of TUNEL+ nuclei co-localized to vWF+ regions as a percentage of the total number of TUNEL+ nuclei of the cardiac organoids for each of the testing conditions. n= at least 5 biological replicates. Mann-Whitney nonparametric test and two tailed confidence interval. (**) represents p<0.005.
Supplemental Figure 10. Molidustat treatment improves survival of cardiac organoids when exposed to hypoxic and ischemic conditions for 72 hours. Viability assay shows that after 72 hours of exposure to (A) Hypoxia (1% O2) and (B) Ischemia (1% O2 and 1/10th media dilution), there is a (C) qualitative improvement in the number of TUNEL negative nuclei in Molidustat treated organoids when compared to control. n= at least 2 biological replicates. Mann-Whitney nonparametric test and two tailed confidence interval.