

Abstract
Spinocerebellar ataxia type 1 (SCA1) is a fatal, dominantly inherited neurodegenerative disease caused by the expansion of CAG repeats in the Ataxin-1 (ATXN1) gene. SCA1 is characterized by balance and coordination deficits due to the predominant loss of Purkinje neurons in the cerebellum. We previously demonstrated that cerebellar astrogliosis beings during the early stages of SCA1, prior to onset of motor deficits and loss of Purkinje neurons. We communicate here that cerebellar astrogliosis contributes to SCA1 pathogenesis in a biphasic, stage of disease dependent manner. We modulated astrogliosis by selectively reducing pro-inflammatory transcriptional regulator nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) signaling in astroglia via a Cre-lox mouse genetic approach. Our results indicate that inhibition of astroglial NF-κB signaling, prior to motor deficit onset, exacerbates disease severity. This is suggestive of a neuroprotective role mediated by astroglia during early stage SCA1. In contrast, inhibition of astroglial NF-κB signaling during late stage of disease ameliorated motor deficits, indicating a potentially harmful role of astroglia late in SCA1. These results indicate that astrogliosis may have a critical and dual role in disease. If so, our results imply that anti-inflammatory astroglia-based therapeutic approaches may need to consider disease progression to achieve therapeutic efficacy.
Keywords: ATAXIN-1, astroglia, neuroprotective, neurodestructive, nuclear factor kappa b, cerebellum, neurodegeneration
Introduction
Spinocerebellar ataxia type 1 (SCA1) is a fatal, dominantly inherited neurodegenerative disease caused by the abnormal expansion of CAG trinucleotide repeats within the Ataxin-1 (ATXN1) gene (Zoghbi & Orr 2009)(Banfi et al. 1994)(Banfi et al. 1996). Since CAG repeats are in the coding region of ATXN1, this mutation results in expanded polyglutamine (polyQ) tract in the ATXN1 protein. There are eight other diseases that are also caused by polyglutamine expansion in distinct proteins, including SCA 2, 3, 6, 7, 17, spino-bulbar muscular atrophy (Kennedy disease), Huntington’s disease, and dentatorubropallidoluysian atrophy, and together they form the group of polyglutamine disorders (Gusella & Macdonald 2000)(La Spada & Taylor 2010)(Paulson et al. 2017).
Ataxia, the loss of motor coordination and balance, defines the onset of disease in SCA1 patients that usually occurs in their mid-thirties (Matilla-Dueñas et al. 2008). The onset and severity of motor deficits are inversely correlated with the number of CAG repeats in ATXN1 gene, with longer CAG repeats causing earlier onset and more severe symptoms (Orr & Zoghbi 2007)(Orr & Zoghbi 2007). Moreover, like other polyQ diseases, SCA1 is characterized by anticipation, meaning that the number of repeats in ATXN1 gene can expand and cause earlier onset and increasingly severe disease in the successive generations. Disease progression for SCA1 patients is characterized by the worsening of motor dysfunction followed by death in 10 to 20 years after disease onset due to dysphagia and dyspnea (Matilla-Dueñas et al. 2008)(Orr & Zoghbi 2007). Currently, no FDA-approved treatments exist for SCA1, but various therapies are under development (Rüb et al., 2013)(Paulson et al. 2017).
Despite the widespread expression of mutant ataxin-1 within the CNS, SCA1 is pathologically characterized by the loss of Purkinje neurons (PN) in the cerebellum (Servadio et al. 1995)(Burright et al. 1995)(Seidel et al. 2012)(Nino et al. 1980). An explanation for the selective vulnerability exhibited by Purkinje neurons could be due to their intrinsic dependence on ATXN1 for cellular function, and toxicity induced by mutant ATXN1. For this reason, research has been centered around the Purkinje-intrinsic mechanisms of ATXN1 toxicity and the involvement of the surrounding glia remained little understood. However, increasing evidence indicates that astroglia contribute to the pathogenesis of many neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS) (Yamanaka et al. 2008) (Boillée et al. 2006) (Ilieva et al. 2009), SCA3 (Kretzschmar et al. 2005), spinocerebellar ataxia 7 (SCA7) (Custer et al. 2006) (Furrer et al. 2011) (Garden et al. 2002), Huntington’s disease (HD) (Meunier et al. 2016)(Hsiao et al. 2013) (Shin et al. 2005)(Jiang et al. 2016), Parkinson’s disease, and multiple system atrophy (MSA). Astroglia, the most common glial cell type in the mammalian brain, are essential for neuronal function (Herculano-Houzel 2014)(Barres 2008) by ensuring neuroprotection: they maintain the blood–brain barrier, regulate regional blood flow, and provide trophic and metabolic support to neurons. In addition, astroglia monitor and modulate neuronal activity (via regulation of extracellular ion and glutamate concentrations), synaptogenesis, and synapse maintenance (Schipke and Kettenmann 2004)(Perea et al. 2014)(Kofuji & Newman 2004)(Nippert et al. 2017)(Yang et al. 2009)(Sloan & Barres 2014). In the cerebellum, these functions are performed by at least three different types of astroglia: Bergmann glia (BG), velate astrocytes, and astroglia in the cerebellar white matter (Bellamy 2006)(Farmer et al. 2016). Bergman glia reside in the Purkinje cell layer, where their cell bodies encircle soma of Purkinje neurons. Processes of Bergmann glia extend alongside dendrites of Purkinje neurons into the molecular layer, where they envelop and regulate excitatory synapses on Purkinje neurons (Bellamy 2006) (Grosche et al. 1999). Because of their location and intimate interactions, Bergmann glia are critical for the function of Purkinje neurons (Rudolph et al. 2016)(Rudolph et al. 2016)(Rothstein et al. 1996)(Djukic et al., 2007). Perturbing Bergmann glial function through altered calcium signaling or potassium and glutamate uptake, for example, can lead to dysfunction of adjacent Purkinje neurons (Custer et al. 2006)(Iino et al., 2001)(Wang et al.,2012)(Djukic et al., 2007). Such findings emphasize the importance of Bergmann glial for Purkinje neuron homeostasis (Balakrishnan & Bellamy 2009). Employing several SCA1 mouse models, we have previously communicated that Bergmann glia, and other cerebellar astroglia, exhibit signs of reactive astrogliosis, such as increased glial fibrillary acidic protein (GFAP) expression, hypertrophy of astroglial cell bodies and processes, and expression of pro-inflammatory mediators, prior to Purkinje neuron loss and onset of motor deficits (Cvetanovic et al. 2015). Astrogliosis is a process through which astrocytes undergo morphological, molecular and functional changes in response to neuronal dysfunction or injury (Ben Haim et al. 2015)(Burda & Sofroniew 2014). However, the functional consequences of astrogliosis in disease remain controversial (Cekanaviciute et al. 2014)(Pardo et al. 2016)(Avdoshina et al. 2010)(Brambilla et al. 2005)(Furman et al. 2012)(Liddelow et al. 2017). Although our previous results indicated occurrence of early disease stage cerebellar astrogliosis (Cvetanovic et al. 2015), the mechanism and consequences of cerebellar astrogliosis on SCA1 pathogenesis remain unknown.
A clue to understanding the role of cerebellar astrogliosis may lie in the study of nuclear factor κB (NF-κB). As one of the key transcriptional factors implicated in neuroinflammation, NF-κB is activated in many neurodegenerative disorders (Hsiao et al. 2013)(Choi et al. 2014)(Mincheva-Tasheva & Soler 2012). Recent studies suggest that NF-κB is an important regulator of gene expression in human astrocytes under both physiological and inflammatory conditions (Choi et al. 2014)(Delhase et al. 1999)(Dunn et al. 2002). Under normal conditions, most of NF-κB is cytoplasmically-sequestered and kept inactive by the inhibitor of nuclear factor κB protein (IκB) proteins (Zandi et al. 1997). During inflammation, NF-κB is canonically activated following phosphorylation of the catalytic unit of IκB kinase (IKK) complex (IKKβ) at Ser180 (Hayden & Ghosh 2008)(Zhong et al. 2002). Activated IKKβ in turn phosphorylates IκB proteins, to mark them for ubiquitination and degradation (Zhong et al. 2002), and allow for release of NF-κB into the nucleus. NF-κB dimers are further activated through post-translational modifications, including phosphorylation at Ser536 to enable promotion of target gene transcription. It is generally accepted that NF-κB activation in astroglia induces the transcription of neurotoxic pro-inflammatory genes to drive disease pathology (Brambilla et al. 2005)(Fu et al. 2010)(Brambilla et al. 2009)(H. Lian et al. 2016).
We characterized NF-κB’s role at two stages of SCA1 disease progression through a novel SCA1 mouse model enabling an astroglia- and stage of disease-specific IKKβ conditional depletion. Our results suggest that astroglial NF-κB signaling may exert novel neuroprotective role early in disease and become detrimental during late stages of SCA1.
Methods and Materials
Generation of ATXN1[82Q];IKKβ flox/flox;Slc1a3-cre/ERT mice
The genetic background of the ATXN1[82Q] mice has been previously described (Burright et al. 1995). These mice were originally generated in FVB/N background and were backcrossed into C57BL/6 mice. In this mouse model, the expression of mutant ataxin-1 with an expanded 82 CAG repeat in its coding sequence, is driven by a Purkinje cell specific (Pcp2/L7) promoter. We received these mice as a gift from Dr. Harry Orr. In order to achieve selective inhibition of astroglial NF-κB signaling, ATXN1[82Q] mice were crossed with conditional IKKβ line IKKβ flox/flox, in which exon 3 of the IKKβ gene is flanked by loxP sites, and additionally with Slc1a3-cre/ERT mice which express astroglia-specific tamoxifen(TMX)-regulated Cre-ERT recombinase under transcriptional control of Slc1a3/GLAST promoter. The IKKB flox/flox line is on C57BL/6 background and was a gift from Dr. Paul Petterson, at the California Institute of Technology. Slc1a3-cre/ERT mice on C57BL/6 background were obtained from the Jackson laboratory (#012586; Tg(Slc1a3-cre/ERT)1Nat/J). We used equal number of the mice of both sex as there is no evidence to suggest sex differences in either SCA1 patients or mouse models of SCA1. Mice were randomly allocated to TMX-treated group ( i.e. to be injected with a synthetic agonist tamoxifen (TMX) to activate the Cre recombinase and thus induce the depletion of IKKβ) or oil–injected control group. We injected ATXN1[82Q];IKKβ flox/flox;Slc1a3-cre/ERT mice with TMX at 8 weeks of age for the early disease stage time point, and at 16 weeks, for the late stage time point. At 8 weeks of age, the mutant ataxin-1 is actively being expressed in Purkinje neurons (postnatal day 10), the astrogliosis already initiated (at 3 weeks), but there is no detectable change in the motor behavior of ATXN1[82Q] mice (i.e. the mice were at the pre-symptomatic stage) (Ebner et al. 2013)(Cvetanovic et al. 2015)(Qu et al. 2017). At 16 weeks, ATXN1[82Q] exhibit motor deficits and atrophy of Purkinje neuron dendrites (Duvick et al. 2010). In control experiments, TMX injection by itself did not exert any significant effect on mouse behavior in wild-type or SCA1 mice. In all the experiments on these mice, investigators were blinded to the genotype/treatment until data was fully collected.
Animal experimentation was approved by University of Minnesota and was conducted in accordance with the National Institutes of Health’s Principles of Laboratory Animal Care (86–23, revised 1985), and the American Physiological Society’s Guiding Principles in the Use of Animals.
Rotarod analysis
An accelerating rotating rod test allowed us to evaluate coordination and motor skill acquisition (#47600; Ugo Basile). Four month-old animals were placed on the rod (3cm diameter, 5.7cm lane, 16cm height to fall) and subjected to four trials per day for a period of 4 days. Each trial lasted for 10 minutes maximum. Rotarod paradigm consisted of acceleration from 5 to 40 rpm over minutes 0 to 5, followed by 40 rpm constant speed from 5 to 10 min. Latency to fall was recorded. Additionally, two consecutive rotation events, if separated by a span of 10 seconds, was also considered a fall. Mice were given 10 minutes of rest between trials. Behavioral scores were subject to statistical analysis using one-way ANOVA with post-hoc Bonferroni testing.
Immunofluorescent (IF) staining
Mouse brains were fixed overnight in 4% paraformaldehyde, cryoprotected by immersing in 30% sucrose, stored in OCT at −80 °C and then sectioned on cryostat (Leica, CM 1850) into 45 μm sections. The sections were washed in cold phosphate buffered saline (PBS) and incubated in blocking buffer (5% normal goat serum in 1% PBS-Triton X) for 1 hour at room temperature. For anti-VGLUT2 primary antibody, prior to blocking, tissues were treated with sodium citrate buffer (10mM sodium citrate; 0.05% Tween-20; pH 6.0) for antigen retrieval for 20 minutes at 95–100 °C followed by 30 minutes of cooling at room temperature. The tissues were then incubated in the blocking buffer overnight at room temperature containing appropriate dilutions of primary antibodies (Calbindin #C9848, Sigma-Aldrich; Iba-1 #019–19741, Wako; GFAP #Z0334, Dako; VGLUT2 #MAB5504, Millipore; SOX2 #sc-17320, Santa Cruz). The tissues were washed three times for 15 minutes in PBS, and incubated for 3 hours at room temperature in appropriate secondary antibodies conjugated with Alexa fluorophores (Alexa Fluor® Dyes; Life technologies) diluted in blocking buffer (1:400). Following three PBS washes for 15 minutes each, the tissues were mounted on slides with Vectashield mounting medium containing 4’, 6’-diamino-2-phenylindole (DAPI) (#H-1500; Vector Laboratories, Burlingame, CA, USA) for observation under the microscope (Olympus FluoView™ FV1000). At least six different z-stacks of 20μm from each mouse were studied and at least 3 mice from each genotype and treatment were examined. All images were taken at 20x magnification.
Quantitative Analysis of immunofluorescent staining
Quantitative analysis of immunofluorescent-stained tissues was performed by using ImageJ (National Institutes of Health). The intensity of staining was determined by measuring mean gray value in the molecular layer of the cerebellum. Microglia density was determined by dividing the number of Iba-1-positive microglia found within the cerebellar cortex by the region of interest (ROI) area. Cerebellar molecular layer thickness was determined from three primary fissures from each mouse brain. Six to eight measurements were taken from each of the primary fissures. The distance from the base of the Purkinje cell bodies to the end of the dendrite (as marked by calbindin staining) was measured. For assessing the height of climbing fibers extending along Purkinje neuron dendrites, the distance from the Purkinje neuron soma to the end of VGLUT2 staining was measured, and the extension of climbing fibers was depicted relative to the molecular layer thickness (measured as described with calbindin staining). SOX-2 labelled Bergmann glia were quantified using FIJI, a distribution of ImageJ. The images were processed by removing background and applying Laplacian smoothing. The intensity threshold for counting was minimally adjusted between 95 and 105 to avoid misrepresentation and maintain consistency. The number of SOX-2 stained cells were counted by 3D Objects Counter Plugin to enable spatial segregation of cells in 3D space. Data was analyzed using one-way ANOVA with Bonferroni’s multiple comparison post-hoc tests.
Isolation of astrocytes from cultured mouse cerebellum
Brains were separated from mice and immersed in ice cold HBSS. Cerebella were separated and meninges removed to eliminate endothelial cells. The cerebella were cut into smaller pieces, washed twice with cold HBSS and digested in 0.05% trypsin/EDTA (0.25% trypsin/EDTA diluted in DMEM) for 20 minutes at 37 °C. Cerebellar cells were then mechanically dissociated by trituration in cell culture medium (DMEM Glutamax™ + 10% FBS + N2 supplement) and passed through a 40 μm cell strainer. The tissues were centrifuged for 10 minutes at 1000 rpm, re-suspended in the culture medium, and plated in poly-D-lysine (PDL)-coated T25 flasks. The culture medium was changed after 24 hours, and then every 3 days until the cells reached confluency after 7–10 days. The T25 flasks were agitated at 250 rpm at 37 °C for 4 hours to remove microglia and oligodendrocytes. The supernatant containing microglia and oligodendrocytes was removed and the remaining astrocytes attached to the surface were trypsinized and re-plated in new PDL-coated T25 flasks. The isolated astrocytes were cultured for 5–7 days and lysed in RIPA buffer (25mM Tris, 150mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100) for further western blot analysis.
Western blot
The concentration of protein lysate prepared from isolated astrocytes was determined using Bio-rad® DC™ Protein Assay Reagents (#500–0116; Bio-rad). Sample loading buffer (SLB, 62.5 mM Tris-HCl pH 6.8, 2.5 % SDS, 0.002 % Bromophenol Blue, 0.7135 M (5%) β-mercaptoethanol and 10 % glycerol) was added to 10–20 μg proteins and loaded into a 12% agarose gel. The proteins resolved for 2 hours at 100V prior to being transfer onto nitrocellulose membrane for 1 hour at 100V. The membrane was incubated in blocking buffer [5% non-fat dry milk diluted in TBS-T (0.1% TBS-Triton X)] for 1 hour at room temperature, followed by overnight incubation at 4 °C in primary antibody diluted in the blocking buffer (GFAP #3670S, CST; NeuN #Mab377, Millipore; Iba-1 #016–20001, Wako; pNF-κB #39675, Active Motif; tNF-κB #39369, Active Motif; IKKβ #2370S, CST). For pNF-κB, 2% BSA diluted in TBS-T was used as an alternative for milk blocking buffer. The membranes were washed in TBS-T three times for 10 minutes each and incubated in HRP-conjugated secondary antibodies diluted in blocking buffer for 1 hour at room temperature. The target protein bands were visualized by adding HRP substrate (Supersignal Westpico #34080, Thermo Scientific) and imaged by using ImageQuant LAS4000 (GE Healthcare).
RT-qPCR
Total RNA was extracted from dissected mouse cerebella using TRIzol (Life Technologies) according to the manufacturer’s instructions. RNA was then treated with DNase to remove any contaminating genomic DNA (TURBO DNA-free™ Kit #AM1907, Thermo Scientific), and reverse transcribed in independent duplicate reactions using random hexamers and SuperScript® III First-Strand Synthesis System (#18080051, Thermofisher Scientific). Quantitative PCR was performed in a Light Cycler®480 II (Roche) by using SYBR Green Master Mix (Roche), and appropriate primers that target genes abundantly expressed in Purkinje neurons and astroglia (IDT Primetime types of primers used; (summarized in table 1) which correlated with disease progression (identified as ‘Magenta cluster of genes’ from Ingram et al., 2016). The mRNA levels were determined with 2-ΔΔCt (Ct = threshold cycle) formula normalized to 18S RNA and using wild-type mice as a reference (E N Burright et al. 1995), and data was analyzed using two-way ANOVA.
Table 1.
List of primers used for RT-qPCR of cerebellar cell type-specific gene expression
| Gene Group | Gene | Forward Sequence (5’→3’) | Reverse Sequence (5’→3’) |
|---|---|---|---|
| Neuron | Inpp51 | ATT CGG ACA CTT TGG AGA GC | CCT TTT CTT GAC CAT TTG CAC |
| ITPR | GAA GGC ATC TTT GGA AGT | ACC CTG AGG AAG GTT CTG | |
| Garnl3 | TCA TGA AGC CGT GTG TGC | CAC GGA TGG GAG GTC ATC | |
| Calbindin | AAG GCT TTT GAG TTA TAT GAT CAG | TTC TCA CAC AGA TCT TTC AGC | |
| Homer 3 | TGA AGA TGC TGT CAG AAG G | CTG TCC TGA AGC GCG AAG | |
| Rgs8 | CTG TCA CAC AAA TCA GAC TCC TG | TGC TTC CGT GCA GAG TC | |
| PCP4 | CCA ACG GAA AAG ACA AGA CG | TGT CGA TAT CAA ATT CTT GGA | |
| Astroglia | Kir4.1 | CCG CGA TTT ATC AGA GC | AGA TCC TTG AGG TAG AGG AA |
| BDNF | AGG ACG CGG ACT TGT ACA CT | GCT GTG ACG CAC TCG CTA AT | |
| EAAT1 | Obtained as a pre-designed primer (IDT; Mm.PT.51.6069702) | ||
| AQP4 | Obtained as a pre-designed primer (IDT; Mm.PT.58.9080805) | ||
| C3 | AAG CAT CAA CAC ACC CAA CA | CTT GAG CTC CAT TCG TGA CA | |
| P2RY | Obtained as a pre-designed primer (IDT; Mm.PT.58.33326673) | ||
| Glul | Obtained as a pre-designed primer (IDT; Mm.PT.49A.5427203) | ||
Statistical analysis
Wherever possible, sample sizes are calculated using power analyses based on the standard deviations from our previous studies, significance level of 5%, and power of 90%. Statistical tests were performed with GraphPad Prism or R. For rotarod, IHC, qRT-PCR and Western blot quantification we have used one- or two-way ANOVA followed by Bonferroni post-hoc tests or when we had only two groups to compare, we had used two-tailed Student’s t-test.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Results
Early in disease phosphorylation of NF-κB and its activating kinase IKKβ are increased in SCA1 cerebellar astroglia
The transgenic ATXN1[82Q] mice that express mutant ATXN1[82Q] protein selectively in Purkinje neurons, are one of the best-characterized mouse models of SCA1 (Burright et al. 1995). ATXN1[82Q] mice closely mirror the cerebellar aspects of the human disease at cellular and behavioral levels (Paulson et al. 2017). Motor coordination deficits and dendritic atrophy of Purkinje neurons begin around 12 weeks of age, with loss of Purkinje neurons following around 24 weeks (Qu et al. 2017). We previously demonstrated that astrogliosis is present in ATXN1[82Q] mice at three weeks of age (Cvetanovic et al. 2015) and thus precedes motor deficits.
Since transcriptional factor NF-κB is a well-known regulator of pro-inflammatory gene expression in astroglia (Choi et al. 2014)(Dunn et al. 2002), we tested for NF-κB activation in ATXN1[82Q] cerebellar astroglia. We observed increased phosphorylation of IKKβ (at Ser176/180) in astroglia isolated from the cerebella of ATXN1[82Q] mice (Figure 1B, 1.8-fold compared to wild-type littermates, N = 3 independent isolates, Student’s t-test P ≤ 0.05). Optimal NF-κB activation relies on the post-translational modifications of NF-κB subunits, such as phosphorylation of p65 at Ser 536 (Kwon et al. 2016)(Viatour et al. 2005). We detected a twofold increase in NF-κB phosphorylation in cerebellar astrocytes from pre-symptomatic SCA1 mice, when compared to their wild-type littermates (8 weeks of age, Figure 1A, N = 4 independent isolates, Student’s t-test P ≤ 0.05). Moreover, this increased phosphorylation persisted into the late post-symptomatic stage of disease (16 weeks, Figure 3B).
Figure 1. Phosphorylation of transcriptional regulator NF-κB and its activating kinase IKKβ in cerebellar astroglia isolated from 8 weeks old SCA1 mice.

Cerebellar astroglia were isolated from 8-week-old mice and protein lysates were analyzed with western blotting (WB). A. WB for IKKβ phosphorylated at Ser 180 (pIKKβ) normalized to total IKKβ (tIKKβ) (N = 3 independent biological isolates). B. WB with antibodies against NF-κB phosphorylated at Ser 536 (pNF-κB) and total NF-κB (tNF-κB, used for normalization) (N = 4 independent biological isolates). C. Astroglial marker GFAP and BG marker Kir4.1 were enriched in isolated astroglia, while markers of neurons (neuronal nuclei (NeuN)) and microglia [ionic calcium binding protein −1 (Iba1)] were not, indicating enrichment of astroglia, including Bergmann Glia. Tubulin served as a loading control. Error bars = S.E.M. Student’s two-tailed t-test * P < 0.05.
Figure 3. TMX induced inhibition of NF-κB signaling in SCA1.

ATXN1[82Q];IKKβF/F;Slc1a3-cre/ERT and IKKβF/F;Slc1a3-cre/ERT mice were injected with TMX or oil at 8 weeks of age. Cerebellar astroglia were isolated two weeks after injection and analyzed on WB with antibodies against IKKβ and tubulin (A, N = 3 independent biological samples, * P< 0.05 using two-tailed Student’s t-test) or active (pNF-κB) and total NF-κB (B, N = 3 independent biological samples, * P < 0.05 using one-way ANOVA with Bonferroni’s multiple comparison test). Error bars = S.E.M.
To validate our method of astroglial isolation, we examined the presence of the markers of astroglia (GFAP), neurons (neuronal nuclei, NeuN) and microglia (ionic calcium binding protein-1, Iba1) in the protein lysates of isolated cells. We have found increased presence of GFAP, while NeuN and Iba-1 were decreased in astroglial lysates compared to the whole cerebellum, confirming successful enrichment in cerebellar astroglia with our method (Figure 1C). Similarly, increase in potassium inward rectifier channel Kir4.1 that is predominantly expressed by Bergmann glia (Farmer et al. 2016), indicates that Bergmann glia constitute significant fraction of astroglial isolates (Figure 1C). Thus, these results indicate that classical NF-κB signaling pathway is active in cerebellar astroglia from the early stages of SCA1.
Creation of mice with conditional, astroglia-selective and TMX-dependent, depletion of IKKβ
NF-κB can be activated in many cell types in the brain with distinct effect (Yates 2015). To characterize the contribution of astroglial NF-κB activation to SCA1 pathogenesis, we created a conditional triple transgenic SCA1 mouse line: ATXN1[82Q];IKKβ flox/flox;Slc1a3-cre/ERT. These mice enabled for selective inhibition of classical NF-κB signaling in astroglia via TMX injection (Figure 2A). To create these transgenic mice, three separate mouse lines were crossed. The three single-transgenic lines were: 1) a conditional IKKβ mouse line (containing loxP-flanked exon 3 of IKKβ; IKKβ flox/flox mice) (Cho et al. 2008), 2) a Slc1a3-cre/ERT mouse line that utilizes the astroglial-specific promoter ‘solute carrier family 1 member 3’ (Slc1a3) to drive tamoxifen (TMX)-inducible CreERT recombinase expression (Madisen et al. 2015)(Paukert et al. 2014), and 3) the mutant ATXN1[82Q] mouse line that selectively expresses mutant ATXN1 in Purkinje neurons (Burright et al. 1995). This system allows for temporal control of astroglial NF-κB activity. Following intraperitoneal (lP) injections of TMX, astroglia-specific CreERT recombinase becomes functional and deletes IKKβ exon 3, which leads to decreased IKKβ activity and ultimate reduction of NF-κB activation in SCA1 astroglia.
Figure 2. Generation of the novel SCA1 transgenic mouse line to reduce astroglial NF-κB signaling in a TMX dependent manner.

A. Schematics illustrating that these SCA1 mice express tamoxifen (TMX)-activated Cre-recombinase specifically in astroglia under the control of Slc1a3 (GLAST) promoter that will remove loxP enclosed exon 3 of IKKβ from astroglia upon the injection of tamoxifen (TMX). B. –E. Immunofluorescence of cerebellar slices from GFPflox;Slc1a3-cre/ERT mice two weeks after TMX injections. B. GFP and astroglial marker GFAP. PCL=Purkinje cell and molecular layer, GCL = granule cell layer, WM= white matter. C. GFP and Purkinje cell marker calbindin. D. GFP and microglial marker Iba1. E. GFP on wild-type and ATXN1[82Q] background. F. ATXN1[82Q] mice and their wild-type littermates were injected with TMX or oil at eight weeks (left side of histogram) or at sixteen weeks. Motor performance was tested on rotarod eight weeks after the injections. Number of mice used for quantification is present in bars above each mouse line. One-way ANOVA with Bonferroni’s multiple comparison did not show statistical difference. Error bars = S.E.M
To assess the astroglial specificity of the Slc1a3-CreERT line, we crossed Slc1a3-CreERT mice with the TMX-inducible and genetically-encoded calcium indicator line GCaMP6(f); the GFP domain of the GCaMP6(f) protein functions as a reporter protein (Paukert et al. 2014). Our data demonstrated that TMX injections into GCaMP6; Slc1a3-cre/ERT mice induces astroglia-specific Cre activation in the cerebellum, as illustrated by the co-localization of the astroglial marker GFAP with the reporter GCAMP6-GFP (Figure 2B). GCaMP6 expression was low in neurons and microglia as shown by the absence of GFP co-localization with calbindin, a marker of Purkinje neurons, and Iba-1, a marker of microglia (Figures 2C and D) (Paukert et al. 2014). We did not detect GFP expression in oil-injected controls (data not shown). Moreover, in line with previous characterization of these mice (Paukert et al. 2014), Bergmann glia demonstrated the strongest GFP signal among all cerebellar astroglia, as shown by the GFP immunoreactivity in Bergmann glia residing in Purkinje cell layer (PCL) compared to velate astroglia residing in granule cell layer (GCL) and astroglia residing in white matter (WM) (Figure 2A). Notably, we observed a similar pattern of Cre activity in SCA1 mice (Figure 2D), indicating that the specificity of Slc1a3-cre/ERT line was not altered with disease.
Importantly, two weeks after TMX administration, we detected a 66.1% decrease in IKKβ protein in the astroglia isolated from the cerebella of TMX-injected IKKβ flox/flox;Slc1a3-cre/ERT mice when compared to control oil-injected IKKβ flox/flox;Slc1a3-cre/ERT mice (Figure 3A, N = 3, Student’s t-test, P < 0.05). We also found a 70.6% inhibition of NF-κB phosphorylation in cerebellar astrocytes from TMX-injected ATXN1[82Q];IKKβflox/flox;Slc1a3-cre/ERT mice compared to control oil-injected ATXN1[82Q];IKKβflox/flox;Slc1a3-cre/ERT mice. No significant difference was detected in NF-κB phosphorylation between TMX and oil injected IKKβ flox/flox;Slc1a3-cre/ERT mice (Figure 3B, N = 3 independent isolates, P < 0.05). These results suggest that we can successfully reduce NF-κB phosphorylation (i.e. activity) in SCA1 astroglia in a time dependent manner.
Inhibition of astroglial NF-κB phosphorylation reduces GFAP reactivity
Increased GFAP immunoreactivity and hypertrophy of cell bodies and processes are commonly used indicators of astrogliosis (Pekny et al. 2014). Additionally, these hallmarks can also be accompanied by the proliferation of astroglia. Indeed, using Bergmann glia-specific marker SRY-box 2 (Sox2), we have found increased number of Bergmann glia in ATXN1[82Q] mice (Figures 4A and C). Next we tested whether decreasing astroglial NF-κB phosphorylation in SCA1 mice has a functional impact on these hallmarks of cerebellar astrogliosis. Our data indicated a significant decrease in GFAP immunoreactivity in TMX-injected ATXN1[82Q];IKKβflox/flox;Slc1a3-cre/ERT mice compared to oil-injected controls, in all three types of cerebellar astroglia: 50.7% decrease in Bergmann glia, 43% decrease in velate astroglia, and 50% decrease in white matter astroglia (Figures 4A and B and data not shown, one-way ANOVA, post-hoc Bonferroni test, P< 0.05, N ≥ 3). Inhibition of NF-κB also caused a decrease in Bergmann glia proliferation (Figure 3C, N ≥ 3, P< 0.05). These results suggest that NF-κB activation contributes to cerebellar astrogliosis in SCA1 mice.
Figure 4. GFAP intensity and number of Bergmann glia.
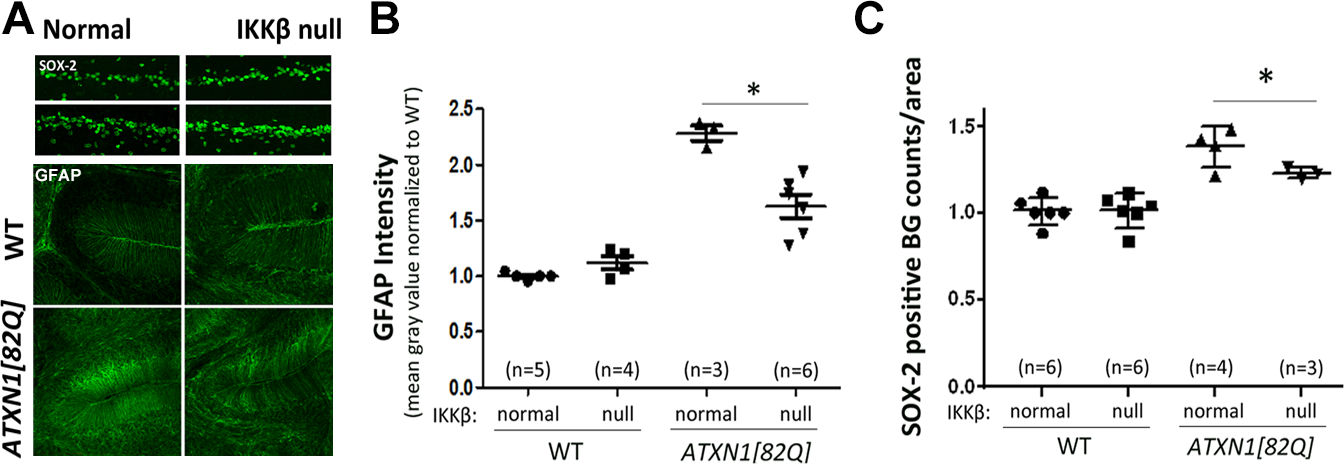
Cerebellar slices from 16-week-old oil- or TMX-injected IKKβF/F;Slc1a3-cre/ERT and ATXN1[82Q];IKKβF/F;Slc1a3-cre/ERT mice were stained with antibodies specific for (A) GFAP, (B) Bergmann glia specific Sex Determining Region Y-Box-2 (SOX-2). Image J was used to quantify the (C) intensity of GFAP staining, (D) number of SOX-2 positive Bergmann glia in the Purkinje layer. Number of mice used for quantification is present in brackets above each mouse line. * P < 0.05 using one-way ANOVA with Bonferroni’s multiple comparison test. Error bars = S.E.M.
Inhibition of astroglial NF-κB signaling early in SCA1 worsens motor deficits
Appearance of motor symptoms defines disease onset in SCA1 patients. Therefore, we refer to mice younger than 12 weeks to be in the ‘early ‘ or ‘pre-symptomatic stage’, and those that are older than 12 weeks to be in the ‘late’ or ‘symptomatic stage.’ Our previous results indicated presence of early astrogliosis indicating it may actively contribute to the pathogenesis of SCA1 (Cvetanovic et al. 2015). To determine whether astroglial NF-κB pathway contributes to pathogenesis of SCA1 early in disease, ATXN1[82Q];IKKβ flox/flox;Slc1a3-cre/ERT mice were injected with TMX at 8 weeks (Figure 5A). We examined mouse motor deficits via rotarod at 16 weeks because 1) ATXN1[82Q] mice showed robust motor deficits at this age, and it allowed sufficient time (8 weeks) for the mice to 2) recover from IP injections, which might affect rotarod performance, and 3) for a decrease in astroglial NF-κB signaling to impact disease. Four groups of mice were tested for the rotarod: (1) TMX-injected ATXN1[82Q];IKKβ flox/flox;Slc1a3-cre/ERT, (2) Oil-injected ATXN1[82Q];IKKβ flox/flox;Slc1a3-cre/ERT, (3) TMX-injected IKKβ flox/flox;Slc1a3-cre/ERT, and (4) Oil-injected IKKβ flox/flox;Slc1a3-cre/ERT mice.
Figure 5. Rotarod performance of ATXN1[82Q];IKKβF/F;Slc1a3-cre/ERT and IKKβ flox/flox;Slc1a3-cre/ERT mice injected with TMX or oil at 8 weeks.
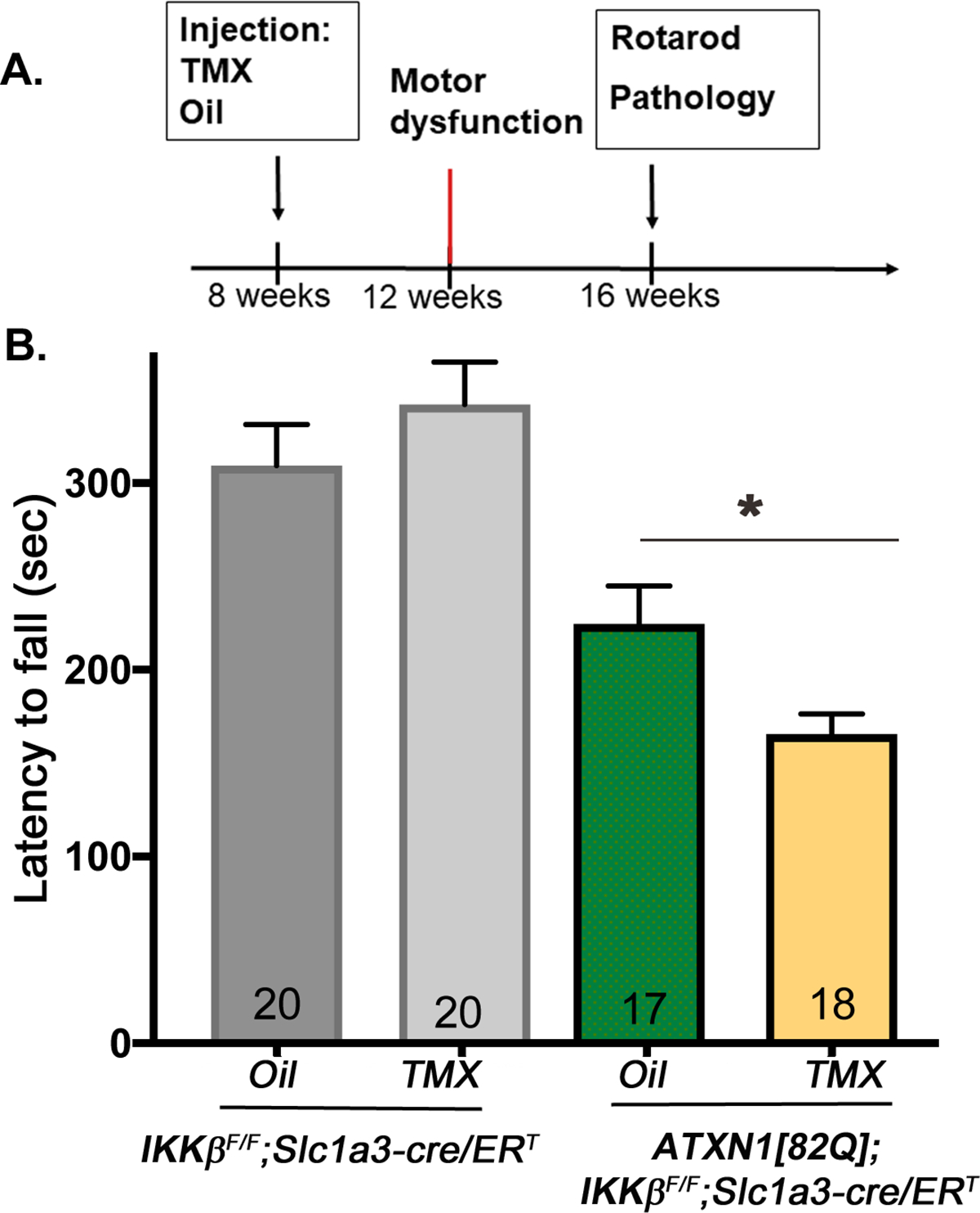
A. Experimental scheme to test the role of astroglial NF-κB early in SCA1. B. Rotarod performance of mice eight weeks after TMX injection at early stage of disease. Error bars = S.E.M., * P < 0.05 using one-way ANOVA with Bonferroni’s multiple comparison test. Number of mice used for quantification is present in bars.
Oil- and TMX-injected IKKβ flox/flox;Slc1a3-cre/ERT mice were undistinguishable on rotarod. TMX-injected ATXN1[82Q];IKKβ flox/flox;Slc1a3-cre/ERT mice showed a significant reduction in latency to fall (average 165.8 sec, N = 18) when compared to oil-injected ATXN1[82Q];IKKβ flox/flox;Slc1a3-cre/ERT mice (average 226.5 sec, N = 17, one-way ANOVA, post-hoc Bonferroni test P < 0.05) (Figure 5B). This suggests that inhibition of astroglial NF-κB early in disease exacerbates SCA1 motor deficits.
Because TMX itself has been shown to have neuroprotective effects in brain disease (Colón et al. 2016)(Duvick et al. 2010), we additionally tested whether TMX alone can affect motor behavior in SCA1 mice. TMX injected ATXN1[82Q] mice and their wild-type littermates performed similarly on rotarod as their oil-injected controls (Figure 2F), suggesting that TMX by itself did not affect pathogenesis of SCA1. This data implicates that astrogliosis during early stage of disease may be beneficial.
Early disease stage inhibition of astroglial NF-κB signaling worsens the pathology of Purkinje neurons
We next determined to what extent in early stages of disease, does astroglial NF-κB signaling contribute to Purkinje neuron dendrite atrophy and glutamatergic synapse loss, a well-characterized hallmarks of SCA1 pathology in mice (Duvick et al. 2010)(Gennarino et al. 2015). To quantify dendritic atrophy and synaptic loss we used antibodies against calbindin, a marker of Purkinje neuron soma and processes, and vesicular glutamate transporter 2 (VGLUT2), a marker of climbing fiber synapses onto Purkinje neurons. In addition, quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) was used to characterize Purkinje neuron gene expression levels as an additional marker of disease progression (Ingram et al. 2016).
We found further decrease in molecular layer width, calbindin intensity, and length of climbing fiber VGLUT2 positive synaptic terminals on calbindin-labeled Purkinje neuron dendrites in TMX-injected ATXN1[82Q]; IKKβ flox/flox;Slc1a3-cre/ERT mice when compared to oil-injected ATXN1[82Q];IKKβ flox/flox;Slc1a3-cre/ERT mice (Figure 6A–E, one-way ANOVA, post-hoc Bonferroni test, P < 0.05, N ≥ 4). These results show that dendritic atrophy and synaptic loss of Purkinje neurons are worsened when astroglial NF-κB is reduced early in disease. Transcriptional levels of Purkinje neuron genes, which have been previously shown to decrease during disease progression in SCA1 mice (Ingram et al. 2016), showed a trend to further suppression in TMX-injected ATXN1[82Q];IKKβ flox/flox;Slc1a3-cre/ERT mice compared to oil-injected ATXN1[82Q];IKKβ flox/flox;Slc1a3-cre/ERT controls (data not shown). Together, these results indicate that astroglial NF-κB signaling may be neuroprotective during early stage SCA1.
Figure 6. Selective depletion of IKKβ in astroglia worsens Purkinje neuron pathology in ATXN1[82Q] mice.

Cerebellar slices from 16-week-old oil- or TMX- injected IKKβF/F Slc1a3-cre/ERT and ATXN1[82Q];IKKβF/F; Slc1a3-cre/ERT mice were stained with antibody specific for Purkinje neuron-marker calbindin (A) or with antibodies against calbindin and vesicular glutamate transporter 2 (VGLUT2) (B) to label climbing fiber synapses on PNs. ImageJ was used to quantify (C) calbindin intensity in the Purkinje neurons, (D) molecular layer thickness and (E) length of climbing fiber synapses (VGLUT2 puncta) on Purkinje neuron dendrites (VGLUT2/calbindin). Number of mice used for quantification is present above each mouse line. * P < 0.05 using one-way ANOVA with Bonferroni’s multiple comparison test. Error bars = S.E.M.
Inhibition of astroglial NF-κB signaling early in disease increases microglial density.
Microglia are widely-accepted as the resident immune cells of the brain that become activated upon insult (Prinz and Mildner 2011)(Aguzzi et al. 2013). Moreover, there is mounting evidence supporting the importance of astroglia-microglia crosstalk in neurodegenerative diseases (Chen et al. 2015)(Lian et al. 2016)(Aguzzi et al. 2013). One way to assess microglial activation is to quantify increase in microglial density using immunofluorescence with microglial marker ionized calcium-binding adapter molecule 1 (Iba1). We previously reported increased microglial density in the cerebella of pre-symptomatic SCA1 mice co-ocurring with cerebellar astrogliosis, but whether they are functionally connected was not known (Cvetanovic et al. 2015). Moreover, we demonstrated that pharmacological depletion of microglia early in disease did not significantly alter astrogliosis (Qu et al. 2017). These results indicated that astrogliosis may occur independently of microglial activation. We now wanted to examine whether astrogliosis, and in particular astroglial NF-κB signaling, affects microglial activation in SCA1, measured as increase in microglial density. Intriguingly, we observed an increase in the density of Iba-1-positive microglia in TMX-treated ATXN1[82Q];IKKβ flox/flox;Slc1a3-cre/ERT mice (Figures 7A and B, N ≤ 4, one-way ANOVA, post hoc Bonferroni test P < 0.05). This suggests that astroglial NF-κB may suppress increase in microglial density early in SCA1. Alternatively, it is possible that the observed increase in microglial density is a consequence of exacerbated neuronal pathology.
Figure 7. Inhibition of astroglial NF-κB signaling increases microglial density.

Cerebellar slices from 16-week-old oil- or TMX-injected IKKβF/F;Slc1a3-cre/ERT and ATXN1[82Q];IKKβF/F;Slc1a3-cre/ERT mice were stained with antibodies specific for Iba1 (A). Image J was used to quantify the density of Iba1 positive microglia (B). Number of mice used for quantification is present in brackets above each mouse line. * P < 0.05 using one-way ANOVA with Bonferroni’s multiple comparison test. Error bars = S.E.M.
Inhibiting astroglial NF-κB signaling during late stages of disease ameliorates SCA1
Astroglial NF-κB signaling is considered harmful (Ilieva et al. 2009)(Hsiao et al. 2013) as suggested by evidence that reducing NF-κB signaling is beneficial in several neurodegenerative diseases (Lian et al. 2015)(Yamamoto & Gaynor 2001). We reasoned that prolonged activation of astroglia may indeed cause them to become detrimental in the post-symptomatic stage of disease. Therefore, we also characterized the contribution of astroglial NF-κB signaling to SCA1 pathogenesis after the onset of motor deficits. To reduce astroglial NF-κB signaling post-symptomatically, we injected ATXN1[82Q];IKKβ flox/flox;Slc1a3-cre/ERT mice with TMX at 16 weeks, one month after the onset of motor deficits at 12 weeks (Figure 8A). We examined motor performance 8 weeks after TMX injection and found that TMX-treated ATXN1[82Q];IKKβ flox/flox;Slc1a3-cre/ERT mice (Figure 8B, latency to fall 237.1 sec, N = 19) performed significantly better on rotarod compared to oil-injected controls (latency to fall 172 sec, N = 19, one-way ANOVA, post-hoc Bonferroni test P < 0.05). These results indicate that inhibiting astroglial NF-κB during the late stage of SCA1 is beneficial. In addition, a modest but significant increase in calbindin intensity was detected, but there was no change in the width of molecular layer, synaptic loss, and the expression of Purkinje neurons disease-associated genes (Figure 8C, one-way ANOVA, post hoc Bonferroni test, P < 0.05, N ≥ 7, and data not shown). This indicates that while the inhibition of astroglial NF-κB signaling in late stage SCA1 provided protection against calbindin loss, it did not significantly rescue most of the pathological changes observed in SCA1 Purkinje neurons.
Figure 8. Selective depletion of IKKβ in astroglia ameliorates Purkinje neuron pathology and motor functions during the late stage of SCA1.

A. Experimental scheme. B. Rotarod testing. Number of mice used for quantification is written inside the bars. C. Cerebellar slices from 6–8 month old oil- or TMX- injected IKKβF/F; Slc1a3-cre/ERT and ATXN1[82Q];IKKβF/F; Slc1a3-cre/ERT mice were stained with antibody specific for Purkinje neuron-marker calbindin. Image J was used to quantify intensity of calbindin staining, showing a significant rescue as characterized by increased calbindin intensity. Number of mice used for quantification is present in brackets above each mouse line. * P < 0.05 using one-way ANOVA with Bonferroni’s multiple comparison test. Error bars = S.E.M.
Surprisingly, reducing astroglial NF-κB activation in late stage SCA1 resulted in increased GFAP reactivity (Figures 9A–B). In addition, a significant decrease in Iba1-positive microglia was also concurrently observed (Figure 9C–D). Together, these results suggest that astroglial NF-κB signaling during late stage SCA1 decreases astrogliosis and increases microglial activation.
Figure 9. Enhanced expression of GFAP in astroglia and decreased density of microglia in ATXN1[82Q];IKKβF/F; Slc1a3-cre/ERT mice with late stage TMX injections.

Cerebellar slices from 6–8 month old oil- or TMX- injected IKKβF/F; Slc1a3-cre/ERT and ATXN1[82Q];IKKβF/F; Slc1a3-cre/ERT mice were stained with antibody specific for (A) GFAP, and (C) Iba-1. Image J was used to quantify intensity of (B) GFAP staining and (D) density of Iba1 positive microglia. Number of mice used for quantification is present in brackets above each mouse line. (An outlier defined as +2SD from the mean is indicated with an arrow in B) showing a significant increase in GFAP, as well as decrease in microgliosis. * P < 0.05 using one-way ANOVA with Bonferroni’s multiple comparison. Error bars = S.E.M.
Discussion
It is generally thought that NF-κB activation in neurons is beneficial, while NF-κB activation in glial cells is detrimental (Lawrence, 2009). However, our study suggests that the activation of canonical pro-inflammatory NF-κB signaling in astroglia has biphasic effect in SCA1: it is neuroprotective during the early stage of disease, and becomes detrimental during the late stages of the disease. A similar bimodal effect of NF-κB signaling was reported in leukemia (Ettou et al. 2013)(Ettou et al. 2012)(Claessens et al. 2005), where it was linked with disease-stage-specific regulation of gene expression by NF-κB in leukocytes.
There are five protein members in NF-κB family: RelA (p65), RelB, and c-Rel that have transcription activation domain, and p50 and p55 that do not. NF-κB proteins can form homo and heterodimers with distinct dimer-specific DNA-binding preferences and transcription inducing or inhibiting activities (Siggers et al. 2012)(Kögel et al. 2004)(Pizzi and Spano 2006). Thus different subunit composition may be one mechanism by which NF-κB exerts stage-of –disease specific effect on astroglia. In addition, NF-κB can activate or repress gene expression depending on its post-translational modification. For example, phosphorylated p65 activates transcription through increased interaction with co-activator CREB binding Protein (CBP/P300) (Gao et al. 2005), while non-phosphorylated p65 interacts with co-repressors Histone deacetylases 1, 2 and 3 (HDAC1–3) (Ashburner et al. 2001)(Bhat et al. 2008)(Robertson et al. 2000) leading to decreased histone acetylation and reduced gene expression. Thus, different post-translational modifications of NF-κB may cause distinct interactions with co-activators or co-repressors that in turn exert differential effect on astroglial NF-κB regulated gene expression at early and late stages of SCA1.
These results have several interesting implications. First, we provide evidence that cerebellar astrogliosis actively contributes to the pathogenesis of SCA1. These results suggest that degeneration of Purkinje neurons in SCA1 is likely due to a combination of intrinsic neural vulnerability and astrogliosis. Thereby, our results indicate that modulation of astroglial functions, and in particular those regulated by NF-κB signaling, may be a promising therapeutic approach. Since NF-κB is activated in astroglia in many neurodegenerative diseases, these findings may be of wider interest.
Second, our results suggest that astroglial NF-κB pathway is beneficial during an early, pre-symptomatic stage of disease. NF-κB has been studied extensively as a target for anti-inflammatory drug development because of its widely known pro-inflammatory functions (Kaltschmidt et al. 1997)(Hunot et al. 1997). Astroglial NF-κB is generally thought to be harmful in many neurological diseases such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease and encephalomyelitis (Hsiao et al. 2013)(Lawrence 2009)(Khoshnan 2004)(Lian et al. 2016)(Hsiao et al. 2013)(Brambilla et al. 2009)(Brambilla et al. 2005)(Lian et al. 2016). In fact, reports of a beneficial role of glial NF-κB in chronic neurodegenerative diseases are scarce. Perhaps its beneficial role was missed due to the limitations in approaches used to manipulate NF-κB signaling. For example, some studies used IκB knock-out mice that may increase the NF-κB signaling to non-physiological levels. Moreover, in none of the previous studies was the role of NF-κB signaling examined at different stages of disease (Hsiao et al. 2013) (Brambilla et al. 2009)(Pekny et al. 2014) (Pekny et al. 2014). Thereby, the biphasic effect of astroglial NF-κB signaling in SCA1 broadens our understanding of the astroglial role in disease and indicates that NF-κB signaling regulates both beneficial and harmful phenotypes of astroglia.
Previous in vitro studies suggest that astroglia may have varying roles that are dependent upon acute and chronic pathological conditions, or degree of neural insult (Pekny et al. 2014). We propose that astroglial may similarly have varying roles that depend on disease stage in vivo. Astroglial NF-κB activation during early stage SCA1 may be a beneficial compensatory response to mild Purkinje neurons dysfunction (Dell’Orco et al. 2015). Activated NF-κB may boost astroglial support of Purkinje neurons and combat their early dysfunction, thus allowing for maintenance of functional cerebellar network and delayed onset of motor deficits. Compensatory homeostatic roles of astroglia eventually fail and dysfunction of Purkinje neurons becomes evident causing motor symptoms to appear. Indeed, Purkinje neurons membrane potential and pacemaker firing are initially normal but, with the onset of motor dysfunction, become disrupted (Dell’Orco et al. 2015). After the onset of symptoms, decreasing astroglial NF-κB activity ameliorated SCA1 motor deficits, indicating that astroglia may become harmful late in disease. However, this did not significantly alter pathology of Purkinje neurons. One simple explanation is that our methods are not sensitive enough to detect a slight rescue in pathology. However, it is also possible that Purkinje neuron pathology cannot be reversed during late stages of SCA1 as previously shown (Zu et al., 2004). Reducing NF-κB signaling may instead rescue glial neuroprotective activities to compensate for neuronal dysfunction and ameliorate motor deficits. For example, we previously reported that the expression of astroglial glutamate transporter GLAST, which functions to remove synaptic glutamate, is decreased in Bergmann glia during a late stage of SCA1 (Cvetanovic 2015). Decreased expression of GLAST in Bergmann glia could lead to increases in extracellular glutamate and promote dysfunction and excitotoxicity of Purkinje neurons (Muller et al. 1996)(Matute, Domercq, and Sánchez-Gómez 2006). Similar decreases in glutamate transporter levels have been detected in other diseases including SCA7 and HD (Custer et al. 2006)(Hsiao et al. 2013)(Jiang et al. 2016). Another potential neuroprotective turned harmful role of astroglia is through disrupted homeostasis of potassium (K+). Kir4.1 is an astroglial potassium channel that plays a critical role in maintaining potassium (K+) homeostasis (Farmer et al. 2016)(Butt and Kalsi 2006)(Bay and Butt 2012). In the cerebellum, Kir4.1 is predominantly expressed by Bergmann glia where it regulates excitability of Purkinje neurons (Butt & Kalsi 2006). In addition, Kir4.1 determines membrane potential and thereby the activity of glutamate transporters (Kucheryavykh et al. 2007)(Power & Empson 2014)(Jiang et al. 2016)(Tong et al. 2014). Recently, it was reported that striatal astroglia in a mouse model of HD downregulate Kir4.1 during disease progression, which causes substantial increases in the potassium and glutamate concentration in striatal extracellular space and significantly contributes to neuronal dysfunction and loss (Tong et al. 2014). Both extracellular K+ and glutamate are elevated during neuronal activity, and can cause neuronal hyperactivity and excitotoxicity. Excitotoxicity has been proposed to be important contributors to neuronal death in many neurodegenerative diseases, including SCA1 (Matute et al. 2006)(Serra et al. 2004)(Power & Empson 2014). It is unknown whether NF-κB regulates expression of these supportive astroglial genes but it is possible that blocking NF-κB may rescue their expression or activity.
Based on our results, it is also possible that astroglial NF-κB signaling regulates microglial activation and their effect on Purkinje neurons. In the future, it will be important to understand the underlying mechanisms behind NF-κB’s supportive and destructive effect on astroglial phenotype, with respect to disease progression.
It is important to note that astroglial NF-κB can be activated in response to the neuronal dysfunction, and to the expression of mutant protein in astroglia. For example, in a mouse model of ALS, the expression of a mutant protein in astroglia and the astroglial reaction to neuronal dysfunction both contribute to the pathogenesis (Yamanaka et al. 2008). In this study, we have used ATXN1[82Q] that express mutant ATXN1 only in Purkinje neurons. Thereby, in these mice NF-κB and astrogliosis are induced solely in response to neuronal dysfunction. We have chosen this model because of its broader relevance to other diseases and because previous studies indicated that Bergmann glia involvement in another polyQ disease SCA7, is predominantly driven by a response to neuronal dysfunction (Furrer et al. 2011)(Lobsiger & Cleveland 2007). However, it is possible that expression of mutant ATXN1 in Bergmann glia could directly impact their neuroprotective functions and contribute to the pathogenesis of SCA1. While expression of ATXN1 in Bergmann glia was previously suggested (Shiwaku et al. 2010), we were not able to confirm it (Figure 10). In addition, timing and extent of astrogliosis and NF-κB activation in the knock-in Atxn1154Q/2Q mice that express ATXN1 with a 154Q expansion, under the endogenous Atxn1 promoter (Watase et al. 2002) is very similar to astrogliosis in Purkinje neuron specific ATXN1[82Q] mice (Cvetanovic et al. 2015). Together, these results suggest that response of Bergmann glia to dysfunctional Purkinje neurons may be the predominant mechanism of astrogliosis in SCA1.
Figure 10. Ataxin-1 expression in cerebellar slices and in in vitro cultures of cerebellar astroglia.
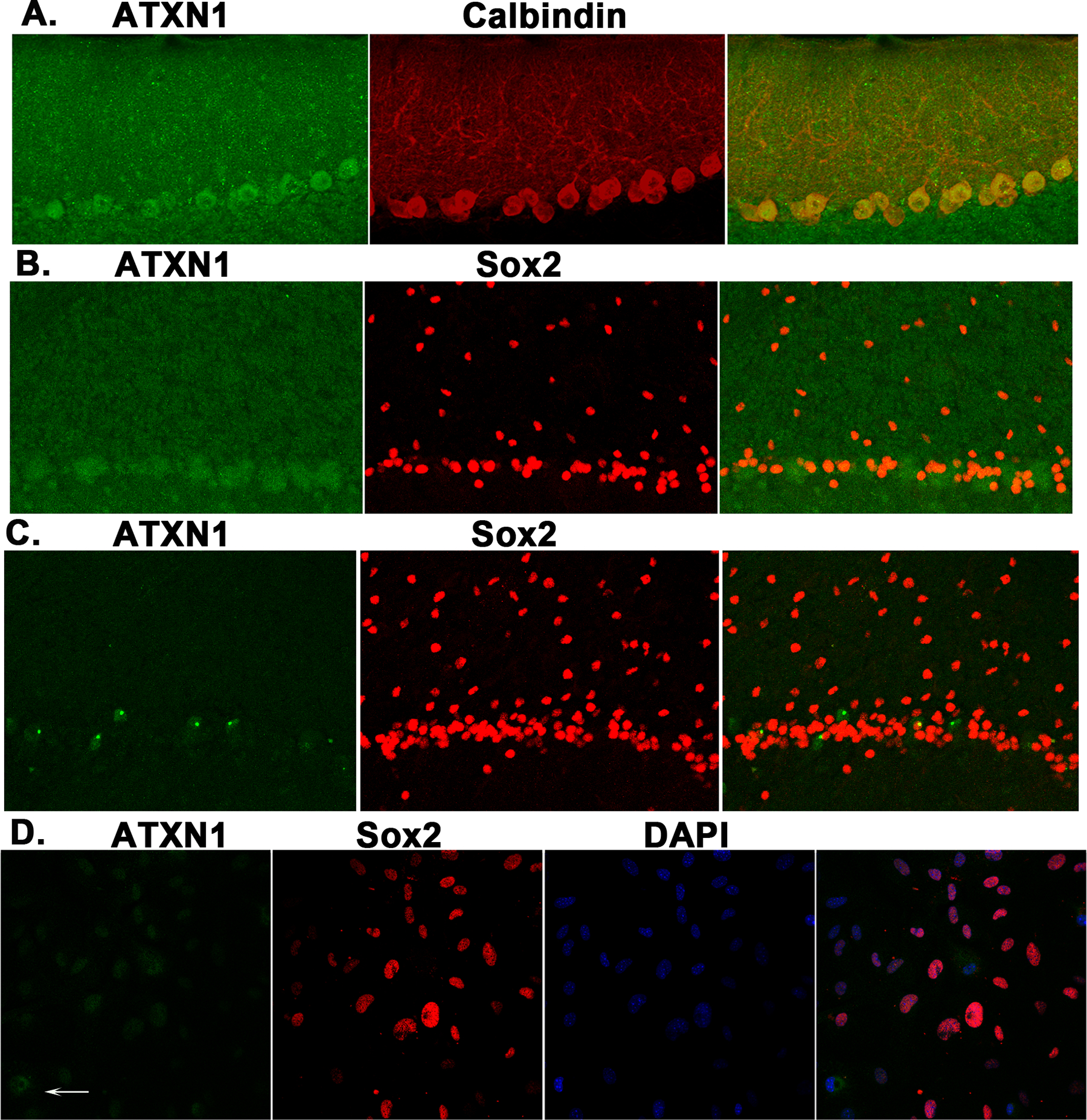
The expression of ataxin-1 protein was prominent in calbindin-positive PN (A) in cerebellar tissue slices from wild-type mice. In contrast, ataxin-1 expression was not detectable in SOX-2-positive BG in cerebellar slices from wild-type (B), ATXN1[82Q] mice (C) and in vitro astroglial cell cultures (D, arrow indicates cell that was positive for ataxin-1 but was Sox-2 negative).
The goal of effectively treating neurodegenerative disorders will likely require an understanding of not only intrinsic neuronal dysfunction, but also of how non-neuronal cells contribute to neuronal dysfunction. Our results suggest that astroglial NF-κB signaling, early in disease,, ameliorates Purkinje neuron pathology, with sustained signaling during latter stages of disease promoting Purkinje neuron dysfunction. By demonstrating active and biphasic involvement of astroglia in SCA1, this study alters our understanding of SCA1 etiology and serves as a reminder for considering disease progression in development of efficacious disease cures.
Supplementary Material
Main Points:
Astroglia play an active, biphasic role in the pathogenesis of inherited neurodegenerative disease Spinocerebellar ataxia type 1.
This biphasic role of astroglia in disease is contingent upon disease stage and regulated by NF-κB pathway.
Astroglia-based anti-inflammatory therapies may need to be timed to disease progression to achieve optimal therapeutic benefits and prevent detrimental effects.
Acknowledgements
We are grateful to Drs. Orr, Khoshnan and Petterson for the generous gift of mouse lines and to all the members of Cvetanovic and Orr laboratories for suggestions, and Aaron Mellesmoen for help with editing. The authors have no conflict of interest to declare.
References:
- Aguzzi Adriano, Barres Ben A, and Bennett Mariko L. 2013. “Microglia: Scapegoat, Saboteur, or Something Else?” Science 339 (6116): 156–61. doi: 10.1126/science.1227901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner Brian P, Westerheide Sandy D, Albert S, and Baldwin Jr. 2001. “The p65 ( RelA ) Subunit of NF- κ B Interacts with the Histone Deacetylase ( HDAC ) Corepressors HDAC1 and HDAC2 To Negatively Regulate Gene Expression.” Molecular and Cellular Biology 21 (20): 7065–77. doi: 10.1128/MCB.21.20.7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avdoshina Valeriya, Biggio Francesca, Palchik Guillermo, Campbell Lee A., and Mocchetti Italo. 2010. “Morphine Induces the Release of CCL5 from Astrocytes: Potential Neuroprotective Mechanism against the HIV Protein gp120.” Glia 58 (13): 1630–39. doi: 10.1002/glia.21035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bay Virginia, and Butt Arthur M.. 2012. “Relationship between Glial Potassium Regulation and Axon Excitability: A Role for Glial Kir4.1 Channels.” Glia 60 (4): 651–60. doi: 10.1002/glia.22299. [DOI] [PubMed] [Google Scholar]
- Bhat Krishna P., Pelloski Christopher E., Zhang Yujian, Kim Se Hoon, deLaCruz Clarissa, Rehli Michael, and Aldape Kenneth D.. 2008. “Selective Repression of YKL-40 by NF-κB in Glioma Cell Lines Involves Recruitment of Histone Deacetylase-1 and −2.” FEBS Letters 582 (21–22): 3193–3200. doi: 10.1016/j.febslet.2008.08.010. [DOI] [PubMed] [Google Scholar]
- Brambilla Roberta, Bracchi-Ricard Valerie, Hu Wen-Hui, Frydel Beata, Bramwell Annmarie, Karmally Shaffiat, Green Edward J, and Bethea John R. 2005. “Inhibition of Astroglial Nuclear Factor kappaB Reduces Inflammation and Improves Functional Recovery after Spinal Cord Injury.” The Journal of Experimental Medicine 202 (1): 145–56. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla Roberta, Persaud Trikaldarshi, Hu Xianchen, Karmally Shaffiat, Shestopalov Valery I., Dvoriantchikova Galina, Ivanov Dmitry, Nathanson Lubov, Barnum Scott R., and Bethea John R.. 2009. “Transgenic Inhibition of Astroglial NF-κB Improves Functional Outcome in Experimental Autoimmune Encephalomyelitis by Suppressing Chronic Central Nervous System Inflammation.” The Journal of Immunology 182 (5): 2628–40. doi: 10.4049/jimmunol.0802954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burright Eric N., Clark H. Brent, Servadio Antonio, Matilla Toni, Feddersen Rodney M., Yunis Wael S., Duvick Lisa a., Zoghbi Huda Y., and Orr Harry T.. 1995. “SCA1 Transgenic Mice: A Model for Neurodegeneration Caused by an Expanded CAG Trinucleotide Repeat.” Cell 82 (6): 937–48. doi: 10.1016/0092-8674(95)90273-2. [DOI] [PubMed] [Google Scholar]
- Butt Arthur M., and Kalsi Amanpreet. 2006. “Inwardly Rectifying Potassium Channels (Kir) in Central Nervous System Glia: A Special Role for Kir4.1 in Glial Functions.” Journal of Cellular and Molecular Medicine. doi: 10.1111/j.1582-4934.2006.tb00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cekanaviciute Egle, Fathali Nancy, Doyle Kristian P., Williams Aaron M., and Buckwalter Marion S.. 2014. “Astrocytic Transforming Growth Factor-Beta Signaling Reduces Subacute Neuroinflammation after Stroke in Mice.” GLIA 62 (8): 1227–40. doi: 10.1002/glia.22675.Astrocytic. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Shih Heng, Oyarzabal Esteban A., Sung Yueh Feng, Chu Chun Hsien, Wang Qingshan, Lan Chen Shiou, Lu Ru Band, and Hong Jau Shyong. 2015. “Microglial Regulation of Immunological and Neuroprotective Functions of Astroglia.” Glia 63 (1): 118–31. doi: 10.1002/glia.22738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvetanovic M, Ingram M, Orr H, and Opal P. 2015. “Early Activation of Microglia and Astrocytes in Mouse Models of Spinocerebellar Ataxia Type 1.” Neuroscience 289. IBRO: 289–99. doi: 10.1016/j.neuroscience.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn Sarah L., Young Elizabeth A., Hall Matthew D., and McNulty Shaun. 2002. “Activation of Astrocyte Intracellular Signaling Pathways by Interleukin-1 in Rat Primary Striatal Cultures.” Glia 37 (1): 31–42. doi: 10.1002/glia.10010. [DOI] [PubMed] [Google Scholar]
- Duvick Lisa, Barnes Justin, Ebner Blake, Agrawal Smita, Andresen Michael, Lim Janghoo, Giesler Glenn J., Zoghbi Huda Y., and Orr Harry T.. 2010. “SCA1-like Disease in Mice Expressing Wild-Type Ataxin-1 with a Serine to Aspartic Acid Replacement at Residue 776.” Neuron 67 (6). Elsevier Inc.: 929–35. doi: 10.1016/j.neuron.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebner B a, Ingram Melissa A, Barnes Justin A, Duvick Lisa A, Frisch Jill L, Clark H Brent, Zoghbi Huda Y, Ebner Timothy J, and Orr Harry T. 2013. “Purkinje Cell Ataxin-1 Modulates Climbing Fiber Synaptic Input in Developing and Adult Mouse Cerebellum.” Journal of Neuroscience 33 (13): 5806–20. doi: 10.1523/JNEUROSCI.6311-11.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furrer SA, Mohanachandran MS, Waldherr SM, Chang C, Damian VA, Sopher BL, Garden GA, and La Spada AR. 2011. “Spinocerebellar Ataxia Type 7 Cerebellar Disease Requires the Coordinated Action of Mutant Ataxin-7 in Neurons and Glia, and Displays Non-Cell-Autonomous Bergmann Glia Degeneration.” Journal of Neuroscience 31 (45): 16269–78. doi: 10.1523/JNEUROSCI.4000-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Zhanguo, Chiao Paul, Zhang Xia, Zhang Xiaohong, Lazar Mitchell a, Edward Seto, Young Howard a, and Ye Jianping. 2005. “Coactivators and Corepressors of NF-kappaB in IkappaB Alpha Gene Promoter.” The Journal of Biological Chemistry 280 (22): 21091–98. doi: 10.1074/jbc.M500754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao Han Yun, Chen Yu Chen, Chen Hui Mei, Tu Pang Hsien, and Chern Yijuang. 2013. “A Critical Role of Astrocyte-Mediated Nuclear Factor-κB-Dependent Inflammation in Huntington’s Disease.” Human Molecular Genetics 22 (9): 1826–42. doi: 10.1093/hmg/ddt036. [DOI] [PubMed] [Google Scholar]
- Hunot S, Brugg B, Ricard D, Michel PP, Muriel MP, Ruberg M, Faucheux B a, Agid Y, and Hirsch EC. 1997. “Nuclear Translocation of NF-kappaB Is Increased in Dopaminergic Neurons of Patients with Parkinson Disease.” PNAS 94 (14): 7531–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram Melissa, Wozniak Emily A.L., Duvick Lisa, Yang Rendong, Bergmann Paul, Carson Robert, O’Callaghan Brennon, Zoghbi Huda Y., Henzler Christine, and Orr Harry T.. 2016. “Cerebellar Transcriptome Profiles of ATXN1 Transgenic Mice Reveal SCA1 Disease Progression and Protection Pathways.” Neuron 89 (6). Elsevier Inc.: 1194–1207. doi: 10.1016/j.neuron.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltschmidt B, Uherek M, Volk B, Baeuerle P a, and Kaltschmidt C. 1997. “Transcription Factor NF-kappaB Is Activated in Primary Neurons by Amyloid Beta Peptides and in Neurons Surrounding Early Plaques from Patients with Alzheimer Disease.” PNAS 94 (6): 2642–47. doi: 10.1073/pnas.94.6.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshnan A 2004. “Activation of the I B Kinase Complex and Nuclear Factor- B Contributes to Mutant Huntingtin Neurotoxicity.” Journal of Neuroscience 24 (37): 7999–8008. doi: 10.1523/JNEUROSCI.2675-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kögel D, Peters M, König HG, Hashemi SMA, Bui NT, Arolt V, Rothermundt M, and Prehn JHM. 2004. “S100B Potently Activates p65/c-Rel Transcriptional Complexes in Hippocampal Neurons: Clinical Implications for the Role of S100B in Excitotoxic Brain Injury.” Neuroscience 127 (4): 913–20. doi: 10.1016/j.neuroscience.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Kretzschmar Doris, Tschäpe Jakob, Da Cruz Alexandre Bettencourt, Asan Esther, Poeck Burkhard, Strauss Roland, and Pflugfelder Gert O.. 2005. “Glial and Neuronal Expression of Polyglutamine Proteins Induce Behavioral Changes and Aggregate Formation in Drosophila.” Glia 49 (1): 59–72. doi: 10.1002/glia.20098. [DOI] [PubMed] [Google Scholar]
- Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Reichenbach A, Skatchkov SN, and Eaton Misty J.. 2007. “Downregulation of Kir4.1 Inward Rectifying Potassium Channel Subunits by RNAi Impairs Potassium Transfer and Glutamate Uptake by Cultured Cortical Astrocytes.” GLIA. doi: 10.1002/glia.20455. [DOI] [PubMed] [Google Scholar]
- Lawrence Toby. 2009. “The Nuclear Factor NF-kappaB Pathway in Inflammation.” Cold Spring Harbor Perspectives in Biology 1 (6): 1–10. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian Hong, Yang Li, Cole Allysa, Sun Lu, Chiang Angie C, Fowler Stephanie W, Shim David J, Rodriguez-rivera Jennifer, Taglialatela Giulio, and Jankowsky Joanna L. 2016. “NFκB-Activated Astroglial Release of Complement C3 Compromises Neuronal Morphology and Function Associated with Alzheimer’s Disease.” Neuron 85 (1): 101–15. doi: 10.1016/j.neuron.2014.11.018.NF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matute Carlos, Domercq María, and Sánchez-Gómez María-Victoria. 2006. “Glutamate-Mediated Glial Injury: Mechanisms and Clinical Importance.” Glia 53 (2): 212–24. doi: 10.1002/glia.20275. [DOI] [PubMed] [Google Scholar]
- Meunier Cécile, Merienne Nicolas, Jollé Charlotte, Déglon Nicole, and Pellerin Luc. 2016. “Astrocytes Are Key but Indirect Contributors to the Development of the Symptomatology and Pathophysiology of Huntington’s Disease.” Glia 64 (11): 1841–56. doi: 10.1002/glia.23022. [DOI] [PubMed] [Google Scholar]
- Muller T, Moller T, Neuhaus J, and Kettenmann H. 1996. “Electrical Coupling among Bergmann Glial Cells and Its Modulation by Glutamate Receptor Activation.” Glia 17 (4): 274–84. doi: 10.1002/(SICI)1098-1136(199608). [DOI] [PubMed] [Google Scholar]
- Pardo Luis, Schlüter Agatha, Valor Luis M., Barco Angel, Giralt Mercedes, Golbano Arantxa, Hidalgo Juan, et al. 2016. “Targeted Activation of CREB in Reactive Astrocytes Is Neuroprotective in Focal Acute Cortical Injury.” Glia 64 (5): 853–74. doi: 10.1002/glia.22969. [DOI] [PubMed] [Google Scholar]
- Pizzi Marina, and Spano PierFranco. 2006. “Distinct Roles of Diverse Nuclear Factor-kB Complexes in Neuropathological Mechanisms.” European Journal of Pharmacology 545 (1): 22–28. doi: 10.1016/j.ejphar.2006.06.027. [DOI] [PubMed] [Google Scholar]
- Prinz Marco, and Mildner Alexander. 2011. “Microglia in the CNS: Immigrants from Another World.” Glia 59 (2): 177–87. doi: 10.1002/glia.21104. [DOI] [PubMed] [Google Scholar]
- Qu Wenhui, Johnson Andrea, Kim Joo Hyun, Lukowicz Abigail, Svedberg Daniel, and Cvetanovic Marija. 2017. “Inhibition of Colony-Stimulating Factor 1 Receptor Early in Disease Ameliorates Motor Deficits in SCA1 Mice.” Journal of Neuroinflammation 14. Journal of Neuroinflammation: 1–11. doi: 10.1186/s12974-017-0880-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson KD, Ait-Si-Ali S, Yokochi T, Wade P a, Jones PL, and Wolffe a P. 2000. “DNMT1 Forms a Complex with Rb, E2F1 and HDAC1 and Represses Transcription from E2F-Responsive Promoters.” Nature Genetics 25 (3): 338–42. doi: 10.1038/77124. [DOI] [PubMed] [Google Scholar]
- Rothstein Jeffrey, Dykes-Hoberg Margaret, Pardo Carlos, Bristol Lynn, Jin Lin, Kuncl Ralph, Kanai Y, et al. 1996. “Antisense Knockout of Glutamate Transporters Reveals a Predominant Role for Astroglial Glutamate Transport in Excitotoxicity and Clearance of Extracellular Glutamate.” Neuron 16: 675–86. papers2://publication/uuid/327F682D-2D03-48C7-A0E6-A57E2CD92157. [DOI] [PubMed] [Google Scholar]
- Rudolph Ramona, Jahn Hannah M., Courjaret Raphael, Messemer Nanette, Kirchhoff Frank, and Deitmer Joachim W.. 2016. “The Inhibitory Input to Mouse Cerebellar Purkinje Cells Is Reciprocally Modulated by Bergmann Glial P2Y1 and AMPA Receptor Signaling.” Glia 64 (7): 1265–80. doi: 10.1002/glia.22999. [DOI] [PubMed] [Google Scholar]
- Schipke Carola G., and Kettenmann Helmut. 2004. “Astrocyte Responses to Neuronal Activity.” Glia 47 (3): 226–32. doi: 10.1002/glia.20029. [DOI] [PubMed] [Google Scholar]
- Siggers Trevor, Chang Abraham B, Ana Teixeira, Wong Daniel, Williams Kevin J, Ahmed Bilal, Ragoussis Jiannis, Udalova Irina A, Smale Stephen T, and Bulyk Martha L. 2012. “Principles of Dimer-Specific Gene Regulation Revealed by a Comprehensive Characterization of NF-κB Family DNA Binding.” Nature Immunology 13 (1): 95–102. doi: 10.1038/ni.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Duvick LA, Kaytor MD, Berlinger MS, Zoghbi HY, Clark HB, Orr HT. 2004. “Recovery from Polyglutamine-Induced Neurodegeneration in Conditional SCA1 Transgenic Mice.” Journal of Neuroscience 24 (40): 8853–61. doi: 10.1523/JNEUROSCI.2978-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.