

Abstract
Emerging and re-emerging viruses periodically cause outbreaks and epidemics all over the world, eventually leading to global events such as the current pandemic of the novel SARS-CoV-2 coronavirus infection COVID-19. Therefore, an urgent need for novel antivirals is crystal clear. Here we present the synthesis and evaluation of an antiviral activity of phenoxazine-based nucleoside analogs divided into three groups: (1) 8-alkoxy-substituted, (2) acyclic, and (3) carbocyclic. The antiviral activity was assessed against a structurally and phylogenetically diverse panel of RNA and DNA viruses from 25 species. Four compounds (11a-c, 12c) inhibited 4 DNA/RNA viruses with EC50 ≤ 20 μM. Toxicity of the compounds for the cell lines used for virus cultivation was negligible in most cases. In addition, previously reported and newly synthesized phenoxazine derivatives were evaluated against SARS-CoV-2, and some of them showed promising inhibition of reproduction with EC50 values in low micromolar range, although accompanied by commensurate cytotoxicity.
Keywords: Antivirals, Phenoxazine, Nucleoside analogs, SARS-CoV-2, RNA viruses, DNA viruses
Graphical abstract

1. Introduction
Historically, naturally occurring nucleoside and nucleotide analogs have undergone different modifications during the search for drug candidates and tools for molecular biology, bioorganic chemistry, and medicine. Several effective drugs for the treatment of viral diseases and cancers have been found among these compounds [[1], [2], [3]], and some possess multiple activities [4]. Therefore, nucleoside and nucleotide analogs were among the first molecules to test against novel emerging viruses, such as SARS-CoV-2 [5].
Nucleosides, consisting of a heterocyclic base and a pentafuranose residue, usually get modifications in one of these structural fragments or both of them at the same time. Exemplary modifications of the carbohydrate moiety, which led to the compounds used in the medical practice, are present in the acyclic nucleosides acyclovir 1a and ganciclovir 1b (Fig. 1 ), specific and potent inhibitors of infections caused by herpes simplex viruses HSV-1 and HSV-2, as well as by varicella-zoster virus (VZV), all belonging to Herpesviridae family [6,7]. Another example of sugar fragment modification, leading to carbocyclic nucleoside analogs, is the replacement of the furanose oxygen atom by a methylene group [8]. Among these molecules, several drugs were found, including the ones against the diseases caused by the viruses utilizing reverse transcription: abacavir 2 [9] targeting the RNA-containing human immunodeficiency virus (HIV) and entecavir 3 [10] targeting the DNA-containing hepatitis B virus (HBV).
Fig. 1.

Nucleoside analogs with acyclic and carbocyclic carbohydrate modifications (1–4) and with expanded heterocyclic bases (4–7).
Antiviral activity of a nucleoside analog can be achieved by changing the size and shape of not only a sugar residue, but also the aromatic heterocyclic base. For example, acyclic nucleoside analogs with expanded heterocyclic bases, ethenonucleosides 4a-b, are active against various herpesviruses, namely HSV-1 and HSV-2 (MIC reached 1.52 and 0.07 μM for 4a and 4b, respectively), VZV and cytomegalovirus (CMV) (4b MIC 14.3 μM for both viruses) [11,12]. Bicyclic pyrimidine nucleoside analogs (BCNAs) demonstrated potent and specific inhibition of VZV reproduction. Compounds 5 and 6 from this series inhibited VZV replication with EC50 of 0.027 and 0.0002 μM, respectively (clinically used drugs brivudine and acyclovir showed EC50 values of 0.009 μM and 3.4 μM, respectively, in that assay) [13,14]. In addition, the analogs of 5 with longer C8–C10 chains were even more active than the parent derivative with the C8 being the most favorable length for maximal activity [13]. Similar ribonucleoside derivatives inhibited reproduction of enterovirus A71 and coxsackievirus A16 on a single-digit micromolar level [15].
We have recently reported a group of ribo- and deoxyribonucleoside analogs bearing an expanded heterocyclic system – 1,3-diaza-2-oxophenoxazine – and showing inhibitory activity against VZV and CMV reproduction [16]. EC50 for the most potent compound 7a (Fig. 1) against the wild type and thymidine kinase (TK) deficient VZV strains was 0.06 and 10 μM, respectively. Thus, to expand the repertoire of phenoxazine-based antiviral candidates, we mainly used experience in anti-VZV drug design. Indeed, acyclic nucleoside analogs and BCNA are substrates of HSV-1 and VZV TKs, which phosphorylate them, providing the first step of the activation process [17]. Dimetoxytritylated analogs of 7b showed an efficient inhibition of RNA-containing tick-borne encephalitis virus (TBEV) in the range of 0.4–3.4 μM [16].
Accordingly, we present a synthesis of a new series of 1,3-diaza-2-oxophenoxazine nucleosides with fatty C8-10 alkoxy substituents in the aromatic moiety, as well as derivatives with acyclic and carbocyclic moieties instead of the ribose residue. Antiviral activity of the synthesized compounds was evaluated against not only DNA, but additionally a broad panel of RNA viruses, including TBEV, Powassan virus (POWV), and Omsk hemorrhagic fever virus (OHFV) (flaviviruses), chikungunya virus (CHIKV, alphavirus), respiratory syncytial virus (RSV, pneumovirus), influenza viruses H1N1 and H3N2. While this work was in preparation, COVID-19 pandemic emerged, and Russian isolate of SARS coronavirus 2 (SARS-CoV-2) was added to the panel [18]. One compound (11a) showed a multiple activity and inhibited both RNA (flavi- and alpha-) and DNA (herpes-) viruses in micromolar concentrations. All the available phenoxazine derivatives were assessed against SARS-CoV-2, and compounds with activities in low micromolar range were identified among them.
2. Results and discussion
2.1. Synthesis of the compounds
The compounds belonging to three classes were synthesized in the current work: (1) seven 8-alkoxy-substituted derivatives (11a-c, 12a-d); (2) five acyclic derivatives (16a-b, 17, 19, and 20) along with their synthetic precursors (13, 14, and 18), and (3) one phenoxazine-containing representative (23) of carbocyclic nucleoside analogs.
The series of phenoxazine nucleoside analogs with fatty alkoxy substituents in position 8 of phenoxazine system was synthesized in the following way. 4-(Alkoxy)-2-nitrophenols 9 were prepared from previously reported 4-alkoxyphenols 8 [19] by nitration in benzene (Scheme 1 ). Then, the reduction of the nitro group afforded anilines 10, which were used without purification in the reaction with 3′,5′-O-acetyl-4-N-(1,2,4-triazol-1-yl)-5-bromo-2′-deoxycytidine in the presence of DIPEA. Subsequent cyclization by refluxing in the mixture of TEA/C2H5OH and aqueous ammonolysis gave desired derivatives 11. Since 1,3-diaza-2-oxophenoxazine is an analog of cytosine and maintains its hhydrogen bonding pattern, we also prepared N 10-methyl substituted derivatives 12 in order to study the effect of the pattern disturbance. Additionally, we also carried out N 10-methylation of 7, the most potent compound against VZV [16], to obtain 12d.
Scheme 1.

Preparation of 8-alkoxy-substituted phenoxazine nucleoside analogs. Reagents and conditions: (a) HNO3, benzene; (b) H2, Pd/C, MeOH; (c) 3′,5′-O-acetyl-4-N-(1,2,4-triazol-1-yl)-5-bromo-2′-deoxycytidine, DIPEA, CH2Cl2; (d) TEA, C2H5OH, reflux; (e) aq NH3, 40 °C; (f) CH3I, DBU, CH2Cl2.
Acyclic uracil and phenoxazine nucleoside analogs and a carbocyclic phenoxazine nucleoside analog were synthesized according to Scheme 2 . 1-(2-Hydroxyethoxymethyl)-5-bromouracil 13 [20] was acetylated to yield 14. Then, C4 of 14 was activated by the Appel reaction followed by the substitution of Cl with either 2-aminophenol or 4-(nonyloxy)-2-aminophenol 15, prepared by catalytic hydrogenation of 9b. The cyclization under refluxing conditions in the mixture of TEA/C2H5OH followed by aq NH3 treatment afforded derivatives 16a (R = H) and 16b (R = OC9H19). Again, to disturb hydrogen bonding pattern, 16a was N 10-methylated by iodomethane in the presence of DBU, yielding 17. Finally, O-dimethoxytritylation of 13, 16a, and 17 gave 18, 19, and 20, respectively. Carbocyclic phenoxazine nucleoside analog 23 was prepared in four steps starting from uracil derivative 21 [21]. First, hydroxyl group of 21 was protected with acetyl, affording 22. Further Appel reaction, 2-aminophenol substitution, cyclization, and ammonia deprotection gave the target compound 23.
Scheme 2.

Preparation of acyclic uracil and phenoxazine nucleoside analogs and a carbocyclic phenoxazine nucleoside analog. Reagents and conditions: (a) Ac2O, Py (b) H2, Pd/C, MeOH; (c) PPh3, CCl4, CH2Cl2, reflux, then 2-aminophenol (for 16a and 23) or 15 (for 16b), DIPEA, CH2Cl2; (d) TEA, C2H5OH, reflux; (e) aq NH3, 40 °C; (f) CH3I, DBU, CH2Cl2; (g) DMTr-Cl, Py.
2.2. Broad-spectrum antiviral activity screening
Antiviral activity and cytotoxicity were assessed for all new compounds in cell-based assays against a wide variety of DNA viruses: alphaherpesviruses (HSV-1, -2, and VZV), betaherpesvirus (CMV); orthopoxvirus (vaccinia virus) and adenovirus (AdV-2), as well as against a wide range of RNA viruses: coronaviruses (HCoV 229E and SARS-CoV-2), alphaviruses (Sindbis and Chikungunya viruses), flaviviruses (YFV, ZIKV, TBEV, POWV, OHFV), enteroviruses (EVs A-C), vesiculovirus (VSV), phlebovirus (Punta Toro virus), influenza viruses (H1N1, H3N2, and B) and orhtopneumovirus (RSV) (Table S1). Assessment results for the active compounds are shown in Table 1 -3, whereas data for the inactive ones are given in the Supplementary Materials (Tables S2 and S3).
Table 1.
Cytotoxicity and antiviral activity of studied compounds against enveloped dsDNA viruses (M ± SD).
| Cmpd | EC50, μMa |
CC50, μM |
|||
|---|---|---|---|---|---|
| VZV, TK+ strain | VZV, TK– strain | CMV, AD-169 | CMV, Davis |
HEL cells | |
| 11a | 0.42 ± 0.08 (>238)b | 15.9 ± 9.8 (>6) | 54.69 (>2) | 42.3 ± 3.4 (>2) | >100 |
| 11b | 0.200 ± 0.099 (>500) | 11.3 ± 14.2 (>8) | >100 | >100 | >100 |
| 11c | 0.68 ± 0.11 (74) | 15.2 ± 2.5 (3) | >100 | 10.4 ± 0.8 (5) | 50.3 ± 2.4 |
| 12a | 8.7 ± 6.7 (>11) | >100 | >100 | >100 | >100 |
| 12b | >20 | >20 | >100 | >100 | >100 |
| 12c | 10.7 ± 1.3 (>9) | 15.1 ± 7.0 (>6) | >20 | 20 | >100 |
| 12d | >100 | >100 | >100 | >100 | >100 |
| 13 | >100 | >100 | >100 | >100 | >100 |
| 14 | >100 | >100 | >100 | >100 | >100 |
| 16a | >100 | >100 | >100 | >100 | >100 |
| 16b | >100 | >100 | >100 | >100 | >100 |
| 17 | >100 | >100 | >100 | >100 | >100 |
| 18 | >20 | 13.3 ± 0.9 (>8) | >100 | 9.2 ± 1.7 (>11) | >100 |
| 19 | >100 | >100 | >100 | >100 | >100 |
| 20 | >20 | >20 | 48.9 (>2) | 44.72 (>2) | >100 |
| 23 | >100 | 64.4 (>1.5) | >20 | >100 | >100 |
| Acyclovir | 12.0 ± 9.7 | 56 ± 33 | – | – | >300 |
| Brivudine | 0.034 ± 0.015 | 0.42 ± 0.26 | – | – | >300 |
| Ganciclovir | – | – | 19 ± 14 | 1.79 ± 0.75 | >300 |
| Cidofovir | – | – | 1.31 ± 0.1 | 0.61 ± 0.29 | >300 |
Effective concentration required to reduce virus plaque formation by 50%.
Selectivity index (SI) is shown in brackets.
Of all the studied DNA viruses, reproduction of representatives of Herpesviridae family only was inhibited by some of the compounds (Table 1) with EC50 values comparable with the reference compounds (acyclovir/brivudine or ganciclovir/cidofovir). Compounds 11b, 12a, and 23 were active against VZV only, 20 against CMV only, and 11a, 11c, and 12c inhibited reproduction of both VZV and CMV.
Reproduction of RNA tick-borne flaviviruses (TBEV, POWV, OHFV) was inhibited in plaque-reduction assay by several compounds, but for mosquito-borne flaviviruses (YFV, ZIKV) such an effect was not observed in cytopathic effect inhibition assay (Table 2 ). The most potent ones were 11a, 11b, 12b, and 12c with EC50 values less than 10 μM. Similarly, the same compounds showed promising activity against CHIKV in plaque-reduction assay, but not against Sindbis virus in cytopathic effect inhibition assay, both from Alphavirus genus. Several compounds (11c, 16a, 17) inhibited reproduction of H1N1 in a yield reduction assay with EC50 less than 10 μM. Moreover, carbocyclic compound 23 was active against RSV. Toxicity of the compounds in the cell lines used for virus cultivation was negligible in most cases.
Table 2.
Cytotoxicity and antiviral activity of studied compounds against enveloped RNA viruses (M ± SD).
| EC50, μMa |
CC50, μM |
EC50, μMa |
CC50, μM |
EC50, μMb |
CC50, μM |
EC50, μMc |
CC50, μM |
|||
|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | TBEV | POWV | OHFV | PEK | CHIKV | Vero | RSV | Hep | H1N1 | MDCK |
| 11a | 2.8 ± 0.4 (>35)d | 1.5 ± 0.5 (>66) | ND | >100 | 2.5 ± 0.5 (>40) | >100 | >100 | >100 | 82 ± 17 | >100 |
| 11b | 5.3 ± 0.2 (>18) | 7.7 ± 1.2 (>13) | ND | >100 | 7.4 ± 1.2 (>13) | >100 | >100 | >100 | >40 | >100 |
| 11c | 40 | 4.4 ± 3.2 (>22) | ND | >100 | ND | >100 | >100 | >100 | 3.4 ± 2.5 (>29) | >100 |
| 12a | 22 ± 6 (>4) | 13.3 ± 0.9 (>7) | ND | >100 | 23.8 ± 1.2 (>4) | >100 | >100 | >100 | 104 ± 42 | >100 |
| 12b | 2.53 ± 0.18 (>40) | 2.4 ± 0.4 (>42) | ND | >100 | 6.3 ± 1.1 (>15) | >100 | >100 | >100 | 35 ± 8 | >100 |
| 12c | 5.4 ± 0.3 (14) | 0.45 ± 0.03 (175) | ND | 79 ± 3 | 0.71 ± 0.02 (83) | 59 ± 2 | >100 | >100 | 23 ± 7 | >40 |
| 12d | 40 | >50 | ND | >100 | >50 | >100 | >100 | >100 | ND | >100 |
| 13 | >50 | >50 | >50 | >100 | >50 | >100 | >100 | >100 | >100 | >100 |
| 14 | >50 | >50 | 33.0 ± 5.3 (>3) | >100 | >50 | >100 | >100 | >100 | 44 ± 24 | >100 |
| 16a | 23.6 ± 0.4 (>4) | >50 | 7.2 ± 0.7 (>13) | >100 | 28 ± 8 (>3) | >100 | >100 | >100 | 25 ± 6 | >100 |
| 16b | ND | ND | ND | ND | ND | ND | ND | ND | >100 | >100 |
| 17 | 16 ± 9 (>6) | 13.5 ± 1.6 (>7) | 4.5 (>22) | >100 | 15 ± 6 (>6) | >100 | >100 | >100 | 35 ± 14 | >100 |
| 18 | 15 ± 4 (3) | 22.0 ± 0.5 (2) | 5 ± 2 (>9) | 46 ± 1 | 5.9 ± 1.4 (>17) | >100 | >100 | >100 | >100 | >100 |
| 19 | 36 ± 3 (>2) | 21 (>5) | 21.4 ± 1.4 (>4) | >100 | >50 | >100 | >100 | >100 | 11 ± 14 | >100 |
| 20 | 16 ± 7 (>6) | 8 ± 2 (>12) | 3.1 ± 1.5 (>32) | >100 | 6.1 ± 1.5 (>16) | >100 | >100 | >100 | 79 ± 44 | >100 |
| 23 | ND | ND | ND | ND | ND | ND | 11 ± 2 (7) | 80 | ND | ND |
| 1-Adamantylmethyl 5-amino-4-(2-phenylethyl)isoxazole-3-carboxylate [28] | 4.0 ± 0.9 | 15 ± 5 | 15 ± 5 | >100 | ND | ND | ND | ND | ND | ND |
| Ribavirin | ND | ND | ND | ND | ND | ND | 2 | 250 | ND | ND |
| Oseltamivir carboxylate | ND | ND | ND | ND | ND | ND | ND | ND | 2.9 ± 0.6 | >100 |
Effective concentration required to reduce virus plaque formation by 50%.
Effective concentration required to inhibit virus-induced cytopathic effect by 50% by measuring the cell viability with the colorimetric formazan-based MTS assay.
Effective concentration required to reduce virus yields by 50%.
Selectivity index (SI) was calculated relative to the corresponding cell line and shown in brackets.
Therefore, several new 1,3-diaza-2-oxophenoxazine nucleoside analogs inhibited reproduction of both DNA and RNA viruses. Moreover, at least four of them inhibited 4 DNA/RNA viruses with EC50 < 20 μM, whereas uracil derivative 18 inhibited 2 DNA and 3 RNA viruses with EC50 ≤ 20 μM.
2.3. Inhibition of SARS-CoV-2 reproduction
Emergence of a novel pandemic RNA virus SARS-CoV-2 urged us to isolate the new virus and adapt a new method for cell-based screening. The strain PIK35 was isolated from nasopharyngeal swab sample of a COVID-19 patient [18]. Virus reproduction caused a pronounced cytopathic signs in Vero cells that allowed us to establish a cytopathic effect inhibition assay. The assay was calibrated using convalescent sera and N-hydroxycytidine (NHC), well-known inhibitor of SARS-CoV-2 reproduction [22]. Serum antibodies and NHC showed a dose-depended inhibition of the strain PIK35 cytopathic effect with EC50 < 10 μM. To expand the activity spectrum study for the new compounds, the series was enriched with the previously reported phenoxazine nucleosides [16]. The results are summarized in Table 3.
Table 3.
Cytotoxicity and antiviral activity of newly synthesized and previously reported phenoxazine derivatives against SARS-CoV-2 (enveloped RNA virus of genus Betacoronavirus family Coronaviridae) in Vero cells.
 |
 |
|||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound |
 |
compound |
 |
compound |
 |
|||||||||||||
| R1 | R2 | R3 | R4 | R5 | EC50, μMa (M ± SD) | CC50, μM (M ± SD) | R1 | R2 | R3 | R5 | EC50, μMa (M ± SD) | CC50, μM (M ± SD) | R5 | EC50, μMa (M ± SD) | CC50, μM (M ± SD) | |||
| 7a | H | H | H | H | H | >100 | >100 | 16a | H | H | H | H | >100 | >100 | 13 | H | >100 | >100 |
| 7b | H | H | H | H | DMTr | >100 | 13.4 ± 6.3 | 16b | OC9H19 | H | H | H | >100 | >100 | 14 | Ac | >100 | >100 |
| 11a | OC8H17 | H | H | H | H | >100 | >100 | 17 | H | H | CH3 | H | >100 | >100 | 18 | DMTr | >100 | >100 |
| 11b | OC9H19 | H | H | H | H | >100 | >100 | 19 | H | H | H | DMTr | >100 | >100 | ||||
| 11c | OC10H21 | H | H | H | H | >100 | >100 | 20 | H | H | CH3 | DMTr | >100 | >100 | ||||
| 12a | OC8H17 | H | CH3 | H | H | >100 | >100 |  |
||||||||||
| 12b | OC9H19 | H | CH3 | H | H | >100 | >100 | |||||||||||
| 12c | OC10H21 | H | CH3 | H | H | >100 | 59 ± 2 | |||||||||||
| 12d | H | H | CH3 | H | H | >100 | >100 | R1 | R2 | R3 | ||||||||
| 25 | H | H | H | OH | H | >100 | >100 | 23 | H | H | H | >100 | ND | |||||
| 26 | H | H | H | OCH3 | H | >100 | >100 | H | ||||||||||
| 27 | H | H | H | OH | DMTr | 11.5 ± 3.7 | 15.9 ± 2.5 | |||||||||||
| 28 | H | H | H | OCH3 | DMTr | 12.4 ± 7.5 | 26.5 ± 2.5 | |||||||||||
| 29 | H | OCH2CH2NH2 | H | H | H | 1.12 ± 0.03 | 1.81 ± 0.56 | R1 | R2 | R3 | ||||||||
| 30 | H | OCH2CH2NH2 | H | OH | H | 2.52 ± 0.44 | 2.92 ± 2.13 | 24 | H | H | H | >100 | >100 | |||||
| 31 | H | OCH2CH2NH2 | H | H | DMTr | >100 | >100 | |||||||||||
| 32 | H | OCH2CH2NH2 | H | OH | DMTr | >100 | >100 | |||||||||||
| 33 | H | H | CH2(O)NH2 | H | H | >100 | >100 | |||||||||||
| 34 | H | H | CH2(O)NH2 | H | DMTr | >100 | 85 ± 21 | |||||||||||
| NHC | 7.4 ± 3.6 | >100 | ||||||||||||||||
Effective concentration required to inhibit virus-induced cytopathic effect by 50%.
Only compounds reported earlier showed promising inhibition of SARS-CoV-2 reproduction with EC50 values in the low micromolar range. The activity was accompanied by rather pronounced cytotoxicity, which was in line with the previous findings.
2.4. Structure-activity relationships
Broad-spectrum phenotypic antiviral activity screening is a powerful technique of boosted discovery of both new antiviral scaffolds and their target viruses. In this work we applied this approach to 16 new phenoxazine nucleoside analogs, extending our previous study [16] to employ modifications of the scaffold used in the successful antiviral drugs [1], or addition of aliphatic chains, reported to enhance antiviral activity of certain nucleosides [[13], [14], [15]]. Virus panel consisted of 24 viruses pathogenic for humans, belonging to different realms and Baltimore classes. Additionally, 25th virus, now known as SARS-CoV-2, emerged while this work was in preparation, and the panel was extended to include it. Thus, more than 400 new antiviral activity values were determined here.
Phenoxazine deoxyribonucleoside series was extended to include 8-alkylated (11, 12) and 10-methylated (12) analogs. These compounds appeared to have the widest activity spectrum in this panel, efficiently inhibiting the reproduction of both DNA and RNA viruses. Among DNA viruses, TK+ VZV was the most susceptible to them, with EC50 values on submicromolar level for 11a-c. Notably, in contrast to BCNAs [13,14] these derivatives were less (about 20–60 fold) but still active against TK− VZV strain suggesting TK-independent mechanism of action. 10-Methylation of these compounds decreased their potency, suggesting mechanistical role of mimicking hydrogen bond pattern of parent nucleosides. On the other hand, 10-methylation did not have a pronounced effect on the activity against RNA viruses, suggesting a non-nucleoside-like mechanism of action, consistent with other observations for alkylated nucleosides [15]. 10-Methylation of non-alkylated phenoxazine nucleoside 7, which showed EC50 of 0.06 against TK+ VZV [16], gave 12d, which did not inhibit reproduction of the studied viruses in the promising concentration range.
Acyclic nucleoside analogs were represented in our series by brominated uracil (13, 14, and 18) and phenoxazine (16, 17, 19, and 20) derivatives. Compounds with unprotected distal hydroxyl (13, 14, 16, and 17) were mostly inactive (13, 14) or showed limited efficiency against RNA viruses only. OHFV was the most susceptible to acyclic phenoxazine derivatives 16 and 17 with EC50 in single-digit micromolar range, and reproduction of other arboviruses was inhibited 3-5-fold less efficiently by them. Dimethoxytritylation of the distal hydroxyl (18–20) made antiviral effect more consistent and even emerged it for TK- VZV and CMV Davis. 8-Alkylation was not a viable strategy for acyclic compounds (16b even led to cell monolayer disruption and immeasurability of the EC50 values), but 10-methylation consistently improved their potency (16, 19 vs. 17, 20).
Compound 23, bearing cyclopentene instead of sugar moiety, appeared as an outlier both in the structure and in the activity. It was the only compound in the series to show at least some activity against RSV, although accompanied by a measurable toxicity. Majority of the series’ compounds did not show toxicity in the studied concentration range. 11c, 12c, and 18 were the only ones with measurable CC50, while EC50 values for the corresponding viruses were 9-175-fold lower in the cases of the highest activity.
Rapid virus isolation and assay establishment allowed us to perform a cell-based screening against a novel threat – SARS-CoV-2. Phenoxazine compounds from new series 11–14, 16–20, and 23, did not show measurable inhibition of SARS-CoV-2 reproduction. Thus, previously studied phenoxazine derivatives 7a-b, 24–34 [16] were additionally assessed in this assay. Only compounds 27–30 appeared active, although their antiviral activity was accompanied by substantial toxicity in the Vero cell line, which was not employed in the earlier experiments. 2-Aminoethoxylated phenoxazine nucleoside analogs 29 and 30 were more potent, although more toxic with selectivity indices only slightly more than 1. Dimethoxytritylated ribonucleosides 27 and 28 were less potent, but showed developable selectivity indices of 1.4 and 2.1, respectively, with 2′O-methylation leading to substantial decrease of toxicity. This observation adds to the wide spectrum of activity of 27, which inhibits reproduction of both VZV strains and TBEV with EC50 in the range of 0.9–2.7 μM [16], and further highlights the need to reduce the toxicity of this compound. It should be also noted that dimethoxytritylation of the distal hydroxyl does not have a consistent effect on anti-SARS-CoV-2 activity, contrary to a generally positive effect observed for RNA viruses both in this study and earlier experiments [16].
3. Conclusions
In summary, the synthesis of phenoxazine-based derivatives and their antiviral activity (about 400 activities totally evaluated) against a broad panel of structurally and replicatively diverse DNA and RNA viruses (25 species including recently emerged SARS-CoV-2) were performed. Most of the 8-alkoxy-substituted and acyclic analogs were active against RNA viruses including tick-borne encephalitis virus, Powassan virus, chikungunya virus, and influenza A virus, whereas only 8-alkoxy-substituted ones inhibited the reproduction of DNA viruses - varicella zoster virus and human cytomegalovirus. Derivatives 11a-c and 12c proved to be antivirals with a multiple activity against at least 4 DNA/RNA viruses with EC50 ≤ 20 μM. These compounds did not show pronounced cytotoxicity against all the studied cell lines. Thus, they may be considered as perspective candidates for further development as antivirals.
4. Experimental section
4.1. Chemistry
All reagents were commercially available unless otherwise mentioned and used without further purification. All solvents were purchased from commercial sources. Thin layer chromatography (TLC) was performed on plates (Merck) precoated with silica gel (60 μm, F254) and visualized using UV light (254 and 365 nm). Column chromatography (CC) was performed on silica gel (0.040–0.063 mm, Merck, Germany). 1H and 13C NMR spectra were recorded on the Bruker Avance III 600 spectrometer at 600 and 151 MHz, respectively. Chemical shifts are reported in δ (ppm) units using residual 1H signals from deuterated solvents as references. The multiplicity are reported using the following abbreviations: s (singlet), d (doublet), t (triplet), m (multiplet), br (broad). The coupling constants (J) are given in Hz. ESI HR mass spectra were acquired on a Thermo Scientific LTQ Orbitrap hybrid instrument (Thermo Electron Corp., Bremen, Germany) in continuous flow direct sample infusion (positive or negative ion mode). Purity of the most promising compounds was >95% as determined by HPLC on Agilent 1200 HPLC system (G1379A degasser, G1312A binary gradient pump, G1367B high performance autosampler, G1316A column thermostat and G1314A variable wavelength detector). The compounds were separated on a YMC-Triart C18 (YMC) (50 × 2.1 mm, 1.9 μm particle size) column, maintained at 30 °C. Eluent A was triple-distilled water with LC/MS grade formic acid (0.1%, v/v), eluent B was LC/MS grade acetonitrile with the same acid additive. The column was eluted at a flow rate of 0.35 mL/min: 0–10 min 100:0 → 0:100 (A:B, v/v); 10–12 min 0:100 → 100:0 (A:B, v/v). Detection wavelength was 254 nm. 4-(Octyloxy)phenol 8a, 4-(nonyloxy)phenol 8b, 4-(decyloxy)phenol 8c, 1-(4′-hydroxy-2′-cyclopenten-1′-yl)-5-bromouracil 21 and 3′,5′-O-acetyl-4-N-(1,2,4-triazol-1-yl)-5-bromo-2′-deoxycytidine were prepared according to the literature [19,21,23].
4.1.1. General procedure for the synthesis of 4-alkoxy-2-nitrophenols 9a-c
To a solution of 4-alkoxyphenol 8 (20 mmol) in benzene (100 mL) at 0 °C 56% aqueous nitric acid solution (20 mmol) was added in one portion with vigorous stirring. After 2 min at 0 °C the mixture was washed with water (100 mL), dried over Na2SO4 and concentrated under reduced pressure to brown oil. The residue was purified by column chromatography on silica gel (0–5% EtOAc in hexane) yielding 9 as a yellow solid:
4.1.1.1. 4-Octyloxy-2-nitrophenol (9a) (2.18 g, 8.14 mmol, yield 41%) was prepared starting from 4-(octyloxy)phenol 8a
1H NMR (600 MHz, CDCl3): δ 10.31 (s, 1H, OH), 7.49 (d, J = 3.0 Hz, 1H, H3), 7.21 (dd, J = 9.2, 3.0 Hz, 1H, H5), 7.07 (d, J = 9.2 Hz, 1H, H6), 3.94 (t, J = 6.5 Hz, 2H, -O–CH2–), 1.81–1.75 (m, 2H, –CH2-), 1.48–1.41 (m, 2H, –CH2-), 1.38–1.25 (m, 8H, 4 × –CH2-), 0.89 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (151 MHz, CDCl3): δ 152.17, 149.88, 133.01, 127.60, 120.70, 106.50, 69.01, 31.77, 29.27, 29.18, 29.02, 25.94, 22.62, 14.04. HRMS (ESI) m/z: calcd for C14H22NO4 + [M+H]+: 268.1543; found 268.1535.
4.1.1.2. 4-Nonyloxy-2-nitrophenol (9b) (1.75 g, 6.98 mmol, yield 35%) was prepared starting from 4-(nonyloxy)phenol 8b
1H NMR (600 MHz, CDCl3): δ 10.31 (s, 1H, OH), 7.50 (d, J = 3.1 Hz, 1H, H3), 7.21 (dd, J = 9.2, 3.1 Hz, 1H, H5), 7.08 (d, J = 9.2 Hz, 1H, H6), 3.95 (t, J = 6.5 Hz, 2H, -O–CH2–), 1.81–1.75 (m, 2H, –CH2-), 1.48–1.42 (m, 2H, –CH2-), 1.34–1.23 (m, 10H, 5 × –CH2-), 0.89 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (151 MHz, CDCl3): δ 152.18, 149.89, 133.02, 127.62, 120.71, 106.51, 69.01, 31.84, 29.48, 29.32, 29.22, 29.02, 25.94, 22.64, 14.06. HRMS (ESI) m/z: calcd for C15H24NO4 + [M+H]+: 282.1700; found 282.1691.
4.1.1.3. 4-Decyloxy-2-nitrophenol (9c) (2.26 g, 7.66 mmol, yield 38%) was prepared starting from 4-(decyloxy)phenol 8c
1H NMR (600 MHz, CDCl3): δ 10.31 (s, 1H, OH), 7.49 (d, J = 3.0 Hz, 1H, H3), 7.21 (dd, J = 9.2, 3.0 Hz, 1H, H5), 7.07 (d, J = 9.2 Hz, 1H, H6), 3.94 (t, J = 6.5 Hz, 2H, -O–CH2–), 1.81–1.75 (m, 2H, –CH2-), 1.48–1.41 (m, 2H, –CH2-), 1.34–1.23 (m, 12H, 6 × –CH2-), 0.88 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (151 MHz, CDCl3): δ 152.18, 149.89, 133.02, 127.63, 120.71, 106.51, 69.01, 31.87, 29.52 (2C), 29.31, 29.29, 29.02, 25.94, 22.65, 14.07. HRMS (ESI) m/z: calcd for C16H25NO4 + [M]+: 295.1784; found 295.1793.
4.1.2. General procedure for the synthesis of 8-alkoxy-1,3-diaza-2-oxophenoxazine nucleosides 11a-c
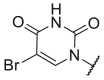
A suspension of 9 (3.00 mmol) and 10% Pd/C (30 mg) in MeOH (30 mL) was stirred for 2 h under hydrogen atmosphere. The catalyst was filtered off and the solution was concentrated under reduced pressure, yielding intermediate 10 as a brown solid. Then, it was dissolved in dry CH2Cl2 (30 mL) and 3′,5′-O-acetyl-4-N-(1,2,4-triazol-1-yl)-5-bromo-2′-deoxycytidine (0.75 equiv, 2.25 mmol) and DIPEA (0.75 equiv, 2.25 mmol) were added under nitrogen atmosphere. The solution was stirred at room temperature overnight, washed successively with saturated aqueous citric acid (30 mL), saturated aqueous NaHCO3 (30 mL) and saline (30 mL), followed by drying over Na2SO4 and concentration under reduced pressure. The foam obtained was dissolved in a mixture of absolute C2H5OH (32 mL) and TEA (3.2 mL) and refluxed under N2 for 48 h. Then, aqueous NH3 (2.5 mL) was added and the mixture was heated at 40 °C overnight. After concentration in vacuo and co-evaporation with acetonitrile, the residue was purified by column chromatography on silica gel (0–3% MeOH in CH2Cl2) yielding 11 as light brown solid.
4.1.2.1. 3-(β-D-2-deoxyribofuranosyl)-8-octyloxy-1,3-diaza-2-oxophenoxazine (11a) (0.30 g, 0.67 mmol, yield 22%)
1H NMR (600 MHz, DMSO‑d 6): δ 10.53 (br s, 1H, NH), 7.54 (s, 1H, H4), 6.70 (d, J = 8.5 Hz, 1H, H6), 6.41–6.36 (m, 2H, H7/H9), 6.12 (t, J = 6.7 Hz, 1H, H1′), 5.18 (d, J = 4.1 Hz, 1H, 3′-OH), 5.05 (t, J = 5.0 Hz, 1H, 5′-OH), 4.24–4.20 (m, 1H, H3′), 3.84 (t, J = 6.4 Hz, 2H, -O–CH2–), 3.79–3.76 (m, 1H, H4′), 3.62–3.54 (m, 2H, H5′), 2.11–2.05 (m, 1H, H2′a), 2.03–1.97 (m, 1H, H2′b), 1.68–1.62 (m, 2H, –CH2-), 1.40–1.33 (m, 2H, –CH2-), 1.32–1.21 (m, 8H, 4 × –CH2-), 0.86 (t, J = 6.9 Hz, 3H, –CH3). 13C NMR (151 MHz, DMSO‑d 6): δ 154.55, 153.13, 152.39, 135.91, 128.16, 126.96, 120.94, 115.20, 108.31, 103.60, 87.19, 84.76, 70.27, 67.68, 61.10, 39.96, 31.09, 28.58, 28.50 (2C), 25.35, 21.93, 13.80. HRMS (ESI) m/z: calcd for C23H32N3O6 + [M+H]+: 446.2286; found 446.2282.
4.1.2.2. 3-(β-D-2-deoxyribofuranosyl)-8-nonyloxy-1,3-diaza-2-oxophenoxazine (11b) (0.26 g, 0.57 mmol, yield 19%)
1H NMR (600 MHz, DMSO‑d 6): δ 10.32 (br s, 1H, NH), 7.53 (s, 1H, H4), 6.70 (d, J = 8.6 Hz, 1H, H6), 6.41–6.36 (m, 2H, H7/H9), 6.12 (t, J = 6.7 Hz, 1H, H1′), 5.18 (d, J = 3.8 Hz, 1H, 3′-OH), 5.05 (t, J = 4.4 Hz, 1H, 5′-OH), 4.25–4.19 (m, 1H, H3′), 3.84 (t, J = 6.4 Hz, 2H, -O–CH2–), 3.79–3.76 (m, 1H, H4′), 3.62–3.53 (m, 2H, H5′), 2.11–2.05 (m, 1H, H2′a), 2.03–1.97 (m, 1H, H2′b), 1.68–1.61 (m, 2H, –CH2-), 1.40–1.33 (m, 2H, –CH2-), 1.31–1.20 (m, 10H, 5 × –CH2-), 0.85 (t, J = 6.9 Hz, 3H, –CH3). 13C NMR (151 MHz, DMSO‑d 6): δ 154.56, 153.11, 152.39, 135.92, 128.16, 126.97, 120.85, 115.20, 108.33, 103.63, 87.19, 84.76, 70.27, 67.68, 61.10, 39.95, 31.13, 28.80, 28.62, 28.49 (2C), 25.33, 21.95, 13.80. HRMS (ESI) m/z: calcd for C24H34N3O6 + [M+H]+: 460.2442; found 460.2431.
4.1.2.3. 3-(β-D-2-deoxyribofuranosyl)-8-decyloxy-1,3-diaza-2-oxophenoxazine (11c) (0.30 g, 0.63 mmol, yield 21%)
1H NMR (600 MHz, DMSO‑d 6): δ 10.49 (br s, 1H, NH), 7.52 (s, 1H, H4), 6.70 (d, J = 8.4 Hz, 1H, H6), 6.41–6.36 (m, 2H, H7/H9), 6.12 (t, J = 6.7 Hz, 1H, H1′), 5.21–5.08 (m, 1H, 3′-OH), 5.06–4.96 (m, 1H, 5′-OH), 4.26–4.19 (m, 1H, H3′), 3.84 (t, J = 6.5 Hz, 2H, -O–CH2–), 3.80–3.76 (m, 1H, H4′), 3.63–3.54 (m, 2H, H5′), 2.12–2.06 (m, 1H, H2′a), 2.03–1.98 (m, 1H, H2′b), 1.68–1.61 (m, 2H, –CH2-), 1.41–1.32 (m, 2H, –CH2-), 1.32–1.19 (m, 12H, 6 × –CH2-), 0.85 (t, J = 6.9 Hz, 3H, CH3). 13C NMR (151 MHz, DMSO‑d 6): δ 154.53, 153.04, 152.33, 135.91, 128.21, 126.92, 120.79, 115.09, 108.34, 103.79, 87.16, 84.74, 70.21, 67.69, 61.06, 40.69, 31.06, 28.75, 28.71, 28.52, 28.45, 28.43, 25.25, 21.85, 13.68. HRMS (ESI) m/z: calcd for C25H36N3O6 + [M+H]+: 474.2599; found 474.2589.
4.1.3. General procedure for N-methylation of 11a-c and 7
To a solution of 11 or 7 (0.22 mmol) in CH2Cl2 (10 mL) DBU (2 equiv, 0.44 mmol) was added at rt. After 15 min iodomethane (2 equiv, 0.44 mmol) was added in one portion and the resulting mixture was stirred at rt for 1 h and then poured into 5% aqueous citric acid solution (10 mL). The organics were extracted with 10% 1-butanol in CH2Cl2 (3 × 10 mL), dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by column chromatography on silica gel (0–3% MeOH in CH2Cl2) yielding 12 as light brown solid.
4.1.3.1. 3-(β-D-2-deoxyribofuranosyl)-8-octyloxy-10-methyl-1,3-diaza-2-oxophenoxazine (12a) (77 mg, 0.17 mmol, yield 76%)
1H NMR (600 MHz, DMSO‑d 6): δ 7.66 (s, 1H, H4), 6.77 (d, J = 8.7 Hz, 1H, H6), 6.59 (d, J = 2.3 Hz, 1H, H9), 6.49 (dd, J = 8.7 Hz, J = 2.3 Hz, 1H, H7), 6.12 (t, J = 6.5 Hz, 1H, H1′), 5.19 (d, J = 4.0 Hz, 1H, 3′-OH), 5.07 (t, J = 4.8 Hz, 1H, 5′-OH), 4.25–4.20 (m, 1H, H3′), 3.92 (t, J = 6.4 Hz, 2H, -O–CH2–), 3.80–3.77 (m, 1H, H4′), 3.64–3.54 (m, 2H, H5′), 3.28 (s, 3H, N–CH3), 2.14–2.09 (m, 1H, H2′a), 2.02–1.97 (m, 1H, H2′b), 1.71–1.64 (m, 2H, –CH2-), 1.42–1.36 (m, 2H, –CH2-), 1.33–1.22 (m, 8H, 4 × –CH2-), 0.86 (t, J = 6.4 Hz, 3H, –CH3). 13C NMR (151 MHz, DMSO‑d 6): δ 154.77, 153.32, 152.86, 136.45, 129.13, 127.04, 122.15, 115.17, 107.84, 102.72, 87.28, 85.00, 70.16, 67.86, 61.01, 40.21, 31.10, 28.60, 28.56, 28.52, 28.45, 25.38, 21.94, 13.81. HRMS (ESI) m/z: calcd for C24H34N3O6 + [M+H]+: 460.2442; found 460.2421.
4.1.3.2. 3-(β-D-2-deoxyribofuranosyl)-8-nonyloxy-10-methyl-1,3-diaza-2-oxophenoxazine (12b) (74 mg, 0.16 mmol, yield 71%)
1H NMR (600 MHz, DMSO‑d 6): δ 7.67 (s, 1H, H4), 6.78 (d, J = 8.7 Hz, 1H, H6), 6.60 (d, J = 2.7 Hz, 1H, H9), 6.50 (dd, J = 8.7 Hz, J = 2.7 Hz, 1H, H7), 6.13 (t, J = 6.6 Hz, 1H, H1′), 5.19 (d, J = 4.2 Hz, 1H, 3′-OH), 5.08 (t, J = 4.9 Hz, 1H, 5′-OH), 4.25–4.21 (m, 1H, H3′), 3.93 (t, J = 6.5 Hz, 2H, -O–CH2–), 3.80–3.78 (m, 1H, H4′), 3.65–3.55 (m, 2H, H5′), 3.29 (s, 3H, N–CH3), 2.15–2.09 (m, 1H, H2′a), 2.03–1.96 (m, 1H, H2′b), 1.72–1.65 (m, 2H, –CH2-), 1.43–1.36 (m, 2H, –CH2-), 1.32–1.22 (m, 10H, 5 × –CH2-), 0.86 (t, J = 6.5 Hz, 3H, –CH3). 13C NMR (151 MHz, DMSO‑d 6): δ 154.77, 153.33, 152.85, 136.45, 129.13, 127.04, 122.15, 115.18, 107.85, 102.73, 87.28, 85.00, 70.16, 67.86, 61.01, 40.21, 31.14, 28.82, 28.63, 28.55, 28.51, 28.45, 25.36, 21.95, 13.81. HRMS (ESI) m/z: calcd for C25H36N3O6 + [M+H]+: 474.2599; found 474.2588.
4.1.3.3. 3-(β-D-2-deoxyribofuranosyl)-8-decyloxy-10-methyl-1,3-diaza-2-oxophenoxazine (12c) (82 mg, 0.17 mmol, yield 76%)
1H NMR (600 MHz, DMSO‑d 6): δ 7.66 (s, 1H, H4), 6.77 (d, J = 8.7 Hz, 1H, H6), 6.59 (d, J = 2.7 Hz, 1H, H9), 6.49 (dd, J = 8.7 Hz, J = 2.7 Hz, 1H, H7), 6.12 (t, J = 6.6 Hz, 1H, H1′), 5.19 (d, J = 4.2 Hz, 1H, 3′-OH), 5.07 (t, J = 5.0 Hz, 1H, 5′-OH), 4.24–4.20 (m, 1H, H3′), 3.92 (t, J = 6.5 Hz, 2H, -O–CH2–), 3.80–3.77 (m, 1H, H4′), 3.63–3.54 (m, 2H, H5′), 3.28 (s, 3H, N–CH3), 2.14–2.09 (m, 1H, H2′a), 2.03–1.97 (m, 1H, H2′b), 1.71–1.64 (m, 2H, –CH2-), 1.42–1.36 (m, 2H, –CH2-), 1.32–1.19 (m, 12H, 6 × –CH2-), 0.85 (t, J = 6.5 Hz, 3H, –CH3). 13C NMR (151 MHz, DMSO‑d 6): δ 154.76, 153.31, 152.84, 136.44, 129.12, 127.03, 122.14, 115.16, 107.85, 102.72, 87.27, 84.99, 70.15, 67.85, 61.00, 40.20, 31.14, 28.85, 28.80, 28.65, 28.60, 28.54, 28.44, 25.34, 21.94, 13.79. HRMS (ESI) m/z: calcd for C26H38N3O6 + [M+H]+: 488.2755; found 488.2749.
4.1.3.4. 3-(β-D-2-deoxyribofuranosyl)-10-methyl-1,3-diaza-2-oxophenoxazine (12d) (58 mg, 0.17 mmol, yield 80%)
1H NMR (600 MHz, DMSO‑d 6): δ 7.68 (s, 1H, H4), 7.05 (dd, J = 1.4 Hz, J = 8.0 Hz, 1H, H6), 6.99 (ddd, J = 1.4 Hz, J = 7.7 Hz, J = 8.0 Hz, 1H, H7), 6.95 (ddd, J = 1.4 Hz, J = 7.7 Hz, J = 8.0 Hz, 1H, H8), 6.85 (dd, J = 1.4 Hz, J = 8.0 Hz, 1H, H9), 6.13 (dd, J = 6.9 Hz, J = 6.4 Hz, 1H, H1′), 5.19 (d, J = 4.2 Hz, 1H, 3′-OH), 5.08 (t, J = 5.1 Hz, 1H, 5′-OH), 4.25–4.21 (m, 1H, H3′), 3.80–3.77 (m, 1H, H4′), 3.64–3.59 (m, 1H, H5′a), 3.59–3.55 (m, 1H, H5′b), 3.29 (s, 3H, N–CH3), 2.14–2.09 (m, 1H, H2′a), 2.03–1.97 (m, 1H, H2′b). 13C NMR (151 MHz, DMSO‑d 6): δ 153.33, 152.88, 142.69, 128.40, 126.91, 123.95, 123.49, 122.29, 114.92 (2C), 87.27, 84.98, 70.16, 61.01, 40.20, 28.32. HRMS (ESI) m/z: calcd for C16H18N3O5 + [M+H]+: 332.1241; found 332.1234.
4.1.4. 1-(4-Hydroxy-2-oxabutyl)-5-bromouracil (13)
This derivative was synthesized according to the literature [20]. 1H NMR data correspond to the literature. 1H NMR (600 MHz, DMSO‑d 6): δ 11.73 (br s, 1H, NH), 8.26 (s, 1H, H6), 5.09 (s, 2H, N–CH2–O), 4.65 (t, J = 5.2 Hz, 1H, OH), 3.54–3.51 (m, 2H, O–CH2-), 3.50–3.46 (m, 2H, –CH2-O). 13C NMR (151 MHz, DMSO‑d 6): δ 159.45, 150.32, 144.42, 95.29, 76.74, 70.63, 59.87. HRMS (ESI) m/z: calcd for C7H10BrN2O4 + [M+H]+: 264.9818; found 264.9801.
4.1.5. 1-(4-Acetoxy-2-oxabutyl)-5-bromouracil (14)
To a solution of 13 (3.10 g, 11.7 mmol) in dry pyridine (60 mL) acetic anhydride (2.2 mL, 23.4 mmol) was added. After 3 h at room temperature the reaction mixture was poured into saturated aqueous NaHCO3 (50 mL) and extracted with EtOAc (2 × 50 mL). The oily residue was co-evaporated with toluene (2 × 10 mL) and the residue was purified by column chromatography on silica gel (75% EtOAc in hexane) yielding 14 (2.98 g, 9.7 mmol, 82.9%). 1H NMR (600 MHz, DMSO‑d 6): δ 11.83 (br s, 1H, NH), 8.27 (s, 1H, H6), 5.10 (s, 2H, N–CH2–O), 4.11–4.08 (m, 2H, –CH2-O), 3.73–3.70 (m, 2H, O–CH2-), 1.99 (s, 3H, CH3). 13C NMR (151 MHz, DMSO‑d 6): δ 170.13, 159.45, 150.40, 144.34, 95.50, 76.56, 66.75, 62.79, 20.49. HRMS (ESI) m/z: calcd for C9H12BrN2O5 + [M+H]+: 306.9924; found 306.9908. The NMR data correspond to the literature [24].
4.1.6. 4-Nonyloxy-2-aminophenol (15)
A suspension of 4-(nonyloxy)-2-nitrophenol 9b (1.46 g, 5.2 mmol) and 10% Pd/C (52 mg) in MeOH (25 mL) was stirred for 2 h under hydrogen atmosphere. The catalyst was filtered off and the solution was concentrated under reduced pressure, yielding 15 (1.26 g, 5.0 mmol, 96%) as beige solid. 1H NMR (600 MHz, DMSO‑d 6): δ 8.39 (s, 1H, OH), 6.49 (d, J = 8.5 Hz, 1H, H6), 6.19 (d, J = 2.9 Hz, 1H, H3), 5.93 (dd, J = 8.5, 2.9 Hz, 1H, H5), 4.49 (br s, 2H, NH2), 3.76 (t, J = 6.5 Hz, 2H, -O–CH2–), 1.65–1.59 (m, 2H, –CH2-), 1.39–1.22 (m, 2H, –CH2-), 1.31–1.21 (m, 10H, 5 × –CH2-), 0.86 (t, J = 7.0 Hz, 3H, CH3). 13C NMR (151 MHz, DMSO‑d 6): δ 152.28, 137.85, 137.31, 114.43, 101.55, 101.16, 67.33, 31.15, 28.86, 28.80, 28.70, 28.53, 25.47, 21.96, 13.80. HRMS (ESI) m/z: calcd for C15H26NO2 + [M+H]+: 252.1958; found 252.1950.
4.1.7. 3-(4-Hydroxy-2-oxabutyl)-1,3-diaza-2-oxophenoxazine (16a)
To a solution of 14 (2.76 g, 9.0 mmol) and PPh3 (4.72 g, 18 mmol) in CH2Cl2 (50 mL) carbon tetrachloride (10 mL) was added and the resulting solution was refluxed for 5 h. Then, the reaction mixture was evaporated to a foam in vacuo, dissolved in CH2Cl2 (50 mL) and 2-aminophenol (1.47 g, 13.5 mmol) was added, followed by TEA (1.9 mL, 13.5 mmol). After 3 h at room temperature the reaction mixture was concentrated to brown foam. The foam was dissolved in a mixture of absolute C2H5OH (60 mL) and TEA (12 mL) and refluxed under N2 for 48 h. After cooling to room temperature aqueous NH3 (10 mL) was added and the mixture was heated at 40 °C overnight followed by concentration and then co-evaporation with acetonitrile (2 × 10 mL) in vacuo. The residue was purified by column chromatography on silica gel (0–4% MeOH in CH2Cl2) yielding 16a (0.57 g, 2.07 mmol, 23%) as light brownish solid. 1H NMR (600 MHz, DMSO‑d 6): δ 10.68 (br s, 1H, NH), 7.35 (s, 1H, H4), 6.90–6.78 (m, 4H, H6/H7/H8/H9), 5.01 (s, 2H, N–CH2–O), 4.64 (t, J = 5.2 Hz, 1H, OH), 3.52–3.47 (m, 4H, 2 × –CH2-). 13C NMR (151 MHz, DMSO‑d 6): δ 153.42, 153.02, 142.16, 127.93, 126.76, 124.55, 123.87, 123.46, 117.51, 114.85, 77.13, 70.31, 59.93. HRMS (ESI) m/z: calcd for C13H14N3O4 + [M+H]+: 276.0979; found 276.0962.
4.1.8. 3-(4-Hydroxy-2-oxabutyl)-8-nonyloxy-1,3-diaza-2-oxophenoxazine (16b)
This derivative (0.67 g, 1.62 mmol, yield 18%, light brownish solid) was prepared as described for the preparation of 16a using 4-(nonyloxy)-2-aminophenol 15 instead of 2-aminophenol. 1H NMR (600 MHz, DMSO‑d 6): δ 10.64 (br s, 1H, NH), 7.33 (s, 1H, H4), 6.71 (d, J = 8.6 Hz, 1H, H6), 6.42–6.37 (m, 2H, H7/H9), 5.01 (s, 2H, N–CH2–O), 4.64 (t, J = 5.2 Hz, 1H, OH), 3.84 (t, J = 6.4 Hz, 2H, O–CH2-), 3.53–3.45 (m, 4H, 2 × –CH2-), 1.69–1.61 (m, 2H, –CH2-), 1.40–1.33 (m, 2H, –CH2-), 1.31–1.20 (m, 10H, 5 × –CH2-), 0.85 (t, J = 6.4 Hz, 3H, CH3). 13C NMR (151 MHz, DMSO‑d 6): δ 154.59, 153.53, 153.11, 135.91, 128.58, 126.86, 124.71, 115.23, 108.54, 104.23, 77.17, 70.32, 67.70, 59.94, 31.14, 28.82, 28.63, 28.51 (2C), 25.34, 21.96, 13.82. HRMS (ESI) m/z: calcd for C22H32N3O5 + [M+H]+: 418.2336; found 418.2324.
4.1.9. 3-(4-Hydroxy-2-oxabutyl)-10-methyl-1,3-diaza-2-oxophenoxazine (17)
To a solution of 16a (0.14 g, 0.5 mmol) in CH2Cl2 (10 mL) DBU (150 μL, 1.0 mmol) was added at rt. After 15 min iodomethane (65 μl, 1.0 mmol) was added in one portion and the resulting mixture was stirred at rt overnight and then poured into 5% aqueous citric acid solution (10 mL). The organics were extracted with 10% 1-butanol in CH2Cl2 (3 × 10 mL), dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by column chromatography on silica gel (0–3% MeOH in CH2Cl2) yielding 17 as light brownish solid (0.10 g, 0.34 mmol, 68%). 1H NMR (600 MHz, DMSO‑d 6): δ 7.52 (s, 1H, H4), 7.07 (dd, J = 1.2, 7.9 Hz, 1H, H6), 7.01 (ddd, J = 1.3, 7.7, 7.9 Hz, 1H, H7), 6.97 (ddd, J = 1.2, 7.5, 7.7 Hz, 1H, H8), 6.87 (dd, J = 1.3, 7.5 Hz, 1H, H9), 5.06 (s, 2H, N–CH2–O), 4.63 (t, J = 5.1 Hz, 1H, OH), 3.53–3.46 (m, 4H, 2 × –CH2-), 3.30 (s, 3H, CH3). 13C NMR (151 MHz, DMSO‑d 6): δ 153.90, 153.78, 142.57, 128.26, 126.67, 126.30, 124.05, 123.52, 115.00, 114.99, 77.33, 70.40, 59.92, 28.37. HRMS (ESI) m/z: calcd for C14H16N3O4 + [M+H]+: 290.1135; found 290.1119.
4.1.10. 1-[4-(4,4′-Dimethoxytrityloxy)-2-oxabutyl]-5-bromouracil (18)
Compound 13 (80 mg, 0.3 mmol) was co-evaporated with anhydrous pyridine (5 mL), then dissolved in anhydrous pyridine (5 mL) and 4,4′-dimethoxytrityl chloride (120 mg, 0.36 mmol) was added in one portion at rt. After 4 h at rt the reaction was stopped by the addition of 5% aqueous NaHCO3 solution (10 mL) and the organics were extracted with DCM (2 × 15 mL). The combined organic layers were dried over Na2SO4, filtered, concentrated and co-evaporated with toluene (3 × 10 mL). Purification was performed by silica gel column chromatography on silica gel (0–0.5% MeOH in CH2Cl2 with 0.1% TEA) yielding 17 (129 mg, 0.23 mmol, 76%) as yellowish foam. 1H NMR (600 MHz, DMSO‑d 6): δ 11.84 (br s, 1H, NH), 8.32 (s, 1H, H6), 7.38–7.35 (m, 2H, H DMTr), 7.31–7.27 (m, 2H, H DMTr), 7.25–7.19 (m, 5H, H DMTr), 6.89–6.85 (m, 4H, H DMTr), 5.16 (s, 2H, N–CH2–O), 3.73 (s, 6H, 2 × OCH3), 3.71–3.67 (m, 2H, –CH2-), 3.06–3.02 (m, 2H, –CH2-). 13C NMR (151 MHz, DMSO‑d 6): δ 159.45, 157.91 (2C), 150.42, 144.77, 144.33, 135.56 (2C), 129.48 (4C), 127.67 (2C), 127.53 (2C), 126.48, 113.04 (4C), 95.51, 85.19, 76.84, 68.36, 62.48, 54.89 (2C). HRMS (ESI) m/z: calcd for C28H28BrN2O6 + [M+H]+: 567.1125; found 567.1105.
4.1.11. 3-[4-(4,4′-Dimethoxytrityloxy)-2-oxabutyl]-1,3-diaza-2-oxophenoxazine (19)
This derivative (yield 71%, yellowish foam) was prepared starting from 16a as described for the preparation of 18. 1H NMR (600 MHz, CD2Cl2): δ 7.78–7.73 (m, 1H, H6), 7.48–7.43 (m, 2H, H DMTr), 7.36–7.31 (m, 4H, H DMTr), 7.31–7.26 (m, 2H, H DMTr), 7.22–7.18 (m, 1H, H DMTr), 6.98 (s, 1H), 6.90–6.85 (m, 2H, H7/H8), 6.85–6.80 (m, 4H), 6.76–6.72 (m, 1H, H9), 5.15 (s, 2H, N–CH2–O), 3.78–3.72 (m, 8H, 2 × OCH3/–CH2–), 3.26–3.20 (m, 2H). 13C NMR (151 MHz, CD2Cl2): δ 159.02 (2C), 155.79, 154.89, 145.47, 142.93, 136.57 (2C), 130.39 (4C), 128.59, 128.56 (2C), 128.20 (2C), 127.12, 127.09, 124.80, 124.21, 124.12, 119.06, 115.36, 113.47 (4C), 86.47, 78.24, 69.34, 63.34, 55.58 (2C). HRMS (ESI) m/z: calcd for C34H30N3O6 − [M − H]-: 576.2140; found 576.2116.
4.1.12. 3-[4-(4,4′-Dimethoxytrityloxy)-2-oxabutyl]-10-methyl-1,3-diaza-2-oxophenoxazine (20)
This derivative (yield 73%, yellowish foam) was prepared starting from 17 as described for the preparation of 18. 1H NMR (600 MHz, DMSO‑d 6): δ 7.55 (s, 1H, H4), 7.39–7.35 (m, 2H, H DMTr), 7.30–7.25 (m, 2H, H DMTr), 7.25–7.21 (m, 4H, H DMTr), 7.20–7.16 (m, 1H, H DMTr), 7.07 (dd, J = 1.3, 8.0 Hz, 1H, H6), 7.01 (ddd, J = 1.5, 7.8, 8.0 Hz, 1H, H7), 6.97 (ddd, J = 1.3, 7.6, 7.8 Hz, 1H, H8), 6.88–6.84 (m, 5H, H DMTr/H9), 5.12 (s, 2H, N–CH2–O), 3.71–3.66 (m, 8H, 2 × OCH3/–CH2–), 3.32 (s, 3H, CH3), 3.05–3.02 (m, 2H, –CH2-). 13C NMR (151 MHz, DMSO‑d 6): δ 157.89 (2C), 153.88, 153.84, 144.71, 142.56, 135.61 (2C), 129.48 (4C), 128.21, 127.63 (2C), 127.59 (2C), 126.78, 126.46, 126.20, 124.09, 123.54, 115.02, 114.99, 112.99 (4C), 85.19, 77.30, 68.06, 62.50, 54.85 (2C), 28.38.HRMS (ESI) m/z: calcd for C35H34N3O6 + [M+H]+: 592.2442; found 592.2421.
4.1.13. 1-(4′-acetoxy-2′-cyclopenten-1′-yl)-5-bromouracil (22)
This derivative (yield 92%, off-white solid) was prepared starting from 1-(4′-hydroxy-2′-cyclopenten-1′-yl)-5-bromouracil 21 as described for the preparation of 14. 1H NMR (600 MHz, CDCl3): δ 9.15 (br s, 1H, NH), 7.57 (s, 1H, H6), 6.34 (dd, J = 2.0, 5.5 Hz, 1H, H2′), 6.01–5.96 (m, 1H, H3′), 5.71–5.62 (m, 2H, H1’/H4′), 2.97 (ddd, J = 7.7, 8.3, 15.8 Hz, 1H, H5′a), 2.10 (s, 3H, CH3), 1.68 (ddd, J = 3.2, 3.4, 15.8 Hz, 1H, H5′b). 13C NMR (151 MHz, DMSO‑d 6): δ 170.11, 158.76, 150.13, 140.38, 137.07, 133.75, 97.14, 76.56, 58.98, 37.38, 20.98.
4.1.14. 3-(4′-hydroxy-2′-cyclopenten-1′-yl)-1,3-diaza-2-oxophenoxazine (23)
This derivative (yield 19%, light brown solid) was prepared starting from 22 as described for the preparation of 16a. 1H NMR (600 MHz, CDCl3-CD3OD = 4:1 (v/v)): δ 6.99 (s, 1H, H4), 6.77–6.68 (m, 2H, H6/H7), 6.77–6.54 (m, 2H, H8/H9), 6.11–6.02 (m, 1H, H2′), 5.71–5.62 (m, 1H, H3′), 5.44–5.35 (m, 1H, H1′), 4.70–4.61 (m, 1H, H4′), 2.82–2.68 (m, 1H, H5′a), 1.40 (ddd, J = 3.9, 3.9, 14.6 Hz, 1H, H5′b). 13C NMR (151 MHz, CDCl3-CD3OD = 4:1 (v/v)): δ 157.44, 156.20, 144.75, 141.76, 133.78, 128.48, 126.67, 126.04, 125.61, 118.65, 117.64, 76.35, 72.50, 62.66, 42.38. HRMS (ESI) m/z: calcd for С15Н13N3O3 + [М+H]+: 284.1030; found 284.1026.
4.2. Biology
4.2.1. Cells and viruses
Cell lines used in the Rega Institute for Medical Research: human embryonic lung fibroblasts (HEL299), human epithelial (Hep2), green monkey kidney (Vero) and Madin-Darby canine kidney (MDCK) – were obtained from American Type Culture Collection (ATCC). Cell lines used in FSBSI “Chumakov FSC R&D IBP RAS”: porcine embryo kidney (PEK), green monkey kidney (Vero) and rhabdomyosarcoma (RD) – originated from the Center’s own collection or National Institute for Biological Standards and Control (NIBSC). Viruses and strains, used in the present work, are specified in Table S1.
4.2.2. Methods
4.2.2.1. Cell toxicity evaluation in HEL, Vero, Hep2 and MDCK cells (Rega Institute for Medical Research)
Cells were seeded at a rate of 5 × 103 cells/well into 96-well plates and allowed to proliferate for 24 h. Then, medium containing different concentrations of the test compounds starting at 100 μM was added. After 3 days of incubation at 37 °C, the cell number was determined with a Beckman Coulter counter. The cytostatic concentrations or CC50 (compound concentration required reducing cell proliferation by 50%) were estimated from graphic plots of the number of cells (percentage of control) as a function of the concentration of the test compounds.
4.2.2.2. Cell viability assay in PEK, RD and Vero cells (FSBSI “Chumakov FSC R&D IBP RAS”)
Two-fold dilutions of studied compounds and DMSO as a negative control were prepared in cultural medium (FSBSI “Chumakov FSC R&D IBP RAS”, Russia) appropriate for a chosen cell line. Cell suspensions were added to the wells with compound dilutions and DMSO control (approx. 105 cells per well) in the appropriate cultural medium supplemented with 5% FBS (Invitrogen, South America). The final concentration series of eight dilutions started from 100 μM. After incubation at 36.5 °C in a CO2-incubator for 5 days cultural medium was substituted with resazurin solution (25 μg/mL). Cells were incubated at 36.5 °C in a CO2-incubator for 4 h. Then 20 μL of 10% SDS was added to stop the reaction. Fluorescence was measured with Promega GloMax-Multi Detection System 525 nm Ex and 580–640 nm Em. As additional controls we used the same series of cells treated with compounds and DMSO dilutions but without resazurin solution to subtract the background fluorescence; and a medium with resazurin solution to set up a minimal value of non-reduced resazurin. All experimental procedures were performed in two replicates. Statistical analysis and fluorescence curves were prepared with MS Excel 2013. The 50% cytotoxic concentration (CC50) was calculated (compound concentration required to induce cytopathic effect in 50% of the cells in monolayer).
4.2.2.3. Cytopathicity or plaque reduction test (Rega Institute for Medical Research)
Confluent cell cultures in 96-well plates were inoculated with 100 CCID50 of virus (CCID50 is a virus dose to infect 50% of the cell cultures) or with 20 plaque forming units (PFU). Following a 2 h adsorption period, viral inoculum was removed and the cell cultures were incubated in the presence of varying concentrations of the test compounds starting at 100 μM. Viral cytopathicity or plaque formation was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds. Antiviral activity was expressed as the EC50 (compound concentration required reducing virus-induced cytopathicity or viral plaque formation by 50%).
4.2.2.4. TBEV, OHFV, POWV and CHIKV plaque reduction test (FSBSI “Chumakov FSC R&D IBP RAS”)
Five-fold dilutions of studied compounds and DMSO as a control were prepared in cell culture medium (FSBSI “Chumakov FSC R&D IBP RAS”, Russia) and added to the virus (30–60 PFU/well) at final concentration series starting from 4 or 50 μM and incubated at 37 °C for 1 h. Then compound+virus mixtures were added to the cell monolayers (Vero for CHIKV, PEK for tick-borne flaviviruses) and incubated at 37 °C for 1 h for infectious virus adsorption. Then, each well was overlaid with 1 mL of 1.26% methylcellulose (Sigma) containing 2% FBS (Invitrogen, South America). After incubation at 37 °C for 4 (CHIKV) or 6 (tick-borne flaviviruses) days, the cells were fixed with 96% ethanol. Plaques were stained with 0.4% crystal violet and counted. EC50 were calculated according to the Reed-and-Muench method [25].
4.2.2.5. Enteroviruses cytopathic effect inhibition test (FSBSI “Chumakov FSC R&D IBP RAS”)
Cytopathic effect inhibition test against representatives of Enterovirus genus was performed as described previously [15]. In brief, eight 2-fold dilutions of stock solutions of the compounds in 4 replicates were prepared in 2 × EMEM (FSBSI “Chumakov FSC R&D IBP RAS”, Russia). Compound dilutions were mixed with equal volumes of the enterovirus suspension containing 100 CCID50 (50% tissue culture infectious dose). Control cells were treated with the same sequential concentrations of DMSO as in compound dilutions. After 1 h incubation at 36.5 °C the RD cell suspension (approx.105 cells per well) in 2 × EMEM containing 5% FBS (Invitrogen, South America) was added to experimental mixtures. A final concentration series started from approx. 100 μM. Each experiment contained virus dose titration in the inoculate to assure the acceptable dose-range. After a 5-day incubation at 37 °C, cytopathic effect (CPE) was visually accessed via microscope. EC50 values were calculated according to the Karber method [26].
4.2.2.6. SARS-CoV-2 cytopathic effect inhibition test (FSBSI “Chumakov FSC R&D IBP RAS”)
Eight 2-fold dilutions of 5 mM stock solutions of the compounds were prepared in DMEM (FSBSI “Chumakov FSC R&D IBP RAS”, Russia). Compound dilutions were mixed with equal volumes of the virus suspension containing 100 CCID50 per well. A final concentration series started from approx. 100 μM. After 1 h incubation at 37 °C, virus-compound mixes were added to the confluent Vero cell monolayers in 2 replicates. Control cells were treated with the same sequential concentrations of DMSO as in compound dilutions. After a 5-day incubation at 37 °C, cytopathic effect (CPE) was visually accessed via microscope. EC50 values were calculated according to the Karber method [26]. Experiment was repeated at least 2 times for each compound. Each experiment contained a positive control compound NHC and virus dose titration to assure the acceptable dose-range.
4.2.2.7. Virus yield reduction assay (Smorodintsev Research Institute of influenza)
The assay was performed as described before [27]. In brief, a series of 3-fold dilutions were prepared from DMSO solution of the compound (2 mg/mL), added to MDCK cell monolayer and incubated for 1 h in CO2-incubator at 37 °C. Then, the virus was added (MOI 1) and then incubated for 24 h in a CO2-incubator at 37 °C. At the end of the incubation period, a series of consecutive 10-fold dilutions in a supporting medium was prepared from culture supernatant and added to the cell monolayers incubated for 72 h in CO2-incubator at 37 °C. The virus titers were determined by hemagglutination reaction in U-bottom immunological 96-well plates with the equal volume of 1% suspension of chicken red blood cells in physiological solution. The virus titer was calculated using the Reed-and-Muench method [25] and expressed in 50% tissue infection doses (TID50) per 100 μL volume. The antiviral activity of the drug was estimated as EC50 – compound dilution decreasing virus titer by 50%.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Financial support from Ministry of Science and Higher Education of the Russian Federation [АААА-А20-120081790043-5, in vitro testing against SARS-CoV-2; 121031300275-3, in vitro testing against flaviviruses] and RFBR [project number 20-34-70143, chemical synthesis] is gratefully acknowledged. NHC was kindly provided by Dr. Liudmila Alexandrova (IMB RAS). 1-Adamantylmethyl 5-amino-4-(2-phenylethyl)isoxazole-3-carboxylate was kindly provided by Drs. Dmitry Vasilenko and Elena Averina (Dept. Chem., Lomonosov MSU)
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2021.113467.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.De Clercq E., Li G.D. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016;29:695–747. doi: 10.1128/CMR.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seley-Radtke K.L., Yates M.K. The evolution of nucleoside analogue antivirals: a review for chemists and non-chemists. Part 1: early structural modifications to the nucleoside scaffold. Antivir. Res. 2018;154:66–86. doi: 10.1016/j.antiviral.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yates M.K., Seley-Radtke K.L. The evolution of antiviral nucleoside analogues: a review for chemists and non-chemists. Part II: complex modifications to the nucleoside scaffold. Antivir. Res. 2019;162:5–21. doi: 10.1016/j.antiviral.2018.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bekerman E., Einav S. Combating emerging viral threats. Science. 2015;348:282–283. doi: 10.1126/science.aaa3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang M.L., Cao R.Y., Zhang L.K., Yang X.L., Liu J., Xu M.Y., Shi Z.L., Hu Z.H., Zhong W., Xiao G.F. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.King D.H. History, pharmacokinetics, and pharmacology of acyclovir. J. Am. Acad. Dermatol. 1988;18:176–179. doi: 10.1016/s0190-9622(88)70022-5. [DOI] [PubMed] [Google Scholar]
- 7.De Clercq E. Another ten stories in antiviral drug discovery (Part C): "old" and "new" antivirals, strategies, and perspectives. Med. Res. Rev. 2009;29:611–645. doi: 10.1002/med.20153. [DOI] [PubMed] [Google Scholar]
- 8.Matyugina E.S., Khandazhinskaya A.L., Kochetkov S.N. Carbocyclic nucleoside analogues: classification, target enzymes, mechanisms of action and synthesis. Russ. Chem. Rev. 2012;81:729–746. [Google Scholar]
- 9.Daluge S.M., Good S.S., Faletto M.B., Miller W.H., StClair M.H., Boone L.R., Tisdale M., Parry N.R., Reardon J.E., Dornsife R.E., Averett D.R., Krenitsky T.A. 1592U89, a novel carbocyclic nucleoside analog with potent, selective anti-human immunodeficiency virus activity. Antimicrob. Agents Chemother. 1997;41:1082–1093. doi: 10.1128/aac.41.5.1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bisacchi G.S., Chao S.T., Bachard C., Daris J.P., Innaimo S., Jacobs G.A., Kocy O., Lapointe P., Martel A., Merchant Z., Slusarchyk W.A., Sundeen J.E., Young M.G., Colonno R., Zahler R. BMS-200475, a novel carbocyclic 2’-deoxyguanosine analog with potent and selective anti-hepatitis B virus activity in vitro. Bioorg. Med. Chem. Lett. 1997;7:127–132. [Google Scholar]
- 11.Boryski J., Golankiewicz B., Declercq E. Synthesis and antiviral activity of 3-substituted derivatives of 3,9-Dihydro-9-Oxo-5h-Imidazo[1,2-a]Purines, tricyclic analogs of acyclovir and ganciclovir. J. Med. Chem. 1991;34:2380–2383. doi: 10.1021/jm00112a010. [DOI] [PubMed] [Google Scholar]
- 12.Golankiewicz B., Ostrowski T., Andrei G., Snoeck R., Declercq E. Tricyclic analogs of acyclovir and ganciclovir - influence of substituents in the heterocyclic moiety on the antiviral activity. J. Med. Chem. 1994;37:3187–3190. doi: 10.1021/jm00045a025. [DOI] [PubMed] [Google Scholar]
- 13.McGuigan C., Barucki H., Blewett S., Carangio A., Erichsen J.T., Andrei G., Snoeck R., De Clercq E., Balzarini J. Highly potent and selective inhibition of varicella-zoster virus by bicyclic furopyrimidine nucleosides bearing an aryl side chain. J. Med. Chem. 2000;43:4993–4997. doi: 10.1021/jm000210m. [DOI] [PubMed] [Google Scholar]
- 14.Balzarini J., McGuigan C. Bicyclic pyrimidine nucleoside analogues (BCNAs) as highly selective and potent inhibitors of varicella-zoster virus replication. J. Antimicrob. Chemother. 2002;50:5–9. doi: 10.1093/jac/dkf037. [DOI] [PubMed] [Google Scholar]
- 15.Kozlovskaya L.I., Golinets A.D., Eletskaya A.A., Orlov A.A., Palyulin V.A., Kochetkov S.N., Alexandrova L.A., Osolodkin D.I. Selective inhibition of Enterovirus A species members’ reproduction by furano[2,3-d]pyrimidine nucleosides revealed by antiviral activity profiling against (+)ssRNA viruses. ChemistrySelect. 2018;3:2321–2325. doi: 10.1002/slct.201703052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kozlovskaya L.I., Andrei G., Orlov A.A., Khvatov E.V., Koruchekov A.A., Belyaev E.S., Nikolaev E.N., Korshun V.A., Snoeck R., Osolodkin D.I., Matyugina E.S., Aralov A.V. Antiviral activity spectrum of phenoxazine nucleoside derivatives. Antivir. Res. 2019;163:117–124. doi: 10.1016/j.antiviral.2019.01.010. [DOI] [PubMed] [Google Scholar]
- 17.Deville-Bonne D., El Amri C., Meyer P., Chen Y., Agrofoglio L.A., Janine J. Human and viral nucleoside/nucleotide kinases involved in antiviral drug activation: structural and catalytic properties. Antivir. Res. 2010;86:101–120. doi: 10.1016/j.antiviral.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 18.Kozlovskaya L., Piniaeva A., Ignatyev G., Selivanov A., Shishova A., Kovpak A., Gordeychuk I., Ivin Y., Berestovskaya A., Prokhortchouk E., Protsenko D., Rychev M., Ishmukhametov A. Isolation and phylogenetic analysis of SARS-CoV-2 variants collected in Russia during the COVID-19 outbreak. Int. J. Infect. Dis. 2020;99:40–46. doi: 10.1016/j.ijid.2020.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pfletscher M., Mezger M., Giese M. On the impact of linking groups in hydrogen-bonded liquid crystals - a case study. Soft Matter. 2018;14:6214–6221. doi: 10.1039/c8sm00802g. [DOI] [PubMed] [Google Scholar]
- 20.Semaine W., Johar M., Tyrrell D.L.J., Kumar R., Agrawal B. Inhibition of hepatitis B virus (HBV) replication by pyrimidines bearing an acyclic moiety: effect on wild-type and mutant HBV. J. Med. Chem. 2006;49:2049–2054. doi: 10.1021/jm058271d. [DOI] [PubMed] [Google Scholar]
- 21.Matyugina E.S., Logashenko E.B., Zenkova M.A., Kochetkov S.N., Khandazhinskaya A.L. 5 ’-Norcarbocyclic analogues of furano[2,3-d]pyrimidine nucleosides. Heterocycl. Commun. 2015;21:259–262. [Google Scholar]
- 22.Sheahan T.P., Sims A.C., Zhou S.T., Graham R.L., Pruijssers A.J., Agostini M.L., Leist S.R., Schafer A., Dinnon K.H., Stevens L.J., Chappell J.D., Lu X.T., Hughes T.M., George A.S., Hill C.S., Montgomery S.A., Brown A.J., Bluemling G.R., Natchus M.G., Saindane M., Kolykhalov A.A., Painter G., Harcourt J., Tamin A., Thornburg N.J., Swanstrom R., Denison M.R., Baric R.S. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Transl. Med. 2020;12 doi: 10.1126/scitranslmed.abb5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Varizhuk A.M., Zatsepin T.S., Golovin A.V., Belyaev E.S., Kostyukevich Y.I., Dedkov V.G., Shipulin G.A., Shpakovski G.V., Aralov A.V. Synthesis of oligonucleotides containing novel G-clamp analogue with C8-tethered group in phenoxazine ring: implication to qPCR detection of the low-copy Kemerovo virus dsRNA. Bioorg. Med. Chem. 2017;25:3597–3605. doi: 10.1016/j.bmc.2017.03.062. [DOI] [PubMed] [Google Scholar]
- 24.Boncel S., Gondela A., Maczka M., Tuszkiewicz-Kuznik M., Grec P., Hefczyc B., Walczak K. Synthesis; 2011. Novel 5-(n-Alkylaminouracil) Acyclic Nucleosides; pp. 603–610. [Google Scholar]
- 25.Reed L.J., Muench H. A simple method of estimating fifty percent endpoints. Am. J. Epidemiol. 1938;27:493–497. [Google Scholar]
- 26.Karber G. Beitragzurkollecktiven behandlungpharmakologischer reihenversuche. Arch. Exptl Pathol Pharmakol. 1931;162:480–483. [Google Scholar]
- 27.Nikolayeva Y.V., Ulashchik E.A., Chekerda E.V., Galochkina A.V., Slesarchuk N.A., Chistov A.A., Nikitin T.D., Korshun V.A., Shmanai V.V., Ustinov A.V., Shtro A.A. 5-(Perylen-3-ylethynyl)uracil derivatives inhibit reproduction of respiratory viruses. Russ. J. Bioorg. Chem. 2020;46:315–320. doi: 10.1134/S1068162020030139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vasilenko Dmitry, AA, Dueva Evgenia, V, Kozlovskaya Liubov, I, Zefirov Nikolay, A, Grishin Yuri, K, Butov Gennady, M, Palyulin Vladimir, A, Kuznetsova Tamara, S, Karganova Galina, G, Zefirova Olga., N Tick-borne flavivirus reproduction inhibitors based on isoxazole core linked with adamantane. Bioorganic chemistry. 2019;87:629–637. doi: 10.1016/j.bioorg.2019.03.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.