

Abstract
Through the tumor necrosis factor (TNF) receptor type II (TNFR2), TNF preferentially activates, expands, and promotes the phenotypic stability of CD4+Foxp3+ regulatory T (Treg) cells. Those Treg cells that have a high abundance of TNFR2 have the maximal immunosuppressive capacity. We investigated whether targeting TNFR2 could effectively suppress the activity of Treg cells and consequently enhance the efficacy of cancer immunotherapy. We found that, relative to a suboptimal dose of the immunostimulatory Toll-like receptor 9 ligand CpG oligodeoxynucleotide (ODN), the combination of the suboptimal dose of CpG ODN with the TNFR2-blocking antibody M861 more markedly inhibited the growth of subcutaneously grafted mouse CT26 colon tumor cells. This resulted in markedly fewer TNFR2+ Treg cells and more interferon-γ–positive (IFN-γ+) CD8+ cytotoxic T lymphocytes infiltrating the tumor and improved long-term tumor-free survival in the mouse cohort. Tumor-free mice were resistant to rechallenge by the same but not unrelated (4T1 breast cancer) cells. Treatment with the combination of TNFR2-blocking antibody and a CD25-targeted antibody also resulted in enhanced inhibition of tumor growth in a syngeneic 4T1 mouse model of breast cancer. Thus, the combination of a TNFR2 inhibitor and an immunotherapeutic stimulant may represent a more effective treatment strategy for various cancers.
INTRODUCTION
Overcoming the immunosuppressive tumor microenvironment is key to achieving effective cancer immunotherapy (1, 2). Tumor-infiltrating CD4+Foxp3+ regulatory T (Treg) cells are potent immunosuppressive cells that represent a major cellular mechanism of tumor immune evasion and play a major role in dampening naturally occurring and therapeutically induced antitumor immune responses (3). Accumulation of Treg cells within tumor tissues, and the resultant high ratio of Treg cells to effector T (Teff) cells, is correlated with poor prognosis of cancer patients, including those with lung cancer (4), breast cancer (5), colorectal cancer (6), pancreatic cancer (7), and other malignancies. Elimination of Treg activity, by either reducing their number or down-regulating their immunosuppressive function using checkpoint inhibitors, has become an effective strategy to enhance the efficacy of cancer therapy (8, 9).
Tumor necrosis factor (TNF) receptor type II (TNFR2) is predominantly present on the maximally suppressive subset of mouse and human Treg cells (10, 11). There is now compelling evidence that the interaction of TNF with TNFR2 promotes the proliferative expansion, suppressive function, and phenotypical stability of Treg cells (12–18). In mouse Lewis lung carcinoma and 4T1 breast tumor model, most of the tumor-infiltrating Treg cells are highly suppressive TNFR2+ Treg cells (10, 19). In humans, the proportion of TNFR2+ Treg cells is also increased in the peripheral blood of lung cancer patients and in the tumor-associated ascites of ovarian cancer patients (20, 21). Recent analysis of single-cell RNA sequencing showed that the expression of TNFR2 is one of the most markedly increased genes on Treg cells, as compared with CD4+ Teff cells and CD8+ cytotoxic T lymphocytes (CTLs) in metastatic melanoma patients, and increased TNFR2 gene expression is associated with exhaustion of CD8+ CTLs (22). Furthermore, the amount of TNFR2 present on the surface of Treg cells is associated with greater lymphatic invasion, a higher incidence of tumor metastasis, a higher clinical stage, and poorer response to treatment in patients with lung cancer and acute myeloid leukemia (AML) (20, 23, 24). This clinical and experimental evidence suggests that the highly suppressive TNFR2+ Treg cells associated with tumors play a major role in tumor immune evasion. Meanwhile, TNFR2 is also found on several tumor cells, including colon cancer (25), Hodgkin lymphoma (26), myeloma (27), renal carcinoma (28), and ovarian cancer (29), leading many to consider TNFR2 an oncogene. Antagonistic antibody targeting TNFR2 induces the death of both Treg cells and OVCAR3 ovarian cancer cells, which have abundant surface TNFR2 (29). On the basis of these observations, we proposed that TNFR2 behaves as an immune checkpoint activator and oncoprotein (30).
TNF can be induced by various immunotherapies, including dendritic cell (DC)–based interventions, tumor vaccines, and Toll-like receptor (TLR) agonists (31–33). Such immunotherapy-induced TNF may, in turn, increase TNFR2 on Treg cells (34), resulting in the expansion and activation of tumor-associated Treg cells through TNFR2. For example, by activating DCs, the TLR9 agonistic CpG oligodeoxynucleotides (ODNs) have the capacity to induce antitumor immune responses in mouse models (35–37). CpG ODNs promote the maturation and improve the function of professional antigen-presenting cells while supporting the generation of antigen-specific B cells and CTLs (38). Intratumoral injection of CpG ODN also induces the differentiation and reduces the immunosuppressive activity of myeloid-derived suppressor cells (MDSCs) (39), therefore enhancing the host’s response to cancer (40). However, treatment with CpG ODN can also induce human or mouse Treg cells that have potent immunosuppressive function, which, in turn, dampens host immune responses against the tumor (41, 42). CpG ODN treatment can increase the production of TNF in cultured murine DCs (43) or human peripheral blood mononuclear cells (44), which is likely responsible for the activation of Treg cells. Therefore, by reducing Treg activity, blockade of TNFR2 may enhance the antitumor effect of an immunotherapeutic such as CpG ODN.
Here, we tested this hypothesis and found that the combination of a blocking antibody (M861) recognizing TNFR2 and CpG ODN more potently inhibited mouse CT26 colon tumor development and induced greater tumor regression in syngeneic mice, resulting in greater long-term tumor-free survival of the mice. In addition, in a mouse 4T1 breast cancer model, a better antitumor effect was observed by simultaneously blocking TNFR2 and CD25. Furthermore, tumor antigen-specific immunity developed in the mice that survived CT26 tumor graftment; they completely (over the time course of the experiment) and selectively resisted a rechallenge by CT26, but not 4T1, tumor cells. This effect was associated with a decrease in the proportion of Treg cells among tumor-infiltrating leukocytes, a reduced surface abundance of TNFR2 on Treg cells, and increased tumor infiltration of interferon-γ (IFN-γ)–producing CD8+ T cells. Thus, the combination of TNFR2 antagonism and immunotherapy may be a promising cancer treatment strategy.
RESULTS
M861 inhibits the stimulatory effect of TNF on Treg cells
Previously, we showed that treatment with TNF preferentially promoted the proliferative expansion of Treg cells, accompanied by increased abundance of TNFR2 on Treg cells in vitro and in lipopolysaccharide (LPS)–treated mice (18, 34). In CD4+ T cells cultured with interleukin-2 (IL-2), treatment with the TNFR2 antibody M861 significantly inhibited TNF-induced proliferation (Fig. 1A) and expansion (Fig. 1B) of Treg cells. M861 also significantly blocked TNF-induced increases in the cell surface abundance of TNFR2 on Treg cells (Fig. 1C). In LPS-challenged mice, although administration of M861 failed to reduce the number of Treg cells in the spleens and lymph nodes within 24 hours, the proportion of TNFR2+ Treg cells was significantly reduced by 64% and the abundance of TNFR2 on splenic Treg cells was significantly reduced by >56% (Fig. 1D). The decrease in TNFR2+ Treg cells was not due to cell death (fig. S1). Therefore, unlike two other antibodies recognizing human TNFR2 described in a recent study (29), M861 was not a Treg-depleting antibody. Furthermore, its capacity to reduce TNFR2 abundance appears to be more potent than its inhibition of the proliferative expansion of Treg cells induced by LPS treatment (Fig. 1). Overall, our data favor the idea that M861 is a blocking antibody that inhibits ligand-induced TNFR2 signaling.
Fig. 1. The in vitro and in vivo effects of an antibody recognizing TNFR2 (M861) on Treg cells.

(A to C) Magnetic-activated cell sorting–purified CD4+ T cells were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE). The cells were cultured with interleukin-2 (IL-2; 10 ng/ml) alone or with tumor necrosis factor (TNF; 20 ng/ml) and M861 (10 μg/ml), as indicated, for 72 hours. Proliferation of regulatory T (Treg) cells shown by CFSE dilution (A), the proportion of Treg cells in CD4+ cell cultures (B), and the surface abundance of TNF receptor type II (TNFR2) on Treg cells (C) were analyzed with fluorescence-activated cell sorting (FACS), gating for Foxp3+ staining (A and C) or not gated (total cells; B). (D) Wild-type Balb/c mice were injected intraperitoneally with phosphate-buffered saline (PBS), lipopolysaccharide (LPS; 200 μg), M861 (100 μg), mouse immunoglobulin G (muIgG), or a combination thereof, as indicated, for 24 hours. The abundance of TNFR2 on the surface of Treg cells in the spleen was analyzed by FACS, gating for CD4+Foxp3+ staining. FACS plots are representative of three independent experiments. Data are means ± SEM of n = 5 mice. Number in each FACS plot indicates the percentage of gated cells. *P < 0.05, **P < 0.01, ***P < 0.001 by Student’s t test (A to D).
Combination therapy with TNFR2-blocking antibody and CpG ODN potently inhibits the growth of CT26 tumors and generated tumor-specific immunity
To examine the effect of TNFR2 blockade on the efficacy of tumor immunotherapy, we treated female CT26 tumor-bearing Balb/c mice with M861 and CpG ODN or various controls (Fig. 2A). Treatment was started when the tumor reached 5 to 6 mm in diameter (day 0). CpG ODNs were administered by intratumoral injection, which was previously reported to achieve an optimal antitumor effect (39). To reveal the beneficial effect of combination therapy, we administered M861 with a suboptimal dose of CpG ODN, neither of which markedly inhibited tumor growth alone (Fig. 2B). The combination of M861 and CpG ODN potently inhibited the growth of primary CT26 tumors (Fig. 2B). Eighty percent of mice were tumor-free and survived up to the end of the experiment at 60 days, whereas mice in other groups died from tumor burden within 50 days after tumor inoculation (Fig. 2C). The individual tumor growth curves varied; although a few mice had slow tumor growth with phosphate-buffered saline (PBS), CpG, or M861 alone, the antitumor effect of the M861 and CpG ODN combination is clear (Fig. 2, D to G).
Fig. 2. M861 in combination with CpG ODN potently inhibits the development of mouse CT26 colon tumors.
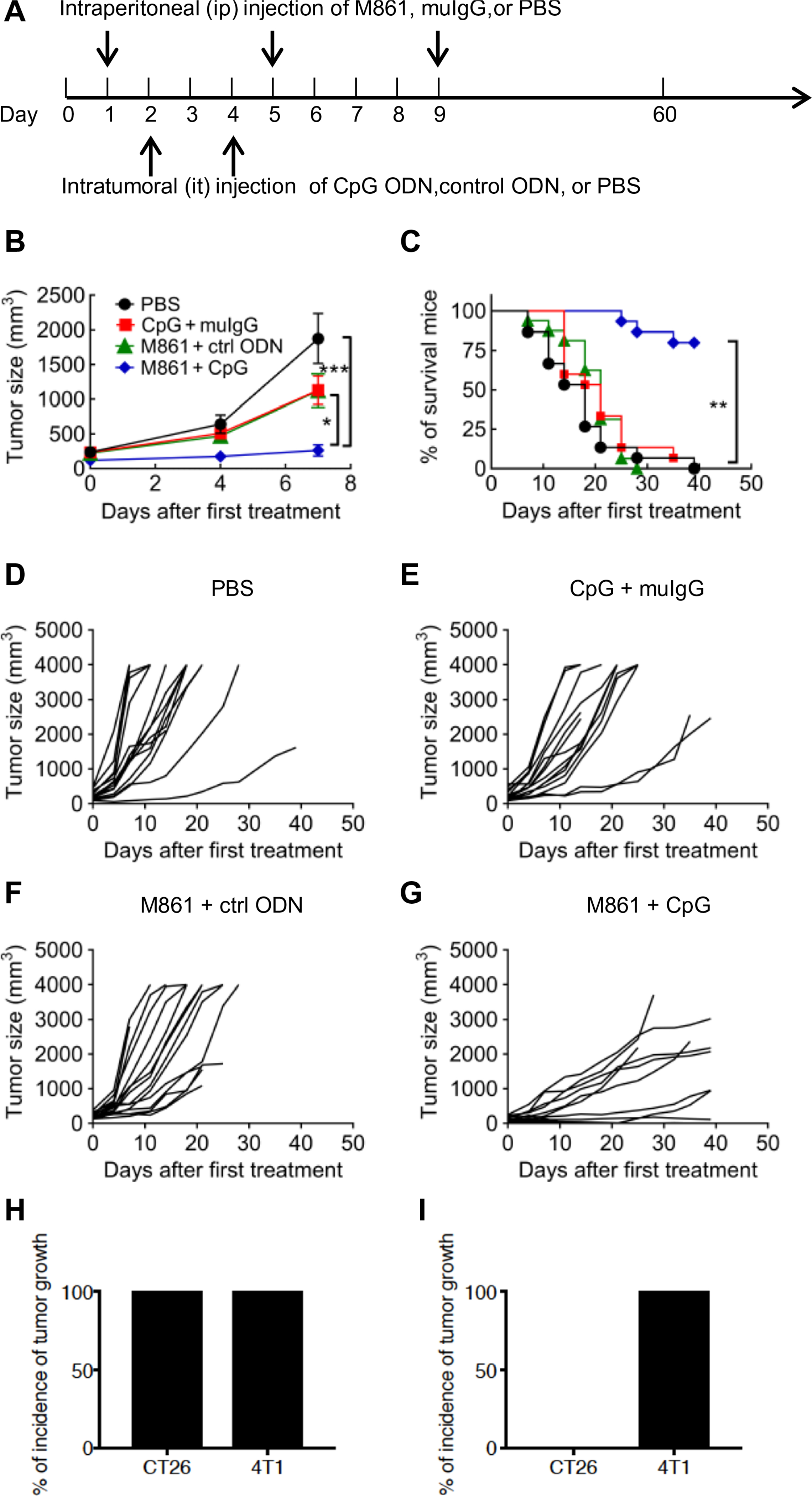
(A) Schematic of the experimental protocol. Balb/c mice were inoculated in the right flank with CT26 tumor cells (2 × 105 cells in 0.2 ml of PBS). When tumor reached 5 to 6 mm in diameter (day 0), mice were then treated with PBS, CpG oligodeoxynucleotide (ODN) plus control (ctrl) IgG, M861 plus control ODN, or CpG ODN plus M861. (B) Mean growth curves of CT26 tumors in mice treated as described in (A). Data are means ± SEM of 15 mice. (C) Survival curves of the CT26 tumor-bearing mice treated as described in (A). (D to G) CT26 tumor growth curves in each individual CT26 tumor-bearing mouse treated with PBS (D), CpG ODN plus control IgG (E), M861 plus control ODN (F), or M861 plus CpG ODN (G). Data are summary of results pooled from three independent experiments (n = 15 mice). The tumor-free mice were reinoculated with CT26 tumor cells into the right flank and 4T1 tumor cells into the left flank 8 weeks after the mice became tumor-free. As a control, normal mice were also inoculated with CT26 tumor cells into the right flank and 4T1 tumor cells into the left flank in the same manner. Data (n = 8 mice) are the percentage of tumor incidence on (H) normal mice and (I) surviving mice on day 26 after (re-)challenge. *P < 0.05, **P < 0.01, ***P < 0.001 by one-way analysis of variance (ANOVA) test (B), log-rank test (C), or Student’s t test (H and I).
To investigate whether the tumor-free mice developed long-term CT26 tumor-specific immunity, the surviving mice were reinoculated subcutaneously with CT26 tumor cells into the right flanks, and 4T1 tumor cells were inoculated into their left flanks. As controls, both 4T1 tumor cells and CT26 tumor cells were inoculated into naïve mice at the same manner, and as expected, both tumors developed in all naïve mice (Fig. 2H). Whereas all of the CT26 (intratumoral)–surviving mice developed measurable 4T1 tumors by day 26 after inoculation, none of these mice developed CT26 colon tumors (Fig. 2I). These results indicate that the treatment with combination of M861 and CpG ODN induced the development of long-term tumor antigen-specific immunity.
M861 did not induce the death of TNFR2+ Treg cells (fig. S1), indicating that its effect was caused by binding and blocking TNFR2 signaling. The possibility that this antibody also binds and neutralizes soluble shed TNFR2 should be addressed in a future study. It was recently reported that antibodies recognizing human TNFR2, in addition to eliminating Treg cells, could also directly act on TNFR2-expressing tumor cells (29). A considerable proportion of CT26 cancer cells are TNFR2+ cells (fig. S2A). However, treatment with M861 (up to 20 μg/ml) did not inhibit the growth of CT26 cells (fig. S2B), indicating that the in vivo inhibitory effect of this antibody on CT26 tumor was not due to any direct effect on tumor cells.
Combination therapy with TNFR2-blocking antibody and CpG ODN reduces the proportion of tumor-infiltrating TNFR2+ Treg cells and increases IFN-γ+ CD8+ CTLs
Because combination therapy resulted in inhibited CT26 tumor growth and marked tumor regression in many of the mice, we were unable to examine the immune cell profile present in the tumor environment. To obtain a tumor mass for further study, we delayed the treatment until tumors reached ~10 mm in diameter, and the effect of treatments on tumor-infiltrating Treg cells was examined by fluorescence-activated cell sorting (FACS) analysis (Fig. 3, A to C). The proportion of intratumoral Treg cells was increased to ~20% by treatment with CpG ODN alone (Fig. 3D). Furthermore, CpG ODN treatment also significantly increased the amount of TNFR2 on Treg cells by ~20% (Fig. 3E). These results suggest that, in addition to stimulating the expansion of Treg cells as previously reported (41, 42), CpG ODN can also increase the function of Treg cells, as indicated by the increased abundance of TNFR2 (10, 45). Both the proportion of TNFR2+ Treg cells and the abundance of TNFR2 in Treg cells (measured as per cell) were markedly reduced by treatment with the TNFR2-blocking antibody (Fig. 3, E and F). Notably, the CpG ODN–induced expansion of Treg cells and increased abundance of TNFR2 on Treg cells were completely abrogated by M861 treatment (Fig. 3, D and E). The combination therapy markedly increased the production of IFN-γ by CD8+ CTLs, and the proportion of IFN-γ–producing CD8+ T cells was greater than threefold as compared with CpG ODN treatment alone (Fig. 3, C and G). Thus, our data indicate that combination therapy reduced Treg activity and promoted the induction of potent type 1 helper T cell antitumor immune responses.
Fig. 3. Effects of M861 in combination with CpG ODN on tumor-infiltrating TNFR2+ Treg cells and IFN-γ+ CD8 CTLs.
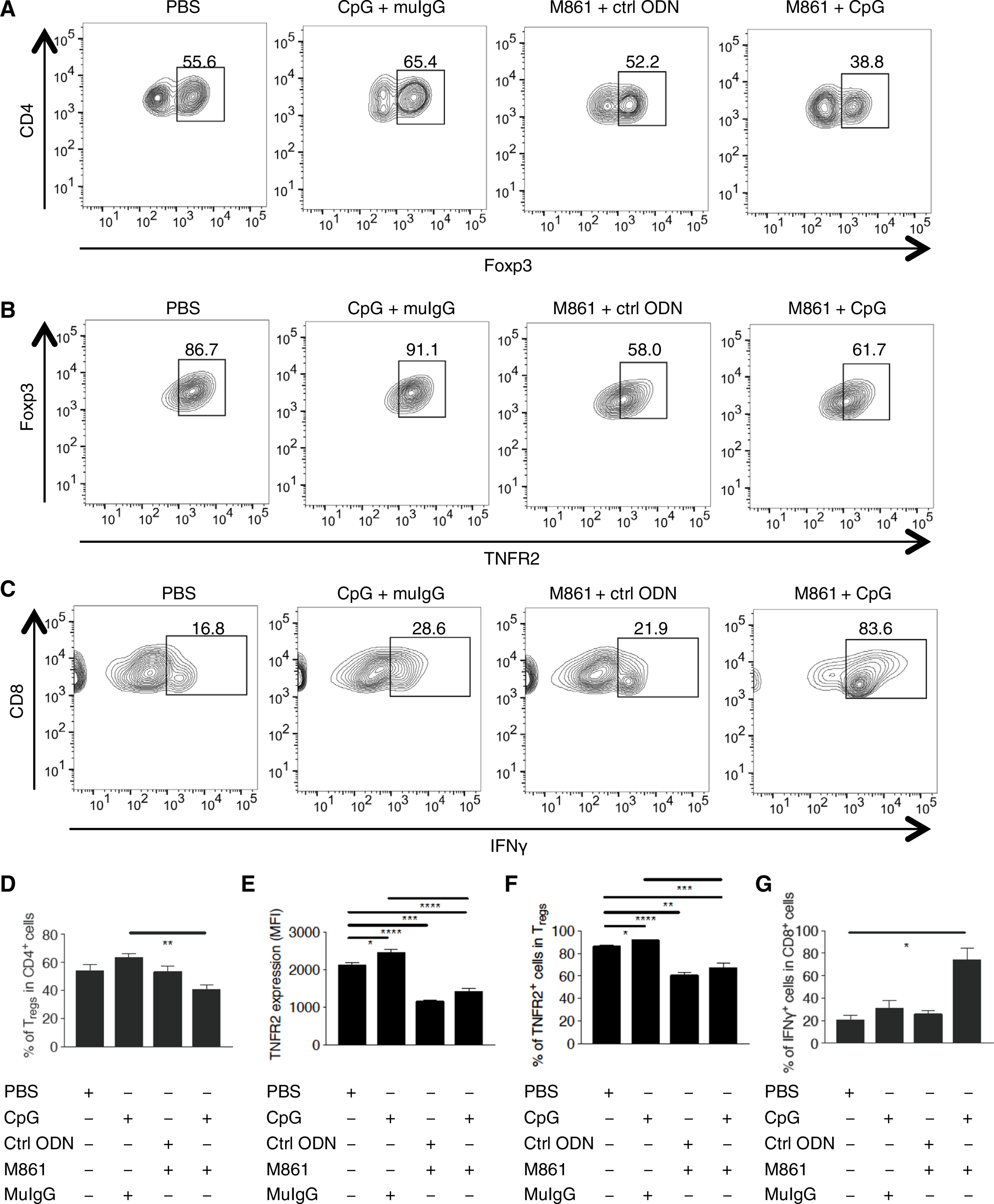
(A) Representative FACS analysis of Treg cells in CD4+ cells from Balb/c mice inoculated with CT26 tumor cells (as described in Fig. 2), treated as indicated when tumor diameter reached 10 mm, and sacrificed for tumor tissue isolation 1 day after the final treatment. The number indicates the percentage of Treg cells in CD4+ cells. (B) Representative FACS analysis of TNFR2+ cells in Treg cells from mice described in (A). The number indicates the percentage of TNFR2+ cells in CD4+Foxp3+ cells. (C) Representative FACS analysis of IFN-γ+ cells in CD8+ cells from mice described in (A). The number indicates the percentage of interferon-γ–positive (IFN-γ+) cells in CD8+ cells. (D to G) Summary of the proportion of Treg cells in intratumoral CD4+ cells (D), the mean fluorescence intensity (MFI) of TNFR2 on CD4+Foxp3+ cells (E), the proportion of TNFR2+ cells in CD4+Foxp3+ cells (F), and the proportion of IFN-γ+ cells in CD8+ cells (G), each from mice described in (A). Flow analysis was gated on live CD45+CD3+ cells. Data were quantified from, or are representative of, at least three independent experiments; n = 5 mice (A, B, and D to F) or 3 mice (C and G). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by Student’s t test.
Combination of TNFR2 antagonistic antibody and CD25 antagonistic antibody inhibits the development of mouse 4T1 breast tumors
CD25, the IL-2 receptor α chain, was the first identified surface marker of Treg cells (46), and targeting of CD25 has been a conventional approach for Treg elimination (47, 48). PC61 is a CD25-recognizing monoclonal antibody that blocks IL-2 binding to the receptor (49), and it was shown that pretreatment with PC61 led to tumor rejection in mouse tumor models (50, 51). We thus compared the effect of TNFR2 antagonistic antibody and CD25 antagonistic antibody on the tumor development. To this end, TR75–54.7, a well-characterized functional blocking TNFR2 antibody (52), and PC61 were administered separately or in combination into mice. Three days later, the mice were inoculated with 4T1 tumor cells (Fig. 4A). The results showed that TR75–54.7 more potently inhibited the development of 4T1 tumor than PC61 (Fig. 4, B and C). Although the difference in tumor size was not statistically different between mice treated with two antibodies (Fig. 4B), only TR75–54.7 markedly enhanced the survival of tumor-bearing mice (Fig. 4C), whereas PC61 did not have such effect (Fig. 4C). Moreover, the combination of TR75–54.7 and PC61 was superior to the monotherapy in the inhibition of tumor growth (Fig. 4B). Consequently, the median survival of tumor-bearing mice was longer after combination therapy (38 days), as compared with monotherapy of TR75–54.7 (34 days) or PC61 (27 days) (Fig. 4, C and D; median survival of PBS control group was 24 days). Therefore, TR75–54.7 has more potent antitumor effect than PC61 in mouse 4T1 breast cancer model, whereas the combination of both resulted in the optimal antitumor effect.
Fig. 4. Effect of TNFR2 antagonistic antibody and CD25 antagonistic antibody on mouse 4T1 breast cancer model.

(A) Schematic of the experimental protocol. TR75–54.7 and/or PC61 were intraperitoneally injected into Balb/c mice 3 days before inoculation of 4T1 tumor cells (1 × 105 cells in 0.1 ml of PBS). (B) Growth kinetics of 4T1 tumors in mice. Data are means ± SEM of five mice. (C) Survival curves of the 4T1 tumor-bearing mice. (D) Median survival of the 4T1 tumor-bearing mice. *P < 0.05 by one-way ANOVA test (B) or log-rank test (C).
DISCUSSION
Our data presented in this study showed that blockade of TNFR2 potently enhanced the effect of immunotherapy with CpG ODN against CT26 tumors, which was attributable to the reduction in Treg activity and consequent mobilization of CD8+ CTLs. Because of the paucity of Treg cells recovered from tumor tissue, we could not determine the functionality of the tumor-infiltrating Treg cells in this study directly. Nevertheless, the marked reduction in TNFR2 abundance on Treg cells, which correlates with Treg function, suggests that M861 reduced the suppressive capacity of Treg cells in the tumor environment. This effect of M861 treatment appears to be specific for tumor-infiltrating Treg cells, given that the proportion of Treg cells, the abundance of TNFR2 on Treg cells, and the suppressive function of Treg cells present in the peripheral lymphoid tissues were not decreased by treatment with the TNFR2-blocking antibody. Although this antibody did not attenuate TNFR2 signaling in unstimulated mice, it potently inhibited the increased abundance of TNFR2 induced by LPS (Fig. 1), suggesting that increased TNFR2 abundance both in the tumor environment and in LPS-challenged mice is similarly induced by the interaction of TNF and TNFR2.
CD25 has been used as an exclusive target for physical depletion of Treg cells to elicit antitumor immunity in both murine cancer models (47) and human cancer patients (48). Depletion of Treg cells by administration of CD25-blocking antibody (PC61) before tumor challenge provokes effective immune response to syngeneic tumors in otherwise unresponsive mice (50, 51). Our previous study showed that TNFR2 was more closely associated with the suppressive function of Treg cells and the phenotype of tumor-infiltrating Treg cells, as compared with CD25 (10, 20, 45). Thus, we directly compared the antitumor effect of a better characterized and commercially available TNFR2 antagonistic antibody (for example, TR75–54.7) (52) and PC61 on mouse 4T1 breast cancer model. The results show that pretreatment with TR75–54.7 is superior to PC61 in the inhibition of tumor growth (Fig. 4). The simultaneous blockade of TNFR2 and CD25 appears to have better effect as compared with monotherapy (Fig. 4). This intriguingly aligned well with our previous observation that, in mice, the sequence of suppressive capacity of Treg subsets was TNFR2+CD25+ cells > TNFR2+CD25− cells > TNFR2−CD25+ cells. Presumably, the maximal suppressive subset of Treg cells was inhibited by the combination treatment of TR75–54.7 and PC61. This possibility will be experimentally examined in a future study.
CD11b+Gr1+ MDSCs also contribute to tumor immune evasion. Recent studies indicate that the generation, accumulation, and function of MDSCs also require TNF-TNFR2 signaling (53, 54). Furthermore, CpG ODNs reportedly reduce the number of MDSCs in tumor-bearing mice (55). Nevertheless, the proportion of MDSCs in the spleen and tumor of CT26 tumor-bearing mice was not decreased by M861 treatment in our study.
Activation and expansion of Treg cells through TNF-TNFR2 interaction represent a general negative feedback loop triggered by many immunological stimulators, including immunotherapeutic agents (56). For example, in addition to CpG ODN, in vivo treatment with the TLR4 ligand LPS (34) or the TLR2 ligand Pam3CSK4 (57) also promotes the activation and expansion of Treg cells, accompanied by the increased abundance of TNFR2 on Treg cells (34). Furthermore, cancer immunotherapy with IL-2, a cytokine that increases the abundance of TNFR2 on Treg cells (18), results in substantial activation and expansion of Treg cells (58, 59). Even the stimulation of immune checkpoint inhibitors can also expand Treg cells. For example, proliferative expansion of Treg cells is induced by PD-1 blockade in melanoma patients (60) and by PD-L1 blockade in HIV patients (61). In this proof-of-principle study, we have shown that the antitumor effect of CpG ODN was markedly enhanced, whereas its stimulation of Treg cells was prevented, by TNFR2 blockade. Whether the effect of TNFR2 blockade can augment other types of immunotherapeutics needs to be determined.
Because of their clear therapeutic value in cancer treatment, strategies have been developed to deplete Treg cells or reduce their suppressive functions by targeting CD25 or other immune checkpoint proteins that are preferentially present on tumor-infiltrating Treg cells (such as CTLA-4, TIM-3, LAG-3, PD-1, and GITR) (62, 63). Because a large abundance of TNFR2 is associated with the maximally suppressive Treg cells in tumor-infiltrating lymphocytes (10, 19–21), targeting of TNFR2 may have an advantage in eliminating the more functional Treg cells. This idea is supported by studies of ovarian cancer and AML that show that patients treated with reagents that reduce TNFR2 abundance on Treg cells have beneficial antitumor effects (21, 23, 24, 29). Further investigation is needed to directly compare the efficacy of targeting of TNFR2 with that of other Treg checkpoint inhibitors.
Together, our findings reveal that an antibody blocking ligand-induced activation of TNFR2 markedly enhances the antitumor efficacy of immunotherapy with CpG ODN in mouse models of colon and breast cancer by reducing the number of tumor-infiltrating TNFR2+ Treg cells while increasing the number of IFN-γ+ CD8+ CTLs. Thus, combining a TNFR2 antagonist with an immunostimulant may represent a novel and more effective treatment strategy for patients with various cancers.
MATERIALS AND METHODS
Mice
Female wild-type 8- to 12-week-old Balb/c mice were obtained from the Animal Production Area of the National Cancer Institute at Frederick (NCI-Frederick). NCI-Frederick is accredited by the American Association for the Accreditation of Laboratory Animal Care International and follows the Public Health Service Policy for the Care and Use of Laboratory Animals. The animal study was approved by Institutional Animal Care and Use Committee of NCI-Frederick. Animal care was provided in accordance with the procedures outlined in the Guide for the Care and Use of Laboratory Animals (National Research Council, revised 1996).
Cells and reagents
The CT26 colon cancer and 4T1 breast cancer cell lines were purchased from the American Type Culture Collection and examined with Molecular Testing of Biological Materials by Animal Health Diagnostic Laboratory (NCI-Frederick) and Luminescence Mycoplasma Test by Animal Molecular Diagnostics Laboratory (NCI-Frederick). Cell lines were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum and 2 mM glutamine at 37°C in a humidified incubator with 5% CO2. CpG ODN 1668 was purchased from InvivoGen. Anti-mouse antibodies, including anti-mouse CD45, CD4, CD8a, and TNFR2 antibodies, were purchased from BD Biosciences. LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit was purchased from Thermo Fisher Scientific. An antibody recognizing mouse TNFR2 (M861), as well as its control immunoglobulin G (IgG) (mouse IgG1), was a gift from Amgen Inc. Anti-mouse TNFR2 antibody TR75–54.7 and anti-mouse CD25 antibody PC61 were gifts from G. Trinchieri [NCI, National Institutes of Health (NIH)].
CT26 tumor cell inoculation and separation of tumor-infiltrating leukocytes
CT26 tumor cells were subcutaneously injected into the right flank of recipient mice in single-cell suspension with 2 × 105 cells in 0.2 ml of PBS per mouse. After indicated times, tumors were excised, minced, and digested in RPMI 1640 supplemented with collagenase IV (1 mg/ml) and deoxyribonuclease I (0.1 mg/ml). The fragments were pushed through a 70-um pore size cell strainer to create a single-cell suspension. In some experiments, tumor-free mice 8 weeks after anti-TNFR2 and CpG ODN treatment were reinoculated with CT26 cells (2 × 105) into the right flank, and the same number of 4T1 cells was injected subcutaneously into the left flank. Tumor size was calculated by the following formula: (length × width2)/2. “Survival” represents the time to develop a 4-cm3 tumor or a moribund state, a humane end point that triggers euthanasia. Mice were monitored daily and were euthanized when signs of morbidity from metastatic disease burden became evident.
Treatment of mouse model of CT26 colon cancer
When the diameter of tumor reached 5 to 6 mm, mice were treated with the following dose schedule: An antibody recognizing TNFR2 (M861) or mouse IgG1 was administered at days 1, 5, and 9 intraperitoneally at 100 μg in 0.2 ml of PBS. CpG ODN or control ODN was administered intratumorally at days 2 and 4 at 20 μg in 0.1 ml of PBS. The same quantity of PBS was administered intraperitoneally or intratumorally as a control. In some experiments, the treatment was started when the diameter of tumor reached 10 mm. One day after the last treatment, mice were sacrificed and tumor and lymphoid tissues were harvested for study.
Treatment of mouse 4T1 breast cancer model
An antibody recognizing TNFR2 (TR75–54.7) and/or an antibody recognizing CD25 (PC61) were administered at 200 μg in 0.2 ml of PBS 3 days before the inoculation of 1 × 105 4T1 breast tumor cells in 0.1 ml of PBS per mouse subcutaneously injected into right mammary fat pads (thoracic No. 2 mammary glands) of recipient female Balb/c mice.
In vitro culture of CD4+ T cells
CD4+ cells were purified from lymphocytes with Mouse CD4 (L3T4) MicroBeads and LS Columns (Miltenyi Biotec). CD4+ cells were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE) and cultured at 4 × 105 cells per well in a 96-well plate with medium (12) alone or IL-2 (10 ng/ml), or with or without TNF (20 ng/ml), or in the presence of M861 (10 μg/ml). After 72 hours, the proliferation of Treg cells (as indicated by CFSE dilution), the proportion of Treg cells, and TNFR2 abundance on Treg cells were analyzed by FACS, gating for Foxp3+ cells.
In vivo treatment with LPS and M861
Normal Balb/c mice were intraperitoneally injected with 200 μg of LPS (Sigma-Aldrich, catalog no. L9764) in PBS. Some mice were injected intraperitoneally with 200 μg of M861 or control Mu IgG1 1 hour before injection of LPS. Mouse spleens and mesenteric lymph nodes were harvested at 24 hours after injection for the FACS analysis of phenotype.
In vitro proliferation of CT26 and 4T1 tumor cells
CT26 and 4T1 tumor cells were seeded into 96-well plate at 5 × 104 cells per well. The cells were cultured with media alone or with M861 (10 or 20 μg/ml). After 72 hours, cells were treated with pulsed 1 mCi [3H]thymidine (PerkinElmer Life Sciences) per well for the last 6 hours of the culture period. The proliferation was evaluated by [3H]thymidine incorporation.
Flow cytometry
After blocking FcR, cells were incubated with appropriately diluted antibodies. The acquisition was performed using a Fortessa cytometer (BD Biosciences), and data analysis was conducted using FlowJo software (Tree Star Inc.). FACS analysis was gated on the live cells only by using a LIVE/DEAD Fixable Dead Cell Stain kit.
Statistical analysis
Two-tailed Student’s t test was used for the comparison of two indicated groups. One-way analysis of variance (ANOVA) test was used for the comparison of tumor growth between groups at the same day shown in Figs. 2B and 4B. Log-rank test was used for the comparison of survival shown in Figs. 2C and 4C. All statistical analysis was performed with GraphPad Prism 7.0.
Supplementary Material
Acknowledgments:
We thank O. M. Z. Howard, K. T. Czarra, T. He, and P. Li for their help in this study. We thank NCI-Frederick Cancer Inflammation Program Fluorescence Cytometry core (K. B. Noer and R. M. Matthai) for expert technical assistance with flow cytometry, and the excellent administrative support from S. D. Livingstone. Funding: This work was supported (in part) by the Intramural Research Program of the NIH, NCI, Center for Cancer Research; Department of Science and Technology, Guizhou Province, under grants G[2014]7020 and J[2015]2003; University of Macau under grants SRG2014–00024-ICMS-QRCM, MYRG2016–00023-ICMS-QRCM, and MYRG2017–00120-ICMS; and the Science and Technology Development Fund of Macao SAR (FDCT) under grant 014/2015/A1.
Footnotes
Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Motz GT, Coukos G, Deciphering and reversing tumor immune suppression. Immunity 39, 61–73 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pitt JM, Marabelle A, Eggermont A, Soria J-C, Kroemer G, Zitvogel L, Targeting the tumor microenvironment: Removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol 27, 1482–1492 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Tanaka A, Sakaguchi S, Regulatory T cells in cancer immunotherapy. Cell Res. 27, 109–118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tao H, Mimura Y, Aoe K, Kobayashi S, Yamamoto H, Matsuda E, Okabe K, Matsumoto T, Sugi K, Ueoka H, Prognostic potential of FOXP3 expression in non-small cell lung cancer cells combined with tumor-infiltrating regulatory T cells. Lung Cancer 75, 95–101 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Shou J, Zhang Z, Lai Y, Chen Z, Huang J, Worse outcome in breast cancer with higher tumor-infiltrating FOXP3+ Tregs: A systematic review and meta-analysis. BMC Cancer 16, 687 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin Y-C, Mahalingam J, Chiang J-M, Su P-J, Chu Y-Y, Lai H-Y, Fang J-H, Huang C-T, Chiu C-T, Lin C-Y, Activated but not resting regulatory T cells accumulated in tumor microenvironment and correlated with tumor progression in patients with colorectal cancer. Int. J. Cancer 132, 1341–1350 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Liu L, Zhao G, Wu W, Rong Y, Jin D, Wang D, Lou W, Qin X, Low intratumoral regulatory T cells and high peritumoral CD8+ T cells relate to long-term survival in patients with pancreatic ductal adenocarcinoma after pancreatectomy. Cancer Immunol. Immunother. 65, 73–82 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishikawa H, Sakaguchi S, Regulatory T cells in cancer immunotherapy. Curr. Opin. Immunol 27, 1–7 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Liu C, Workman CJ, Vignali DAA, Targeting regulatory T cells in tumors. FEBS J. 283, 2731–2748 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Chen X, Subleski JJ, Kopf H, Howard OMZ, Männel DN, Oppenheim JJ, Cutting edge: Expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+ T regulatory cells: Applicability to tumor-infiltrating T regulatory cells. J. Immunol 180, 6467–6471 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen X, Subleski JJ, Hamano R, Howard OMZ, Wiltrout RH, Oppenheim JJ, Co-expression of TNFR2 and CD25 identifies more of the functional CD4+FOXP3+ regulatory T cells in human peripheral blood. Eur. J. Immunol 40, 1099–1106 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grinberg-Bleyer Y, Saadoun D, Baeyens A, Billiard F, Goldstein JD, Gregoire S, Martin GH, Elhage R, Derian N, Carpentier W, Marodon G, Klatzmann D, Piaggio E, Salomon BL, Pathogenic T cells have a paradoxical protective effect in murine autoimmune diabetes by boosting Tregs. J. Clin. Invest 120, 4558–4568 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen X, Wu X, Zhou Q, Howard OMZ, Netea MG, Oppenheim JJ, TNFR2 is critical for the stabilization of the CD4+Foxp3+ regulatory T. cell phenotype in the inflammatory environment. J. Immunol 190, 1076–1084 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zaragoza B, Chen X, Oppenheim JJ, Baeyens A, Gregoire S, Chader D, Gorochov G, Miyara M, Salomon BL, Suppressive activity of human regulatory T cells is maintained in the presence of TNF. Nat. Med 22, 16–17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chopra M, Biehl M, Steinfatt T, Brandl A, Kums J, Amich J, Vaeth M, Kuen J, Holtappels R, Podlech J, Mottok A, Kraus S, Jordán-Garrote A-L, Bäuerlein CA, Brede C, Ribechini E, Fick A, Seher A, Polz J, Ottmüller KJ, Baker J, Nishikii H, Ritz M, Mattenheimer K, Schwinn S, Winter T, Schäfer V, Krappmann S, Einsele H, Müller TD, Reddehase MJ, Lutz MB, Männel DN, Berberich-Siebelt F, Wajant H, Beilhack A, Exogenous TNFR2 activation protects from acute GvHD via host T reg cell expansion. J. Exp. Med 213, 1881–1900 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leclerc M, Naserian S, Pilon C, Thiolat A, Martin GH, Pouchy C, Dominique C, Belkacemi Y, Charlotte F, Maury S, Salomon BL, Cohen JL, Control of GVHD by regulatory T cells depends on TNF produced by T cells and TNFR2 expressed by regulatory T cells. Blood 128, 1651–1659 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Pierini A, Strober W, Moffett C, Baker J, Nishikii H, Alvarez M, Pan Y, Schneidawind D, Meyer E, Negrin RS, TNF-α priming enhances CD4+FoxP3+ regulatory T-cell suppressive function in murine GVHD prevention and treatment. Blood 128, 866–871 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen X, Bäumel M, Männel DN, Howard OMZ, Oppenheim JJ, Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J. Immunol 179, 154–161 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Chen X, Yang Y, Zhou Q, Weiss JM, Howard OMZ, McPherson JM, Wakefield LM, Oppenheim JJ, Effective chemoimmunotherapy with anti-TGFβ antibody and cyclophosphamide in a mouse model of breast cancer. PLOS ONE 9, e85398 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan F, Du R, Wei F, Zhao H, Yu J, Wang C, Zhan Z, Ding T, Ren X, Chen X, Li H, Expression of TNFR2 by regulatory T cells in peripheral blood is correlated with clinical pathology of lung cancer patients. Cancer Immunol. Immunother. 64, 1475–1485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Govindaraj C, Scalzo-Inguanti K, Madondo M, Hallo J, Flanagan K, Quinn M, Plebanski M, Impaired Th1 immunity in ovarian cancer patients is mediated by TNFR2+ Tregs within the tumor microenvironment. Clin. Immunol 149, 97–110 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Tirosh I, Izar B, Prakadan SM, Wadsworth II MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, Fallahi-Sichani M, Dutton-Regester K, Lin J-R, Cohen O, Shah P, Lu D, Genshaft AS, Hughes TK, Ziegler CGK, Kazer SW, Gaillard A, Kolb KE, Villani A-C, Johannessen CM, Andreev AY, Van Allen EM, Bertagnolli M, Sorger PK, Sullivan RJ, Flaherty KT, Frederick DT, Jané-Valbuena J, Yoon CH, Rozenblatt-Rosen O, Shalek AK, Regev A, Garraway LA, Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Govindaraj C, Tan P, Walker P, Wei A, Spencer A, Plebanski M, Reducing TNF receptor 2+ regulatory T cells via the combined action of azacitidine and the HDAC inhibitor, panobinostat for clinical benefit in acute myeloid leukemia patients. Clin. Cancer Res. 20, 724–735 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Govindaraj C, Madondo M, Kong YY, Tan P, Wei A, Plebanski M, Lenalidomide-based maintenance therapy reduces TNF receptor 2 on CD4 T cells and enhances immune effector function in acute myeloid leukemia patients. Am. J. Hematol 89, 795–802 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Hamilton KE, Simmons JG, Ding S, Van Landeghem L, Lund PK, Cytokine induction of tumor necrosis factor receptor 2 is mediated by STAT3 in colon cancer cells. Mol. Cancer Res. 9, 1718–1731 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakayama S, Yokote T, Tsuji M, Akioka T, Miyoshi T, Hirata Y, Hiraoka N, Iwaki K, Takayama A, Nishiwaki U, Masuda Y, Hanafusa T, Expression of tumour necrosis factor-α and its receptors in Hodgkin lymphoma. Br. J. Haematol 167, 574–577 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Rauert H, Stühmer T, Bargou R, Wajant H, Siegmund D, TNFR1 and TNFR2 regulate the extrinsic apoptotic pathway in myeloma cells by multiple mechanisms. Cell Death Dis. 2, e194 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Al-Lamki RS, Tumor necrosis factor receptor 2: Its contribution to acute cellular rejection and clear cell renal carcinoma. Biomed. Res. Int 2013, 821310 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Torrey H, Butterworth J, Mera T, Okubo Y, Wang L, Baum D, Defusco A, Plager S, Warden S, Huang D, Vanamee E, Foster R, Faustman DL, Targeting TNFR2 with antagonistic antibodies inhibits proliferation of ovarian cancer cells and tumor-associated Tregs. Sci. Signal 10, eaaf8608 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Chen X, Oppenheim JJ, Targeting TNFR2, an immune checkpoint stimulator and oncoprotein, is a promising treatment for cancer. Sci. Signal 10, eaal2328 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Jung ID, Shin SJ, Lee M-G, Kang TH, Han HD, Lee SJ, Kim WS, Kim HM, Park WS, Kim HW, Yun C-H, Lee EK, Wu T-C, Park Y-M, Enhancement of tumor-specific T cell-mediated immunity in dendritic cell-based vaccines by Mycobacterium tuberculosis heat shock protein X. J. Immunol 193, 1233–1245 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weir C, Hudson AL, Moon E, Ross A, Alexander M, Peters L, Langova V, Clarke SJ, Pavlakis N, Davey R, Howell VM, Streptavidin: A novel immunostimulant for the selection and delivery of autologous and syngeneic tumor vaccines. Cancer Immunol. Res. 2, 469–479 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Tacken PJ, Zeelenberg IS, Cruz LJ, van Hout-Kuijer MA, van de Glind G, Fokkink RG, Lambeck AJA, Figdor CG, Targeted delivery of TLR ligands to human and mouse dendritic cells strongly enhances adjuvanticity. Blood 118, 6836–6844 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Hamano R, Huang J, Yoshimura T, Oppenheim JJ, Chen X, TNF optimally activatives regulatory T cells by inducing TNF receptor superfamily members TNFR2, 4–1BB and OX40. Eur. J. Immunol 41, 2010–2020 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nierkens S, den Brok MH, Sutmuller RPM, Grauer OM, Bennink E, Morgan ME, Figdor CG, Ruers TJM, Adema GJ, In vivo colocalization of antigen and CpG within dendritic cells is associated with the efficacy of cancer immunotherapy. Cancer Res. 68, 5390–5396 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Furumoto K, Soares L, Engleman EG, Merad M, Induction of potent antitumor immunity by in situ targeting of intratumoral DCs. J. Clin. Invest 113, 774–783 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guéry L, Dubrot J, Lippens C, Brighouse D, Malinge P, Irla M, Pot C, Reith W, Waldburger J-M, Hugues S, Ag-presenting CpG-activated pDCs prime Th17 cells that induce tumor regression. Cancer Res. 74, 6430–6440 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Klinman DM, Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat. Rev. Immunol 4, 249–259 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Shirota Y, Shirota H, Klinman DM, Intratumoral injection of CpG oligonucleotides induces the differentiation and reduces the immunosuppressive activity of myeloid-derived suppressor cells. J. Immunol 188, 1592–1599 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vollmer J, Krieg AM, Immunotherapeutic applications of CpG oligodeoxynucleotide TLR9 agonists. Adv. Drug Deliv. Rev. 61, 195–204 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Moseman EA, Liang X, Dawson AJ, Panoskaltsis-Mortari A, Krieg AM, Liu Y-J, Blazar BR, Chen W, Human plasmacytoid dendritic cells activated by CpG oligodeoxynucleotides induce the generation of CD4+CD25+ regulatory T cells. J. Immunol 173, 4433–4442 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, Mellor AL, IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J. Immunol 183, 2475–2483 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sparwasser T, Koch E-S, Vabulas RM, Heeg K, Lipford GB, Ellwart JW, Wagner H, Bacterial DNA and immunostimulatory CpG oligonucleotides trigger maturation and activation of murine dendritic cells. Eur. J. Immunol 28, 2045–2054 (1998). [DOI] [PubMed] [Google Scholar]
- 44.Ågren J, Thiemermann C, Foster SJ, Wang JE, Aasen AO, Cytokine responses to CpG DNA in human leukocytes. Scand. J. Immunol 64, 61–68 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Chen X, Hamano R, Subleski JJ, Hurwitz AA, Howard OMZ, Oppenheim JJ, Expression of costimulatory TNFR2 induces resistance of CD4+FoxP3− conventional T cells to suppression by CD4+FoxP3+ regulatory T cells. J. Immunol 185, 174–182 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M, Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol 155, 1151–1164 (1995). [PubMed] [Google Scholar]
- 47.Setiady YY, Coccia JA, Park PU, In vivo depletion of CD4+FOXP3+ Treg cells by the PC61 anti-CD25 monoclonal antibody is mediated by FcγRIII+ phagocytes. Eur. J. Immunol 40, 780–786 (2010). [DOI] [PubMed] [Google Scholar]
- 48.Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ Jr., Colligon TA, Trosko JA, Leinbach LI, Pletcher CH, Tweed CK, DeMichele A, Fox KR, Domchek SM, Riley JL, Vonderheide RH, CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci. Transl. Med 4, 134ra62 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lowenthal JW, Corthésy P, Tougne C, Lees R, MacDonald HR, Nabholz M, High and low affinity IL 2 receptors: Analysis by IL 2 dissociation rate and reactivity with monoclonal anti-receptor antibody PC61. J. Immunol 135, 3988–3994 (1985). [PubMed] [Google Scholar]
- 50.Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E, Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor α) monoclonal antibody. Cancer Res. 59, 3128–3133 (1999). [PubMed] [Google Scholar]
- 51.Shimizu J, Yamazaki S, Sakaguchi S, Induction of tumor immunity by removing CD25+CD4+ T cells: A common basis between tumor immunity and autoimmunity. J. Immunol 163, 5211–5218 (1999). [PubMed] [Google Scholar]
- 52.Sheehan KC, Pinckard JK, Arthur CD, Dehner LP, Goeddel DV, Schreiber RD, Monoclonal antibodies specific for murine p55 and p75 tumor necrosis factor receptors: Identification of a novel in vivo role for p75. J. Exp. Med 181, 607–617 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, Wu H, Xu X, Erben U, Wu P, Syrbe U, Sieper J, Qin Z, TNF signaling drives myeloid-derived suppressor cell accumulation. J. Clin. Invest 122, 4094–4104 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu X, Li B, Li X, Zhao X, Wan L, Lin G, Yu M, Wang J, Jiang X, Feng W, Qin Z, Yin B, Li Z, Transmembrane TNF-α promotes suppressive activities of myeloid-derived suppressor cells via TNFR2. J. Immunol 192, 1320–1331 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Zoglmeier C, Bauer H, Nörenberg D, Wedekind G, Bittner P, Sandholzer N, Rapp M, Anz D, Endres S, Bourquin C, CpG blocks immunosuppression by myeloid-derived suppressor cells in tumor-bearing mice. Clin. Cancer Res. 17, 1765–1775 (2011). [DOI] [PubMed] [Google Scholar]
- 56.Chen X, Oppenheim JJ, TNF-α: An activator of CD4+FoxP3+TNFR2+ regulatory T cells. Curr. Dir. Autoimmun 11, 119–134 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen Q, Davidson TS, Huter EN, Shevach EM, Engagement of TLR2 does not reverse the suppressor function of mouse regulatory T cells, but promotes their survival. J. Immunol 183, 4458–4466 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sim GC, Martin-Orozco N, Jin L, Yang Y, Wu S, Washington E, Sanders D, Lacey C, Wang Y, Vence L, Hwu P, Radvanyi L, IL-2 therapy promotes suppressive ICOS+ Treg expansion in melanoma patients. J. Clin. Invest 124, 99–110 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lemoine FM, Cherai M, Giverne C, Dimitri D, Rosenzwajg M, Trebeden-Negre H, Chaput N, Barrou B, Thioun N, Gattegnio B, Selles F, Six A, Azar N, Lotz JP, Buzyn A, Sibony M, Delcourt A, Boyer O, Herson S, Klatzmann D, Lacave R, Massive expansion of regulatory T-cells following interleukin 2 treatment during a phase I-II dendritic cell-based immunotherapy of metastatic renal cancer. Int. J. Oncol 35, 569–581 (2009). [DOI] [PubMed] [Google Scholar]
- 60.Woods DM, Rannakrishnan R, Sodré AL, Berglund A, Weber J, PD-1 blockade induces phosphorylated STAT3 and results in an increase of Tregs with reduced suppressive function. J. Immunol 198 (2017). [Google Scholar]
- 61.Peligero C, Argilaguet J, Güerri-Fernandez R, Torres B, Ligero C, Colomer P, Plana M, Knobel H, García F, Meyerhans A, PD-L1 blockade differentially impacts regulatory T cells from HIV-infected individuals depending on plasma viremia. PLOS Pathog. 11, e1005270 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Whiteside TL, The role of regulatory T cells in cancer immunology. Immunotargets Ther. 4, 159–171 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pere H, Tanchot C, Bayry J, Terme M, Taieb J, Badoual C, Adotevi O, Merillon N, Marcheteau E, Quillien V, Banissi C, Carpentier A, Sandoval F, Nizard M, Quintin-Colonna F, Kroemer G, Fridman WH, Zitvogel L, Oudard S, Tartour E, Comprehensive analysis of current approaches to inhibit regulatory T cells in cancer. Oncoimmunology 1, 326–333 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.