

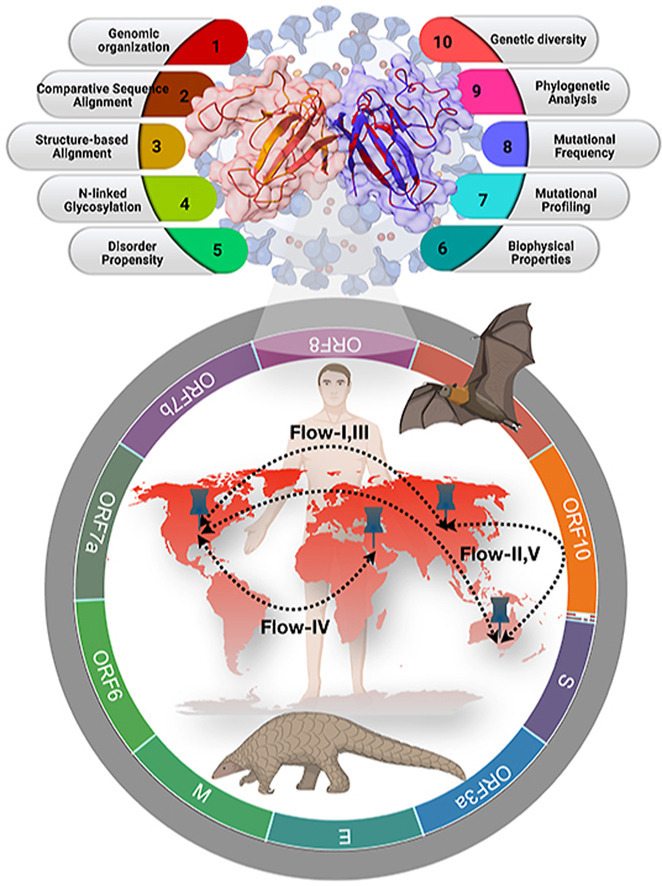
Abstract
Immune evasion is one of the unique characteristics of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) attributed to its ORF8 protein. This protein modulates the adaptive host immunity through down-regulation of MHC-1 (Major Histocompatibility Complex) molecules and innate immune responses by surpassing the host's interferon-mediated antiviral response. To understand the host's immune perspective in reference to the ORF8 protein, a comprehensive study of the ORF8 protein and mutations possessed by it have been performed. Chemical and structural properties of ORF8 proteins from different hosts, such as human, bat, and pangolin, suggest that the ORF8 of SARS-CoV-2 is much closer to ORF8 of Bat RaTG13-CoV than to that of Pangolin-CoV. Eighty-seven mutations across unique variants of ORF8 in SARS-CoV-2 can be grouped into four classes based on their predicted effects (Hussain et al., 2021) [1]. Based on the geo-locations and timescale of sample collection, a possible flow of mutations was built. Furthermore, conclusive flows of amalgamation of mutations were found upon sequence similarity analyses and consideration of the amino acid conservation phylogenies. Therefore, this study seeks to highlight the uniqueness of the rapidly evolving SARS-CoV-2 through the ORF8.
Keywords: ORF8, Phylogenetics, Mutational hotspots, Physicochemical properties, ORF8 evolution, SARS-CoV-2
Graphical abstract
1. Introduction
Severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) is a novel coronavirus whose first outbreak was reported in December 2019 in Wuhan, China, where a cluster of pneumonia cases was detected. In March 11, 2020, WHO declared this outbreak a pandemic [[2], [3], [4], [5]]. As of March 30, 2021, a total of 127.8 million confirmed COVID-19 cases had been reported worldwide, with 2.8 million deaths (World Health Organization. Coronavirus disease 2019 (COVID-19) situation). SARS-CoV-2 belongs to the family Coronaviridae and has 55% nucleotide similarity and 30% protein sequence similarity with SARS-CoV, which caused the outbreak of SARS in 2002 [[6], [7], [8]]. SARS-CoV-2 is an enveloped, single-stranded RNA virus of positive polarity whose genome is approximately 30 kb in length and encodes 16 non-structural proteins, four structural, and six accessory proteins [[9], [10], [11]], ORF3a, ORF6, ORF7a, ORF7b, ORF8, and ORF10 (Fig. 1 A) [[12], [13], [14], [15], [16]]. Among these accessory proteins, SARS-CoV-2 ORF8 is a complete protein, as it is different from any other known coronavirus ORF8 and thereby can be associated with SARS-CoV-2 pathogenicity [17,18]. The SARS-CoV-2 ORF8 displays arrays of functions; inhibition of interferon 1, promotion of viral replication, induction of apoptosis, and modulation of the ER stress [[19], [20], [21]].
Fig. 1.

Landscape of ORF8 protein. (A) Genomic organization of SARS-CoV-2 genome highlighting the ORF8 region with vdW and cartoon representation of the ORF8 structure. (B) The SARS-CoV-2 ORF8 protein structure (surface representation) showing 2.04 Å resolution by X-ray crystallography bearing PDB ID: 7JTL. (Below) Schematic illustration of the ORF8 dimer structure (Chain A (blue) and B (turquoise)) depicting disulfide bonds showing both intermolecular and intramolecular bond pairing. (C) Schematic illustration of immune invasion mechanism of ORF8 overexpression that modulates downregulation MHC-1 complex. This figure was created with Biorender.com.
The SARS-CoV-2 ORF8 is a 121 amino acid (aa) long protein, which has an N-terminal hydrophobic signal peptide (1–15 aa), and an ORF8 chain (16–121 aa) bearing dimer crystallography determined to 2.04 Å (PDB-ID:7JTL) (Fig. 1B) [22,23]. The functional motif (VLVVL) of SARS-CoV ORF8b, responsible for the induction of cell stress pathways and activation of macrophages, is absent from the SARS-CoV-2 ORF8 protein [24]. In the later stages of the SARS-CoV epidemic, it was found that a 29 nucleotide deletion in the ORF8 protein caused it to split into ORF8a (39 aa) and ORF8b (84aa), rendering it functionless [25]. Although such deletions have not been reported for SARS-CoV-2, a 382-nucleotide deletion variant (Δ382) was identified in Singapore and other countries, which caused the deletion of the entire ORF8 protein [26]. Patients with the Δ382 variant exhibited less severe symptoms, including milder hypoxic conditions and low cytokine activity compared to patients infected with the wildtype virus [26]. Also, the SARS-CoV-2 ORF8 functions in interspecies transmission and viral replication efficiency as the Δ382 deletion variant resulted in a reduced viral replication ability in human cells [27]. However, the SARS-CoV-2 ORF8 mainly acts as an immune-modulator by down-regulating MHC class I molecules, thereby shielding the infected cells against cytotoxic T cells, killing the target cells (Fig. 1C). Simultaneously, it is a potent inhibitor of the type 1 interferon signaling pathway, a key component of antiviral host immune response [28,29]. The ORF8 also regulates unfolded protein response (UPR) induced due to the ER stress by triggering the ATF-6 activation, thus enhancing the survivability of infected cells [30]. Since this protein impacts various host processes and develops various strategies for evading the host immune responses, it is essential to study the ORF8 mutations (natural variability) to understand better the viral infectivity and development of potent antiviral drugs against SARS-CoV-2 [31].
The present study identified a set of distinct mutations across unique variants of the SARS-CoV-2 ORF8 and classified them according to their predicted effect on the host (i.e., disease or neutral) and their consequences for protein structural stability. Furthermore, a comparison of the ORF8 protein of SARS-CoV-2 with Bat-RaTG13-CoV and Pangolin-CoV ORF8 was conducted to determine the evolutionary relationships regarding sequence similarity and originality of these paralogues. Similarly, a hydropathy and charge examination of the SARS-CoV-2 ORF8 mutations in distinct domains were executed to explore the possible effect on functionality changes. A possible flow of mutations scales simultaneously concerning the different geographical locations and chronological time has been depicted through phylogenetic analysis. Hence, validating the proposed sequence-based and amino acid conservation-based phylogeny is critical.
2. Results
2.1. Structural view of SARS-CoV-2 ORF8 protein in comparison to SARS-CoV
The SARS-CoV-2 ORF8 protein (YP 009724396) is a 121-amino-acid-long protein, which has an N-terminal hydrophobic signal peptide (1–15 aa) and an ORF8 chain (16–121 aa). Fig. S1 shows a schematic representation of ORF8 (SARS-CoV-2). In this protein, the total number of hydrophilic residues (63) was more extensive than that of the hydrophobic residues (58). The ORF8 protein of SARS-CoV-2 has only 55.4% nucleotide and 30% amino acid similarity with SARS-CoV, as shown in Fig. S2. Although the SARS-CoV-2 ORF8 has different genome characteristics, it exhibits high functional similarity with SARS-CoV ORF8ab. The ORF8 of SARS-CoV-2 consists of a 60-residue core similar to SARS-CoV-2 ORF7a (PDB-ID:6W37), with the addition of two dimerization interfaces unique to SARS-CoV-2 ORF8 (Fig. 2 A). The superimposition of ORF7a, ORF8a, and ORF8b of SARS-CoV revealed significant insights into the SARS-CoV-2 ORF8 protein architecture. The Root Mean Square Deviation and (Secondary structure matching (Q score) [32] depicted a deviation of 3.206 Å, 2.301 Å, 1.007 Å and 0.078, 0.036, and 0.621 when the SARS-CoV proteins were superimposed with ORF8 protein of SARS-CoV-2. From the aforementioned analysis, SARS-CoV ORF8b showed a high degree of similarity as a greater Q score represents high similarity, whereas ORF7a and ORF8a consist of less similarity with the ORF8 of SARS-CoV-2. The SARS-CoV ORF8ab original protein possesses an N-terminal hydrophobic signal sequence, which directs its transport to the endoplasmic reticulum (ER). However, after deleting the 29 nucleotides, which splits the ORF8ab protein into ORF8a and ORF8b, only ORF8a can translocate to the ER, and ORF8b remains distributed throughout the cell. Likewise, the SARS-CoV-2 ORF8 protein also contains an N-terminal hydrophobic signal peptide (1–15 aa), which is involved in the same function. The ER has an internal oxidative environment akin to other organelles, necessary for correct protein folding and oxidation processes. Due to this oxidative environment, the formation of intra or intermolecular disulfide bonds between unpaired cysteine residues can occur as the SARS-CoV ORF8ab protein is an ER-resident protein. There are ten cysteine residues present in ORF8 of SARS-CoV, which can be involved in disulfide linkages leading to the formation of homomultimeric complexes in the ER. Similarly, the ORF8 of SARS-CoV-2 also has seven cysteines, which may be expected to form these types of disulfide linkages.
Fig. 2.

Structure-based alignment of SARS-CoV-2 ORF8. (A) Superimposition of SARS-CoV-2 ORF8 with SARS-CoV ORF7a, SARS-CoV ORF8a and SARS-CoV ORF8b protein structures illustrating Q score and RMSD. (B) SARS-CoV-2 ORF8 surface structure bearing conserved region and D2 domain (protein dimer chains A (violet) and B (red)). (Below) Schematic illustration of the SARS-CoV-2 ORF8 structure depicting anti-parallel β sheets (β1-β8). N and C termini are labeled accordingly. The green color shows the conserved region of the SARS-CoV-2 ORF8 protein. (C) Interaction between the interface residues between the two chains A and B (ORF8 dimer) showing bonding patterns.
Upon inspection of the SARS-CoV-2 ORF8 protein, it was found that it consists of two domains, named D1 and D2, in which D1 consists of a signal peptide and D2 consists of the ORF8 domain. The most conserved region in the ORF8 protein of SARS-CoV-2 is “PFTINCQE” (highlighted in green) which is present in the catalytic core of the protein (Fig. 2B). The dimeric form of the protein consists of intermolecular disulfide bonds formed by Cys20 (yellow color) of each monomer (Chain A and B) (Fig. 2C). The ORF8 monomer also comprises of two-antiparallel β-sheets (smallest sheet with β2, β5, and β6 and the larger one with β3, β4, β7, and β8 where β8 is linked to β1). The dimer structure of SARS-CoV-2 ORF8 i.e., Chain A, interfaces with Chain B involving 1 disulfide bond, 4 salt bridges, 12 hydrogen bonds, and 70 non-bonded contacts. Most of the interface residues that are involved in interactions are aliphatic amino acids (grey color) followed by positive amino acids (blue color) (Fig. 2C). The SARS-CoV ORF8ab is characterized by an asparagine residue at position 81 with the Asn-Val-Thr motif responsible for the N-linked glycosylation SARS-CoV-2 ORF8 has an N-linked glycosylation site at Asn78, and its glycosylation motif is Asn-Tyr-Thr (Fig. 3 A). Val77 in SARS-CoV ORF8b has been pointed out to play a critical role in the induction of the intracellular aggregation, lysosomal stress, and interleukin-mediated inflammatory responses by activating NLRP3 inflammasomes [33]. Furthermore, Val77 is conserved in SARS-CoV-2 (Wuhan strain) and the recently identified British variation SARS-CoV-2/B.1.1.7 [1,34], contributing to the pathological manifestation of infections. SARS-CoV-2/B.1.1.7 may have an evolutionary advantage over SARS-CoV-2/Wuhan based not only on antigenic changes in the spike and ORF8 proteins but also on enhanced cytokine-mediated inflammatory responses because of intracellular aggregation in host cells. The SARS-CoV-2 ORF8 is engaged in protein-protein and protein-DNA interactions, while SARS-CoV ORF8ab shows only protein-protein interactions [35]. Most conserved regions in SARS-CoV-2 ORF8 lie around the helix-coil and strand-coil junctions, signifying these regions' functional importance. Predicted α-helical regions were also found to have conserved amino acids. It could be hypothesized that these junctions are involved in protein-protein interactions, and consequently, these regions are naturally conserved (Table S1 and Fig. S3). Our findings agree with recently published data on conserved regions in the SARS-CoV-2 ORF8 [36].
Fig. 3.

(A) SARS-CoV-2 ORF8 monomer and SARS-CoV ORF8b showing N-linked glycosylation sites analyzed through NetNGlyc 1.0. The N-linked glycosylation sites are marked red. (B) Structural alignment of two ORF8 sequences (116 among 121 residues was identical) of Bat-RaTG13-CoV QHR63307.1 and AVP78048.1,illustrating mutations at particular sites. The same presentation using Web Logo server.
2.2. Proximal evolutionary origin of the SARS-CoV-2 ORF8 in comparison to bat-RaTG13-CoV-ORF8 and pangolin-CoV-ORF8
The ORF8 protein sequences of Pangolin-CoV and Bat-RaTG13-CoV were aligned against SARS-CoV-2 ORF8 amino acid sequence to understand the proximal evolutionary origin of the SARS-CoV-2 ORF8 protein. The unique ORF8 variants of Pangolin-CoV sequences (QIA48620.1, QIA48638.1, QIA48647.1, and QIQ54055.1) were aligned, which suggested that the Pangolin-CoV ORF8 protein is conserved. There are three available ORF8 sequences of Bat-RaTG13-CoV (AVP78048.1, AVP78037.1, and QHR63307.1), of which two variants (QHR63307.1 and AVP78048.1) have turned out to be characterized by 96% sequence similarity, where mutations L3F, T14A, K44R, F104Y, and V114I were embedded in the ORF8 sequences of Bat-RaTG13-CoV (Fig. 3B and Fig. S4). Furthermore, the ORF8 protein of SARS-CoV-2 is very much similar (95%) to that of Bat-RaTG13-CoV based on sequence similarity as well as phylogenetic relationships (Fig. 6). Sequence alignment indicates six amino acid differences between the ORF8 from SARS-CoV-2 and Bat-RaTG13-CoV (Fig. S5).
Fig. 6.

Mutational profiling and their amino acid positions in ORF8 proteins of SARS-CoV-2 (96 variants). (Upper left) Frequency distribution of various mutations in the ORF8 protein variants (96 variants) of SARS-CoV-2. A red circle marked the mutations (Single Nucleotide Polymorphism (SNP)), Deletions with green/black, and Insertions with cyan/blue. (Upper left) Structural representation of SARS-CoV-2 ORF8 monomer showing high-frequency mutations. Below each sequence, a 2D presentation of the secondary structure plot has been depicted using PDBsum. (β Strands are presented in blue with naming convention as A (long β strands) and B (short β strands)).
We have also aligned the Pangolin-CoV ORF8 (QIQ54055.1 ORF8 protein) sequence with SARS-CoV-2 (YP 009724396.1 ORF8 protein) and found that there is a sequence similarity of 88%, as depicted in Fig.S6. We observed a difference of 15 amino acid residues between the Pangolin-CoV and SARS-CoV-2 ORF8. It was established that in both the Bat-RaTG13-CoV and Pangolin-CoV ORF8 proteins, the mutations L10I, V65A, and S84L were present. So, it can be hypothesized that the SARS-CoV-2 ORF8 may have originated from Pangolin-CoV or Bat-RaTG13-CoV ORF8.
In terms of structural alignment, when the SARS-CoV-2 ORF8 protein structure was superimposed against Bat-RaTG13-CoV and Pangolin-CoV ORF8, it was observed that both Bat-RaTG13-CoV and Pangolin-CoV ORF8 structure contains a small helix which was not observed in the SARS-CoV-2 ORF8 structure (PDB-ID:7JTL) (Fig. 4 A). The helix region's difference consists of Val49, Gly50, and Ala51, which has evolved as a beta-hairpin structure in the SARS-CoV-2 ORF8 structure. The amino acid difference between all three sequences depicted in Fig. 4B shows that the conserved region PFTINCQE has been conserved in all three species. Comparison of the secondary structure of ORF8 proteins from SARS-CoV-2, Bat-RaTG13-CoV, and Pangolin-CoV indicates changes at four different locations (Table S2). From Table S2, it is inferred that the secondary structures of ORF8 (SARS-CoV-2) and Bat-RaTG13-CoV are closely related compared to the ORF8 of Pangolin-CoV. Based on the sequence alignment, the ORF8 of SARS-CoV-2 differs substantially from the ORF8 of Pangolin-CoV in terms of a greater number of amino acid differences (mutations). It can be hypothesized that the SARS-CoV-2 ORF8 protein is using ORF8 of Bat RaTG13-CoV as a blueprint of its structure.
Fig. 4.

Comparative sequence and structural analysis of SARS-CoV-2, Bat-CoV-RaTG13, and Pangolin-CoV ORF8. (A) Secondary structure analysis of ORF8 protein structures. Rounded circle represents the helix region (green color). The β sheets are illustrated using ChimeraX (violet color). (B) Web logo presentation of the species' aligned sequences mentioned above of ORF8 amino acid sequences depicting the mismatches (arrows). The dotted arrows indicates all mismatches across the aligned sequences. (C) Comparison of the intrinsic disorder predisposition of the reference ORF8 protein (YP 009724396.1) of the NC 045512 SARS-CoV-2 genome from Wuhan, China (bold red curve) with disorder predispositions of ORF8 from the Pangolin-CoV (QIA48620.1) and Bat-RaTG13-CoV (QHR63307.1). (D). Analysis of the intrinsic disorder predisposition of the unique variants of the SARS-CoV-2 ORF8 in comparison with the reference ORF8 protein (YP 009724396.1) from the NC 045512 SARS-CoV-2 genome from Wuhan, China (bold red curve). Analysis was conducted using the PONDR® VSL2 algorithm [37], one of the more accurate standalone disorder predictors [[37], [38], [39], [40]]. A disorder threshold is indicated as a thin line (at score = 0.5). Residues/regions with disorder scores >0.5 are considered as disordered. Light cyan vertical bars represent positions of β-strands, whereas light pink and light-yellow vertical bars show regions with missing electron density in the crystal structures of the ORF8 protein from SARS-CoV-2 (PDB ID: 7JILB and 7JX6, respectively).
2.3. Evaluating the propensity of various ORF8 proteins for intrinsic disorder
The differences between the ORF8 of SARS-CoV-2, Bat-RaTG13-CoV, and Pangolin-CoV can be further demonstrated by analyzing the per-residue intrinsic disorder predispositions of these proteins. Results of this analysis shows the results in Fig. 4C, which illustrates that the intrinsic disorder propensity of the ORF8 from SARS-CoV-2 is closer to that of the ORF8 from Bat-RaTG13-CoV than to the disorder potential of ORF8 from Pangolin-CoV. This agrees with the results of the analyses mentioned above conducted in this study. Because SARS-COV-2 ORF8 is closer to Bat-RaTG13-CoV, we then analyzed the variants of SARS-CoV-2 ORF8 itself (96 variants) to shed light on the possible effect of mutations on disorder profiles within the variants of ORF8. When all the 96 variant sequences were aligned, it was observed that 6.6% of the region is 100% evolutionary conserved across all 96 distinct variants of the 121-amino-acid-long ORF8 protein, including the largest conserved region PFTINCQE’ (in D2 domain) (Fig. S2) in the ORF8 protein of SARS-CoV-2. The intrinsic disorder profile analysis revealed that the intrinsic disorder predispositions could vary significantly, especially in highly and moderately flexible regions (among 96 variants) (Fig. 4D). Although many mutations are disorder-silent, some increase the local disorder propensity, whereas others cause a noticeable decrease in disorder predisposition. For example, local disorder predisposition in the vicinity of residue 20 was increased in the variants QKV39247.1 and QLC47867.1. Variants QMT50144.1 and QLH01532.1 have a prominent new peak in the vicinity of residue 30, where most other variants have a shoulder. Although variant QJS56890.1 also has a prominent peak in residue 30 and has one of the highest peaks in the vicinity of residue 50, the intensity of its peak in the vicinity of residue 20 is noticeably decreased. Variant QMT96539.1 showed higher disorder propensity in the vicinity of residue 110. Variants QMT49652.1 and QMT54388.1 have the lowest disorder predisposition in residue 50, whereas the lowest disorder propensity in the vicinity of residue 70 is found in variants QKV06506.1 and QKQ29929.1. Finally, although variant QMU91370.1 is almost indistinguishable from variant QJS56890.1 within the first 40 residues, its intrinsic disorder predisposition in the vicinity of residue 70 is one of the lowest among all proteins analyzed in this study. Comparison of Fig. 4C and D shows that the variability in the disorder predisposition between many variants of the ORF8 protein from SARS-CoV-2 isolates is noticeably greater than that between the reference ORF8 from SARS-CoV-2 and ORF8 proteins from Bat-RaTG13-CoV and Pangolin-CoV.
One could ask how these disorder predictions would correlate with the actual structure of the ORF8 protein from SARS-CoV-2. Recently, two crystal structures of this protein's dimeric form were reported, PDB ID: 7JTL and PDB ID: 7JX6 [41] Fig. S7 represents the results of multiple structure alignments of four ORF8 chains found in these structures, chains A and B of 7JTL and chains A and B of 7JX6. Despite high overall structural similarity (87 residues are aligned with the root mean square deviation, RMSD, of 0.57 Å), Fig.S7 shows that several loop regions are characterized by relatively high structural flexibility, with the highest flexibility being found in the 63–78 loop. Curiously, this long loop is characterized by high structural plasticity, being very differently present in different ORF8 chains. In fact, in the 7JX6 dimer, it is entirely missing in the chain B structure but exists as a loop with two short β-strands (residues 61–63 and 68–70) in the chain A structure. In the 7JTL dimer, residues 63–68 are missing. Note that residues 15–18 are missing in this structure as well. Fig. 4D provides an outlook of the correlation between the intrinsic disorder predisposition of ORF8 protein and its structure. It is seen that most stable secondary structure elements (8 β-strands shown by light cyan vertical bars) are preferentially located within regions with low intrinsic disorder propensity, whereas the structurally mobile 63–78 loop is predicted to be highly flexible.
The conformational flexibility of the ORF8 protein could be responsible for or mediating unique immune suppression and immune-evasion capabilities of SARS-CoV-2, which may contribute to the high transmissibility and vigorous pathogenesis of this virus [42]. Although at early stages of pandemics, the ORF8 protein was shown to be an immunogenic secreted protein that induces neutralizing antibodies and can be utilized for the accurate diagnosis of COVID-19 [43], the presence of multiple ORF8 mutations requires a systematic analysis of its peptide map to determine the effects of these mutations on the neutralization potential of the ant-ORF8 antibodies, which may have therapeutic and/or diagnostic values.
2.4. Shedding light on physicochemical properties of ORF8 across SARS-CoV-2, bat-RaTG13-CoV, and Pangolin-CoV
As the analyses mentioned above showed the similarity profile based on sequence and structure alignments, awareness of the physicochemical properties of the SARS‐CoV‐2 ORF8 protein is required to understand the composition of these viral proteins to develop subunit vaccines or for designing drugs targeting these specific proteins [44]. Physicochemical analysis revealed that the total number of hydrophilic residues in the SARS-CoV-2 ORF8 protein was higher than that of the hydrophobic residues [24]. However, the predicted secondary structure and solvent accessibility analysis (Fig. S8) indicated that the highest solubility score for this protein is four, indicating that although hydrophilic residues are higher in number, they are insufficient to ensure high protein solubility. Fig. S8 shows the predicted secondary structure and solvent accessibility of the ORF8 proteins of SARS-CoV-2, Bat-RaTG13-CoV, and Pangolin-CoV obtained using the ab-initio web server QUARK to perceive the differences. The frequencies of the hydrophobic, hydrophilic, and charged amino acids were compared among the four ORF8 proteins of SARS-CoV, SARS-CoV-2, Bat-RaTG13-CoV, and Pangolin-CoV. As seen in Table S3, SARS-CoV-2 ORF8, Bat-RaTG13-CoV ORF8, and Pangolin-CoV ORF8 are all similar in terms of hydrophobicity and hydrophilicity, and it is known that hydrophobicity and hydrophilicity play an essential role in protein folding, which determines the tertiary structure of the protein and thereby affects the functions of ordered proteins. The ORF8 sequences of SARS-CoV-2, Bat-RaTG13-CoV, and Pangolin-CoV have almost the exact content of positive and negative charged amino acids. Therefore, we can hypothesize that these proteins probably have similar electrostatic and hydrophobic interactions, contributing to their functionality. Again, for the SARS-CoV ORF8ab, it was found that the number of positively and negatively charged amino acids are similar to those of the SARS-CoV-2 ORF8. Although the SARS-CoV sequence bears less similarity with the SARS-CoV-2, these proteins are likely similar in terms of electrostatic and hydrophobic interactions as well.
Moreover, a pairwise matrix of correlation coefficients between SARS-CoV-2 ORF8, Bat-RaTG13-CoV ORF8, and Pangolin-CoV ORF8 has been illustrated based on default parameters, the Pearson correlation coefficient R and the Spearman rank correlation coefficient using VOLPES [24] in terms of the level of similarity between physicochemical properties i.e., hydrophobicity [45] (Fig. 5 A), pH7.0 (Fig. 5B), beta propensity (Fig. 5C) and relative mutability [46] (Fig. 5D). The analysis revealed that in most properties, SARS-CoV-2 ORF8 has a high correlation with BatRatG13-CoV ORF8 protein and shows less correlation with Pangolin-CoV ORF8. However, BatRatG13-CoV ORF8 and Pangolin-CoV ORF8 share an excellent correlation. Since we have previously explored the amino acid changes based on multiple sequence alignments, the relative mutability factor may shed light on the similarity correlation between SARS-CoV-2 and BatRatG13-CoV, proving that SARS-CoV-2 is closer to BatRatG13-CoV.
Fig. 5.

Comparison of global biophysical properties of ORF8 proteins of SARS-CoV-2, Bat-RaTG13-CoV, and Pangolin-CoV. (A–D) A pairwise matrix of correlation coefficients between SARS-CoV-2 ORF8, Bat-RaTG13-CoV ORF8, and Pangolin-CoV ORF8 has been illustrated based on default parameters, the Pearson correlation coefficient R and the Spearman rank correlation coefficient using VOLPES in terms of the level of similarity between physicochemical properties i.e., hydrophobicity, pH7.0, beta propensity, and relative mutability, respectively. High values (cyan) and lower values (green) color represent correlation coefficient values. The individual panel across rows was compared against each other, and the coefficients were calculated. Similarly, each panel across rows was calculated. For instance, in Fig. 5A, R = 1 represents the hydrophobicity correlation of SARS-CoV-2 ORF8 with SARS-CoV-2 ORF8. Similarly, R = 0.97 represents the similarity between SARS-CoV-2 ORF8 with BatRatG13-CoV ORF8 across the same row. (E–H) Physicochemical properties, i.e., Theoretical pI, Instability Index, GRAVY, and Aliphatic index of ORF8, were calculated using ProtParam, respectively.
Furthermore, we analyzed three ORF8 sequences and checked their molecular weights, isoelectric points (pIs) (Fig. 5E), Instability index (Fig. 5F), hydropathy (GRAVY) (Fig. 5G), Aliphatic Index (Fig. 5H), net charge, and extinction coefficient using a peptide property calculator (https://pepcalc.com/) (Fig. S9) and ProtParam. We found that all properties are almost similar between the SARS-CoV-2 and the BatRatG13-CoV ORF8 protein. While inspecting the chemical aspects, e.g., molecular weight, the ORF8 protein of Bat-RaTG13-CoV, Pangolin-CoV, and SARS-CoV-2 are very closely related based on their chemical aspects of amino acid residues (Fig. S9). The isoelectric point (pI) and the protein's molecular weight tell us about the protein's biochemical and functional aspects. Since the ORF8 sequences of Bat-RaTG13-CoV and SARS-CoV-2 have the same pI and molecular weights, they can be grouped under a single functional header. The pI of the Pangolin-CoV ORF8 is higher than that of the SARS-CoV-2 ORF8, indicating that the ORF8 of the Pangolin-CoV is more negatively charged than the SARS-CoV-2 ORF8.
2.5. Natural variants of SARS-CoV-2 ORF8 protein
Each of the ORF8 amino acid sequences (96 variants) was aligned concerning the ORF8 protein (YP 009724396.1) from Wuhan, China, using the multiple sequence alignment tools (NCBI Blastp suite), and the corresponding results were used to identify mutations and their associated positions [47]. It is noted that a mutation from an amino acid A1 to A2 at a position p is denoted by A1pA2 or A1(p)A2. Fig. 6 and Table S4 describe various mutations with their respective locations. The missense mutations were found within the entire ORF8 sequence starting from the amino acid position 3 to 121, and some insertion mutations occurred at the end of the C-terminal region. Furthermore, it was discovered that several positions within the amino acid sequence of SARS-CoV-2 ORF8 protein might have more than one mutation [36]. For example, at position 11, which is a threonine (T) in a reference ORF8 protein, one might find isoleucine (I), alanine (A), or lysine (K) in some SARS-CoV-2 ORF8 natural variants. Based on the observed mutations in different ORF8 variants, it is noticed that threonine (T) and tryptophan (W) are the most vulnerable for mutations. It is noteworthy that the SARS-CoV-2 ORF8 is rapidly undergoing mutational changes, indicating that it is a highly evolving protein, whereas the Bat-RaTG13-CoV ORF8 (Fig. S5) and the Pangolin-CoV ORF8 are highly conserved (Fig. S6). A list of mutations and their frequency distributions is presented in Table S5, along with a histogram plot (Fig. 6 (upper left)). Structural representation of a single chain ORF8 protein depicting the high-frequency mutation has been shown in (Fig. 6 (upper right)).
The N-terminal signal peptide of ORF8 (D1) of SARS-CoV-2 is hydrophobic. We further analyzed mutations within this region and observed that hydrophobic to hydrophobic mutations were dominating, indicating that the domain's hydrophobicity is maintained (Table S6). Therefore, we can postulate that there are probably no functional changes in the hydrophobic N-terminal signal peptide associated with the evolutionary variability. Furthermore, it was found that there was a change from hydrophilic to hydrophobic residues in two positions of the D1 region, thereby further enhancing its hydrophobic nature. Although hydrophobic to hydrophilic and hydrophilic to hydrophilic mutations were also observed, they were not expected to have significant effects when compared to hydrophobicity changes, as hydrophobic mutations were observed at eight positions.
In contrast, hydrophilic mutations were only present at five positions. The ORF8 chain (D2) was demonstrated to be a region enriched in mutations affecting hydrophilic residues, with corresponding mutations being found in thirty-eight positions. Out of all mutations, only twenty-three mutations were affecting hydrophobic residues in D2.
2.6. Mutation profiling of SARS-CoV-2 ORF8 natural variants
Distinct, non-synonymous mutations and the associated frequency of mutations predicted effects (using Meta-SNP), as well as the predicted changes in the structural stability (using I-MUTANT) due to mutation(s), are presented in (supplementary table S1). The most frequent mutation in the ORF8 proteins turned out to be L84S (hydrophobic (L) to non-charged hydrophilic (S)), which is a clade (S) determining mutation with the frequency of 23 (Hosseini Rad Sm and McLellan 2020). The results suggest that the L84S mutation decreases structural stability and can change the ORF8 functions. Based on the predicted effects and changes of stability, we classified the mutations into four types Table S7: Disease-Decreasing: This class includes disease mutations that are decreasing the stability of the protein, with most of them occurring in D2; Neutral-Decreasing: Although the mutations are of a neutral type and supposedly are not harmful to the host, they cause protein structure stability to decrease; Disease-Increasing: These mutations lie within the D1; they increase the protein's stability, making the hydrophobic N-terminal more stable and thereby making the localization of ORF8 to ER more efficient; Neutral-Increasing: The frequency of mutations is very low in this class, although the mutations are neutral, they increase the protein's stability effectively, and they all occur in the D2 domain (supplementary table S2). Supplementary tables S3 and S4 list unique ORF8 protein IDs and their associated mutations with domain(s), and the predicted effects and changes of structural stability are presented.
Furthermore, based on the three different types of mutations viz. neutral, disease, and mix of neutral & disease, all ORF8 proteins are classified into three groups, which are presented in (supplementary table S5). It was concluded that most mutations examined in the distinct variants of the ORF8 proteins of SARS-CoV-2 turned up to be neutral, while 42% of the mutations become disease-causing as predicted. Furthermore, based on their abundance in several SARS-CoV-2 ORF8 variants, mutations S24L (which is present in 27 variants) and L84S (which was found in 23 variants) can be classified as strain-determining. Notably, one of them (L84S) was already defined in the literature [48]. Tables S6 and S7 represent the lists of ORF8 protein IDs and associated details on sequences with these two strain-determining mutations. Note that there are 64 ORF8 sequences, which do not possess strain-determining mutations. This high mutational variability suggests that the ORF8 protein is undoubtedly one of the essential proteins, which directs the pathogenicity of a variety of strains of SARS-CoV-2.
2.7. Remarks based on mutations over ORF8 proteins of SARS-CoV-2, bat-RaTG13-CoV, and Pangolin-CoV
Next, we compared the SARS-CoV-2 ORF8 with the Bat-RaTG13-CoV and the Pangolin-CoV ORF8 to study mutation evolution. The mutations in the ORF8 protein regarding the reference ORF8 sequence of Bat-RaTG13-CoV were found to be of the neutral type as predicted through the webserver Meta-SNP. All of them are expected to cause a decrease in ORF8 stability as determined using the server I-MUTANT (Fig. S5). The detailed analysis of all mutations is presented in Table S8. Based on these data, it can be suggested that several mutations in SARS-CoV-2 ORF8 can be considered as a reversal mutation. These are mutations at specific positions in the SARS-CoV-2 ORF8, which are substituted by the residues present in the Bat-RaTG13-CoV and the Pangolin-CoV ORF8 proteins. The results indicate that some positions in the SARS-CoV-2 ORF8 represent a kind of reverse genetic engineering compared with the Bat-RaTG13-CoV and the Pangolin-CoV ORF8 (Table S9).
2.8. Possible flow of mutations in ORF8 evolution
Here we present five different possible mutation flows according to data collection of the virus samples from patients [49,50]. Sequence homology and amino acid composition-based phylogenies have been drawn for the associated ORF8 proteins in each flow. Note that the ORF8 sequence QLJ93922.1 (USA) possesses consecutive (38–46 aa) deletion mutations. The other sequence, QKI36860.1 (Guangzhou, China) accommodates four insertion mutations at the end of the C-terminal region (122–125) along with two other mutations, S84L and D119E.
2.9. Flow-I
In this flow of mutations, we have described the occurrence of mutations in the US sequences based on chronological order, considering the Wuhan ORF8 sequence YP 009724396 as the reference sequence (Fig. 7 ). The protein sequence QMI92505.1 possesses a mutation L4F of neutral type with no change in hydropathy. However, it showed a decreasing effect on the stability of the protein. Following this sequence, another sequence, QMT48896.1, was identified following the time scale, in which a second mutation located at D63 N emerged. This mutation is of neutral type, and no change in hydropathy was observed. Therefore, this sequence accumulated two neutral mutations, which may affect the protein's function as both mutations cause a decrease in protein stability. The QMT96239.1 sequence harbors another mutation, G8R, which is of the disease-increasing type, and the hydropathy changed from hydrophobic to hydrophilic. Another mutation, D35Y, occurred as a second-order mutation in the QMU92030.1 sequence in addition to the G8R mutation. As D35Y is neutral and G8R is of the disease-increasing type, their combination may alter both the protein's structure and function. To support these mutation flows, we analyzed the protein sequence similarity based on phylogeny and amino acid composition. The reference ORF8 sequence YP 009724396 was found to be much more like the variants QMT48896.1 and QMI92505.1, which are more like each other as depicted in the sequence-based phylogeny (Fig. 7A). This sequence-based similarity of the QMT48896.1 and QMI92505.1 ORF8 proteins is illustrated in the chronology of mutations. Similarly, the mutation flow of the sequences QMT96239.1 and QMU92030.1 is supported by the respective sequence-based similarity. The network of five ORF8 protein variants from the US is justified based on the similar amino acid compositions/conservations across the five sequences.
Fig. 7.

Possible flow of mutations in ORF8 evolution. (A–E) The possible flow of mutations in the ORF8 (SARS-CoV-2) sequences isolated in the US, Australia, US, US and Saudi Arabia, Australia. (Right panel) Phylogenetic relationship based on amino acid sequence similarity and amino acid composition (right) of ORF8 proteins of SARS-CoV-2 in US, Australia, US, US and Saudi Arabia, Australia.
2.10. Flow-II
We observed one sequence with first-order mutations in this flow of mutations, where only one mutation accumulated in the sequence (Fig. 7B). Additionally, four sequences (all are from the US) were identified with second-order mutations, suggesting that four sequences were found to have two mutations. The protein sequence QLY90504.1 possesses a second mutation at position 67, which changed the hydrophilic serine (S) to the hydrophobic phenylalanine (F). Therefore, it may account for disrupting the ionic interactions as it is a neutral mutation, the corresponding sequence accumulated two neutral mutations. The protein sequence QLH58953.1 acquired a second mutation, P38S, which was found to be of the disease-increasing type, and the hydropathy also changed from hydrophobic to hydrophilic, indicating that these mutations may be of some importance. The protein sequence QLH58821.1 possesses a second mutation, V62L, which was found to be of the disease-neutral type with no hydropathy change. Here, this sequence accumulated two neutral mutations, which may account for some functional changes. By comparing both the sequence-based phylogeny and amino acid conservation-based phylogeny, we found that according to sequence-based phylogeny, the Australian sequence is closely related to the ORF8 Wuhan sequence. However, according to the pathway, it should be closely related to both the Wuhan sequence and second-order mutations. This can be attributed to the presence of 119 amino acid residues instead of 121 amino acid residues. In this case, the sequence has two amino acid deletions. Therefore, it is present at the first node.
2.11. Flow-III
We analyzed the US sequences considering the Wuhan sequence (YP 009724396.1) as the reference and found one sequence, QKC05159.1, with a single mutation and seven sequences with two mutations each (Fig. 7C). The first sequence, QKC05159.1, contained the L84S mutation (strain-determining mutation), neutral. However, the hydropathy changed from hydrophobic to hydrophilic, which may account for some significant change of a function. The sequences that accumulated a second mutation along with L84S are as follows:
QMT28672.1: This sequence possesses a second mutation V5F, which was predicted to be of neutral type with no hydropathy change. Hence this sequence acquired two neutral mutations, and together these mutations may alter the protein's function.
QMS53022.1: This protein sequence acquired a second mutation at position 11, which changed the hydrophilic threonine (T) to the hydrophobic isoleucine (I), affecting the ionic interactions. This mutation was found to be a disease-increasing type, so it may affect the protein structure.
QMT50804.1: This sequence gained a second mutation, E19D, which was predicted to be of the disease-increasing type with no hydropathy change. The sequence first accumulated a neutral mutation then a disease-increasing mutation, signifying that these mutations may have some functional importance.
QJD48694.1: H112Q occurred as a second mutation in this sequence, which was found to be of the disease-increasing type with no hydropathy change. Consequently, these mutations may contribute to the immune evasion property of the virus.
QKV06506.1: This sequence possesses the S67F mutation, which was predicted to be of neutral type, and changed the hydrophilic serine (S) to the hydrophobic phenylalanine (F), thus interfering with the ionic interactions that potentially increase or decrease the affinity of the viral protein for a particular host cell protein.
QKV40062.1: This sequence acquired a second mutation at Q72H, which was found to be a neutral mutation, and no change in hydropathy was observed. As this sequence accumulated two neutral mutations, it can be assumed that neutral mutations also have significant importance.
QKV07730.1: The T11A mutation occurred as the second mutation in this sequence, which was predicted to be of the disease-increasing type, and the hydropathy was changed from hydrophilic to hydrophobic. Hence the structure and function of the protein are expected to differ.
From the sequence-based phylogeny, it was observed that the Wuhan sequence was the first to originate. Although QKC05159.1 is the first sequence in our flow considering the time, it was found that in the phylogenetic tree, it is present at the fourth node instead of the second node, which is probably due to the presence of ambiguous mutations in this sequence. It was also determined that QKV07730.1 is very similar to QMT50804.1, and QMT28672.1 was observed to be similar to QKV07730.1 and QMT50804.1. All the other sequences have second-order mutations and are closely related to each other and follow the chronology. From the amino acid-based analysis, the Wuhan sequence has a high sequence similarity to QKV06506.1, thus proving that this sequence was identified chronologically after the Wuhan sequence QKC05159.1 and QMT28672.1, which again are very similar to each other.
2.12. Flow-IV
In another possible flow of mutations (Fig. 7D), we have found one sequence with a single mutation, six sequences with two mutations, and another two sequences with three mutations. The US sequence QKC05159.1 was identified to have the L84S mutation, which is a strain determining mutation, and was predicted to be a neutral mutation where hydropathy was changed from hydrophobic to hydrophilic. The sequences that accumulated second mutations along with L84S are the following, and it should be noted that the mutational accumulation occurred in a single strain:
QMS54342.1: This US sequence acquired the E110Q mutation, which was predicted to be of the disease-increasing type, where no change in hydropathy was observed, and consequently, it may contribute to virulence properties of the virus.
QLH01196.1: The A65S mutation occurred as a second mutation in this US sequence, which was found to be of neutral type. However, the hydropathy changed from hydrophobic to hydrophilic, thus potentially influencing the function of the protein.
QMU91334.1: This US sequence possesses the D63G mutation, which was predicted to be of neutral type. However, the hydropathy changed from hydrophilic to hydrophobic so, this sequence accumulated two neutral mutations, which may allow the virus to evolve in terms of virulence.
QMU91550.1: This US sequence mutated at position 62, which changed valine (V) to leucine (L), thereby mostly keeping the protein's hydrophobicity unchanged. It was a neutral mutation even though it may influence the virulence properties of the protein. This sequence was followed by another sequence, QKG86865.1, with a third mutation at position 36, which changed the hydrophobic proline (P) to the hydrophilic serine (S). Thus, the mutation was neutral, thus accumulating two neutral and one disease-increasing mutation, being significant for the evolution of the virus. We identified one more sequence, QLH57924.1, which possesses a third mutation, F16L, which was predicted to be neutral, and no hydropathy change was observed. This sequence acquired three neutral mutations that may promote virus survival.
QKU37052.1: This sequence with the W45L mutation was reported in Saudi Arabia, which was found to be of the disease-increasing type with no hydropathy change. Therefore, this sequence also accumulated one neutral and one disease-increasing mutation, affecting both the protein's structure and function.
QMT96539.1: The F104S mutation was reported in the US sequence, which was found to be of a disease-increasing type, and the hydropathy changed from hydrophobic to hydrophilic. Altogether, the sequence possesses one neutral and one disease-increasing mutation that may allow the virus to acquire new properties for better survival strategies. Sequence-based phylogeny suggested that the Wuhan sequence originated first.
Due to ambiguous amino acids, the sequence QKC05159.1 was not observed close to be close to the Wuhan sequence. QKG86865.1 and QLH57924.1 were found to have third-order mutations, and they are assumed to be closely related by the flow, and the same has been supported by amino acid conservation-based phylogeny.
2.13. Flow-V
QJR88780.1 (Australia) possesses the mutation L84S compared to the Wuhan ORF8 sequence YP 009724396.1 (Fig. 7E). Another sequence, QJR88936.1, was reported, which possesses a second mutation, V62L. This mutation was predicted to be neutral with no change in hydropathy. However, the hydrophobicity increased. This sequence belongs to a particular strain and acquires two neutral mutations, indicating that these mutations may play some vital role in the function of ORF8a. As shown in both the sequence-based phylogeny and amino acid conservation-based phylogeny, the Wuhan sequence originated earlier, and the sequences QJR88780.1 and QJR88936.1 are more closely related to each other than to the Wuhan sequence as both sequences have one common mutation not present in the Wuhan sequence.
3. Discussion
Among SARS-CoV-2 proteins, the ORF8 accessory protein is unique because it plays a vital role in bypassing the host immune surveillance mechanism. This protein is found to have a wide variety of mutations, and among them, L84S (23) and S24L (7) have the highest frequency of occurrence, which bears distinct functional significance. It has been reported that L84S and S24L show antagonistic effects on protein folding stability of the SARS-CoV-2 ORF8 [48]. L84S destabilizes protein, thereby up-regulating host-immune activity and S24L positively favors folding stability, thus enhancing the functionality of the ORF8 protein. L84S is already established as a strain-determining mutation, and since, according to our studies, both L84S and S24L do not occur together in a single sequence of the SARS-CoV-2 ORF8 protein, it is proposed that virus with the S24L mutation is a new strain altogether. We also observed that hydrophobic to hydrophobic mutations are dominant in the D1 domain. Therefore, hydrophobicity is an essential property for the N-terminal signal peptide. However, in the D2 domain, hydrophobic to hydrophilic mutations are observed more frequently, consequently making the ionic interactions more favorable and allowing the protein to evolve, providing better pathogenicity efficacy.
The ORF8 sequence of SARS-CoV-2 shows 93% similarity with the Bat-RaTG13-CoV and 88% similarity with that of the Pangolin-CoV ORF8. Thus, the ORF8 protein of SARS-CoV-2 can be considered a valuable candidate for deterministic evolutionary studies and the determination of the origin of SARS-CoV-2. We also analyzed a wide variety of mutations in the SARS-CoV-2 ORF8, where we compared them with the ORF8 of Bat-RaTG13-CoV and the Pangolin-CoV in relation to charge and hydrophobicity. We found that the Bat-RaTG13-CoV ORF8 protein exhibits precisely the same properties as that of the SARS-CoV-2 ORF8 protein, whereas the properties of the Pangolin-CoV ORF8 are relatively less similar to the SARS-CoV-2 ORF8. Furthermore, to study the evolutionary nature of mutations in the ORF8, we aligned three bat sequences and found that two of them were the same, and there were only six amino acid differences in the third compared to the other two sequences. So, only two variants were identified for the Bat-RaTG13-CoV ORF8. Therefore, it shows that the mutation rate is slow in the Bat-RaTG13-CoV ORF8.
However, for pangolins, no differences were observed among four Pangolin-CoV ORF8 sequences, and therefore, only a single variant of ORF8 was identified. The Bat-RaTG13-CoV, the Pangolin-CoV, and the SARS-CoV-2 ORF8 displayed a high similarity index based on sequence alignment, biochemical characteristics, and secondary structure analysis [51]. Additionally, in the ORF8 of SARS-CoV-2, specific mutations were found to exhibit exact reversal regarding bats and pangolins and, therefore, point towards the genomic origin of SARS-CoV-2. However, unlike Bat-RaTG13-CoV and Pangolin-CoV, the mutational distribution of SARS-CoV-2 ORF8 is widespread, ranging from position 3 to 121, having no defined conserved region. This is a rather surprising observation. Furthermore, this property differentiates the SARS-CoV-2 ORF8 from Bat-RaTG13-CoV and Pangolin-CoV, raising the question of the natural path of evolution of mutations in SARS-CoV-2.
We further predicted the types and effects of mutations of 95 sequences and grouped them into four clusters, and found that the disease-decreasing type mutations with decreasing effect on stability are more prominent. Consequently, it is hypothesized that these mutations are promoting viral survival. Furthermore, we tracked the possible flow of mutations following time and geographic locations and validated our proposal concerning sequence-based and amino acid conservation-based phylogeny and therefore putting forward the order of accumulation of mutations. We are aware of the need to confirm our prediction by structural biology data and experimental observations from in vitro and in vivo studies. However, the powerful tools in bioinformatics have allowed us to generate a reasonable basis for the potential effect of SARS-CoV-2 ORF8 mutations.
4. Conclusions
This study represents the results of a comprehensive analysis of the uniqueness of the pandemic-causing SARS-CoV-2 by focusing on one of its accessory proteins, the ORF8 protein. ORF8 is involved in modulating the adaptive host immunity and innate immune responses by surpassing interferon-mediated antiviral host responses. Our study relies heavily on bioinformatics to analyze the 87 unique mutations identified in the SARS-CoV-2 ORF8 protein, the phylogenetic comparison to bat and pangolin CoV ORF8 proteins and evaluation of the potential effect of the identified mutations on SARS-CoV-2 virulence. We acknowledge that our findings lack support from direct structural analyses and cell-based confirmations but believe that the data presented on ORF8 can provide a suitable basis for further explorations to improve our understanding of the essential functions of providing all potential means to tackle the current pandemic. In future endeavors, more critical studies on the ORF8 protein of SARS-CoV-2 are necessary for a better understanding of the importance of high-frequency mutations and their role related to the host immune system and to validate the origin of SARS-CoV-2 more precisely.
5. Materials and methods
5.1. Dataset of the ORF8 of SARS-CoV-2
As of February 14, 2021, - 29,881 complete genomes of SARS-CoV-2 were available on the NCBI (National Center for Biotechnology Information) database. Each genome contains one gene for the accessory protein ORF8, and among them, only 127 sequences were found to be unique. The amino acid sequences of the ORF8 variants were exported in FASTA format using the file operations through MATLAB (version 9.3.0.713579 (R2020a)). Among these 127 unique ORF8 sequences, only 96 ORF8 protein sequences contain various mutations, and the remaining sequences either do not possess any mutations or only ambiguous mutations. The present study focused on 96 ORF8 proteins. An ORF8 protein sequence (YP 009724396.1) of the SARS-CoV-2 genome (NC 045512) from Wuhan, China, was used as the reference to identify mutations [52].
The ORF8 protein sequences of SARS-CoV, Pangolin-CoV, and Bat-RaTG13-CoV were retrieved from the NCBI as reference sequences for understanding the proximal evolutionary origin of the SARS-CoV-2 ORF8 protein. The unique ORF8 variant was obtained among the four available Pangolin-CoV sequences (QIA48620.1, QIA48638.1, QIA48647.1, and QIQ54055.1), were retrieved along with three available ORF8 sequences of Bat-RaTG13-CoV (AVP78048.1, AVP78037.1, and QHR63307.1), of which two variants (QHR63307.1 and AVP78048.1) showed 96% sequence similarity.
5.2. Structural modeling and visualization
The structures of the SARS-CoV-2 ORF8 and the SARS-CoV ORF7a have been retrieved from Protein Data Bank bearing with the accession number ID:7JTL (www.rcsb.org) and the accession number 6W37 (www.rcsb.org), respectively. All other ORF8 structures i.e., ORF8a and ORF8b of SARS-CoV, ORF8 of Bat-RaTG13-CoV, and Pangolin-CoV were modeled using the Swiss Model server [53] since ITASSER was not available due to server maintenance. The structures were analyzed and visualized using the UCSF ChimeraX tool [54]. The plotting was done by using ORIGIN8 software. PDBsum database has been used to retrieve the SARS-CoV-2 ORF8 protein structure information, including the 2D secondary structure plot and interface regions [55].
5.3. Mutation identification
Mutations are responsible for several genetic orders/disorders. Identifying these mutations requires novel detection methods, which have been reported in the literature [56]. In this study, each unique ORF8 sequence was aligned using NCBI protein p-BLAST and omega-blast suites to determine mismatches, and thereby missense mutations (amino acid changes) were identified [57,58]. For the effect of identified mutations, a web server Meta-SNP was used, and for the structural effects of mutations, another web server I-MUTANT was used [59,60]. The web server QUARK was used to predict the secondary structure of ORF8 proteins [61,62]. The mutation profiles have been presented using the Web-Logo3 server [63].
5.4. Amino acids compositions and phylogeny
The frequency of occurrence of each amino acid Ai was determined for each primary sequence of ORF8 proteins. For all 96 ORF8 proteins, a twenty-dimensional frequency vector of amino acids was obtained. A distance matrix (Euclidean distance) was formed by measuring the distance (pairwise) between the twenty-dimensional frequency vectors for each ORF8 protein [64,65]. Thereby, applying the nearest neighbor-joining method, a phylogeny was derived from the distance matrix formed for each ORF8 protein of interest [[66], [67], [68]]. The following method was used to compute the distances of the new nodes to all other nodes at every iteration; the equation to calculate the distances between the new node, n, after joining i and j and all nodes (k), was the following:
| D(n, k) = a ∗ D(i, k) + (1 − a) ∗ D(j, k) − a ∗ D(n, i) − (1 − a) ∗ D(n, j) |
This equation could help us find the correct tree with additive data (minimum variance reduction). Note that, typically, equal variance and independence of evolutionary distance estimates (a = 1/2) are assumed [69]. The fundamental physicochemical properties were obtained by VOLPES, allowing unprecedented insights into the amino acid sequences of ORF8 variants among all species and in-depth exploration.
5.5. The propensity of intrinsic disorder
Per-residue disorder distribution in sequences of query proteins was evaluated by PONDR® VSL2 [70], one of the most accurate standalone disorder predictors [[71], [72], [73], [74]]. The per-residue disorder predisposition scores are on a scale from 0 to 1, where 0 and 1 indicate fully ordered and disordered residues, respectively. Values above the threshold of 0.5 are considered to correspond to disordered residues, whereas residues with disorder scores between 0.25 and 0.5 are considered highly flexible, and residues with disorder scores between 0.1 and 0.25 are regarded as moderately flexible.
Author contributions
SSH conceived the project. SG, DA, SSH, and VNU examined the mutations and performed Analysis. SSH, PPC, SG, DA, MMT and VNU analyzed the results. SH, MMT and AAAA, wrote the initial draft. SSH, SG, DA, PPC, MS, DP, PA, TMAEA, ASA, RK, KL, MT, AL, GKA, VNU, SPS, WBC, BDU, NR, and AMB reviewed and edited the manuscript. PKP made all images. All the authors checked, reviewed, and approved the final version of the manuscript.
Data and materials availability
The following are available online as supplementary information/material, 96 unique ORF8 protein IDs with associated information.
Mutations across ORF8 proteins of SARS-CoV-2, and their predicted effects, ORF8 protein IDs and corresponding mutations, predicted effects, list of the three types of mutations possessed by 93 distinct SARS-CoV-2 ORF8 proteins, and Observations on mutations in ORF8 concerning ORF8 of Pangolin-CoV, Bat-RaTG13-CoV. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
Declaration of competing interest
All authors declare no conflict of interest.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.compbiomed.2021.104380.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Hussain M., Shabbir S., Amnaullah A., Raza F., Imdad M.J., S.J.J.o.M.V. Zahid . vol. 7. 2021. (Immunoinformatic Analysis of Structural and Epitope Variations in Spike and Orf8 Proteins of SARS‐CoV‐2/B. 1.1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rothan H.A., Byrareddy S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020;109:102433. doi: 10.1016/j.jaut.2020.102433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kraemer M.U.G., Yang C.H., Gutierrez B., Wu C.H., Klein B., Pigott D.M., Open C.-D.W.G., du Plessis L., Faria N.R., Li R., Hanage W.P., Brownstein J.S., Layan M., Vespignani A., Tian H., Dye C., Pybus O.G., Scarpino S.V. The effect of human mobility and control measures on the COVID-19 epidemic in China. Science. 2020;368:493–497. doi: 10.1126/science.abb4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zumla A., Niederman M.S., Editorial The explosive epidemic outbreak of novel coronavirus disease 2019 (COVID-19) and the persistent threat of respiratory tract infectious diseases to global health security. Curr. Opin. Pulm. Med. 2020;26:193–196. doi: 10.1097/MCP.0000000000000676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amawi H., Abu Deiab G.a.I., Aljabali A.A.A., Dua K., Tambuwala M.M.J.T.d. COVID-19 pandemic: an overview of epidemiology, pathogenesis. diagnostics and potential vaccines and therapeutics. 2020;11:245–268. doi: 10.4155/tde-2020-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ceccarelli M., Berretta M., Venanzi Rullo E., Nunnari G., Cacopardo B. Differences and similarities between Severe Acute Respiratory Syndrome (SARS)-CoronaVirus (CoV) and SARS-CoV-2. Would a rose by another name smell as sweet? Eur. Rev. Med. Pharmacol. Sci. 2020;24:2781–2783. doi: 10.26355/eurrev_202003_20551. [DOI] [PubMed] [Google Scholar]
- 7.Xu J., Zhao S., Teng T., Abdalla A.E., Zhu W., Xie L., Wang Y., Guo X. Systematic comparison of two animal-to-human transmitted human coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses. 2020:12. doi: 10.3390/v12020244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aljabali A.A., Bakshi H.A., Satija S., Metha M., Prasher P., Ennab R.M., Chellappan D.K., Gupta G., Negi P., Goyal R.J.P.n. COVID-19: underpinning research for detection. Therapeutics, and Vaccines Development. 2020;8:323–353. doi: 10.2174/2211738508999200817163335. [DOI] [PubMed] [Google Scholar]
- 9.Giri R., Bhardwaj T., Shegane M., Gehi B.R., Kumar P., Gadhave K., Oldfield C.J., Uversky V.N. Understanding COVID-19 via comparative analysis of dark proteomes of SARS-CoV-2, human SARS and bat SARS-like coronaviruses. Cell. Mol. Life Sci. 2020 doi: 10.1007/s00018-020-03603-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y.Z., Holmes E.C. A genomic perspective on the origin and emergence of SARS-CoV-2. Cell. 2020;181:223–227. doi: 10.1016/j.cell.2020.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shen Z., Xiao Y., Kang L., Ma W., Shi L., Zhang L., Zhou Z., Yang J., Zhong J., Yang D., Guo L., Zhang G., Li H., Xu Y., Chen M., Gao Z., Wang J., Ren L., Li M. Genomic diversity of severe acute respiratory syndrome-coronavirus 2 in patients with coronavirus disease 2019. Clin. Infect. Dis. 2020;71:713–720. doi: 10.1093/cid/ciaa203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shang J., Han N., Chen Z., Peng Y., Li L., Zhou H., Ji C., Meng J., Jiang T., Wu A. Brief Bioinform; 2020. Compositional Diversity and Evolutionary Pattern of Coronavirus Accessory Proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu J., Deng W., Li S., Yang X. Cell Mol Life Sci; 2020. Advances in Research on ACE2 as a Receptor for 2019-nCoV. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J.Y., Liao C.H., Wang Q., Tan Y.J., Luo R., Qiu Y., Ge X.Y. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. 2020;286:198074. doi: 10.1016/j.virusres.2020.198074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim D., Lee J.Y., Yang J.S., Kim J.W., Kim V.N., Chang H. The architecture of SARS-CoV-2 transcriptome. Cell. 2020;181:914–921 e910. doi: 10.1016/j.cell.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu D.X., Yuan Q., Liao Y. Coronavirus envelope protein: a small membrane protein with multiple functions. Cell. Mol. Life Sci. 2007;64:2043–2048. doi: 10.1007/s00018-007-7103-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y., Zhang J., Chen Y., Luo B., Yuan Y., Huang F., Yang T., Yu F., Liu J., Liu B., Song Z., Chen J., Pan T., Zhang X., Li Y., Li R., Huang W., Xiao F., Zhang H. bioRxiv; 2020. The ORF8 Protein of SARS-CoV-2 Mediates Immune Evasion through Potently Downregulating MHC-I. 2020.2005.2024.111823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mohammad S., Bouchama A., Mohammad Alharbi B., Rashid M., Saleem Khatlani T., Gaber N.S., Malik S.S. SARS-CoV-2 ORF8 and SARS-CoV ORF8ab: genomic divergence and functional convergence. Pathogens. 2020;9 doi: 10.3390/pathogens9090677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan Y., Schneider T., Leong M., Aravind L., Zhang D. Novel immunoglobulin domain proteins provide insights into evolution and pathogenesis of SARS-CoV-2-related viruses. mBio. 2020:11. doi: 10.1128/mBio.00760-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen S., Zheng X., Zhu J., Ding R., Jin Y., Zhang W., Yang H., Zheng Y., Li X., Duan G. Extended ORF8 gene region is valuable in the epidemiological investigation of severe acute respiratory syndrome-similar coronavirus. J. Infect. Dis. 2020;222:223–233. doi: 10.1093/infdis/jiaa278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khailany R.A., Safdar M., Ozaslan M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020;19:100682. doi: 10.1016/j.genrep.2020.100682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alam I., Kamau A., Kulmanov M., Arold S.T., Pain A., Gojobori T., Duarte C.M. bioRxiv; 2020. Functional Pangenome Analysis Provides Insights into the Origin, Function and Pathways to Therapy of SARS-CoV-2 Coronavirus; p. 2020. 2002.2017.952895. [Google Scholar]
- 23.Alam I., Kamau A.A., Kulmanov M., Jaremko L., Arold S.T., Pain A., Gojobori T., Duarte C.M. Functional pangenome analysis shows key features of E protein are preserved in SARS and SARS-CoV-2. Front Cell Infect Microbiol. 2020;10:405. doi: 10.3389/fcimb.2020.00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu J., Yuan X., Wang B., Gu R., Li W., Xiang X., Tang L., Sun H. Severe acute respiratory syndrome coronavirus 2: from gene structure to pathogenic mechanisms and potential therapy. Front. Microbiol. 2020;11:1576. doi: 10.3389/fmicb.2020.01576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schaecher S.R., Pekosz A. In: Molecular Biology of the SARS-Coronavirus. Lal S.K., editor. Springer Berlin Heidelberg; Berlin, Heidelberg: 2010. SARS coronavirus accessory gene expression and function; pp. 153–166. [Google Scholar]
- 26.Young B.E., Fong S.W., Chan Y.H., Mak T.M., Ang L.W., Anderson D.E., Lee C.Y., Amrun S.N., Lee B., Goh Y.S., Su Y.C.F., Wei W.E., Kalimuddin S., Chai L.Y.A., Pada S., Tan S.Y., Sun L., Parthasarathy P., Chen Y.Y.C., Barkham T., Lin R.T.P., Maurer-Stroh S., Leo Y.S., Wang L.F., Renia L., Lee V.J., Smith G.J.D., Lye D.C., Ng L.F.P. Effects of a major deletion in the SARS-CoV-2 genome on the severity of infection and the inflammatory response: an observational cohort study. Lancet. 2020;396:603–611. doi: 10.1016/S0140-6736(20)31757-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parlikar A., Kalia K., Sinha S., Patnaik S., Sharma N., Vemuri S.G., Sharma G. Understanding genomic diversity, pan-genome, and evolution of SARS-CoV-2. PeerJ. 2020;8 doi: 10.7717/peerj.9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park M.D. Immune evasion via SARS-CoV-2 ORF8 protein? Nat. Rev. Immunol. 2020;20:408. doi: 10.1038/s41577-020-0360-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar S., Nyodu R., Maurya V.K., Saxena S.K. In: Coronavirus Disease 2019 (COVID-19) Saxena S.K., editor. Springer; 2020. Host immune response and immunobiology of human SARS-CoV-2 infection; pp. 43–53. [Google Scholar]
- 30.Sung S.C., Chao C.Y., Jeng K.S., Yang J.Y., Lai M.M. The 8ab protein of SARS-CoV is a luminal ER membrane-associated protein and induces the activation of ATF6. Virology. 2009;387:402–413. doi: 10.1016/j.virol.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frieman M., Baric R. Mechanisms of severe acute respiratory syndrome pathogenesis and innate immunomodulation. Microbiol. Mol. Biol. Rev. 2008;72:672–685. doi: 10.1128/MMBR.00015-08. Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krissinel E., K.J.A.C.S.D.B.C. Henrick . vol. 60. 2004. pp. 2256–2268. (Secondary-structure Matching (SSM), a New Tool for Fast Protein Structure Alignment in Three Dimensions). [DOI] [PubMed] [Google Scholar]
- 33.Shi C.-S., Nabar N.R., Huang N.-N., J.H.J.C.d.d. Kehrl SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates. NLRP3 inflammasomes. 2019;5:1–12. doi: 10.1038/s41420-019-0181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mohammad S., Bouchama A., Mohammad Alharbi B., Rashid M., Saleem Khatlani T., Gaber N.S., Malik S.S.J.P. vol. 9. 2020. p. 677. (SARS-CoV-2 ORF8 and SARS-CoV ORF8ab: Genomic Divergence and Functional Convergence). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meng F., Uversky V., Kurgan L. Computational prediction of intrinsic disorder in proteins. Curr. Protocols Protein Sci. 2017;88 doi: 10.1002/cpps.28. 2.16. 11-16.12.16. 14. [DOI] [PubMed] [Google Scholar]
- 36.Alkhansa A., Lakkis G., El Zein L.J.G.r. Mutational analysis of SARS-CoV-2 ORF8 during six months of COVID-19. Pandemic. 2021;23:101024. doi: 10.1016/j.genrep.2021.101024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peng K., Vucetic S., Radivojac P., Brown C.J., Dunker A.K., Obradovic Z.J.J.o.b., biology c. vol. 3. 2005. pp. 35–60. (Optimizing Long Intrinsic Disorder Predictors with Protein Evolutionary Information). [DOI] [PubMed] [Google Scholar]
- 38.Fan X., L.J.J.o.B.S. Kurgan . vol. 32. 2014. pp. 448–464. (Dynamics, Accurate Prediction of Disorder in Protein Chains with a Comprehensive and Empirically Designed Consensus). [DOI] [PubMed] [Google Scholar]
- 39.Meng F., Uversky V.N., Kurgan L.J.C., Sciences M.L. Comprehensive review of methods for prediction of intrinsic disorder and its molecular functions. 2017;74:3069–3090. doi: 10.1007/s00018-017-2555-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peng Z.-L., Kurgan L.J.C.P., Science P. Comprehensive comparative assessment of in-silico predictors of disordered regions. 2012;13:6–18. doi: 10.2174/138920312799277938. [DOI] [PubMed] [Google Scholar]
- 41.Flower T.G., Buffalo C.Z., Hooy R.M., Allaire M., Ren X., Hurley J.H. bioRxiv; 2020. Structure of SARS-CoV-2 ORF8, a Rapidly Evolving Coronavirus Protein Implicated in Immune Evasion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Flower T.G., Buffalo C.Z., Hooy R.M., Allaire M., Ren X., Hurley J.H.J.B. 2020. Structure of SARS-CoV-2 ORF8, a Rapidly Evolving Coronavirus Protein Implicated in Immune Evasion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang X., Lam J.-Y., Wong W.-M., Yuen C.-K., Cai J.-P., Au S.W.-N., Chan J.F.-W., To K.K., Kok K.-H., Yuen K.-Y.J.M. 2020. Accurate Diagnosis of COVID-19 by a Novel Immunogenic Secreted SARS-CoV-2 Orf8 Protein; p. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shahcheraghi S.H., Ayatollahi J., Aljabali A.A., Shastri M.D., Shukla S.D., Chellappan D.K., Jha N.K., Anand K., Katari N.K., Mehta M.J.T.D. An overview of vaccine development for COVID-19. 2021;12:235–244. doi: 10.4155/tde-2020-0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kyte J., R.F.J.J.o.m.b. Doolittle A simple method for displaying the hydropathic character of a protein. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 46.M.O.J.A.o.p.s. Dayhoff structure, A model of evolutionary change in proteins. 1972;5:89–99. [Google Scholar]
- 47.Parlikar A., Kalia K., Sinha S., Patnaik S., Sharma N., Vemuri S.G., Sharma G.J.P. Understanding genomic diversity, pan-genome. and evolution of SARS-CoV-2. 2020;(8) doi: 10.7717/peerj.9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hosseini Rad Sm A., McLellan A.D. Implications of SARS-CoV-2 mutations for genomic RNA structure and host microRNA targeting. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21134807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hassan S.S., Attrish D., Ghosh S., Choudhury P.P., Roy B.J.b. 2020. Pathogenetic Perspective of Missense Mutations of Orf3a Protein of Sars-Cov2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seyran M., Hassan S., Uversky V.N., Pal Choudhury P., Uhal B.D., Lundstrom K., Attrish D., Rezaei N., Aljabali A.A., Ghosh S. Multidisciplinary Digital Publishing Institute; 2021. Urgent Need for Field Surveys of Coronaviruses in Southeast Asia to Understand the SARS-CoV-2 Phylogeny and Risk Assessment for Future Outbreaks. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seyran M., Takayama K., Uversky V.N., Lundstrom K., Palù G., Sherchan S.P., Attrish D., Rezaei N., Aljabali A.A., Ghosh S.J.T.F.j. 2020. The Structural Basis of Accelerated Host Cell Entry by SARS‐CoV‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang X., Zhou Q., He Y., Liu L., Ma X., Wei X., Jiang N., Liang L., Zheng Y., Ma L., Xu Y., Yang D., Zhang J., Yang B., Jiang N., Deng T., Zhai B., Gao Y., Liu W., Bai X., Pan T., Wang G., Chang Y., Zhang Z., Shi H., Ma W.L., Gao Z. Nosocomial outbreak of COVID-19 pneumonia in Wuhan, China. Eur. Respir. J. 2020;55 doi: 10.1183/13993003.00544-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L.J.N.a.r. SWISS-MODEL: homology modelling of protein structures and complexes. 2018;46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pettersen E.F., Goddard T.D., Huang C.C., Meng E.C., Couch G.S., Croll T.I., Morris J.H., Ferrin T.E.J.P.S., ChimeraX U.C.S.F. Structure visualization for researchers, educators, and developers. 2021;30:70–82. doi: 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Laskowski R.A., Jabłońska J., Pravda L., Vařeková R.S., Thornton J.M.J.P.s. PDBsum: Structural summaries of PDB entries. 2018;27:129–134. doi: 10.1002/pro.3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cotton R.G. Current methods of mutation detection. Mutat. Res. 1993;285:125–144. doi: 10.1016/0027-5107(93)90060-s. [DOI] [PubMed] [Google Scholar]
- 57.Boratyn G.M., Thierry-Mieg J., Thierry-Mieg D., Busby B., Madden T.L. Magic-BLAST, an accurate RNA-seq aligner for long and short reads. BMC Bioinf. 2019;20:405. doi: 10.1186/s12859-019-2996-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Madeira F., Park Y.M., Lee J., Buso N., Gur T., Madhusoodanan N., Basutkar P., Tivey A.R.N., Potter S.C., Finn R.D., Lopez R. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019;47:W636–W641. doi: 10.1093/nar/gkz268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Capriotti E., Altman R.B., Bromberg Y. Collective judgment predicts disease-associated single nucleotide variants. BMC Genom. 2013;14(Suppl 3):S2. doi: 10.1186/1471-2164-14-S3-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Capriotti E., Fariselli P., Casadio R. I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005;33:W306–W310. doi: 10.1093/nar/gki375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu D., Zhang Y. Toward optimal fragment generations for ab initio protein structure assembly. Proteins. 2013;81:229–239. doi: 10.1002/prot.24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu D., Zhang Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins. 2012;80:1715–1735. doi: 10.1002/prot.24065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crooks G.E., Hon G., Chandonia J.-M., Brenner S.E.J.G.r. WebLogo: a sequence logo generator. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Irving J.A., Pike R.N., Lesk A.M., Whisstock J.C. Phylogeny of the serpin superfamily: implications of patterns of amino acid conservation for structure and function. Genome Res. 2000;10:1845–1864. doi: 10.1101/gr.gr-1478r. [DOI] [PubMed] [Google Scholar]
- 65.Peacock D., Boulter D. Use of amino acid sequence data in phylogeny and evaluation of methods using computer simulation. J. Mol. Biol. 1975;95:513–527. doi: 10.1016/0022-2836(75)90314-9. [DOI] [PubMed] [Google Scholar]
- 66.Hassan S.S., Choudhury P.P., Roy B., Jana S.S. Genomics; 2020. Missense Mutations in SARS-CoV2 Genomes from Indian Patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hassan S.S., Choudhury P.P., Roy B. Genomics; 2020. SARS-CoV2 Envelope Protein: Non-synonymous Mutations and its Consequences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Austerlitz F., David O., Schaeffer B., Bleakley K., Olteanu M., Leblois R., Veuille M., Laredo C. DNA barcode analysis: a comparison of phylogenetic and statistical classification methods. BMC Bioinf. 2009;10(Suppl 14):S10. doi: 10.1186/1471-2105-10-S14-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saitou N., Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 70.Obradovic Z., Peng K., Vucetic S., Radivojac P., Dunker A.K. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins. 2005;61(Suppl 7):176–182. doi: 10.1002/prot.20735. [DOI] [PubMed] [Google Scholar]
- 71.Fan X., Kurgan L. Accurate prediction of disorder in protein chains with a comprehensive and empirically designed consensus. J. Biomol. Struct. Dyn. 2014;32:448–464. doi: 10.1080/07391102.2013.775969. [DOI] [PubMed] [Google Scholar]
- 72.Meng F., Uversky V.N., Kurgan L. Comprehensive review of methods for prediction of intrinsic disorder and its molecular functions. Cell. Mol. Life Sci. 2017;74:3069–3090. doi: 10.1007/s00018-017-2555-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Peng Z.L., Kurgan L. Comprehensive comparative assessment of in-silico predictors of disordered regions. Curr. Protein Pept. Sci. 2012;13:6–18. doi: 10.2174/138920312799277938. [DOI] [PubMed] [Google Scholar]
- 74.Meng F., Uversky V., Kurgan L. Computational prediction of intrinsic disorder in proteins. Curr. Protein Pept. Sci. 2017;88:2 16 11–12 16 14. doi: 10.1002/cpps.28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.