

Abstract
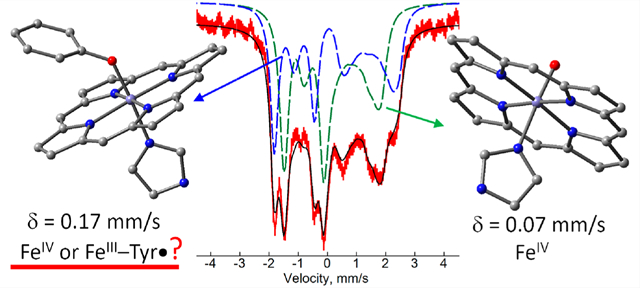
The BthA protein from the microorganism Burkholderia thailandensis contains two hemes with axial His/OH2 and His/Tyr coordinations separated by the closest interheme distance of 14 Å. BthA has a similar structure and belongs to the same family of multiheme cytochrome c peroxidases as MauG, which performs long-range oxidation of the partner protein methylamine dehydrogenase. Magnetic Mössbauer spectroscopy of the diferric state of BthA corroborates previous structural work identifying a high-spin (His/OH2) peroxidatic heme and a low-spin (His/Tyr) electron transfer heme. Unlike MauG, addition of H2O2 fully converts the diferric form of BthA to a stable 2e− oxidized state, allowing a new assessment of this state. The peroxidatic heme is found to be oxidized to a canonical compound II, S = 1 oxoiron(IV) heme. In contrast, the electronic properties of the oxidized His/Tyr heme are puzzling. The isomer shift of the His/Tyr heme (0.17 mm/s) is close to that of the precursor S = 1/2 Fe3+ heme (0.21 mm/s) which suggests oxidation of the Tyr. However, the spin-dipolar hyperfine coupling constants are found here to be the same as those for the ferryl peroxidatic heme, indicating that the His/Tyr heme is also a compound II, S = 1 Fe4+ heme and ruling out oxidation of the Tyr. DFT calculations indicate that the unusually high isomer shift is not attributable to the rare axial His/Tyr heme coordination. The calculations are only compatible with spectroscopy for an unusually long Fe4+–OTyr distance, which is presumably under the influence of the protein environment of the His/Tyr heme moiety in the H2O2 oxidized state of the protein. The results offer new insights into how high valence intermediates can be tuned by the protein environment for performing long-range oxidation.
Graphical Abstract
INTRODUCTION
Bacterial cytochrome c peroxidases (bCCPs) reduce H2O2 to H2O in the periplasm of Gram-negative bacteria, using electrons delivered by small electron transfer proteins.1 bCCPs are structurally and mechanistically distinct from well-studied monoheme plant and fungal peroxidases in that they house two c-type heme prosthetic groups covalently bound to the protein backbone by a CXXCH binding motif.2 The bCCP superfamily also includes MauG, an enzyme that shares the same canonical bCCP fold and many similarities in key amino acid positions, yet catalyzes different chemistry. Where bCCPs only appear to be capable of oxidizing a redox partner protein of relatively low potential (~250 mV), MauG is responsible for the oxidation of specific Trp residues (~1 V) in the precursor protein of methylamine dehydrogenase.3–5 A molecular perspective of how bCCPs and MauG differ functionally has proven elusive, as the structural and biophysical properties of the two diheme enzymes are similar.
Burkholderia is an opportunistic bacterium, with known implications in diseases such as cystic fibrosis. Strains of the Burkholderia cenocepacia complex (Bcc) are pathogenic and have become resistant to antibiotics. Previously, we characterized the class A diheme enzyme BthA (gene ID: BTH_II1092) from B. thailandensis to reveal that BthA is a cryptic peroxidase, capable of H2O2 turnover when in the presence of an external electron source, generating a rare bis-Fe4+ state of the hemes.6 The crystal structure of the as-isolated bis-Fe3+ state of BthA shows that the axial ligands to the two hemes are the same as in MauG (Figure 1).
Figure 1.
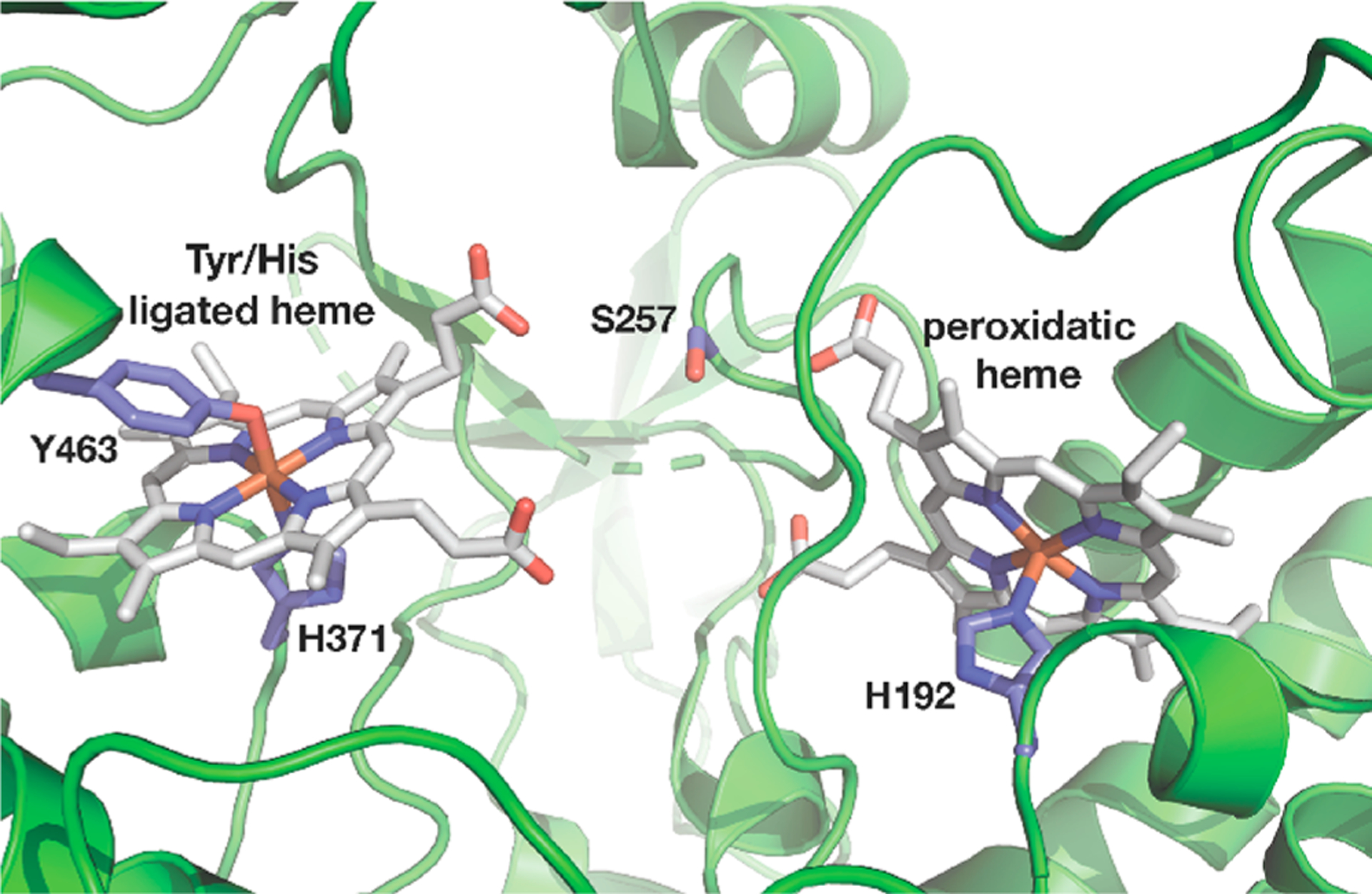
Diheme center in the XRD structure of BthA (PDB: 6NX0) showing protein-bound ligands.
MauG is responsible for the last step in the biosynthesis of tryptophan tryptophylquinone (TTQ), the catalytic cofactor of methylamine dehydrogenase (MADH).5 The biosynthesis of TTQ involves a six-electron oxidation of two tryptophan residues (βTrp57 and βTrp107) present in the protein precursor preMADH, achieved by long-range ET through the two heme sites of MauG, a precise process that earned MauG recognition as “Nature’s sniper”.7–9 Studies of MauG have focused on the characterization of the so-called bis-Fe4+ species which is formed upon reaction with H2O2 in the absence of substrate (preMADH) and is stable for up to 10 min at room temperature.10–12 The same rare oxidation state is also observed for BthA, but it has a mean lifetime of 1 h.6 The proposed mechanism of MauG involves H2O2 binding to the peroxidatic heme to generate an unstable Cpd I species which is rapidly reduced to compound II (Cpd II) by oxidation of the His/Tyr heme.10 The bis-Fe4+ state is a unique alternative to the extensively studied compound I (Cpd I) of monoheme cytochrome P450 enzymes and monoheme plant peroxidases.13,14 Mutation of the His/Tyr heme in MauG has been shown to disrupt the interheme ET network, thereby trapping the unstable Cpd I species. While this state contained the same oxidizing equivalents as the bis-Fe4+ species, the MauG mutant did not react with preMADH. As a result, it was demonstrated that only the bis-Fe4+ species is uniquely capable of TTQ maturation in MauG.
The unique stability of the bis-Fe4+ species has been the subject of numerous papers, investigating possible mechanisms for how a ferryl His/Tyr heme may be stabilized. Spin delocalization between the Fe4+ and bound Tyr residue has been attributed to the unique stability of this heme in MauG.15 The ability to generate the bis-Fe4+ state in near full-yield in BthA provides an excellent opportunity for further study of this intermediate by spectroscopy. Here we resolve the electronic properties of the hemes in BthA based on spectroscopic data for the electric and magnetic hyperfine interactions of 57Fe assisted by density functional theory (DFT). New spectroscopic data are presented from variable field Mössbauer for the first time with MauG or BthA, which can determine the spin states and hyperfine A tensors for the two hemes. Although the bis-Fe4+ states in MauG and BthA represent a phase in the redox chemistry of these enzymes comparable to Cpd I in monoheme enzymes, the electronic structures of both hemes are similar, as we shall show, to those of canonical Cpd II S = 1 Fe4+ hemes, ruling out oxidation of Tyr. Initial DFT calculations for the His/Tyr Fe4+ heme predict an isomer shift typical of the Cpd II state of the well characterized hemes of Table 1. Thus, the presence of axial His/Tyr ligands to the heme is an insufficient condition for acquiring the large isomer shift, suggesting that factors related to the protein structure at large may be in play. We propose, with support from DFT calculations, that the large isomer shift for the Fe4+ state of the His/Tyr heme in both BthA and MauG is the consequence of a protein induced constraint which causes a long Fe–OTyr bond. The long bond may tune the reduction potential of the iron for performing targeted long-range ET chemistry.
Table 1.
Electric Hyperfine Parameters and Axial Ligands in Ferryl Porphyrin Proteins and Model Compoundsa
| systemc | axial ligands | δ (mm/s) | ΔEQ (mm/s) | ref |
|---|---|---|---|---|
| JRP-II | His/O | 0.03, 0.11b | 1.59, 1.46b | 18, 19 |
| HRP-II | His/O | 0.03 | 1.61 | 20 |
| CCP-I | His/O | 0.05 | 1.55 | 21 |
| Mb-II | His/O | 0.09 | 1.44 | 18 |
| Cyt-c’β | His/O | 0.07 | 1.72 | 22 |
| MauG | His/O | 0.06 | 1.70 | 10 |
| BthA | His/O | 0.07 | 1.68 | 6 |
| MauG | His/Tyr | 0.17 | 2.54 | 10 |
| BthA | His/Tyr | 0.17 | 2.60 | 6 |
| CPO-II | Cys/O | 0.11 | 1.59 | 23 |
| CPO-II | Cys/OH | 0.10 | 2.06 | 23 |
| Catalase-II | Tyr/O | 0.07 | 1.47 | 24 |
| Catalase-II | Tyr/OH | 0.03 | 2.29 | 24 |
| Fe(O)TPPc | pyridine/O | 0.10 | 1.56 | 20 |
| Fe(O)TMPc | Cl/O | 0.07 | 1.35 | 25 |
Isomer shifts refer to samples at 4.2 K unless indicated otherwise and are defined relative to iron metal at room temperature.
Data referring to a sample at 77 K. The δ at 4.2 K is typically ~0.01 mm/s higher than at 77 K due to the second-order Doppler shift.
Abbreviations: Japanese radish peroxidase (JRP), horse radish peroxidase (HRP), myoglobin (Mb), cytochrome cβ (Cyt-c′β), chloroperoxidase Cpd II (CPO-II), tetraphenylporphyrin (TPP), tetra(mesityl)porphyrin (TMP).
In MauG, BthA, and bCCPs, the Fe3+ heme site which reacts with H2O2 has an axial ligation consisting of His and in some structures a water. This site has been called the five-coordinate heme, but given the presence of water in some structures, we will refer to it as the peroxidatic heme. The second nearby Fe3+ heme is six-coordinate and assists in electron transfer (ET) to and from the peroxidatic heme. In canonical bCCPs, the six-coordinate heme has axial His/Met ligation, while in MauG and BthA the axial ligands are His/Tyr. This second heme site will be referred to as the His/Tyr heme. These ligand changes confer changes in the redox potentials of the two hemes, which has implications in the peroxidase reactivity of BthA relative to canonical bCCPs. In bCCPs, the His/Met heme has a higher reduction potential than the peroxidatic heme, and consequently, the enzyme can be poised in a semireduced state in which the six-coordinate heme is Fe2+ and the peroxidatic heme is Fe3+. In contrast, the two hemes of MauG and BthA are close in redox potential, and the semireduced form of these enzymes cannot be formed without simultaneously reducing the second site.16
In the as-isolated (resting) form of MauG and BthA, the optical, EPR, and Mössbauer spectroscopies of the peroxidatic heme are typical of S = 5/2 Fe3+ hemes with His ligation. These spectroscopies also show that the His/Tyr heme is in a S = 1/2 Fe3+ oxidation state as expected for six-coordinate hemes. EXAFS results for MauG show a short Fe–O/N distance of 1.89 Å which was assigned to the Fe–OTyr bond.15 Upon reaction with H2O2, for both MauG and BthA, these spectroscopies show that the two heme sites are oxidized. The peroxidatic site in this state has a Mössbauer isomer shift (δ = 0.07 mm/s) that is in the range of S = 1 Fe4+=O heme species, such as observed from myoglobin (Mb), horseradish peroxidase (HRP), and yeast CCP in which there is no radical in the porphyrin, a state known as compound II (Cpd II). The Mössbauer parameters for characterized Fe4+–O(H) porphyrin Cpd II complexes are given in Table 1. The complexes include the axial heme coordinations His/O, Cys/O, Cys/OH, Tyr/O, and Tyr/OH, which span a range 0.03 < δ < 0.11 mm/s. Further evidence for the Fe4+=O heme species in MauG comes from EXAFS data which show a short Fe–O bond length of 1.69 Å.15
In contrast, the value δ = 0.17 mm/s for the oxidized His/Tyr heme in MauG and BthA lies far above the range of Cpd II hemes and is close to the value prior to oxidation of the His/Tyr heme, δ = 0.21 mm/s, which is typical of S = 1/2 Fe3+ hemes. It is clear from EPR and Mössbauer spectroscopy that the spin of the oxidized His/Tyr heme site is integer, but it is not clear if the oxidation is predominately iron based. Oxidation of the Tyr and spin coupling to an Fe3+ heme could also result in an integer spin state. Previously, the large isomer shift was attributed to a covalent Fe–Tyr bond with significant transfer of electron density from Tyr to Fe and the attending presence of unpaired aromatic spin density on the Tyr.17 While the two hemes of the H2O2 oxidized state are formally Fe4+, it has yet to be demonstrated that both irons are truly oxidized.
The DFT calculations for the peroxidatic and His/Tyr heme sites of BthA in both the di-Fe3+ and di-Fe4+ states were performed with the program suite Gaussian ’09 using the pure density functional BP86 unless otherwise noted. The two heme sites in BthA are of heme c type and contain one or two axial ligands. Four structural models for the His/Tyr c-heme active site of BthA have been used in the DFT calculations (Figure 2). These structures use either proto-heme or c-heme and increasingly resemble the active site with changes in the axial ligand substituents. The structures will be referred to using the labels in Figure 2. Unless signified as c-heme, the calculations use proto-heme. Initial calculations were simplified using imidazole (Im) and phenoxide (Ph) as axial ligands, either unconstrained, or fixed in place by the H atoms in blue. Subsequent calculations to better resemble His or Tyr were performed by including a CH3 where Cβ would be in the respective amino acid. The atoms of which the positions have been frozen in the geometry optimization based on the XRD structure of BthA are shown in blue in Figure 2. Although the carboxylate groups in the side chains of heme c are deprotonated at neutral pH, they were kept in the protonated state in the DFT calculations to mimic the effect of protein hydrogen bonding to these groups. The hyperfine parameters for the various models were evaluated for optimized geometries. Further details are given in the SI.
Figure 2.
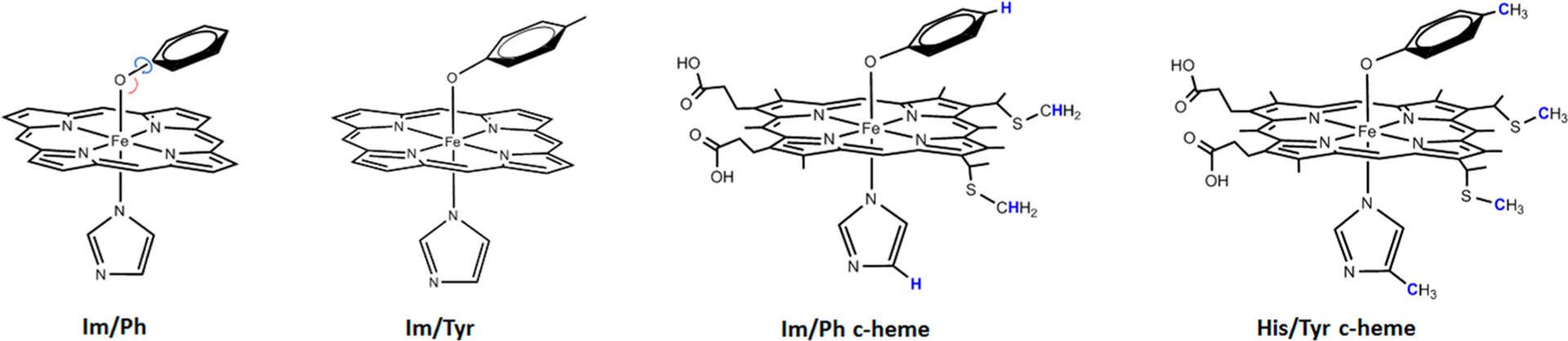
DFT models for His/Tyr heme of BthA. Frozen atoms are shown in blue. Left cartoon shows the bond and dihedral angles described in the text. Ph and Tyr refer to the deprotonated phenol and tyrosine anions. Im and His are electrically neutral imidazole and histidine. Carbons and hydrogens are only shown when part of a restricted group.
RESULTS
Mössbauer Spectroscopy.
Variable magnetic field Mössbauer spectra of 57Fe enriched as-isolated BthA recorded for a sample temperature of 4.2 K are shown in Figure 3 (red traces). The data show an approximately 1:1 superposition of magnetically split spectra of high- and low-spin Fe3+ hemes. At higher temperatures, the spectra develop broad features indicative of intermediate electronic relaxation relative to the 57Fe nuclear Larmor frequency. The black traces are simulations composed of high- and low-spin Fe3+ hemes in a ratio of 45:55, respectively. This ratio agreed with that of EPR samples of as-isolated BthA.6 The ratio of the two species may depend on the method of purification, due to the conversion of the catalytic site heme to a six-coordinate site as a function of pH. The arrows in Figure 3 indicate the position of adventitious Fe3+ features that accounted for less than 10% of the total iron. The simulations (black traces) are the result of global least-squares fitting with a set of parameters for S = 5/2 and S = 1/2 spin centers that achieved a good match to the entire data set. The Mössbauer parameters for the two ferric hemes are given in Table 2. For one simulation, the dashed lines above Figure 3B display the individual spectra of the peroxidatic heme (green, high-spin) and His/Tyr heme (blue, low-spin) that give the simulation (black line).
Figure 3.
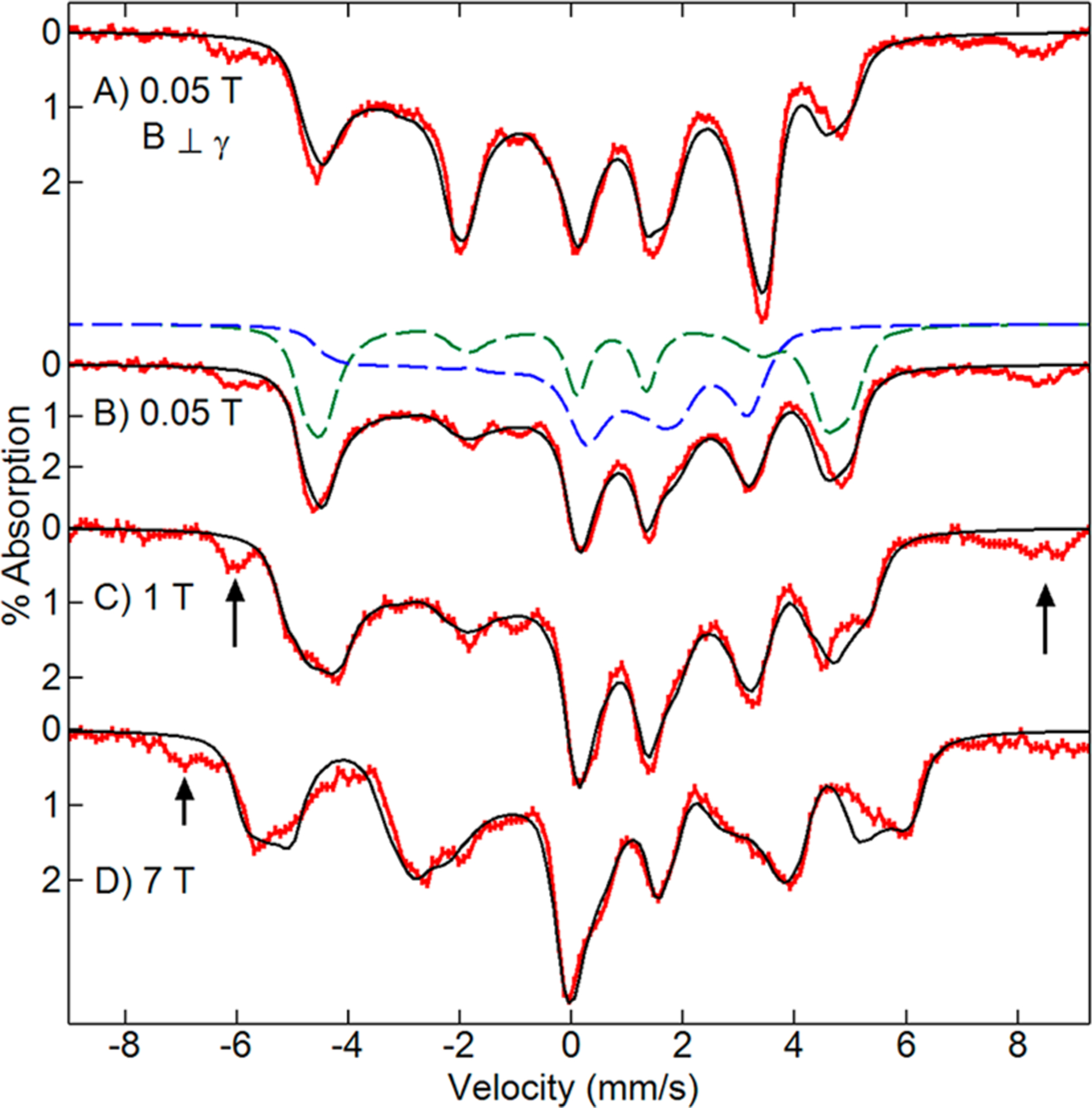
Mössbauer spectra (4.2 K, red traces) of 57Fe-enriched as-isolated 2 mM BthA recorded with magnetic fields (as listed) perpendicular (A) and parallel (B–D) to the incident γ radiation direction. The black traces are global least-squares fits for S = 5/2 and 1/2 Fe3+ species using the parameters in Table 2 (Γ = 0.44 mm/s). For (B), the individual species giving the sum are shown: S = 5/2 peroxidatic heme (green dash), S = 1/2 His/Tyr heme (blue dash). The arrows denote a minor adventitious iron species.
Table 2.
Experimental Parameters for BthA and after Oxidation with H2O2 with Comparisons to Similar Heme Sitesa
| spin | δ (mm/s) | ΔEQ (mm/s) | D (cm−1) | E/D | A (T) | η | |
|---|---|---|---|---|---|---|---|
| as-isolated | |||||||
| BthA, peroxidatic | 5/2 | 0.42 | +1.37 | 11 | 0 | −21, −18, −18b | 0.1 |
| BthA, His/Tyr | 1/2 | 0.21 | −2.67 | −44, −1, +24c | 0 | ||
| MauG, peroxidatic | 5/2 | 0.50 | +2.0 | 10 | 0 | −18, −18, −18 | 0 |
| MauG, His/Tyr | 1/2 | 0.25 | −1.97 | −36, +9, +30 | 0 | ||
| met-Mb | 5/2 | 0.42 | 1.24 | 12 | 0 | −20 | 0 |
| HRP | 5/2 | 0.40 | +1.60 | 14 | 0.01 | −20, −18, −20 | 0 |
| After H2O2 | |||||||
| BthA, peroxidatic | 1 | 0.07(1) | +1.64(3) | 25 | 0 | −23(1), −23(1), −6(2)d
ASD: −6, −6, +12 |
0 |
| BthA, His/Tyr | 1 | 0.17(1) | +2.55(3) | 27 | 0 | −21(1), −21(1), −3(2)d
ASD: −6, −6, +12 |
0.04 |
| MauG, peroxidatic | 1 | 0.06 | 1.70 | ||||
| MauG, His/Tyr | 1 | 0.17 | 2.54 | ||||
| Mb II | 1 | 0.09 | +1.43 | 24 | 0 | −19, −19, −3e ASD: −5, −5, +11 |
0 |
| HRP II | 1 | 0.03 | +1.61 | 22 | 0 | −19, −19, −3 ASD: −5, −5, +11 |
0 |
The parameters for the two hemes of BthA are similar to those reported for MauG (Table 2).10 The analysis of the Mössbauer spectra of MauG was limited to one low-field spectrum, thus the parameters of MauG have higher uncertainty and may, within error, be equal to those for BthA. The parameters of the peroxidatic heme are all close to those of Mb and HRP (Table 2) which also have an axial His coordinated to a high-spin Fe3+ heme. For the His/Tyr heme of BthA, the parameters are all in the range of other low-spin heme complexes with specific characteristics including a large negative value of ΔEQ and large anisotropy in the A tensor.26
Ten equivalents of H2O2 were added to a sample of 57Fe enriched as-isolated BthA and frozen after 1 min. The Mössbauer spectrum after H2O2 addition showed quantitative oxidation of both Fe3+ hemes (Figure 4). The Mössbauer parameters of the quadrupole doublets of both hemes (Table 2) match the values of our previous work given in Table 1.6 This H2O2 oxidized state of the enzyme has a half-life of 50 min at room temperature,6 whereas in MauG the bis-Fe4+ state has nearly fully returned to the diferric state in under 20 min.11 The quantitative conversion allowed us to characterize the H2O2 oxidized state with variable temperature and magnetic field Mössbauer spectroscopy. Figure 4 shows the spectral changes as a function of magnetic field (A–C) and of temperature at high magnetic field (C–F). The resolution of spectra and agreement with simulation indicates that all spectra for temperatures above 4 K are in the fast fluctuation limit, and thus the thermal average of the spin expectation values is used to determine the internal hyperfine field.
Figure 4.
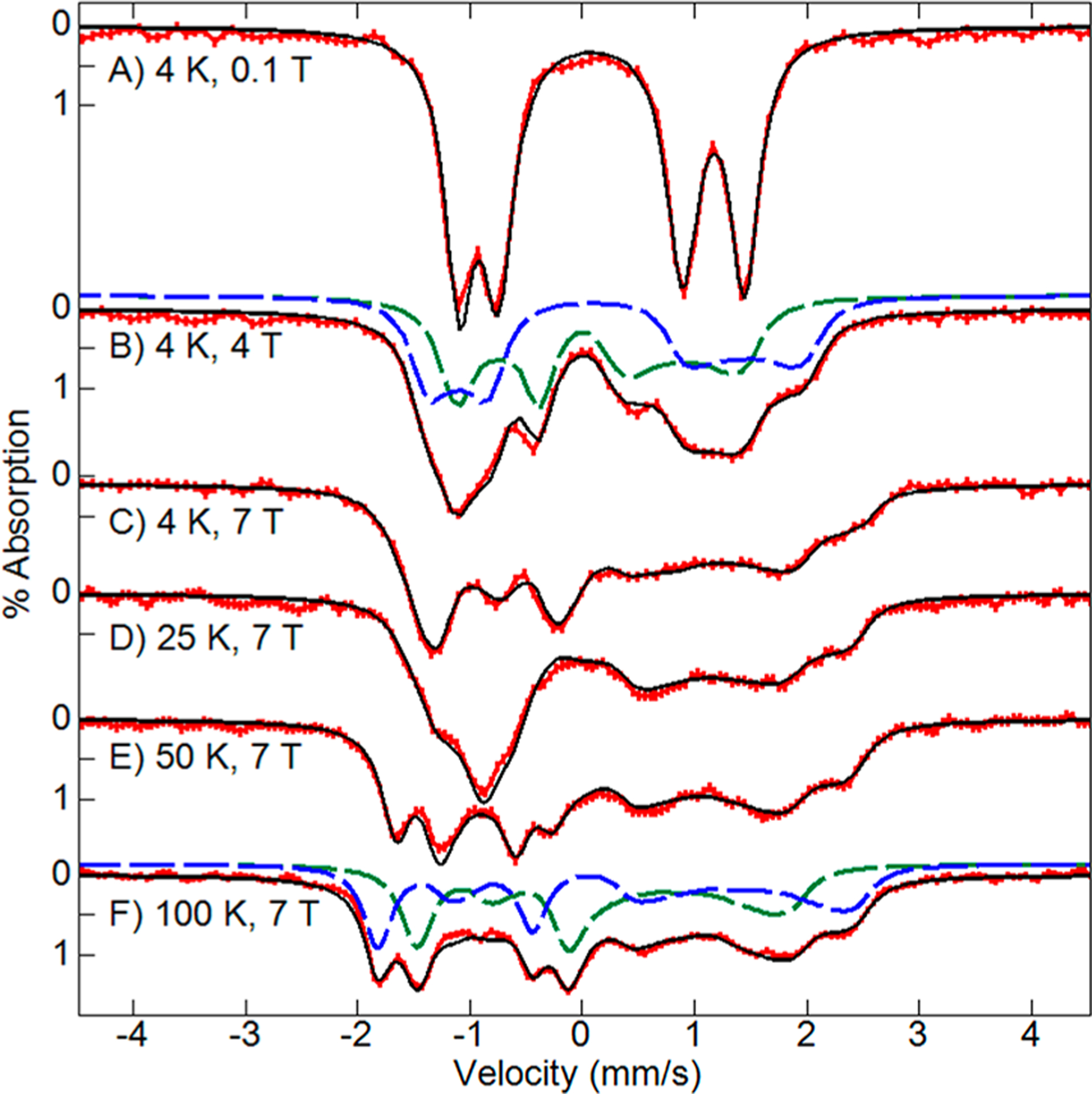
Mössbauer spectra (red traces) of 0.5 mM 57Fe-enriched BthA after addition of 10 equiv of H2O2 recorded at temperatures and magnetic fields (as listed) for a parallel incident γ radiation direction. The black traces are global least-squares fits for two S = 1 Fe4+ species using the parameters in Table 2 (Γ = 0.35 mm/s). For (B and F), the individual species giving the sum are shown: peroxidatic heme (green dash), His/Tyr heme (blue dash).
The simulations (black traces) are the result of global least-squares fitting with two S = 1 species that achieved an excellent match to the entire data set of Figure 4. The relative concentrations of the two S = 1 species were found to be equal. The Mössbauer parameters for the two hemes are given in Table 2. For Figures 4B and F, the dashed lines display the individual spectra of the peroxidatic heme (green dash) and His/Tyr heme (blue dash) that together give the respective simulations (black lines). The high quality of spectra and full conversion to the H2O2 oxidized state allowed a more thorough characterization of the electronic structure of the hemes than previously possible for the rare His/Tyr heme. From the simulations of Figure 4, the full electric and magnetic hyperfine tensors of the two hemes were determined. The simulation of the peroxidatic heme (green dashed trace) has high similarity to the published spectra of the Cpd II states of Mb, JRP, and HRP.26 Consequently, the values of δ, ΔEQ, components of the A tensor, D, and E/D are all similar (Table 2).
The simulations of the His/Tyr heme (Figures 4B, F, blue dashed trace) have a similar shape to those of the peroxidatic heme, but the positions of the peaks are significantly shifted owing to the larger values of ΔEQ and δ. Generally, the magnetic hyperfine tensor has three contributions, A = AFC + ASD + AL, from isotropic Fermi contact, spin-dipolar, and orbital terms, respectively. For Fe4+ complexes, the orbital contribution can be ignored.27 The spin-dipolar contribution can then be calculated by subtracting the trace from the principal values of the A tensor, resulting in the values given in Table 2. Prior to this work, the A tensors for the Cpd II states of most complexes were determined using g tensors with significant deviation from 2 (gx,y ≈ 2.25) based on a t2g4 crystal field model for Fe4+.28 DFT calculations have since demonstrated that this model is too limited and that the g-values are closer to 2.27 We have assumed g = 2.0, and owing to the magnetic hyperfine splitting being proportional to the product gA, the A tensors determined here are approximately 10% higher in magnitude than in the older work referenced. As Table 2 indicates, and critical to the ensuing discussion in this paper, all components of the spin-dipolar contribution to the magnetic hyperfine tensor for the His/Tyr heme, ASD, are within uncertainty the same as those of the peroxidatic heme, both of which are the same as the values from the Cpd II states of Mb and HRP. The principal values of the spin-dipolar tensor are determined by the orbitals containing the unpaired electrons of the iron site. The similarity of values between the peroxidatic heme and His/Tyr heme requires the same d-orbital spin density for both sites. These results unambiguously demonstrate that the electronic configuration of both hemes is S = 1 Fe4+ with insignificant aromatic spin density on the Tyr. The experimental spin-dipolar tensor will provide a powerful criterion for assessing the validity of the DFT calculations discussed below.
Electronic Structure of the Hemes in the Resting State of BthA.
The peroxidatic heme in BthA consists of an Fe3+ c-heme with axial coordination to His and a water molecule.6 This coordination favors a high-spin (S = 5/2) state, which was confirmed by EPR and Mössbauer spectroscopy. DFT calculations with an axial imidazole, both with and without an axial water molecule, gave hyperfine values (Table 3) in good agreement with experiment. Although the influence of the water is minor, the agreement with the experimental isomer shift is slightly better with water, consistent with the presence of an axial water in the crystal structure. The changes in the hyperfine parameters between proto- and c-heme were minor.
Table 3.
Comparison of Hyperfine Fine Parameters and Fe–O Distances from Experiment (Bold Text) and DFT (Plain Text) for the S = 5/2 Peroxidatic and S = 1/2 His/Tyr Hemes of Resting BthA
| structuresa | δ (mm/s) | ΔEQ (mm/s) | ASDb (T) | Fe-O (Å) |
|---|---|---|---|---|
| peroxidatic heme | 0.42 | + 1.37 | ~0 | 2.20d |
| Im/OH2 | 0.39 | +0.99 | ~0 | 2.19 |
| Im/OH2 c-heme | 0.42 | +1.04 | ~0 | 2.21 |
| Im/c-heme | 0.39 | +1.04 | ~0 | |
| His/OH2 c-heme | 0.39 | +1.08 | ~0 | 2.08 |
| His/Tyr heme | 0.21 | −2.67 | 6, 31, −37c | 1.97d |
| Im/Ph | 0.24 | −2.16 | 8, 14, −22 | 1.91 |
| Im/Tyr c-heme | 0.24 | −2.32 | 9, 14, −23 | 1.87 |
| Im−/Tyr c-heme | 0.21 | −1.89 | 8, 15, −23 | 1.92 |
| His/Tyr c-heme | 0.24 | −2.25 | 9, 14, −23 | 1.87 |
The His/Tyr heme consists of an Fe3+ c-heme with axial His and Tyr ligands. This coordination favors a low-spin (S = 1/2) state, which was confirmed by EPR and Mössbauer spectroscopy. The DFT values for the isomer shift and quadrupole splitting of the low-spin Fe3+ ground state are consistent with experiment (Table 3), and the Fe–OTyr bond length is close to the value from EXAFS measurements of MauG.15 The traceless component of the A tensor deduced from experiment contains spin-dipolar (ASD) and orbital (AL) contributions. The orbital contribution is significant for low-spin Fe3+ complexes and has not been accurately determined, consequently, the listed ASD values obtained by DFT and experiment do not agree. The DFT values for the hyperfine parameters of the ferric state show only a minor dependence on the choice of the heme and the deprotonation of the His ligand.
Electronic Structure of Hemes in BthA after H2O2 Addition.
The addition of H2O2 caused a one-electron oxidation of the ferric peroxidatic heme to give an S = 1 Fe4+=O center typical of Cpd II hemes. DFT predictions of isomer shifts have been demonstrated previously for a variety of iron complexes.29–32 As a further test of DFT methods specific to the oxidation states of the heme complexes in this work, Table 4 presents calculations of δ for Cpd II hemes with axial coordination of the following: His/O (Mb), Cys/O and Cys/OH (CPO), Tyr/O, and Tyr/OH (Catalase). All gave values of δ that agreed within an error margin of 0.04 mm/s with experiment. Table 4 shows that protonation of the oxo lowers δ and that Tyr coordination gives values of δ typical of Cpd II species.
Table 4.
Experimental and DFT Values of δ (mm/s) for Cpd II Species
The DFT values of δ, ΔEQ, and the ASD tensor for the Fe4+=O species in the upper half of Table 5 show good agreement with experimental data for the H2O2 oxidized peroxidatic heme of BthA and are typical of ferryl compounds with an S = 1, t2g4 configuration. Table 5 also includes the results from DFT calculations for the case that the axial oxo ligand is protonated. The isomer shift and quadrupole splitting for this Fe4+–OH species are calculated to be well below and well above the experimental values, respectively, ruling out the hydroxo species. The DFT-predicted increase in ΔEQ above 2.0 mm/s upon protonation is consistent with the trend in the experimental data for this quantity in the Cpd II complexes of Catalase and CPO (Table 1).
Table 5.
Comparison of Hyperfine Fine Parameters and Fe–OTyr Distances from Experiment (Bold Text) and DFT Calculations (Plain Text) for the Peroxidatic and His/Tyr S = 1 Hemes of BthA after H2O2 Addition
| structurea | δ (mm/s) | ΔEQ (mm/s) | ASDb (T) | Fe−OTyr (Å) |
|---|---|---|---|---|
| peroxidatic | 0.07 | +1.64 | −6, −6, 12 | 1.69c |
| Im/O | 0.09 | +0.98 | −5, −5, 10 | 1.67 |
| Im/O c-heme | 0.08 | +0.99 | −5, −5, 10 | 1.67 |
| Im/OH | 0.02 | +2.35 | −6, −6, 12 | 1.79 |
| His/O c-heme | 0.08 | +0.96 | −5, −6, 11 | 1.67 |
| His/Tyr heme | 0.17 | +2.55 | -6, −6, 12 | −d |
| Im/Ph | 0.08 | +2.65 | −5, −8, 13 | 1.84 |
| Im/Tyr | 0.10 | +2.52 | −6, −7, 13 | 1.85 |
| Im−/Tyr | 0.06 | +1.85 | −6, −7, 13 | 1.88 |
| Im/TyrH | 0.13 | +3.19 | −2, −9, 11 | 2.07 |
| Im/Tyr | 0.15 | +2.89 | −5, −7, 12 | 1.99e |
| His/Tyr c-heme | 0.10 | +2.46 | −5, −8, 13 | 1.85 |
| His/Tyr c-heme | 0.16 | +2.79 | −5, −8, 13 | 2.00e |
Axial ligands to proto- or c-heme are listed (see Figure 2).
Spin-dipolar A tensor in ΕASD•Ŝ for S = 1.
EXAFS result for MauG.15
EXAFS of MauG suggested 1.86 Å15 but see Discussion.
The Fe–OTyr distance was kept constant during the geometry optimization.
Unlike in the case of the oxidized peroxidatic heme, the DFT solutions for the oxidized His/Tyr heme show a strong dependence on the functional used. This was shown by comparing the results of BP86 and B3LYP calculations. The two functionals gave qualitatively different results for the oxidized S = 1 state of the Tyr/His heme due to the propensity of these functionals to give Fe4+–Tyr (BP86) and Fe3+–Tyr• (B3LYP) electronic configurations. The S = 1 Fe4+ ground state obtained by BP86 agrees with the state determined from our analysis of the experimental Mössbauer data for the oxidized Tyr/His heme in BthA, whereas the B3LYP results do not. Consequently, we have presented the BP86 results (Table 5) and adopted this functional for our analyses.
The oxidation of the low-spin ferric state of the His/Tyr heme site to the S = 1 state can proceed by the removal of an electron from either a 3d orbital of the iron (Figure 5, left) or the redox active π orbital of the tyrosine ligand (Figure 5, middle and right). In the latter case, the spin of the tyrosine radical STyr = 1/2 can be coupled either ferromagnetically (F) to a low-spin Fe3+ (SFe = 1/2, middle) or antiferromagnetically (AF) to an intermediate-spin Fe3+ (SFe = 3/2, right). In all cases the three spin-up electrons in the shell do not contribute to aspherical properties such as the quadrupole splitting (ΔEQ) and the spin-dipolar tensor (ASD). These properties are essentially determined by the spin-down electron in the Fe4+–Tyr configuration (left) and by the spin-down hole in the Fe3+–Tyr• configuration (middle), both of which have been encircled in red in Figure 5. The particle–hole relationship implies that ΔEQ and ASD change sign in passing between the two configurations (see Figure 5 values). Thus, ΔEQ and the principal component of ASD with the largest magnitude (AzSD) both retain the negative sign observed for the ferric state of the Tyr-heme site in the case of the (Fe3+–Tyr•)F state (middle diagram) and have positive signs for Fe4+–Tyr and (Fe3+–Tyr•)AF (left and right diagrams). Analysis of the magnetic Mössbauer spectra provides both the magnitude and the sign of ΔEQ and of the magnetic hyperfine coupling tensor, A, from which the principal values of ASD have been deduced. The experimental results (Table 5) show that both ΔEQ and AzSD are positive, consequently the (Fe3+–Tyr•)F state is ruled out. Both ΔEQ and ASD have the same sign for the Fe4+–Tyr and (Fe3+–Tyr•)AF states; however, the magnitude of the ASD values are 1.5 times larger for (Fe3+–Tyr•)AF. The experimental value of AzSD agrees with the DFT value for Fe4+–Tyr but not with the DFT value for (Fe3+–Tyr•)AF, ruling out the (Fe3+–Tyr•)AF state.
Figure 5.
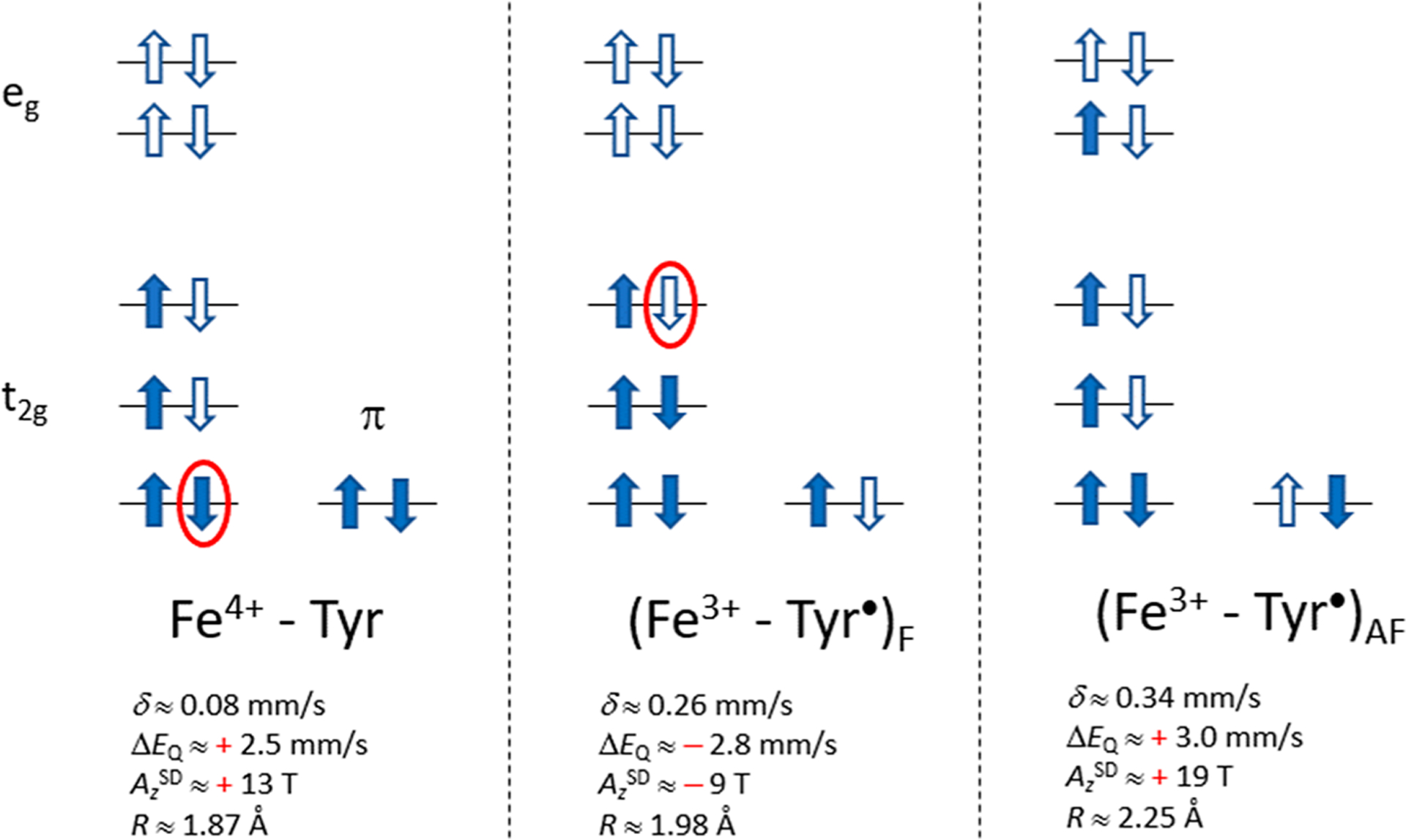
Potential electronic configurations for the S = 1 state of His/Tyr heme of BthA: Fe4+, Tyr• ferromagnetically (F) coupled to S = 1/2 Fe3+, and Tyr• antiferromagnetically (AF) coupled to S = 3/2 Fe3+. Occupied and unoccupied spin–orbitals are represented by shaded and hollow arrows, respectively. The red ellipses enclose an electron (left) and a hole (middle) that are essential for the hyperfine interactions (see text). DFT-based estimates δ, ΔEQ, AzSD (S = 1), and the Fe–OTyr distance (R) are given.
While the above evaluation of the hyperfine parameters across experiment and DFT supports the oxidation of iron to an Fe4+ His/Tyr configuration, an increase of the iron oxidation state usually results in a significantly lower value of the 57Fe isomer shift: δred − δox = Δδ > 0. However, the value Δδ = 0.04 mm/s observed in passing from the Fe3+ to the Fe4+ state of the His/Tyr heme site in BthA is small. For example, the Δδ between high-spin Fe3+=O and Fe4+=O moieties supported by tripodal nonheme ligands is ~0.30 mm/s, and a similar value is found between the intermediate-spin Fe3+–OH2 and Fe4+=O moieties supported by tetra-amido macrocyclic ligands (TAML). These examples have been well described by DFT and demonstrate the capability of DFT to predict values of δ.29,33 Table 4 gives calculations of δ for several Cpd II heme complexes as a further test of DFT methods specific to the oxidation state of the hemes in this work. In contrast, Δδ of complexes in which the oxidation is ligand-centered is near zero.34,35 Although the small change in δ for the His/Tyr heme site of BthA is suggestive of a ligand-based oxidation, this possibility has been ruled out above based on the magnetic Mössbauer data. The quantity Δδ depends not only on the oxidation state but also on alterations in ligation and spin that may occur upon oxidation, which are considered next.
Possibly, the high isomer shift may arise from variable protonation states about the immediate coordination sphere of the His/Tyr heme site. The most probable protonation state of the axial ligands at physiological pH (imidazole and deprotonated phenolic O) has a DFT value of δ = 0.10 mm/s (Im/Tyr, Table 5) in the range typical of Cpd II complexes (δ = 0.03 to 0.11 mm/s) but significantly smaller than observed experimentally (δ = 0.17 mm/s). The DFT Im/Tyr structure has an Fe4+–O length of 1.85 Å, which is typical of crystallographically characterized Fe-Phenol species,36,37 and an ASD typical for S = 1 Fe4+. Also considered was deprotonation of His (Im–/Tyr) which slightly lowered δ, increasing the discrepancy with experiment. The value for Im/TyrH (δ = 0.13 mm/s) is closer to experiment, but a protonated (phenolic O) bound Tyr is chemically implausible for the Fe4+ oxidation state.38 We also considered variations in the Fe–O–CTyr bond angle at the coordinating Tyr oxygen. However, these gave only small changes in the Fe4+ isomer shifts all of which were <0.10 mm/s and unacceptably low relative to experiment (see Table S2).
It was noted above that the hybrid density functional B3LYP predicts the formation of a Tyr radical, notably the (Fe3+–Tyr•)AF state shown in the right diagram of Figure 5. The predicted isomer shifts from these calculations are all much higher (~0.34 mm/s) than for the Fe4+ state (obtained with BP86). In addition, the principal values of ASD obtained with B3LYP are 1.5 times higher than experiment (Figure 5, right diagram). The higher isomer shift and ASD values calculated using B3LYP are typical of intermediate-spin Fe3+ heme complexes and are precluded by our experimental data.
The above analysis shows that DFT calculations with BP86 for models of the oxidized His/Tyr heme of BthA gives an S = 1 Fe4+ ground state with ΔEQ and AzSD values in agreement with experiment but a δ that is 0.10 mm/s below experiment. In contrast, B3LYP gives the (Fe3+–Tyr•)AF state with δ and AzSD values much larger than observed experimentally. Conceivably, the His/Tyr heme oxidation could be partially delocalized through a covalent interaction with the aromatic ring of Tyr, representing an electronic structure between the limits of BP86 and B3LYP. While such a delocalization might increase δ, it will also affect the magnetic properties of the iron. The spin-dipolar contribution ASD to the A tensor can be explicitly determined from the newly obtained magnetic Mössbauer measurements on BthA. ASD is sensitive to the electronic structure and can be directly compared to DFT values. With this in mind, we considered quantum mechanical admixtures of the Fe4+ ground state with the (Fe3+–Tyr•)AF excited state. The mixing of the Fe4+ and (Fe3+–Tyr•)AF configurations (Figure 5, left and right) can be described by DFT as a partial delocalization of the spin-up electron from the redox active tyrosine π orbital into one of the vacant 3d orbitals of iron. An obvious cause for the difference between the BP86 and B3LYP solutions for the oxidized His/Tyr heme is in the definition of the exchange term as a pure functional (0% Hartree–Fock Exchange) in BP86 and a hybrid functional (20% HFE) in B3LYP. Conceivably, by making an adjustment to the HFE percentage, a specific quantum mechanical admixture could be obtained that reproduces δ, ΔEQ, and AzSD simultaneously within the experimentally allowed margins. To perform this analysis, we employed the user defined functional provided by Gaussian. Starting with BP86 (0% HFE), the HFE was stepwise increased to the B3LYP value of 20%. The results of this analysis are given in the Supporting Information. We found that values of HFE (11%) which raised the value of δ by ~0.01 mm/s already resulted in an ASD that was incompatible with experiment. Further increases in HFE to raise δ only exacerbated the differences in ASD with experiment.
Thus far, we have explored structural models consisting of the metal and variations of its ligands to explain the large δ value for the oxidized His/Tyr heme in BthA and MauG. As these explorations have failed to identify the origin of the large δ value, we now direct our attention to external forces enacted by the protein on the His/Tyr heme. A structural factor that may affect the isomer shift is the position of the tyrosine relative to the heme moiety and changes therein imposed by the protein environment. The position of the tyrosine relative to the heme is defined by the bond distance Fe–OTyr, the bond angle Fe–OTyr–CTyr, and the dihedral angle Fe–OTyr–CTyr–CTyr. The results of a DFT analysis of these factors have been summarized in Table S2 for the Im/Ph proto-heme structure (Figure 2). Unconstrained geometry optimization (row 1, Table S2) gives a large bond angle (145°) and a short bond distance (1.81 Å) with the phenolate plane perpendicular to the heme plane (dihedral ~ 0). The structure was also optimized while constraining the bond angle and dihedral to their values in the crystallographic structure for diferric BthA. The DFT result (row 2, Table S2) gave an Fe4+–OTyr bond length that increased to 1.85 Å. This distance is slightly lower than the bond length of 1.89 Å from EXAFS for the low-spin Fe3+ site in MauG15 but significantly smaller than the 1.97 Å in the XRD structure of BthA. The Fe–OTyr distances from DFT resemble those reported for high-spin Fe3+ phenolate complexes, which have distances of 1.83 and 1.85 Å.36,37 The angularly constrained S = 1 Fe4+ has an Fe–OTyr bond distance of 1.86 Å and an energy that is 1600 cm−1 (~4 kcal/mol) higher than for the unconstrained structure (Table S2). Possibly, upon oxidation to the bis-Fe4+ state, changes in protein structure allow a larger Fe–OTyr–CTyr angle which lowers this energy. However, our calculations (Table S2) find that the Fe–OTyr–CTyr angle has little influence on the value of the isomer shift.
Several calculations were performed to examine the effect of the Fe–O bond length by imposing a fixed value for this bond in the geometry optimization. Lengthening this bond by 0.14 Å increases δ by ~0.05 to 0.15 mm/s (Table 5). Constraining the Fe4+–OTyr distance to 2.00 Å during optimization of the His/Tyr c-heme structure raises δ to 0.16 mm/s (last row of Table 5) but causes only minor changes in ΔEQ and ASD. The values of δ, ΔEQ, and ASD obtained for this structure are all in agreement with experiment. The bond stretch increases the electronic energy by 800 cm−1 (~2 kcal/mol) and may be caused by changes in the relative position of the coordinating Tyr, porphyrin, and His residues that occur in passing from the resting bis-Fe3+ state for which the structure has been determined to the oxidized bis-Fe4+ state for which no structure is available for either BthA or MauG.
DISCUSSION
The structure and electronic properties of heme sites in proteins as a function of axial ligands constitute a mature field with a long history. Thus, the recent discovery of a rare axial His/Tyr coordinated heme that can access a high oxidation state has generated much interest.10 High valent iron-oxo species are known key intermediates in the heme catalyzed oxidation of a wide range of substrates. The well-studied mechanisms of peroxidases and cytochrome P450 use H2O2 or O2 to generate the Cpd I intermediate which is two oxidation equivalents above Fe3+. Cpd I is an oxoiron(IV) species with a porphyrin cation radical which performs 1e− substrate oxidation, thereby reducing the porphyrin radical to generate the (hydr)oxoiron(IV) Cpd II intermediate.
In some cases when the oxidation equivalent of the porphyrin radical is rapidly transferred to another site, for example yeast CCP, the overall oxidation state is still referred to as Cpd I, even though the heme has an electronic structure of a Cpd II species. Other such examples are BthA and MauG, in which the presumptive porphyrin radical formed in the 2e− oxidation of the peroxidatic site is rapidly transferred to the His/Tyr heme, resulting in two S = 1 Fe4+ sites, one with His/O and the other with His/Tyr axial coordination. Oxoiron(IV) or hydroxoiron(IV) heme intermediates have been characterized from several types of heme structures where the axial ligand trans to the oxo or hydroxo correlates with important divisions in enzymatic function (Table 1). In peroxidases, such as HRP, the axial His trans to the oxo lowers the oxo pKa, resulting in the oxo adduct, while in halogenating enzymes such as CPO, the axial Cys trans to oxo raises the oxo pKa to give the hydroxo adduct.23,39
The Mössbauer spectra of H2O2 oxidized BthA revealed two species with different parameters. The values of δ = 0.07 mm/s and ΔEQ = 1.64 mm/s are typical of Cpd II species with an oxo coordinated to Fe4+. Since the peroxidatic site is similar in structure to those in HRP, Mb, and CCP, this Mössbauer species has been assigned to the peroxidatic heme. Consequently, the other Mössbauer species with δ = 0.17 mm/s and ΔEQ = 2.55 mm/s has been assigned to the unique His/Tyr heme. A comparison of the changes in ΔEQ due to the difference in the axial ligation of the two sites (=O vs −OTyr) with those brought about by protonation (=O vs −OH, Table 1) reveals the same trend for ΔEQ, namely an increase to a value larger than 2.0 mm/s, suggesting electronic similarity between heme complexes of Fe4+–OTyr and Fe4+–OH. As the double bond of an axial oxo ligand exerts a large trans effect, the singly bonded Tyr/OH coordination in protonated Catalase Cpd II may arguably have the greatest similarity with the His/Tyr heme of BthA among the Cpd II species listed in Table 1. However, the isomer shift for the Tyr/OH in Catalase Cpd II (0.03 mm/s) is at the lower end of the typical range of Cpd II hemes. The isomer shift decreases upon protonation from oxo to hydroxo in ferryl hemes, in contrast to the relationship between the peroxidatic and His/Tyr hemes in BthA.
While the values of δ are significantly different for the two hemes, the ASD tensor of the His/Tyr Fe4+ heme is within experimental uncertainty equal to the ASD tensors of the peroxidatic site of BthA and the Cpd II states of HRP and Mb, indicating the same S = 1 Fe4+ electronic configuration for all these complexes. The value δ = 0.17 mm/s for the His/Tyr heme is close to that of the Fe3+ complex prior to oxidation (0.21 mm/s), but the magnetic Mössbauer data shows that the oxidation is iron based, ruling out aromatic spin density on the Tyr. The DFT models presented in this paper have attempted to bridge these two experimental findings, seemingly at odds with each other, toward one cohesive structural model of the bis-Fe4+ intermediate.
Without the protein constraint, the DFT calculations (Table 5) of the oxidized peroxidatic and His/Tyr hemes in BthA gave isomer shifts that are nearly equal to experiment for the peroxidatic heme but much smaller than experiment for the His/Tyr heme. However, the DFT results for the principal values of the ASD tensor for the two hemes agree with experiment, and the calculations confirm the experimental increase in ΔEQ in passing from the peroxidatic to the His/Tyr heme. We have considered many structural variations of the His/Tyr heme in an effort to bring the DFT calculated isomer shift in range of experiment, including the protonation state of the axial ligands, proto- and c-hemes, and variation of the bond angle at the coordinating Tyr oxygen angle. All these structures gave Fe4+ isomer shifts far below experiment. Importantly, the ASD tensor is incompatible with Fe3+–Tyr• configurations. Even small admixtures of the Fe3+–Tyr• configuration with Fe4+–Tyr, that raise δ by a small amount but still far from experiment, resulted in unacceptably high values for ASD relative to experiment. Thus, these results conclusively confirm the assignment of this intermediate as bis-Fe4+.
Only the elongation of the Fe–OTyr bond length from its unconstrained value of 1.85 Å to 2.00 Å gave isomer shift and hyperfine values in agreement with experiment. A previous EXAFS study of H2O2 oxidized MauG has modeled the data from the two heme sites with Fe–O bond lengths of 1.69 and 1.86 Å and 10 Fe–N bonds with lengths of 2.0 Å.15 Owing to the spectral overlap in EXAFS of two hemes with many similar Fe–N porphyrin distances close to the Fe–OTyr distance and an unknown fraction of the protein in the diferric state, the distance of 1.86 Å, though consistent with the data, may have been prompted by the DFT structure. However, this bond length is not compatible with the experimental isomer shift, and previous attempts to account for the isomer shift are now unsatisfactory when the ASD tensor presented in this paper is an added constraint to the models. We propose that the long Fe–OTyr bond in the bis-Fe4+ oxidation state of BthA and MauG is a consequence of a protein-derived constraint on the axial Tyr. Both MauG and BthA have very similar tertiary structures in the region of the protein with connections to the axial Tyr of the heme, including a long, similarly oriented helical domain which contains the axial Tyr. BthA and MauG exhibit the same isomer shift and could experience a similar constraint on the Fe–OTyr bond length. It is possible that the simultaneous oxidation of the two hemes in BthA have an impact on the protein structure. Protein constraints have been shown previously to influence metal coordination in other metalloproteins.40,41 Despite numerous variations in modeling the coordination environment surrounding the His/Tyr heme, only a constrained long Fe–OTyr bond length of 2.00 Å could match the new experimental constraints added by magnetic Mössbauer spectroscopy. These findings now suggest that similar protein-derived entatic states may be present in BthA (and likewise, possibly MauG). It is conceivable, but still must be demonstrated, that this proposed long Fe–OTyr bond length could contribute to the stability of the bis-Fe4+ state in BthA.
The functional diversity of hemes is further expanded in multiheme systems such as diheme bCCPs. These systems generally have one high potential Met/His and one low potential peroxidatic heme, which have been proposed to keep the system at a low over potential and limit the formation of high valent species, preventing subsequent protein oxidation.42 As noted previously, the axial Tyr lowers the reduction potential of the His/Tyr heme to closely match the His-only peroxidatic heme.9 This constitutes a protection mechanism whereby the Cpd I state, generated at the peroxidatic heme following peroxide heterolytic OO bond cleavage, can immediately distribute an oxidation equivalent to the nearby His/Tyr heme rather than causing a deleterious reaction with a protein side chain. The His/Tyr heme enables BthA and MauG to extend the lifetime of this formal Cpd I state to at least 1 h for BthA. In the case of MauG, the ensuing reaction involves long-range electron transfer to generate radical intermediates at Trp199 (in MauG) and Trp57 and Trp108 in the β-subunit of pre-MADH prior to TTQ formation.7–9,43,44 The unique stability of the bis-Fe4+ in MauG has been previously attributed to charge resonance stabilization with a medial Trp93 residue,4 as well as charge delocalization between the Fe4+ and the bound Tyr residue.15 However, BthA does not possess a medial Trp residue for charge resonance stabilization yet also has the 960 nm NIR feature associated with charge resonance in MauG. While the Trp residue is deemed essential for bis-Fe4+ formation in MauG, the presence of the 960 nm feature in BthA suggests that Trp is not required for electron transfer to the His/Tyr heme in BthA, and the long lifetime of this state in BthA suggests alternate mechanisms can generate and stabilize the bis-Fe4+ intermediate.
CONCLUSIONS
The ability to generate the bis-Fe4+ intermediate in full yield for BthA has allowed characterization of the electronic structure of the iron sites by magnetic Mössbauer spectroscopy. This analysis shows that the two ferric hemes in BthA can be fully converted to stable S = 1 Fe4+ states with magnetic hyperfine couplings typical of Cpd II by addition of H2O2, ruling out oxidation of the Tyr. While the magnetic hyperfine couplings are indicative of S = 1 Fe4+ species, the isomer shift of the axial His/Tyr heme (0.17 mm/s) is out of the range of known Cpd II heme species. Based on DFT modeling and in agreement with all spectroscopic constraints, we propose the high isomer shift is owing to a long Fe4+–OTyr bond under the influence of the protein environment.
The results presented here offer new insights into how high valence intermediates may be stabilized by the protein environment. As long-range tunneling is slow, the lifetime of the electron accepting state must be long lest it preempts this process. Electron tunneling benefits from matching the energies of the donor and acceptor orbitals, suggesting that protein-induced lengthening of the Fe–OTyr bond may play a role in tuning the transfer process. BthA has a similar structure to MauG and shows the same unique high isomer shift. Consequently, BthA may perform similar long-range oxidation of a partner protein which has yet to be identified.
Supplementary Material
ACKNOWLEDGMENTS
The work was funded by National Institute of Health grants R01-GM077387 (M.P.H.) and R01-GM110390 (S.J.E.).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.0c01349.
Experimental methods and additional DFT content related to calculation of isomers shifts (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.inorgchem.0c01349
The authors declare no competing financial interest.
Contributor Information
Andrew C. Weitz, Department of Chemistry, Boston University, Boston, Massachusetts 02215, United States
Saborni Biswas, Department of Chemistry, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, United States.
Emile L. Bominaar, Department of Chemistry, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, United States;.
Michael P. Hendrich, Department of Chemistry, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, United States;.
REFERENCES
- (1).Welinder KG Superfamily of Plant, Fungal and Bacterial Peroxidases. Curr. Opin. Struct. Biol 1992, 2, 388–393. [Google Scholar]
- (2).Poulos TL Heme Enzyme Structure and Function. Chem. Rev 2014, 114, 3919–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wang Y; Graichen ME; Liu A; Pearson AR; Wilmot CM; Davidson VL Maug, a Novel Diheme Protein Required for Tryptophan Tryptophylquinone Biogenesis. Biochemistry 2003, 42, 7318–7325. [DOI] [PubMed] [Google Scholar]
- (4).Jensen LM; Sanishvili R; Davidson VL; Wilmot CM In Crystallo Posttranslational Modification within a Maug/Pre-Methylamine Dehydrogenase Complex. Science 2010, 327, 1392–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Li X; Feng M; Wang Y; Tachikawa H; Davidson VL Evidence for Redox Cooperativity between C-Type Hemes of Maug Which Is Likely Coupled to Oxygen Activation During Tryptophan Tryptophylquinone Biosynthesis. Biochemistry 2006, 45, 821–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rizzolo K; Cohen SE; Weitz AC; Lopez Munoz MM; Hendrich MP; Drennan CL; Elliott SJ A Widely Distributed Diheme Enzyme from Burkholderia That Displays an Atypically Stable Bis-Fe(IV) State. Nat. Commun 2019, 10, 1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Li X; Fu R; Liu A; Davidson VL Kinetic and Physical Evidence That the Diheme Enzyme Maug Tightly Binds to a Biosynthetic Precursor of Methylamine Dehydrogenase with Incompletely Formed Tryptophan Tryptophylquinone. Biochemistry 2008, 47, 2908–2912. [DOI] [PubMed] [Google Scholar]
- (8).Shin S; Abu Tarboush N; Davidson VL Long-Range Electron Transfer Reactions between Hemes of MauG and Different Forms of Tryptophan Tryptophylquinone of Methylamine Dehydrogenase. Biochemistry 2010, 49, 5810–5816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Geng J; Davis I; Liu F; Liu A Bis-Fe(IV): Nature’s Sniper for Long-Range Oxidation. J. Biol. Inorg. Chem 2014, 19, 1057–1067. [DOI] [PubMed] [Google Scholar]
- (10).Li X; Fu R; Lee S; Krebs C; Davidson VL; Liu A A Catalytic Di-Heme Bis-Fe(IV) Intermediate, Alternative to an Fe(IV)=O Porphyrin Radical. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 8597–8600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lee S; Shin S; Li X; Davidson VL Kinetic Mechanism for the Initial Steps in MauG-Dependent Tryptophan Tryptophylquinone Biosynthesis. Biochemistry 2009, 48, 2442–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Yukl ET; Williamson HR; Higgins L; Davidson VL; Wilmot CM Oxidative Damage in MauG: Implications for the Control of High-Valent Iron Species and Radical Propagation Pathways. Biochemistry 2013, 52, 9447–9455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Rittle J; Green MT Cytochrome P450 Compound I: Capture, Characterization, and C-H Bond Activation Kinetics. Science 2010, 330, 933–937. [DOI] [PubMed] [Google Scholar]
- (14).Krest CM; Onderko EL; Yosca TH; Calixto JC; Karp RF; Livada J; Rittle J; Green MT Reactive Intermediates in Cytochrome P450 Catalysis. J. Biol. Chem 2013, 288, 17074–17081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jensen LM; Meharenna YT; Davidson VL; Poulos TL; Hedman B; Wilmot CM; Sarangi R Geometric and Electronic Structures of the His-Fe(IV)=O and His-Fe(IV)-Tyr Hemes of Maug. J. Biol. Inorg. Chem 2012, 17, 1241–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shimizu H; Schuller DJ; Lanzilotta WN; Sundaramoorthy M; Arciero DM; Hooper AB; Poulos TL Crystal Structure of Nitrosomonas Europaea Cytochrome C Peroxidase and the Structural Basis for Ligand Switching in Bacterial Di-Heme Peroxidases. Biochemistry 2001, 40, 13483–13490. [DOI] [PubMed] [Google Scholar]
- (17).Ling Y; Davidson VL; Zhang Y Unprecedented Fe(IV) Species in a Diheme Protein MauG: A Quantum Chemical Investigation on the Unusual Mössbauer Spectroscopic Properties. J. Phys. Chem. Lett 2010, 1, 2936–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Harami T; Maeda Y; Morita Y; Trautwein A; Gonser U Mössbauer Spectroscopic Determination of Electronic-Structure of Highly Oxidized Iron in Hemoproteins. J. Chem. Phys 1977, 67, 1164–1169. [Google Scholar]
- (19).Maeda Y; Higashim T; Morita Y Mössbauer Effect in Peroxidase. Biochem. Biophys. Res. Commun 1967, 29, 362–367. [DOI] [PubMed] [Google Scholar]
- (20).Schulz CE; Rutter R; Sage JT; Debrunner PG; Hager LP Mössbauer and Electron Paramagnetic Resonance Studies of Horseradish Peroxidase and Its Catalytic Intermediates. Biochemistry 1984, 23, 4743–4754. [DOI] [PubMed] [Google Scholar]
- (21).Lang G; Spartalian K; Yonetani T Mössbauer Spectroscopic Study of Compound Es of Cytochrome-C Peroxidase. Biochim. Biophys. Acta, Gen. Subj 1976, 451, 250–258. [DOI] [PubMed] [Google Scholar]
- (22).Liew FN; Brandys MA; Biswas S; Nguyen JN; Rahmawati M; Nevala M; Elmore BO; Hendrich MP; Kim HJ Cytochrome C’β-Met Is a Variant in the P460 Superfamily Lacking the Heme-Lysyl Cross-Link: A Peroxidase Mimic Generating a Ferryl Intermediate. Biochemistry 2020, 59, 704–716. [DOI] [PubMed] [Google Scholar]
- (23).Stone KL; Hoffart LM; Behan RK; Krebs C; Green MT Evidence for Two Ferryl Species in Chloroperoxidase Compound II. J. Am. Chem. Soc 2006, 128, 6147–6153. [DOI] [PubMed] [Google Scholar]
- (24).Horner O; Mouesca JM; Solari PL; Orio M; Oddou JL; Bonville P; Jouve HM Spectroscopic Description of an Unusual Protonated Ferryl Species in the Catalase from Proteus Mirabilis and Density Functional Theory Calculations on Related Models. Consequences for the Ferryl Protonation State in Catalase, Peroxidase and Chloroperoxidase. J. Biol. Inorg. Chem 2007, 12, 509–525. [DOI] [PubMed] [Google Scholar]
- (25).Wolter T; Meyer-Klaucke W; Muther M; Mandon D; Winkler H; Trautwein AX; Weiss R Generation of Oxoiron(IV) Tetramesitylporphyrin Pi-Cation Radical Complexes by M-CPBA Oxidation of Ferric Tetramesitylporphyrin Derivatives in Butyronitrile at −78 Degrees C. Evidence for the Formation of Six-Coordinate Oxoiron(IV) Tetramesitylporphyrin Pi-Cation Radical Complexes Fe-IV=O(Tmp·)X (X= Cl-, Br-), by Mössbauer and X-Ray Absorption Spectroscopy. J. Inorg. Biochem 2000, 78, 117–122. [DOI] [PubMed] [Google Scholar]
- (26).Debrunner PG Mössbauer Spectroscopy of Iron Porphyrins. Physical Bioinorganic Chemistry Series 1989, 4, 137–234. [Google Scholar]
- (27).Sinnecker S; Slep LD; Bill E; Neese F Performance of Nonrelativistic and Quasi-Relativistic Hybrid DFT for the Prediction of Electric and Magnetic Hyperfine Parameters in Fe-57 Mössbauer Spectra. Inorg. Chem 2005, 44, 2245–2254. [DOI] [PubMed] [Google Scholar]
- (28).Oosterhuis WT; Lang G Magnetic Properties of T42g Configuration in Low Symmetry Crystal Fields. J. Chem. Phys 1973, 58, 4757–4765. [Google Scholar]
- (29).Chanda A; Shan X; Chakrabarti M; Ellis WC; Popescu DL; Tiago de Oliveira F; Wang D; Que L Jr.; Collins TJ; Munck E; Bominaar EL (TAML)Fe(IV)=O Complex in Aqueous Solution: Synthesis and Spectroscopic and Computational Characterization. Inorg. Chem 2008, 47, 3669–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Neese F Prediction and Interpretation of the Fe-57 Isomer Shift in Mössbauer Spectra by Density Functional Theory. Inorg. Chim. Acta 2002, 337, 181–192. [Google Scholar]
- (31).Liu TQ; Lovell T; Han WG; Noodleman L DFT Calculations of Isomer Shifts and Quadrupole Splitting Parameters in Synthetic Iron-Oxo Complexes: Applications to Methane Mono-oxygenase and Ribonucleotide Reductase. Inorg. Chem 2003, 42, 5244–5251. [DOI] [PubMed] [Google Scholar]
- (32).Zhang Y; Mao JH; Oldfield E Fe-57 Mössbauer Isomer Shifts of Heme Protein Model Systems: Electronic Structure Calculations. J. Am. Chem. Soc 2002, 124, 7829–7839. [DOI] [PubMed] [Google Scholar]
- (33).Weitz AC; Mills MR; Ryabov AD; Collins TJ; Guo YS; Bominaar EL; Hendrich MP A Synthetically Generated LFe(IV)OHn Complex. Inorg. Chem 2019, 58, 2099–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Snodin MD; Ould-Moussa L; Wallmann U; Lecomte S; Bachler V; Bill E; Hummel H; Weyhermuller T; Hildebrandt P; Wieghardt K The Molecular and Electronic Structure of Octahedral Tris(Phenolato)Iron(III) Complexes and Their Phenoxyl Radical Analogues: A Mössbauer and Resonance Raman Spectroscopic Study. Chem. - Eur. J 1999, 5, 2554–2565. [Google Scholar]
- (35).Bartos MJ; Kidwell C; Kauffmann KE; Gordon-Wylie SW; Collins TJ; Clark GC; Münck E; Weintraub ST A Stable Aquairon(III) Complex with S = 1: Structure and Spectroscopic Properties. Angew. Chem., Int. Ed. Engl 1995, 34, 1216–1219. [Google Scholar]
- (36).Goff HM; Shimomura ET; Lee YJ; Scheidt WR Synthesis, Molecular-Structure, and Solution Properties of a Phenolate-Bridged (Tetraarylporphinato)Iron(III) Dimer. Inorg. Chem 1984, 23, 315–321. [Google Scholar]
- (37).Heistand RH; Roe AL; Que L Dioxygenase Models. Crystal Structures of [N,N′-(1,2-Phenylene)Bis(Salicylideniminato)]-(Catecholato-O)Iron(III) and Mu-(1,4-Benzenediolato-O,O′)-Bis-[N,N′-Ethylenebis(Salicylideniminato)Iron(III)]. Inorg. Chem 1982, 21, 676–681. [Google Scholar]
- (38).Das TK; Couture M; Lee HC; Peisach J; Rousseau DL; Wittenberg BA; Wittenberg JB; Guertin M Identification of the Ligands to the Ferric Heme of Chlamydomonas Chloroplast Hemoglobin: Evidence for Ligation of Tyrosine-63 (B10) to the Heme. Biochemistry 1999, 38, 15360–15368. [DOI] [PubMed] [Google Scholar]
- (39).Stone KL; Behan RK; Green MT Resonance Raman Spectroscopy of Chloroperoxidase Compound II Provides Direct Evidence for the Existence of an Iron(IV)-Hydroxide. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 12307–12310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Berghuis AM; Brayer GD Oxidation State-Dependent Conformational-Changes in Cytochrome-C. J. Mol. Biol 1992, 223, 959–976. [DOI] [PubMed] [Google Scholar]
- (41).Mara MW; Hadt RG; Reinhard ME; Kroll T; Lim H; Hartsock RW; Alonso-Mori R; Chollet M; Glownia JM; Nelson S; Sokaras D; Kunnus K; Hodgson KO; Hedman B; Bergmann U; Gaffney KJ; Solomon EI Metalloprotein Entatic Control of Ligand-Metal Bonds Quantified by Ultrafast X-Ray Spectroscopy. Science 2017, 356, 1276–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Frato KE; Walsh KA; Elliott SJ Functionally Distinct Bacterial Cytochrome C Peroxidases Proceed through a Common (Electro)Catalytic Intermediate. Biochemistry 2016, 55, 125–132. [DOI] [PubMed] [Google Scholar]
- (43).Tarboush NA; Jensen LM; Yukl ET; Geng J; Liu A; Wilmot CM; Davidson VL Mutagenesis of Tryptophan199 Suggests That Hopping Is Required for MauG-Dependent Tryptophan Tryptophylquinone. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 16956–16961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Yukl ET; Liu F; Krzystek J; Shin S; Jensen LM; Davidson VL; Wilmot CM; Liu A Diradical Intermediate within the Context of Tryptophan Tryptophylquinone. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 4569–4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.