

Abstract
Advances in Pd-catalysed cross-coupling reactions have facilitated the development of stereospecific variants enabling the use of configurationally stable, enantioenriched, main-group organometallic nucleophiles to form C(sp3)–C(sp2) bonds. Such stereospecific cross-coupling reactions constitute a powerful synthetic approach to attaining precise 3D control of molecular structure, allowing new stereogenic centres to be readily introduced into molecular architectures. Examples of stereospecific, Pd-catalysed cross-coupling reactions have been reported for isolable enantioenriched alkylboron, alkyltin, alkylgermanium and alkylsilicon nucleophiles. In these reactions, a single, dominant stereospecific pathway of transmetallation to palladium is required to effect efficient chirality transfer to the cross-coupled product. However, the potential for competing stereoretentive and stereoinvertive pathways of transmetallation complicates the stereochemical transfer in these processes and impedes the rational development of new stereospecific cross-coupling variants. In this Review, we describe the use of enantioenriched organometallic nucleophiles in stereospecific, Pd-catalysed cross-coupling reactions. We focus on systems involving well-defined, isolable, enantioenriched nucleophiles in which a clear stereochemical course of transmetallation is followed. Specific modes of electronic activation that influence the reactivity of alkylmetal nucleophiles are described and presented in the context of their impact on the stereochemical course of cross-coupling reactions. We expect that this Review will serve as a valuable resource to assist in deconvoluting the many considerations that potentially impact the stereochemical outcome of Pd-catalysed cross-coupling reactions.
Transition-metal-catalysed cross-coupling reactions have revolutionized the modern approach to organic synthesis1. In standard Pd-catalysed cross-coupling reactions, a main-group organometallic nucleophile undergoes a formal substitution reaction with a carbon electrophile in a Pd(0)–Pd(II) catalytic cycle. Over the past few decades, Pd-catalysed C(sp2)–C(sp2) bond-forming reactions have evolved into routine, reliable, high-yielding processes. More recently, use of C(sp3) cross-coupling partners has been investigated in C(sp3)–C(sp2) and C(sp3)–C(sp3) variants of Pd-catalysed bond-forming reactions2,3. When configurationally stable enantioenriched secondary (or tertiary) alkylmetal nucleophiles are employed in these reactions, new stereogenic centres may be generated by stereospecific cross-coupling reactions, provided that stereochemical integrity can be maintained throughout the course of the cross-coupling reaction4–6 (FIG. 1a). Such stereospecific cross-coupling reactions can facilitate the rapid preparation of enantioenriched libraries of chiral molecules from common enantioenriched organometallic building blocks with precise and predictable stereochemical control. In these reactions, configurational stability of the nucleophile is an important consideration, with isolable enantioenriched main-group organometallic nucleophiles being the most conducive to broad application in stereospecific cross-coupling processes. As such, alkylboron, alkyltin, alkylsilicon and alkylgermanium reagents, which tend to be isolable and configurationally stable under ambient conditions, are most useful in reactions in which transfer of chirality from a main-group organometallic nucleophile is desired7. Stereospecific cross-coupling reactions of enantioenriched alkylboron and alkyltin nucleophiles have emerged as particularly powerful strategies to access structurally diverse libraries of non-racemic molecules4. In this Review, we describe the use of enantioenriched organometallic nucleophiles in stereospecific Pd-catalysed cross-coupling reactions. We focus on systems involving well-defined, isolable, enantioenriched nucleophiles from which a clear stereochemical course of transmetallation is followed. Because net-stereoretentive or net-stereoinvertive transmetallation may be observed in these reactions (FIG. 1b), we categorize cross-coupling reactions by main-group metal and by the predominant pathway of transmetallation, which will facilitate uniformity in the mechanistic discussions. We note that stereospecific cross-coupling reactions of enantioenriched electrophiles3,8,9 and stereospecific 1,2-migrations of enantioenriched alkyl boronate nucleophiles10,11 constitute powerful complementary approaches to stereospecific synthesis. However, such approaches to chirality transfer are beyond the scope of this Review.
Fig. 1 |. Stereochemical considerations in the coupling of enantioenriched nucleophiles.

a | Catalytic cycle for Pd-catalysed cross-coupling reactions using configurationally stable enantioenriched nucleophiles4. b | Potential transmetallation pathways. c | Inverse relationship between configurational stability and nucleophilicity for main-group organometallic nucleophiles7. d | Relative rates of group transfer in Pd-catalysed coupling reactions and possible modes of activation.
The use of alkyl nucleophiles in transition-metal-catalysed cross-coupling reactions is particularly challenging owing to slow alkyl transmetallation to Pd aryl intermediates (1, FIG. 1a), as well as the propensity of alkyl ligands bearing β-hydrogens to undergo competitive β-hydride elimination from intermediates 2. In C(sp2)–C(sp2) cross-coupling reactions, both of these challenges are circumvented — the increased s character of the C–M bond results in faster transmetallation and the lack of β-hydrogens precludes the β-hydride elimination pathway. For cross-coupling reactions involving C(sp3)–M nucleophiles, alkyl transfer can be facilitated by inclusion of an sp2-hybridized α-substituent4. Thus, benzylic and allylic groups undergo more facile transmetallation than unactivated alkyl groups. For allyl–M nucleophiles, α-transmetallation and γ-transmetallation pathways are both possible, although γ-transmetallation is generally faster12. Because the rate of alkyl transfer also decreases with increased steric congestion, transmetallation of secondary and tertiary alkyl groups is particularly problematic. If alkyl transmetallation is successfully achieved, reversible formation of a metal hydride and an olefin through β-hydride elimination/reinsertion sequences commonly results in isomerization of the alkyl group and formation of an intractable mixture of isomeric products. Therefore, not only do challenges associated with slow transmetallation need to be overcome but reductive elimination must also outcompete potential β-hydride elimination. Efforts to develop efficient cross-coupling approaches to transfer chirality from enantioenriched organometallic nucleophiles are additionally hindered by the inverse relationship between the configurational stability and nucleophilicity of the C–M bond at the stereogenic centre of enantioenriched nucleophiles7 (FIG. 1c).Although increased covalent character of a C–M bond tends to coincide with greater configurational stability, increased covalent character also tends to coincide with reduced nucleophilicity. As a result of these combined challenges, the viability of stereospecific cross-coupling reactions commonly relies upon inclusion of a structural component on the nucleophile that can activate (or enhance) nucleophilicity. Commonly employed activation strategies in stereospecific cross-coupling reactions include incorporation of a C(sp2) α-substituent, an α-heteroatom and/or a remote coordinating group (FIG. 1d). Additionally, use of an acyl electrophile in place of an aryl electrophile renders the Pd(II) oxidative addition product 1 more electrophilic, which increases the rate of subsequent transmetallation and reductive elimination steps13,14. Throughout this Review, the specific activation modes required for each cross-coupling reaction will be emphasized and addressed.
Stereoretentive and stereoinvertive transmetallation pathways are both well precedented in Pd-catalysed cross-coupling reactions of enantiomerically enriched main-group organometallic nucleophiles1,4. The stereospecificity of such cross-coupling reactions is, thus, dictated by the dominant transmetallation mechanism of alkyl transfer to palladium. Stereoretentive alkyl transfer may occur via a closed or an open SE2 transition state, as depicted in FIG. 1b. Stereoinvertive transmetallation by a SE2 mechanism requires involvement of the minor bonding lobe of the C–M bond in the transmetallation pathway. For reactions in which stereoretentive and stereoinvertive transmetallation pathways are both energetically viable, partial stereoerosion can be expected, with the net stereochemical transfer determined by the energy difference between these competing pathways. In principle, loss of stereochemical integrity may also occur through the intermediacy of alkyl radicals or through β-hydride elimination/reinsertion sequences. Therefore, care must be exercised when attempting to rationalize the mechanistic underpinnings behind racemization events in these reactions. Because small energy differences may separate the transition states of the stereoretentive and stereoinvertive pathways, stereospecificity is often tied to subtle electronic, steric and ligand properties of the cross-coupling reaction. Throughout this Review, the interplay of these properties, and their cumulative mechanistic impact, will be described, when possible.
Alkylboron nucleophiles
Stereospecific cross-coupling reactions involving enantioenriched alkylboron nucleophiles have been most widely explored and developed. Therefore, stereospecific Suzuki reactions will be addressed independently from cross-coupling reactions involving other main-group organometallic nucleophiles. The majority of stereospecific Suzuki cross-coupling reactions involves the use of electronically activated alkylboron reagents, although the use of unactivated alkylboron nucleophiles has also been established.
Stereospecific coupling of unactivated, enantioenriched alkylborons.
The first investigations of the mechanism of alkylboron transmetallation to palladium were performed separately by Woerpel15 and Soderquist16, using the Whitesides protocol for probing the stereochemical course of organometallic reactions17. In these studies, vicinally deuterated (syn and anti) primary alkyl-9-BBN nucleophiles (3–6) were employed in Suzuki cross-coupling reactions, with the JH-H coupling constants of the vicinal protons of the coupling products used to assign the stereochemistry of the resulting products (FIG. 2a). These studies showed that transmetallation proceeds with retention of configuration for alkylboron nucleophiles 3–6. This work provided an important foundation on which further investigations of the stereochemical course of alkylboron transmetallation could be based, although several important questions and considerations remained. Most importantly, could mechanistic studies of primary alkylboron transmetallation be broadly extrapolated to the transmetallation of secondary alkylboron nucleophiles? Additionally, it was unclear whether results obtained using alkyl-9-BBN would parallel those obtained using less nucleophilic boronic acids and boronic esters. Finally, the requisite inclusion of a bulky β-substituent in the stereochemical probes could, in principle, bias the stereochemical course of the Suzuki reaction away from the stereoinvertive pathway.
Fig. 2 |. Use of unactivated, stereodefined organoboron nucleophiles in Pd-catalysed cross-coupling reactions.

a | Coupling of primary 9-BBN nucleophiles. Left: the Whitesides protocol using deuterium-labelled substrates to determine the stereochemical course of reaction by measuring vicinal proton–proton coupling constants17. Centre and right: examples showing that coupling in these cases occurs with retention of stereochemistry15,16. b | By contrast, coupling of secondary trifluoroborate nucleophiles was initially shown to occur through a stereoinvertive pathway and, later, that the preferred pathway was influenced by the electronic properties of the supporting phosphine ligand and the aryl electrophile18,23. c | Burke’s coupling of N-2-benzyloxycyclopentyl-iminodiacetic acid (BIDA) boronates occurs with near-perfect stereoretention26. d | Substrate steric effects can override previously determined patterns, as shown by Molander and Dreher28. Part b is adapted with permission from reF.23, AAAS.
Biscoe reported the first example of a Suzuki cross-coupling reaction using unactivated, enantioenriched secondary alkylboron nucleophiles18 (FIG. 2b). This study extended the work of Woerpel and Soderquist, who had focused on the use of primary alkylboron nucleophiles, to cross-coupling reactions of secondary alkylboron nucleophiles. Using PtBu3-ligated precatalyst 7 (REFS19–21), Biscoe showed that enantioenriched secondary alkyltrifluoroborates undergo efficient Pd-catalysed cross-coupling reactions with aryl chlorides and that transmetallation of the unactivated secondary alkylboron nucleophile proceeds mainly through a stereoinvertive pathway. This observation was inconsistent with the stereoretentive transmetallation pathway proposed by Woerpel and Soderquist for primary alkyl-9-BBN nucleophiles15,16 and suggested that factors determining the dominant pathway of transmetallation are significantly more complex than previously considered. In Biscoe’s study, secondary alkyltrifluoroborates were employed owing to their increased stability relative to secondary alkylboronic acids, alongside their ability to generate the corresponding active alkylboronic acid in situ under the reaction conditions22. Biscoe and Sigman subsequently undertook a more detailed examination of the aspects of the Suzuki cross-coupling reaction that control the preferred transmetallation pathway23. They observed that selectivity for stereoinversion varied directly with the electronic properties of the aryl electrophile. Electron-rich aryl chlorides showed greater preference for the stereoinvertive pathway than electron-deficient aryl chlorides — a trend paralleling that predicted from Hammett σ-values. These data suggested that subtle electronic effects could influence the mechanism of transmetallation. A screen of phosphine ligands then showed that a net-stereoretentive coupling could be achieved using alternative supporting ligands, with no obvious correlation existing between the steric properties of the ligands and the stereochemical outcomes in these reactions. Because the ligand effects were difficult to deconvolute, predictive statistical models were employed using a variety of molecular descriptors of phosphine ligands24. Using this strategy, Biscoe and Sigman designed new phosphine ligands 8 and 9, which selectively promoted the stereoretentive pathway while suppressing the β-hydride elimination pathway. An improved ligand (10) for the stereoinvertive process was also found during this process25. Thus, a stereodivergent Suzuki cross-coupling reaction was achieved in which both enantiomers of the cross-coupling product could be separately generated in up to 99% es from a single-enantiomer alkylboron nucleophile by employing the appropriate phosphine ligand. Multivariate-linear-regression analysis of stereochemical outputs obtained using 20 different phosphine ligands in a model cross-coupling reaction showed that enantioselectivity could be predicted using two ligand parameters: the average energy of the P–C antibonding orbitals, which is representative of π-backbonding ability (promotes stereoretentive transmetallation), and the energy of the valence lone-pair orbital of phosphorus, which is representative of σ-donation ability (promotes stereoinvertive transmetallation). Therefore, the stereochemical outcome of transmetallation for unactivated alkylboron nucleophiles is prominently tied to the electronic properties of the supporting phosphine ligand. Biscoe and Sigman propose that π-backbonding to the phosphine ligand enhances preference for the stereoretentive pathway through stabilization of the hydroxideligated palladium intermediate involved in the closed, stereoretentive transition state. Conversely, strong σ-donation from the phosphine ligand is proposed to stabilize a two-coordinate, cationic palladium complex that foregoes the stereoretentive pathway in favour of the stereoinvertive transmetallation pathway.
Burke has reported an alternative approach to stereoretentive Suzuki cross-coupling reactions of unactivated alkylboron nucleophiles26 (FIG. 2c). This strategy builds upon conditions originally employed by Crudden in which inclusion of superstoichiometric Ag2O promotes stereospecific cross-coupling reactions using enantioenriched benzylic boronic esters27. Although the precise role of Ag2O remains unclear, it is suspected that Ag2O plays an essential role in facilitating transmetallation. In Burke’s study, the use of bulky triarylphosphine ligands greatly enhanced preference of the stereoretentive transmetallation pathway when used alongside enantioenriched secondary alkylboronic acids, which were generated by hydrolysis of their corresponding N-2-benzyloxycyclopentyl-iminodiacetic acid (BIDA) boronates. Ligand 11 is particularly effective at supporting stereoretentive transmetallation, with near-perfect selectivity generally being achieved. It is proposed that the bulky triarylphosphine on palladium selectively blocks the stereoinvertive pathway of transmetallation, while still allowing stereoretentive transmetallation via a closed, four-membered transition state within the basal plane of the PdII complex. This logic suggests that steric properties of phosphine ligands could be tuned to selectively promote transmetallation. In the stereodivergent reactions developed by Biscoe and Sigman, the stereochemical outcome of transmetallation is intimately tied to the electronic properties of the ligand, while the bulkiness of the supporting phosphines is necessary to suppress competing β-hydride elimination pathways that can result in alkyl isomerization and erosion of enantiospecificity. The ability of the bulky electron-deficient ligand 11 to promote the stereoretentive transmetallation pathway is consistent with the model for electronically controlled transmetallation proposed by Biscoe and Sigman. The Burke study additionally suggests that steric properties may be an important element of ligand design for reinforcing a specific transmetallation pathway — although it can be difficult to deconvolute the stereochemical impact of steric shielding from the potential impact of suppressed β-hydride elimination.
Studies by Molander and Dreher have shown that trans-2-methylcylcohexyl trifluoroborate (12) undergoes transmetallation through a stereoretentive pathway using PtBu3 as a supporting ligand28 (FIG. 2d). This result seemingly contradicts the study conducted by Biscoe and Sigman, in which the strong σ-donating property of PtBu3 was proposed to promote stereoinvertive transmetallation to palladium18,23. However, the work of Molander and Dreher highlights the important influence of substrate steric effects on the net stereochemical course of alkylboron transmetallation. The studies by Biscoe and Sigman clearly indicate that stereoinvertive and stereoretentive pathways of transmetallation are both viable using unactivated secondary alkylboron nucleophiles. As the electronic properties of the supporting ligand are able to dictate the dominant stereochemical pathway, it is likely that substrate steric effects can, likewise, direct transmetallation. For transmetallation to be under steric control, the steric effects of the substrate must transcend the electronic effects of the ligands. This appears to be the case for substrate 12. The stereoinvertive pathway of transmetallation from major conformer 13a is likely impeded by the axial hydrogen atoms, and stereoinvertive transmetallation from minor conformation 13b is likely impeded by the axial methyl group located anti to the C–B bond. In principle, a similar steric influence to the latter could potentially bias the pathway of transmetallation for the primary alkyl-9-BBN nucleophiles (3–6) that were originally used as stereochemical probes by Woerpel and Soderquist15,16.
To facilitate deconvolution of potentially interlaced influences on the pathway of transmetallation, all examples in FIG. 2 involve alkylboron nucleophiles without electronic perturbations, which enables examination of stereochemical influences resulting from stepwise modification of ligand and substrate structure. The studies depicted in FIG. 2 convincingly demonstrate that (i) steric properties of ligands, (ii) electronic properties of ligands, (iii) electronic properties of the aryl electrophile and (iv) steric properties of the alkylboron nucleophile must all be considered when attempting to predict the net stereochemical course of an alkyl Suzuki reaction. Through investigations of these systems, reasonable, albeit rudimentary, mechanistic models can be proposed to rationalize changes in transmetallation mechanisms for alkylboron nucleophiles lacking electronic perturbations.
Stereoretentive coupling of activated, enantioenriched alkylborons.
Certain structural modifications of an alkylboron nucleophile can render the alkyl unit more activated towards transmetallation to palladium. These structural modifications are detailed in FIG. 1b. Cross-coupling reactions involving cyclopropylboron reagents constitute the first examples of Suzuki reactions using activated alkylboron nucleophiles. Cyclopropyl nucleophiles undergo particularly facile transmetallation as a result of cyclopropyl ring strain, which enhances the s character of the C–M bond. Accordingly, cyclopropyl nucleophiles behave more like C(sp2) nucleophiles than like C(sp3) nucleophiles. Several research groups have independently contributed to studies of Suzuki reactions using cyclopropylboron nucleophiles29 (FIG. 3, entries 1 and 2). Use of the corresponding boronic acids30–32, boronate esters33 and trifluoroborates34 have been demonstrated in such reactions. In all instances, stereoretentive transmetallation is exclusively observed. High stereospecificity is observed in these reactions for cis-substituted and trans-substituted cyclopropylboron derivatives. Additional activation imparted by the use of an acyl electrophile does not alter the observed mechanism35. These results suggest that the strained transition state required for cyclopropyl transmetallation by a stereoinvertive mechanism may involve too high an activation barrier to successfully compete with the stereoretentive pathway. Even the additional steric effects that arise from the presence of cis-substituents fail to alter the stereoretentive mechanistic course of this reaction34.
Fig. 3 |. Examples of electronically activated alkylboron nucleophiles in net-stereoretentive suzuki cross-coupling reactions27–45.
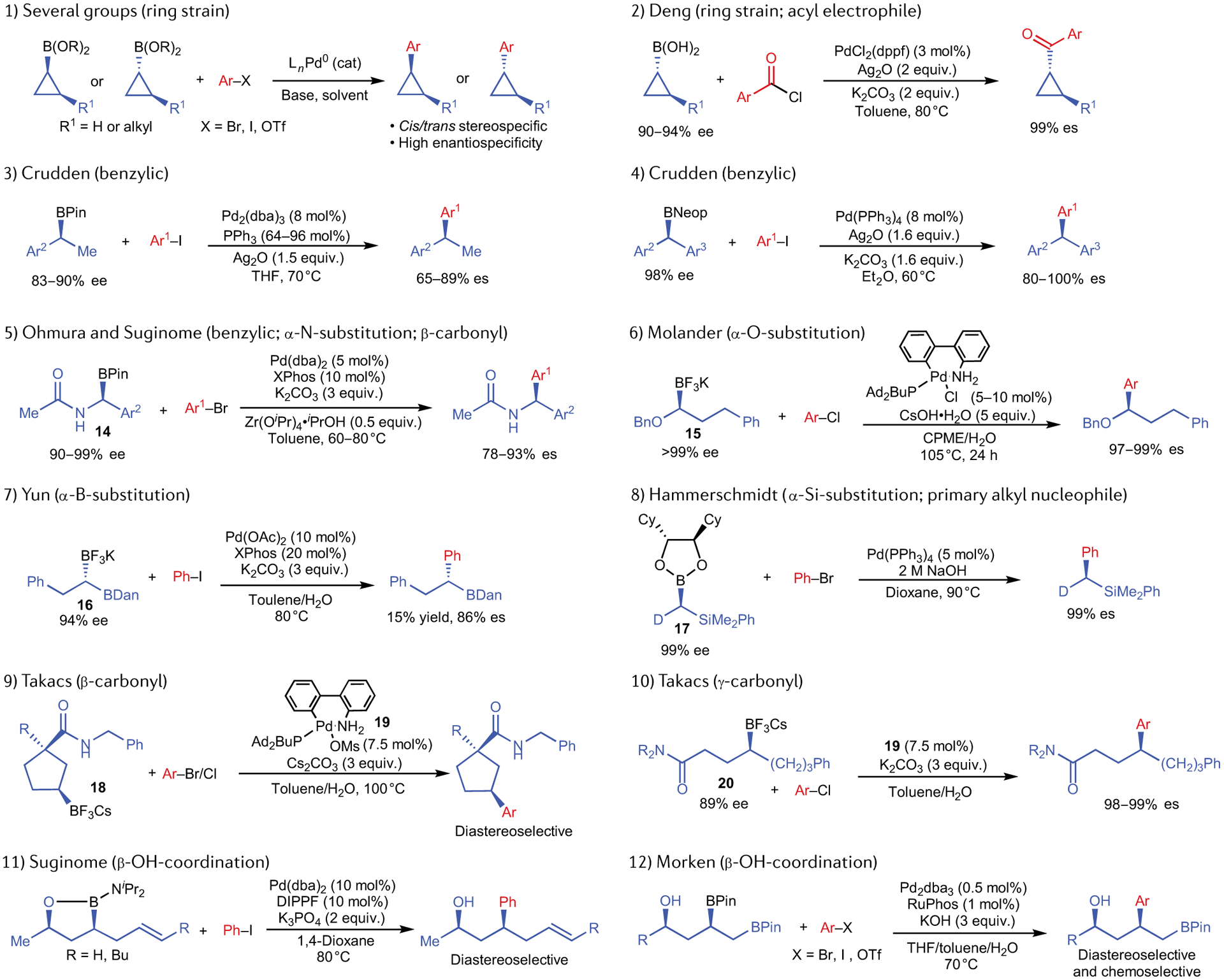
The substrates feature a variety of α- and β-substituents and several different Pd catalysts have been used.
Crudden demonstrated the first stereospecific Pd-catalysed Suzuki cross-coupling reaction that tolerated use of non-cyclopropyl secondary alkylboron nucleophiles27,36 (FIG. 3, entry 3). Using enantioenriched secondary benzylic organoboronic esters alongside catalytic Pd(PPh3)4 and in the presence of additional exogenous PPh3, stereoretentive transmetallation of the benzylic organoboron nucleophile was observed in cross-coupling reactions with aryl iodides. The use of a stoichiometric Ag(I) additive is key to the success of these reactions. Thus, it is possible that a cationic Pd(II) complex intermediate is required to effect efficient transmetallation of the secondary benzylic organoboronic ester. Using modified conditions in which neopentyl glycol boronic esters were employed in place of pinacol boronic esters, Crudden extended this chemistry to stereospecific coupling reactions of enantioenriched gem-diarylmethine boronates, which enable the preparation of highly enantioenriched triarylmethane derivatives37 (FIG. 3, entry 4). Transmetallation of the doubly benzylic gem-diarylmethine boronates again occurs with net retention of absolute stereochemistry in these reactions. Suginome and Ohmura demonstrated that α-nitrogen and benzyl substitution at the stereogenic centre, as well as the presence of a β-coordinating group, could be leveraged to achieve stereospecific cross-coupling reactions of enantioenriched α-(acylamino)benzylboronic ester nucleophiles (14)38. Using a catalytic system comprised of Pd/XPhos in the presence of Zr(OiPr)4, high selectivity for stereoretentive transmetallation can be achieved (FIG. 3, entry 5). It is noteworthy that net-stereoinvertive transmetallation can also be achieved in the absence of the Lewis-acid additive. Thus, additive-controlled stereodivergence was realized for this reaction. The Lewis acid is proposed to bind the oxygen atom of the amide, thus, inhibiting coordination of the amide to boron and facilitating formation of the four-membered closed transition state that results in stereoretentive transmetallation (see FIG. 1b). Molander has reported a stereoretentive Pd-catalysed cross-coupling reaction using enantioenriched 1-(benzyloxy)alkyltrifluoroborates (15)39 (FIG. 3, entry 6). No cross-coupling product was observed when the benzyl protecting group was replaced with a methyl group. Therefore, the benzyl group of 15 either plays a direct role in the transmetallation process or provides important stabilization of the Pd(II) intermediate following transmetallation. Yun has developed an asymmetric Cu-catalysed hydroboration reaction of borylalkenes, which provides access to enantioenriched 1,1-diborylated compounds (16)40. The α-boronyl-substituted stereogenic centre of 16 undergoes chemoselective and enantiospecific transmetallation with net stereoretention of absolute configuration when used in a Pd-catalysed Suzuki reaction, although the cross-coupling product forms only in low yield (FIG. 3, entry 7). Hammerschmidt prepared an enantioenriched, deuterated, primary α-silyl organoboron nucleophile (17) that showed high preference for stereoretentive transmetallation to palladium41 (FIG. 3, entry 8). The stereoretentive course of primary alkylboron transmetallation is consistent with the observations of Woerpel15 and Soderquist16, who have reported stereoretentive transmetallation using unactivated primary alkylboron nucleophiles (see FIG. 2a). Takacs has reported two examples of Pd-catalysed Suzuki cross-coupling reactions in which the presence of a remote carbonyl group appears to promote a stereoretentive alkylboron transmetallation pathway42,43 (FIG. 3, entries 9 and 10). Intramolecular coordination of the carbonyl and boron has previously been proposed to promote stereoinvertive transmetallation4. For 18, it is plausible that the rigid conformation of the five-membered ring impedes such intramolecular coordination and, thereby, provides access to a viable stereoretentive transmetallation pathway. Similarly, the γ-carbonyl group of 20 may be less disposed towards intramolecular coordination than β-carbonyl groups, the latter being present in nearly all examples of stereoinvertive alkylboron transmetallation. Alternatively, the mechanism of transmetallation may be entirely under ligand control. In both examples reported by Takacs, use of Ad2PBu as the supporting phosphine ligand results in the observation of a net-stereoretentive reaction pathway. This is consistent with Molander’s observation of net stereoretention when using Ad2PBu in entry 6 of FIG. 3, although the Molander system involves a OBn-substituted stereocentre, which may impart an element of electronic control to the transmetallation mechanism. Suginome44 (FIG. 3, entry 11) and Morken45 (FIG. 3, entry 12) have independently reported the ability of β-hydroxy (that is, γ-oxygenated) secondary alkylboronates to undergo transmetallation with net retention of stereochemistry. In both of these examples, γ-oxygenation is required to promote selective transmetallation of the secondary alkyl group, which enables diastereoselective cross-coupling reactions to be achieved. Activation imparted by the β-hydroxy group even enables the chemoselective transmetallation and coupling of a secondary C–B bond in the presence of a primary C–B bond (FIG. 3, entry 12).
The powerful activation imparted by α-C(sp2) substitution of main-group alkylmetal nucleophiles has been harnessed by several researchers to develop stereospecific Pd-catalysed cross-coupling reactions of enantioenriched allylic and propargylic organoboron species (FIG. 4a). In principle, transmetallation of allylic and propargylic nucleophiles may occur through an initial σ-transmetallation or γ-transmetallation mechanism (FIG. 4b). Organ46 and Buchwald47 have demonstrated that selectivity for σ-transmetallation or γ-transmetallation is greatly influenced by the choice of supporting ligand. The resulting allylpalladium species formed in each mechanism can also potentially interconvert through a common π-allyl complex. However, based on the regioselectivity of the coupling reactions, a mechanism involving γ-transmetallation of the allyl or propargyl unit via a closed, six-membered cyclic transition state is most commonly proposed for these reactions. In the case of allyl transfer, E/Z stereochemistry of the resulting olefin is dictated by a minimization of A1,3 strain during transmetallation. Aggarwal reported the first example of a Pd-catalysed cross-coupling reaction involving stereochemical transfer from an enantioenriched allylic or propargylic organoboron nucleophile48 (FIG. 4a, entry 1). Using conditions developed by Crudden for stereospecific benzylic Suzuki cross-coupling reactions (see FIG. 3, entry 3), Aggarwal showed that enantioenriched tertiary propargylic boronic esters undergo stereospecific γ-transmetallation in Pd-catalysed Suzuki reactions to afford highly enantioenriched tetrasubstituted allenes. In collaboration, Crudden and Aggarwal extended these reaction conditions to stereospecific cross-coupling reactions of enantioenriched allylboronic esters49 (FIG. 4a, entry 2). High preference for γ-transmetallation was demonstrated using a regio-specifically deuterated allylboron nucleophile. Correlation of the absolute stereochemistry with the olefin stereochemistry suggests that transmetallation occurred through a syn-SE′ mechanism. Using a catalytic system comprised of Pd/RuPhos, Morken developed an alternative procedure for stereospecific coupling reactions of enantioenriched allylboronic esters50 (FIG. 4a, entry 3). This reaction is highly γ-selective for a broad scope of aryl and heteroaryl electrophiles and enables access to enantioenriched quaternary centres. Hall has developed ligand-controlled, regiodivergent, enantiospecific Suzuki coupling reactions of enantioenriched pyranyl and piperidyl allylic boronates 21 that enable access to enantioenriched 2-substituted and 4-substituted dihydropyrans and dehydropiperidine derivatives51 (FIG. 4a, entry 4). 2-Substituted products are preferentially generated from 21 using XPhos52 as a supporting ligand, which appears to promote γ-selective transmetallation of both pyranyl and piperidyl allylic boronates through a syn-SE′ mechanism. By contrast, selective formation of 4-substituted products is achieved using (4-CF3-C6H4)3P (22) for pyranyl boronates and the N-heterocyclic carbene ligand IPr for piperidyl boronates. Hall proposes that phosphine ligand 22 promotes an initial γ-selective transmetallation by a syn-SE′ mechanism, followed by rapid σ–π equilibration via a π-allyl intermediate complex to generate the thermodynamically favoured O-conjugated olefin, whereas the N-heterocyclic carbene ligand is proposed to promote σ-selective transmetallation through a direct syn-SE mechanism. Because bulky, strongly σ-donating ligands such as XPhos and IPr are expected to promote rapid reductive elimination while slowing η1–η3 bond slippage, isomerization through a π-allyl intermediate complex is most likely to occur with an electron-deficient phosphine such as 22, which is consistent with the observed ligand-controlled regiodivergence. Subsequently, Hall extended this regiodivergent process to cross-coupling reactions of enantioenriched 2-ethoxydihydropyranyl boronates (24)53 (FIG. 4a, entry 6). Regiospecificity and stereospecificity in this reaction is not biased by the 1,3-syn substitution pattern of 24. Lastly, Aggarwal has recently reported a Pd-catalysed arylation reaction that involves the stereospecific γ-transmetallation of dearomatized enantioenriched alkylboron reagent 23 (REF.54) (FIG. 4a, entry 5). Use of the analogous benzylic boronic ester under identical reaction conditions failed to generate the benzyl cross-coupling product. Therefore, direct γ-transmetallation from 23 is required to access the benzyl-Pd(II) species of the catalytic cycle. For all transformations depicted in FIG. 4a, selective transmetallation through a syn-SE′ mechanism has been proposed. In principle, an anti-SE′ mechanism of transmetallation is also possible via an open transition state55.
Fig. 4 |. Propargylic and allylic organoboron nucleophiles in couplings.

a | Couplings of propargylic and allylic boron nucleophiles often give isomerized products but generally occur with net retention of stereochemistry48–55. b | Direct γ-transmetallation or an α-transmetallation followed by isomerization via a π-allyl intermediate both lead to the same isomerized product46,47.
Stereoinvertive coupling of activated enantioenriched alkylborons.
In 2010, Suginome and Ohmura56 and Molander57 independently reported the first examples of stereoinvertive Pd-catalysed cross-coupling reactions using enantioenriched alkylboron nucleophiles (FIG. 5a, entry 1). In the process described by Suginome and Ohmura, α-(acylamino)benzylboronic ester nucleophiles (25) were employed, which undergo facile transmetallation owing to the presence of α-nitrogen and benzyl substitution at the stereogenic centre, as well as the presence of a β-coordinating group56. Selectivity for stereoinvertive transmetallation was optimal using XPhos as the supporting ligand and boronic esters bearing a bulky pivaloyl group, although inclusion of PhOH as an additive was later shown to improve the enantiospecificity for boronic esters bearing an acetyl group38.More recently, Suginome and Ohmura have extended the substrate scope of this process to non-benzylic α-(acylamino)alkylboron nucleophiles (26) using alternative reaction conditions that include PCy2Ph as the supporting ligand and elevated temperatures58 (FIG. 5a, entry 2). In these reactions, selectivity for the stereoinvertive pathway is sensitive to the steric environment of the stereogenic nucleophilic centre. The presence of larger R groups decreases the preference for stereoinvertive transmetallation in this system, which is consistent with trends observed by Biscoe and Sigman using unactivated alkylboron nucleophiles23. In the process described by Molander, enantioenriched alkyl β-trifluoroboratoamides (27) were employed, which only contain a β-coordinating group as the mode of activation57 (FIG. 5a, entry 3). Using XPhos as the supporting ligand, Molander demonstrated nearly perfect selectivity for the stereoinvertive transmetallation pathway. The ligand XPhos was also employed by Hall in stereoinvertive Pd-catalysed cross-coupling reactions of enantioenriched 3,3-diboronyl carboxyesters (28) (REF.59) (FIG. 5a, entry 4). Use of 3,3-diboronyl carboxyesters (28) comprised of a trifluoroborate unit and a 1,8-diaminonaphthalenyl (dan) boron unit enables chemoselective and enantiospecific transmetallation at the stereogenic centre. In this process, facile transmetallation at the trifluoroborate unit is promoted through the cooperative effects of β-carbonyl coordination and α-boronyl substitution at the C–BF3K bond60. The stereochemical outcome of this reaction is particularly noteworthy because Yun has demonstrated that an analogous system without the β-carbonyl group preferentially undergoes stereoretentive transmetallation40 (see FIG. 3, entry 7). Two stereochemical models, both involving intramolecular carbonyl–boron coordination, have been proposed to explain the overwhelming preference for stereoinvertive transmetallation in these systems (FIG. 5b). The first model invokes α-deprotonation of the alkylboron nucleophile, which should enhance the Lewis basicity of the carbonyl relative to the non-deprotonated variant38,56. The second model involves a dative interaction of the carbonyl and boron, without deprotonation at the α-carbon57. Both models result in the formation of tetracoordinate boron, precluding formation of the closed four-membered transition state that gives access to the stereoretentive transmetallation pathway (see FIG. 1b). Consequently, the stereoinvertive pathway becomes the operative pathway for transmetallation.
Fig. 5 |. Suzuki couplings of activated alkylboron nucleophiles that occur with net inversion of stereochemistry.

a | Examples of electronically activated organoboron nucleophiles in net-stereoinvertive Suzuki cross-coupling reactions56–63. b | Mechanistic models for stereoinvertive transmetallation of alkylboron nucleophiles56–58.
Only two reported examples of net-stereoinvertive Suzuki reactions involving an electronically activated alkylboron nucleophile not containing a β-carbonyl group have been reported. Through the elegant use of 10B labelling, Morken has demonstrated that geminal bis(pinacolboronates) undergo stereoinvertive transmetallation to palladium using monodentate-TADDOL-derived ligand 29 (REFS61,62) (FIG. 5a, entry 5). As the alkylboron nucleophiles employed by Morken and Yun each lack a remote coordinating group, it is unclear from where the divergent stereochemical outcomes originate. In Morken’s process, it is possible that the enhanced hydroxide-binding ability of geminal diboronates60 enables a boron-tethered hydroxide to play the role of the remote carbonyl in the model for stereoinvertive transmetallation. In Yun’s process (FIG. 3, entry 7), the reduced Lewis acidity of B(dan) might impede such intramolecular coordination, thus promoting stereoretentive transmetallation. In the second example, Liao has shown that the PCy3 ligand promotes stereoinvertive transmetallation of enantio enriched gem-diarylmethine boronates (30)63 (FIG. 5a, entry 6). Although the ligand effects have, thus far, not been rigorously examined in this system, it is conceivable that the stereochemical outcome of transmetallation is tied to the electronic properties of the phosphine ligand in a similar manner to that described by Biscoe and Sigman for non-activated alkylboron nucleophiles23.
Other alkylmetal nucleophiles
Copper-assisted coupling of enantioenriched alkyltins.
The use of alkylstannane reagents in Pd-catalysed cross-coupling reactions is complicated by the requirement for four substituents on tin. This problem is circumvented in traditional Stille reactions by exploiting the enhanced propensity for transfer of C(sp) and C(sp2) substituents from tin. Comparatively, unactivated C(sp3) substituents undergo much slower transfer from tin64. Thus, three alkyl substituents can be employed as inert spectator ligands in cases where the selective transfer of an alkynyl, alkenyl or aryl substituent is desired. When tetraalkylstannane nucleophiles are employed in cross-coupling reactions, transfer of only one alkyl unit is typically observed, with three potentially precious alkyl units wasted during this process. Thus, the development of stereospecific Stille cross-coupling reactions poses the additional challenge of achieving selective transfer of an inherently bulky secondary alkyl unit. To facilitate the selective transfer of a single secondary alkyl unit, incorporation of activation modes into the organostannane structure is required. It has been demonstrated that the presence of a C(sp2) α-carbon, α-heteroatom, remote coordinating group, ring strain and/or carbastannatrane backbone can facilitate selective secondary alkyl transfer from a tetraalkylstannane4 (see FIG. 1d). All examples of stereospecific Stille cross-coupling reactions require at least one such mode of activation to promote selective transmetallation of the secondary alkyl unit from a tetraalkylstannane nucleophile. Of these activation modes, the use of carbastannatranes constitutes the only traceless activation strategy not tied to and limited by the electronic properties of the alkyl nucleophile. Additionally, the use of catalytic or stoichiometric Cu(I) additives as co-transmetallating agents is usually required to facilitate alkyl transfer from tin to palladium.
All reported examples of enantiospecific Pd-catalysed Stille cross-coupling reactions using catalytic or stoichiometric Cu(I) are shown in FIG. 6. In contrast to enantiospecific Suzuki cross-coupling reactions, the vast majority of these processes proceed with net retention of stereochemistry. Numerous activation modes have been exploited in these reactions to facilitate selective alkyl transmetallation from organostannanes. Biscoe and Vedejs demonstrated the use of alkylcarbastannatranes in stereospecific Stille reactions. The carbastannatrane backbone selectively labilizes the apical tin substituent, which enables facile transmetallation of otherwise unactivated alkyl units65,66. Biscoe demonstrated that enantioenriched alkylcarbastannatranes bearing secondary alkyl groups undergo stereospecific transmetallation to palladium in a general manner when JackiePhos (31) is included as the supporting ligand67 (FIG. 6, entry 1). When acyl electrophiles are employed in the coupling reaction, Pd(PPh3)4 catalyses the cross-coupling reaction on account of the enhanced electrophilicity of Pd(II)-acyl complexes68 (FIG. 6, entry 2). Vedejs employed carbastannatrane activation alongside α-nitrogen substitution and ring-strain activation to achieve a cis-stereospecific arylation of aziridinyl carbastannatrane 32 (REF.69) (FIG. 6, entry 3). Because 32 is so highly activated, it undergoes undesired protodestannylation reactions, as well as Cu-promoted ring-opening. During investigations of enantioenriched α-carbastannatranyl pyrrolidine derivatives in cross-coupling reactions, Biscoe similarly observed that the combination of carbastannatrane activation and α-nitrogen substitution renders the pyrrolidine fragment overactivated, resulting in competitive destannylation. Replacement of the carbastannatrane backbone with a tricyclohexyltin group results in a significantly more stable α-stannylpyrrolidine nucleophile, which selectively transfers the activated pyrrolidine unit70 (FIG. 6, entry 4). This strategy was successfully extended to enantioenriched azetidine and open-chain α-nitrogen-substituted tricyclohexyltin nucleophiles. In these reactions, RSnnBu3 nucleophiles cannot be used owing to competitive n-butyl groups transfer to palladium. Falck71, Hammerschmidt41 and Walczak72 have demonstrated the use of α-oxygen-substituted alkyltin nucleophiles in stereospecific Pd-catalysed cross-coupling reactions assisted by Cu(I) (FIG. 6, entries 5–8). Selective transfer of the α-oxygen-substituted stereocentres was achieved in the presence of n-butyl spectator ligands, which suggests fast transfer of α-oxygenated alkyl groups relative to n-butyl groups. Finally, Liao demonstrated one example of stereospecific benzyl transfer and coupling using the conditions established by Biscoe for stereospecific Stille reactions of alkylcarbastannatranes73 (FIG. 6, entry 9). There appears to be aspects of mechanistic uniformity in the aforementioned stereospecific Stille reactions because they all proceed with net retention of absolute stereochemistry. It is probable that these reactions proceed through an initial stereoretentive alkyl transmetallation from tin to copper, followed by a second stereoretentive transmetallation from copper to palladium. This proposal is speculative, however, as the possibility of a doubly stereoinvertive transmetallation mechanism has not been conclusively eliminated. Although still circumstantial, better evidence for a doubly stereoretentive transmetallation mechanism comes from the stereochemical outcomes observed in Pd-free, Cu-catalysed enantiospecific Stille reactions. Pioneered by Falck74–77, Liebeskind78 and Hoppe79, these reactions involve α-oxygen-substituted tributyltin reagents also bearing a potentially coordinating β-substituent, and afford net-stereoretentive coupled products using only copper catalysis (FIG. 7a). The observation of net stereoretention in these reactions strongly suggests that alkyl transmetallation from tin to copper proceeds predominately through a stereoretentive pathway. The same logic suggests that alkyl transmetallation from copper to palladium similarly occurs through a stereoretentive pathway (FIG. 7b). Although the abundance of net-stereoretentive Stille reactions involving Pd and Cu co-catalysis provides circumstantial support for a doubly stereoretentive transmetallation pathway, care must still be exercised when predicting the stereochemical course of alkyl transmetallation from Cu to Pd, as ligand-dependent stereodivergent transmetallation has been reported in reactions in which in-situ-generated enantioenriched alkylcopper intermediates undergo Pd-catalysed arylation reactions80. Chong has reported the singular example of a net-stereoinvertive Pd/Cu co-catalysed Stille reaction81 (FIG. 6, entry 10). In this process, transmetallation is promoted with an alkylstannane nucleophile (33) activated by α-C(sp2) and α-heteroatom substitution, as well as by the use of an acyl electrophile. It is interesting that this highly activated system promotes a net-stereoinvertive transmetallation pathway, particularly because each activation mode, separately employed, has been shown to promote net-stereoretentive couplings.
Fig. 6 |. Examples of the coupling of enantioenriched organostannanes in copper-promoted palladium-catalysed stille reactions.

With the exception of example 10, reactions occur with net retention of stereochemistry64–73,81.
Fig. 7 |. Copper-catalysed and copper-mediated cross-couplings of alkylstannanes.
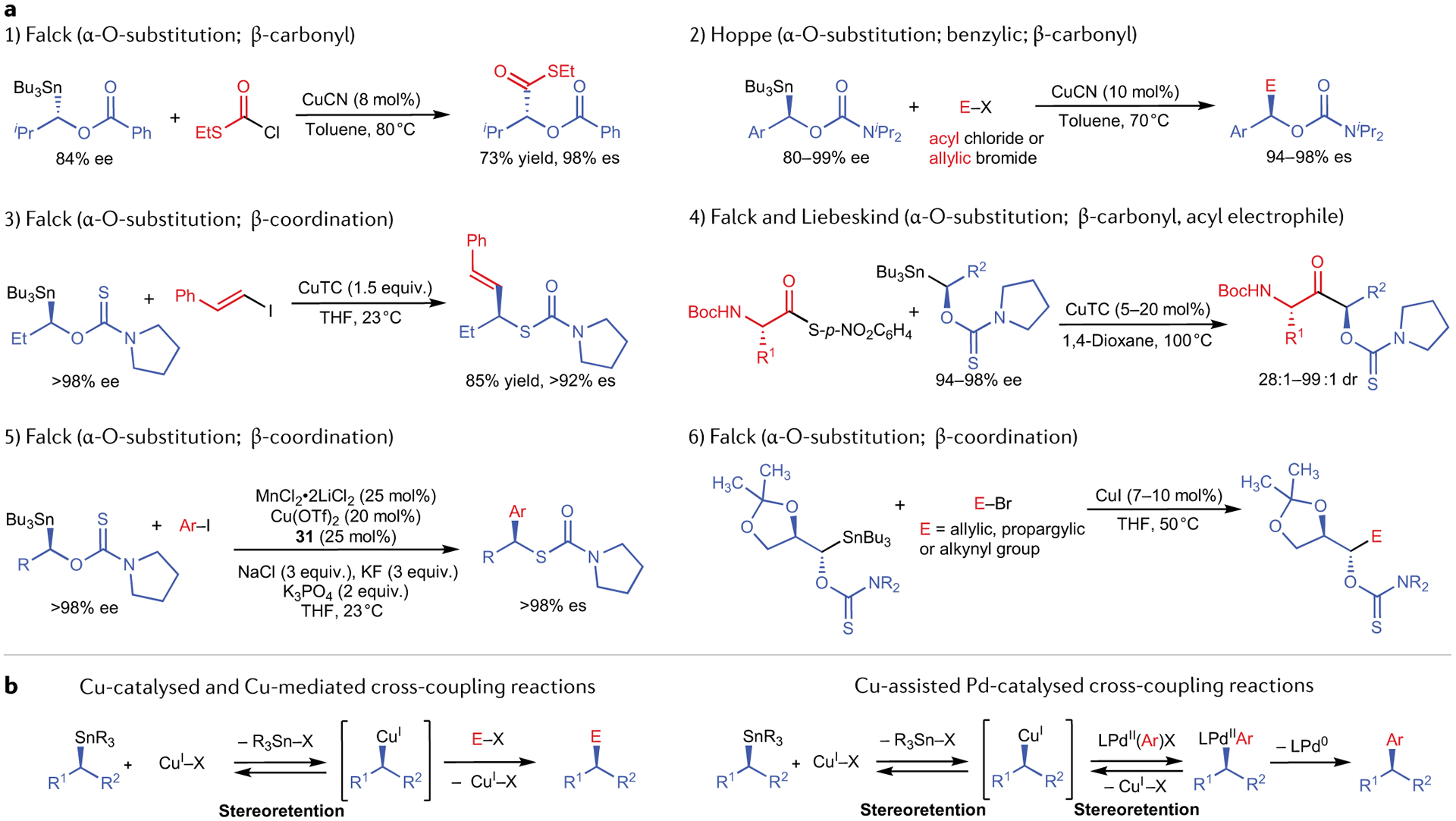
a | Examples of copper-mediated and copper-catalysed Stille reactions — these reactions generally occur with net retention of stereochemistry74–79. b | Mechanisms of transmetallation of C–Sn to form C–Cu in cross-coupling reactions with and without Pd4. Note that, in the second case, there are two transmetallation steps that must both occur with retention in order to obtain the product.
Coupling of enantioenriched alkyltins, alkylgermaniums and alkylsilicons.
Whereas copper-assisted palladium-catalysed Stille cross-coupling reactions of enantioenriched alkylstannanes most commonly exhibit net stereoretention as the stereochemical outcome, in the absence of copper additives, the stereochemical course of transmetallation in the Stille reaction is much less predictable. Pioneering studies by Stille (FIG. 8a, entry 1) used enantiomerically enriched deuterated benzylstannane reagent 34 as a stereochemical probe of transmetallation to palladium64. Stille determined that a primary benzyl group undergoes transmetallation predominately through a stereoinvertive pathway in a Pd-catalysed acylation reaction. Similarly, Hoppe observed a preference for stereoinvertive transmetallation using an enantioenriched allylstannane bearing a remote carbamate (35) in a Pd-catalysed arylation reaction82 (FIG. 8a, entry 2). By con trast, Falck has reported a Pd-catalysed Stille reaction in which an enantioenriched alkylstannane activated by the presence of an α-oxygen atom and β-carbonyl group (36) undergoes transmetallation through a stereoretentive mechanism83 (FIG. 8a, entry 3). Hodgson has also reported an instance where a cyclopropyltin nucleophile undergoes stereoretentive Pd-catalysed arylation84 (FIG. 8a, entry 4). It is likely that the preferred pathway of alkyl transmetallation in these systems is dictated by the combined influence of the specific mode of electronic activation in the alkystannane nucleophile, as well as by the properties of the supporting phosphine ligand and solvent. Indeed, stereospecific Suzuki reactions have exhibited high sensitivity to similar reaction perturbations. However, in contrast to Suzuki reactions, no systematic studies have been conducted to deconvolute the influence of alkylstannane structure, ligand structure and solvent on the stereochemical course of Stille cross-coupling reactions.
Fig. 8 |. Pd-catalysed cross-coupling reactions of enantioenriched alkyltin, alkylgermanium and alkylsilicon nucleophiles.

a | Examples of copper-free Stille (alkyltin) cross-couplings — both retention and inversion of stereochemistry have been observed62,82–84. b | Examples of cross-couplings with organosilicon and organogermanium nucleophiles85–90. c | γ-Transmetallation from silicon to palladium with allylic nucleophiles primarily occurs by a SE′ mechanism and may occur with syn- or anti-stereoselectivity, depending on reaction conditions87–90.
Cross-coupling reactions using alkylgermanium and alkylsilicon nucleophiles (FIG. 8b) have not been as widely investigated as analogous cross-coupling reactions involving alkylboron and alkyltin nucleophiles. Therefore, few examples of Pd-catalysed reactions involving stereospecific alkyl transfer from enantioenriched alkylgermanium or alkylsilicon nucleophiles have been reported. There exists only one example of a Pd-catalysed cross-coupling reaction using an enantioenriched secondary alkylgermanium nucleophile. In this work, Xiao employed the germanium analogue of an alkylcarbastannatrane in Cu-free Pd-catalysed cross-coupling reactions85 (FIG. 8b, entry 1). In comparison to alkylcarbastannatranes, alkylcarbagermatranes exhibit reduced nucleophilicity and greater stability in cross-coupling reactions. Leveraging the reduced nucleophilicity of alkylcarbagermatranes, Xiao developed a general Pd-catalysed cross-coupling reaction of primary alkylcarbagermatranes using 38 as a supporting ligand. Included in this work, a single example of a cross-coupling reaction using an enantioenriched secondary benzylic carbagermatrane (37) was reported. Consistent with the findings of Stille using an enantioenriched primary benzylstannane, the enantioenriched 37 underwent direct transmetallation to palladium (without co-transmetallation to copper) by a stereoinvertive mechanism86. Hiyama has examined an enantioenriched secondary benzyltrifluorosilane (39) in Pd-catalysed cross-coupling reactions with aryl triflates87 (FIG. 8b, entry 2). In this system, it was observed that the stereochemistry of benzylsilane transmetallation correlates directly to the temperature of the reaction. When the coupling reaction was conducted in THF at 50 °C, the stereoretentive transmetallation pathway was strongly favoured; when the reaction temperature was raised to 100 °C, the stereoinvertive transmetallation pathway was strongly favoured. Increased selectivity for the stereoinvertive transmetallation pathway was also observed to coincide with the addition of polar aprotic co-solvents such as HMPA, DMF and DMSO. Thus, the stereochemical outcome of benzylsilane transmetallation appears to be affected by solvent as well as temperature. The authors propose that a cyclic, four-membered transition state is necessary to promote the stereoretentive mechanism and that higher reaction temperatures or the inclusion of polar solvents disfavours formation of the fluoride-bridged intermediate that precedes the transition state for stereoretentive transmetallation (see FIG. 1b). When the fluoride-bridged intermediate is disfavoured, the stereoinvertive pathway is proposed to become operatively dominant. This is a very reasonable mechanistic rationalization of the observed stereochemical outcomes of these reactions, mirroring the competitive pathways that have been demonstrated in Suzuki reactions involving alkylboron nucleophiles.
In comparison to cross-coupling reactions using enantioenriched allylborane nucleophiles (see FIG. 4a), cross-coupling reactions using enantioenriched allylsilane nucleophiles have not been as broadly studied. Hatanaka and Hiyama reported the first example of a Pd-catalysed cross-coupling reaction using an enantioenriched allylsilane88. In this reaction, allylsilane transmetallation occurs primarily through a γ-selective SE′ mechanism, with the absolute configuration resulting from transmetallation being highly dependent on the identity of the fluoride salt and the solvent polarity. When tris(dimethylamino)sulfonium difluorotrimethylsilicate (TASF) and DMF are employed, high selectivity for a stereoinvertive anti-SE′ mechanism is observed (FIG. 8b, entry 3). When CsF and THF are employed, high selectivity for a stereoretentive syn-SE′ mechanism is observed. It is proposed that metal coordination plays an important role in determining the stereochemical pathway of transmetallation. According to this proposal, the use of a polar solvent that binds metal cations and/or the use of a fluoride source with a metal cation incapable of strong anionic coordination biases transmetallation towards the γ-selective anti-SE′ mechanism shown in 42. However, use of a low-polarity solvent alongside a fluoride source bearing a coordinative counterion facilitates the γ-selective syn-SE′ mechanism shown in 43. In an analogous manner to that shown in FIG. 4a for the γ-selective transmetallation of enantioenriched allylboron nucleophiles, minimization of allylic strain during transmetallation of allylsilanes determines the E/Z stereochemistry of the resulting olefin. Hiyama followed these studies with an investigation of enantioenriched 2-cyclohexenylsilane 40 in Pd-catalysed cross-coupling reactions with 4-iodoacetophenone89 (FIG. 8b, entry 4). Using a combination of Pd(PPh3)4 as the catalyst, TBAF as the fluoride source and THF as the solvent, the reaction proceeds in high yield and primarily through a stereoretentive syn-SE′ mechanism. However, the stereochemical and regiochemical outcomes of this reaction were also found to vary greatly, depending upon the leaving group, fluoride source and phosphine ligand employed. Indeed, a net-stereoinvertive transformation could be achieved through the use of CsF as a fluoride source, although the yield of this process was quite low. These combined stereochemical observations suggest that the factors controlling the mechanism of allylsilane transmetallation are complex and deserving of further examination. More recently, Denmark has developed Pd-catalysed cross-coupling reactions of allylic silanolate salts (41) and aryl bromides90 (FIG. 8b, entry 5). The syn-SE′ mechanism of transmetallation is exclusively observed in this reaction. It is proposed that the silanolate facilitates syn transfer through formation of an intermediate arylpalladium silanolate of type 43. Thus, the use of silanolates circumvents the competitive transmetallation pathways previously observed in the work of Hiyama.
Conclusions
Over the past decade, stereospecific Pd-catalysed cross-coupling reactions using configurationally stable enantioenriched organometallic nucleophiles have emerged as powerful tools for the construction of stereodefined organic structures. Significant advances have been made in our mechanistic understanding of the factors governing stereochemical transfer in these reactions. It is now clear that small energy differences may separate the transition states of the stereoretentive and stereoinvertive pathways for alkyl transmetallation. These pathways, and thus stereospecificity, are often tied to subtle electronic, steric and ligand properties of the cross-coupling reaction that can be difficult to deconvolute. Therefore, the effect of discrete electronic and steric perturbations of substrate and ligand structure must be carefully examined. Only through rigorous mechanistic studies will we uncover the parameters that guide stereochemical transfer in these reactions and be able to generate better predictive mechanistic models of stereoselectivity. Thus, we strongly caution against the blind use of precedent to predict or rationalize the stereospecificity of new Pd-catalysed cross-coupling reactions involving modified reaction parameters.
Acknowledgements
The authors thank the City College of New York, the National Institutes of Health (R01GM131079) and the National Science Foundation (CHE-1665189) for support of this work.
Footnotes
Competing interests
The authors declare no competing interests.
Peer review information
Nature Reviews Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work.
References
- 1.de Meijere A, Bräse S & Oestreich M (eds) Metal-Catalyzed Cross-Coupling Reactions and More (Wiley, 2014). [Google Scholar]
- 2.Jana R, Pathak TP & Sigman MS Advances in transition metal (Pd, Ni, Fe)-catalyzed cross-coupling reactions using alkyl-organometallics as reaction partners. Chem. Rev 111, 1417–1492 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cherney AH, Kadunce NT & Reisman SE Enantioselective and enantiospecific transition-metal-catalyzed cross-coupling reactions of organometallic reagents to construct C–C bonds. Chem. Rev 115, 9587–9652 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang C-Y, Derosa J & Biscoe MR The use of stable, optically active organometallic nucleophiles in cross-coupling reactions. Chem. Sci 6, 5105–5113 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rygus JPG & Crudden CM Enantiospecific and iterative Suzuki–Miyaura cross-couplings. J. Am. Chem. Soc 139, 18124–18137 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Leonori D & Aggarwal VK Stereospecific couplings of secondary and tertiary boronic esters. Angew. Chem. Int. Ed 54, 1082–1096 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Boudier A, Bromm LO, Lotz M & Knochel P New applications of polyfunctional organometallic compounds in organic synthesis. Angew. Chem. Int. Ed 39, 4414–4435 (2000). [PubMed] [Google Scholar]
- 8.Swift EC & Jarvo ER Asymmetric transition metal-catalyzed cross-coupling reactions for the construction of tertiary stereocenters. Tetrahedron 69, 5799–5817 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pound SM & Watson MP Asymmetric synthesis via stereospecific C–N and C–O bond activation of alkyl amine and alcohol derivatives. Chem. Commun 54, 12286–12301 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sandford C & Aggarwal VK Stereospecific functionalizations and transformations of secondary and tertiary boronic esters. Chem. Commun 53, 5481–5494 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Namirembe S & Morken JP Reactions of organoboron compounds enabled by catalyst-promoted metalate shifts. Chem. Soc. Rev 48, 3464–3474 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pratihar S & Roy S Reactivity and selectivity of organotin reagents in allylation and arylation: nucleophilicity parameter as a guide. Organometallics 30, 3257–3269 (2011). [Google Scholar]
- 13.Jover J, Fey N, Purdie M, Lloyd-Jones GC & Harvey JN A computational study of phosphine ligand effects in Suzuki–Miyaura coupling. J. Mol. Catal. A Chem 324, 39–47 (2010). [Google Scholar]
- 14.Hartwig JF Electronic effects on reductive elimination to form carbon–carbon and carbon–heteroatom bonds from palladium(II) complexes. Inorg. Chem 46, 1936–1947 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Ridgway BH & Woerpel KA Transmetallation of alkylboranes to palladium in the Suzuki coupling reaction proceeds with retention of stereochemistry. J. Org. Chem 63, 458–460 (1998). [DOI] [PubMed] [Google Scholar]
- 16.Matos K & Soderquist JA Alkylboranes in the Suzuki–Miyaura coupling: stereochemical and mechanistic studies. J. Org. Chem 63, 461–470 (1998). [DOI] [PubMed] [Google Scholar]
- 17.Bock PL, Boschetto DM, Rasmussen JR, Demers JP & Whitesides GM The stereochemistry of reactions at carbon-transition metal σ-bonds. (CH3)3CCHDCHDFe(CO)2C5H5. J. Am. Chem. Soc 96, 2814–2825 (1974). [Google Scholar]
- 18.Li L, Zhao S, Joshi-Pangu A, Diane M & Biscoe MR Stereospecific Pd-catalyzed cross-coupling reactions of secondary alkylboron nucleophiles and aryl chlorides. J. Am. Chem. Soc 136, 14027–14030 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biscoe MR, Fors BP & Buchwald SL A new class of easily activated palladium precatalysts for facile C–N cross-coupling reactions and the low temperature oxidative addition of aryl chlorides. J. Am. Chem. Soc 130, 6686–6687 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kinzel T, Zhang Y & Buchwald SL A new palladium precatalyst allows for the fast Suzuki–Miyaura coupling reactions of unstable polyfluorophenyl and 2-heteroaryl boronic acids. J. Am. Chem. Soc 132, 14073–14075 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bruno NC, Tudge MT & Buchwald SL Design and preparation of new palladium precatalysts for C–C and C–N cross-coupling reactions. Chem. Sci 4, 916–920 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Butter M et al. Aryl trifluoroborates in Suzuki–Miyaura coupling: the roles of endogenous aryl boronic acid and fluoride. Angew. Chem. Int. Ed 49, 5156–5160 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Zhao S et al. Enantiodivergent Pd-catalyzed C–C bond formation enabled through ligand parameterization. Science 362, 670–674 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niemeyer Z, Milo A, Hickey DP & Sigman MS Parameterization of phosphine ligands reveals mechanistic pathways and predicts reaction outcomes. Nat. Chem 8, 610–617 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Chen L, Ren P & Carrow BP Tri(1-adamantyl) phosphine: Expanding the boundary of electron-releasing character available to organo-phosphorus compounds. J. Am. Chem. Soc 138, 6392–6395 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Lehman JW et al. Axial shielding of Pd(II) complexes enables perfect stereoretention in Suzuki-Miyaura cross-coupling of Csp3 boronic acids. Nat. Commun 10, 1263 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Imao D, Glasspoole BW, Laberge VS & Crudden CM Cross coupling reactions of chiral secondary organoboronic esters with retention of configuration. J. Am. Chem. Soc 131, 5024–5025 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Dreher SD, Dormer PG, Sandrock DL & Molander GA Efficient cross-coupling of secondary alkyltrifluoroborates with aryl chlorides – reaction discovery using parallel microscale experimentation. J. Am. Chem. Soc 130, 9257–9259 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rubin M, Rubina M & Gevorgyan V Transition metal chemistry of cyclopropenes and cyclopropanes. Chem. Rev 107, 3117–3179 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Zhou S-M, Deng M-Z, Xia L-J & Tang M-H Efficient Suzuki-type cross-coupling of enantiomerically pure cyclopropylboronic acids. Angew. Chem. Int. Ed 37, 2845–2847 (1998). [DOI] [PubMed] [Google Scholar]
- 31.Luithle JEA & Pietruszka J Synthesis of enantiomerically pure cyclopropanes from cyclopropylboronic acids. J. Org. Chem 64, 8287–8297 (1999). [DOI] [PubMed] [Google Scholar]
- 32.Rubina M, Rubin M & Gevorgyan V Catalytic enantioselective hydroboration of cyclopropenes. J. Am. Chem. Soc 125, 7198–7199 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Lohr S & de Meijere A 2-(Bicyclopropylidenyl)-and 2-(trans-2′-cyclopropylcyclopropyl)-4,4,5,5-tetramethyl-1,3-dioxa-2-borolane and their palladium-catalyzed cross-coupling reactions. Synlett 2001, 489–492 (2001). [Google Scholar]
- 34.Fang G-H, Yan Z-J & Deng M-Z Palladium-catalyzed cross-coupling of stereospecific potassium cyclopropyl trifluoroborates with aryl bromides. Org. Lett 6, 357–360 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Chen H & Deng M-Z A novel stereocontrolled synthesis of 1,2-trans cyclopropyl ketones via Suzuki-type coupling of acid chlorides with cyclopropylboronic acids. Org. Lett 2, 1649–1651 (2000). [DOI] [PubMed] [Google Scholar]
- 36.Glasspoole BW, Oderinde MS, Moore BD, Antoft-Finch A & Crudden CM Highly chemoselective and enantiospecific Suzuki–Miyaura cross-couplings of benzylic organoboronic esters. Synthesis 45, 1759–1763 (2013). [Google Scholar]
- 37.Matthew SC, Glasspoole BW, Eisenberger P & Crudden CM Synthesis of enantiomerically enriched triarylmethanes by enantiospecific Suzuki–Miyaura cross-coupling reactions. J. Am. Chem. Soc 136, 5828–5831 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Awano T, Ohmura T & Suginome M Inversion or retention? Effects of acidic additives on the stereochemical course in enantiospecific Suzuki–Miyaura coupling of α-(acetylamino)benzylboronic esters. J. Am. Chem. Soc 133, 20738–20741 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Molander GA & Wisniewski SR Stereospecific cross-coupling of secondary organotrifluoroborates: potassium 1-(benzyloxy)alkyltrifluoroborates. J. Am. Chem. Soc 134, 16856–16868 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feng X, Jeon H & Yun J Regio- and enantioselective copper(I)-catalyzed hydroboration of borylalkenes: asymmetric synthesis of 1,1-diborylalkanes. Angew. Chem. Int. Ed 52, 3989–3992 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Krizkova PM & Hammerschmidt F On the configurational stability of chiral heteroatom-substituted [D1]methylpalladium complexes as intermediates of Stille and Suzuki–Miyaura cross-coupling reactions. Eur. J. Org. Chem 2013, 5143–5148 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoang GL et al. Enantioselective desymmetrization via carbonyl-directed catalytic asymmetric hydroboration and Suzuki–Miyaura cross-coupling. Org. Lett 17, 940–943 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoang GL & Takacs JM Enantioselective γ-borylation of unsaturated amides and stereoretentive Suzuki–Miyaura cross-coupling. Chem. Sci 8, 4511–4516 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Daini M & Suginome M Palladium-catalyzed, stereoselective, cyclizative alkenylboration of carbon–carbon double bonds through activation of a boron–chlorine bond. J. Am. Chem. Soc 133, 4758–4761 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Blaisdell TP & Morken JP Hydroxyl-directed cross-coupling: a scalable synthesis of debromohamigeran E and other targets of interest. J. Am. Chem. Soc 137, 8712–8715 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Farmer JL, Hunter HN & Organ MG Regioselective cross-coupling of allylboronic acid pinacol ester derivatives with aryl halides via Pd-PEPPSI-IPent. J. Am. Chem. Soc 134, 17470–17473 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Yang Y & Buchwald SL Ligand-controlled palladium-catalyzed regiodivergent Suzuki–Miyaura cross-coupling of allylboronates and aryl halides. J. Am. Chem. Soc 135, 10642–10645 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Partridge BM, Chausset-Boissarie L, Burns M, Pulis AP & Aggarwal VK Enantioselective synthesis and cross-coupling of tertiary propargylic boronic esters using lithiation–borylation of propargylic carbamates. Angew. Chem. Int. Ed 51, 11795–11799 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Chausset-Boissarie L et al. Enantiospecific, regioselective cross-coupling reactions of secondary allylic boronic esters. Chem. Eur. J 19, 17698–17701 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Potter B, Edelstein EK & Morken JP Modular, catalytic enantioselective construction of quaternary carbon stereocenters by sequential cross-coupling reactions. Org. Lett 18, 3286–3289 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ding J, Rybak T & Hall DG Synthesis of chiral heterocycles by ligand-controlled regiodivergent and enantiospecific Suzuki Miyaura cross-coupling. Nat. Commun 5, 5474 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Huang X et al. Expanding Pd-catalyzed C–N bond-forming processes: The first amidation of aryl sulfonates, aqueous amination, and complementarity with Cu-catalyzed reactions. J. Am. Chem. Soc 125, 6653–6655 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Rybak T & Hall DG Stereoselective and regiodivergent allylic Suzuki–Miyaura cross-coupling of 2-ethoxydihydropyranyl boronates: synthesis and confirmation of absolute stereochemistry of diospongin B. Org. Lett 17, 4156–4159 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Rubial B et al. Enantiospecific synthesis of ortho-substituted 1,1-diarylalkanes by a 1,2-metalate rearrangement/anti-SN2′ elimination/rearomatizing allylic Suzuki–Miyaura reaction sequence. Angew. Chem. Int. Ed 58, 1366–1370 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ardolino MJ & Morken JP Congested C–C bonds by Pd-catalyzed enantioselective allyl–allyl cross-coupling, a mechanism-guided solution. J. Am. Chem. Soc 136, 7092–7100 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ohmura T, Awano T & Suginome M Stereospecific Suzuki–Miyaura coupling of chiral α-(acylamino) benzylboronic esters with inversion of configuration. J. Am. Chem. Soc 132, 13191–13193 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Sandrock DL, Jean-Gerard L, Chen C-Y, Dreher SD & Molander GA Stereospecific cross-coupling of secondary alkyl β-trifluoroboratoamides. J. Am. Chem. Soc 132, 17108–17110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ohmura T, Miwa K, Awano T & Suginome M Enantiospecific Suzuki–Miyaura coupling of nonbenzylic α-(acylamino)alkylboronic acid derivatives. Chem. Asian J 13, 2414–2417 (2018). [DOI] [PubMed] [Google Scholar]
- 59.Lee JCH, McDonald R & Hall DG Enantioselective preparation and chemoselective cross-coupling of 1,1-diboron compounds. Nat. Chem 3, 894–899 (2011). [DOI] [PubMed] [Google Scholar]
- 60.Endo K, Ohkubo T, Hirokami M & Shibata T Chemoselective and regiospecific Suzuki coupling on a multisubstituted sp3-carbon in 1,1-diborylalkanes at room temperature. J. Am. Chem. Soc 132, 11033–11035 (2010). [DOI] [PubMed] [Google Scholar]
- 61.Sun C, Potter B & Morken JP A catalytic enantiotopic-group-selective Suzuki reaction for the construction of chiral organoboronates. J. Am. Chem. Soc 136, 6534–6537 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Potter B, Szymaniak AA, Edelstein EK & Morken JP Nonracemic allylic boronates through enantiotopic-group-selective cross-coupling of geminal bis(boronates) and vinyl halides. J. Am. Chem. Soc 136, 17918–17921 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lou Y et al. Copper-catalyzed enantioselective 1,6-boration of para-quinone methides and efficient transformation of gem-diarylmethine boronates to triarylmethanes. Angew. Chem. Int. Ed 54, 12134–12138 (2015). [DOI] [PubMed] [Google Scholar]
- 64.Labadie JW & Stille JK Mechanisms of the palladium-catalyzed couplings of acid chlorides with organotin reagents. J. Am. Chem. Soc 105, 6129–6137 (1983). [Google Scholar]
- 65.Vedejs E, Haight AR & Moss WO Internal coordination at tin promotes selective alkyl transfer in the Stille coupling reaction. J. Am. Chem. Soc 114, 6556–6558 (1992). [Google Scholar]
- 66.Jurkschat K, Tzschach A & Meunier-Piret J Crystal and molecular structure of 1-aza-5-stanna-5-methyltricyclo[3.3.3.01,5]undecane. Evidence for a transannular donor–acceptor interaction in a tetraorganotin compound. J. Organomet. Chem 315, 45–49 (1986). [Google Scholar]
- 67.Li L, Wang C-Y, Huang R & Biscoe MR Stereoretentive Pd-catalyzed Stille cross-coupling reactions of secondary alkyl azastannatranes and aryl electrophiles. Nat. Chem 5, 607–612 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang C-Y, Ralph G, Derosa J & Biscoe MR Stereospecific palladium-catalyzed acylation of enantioenriched alkylcarbastannatranes: a general alternative to asymmetric enolate reactions. Angew. Chem. Int. Ed 56, 856–860 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Theddu N & Vedejs E Stille coupling of an aziridinyl stannatrane. J. Org. Chem 78, 5061–5066 (2013). [DOI] [PubMed] [Google Scholar]
- 70.Ma X et al. A general approach to stereospecific cross-coupling reactions of nitrogen-containing stereocenters. Chem 6, 781–791 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ye J, Bhatt RK & Falck JR Stereospecific palladium/copper cocatalyzed cross-coupling of α-alkoxy- and α-aminostannanes with acyl chlorides. J. Am. Chem. Soc 116, 1–5 (1994). [Google Scholar]
- 72.Zhu F, Rouke MJ, Yang T, Rodriguez J & Walczak MA Highly stereospecific cross-coupling reactions of anomeric stannanes for the synthesis of C-aryl glycosides. J. Am. Chem. Soc 138, 12049–12052 (2016). [DOI] [PubMed] [Google Scholar]
- 73.Jia T, Cao P, Wang D, Lou Y & Liao J Copper-catalyzed asymmetric three-component borylstannation: enantioselective formation of C–Sn bond. Chem. Eur. J 21, 4918–4922 (2015). [DOI] [PubMed] [Google Scholar]
- 74.Falck JR, Bhatt RK & Ye J Tin–copper transmetalation: Cross-coupling of α-heteroatom-substituted alkyltributylstannanes with organohalides. J. Am. Chem. Soc 117, 5973–5982 (1995). [Google Scholar]
- 75.Mohapatra S, Bandyopadhyay A, Barma DK, Capdevila JH & Falck JR Chiral α,β-dialkoxy- and α-alkoxy-β-aminostannanes: preparation and copper-mediated cross-coupling. Org. Lett 5, 4759–4762 (2003). [DOI] [PubMed] [Google Scholar]
- 76.Falck JR, Patel PK & Bandyopadhyay A Stereospecific cross-coupling of α-(thiocarbamoyl) organostannanes with alkenyl, aryl, and heteroaryl iodides. J. Am. Chem. Soc 129, 790–793 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dakarapu R & Falck JR Stereospecific Stille cross-couplings using Mn(II)Cl2. J. Org. Chem 83, 1241–1251 (2018). [DOI] [PubMed] [Google Scholar]
- 78.Li H, He A, Falck JR & Liebeskind LS Stereocontrolled synthesis of α-amino-α′-alkoxy ketones by a copper-catalyzed cross-coupling of peptidic thiol esters and α-alkoxyalkylstannanes. Org. Lett 13, 3682–3685 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lange H, Frohlich R & Hoppe D Cu(I)-catalyzed stereospecific coupling reactions of enantioenriched α-stannylated benzyl carbamates and their application. Tetrahedron 64, 9123–9135 (2008). [Google Scholar]
- 80.Logan KM, Smith KB & Brown MK Copper/palladium synergistic catalysis for the syn- and anti-selective carboboration of alkenes. Angew. Chem. Int. Ed 54, 5228–5231 (2015). [DOI] [PubMed] [Google Scholar]
- 81.Kells KW & Chong JM Stille coupling of stereochemically defined α-sulfonamidoorganostannanes. J. Am. Chem. Soc 126, 15666–15667 (2004). [DOI] [PubMed] [Google Scholar]
- 82.Kalkofen R & Hoppe D First example of an enantiospecific sp3-sp2 Stille coupling of a chiral allylstannane with aryl halides. Synlett 2006, 1959–1961 (2006). [Google Scholar]
- 83.Goli M, He A & Falck JR Pd-catalyzed cross-coupling of α-(acyloxy)-tri-n-butylstannanes with alkenyl, aryl, and heteroaryl electrophiles. Org. Lett 13, 344–346 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hodgson DM et al. Intramolecular cyclopropanation of unsaturated terminal epoxides and chlorohydrins. J. Am. Chem. Soc 129, 4456–4462 (2007). [DOI] [PubMed] [Google Scholar]
- 85.Xu M-Y et al. Alkyl carbagermatranes enable practical palladium-catalyzed sp2–sp3 cross-coupling. J. Am. Chem. Soc 141, 7582–7588 (2019). [DOI] [PubMed] [Google Scholar]
- 86.Ma X, Diane M, Ralph G, Chen C & Biscoe MR Stereospecific electrophilic fluorination of alkylcarbastannatrane reagents. Angew. Chem. Int. Ed 56, 12663–12667 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hatanaka Y & Hiyama T Stereochemistry of the cross-coupling reaction of chiral alkylsilanes with aryl triflates: a novel approach to optically active compounds. J. Am. Chem. Soc 112, 7793–7794 (1990). [Google Scholar]
- 88.Hatanaka Y, Goda K & Hiyama T Regio- and stereoselective cross-coupling reaction of optically active allylsilanes: stereocontrol of palladium-mediated SE′ reactions. Tetrahedron Lett. 35, 1279–1282 (1994). [Google Scholar]
- 89.Hiyama T et al. Chirality transfer via the palladium-catalyzed cross-coupling reaction of optically active 2-cyclohexenylsilane: stereochemical and mechanistic aspects. Organometallics 15, 5762–5765 (1996). [Google Scholar]
- 90.Denmark SE & Werner NS On the stereochemical course of palladium-catalyzed cross-coupling of allylic silanolate salts with aromatic bromides. J. Am. Chem. Soc 132, 3612–3620 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]