

Abstract
Genome editing using programmable nucleases is revolutionizing life science and medicine. Off-target editing by these nucleases remains a considerable concern, especially in therapeutic applications. Here we review tools developed for identifying potential off-target editing sites and compare the ability of these tools to properly analyze off-target effects. Recent advances in both in silico and experimental tools for off-target analysis have generated remarkably concordant results for sites with high off-target editing activity. However, no single tool is able to accurately predict low-frequency off-target editing, presenting a bottleneck in therapeutic genome editing, because even a small number of cells with off-target editing can be detrimental. Therefore, we recommend that at least one in silico tool and one experimental tool should be used together to identify potential off-target sites, and amplicon-based next-generation sequencing (NGS) should be used as the gold standard assay for assessing the true off-target effects at these candidate sites. Future work to improve off-target analysis includes expanding the true off-target editing dataset to evaluate new experimental techniques and to train machine learning algorithms; performing analysis using the particular genome of the cells in question rather than the reference genome; and applying novel NGS techniques to improve the sensitivity of amplicon-based off-target editing quantification.
Over the past few decades, the emergence of programmable nucleases has revolutionized the field of genome editing. Programmable nucleases, including zinc finger nucleases (ZFNs)1–3, transcription activator-like (TAL) effector nucleases (TALENs)4–6, clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (Cas9) (CRISPR–Cas9) systems and their derivatives, such as base editors7–11, allow for site-specific and permanent alterations to the genomes of a wide variety of organisms. Most of the programmable nucleases function by creating a DNA double-strand break (DSB) at the intended target locus in a cell, which is subsequently repaired by the non-homologous end joining (NHEJ) pathway, resulting in insertion/deletion (indel) mutations at the target site, or by the homology-directed repair (HDR) pathway, leading to the targeted integration of a donor sequence. A glossary of abbreviations used in our review is provided in Box 1.
Box 1 |. Glossary.
Base editor: Cas9 nickase fused to an active deaminase for targeted conversion of cytosine to thymine or adenine to guanine without the generation of a DNA double-strand break.
Cas9 (CRISPR-associated protein 9): nuclease capable of generating DNA double-strand breaks in a sequence-specific manner in combination with a gRNA.
CNN (convolutional neural networks): a specific type of artificial neural network that uses convolution for supervised learning and data classifications. Typically used for image recognition.
CRISPR (clustered regularly interspaced short palindromic repeats): DNA sequences in prokaryotes that play a key role in antiviral defense.
dCas9 (nuclease-dead Cas9): a modified Cas9 enzyme where both nuclease domains have been inactivated to create a DNA-binding protein that does not cut DNA.
DSB (double-strand break): DNA lesion where both strands of the DNA duplex are cleaved.
FokI nuclease domain: non-specific DNA cleavage domain from the type IIS restriction enzyme FokI.
gRNA (single guide RNA): a short RNA sequence (100 nucleotides) that interacts with Cas9 to generate ribonucleoprotein complexes capable of sequence-specific DNA cleavage.
HDR (homology-directed repair): a DNA repair pathway that requires a DNA donor template, resulting in the targeted integration of a donor sequence.
ICE (Inference of CRISPR Edits): Python script and webtool for analyzing Sanger sequence files of CRISPR-edited cells.
Indel (insertion or deletion): DNA sequence mutations arising from imperfect repair of DNA double-strand breaks where bases are inserted or removed.
nCas9 (Nickase Cas9): a modified Cas9 where one of two nuclease domains is disrupted, resulting in a Cas9 protein capable of cleaving one strand of a DNA duplex resulting in DNA nicks.
NHEJ (non-homologous end joining): a DNA repair pathway that results in the direct ligation of DNA break ends in the absence of a homologous template for repair.
NIST (National Institute of Standards and Technology): a measurement standards laboratory that supplies standard reference materials. The NIST Genome Editing Consortium is tasked with establishing standards in genome editing.
PAM (protospacer adjacent motif): a short DNA sequence recognized by Cas9 and essential for DNA binding and cleavage by Cas9.
PRC (precision recall curve): a plot of the precision (y-axis) and the recall (x-axis) where the precision is calculated as the ratio of the number of true positives divided by the sum of the true positives and false positives, and recall is calculated as the ratio of the number of true positives divided by the sum of the true positives and the false negatives.
PWM (position weight matrix): a matrix of weights for distinguishing between true binding sites from non-target sites with similar sequences. This matrix can be used to scan genomes for potential off-target site discovery.
ROC curve (receiver operating characteristic curve): a plot of the true-positive rate (y-axis) versus the false-positive rate (x-axis). The true-positive rate is calculated as the number of true positives divided by the sum of the number of true positives and the number of false negatives. The false-positive rate is calculated as the number of false positives divided by the sum of the number of false positives and the number of true negatives. The area under the curve (AUC) can be used as a summary of the model performance.
RVD (repeat variable diresidue): TAL effectors consist of repeated highly conserved domains of 33–34 amino acids with divergent amino acid residues at the 12th and 13th positions known as the repeat variable diresidue. These RVDs determine the DNA binding specificity of the TAL effector with one RVD binding to one nucleotide.
TAL effector nuclease (TALEN): engineered TAL effectors consisting of 12–31 repeats fused to the FokI nuclease domain. Because the FokI domain requires dimerization to cleave DNA, a pair of TALENs must bind with appropriate spacing and orientation to successfully cleave the DNA target.
TIDE (tracking of indels by decomposition): R code and webtool for analyzing Sanger sequence files of CRISPR-edited cells.
Zinc finger: small protein motif first identified as DNA-binding motifs in transcription factors. Each zinc finger typically recognizes 3 bp of DNA, and tandem arrays of zinc fingers allow for longer sequences of DNA to be recognized.
Zinc finger nuclease (ZFN): engineered zinc finger proteins consisting of three to six zinc finger repeats fused to the FokI nuclease domain. Because the FokI domain requires dimerization to cleave DNA, a pair of ZFNs must bind with appropriate spacing and orientation to successfully cleave the DNA target.
Figure 1 shows four major classes of programmable nucleases: ZFNs, TALENs, CRISPR–Cas9 and base editors. For ZFNs and TALENs, a pair of nucleases is required to generate a DSB at a specific (predetermined) target locus. In the case of ZFNs (Fig. 1a), each ZFN contains a DNA-binding domain (zinc finger protein) fused to the FokI non-specific DNA cleavage domain. With each zinc finger binding to three DNA bases, a zinc finger protein typically consists of an array of 3–6 zinc fingers to recognize 9–18 DNA bases; thus, a ZFN pair targets a DNA sequence of 18–36 bases. For TALENs, the nuclease is formed by fusing a transcription activator-like effector (TALE) DNA-binding domain to the FokI nuclease domain (Fig. 1b). Each DNA-binding domain of TALE contains a variable number of 33–35 amino acid repeats that specify the DNA-binding sequence primarily through their 12th and 13th repeat-variable di-residues (RVDs). As illustrated in Fig. 1c, the CRISPR–Cas9 system targets the site of interest using a single guide RNA (gRNA). The gRNA sequence typically comprises a 5′ 17–20-nucleotide sequence complementary to the target DNA sequence and a 3′ end sequence that interacts with the Cas9 protein. A protospacer-associated motif (PAM) of 2–5 nucleotides on the target DNA is required for Cas9 binding, which is located directly downstream of the target sequence on the non-target DNA strand. Cas9 is guided by the gRNA to the target site and cleaves the DNA sequence it binds, giving rise to a DSB. Base editors generate single-nucleotide changes in DNA12. A base editor typically consists of a Cas9 nickase (nCas9) fused to an adenosine or cytosine deaminase, which is capable of converting A to G or C to T, respectively, in genomic DNA12 (Fig. 1d). Base editing has the potential to make genome editing more versatile and safer. A new class of gene editors, known as prime editors, uses nCas9 fused to an engineered reverse transcriptase, programmed with a prime editing guide RNA (pegRNA) that both specifies the target site and encodes the desired base editing11. Although prime editing has the potential to expand the scope and capability of genome editing13, it is still in an early stage of development and is, thus, not included in our discussion here.
Fig. 1 |. Schematics showing four major classes of programmable nucleases.

a–c, Programmable nucleases for genome editing include (a) ZFNs, (b) TALENs, (c) CRISPR and CRISPR–Cas9 systems. Fn, FokI nuclease domain. d, A base editor is a CRISPR–Cas9 system that directly converts one DNA base or base pair into another without making a DSB. It consists of an nCas9 fused to an adenosine or cytosine deaminase, which is capable of converting A to G or C to T, respectively, in genomic DNA.
Programmable nucleases have a wide range of applications, including genetic modification of bacteria, plants and animals; enhancing understanding of, and regulating, gene functions; establishing human disease models for basic research and drug discovery; and targeted therapeutic intervention14–17. In particular, the potential of programmable nuclease-based genome editing in therapeutic applications has been broadly recognized, and, to date, there are 53 genome editing-based clinical trials registered at clinicaltrials.gov: 15 with ZFNs, 6 with TALENs and 32 with CRISPR–Cas9 systems. However, several major challenges currently affect clinical translation of programmable nuclease-based gene editing, including pre-existing immunity18–21, in vivo delivery efficiency22 and potential off-target effects23–25.
This review article focuses on the analysis of off-target effects, which remain a major safety concern in therapeutic applications of genome editing. An off-target event can be defined as programmable nuclease-induced DNA cleavage at a site anywhere in the genome other than the intended target site. When an off-target cutting event occurs, it can be repaired via the NHEJ pathway, potentially resulting in an indel mutation; or, if it occurs simultaneously with an on-target or a second off-target cutting event, the off-target cutting activity can generate a chromosomal rearrangement, such as an inversion or translocation, or a large deletion between the two break points26.
Several tools, both in silico and experimental, have been developed to identify potential off-target sites for programmable nucleases (Tables 1–4). For researchers performing genome editing experiments with programmable nucleases, it can be difficult to choose among these methods for off-target analysis. Here we outline and analyze the most commonly used methods developed for the identification of off-target sites, evaluating their strengths and weaknesses and highlighting the challenges in accurately identifying off-target sites and quantifying off-target effects. We focus on tools designed for CRISPR–Cas9 systems owing to their widespread use, although methods for ZFNs and TALENs are also briefly reviewed. After discussing both the experimental and computational methods available for off-target site identification, we present a performance comparison of off-target analysis techniques and recommend best practices for evaluating off-target effects of CRISPR–Cas9-based gene editing. Future directions for improving off-target analysis methodologies are also discussed.
Table 1 |.
Comparison of in silico off-target site search tools for ZFNs and TALENs
| Name | Screening approach or algorithm | Off-target ranking/scoring basis | Strengths | Weaknesses |
|---|---|---|---|---|
| ZFN tools | ||||
| PROGNOS78 (website) | Base by base | Weighted homology, conserved G score and polarity effects | Allows user-defined spacing, ZF homodimerization and ambiguous bases. Analyzes ZF subunits. Relatively low false-positive ratios | Limited training set |
| ZFN-site128 (website) | TagScan129 | Sequence homology | Allows user-defined spacing, ZF homodimerization and ambiguous bases | No scoring algorithm |
| Zinc finger tools130 (website) | Search in <10-kb user-defined sequence | N/A | Screens ZF targets in user-supplied DNA sequences | Screens limited to 49 triplets with validated ZF domains |
| TALEN tools | ||||
| PROGNOS78 (website) | Base by base | Modular position weight matrix, binding energy compensation, polarity effects | Allows up to 20 mismatches. Relatively low false-positive ratios (∼11:1)a | Limited training set |
| CHOPCHOP62 (website) | Bowtie | Weighed off-target site number | Allows up to two mismatches | Potentially misses off-target sites |
| TALENgetter/, TALENoffer76 (website, command line) | Base by base with threshold-based speed-up strategy | Machine-learning-based modular position weight matrix | Allows up to ten mismatches; allows the use of rare RVDs | Scoring algorithm not as accurate as PROGNOS78 |
| TALE-NT 2.0 (ref. 75; website) | Base by base | Modular position weight matrix | The first scoring tool for TALEN off-target analysis | Potential worse performance if using custom RVD designs |
The false-positive ratios are defined as ‘the number of screened sites with no detectable activity to the number with detectable activity measured by experimental prediction methods’78.
Table 4 |.
Comparison of experimental off-target site search tools with estimated performance characteristics
| Name | Method | What is detected? | Setting | Cut site enrichment | Cellular false positives |
|---|---|---|---|---|---|
| In vitro | |||||
| Digenome-seq47 | NGS fragment end statistics | DSB | DNA | 170* | 65% |
| CIRCLE-seq49 | DSB end enrichment | DSB | DNA | 821 | 88% |
| SITE-seq48 | DSB end enrichment | DSB | DNA | 178 | 95% |
| DIG-seq51 | NGS fragment end statistics | DSB | Chromatin | 289* | 64% |
| In vivo | |||||
| HTGTS26 | Rearrangement detection | Repair product | Cells | 4,700 | n.d. |
| GUIDE-seq27 | Oligonucleotide integration | Repair product | Cells | 29,000 | 20% |
| DISCOVER-seq55 | DSB end enrichment | DSB | Cells/tissue | 233 | ∼0% |
| BLISS54 | DSB end tagging | DSB | Cells/tissue | 160 | 20% |
Summary of experimental techniques for off-target cutting by programmable nucleases. Because all methods rely on specific enrichment of either DNA close to cut sites or DNA ends close to cut sites, we use the degree to which they achieve enrichment of DNA fragments near the on-target editing site (‘Cut site enrichment’) as a proxy for their sensitivity. For ‘Cellular false positives’, we use the fraction of sites identified by each technique that fail to be edited (indels <0.1%) in the accompanying cellular validation studies; see main text for details.
Cut site enrichment for Digenome-seq and DIG-seq was assessed by enrichment of sequencing fragments whose ends are precisely at the on-target editing site.
n.d., not determined.
Workflow for analysis of off-target editing
In general, the off-target cutting activity at a particular sequence in a genome depends on its homology with the target sequence, molecular interaction with the programmable nuclease and accessibility. To analyze the off-target effects, it is necessary to first identify potential off-target sites in the genome of interest using an in silico tool and/or an experimental genome-wide off-target identification method, such as GUIDE-seq27, and then to quantify the indel rates at the predicted sites using a quantitative assay. Specifically, the loci of interest are amplified from genomic DNA extracted from a pool of cells using PCR, and the resulting amplicons can be analyzed for sequence mismatch incorporations by a variety of methods, including the Surveyor nuclease assay28, digestion by T7 Endonuclease I (ref. 29), Sanger sequencing trace decomposition (tracking of indels by decomposition (TIDE)30 and Inference of CRISPR Edits (ICE)31) or direct detection of mutations using next-generation sequencing (NGS).
Direct detection of mutations using NGS
NGS on PCR amplicons (hereafter referred to as Amp-NGS) remains the gold standard for confirming off-target cutting by programmable nucleases, owing to its high sensitivity and applicability to any sample that has been subjected to gene editing by programmable nucleases. The sensitivity of Amp-NGS is limited by PCR and NGS errors, which confound the detection of true cutting events. NGS read errors are dominated by base substitution errors, whereas NHEJ repair of nuclease-induced DSBs leads to short indels, so Amp-NGS results are typically quantitated on the basis of indel frequency rather than base mutation frequency. The generally recognized sensitivity limit of Amp-NGS is ~0.1%, dictated by the rate at which indels arise during both target amplification and the NGS read process32. For many sites, this 0.1% of off-target cutting activity might be an overestimate, because, at these potential off-target sites, the NGS reads with indels could be a result of PCR and NGS errors. Furthermore, a small number of sites might exhibit apparent indel rates of > 0.1% even without treatment with programmable nucleases. Therefore, negative controls need to be performed for each off-target site being analyzed using Amp-NGS to determine its true background signal. Typical negative controls comprise cells subjected to mock delivery conditions in the absence of Cas9 protein. Additional controls using a non-targeting gRNA should be interpreted with caution as we have previously observed significant gRNA-dependent off-target events in vivo using a non-targeting gRNA33.
Experimental tools for off-target site identification
Several experimental tools have been developed to detect off-target activity of programmable nucleases (Fig. 2). For clarity, we group experimental techniques into three broad categories: (i) detection of nuclease binding, (ii) detection of nuclease-induced DSBs and (iii) detection of repair products arising from nuclease-induced DSBs. Because all of these techniques are intended to be as unbiased as possible, they are in general applicable across all the different programmable nuclease families. Of the techniques surveyed here, several approaches were pioneered in the study of ZFNs, the first truly engineered nuclease platform, and subsequently refined for use with TALENs and CRISPR–Cas9 systems. The performance evaluation for these techniques and best practice recommendations are given in later sections (see ‘Performance comparisons’ and ‘Best practices’ sections below).
Fig. 2 |. Schematics showing in vitro and in vivo experimental techniques most commonly used to characterize off-target cutting by CRISPR–Cas9.

These techniques include those for detection of (i) in vitro (biochemical) tagging of Cas9 cut sites, (ii) in vivo (cellular) tagging of Cas9 cut sites and (iii) targeted sequencing of PCR amplicons. Cas9 binding techniques are not commonly used to characterize off-target cutting and are, therefore, not shown. Targeted amplicon sequencing is routinely used to verify off-target sites identified by these experimental techniques.
Detection of nuclease binding
Initial efforts at analyzing off-target cutting of ZFNs relied on the characterization of the binding specificity of monomeric zinc finger proteins to DNA using assays such as SELEX and its derivatives34–36, bacteria-1-hybrid screening37, ELISA38 and microarrays39. Sequences bound by individual zinc finger proteins could then be used to search the genome of interest for homodimeric or heterodimeric off-target ZFN sites. Similarly, binding of Cas9 to DNA targets has been characterized both in vitro40 and in vivo41,42 using sequencing techniques.
Although nuclease binding is the most straightforward to detect, it is the least informative, because nuclease binding is necessary but not sufficient for cutting. This appears to be true for ZFNs, TALENs and CRISPR–Cas9 systems. Off-target detection techniques that rely solely on nuclease binding thus tend to yield large numbers of false-positive sites and are not in common use.
Detection of nuclease activity
Instead of detecting the binding of nucleases, another approach to discovering nuclease off-target effect is to detect the cutting activity directly. For instance, Pattanayak et al.43 used DNA substrates generated by rolling circle amplification of a random library to directly determine the sequences that could be cleaved in vitro by a pair of ZFNs. These were then used to build a statistical model whereby off-target cutting of genomic locations could be predicted. In vitro off-target sequence identification using synthetic DNA substrates was also used to determine the specificity of CRISPR–Cas9 systems44–46.
Early work to discover DSBs generated in vitro by Cas9 looked at fragmentation patterns in libraries generated from purified genomic DNA (Digenome-seq)47. Recently, two techniques, SITE-seq48 and CIRCLE-seq49, were developed where sequencing adapters are ligated to the DSBs resulting from nuclease activity. These adapters are used to enrich for the fragments that arise from the DSBs to facilitate sequencing. In the case of SITE-seq, the adapters are also biotinylated, and further enrichment is achieved by performing a pulldown of ligated fragments using streptavidin-coated beads. CHANGE-seq, a high-throughput method based on DNA circularization, was also just developed to analyze the genome-wide off-target activities of CRISPR–Cas9 nucleases in vitro50.
All of the aforementioned techniques start with purified DNA as the substrate, with the drawback that the chromatin state of the substrate is not considered. Similarly to the in vitro nuclease binding assays, inability to consider the chromatin state in a living cell, and, therefore, the cut-site accessibility in the assay, gives rise to a large number of false-positive events. DIG-seq, an updated version of Digenome-seq, was developed to perform the same fragmentation pattern assay in nuclease-digested chromatin51. Several additional techniques attempt to detect DSBs being produced in cells. BLESS ligates biotinylated adapters to DSBs in fixed cells and then uses these adapters to capture DNA proximal to DSBs52,53. BLISS ligates indexed adapters to DSBs in fixed cells and then performs in vitro transcription from those adapters, followed by NGS54. DISCOVER-seq enriches DSBs by immunoprecipitation of MRE11, a protein that specifically binds to DSBs in cells and in vivo55.
Detection of DSB repair products
Detection of DSBs generated in living cells might have limited sensitivity because cells can efficiently repair these DSBs. A potentially more sensitive approach would be to specifically enrich the repair products containing mutated sequences, which are expected to accumulate over time. This approach was initially demonstrated for ZFNs using an integrase-deficient lentiviral vector (IDLV). However, IDLV capture is only able to reliably detect off-target sites with >1% activity56 and underperforms compared to the in vitro ZFN cutting assay57. A newer technique, GUIDE-seq, increases the sensitivity by flooding cells with short (34-bp) double-stranded oligodeoxynucleotides (dsODNs) that can be inserted at the DSB sites when nuclease cutting occurs. Detection of dsODN insertion events provides improved sensitivity, and GUIDE-seq is currently the preferred experimental technique by many groups for identifying potential off-target sites. A major drawback of GUIDE-seq is that it requires delivery of dsODN into cells, and not all cell types, especially primary cells, are amenable to dsODN delivery49. In cases where GUIDE-seq is unfeasible for the cell type of interest, a substitute cell type, such as U2OS, is often used. This, however, might lead to false positives and/or false negatives because some off-target effects can be cell type specific.
Instead of detecting exogenous DNA insertion events, HTGTS26 and LAM-HTGTS58 look for endogenous repair products in the form of chromosomal rearrangements with known cutting loci. This allows off-target cutting detection in most cell types.
Bioinformatic prediction of off-target sites
The bioinformatic analysis for identifying off-target sites of programmable nucleases can be divided into two steps. In the first step, site detection, the target genome is scanned for potential sites based on homology to the on-target sequence. Several studies, especially those performed early on, used simple homology search programs, such as BLAST, for off-target screening59,60. Many bioinformatic off-target prediction tools used read mapping programs, such as Bowtie61 (e.g., implemented in CHOPCHOP62 and GT-Scan63) and Bowtie2 (ref. 64; e.g., implemented in E-CRISP website65 and CRISPRscan66), to perform site detection. However, these screening algorithms should be avoided because they were not designed for locating homologous sequences that are short (12–24 bp) and can contain relatively large numbers of sequence mismatches (up to six) or short indels. More recent tools, such as CRISPRitz67, are specifically designed to accomplish this task efficiently and, thus, should be used instead.
A site detection program typically yields tens to hundreds of potential off-target cut sites. Thus, in the second step—site scoring/ranking—potential off-target sites detected in the first step are scored and/or ranked based on either the degree of homology to the target sequence or the expected cutting activity of the programmable nuclease. This allows users to focus on the top-ranked sites for experimental validation using, for example, targeted deep sequencing. In some cases, scoring is accomplished by the application of a pre-defined formula, and, in other cases, the scoring algorithm is obtained using machine learning (ML) based on existing off-target cleavage data as the training set. However, owing to limited datasets of experimentally validated true off-target sites, neither formula-based nor ML-based scoring/rankings are very accurate, and true off-target sites can be missed when taking the top 10 or top 20 sites from the ranked list for validation. This has been a major issue in the off-target analysis of CRISPR–Cas9 systems.
Unlike experimental techniques developed for identifying potential off-target sites, which can be applied to different nucleases that generate DSBs, bioinformatic techniques are typically specific to the nuclease class of interest. We, therefore, discuss different nuclease classes separately, considering heterodimeric programmable nucleases (ZFNs and TALENs) first and then CRISPR–Cas9 systems.
Bioinformatic approaches for off-target evaluation of ZFNs and TALENs
Both ZFNs and TALENs are designed as heterodimers, with two DNA recognition domains (Fig. 1) flanking a short spacer (5–7 nucleotides) that contains the cut site. Off-target sites reflect this design strategy, with perfect and imperfect matches of each domain spaced by a range of distance intervals. Homodimerization has also been observed with both ZFNs and TALENs, and this also contributes to off-target cutting.
Early analyses of potential off-target sites for ZFNs and TALENs, performed for small sets of nuclease designs, used general sequence mapping programs, such as BLAST and Bowtie, to generate lists of candidate sites and performed ranking of off-target sites using the number of mismatches within recognition domains68–74. Several of the top-ranked sites were then assessed experimentally. Strategies for off-target site detection were later codified into ZFN and TALEN design tools, as well as standalone tools that assess off-target specificity. Computational tools capable of identifying potential off-target sites for ZFNs and TALENs are listed in Table 1.
In addition to performing more thorough site detection, some design tools also incorporated new knowledge arising from more thorough characterizations of the programmable nucleases to enable more sophisticated site prioritization. For TALENs, binding specificities of natural TAL effectors were first mined to generate binding frequency matrices between RVDs in TAL effectors and nucleotides at the corresponding positions of the recognition domain. This allowed the Paired Target Finder feature of TAL effector–nucleotide targeter (TALE-NT)75 to sum up the relative score of each RVD–nucleotide association using the frequency matrix for potential target sites. The search tool TALENoffer76 further incorporates the contributions of different RVDs to TALEN cutting activity77.
The off-target prediction tool PROGNOS78 incorporated molecular features of nuclease–DNA interactions and used experimentally confirmed off-target sites as the training set to obtain scoring algorithms for off-target site identification of both ZFNs and TALENs. PROGNOS also factors in ‘polarity’ effects, whereby the location of mismatches within the nuclease target site affects the DNA–protein binding affinity79. PROGNOS has relatively low false-positive rates, and its false-negative rates are similar to experiment-based predictions, making it a robust off-target search method for ZFNs and TALENs.
CRISPR–Cas9 off-target site identification and ranking
Many tools have been developed to identify potential CRISPR–Cas9 off-target sites80. Some of the tools, such as Cas-OFFinder81, Crisflash82 and CasOT83, identify off-target sites without ranking them and, thus, can be used only for screening gRNA designs. Details of screening algorithms designed for CRISPR–Cas9 off-target identification are listed in Table 2. Other tools have the capability of scoring and ranking the potential off-target sites identified (Table 3). For example, E-CRISP65, one of the early approaches for off-target identification, ranks off-target hits by alignment scores. CCTOP84 and COSMID78, on the other hand, rank the potential off-target sites by considering the position of mismatches relative to the PAM sequence, based on the observation that mismatches closer to the PAM are more likely to prevent Cas9 cutting23,85. COSMID also allows input of one-base insertion (DNA bulge) and one-base deletion (RNA bulge) relative to the perfectly matched sequence, because these can be tolerated by Cas9 (ref. 78).
Table 2 |.
Comparison of in silico off-target site screening tools for CRISPR-Cas9 systems
| Name | Strengths | Weaknesses |
|---|---|---|
| BLAST131 (website, command line) | A time-optimized sequence alignment tool (seeding-based algorithm), bulges allowed | Less accurate (can miss potential off-target sites), limited mismatch numbers, no custom PAM |
| TagScan129 (website, command line) | A time-optimized sequence screening tool for queries <60 bases, web support | Limited mismatch numbers, no custom PAM, no bulges allowed |
| Bowtie61 (command line) | A time-optimized alignment tool for queries <50 bases | Less accurate (can miss potential off-target sites), limited mismatch numbers, no custom PAM, no bulges allowed |
| Bowtie2 (ref. 64) (command line) | A time-optimized alignment tool, bulges allowed | Misses off-target sites with low mismatch numbers80, limited mismatch numbers and no custom PAM |
| CasOT83 (command line) | A sequence screening tool for CRISPR-Cas9 system, custom PAM, user-defined mismatch number in seed/non-seed region, paired-gRNA mode allowed | Time-consuming132, no bulges allowed |
| Cas-OFFinder81 (website, command line) | A commonly used sequence screening tool for CRISPR-Cas systems, web support, custom PAM, user-defined mismatch number, bulges allowed | Can miss potential off-target sites with complex DNA/RNA bulges67, moderate speed132 |
| dsNickFury91(Command line) | A sequence screening tool for CRISPR-Cas9 system, custom PAM, user-defined mismatch number | No bulges allowed |
| FlashFry133 (command line) | A time optimized sequence screening tool for CRISPR-Cas systems, custom PAM, user-defined mismatch number. Good for large datasets | No bulges allowed |
| Crisflash82 (command line) | A sequence screening tool for CRISPR-Cas9 system, custom PAM, user-defined mismatch number, accepts genetic variation data on haplotype level | No bulges allowed |
| CRISPRitz67 (command line) | A time-optimized sequence screening tool for CRISPR-Cas9 system, custom PAM, user-defined mismatch number, bulges allowed, accepts genetic variation data | Cannot process genetic variation data on haplotype level |
Table 3 |.
Comparison of in silico off-target site ranking and scoring tools for CRISPR-Cas9
| Name | Main features reported | Strengths | Weaknesses |
|---|---|---|---|
| E-CRISP65 (website); formula based | Mismatch numbers | An early approach for CRISPR-Cas9 off-target identifications | Rankings were outperformed by other algorithms |
| CCTOP84 (website); formula based | Mismatch positions and numbers | Web support | Scorings were outperformed by other algorithms |
| COSMID78 (website); formula based | Mismatch positions and numbers | Web support, bulges allowed | Scorings were outperformed by other algorithms |
| Cropit86 (website); formula based | Mismatch numbers and continuities (optional: chromatin states) | Web support. Better performance than other formula-based algorithms on ChIP-seq data | Scores did not correlate well with cleavage-based genome-wide experimental data |
| MIT23 (websitea); formula-based modular PWM (see Box 1) | Mismatch positions, numbers and mean distances | Web support. The most popular formula-based algorithm. Good ranking performance80 | Scorings were outperformed by CFD88, no bulges allowed |
| Hsu score23 (command linea); normalized modular PWM | Mismatch positions and numbers | A simplified version of MIT | Scores did not correlate with experimental data as well as the MIT score |
| CFD88 (command line); modular PWM | Mismatch positions, numbers and identities | Based on the biggest cleavage dataset to date. Good ranking performance80 | No bulges allowed |
| predictCRISPR90 (command line); machine learning | 281 sequence-related features | All the machine-learning-based tools showed similar to better performances than the algorithms in other categories. However, because most of these models were trained by genome-wide experiment data, which were largely overlapped to most of the training sets, a potential over-fitting issue exists in the comparison (as described in detail in the ‘Performance comparisons’ section and shown in Supplementary Table 1). Elevation is recommended for in silico prediction, and CRISTA is the only option that allows bulges | |
| CRISTA89 (website, command line); machine learning | Nucleotide identities, alignment, thermodynamics and genomic contents | ||
| Elevation91 (website, command line); Machine learning | gRNA spacer sequence and off-target sequence | ||
| CNN_std92 (command line); deep learning | gRNA spacer sequence and off-target sequence | ||
| deepCRISPR93 (command line); deep learning | gRNA spacer sequence and off-target sequence, cell-type-specific features | ||
| Synergizing CRISPR94 (command line); deep learning | Scores from five other algorithms (CFD, MIT Website, MIT score, Cropit, CCTop) and evolutionary conservation | ||
Implemented in the CRISPOR website and reviewed in ref. 80.
Further improvements to ranking potential off-target sites use experimental Cas9 binding and cutting data. CROP-IT86 divides the protospacer sequence into three regions with different weights for mismatches, using Cas9 ChIP-seq data from previous studies41 for weight parameter optimization. CROP-IT further adds a location-based, cell-type-specific accessibility score derived from genome-wide DNAse I-seq data87. The MIT score (also known as crispr.mit.edu or Hsu score) attempts to estimate the off-target cutting rate using a mismatch weight matrix derived from detailed studies of gRNA variants and rescales the final score according to the minimum distance between mismatches23. The original paper by Hsu et al.23 provided several ways of calculating the scores for ranking, and the normalized aggregate frequencies method performed the best80. Finally, cutting frequency determination (CFD)88 uses a position- and base change-specific scoring matrix derived from systematically altering gRNAs targeting the CD33 gene.
The availability of large CRISPR–Cas9 activity datasets, as well as computational tools, has led to the development of ML-based algorithms for off-target prediction. Details of each of these ML-based algorithms, such as structures and training sets, are listed in Supplementary Table 1. CRISTA models off-target cutting data derived from three different genome-wide assays (GUIDE-seq, HTGTS and BLESS)89 with a random forest algorithm using a broad range of features spanning six categories: nucleotide identities; alignment-related features (including bulges); RNA thermodynamics; genomic locations; features from experimental databases (such as DNAse I hypersensitivity and gene expression level); and DNA enthalpy and geometry features. Another ML approach, predictCRISPR90, tested a support vector machine model with a validated dataset. A more recent ML approach, Elevation91, uses a two-layer regression model in which the first layer predicts the off-target activity of a single mismatch between the target DNA and gRNA, and the second layer combines the contribution of each single mismatch to the gRNA target score with that of multiple mismatches. Deep learning has also been applied to off-target prediction. CNN_std92 and deepCRISPR93 are two convolutional neural network (CNN)-based models for CRISPR–Cas9 off-target site prediction. deepCRISPR also integrates several modalities of epigenetic information. However, the architecture of these deep learning models precludes the consideration of insertions and deletions relative to the gRNA target sequence. Finally, SynergizingCRISPR takes a different approach to using ML, whereby prediction scores from five other tools (MIT website, MIT/Hsu score, CFD, Cropit and CCTop) rather than the gRNA and potential off-target sequences are used as inputs to the model94.
Although most of the bioinformatic off-target search tools are designed for CRISPR–Cas9, a recent study reported a CNN-based classifier for CRISPR-Cpf1 activity and specificity prediction95. This algorithm was the first one built for Cpf1 (i.e., Cas12a) and was trained using the dataset of a lentiviral library-based AsCpf1 gRNA target pair established by Kim et al.96.
Off-target analyses of base editors
Base editors use an nCas9 fused to a deaminase or glycosylase inhibitor to directly convert one DNA base or base pair into another without making DSBs97. Whole-genome sequencing (WGS) revealed that third-generation base editors (BE3s) could induce genome-wide off-target effects in mice98 and rice99, showing a significant amount of gRNA-independent single-nucleotide mutations with high frequency in transcribed regions of the genome, suggesting that the off-target effects were caused by the fused rAPOBEC1 deaminase of BE3. Investigation of transcriptome-wide RNA off-target mutations showed that both adenine base editors (ABEs) and cytosine base editors (CBEs) could generate gRNA-independent off-target mutations100–102. In addition, gRNA-dependent off-target editing was observed103,104. Novel in vitro genome-wide off-target detection assays for ABEs and CBEs were established by capturing dCas9-induced DNA nicks using NGS96,105,106. The specificity of BE3 was analyzed using modified USER-Digenome-seq47, indicating that BE3 could tolerate mismatches in gRNA–DNA base pairing, with a different off-target efficiency pattern than that of active Cas9.
Two recent studies established EndoV-seq106 and Endo-Digenome-seq105 assays, respectively, to assess the specificity of ABEs, using EndoV/EndoVIII to generate the second nick after ABE editing and WGS to identify the resulting DSBs. Both studies showed lower gRNA-dependent off-target effects than that of wild-type SpCas9, although gRNA-independent off-target editing remains a critical issue. More recently, gRNA-independent off-target base editing was studied, including the use of sensitive R-loop assays without requiring WGS107,108. Bioinformatics-based algorithms need to be established to better predict the gRNA-dependent off-target effects, and the mechanisms of gRNA-independent off-target effects need to be better established before accurate predictions could be made.
Performance comparisons for CRISPR–Cas9-based techniques
To guide the reader toward a better understanding of the relative strengths and weaknesses of the CRISPR–Cas9 off-target analysis tools, both experimental and in silico, we compared the performance of these techniques. The ideal dataset for these comparisons is difficult to obtain, especially for experimental techniques, which need to be performed under conditions as similar as possible for the same Cas9 and gRNA. Our performance comparison, therefore, comprises several ad hoc analyses intended to discern gross differences between the different approaches.
Comparison of experimental techniques
We assessed the performance of a selection of experimental techniques, including Digenome-seq47, DIG-seq51, CIRCLE-seq49, SITE-seq48, HTGTS26, GUIDE-seq27, DISCOVER-seq55 and BLISS54, in two different ways and summarized the results in Table 4. First, we attempted to determine the relative sensitivities of these techniques—i.e., how often these techniques are able to detect true-positive off-target editing events. We found it difficult to define ‘gold standard’ lists of off-target cutting sites to directly determine false-negative rates for these methods, because they were generally performed for disparate gRNA sequences in disparate experimental systems. To side-step this issue, we used on-target read enrichment as a proxy for sensitivity. We reasoned that each of the experimental techniques considered here relies on some sort of enrichment for the nuclease cut sites, including enrichment for genomic DNA bearing the cut sites, as in HTGTS; enrichment for the precise locations of the cut sites, as in Digenome-seq; or enrichment for both, as in GUIDE-seq. The degree of enrichment over background (i.e., what is expected of randomly fragmented genomic reads) should, therefore, be correlated with how well a given technique is able to detect the rare cutting events that give rise to off-target editing. Because none of the techniques treats on-target editing events differently from off-target events, enrichment over background can be assessed readily for the on-target editing events and extrapolated to off-target editing.
To accomplish this, we downloaded raw reads from entries in the Sequence Read Archive associated with each technique using the SRA Toolkit (entries listed in Supplementary Table 2). We mapped these reads to the hg38 reference genome using BWA-MEM and counted reads within 400 bp of the expected on-target cut sites using SAMtools109. Read counts were then divided by how many random genomic reads would be expected within the same region, given the total number of reads that mapped to the human genome, to yield the on-target enrichment. For the cases of Digenome-seq110 and DIG-seq, where enrichment is for fragment ends rather than fragments themselves, we counted reads whose 5′ ends fell precisely on the on-target cut site and compared those counts to what was expected given random genomic fragmentation.
Second, we assessed the relative specificities of each technique. In general, false-positive rates can be determined by performing Amp-NGS on DNA extracted from gene-edited cells, using primers flanking sites discovered by the technique in question. We, therefore, used Amp-NGS data from the publications reporting each discovery technique to assess their respective specificities, applying the standard 0.1% indel threshold to distinguish true from false positives. As with the sensitivity comparison, this assessment is inevitably imperfect because the underlying datasets are not all directly comparable. The situation is further complicated by the fact that roughly half of the techniques being considered here are performed on purified DNA, which lacks the chromatin structure that can potentially prevent cutting by programmable nucleases within cells. However, the purpose of performing off-target site identification is usually to generate predictions as to which sites will likely be edited in cells. We, therefore, think that the degree to which these predictions are validated as true off-target activity in cells should be used as the measure of specificity, even when that technique is not itself performed with living cells. We labeled the corresponding column in Table 4 ‘Cellular false positives’, to highlight the fact that the false-positive rate is for validation with living cells and might not be relevant to other applications of Cas9 and other programmable nucleases. As noted above, defining false negatives for experimental methods in a way that can be consistently applied is not currently feasible given the paucity of data derived from similar experiments.
Results of these assessments suggest that GUIDE-seq is the best-performing experimental technique: it shows the highest on-target enrichment with a moderate number of false positives. Some caution needs to be taken in interpreting these results: on-target enrichment can be correlated with the number of PCR cycles and is, thus, an imperfect readout of sensitivity, and variations in the gRNAs and cells used to perform off-target identification and validation can potentially cause biases in the observed false-negative rates. Still, the status of GUIDE-seq as the most commonly used experimental off-target technique would appear to be well justified.
Comparison of computational techniques
We used a list of experimentally validated true off-target editing sites to assess the performance of computational techniques. Here, our manually curated true off-target list includes sites having editing rates of >0.1% quantified by Amp-NGS and processed by CRISPResso2 (ref. 111). Not all sites so generated yielded scores in all ranking algorithms. For instance, only COSMID and CRISTA were able to score sites with base insertions (DNA bulge) and deletions (RNA bulge). In cases where an algorithm gave no score, we used a score of zero instead (see below). Supplementary Table 3, and references therein, contain all the information used in this performance assessment. Specifically, experimentally validated off-target sites were collected from nine different studies with true editing rate >0.1% as measured by Amp-NGS (eight studies)47,56,112–117 or T7 Endonuclease I (one study)24. For each gRNA, off-target sites were screened by Cas-OFFinder allowing up to four mismatches and one base DNA/RNA bulge (Supplementary Table 4). As shown in Table 3 and Supplementary Table 1, gRNAs in the training datasets of most of the ML-based algorithms had some overlap with our testing set in the performance comparison. To mitigate the potential for overfitting, we identified gRNAs tested by Amp-NGS in four studies114–117 that were not included in any training or testing set of ML-based algorithms (listed in Supplementary Table 5) and additionally assessed algorithm performances with only these gRNAs. Standard receiver operating characteristic (ROC) curves and precision recall curves (PRCs) were generated using Scikit-learn118 and are shown in Supplementary Figs. 1 and 2. In addition to these curves, which can be difficult to use directly in designing experiments, we used the same underlying data to compute the true-positive rates as a function of the total number of sites (Fig. 3). That is, for each technique and sample size n, we determined the fraction of experimentally validated off-target sites ranked among the top n candidate sites by that technique. This curve, then, can be used to estimate the number of top-ranked sites that need to be assessed experimentally to detect true off-target sites with a given sensitivity.
Fig. 3 |. The ability of various off-target site identification algorithms to correctly rank experimentally confirmed true off-target sites.

For each algorithm, we plot the fraction of true off-target sites found in the set of top-ranked sites as a function of the size of that set, averaged across 27 gRNAs for which validated off-target cutting data exist. These results can be interpreted as estimates of how many computationally predicted off-target sites need to be experimentally assayed to achieve a given level of coverage for true off-target cutting events. Algorithms that are more useful for this task, then, yield curves that are further to the left, because they allow the user to assay fewer sites to achieve the same level of sensitivity for true-positive off-target events. a, Potential off-target sites for each of the 27 gRNAs were computed by Cas-OFFinder and separately ranked by each algorithm. b, Same as in a, except only for the four novel gRNAs whose results were not present in the training datasets of any of the ML algorithms to mitigate potential risk of over-fitting. Source data for a and b are provided in Supplementary Tables 4 and 5, respectively.
To determine off-target scores, CCTOP84 off-target scores were computed based on the formula in the original paper. Code for the MIT score (Hsu score)23 and CROP-IT86 score was adapted from the CRISPOR review80. Code for CFD score88 was obtained from the authors. Elevation91, predictCRISPR90, CNN_std92 and CRISTA89 were implemented based on instructions provided by original authors. Code for COSMID78 was adapted from source code obtained from Peng Qiu at the Georgia Institute of Technology. To keep all the scores positively correlated to editing efficiency, we used 48.4 to subtract the original COSMID score, making zero correlated to no editing efficiency. Default models were used for all ML algorithms without re-training. Algorithms requiring cell-line-specific information were not included owing to the lack of relevant data.
From the plots shown in Fig. 3a and Supplementary Fig. 1, it appears that Elevation is the best performer, both by area under the curve (AUC) of ROC and PRC and by the true-positive rate for reasonable numbers (<200) of top-ranked sites. One caveat here is that Elevation is an ML-based technique whose training dataset overlaps extensively with the assessment dataset that we collected in this study. The risk of over-fitting here is somewhat mitigated by the fact that Elevation’s training dataset incorporates all sites identified by unbiased techniques, instead of only those sites that were validated by Amp-NGS. We further mitigate this risk by redoing the sensitivity analysis using targeted Amp-NGS data from four gRNAs in our dataset not present in the training sets of any of the ML approaches (Fig. 3b and Supplementary Fig. 2). In this re-analysis, Elevation is still among the top three performers (the other two being CFD and CRISTA). Interestingly, this is true despite the fact that Elevation does not consider sites containing DNA or RNA bulges. This is likely because the number of validated true CRISPR–Cas9 off-target sites containing DNA or RNA bulges is still small. Whether the paucity of true off-target sites containing indels reflects the biology of CRISPR–Cas9 or the lack of studies focusing on bulge-containing off-target sites remains to be seen. Given its ability to rank off-target sites with DNA/RNA bulges and its overall performance, CRISTA can be a good alternative for scoring potential off-target sites.
Best practices
As an example of determining off-target effects of programmable nucleases, we describe in Box 2 the analysis of a CRISPR–Cas9 system designed to correct the single-base mutation in the β-globin gene that causes sickle cell disease (SCD)114. The original off-target site prediction was performed using both COSMID and GUIDE-seq, and the NGS quantification of off-target activity was carried out using genomic DNA from gene-edited CD34+ hematopoietic stem and progenitor cells (HSPCs) from patients with SCD. Here we further performed off-target prediction and ranking using Elevation and CRISTA for the same gRNA and compared the results with those using GUIDE-seq and COSMID, as shown in Box 2.
Box 2 |. Off-target determination for a gRNA sequence for SCD.
As a real-world example of characterizing off-target effects of programmable nucleases, we describe here the analysis of a CRISPR–Cas9 system designed to treat SCD114. The CRISPR gRNA R66SCD targets the SCD mutant site in HBB (with the target sequence next to PAM as GTAACGGCAGACTTCTCCACNGG). Co-delivery of the R66SCD/SpCas9 RNP with a short ssODN donor template elicits gene correction of the sickling mutation locus in CD34+ HSPCs from patients with SCD. Injection and engraftment of a sufficient number of these gene-edited HSPCs is potentially curative for SCD.
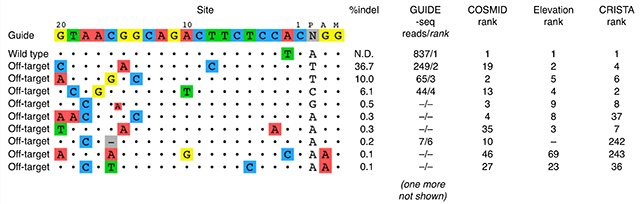
The above figure shows all of the sites at which off-target cutting was detected in CD34+ HSPCs by targeted NGS of ~7,500 cells, along with corresponding editing rates shown as ‘%indel’. To generate the list of potential off-target editing sites, we first performed computational prediction using COSMID63, which identified 57 potential off-target sites. To complement the computational prediction, we also performed experimental off-target site discovery using GUIDE-seq in U2OS cells. This yielded six potential off-target sites, all of which had been predicted using COSMID. Targeted NGS of the 57 potential off-target sites, yielding at least 9,000 total reads per site, showed that nine of them had detectable off-target activity. These are listed in the figure in order of decreasing editing activity seen at that site, as determined by fraction of total sequencing reads from those sites bearing indels (‘% indels’).
In addition to GUIDE-seq and COSMID, we performed off-target prediction using two additional computational prediction tools (Elevation and CRISTA). Both of these techniques identified a large number of potential off-target sites. Because these techniques give scores for each potential off-target site that they identify, we sought to determine whether the scores could aid in prioritizing which sites to assess by targeted NGS. We, therefore, show the rank for each confirmed off-target site within the predictions arising from each discovery method. In some cases, the methods failed to identify a site that was confirmed to have off-target editing; these have a ‘−’ where the rank would otherwise be.
Our results here show remarkable agreement among the various methods for predicting off-target sites at which editing rates are high. Even though it was performed in a different cell line, GUIDE-seq was nevertheless able to discover the top three off-target sites for R66SCD gRNA. Further, all off-target sites at which the indel rate was >0.5% were ranked among the top ten by both Elevation and CRISTA. However, these methods start to diverge at sites with lower off-target editing rates. Therefore, we cannot be assured that any approach to off-target site discovery, either experimental or computational, can predict all off-target sites for which the true editing rate is at least 0.1% without introducing a large number of false positives. As even this low off-target editing rate can potentially compromise the safety of gene-edited therapeutic products (see ‘Clinical consequences’ section), much work remains to improve the quality of off-target prediction.
As can be seen in Box 2, analysis of any given programmable nuclease can be complex, with each different technique giving different results. In our own CRISPR–Cas9 gene editing work, these observations have driven us to follow several principles for the analysis of off-target effects by programmable nucleases. An overview of the recommended workflow, from gRNA design to off-target validation, is shown in Fig. 4. Before extensive off-target analysis as outlined below, the first step is to confirm efficient on-target editing in the cell type of interest. TIDE30 and ICE31 are common tools that quickly estimate the level of editing by analyzing Sanger sequencing traces from CRISPR-treated cells. Once a lead candidate of efficient gRNA (s) has been identified, the following steps give an overview of current best practices for assaying off-target effects.
Fig. 4 |. Recommended workflow for identifying CRISPR–Cas9 off-target editing.

(1) gRNA designs for the target gene are screened using an in silico tool—e.g., Elevation to identify candidate gRNAs with limited potential for off-target editing. (2) Candidate gRNAs are delivered to cells, and (3) Editing efficiency is assessed via Sanger sequencing trace analysis and used to confirm lead candidate gRNAs with high on-target editing. (4) Identification of off-target sites using an experimental tool—e.g., GUIDE-seq with (5) detection of off-target sites by NGS. (6) PCR amplification of bioinformatically predicted and experimentally identified off-target sites for (7) NGS and bioinformatic analysis to generate a (8) final off-target report for lead candidate gRNAs.
Combine experimental and in silico analyses to assemble a list of potential off-target editing sites. Any given methodology has the possibility of missing true off-target editing. Using at least one bioinformatics-based tool and one experimental tool allows these approaches to complement each other. The experimental tool provides an independent assessment of off-target editing rates, allowing one to discern and reject nuclease designs, such as the gRNA designed to target VEGFA site 2 (ref. 49), that cuts in a promiscuous fashion. On the other hand, the in silico tool can be useful in picking up the potential off-target sites that were missed by the experimental tool, especially in cases where the true off-target sites missed by the experimental tool affect the final product for a therapeutic application (e.g., edited stem cells for clinical use). From the above performance comparisons, Elevation is recommended for in silico prediction, and the top ~100 potential off-target sites should be retained for downstream validation. GUIDE-seq is recommended as the experimental tool, especially when it can be performed using the cell type of interest. For well-behaved CRISPR–Cas9 protospacer sequences, GUIDE-seq typically yields 5–10 potential off-target sites, many of which might overlap with computationally identified sites. The inclusion of sites identified by GUIDE-seq, therefore, is not expected to significantly increase the burden of downstream experimental validation.
Use Amp-NGS as the gold standard assay for determining true off-target sites. As many potential off-target sites should be assessed as is practical, to minimize the likelihood of missing important bona fide off-target editing events, because, to date, none of the ranking algorithms is entirely accurate (as shown in the ‘Comparison of computational techniques’ section). The decreasing costs of NGS and fluid-handling robotics allows a laboratory of even modest means to assay tens to hundreds of potential off-target sites for any given gRNA. Matched negative control assays must also be performed using unedited cells, because detection limits vary across different genomic loci. A recent review119 compared three web-based Amp-NGS data analysis tools (CRISPResso2 (ref. 111), Cas-Analyzer120 and CRISPR-GA121), among which CRISPResso2 was recommended because of its detailed output report, functionality of batch analysis and capability to be used in base editing applications.
Concluding remarks
Much progress has been made in both experimental and computational approaches to analyzing off-target effects of programmable nucleases, especially for CRISPR–Cas9-based systems. As the field matures, several key areas of research will improve the accuracy and relevance of off-target editing detection and quantitation technologies.
Clinical consequences
To our knowledge, to date, no clinical trials have reported adverse events arising from off-target effects of gene editing using any programmable nuclease. Although this can partly be ascribed to the attention paid to off-target editing in pre-clinical studies, it also likely reflects the fact that few such studies have been completed, and that these studies typically enroll small numbers of patients.
A simple calculation suggests that the risk of adverse events arising from off-target editing is not necessarily small. For curing SCD, for example, 2–5 million gene-edited CD34+ HSPCs per kilogram of body weight might constitute a potentially curative dose122. Off-target editing at a rate of 0.1% is, thus, expected to give rise to many thousands of cells bearing an off-target edit. Because rare gain-of-function and loss-of-function mutations have led to clonal expansion within virally transduced therapy products123,124, the technological detection limit of 0.1% might be insufficient to identify all potentially dangerous off-target editing events, and the long-term consequences of off-target editing remain largely unknown. More molecular biology, bioinformatics and clinical research will be required to determine what the detection limit should be, and further technology development will be needed to achieve it. Furthermore, to date, most of the off-target analyses focus on small indel mutations at the off-target cut sites; however, owing to simultaneous on- and off-target cutting, intra- and inter-chromosomal rearrangements, such as inversions, large deletions and translocations, might occur114. Although chromosomal rearrangements are likely rare events, even a very small number of stem cells harboring these detrimental events could clonally expand in vivo and cause diseases such as cancer.
Improving quantitation
The 0.1% detection limit for amplicon sequencing reflects current practices and can be improved upon in several ways. Miller et al.125 used oversampling and rigorous statistical analyses to improve upon this limit by approximately tenfold. Further improvements should be possible using unique molecular identifier tagging in initial rounds of PCR, followed by oversampled NGS.
More data, better data
At the moment, experimental data on true off-target effects are scattershot. Each experimental method published so far has been performed on different sets of gRNAs and often in different cell types. This has prevented the field from obtaining a systematic understanding of how these experimental methods compare with each other and necessitated our use of on-target enrichment as an imperfect proxy for how sensitive each method is. More, and better, datasets will improve our understanding of the relative merits of each experimental and computational technique and will also improve the performance of ML tools in predicting potential off-target sites.
Future studies should be performed on consistent sets of programmable nucleases in consistent cell types, and the existing methods should be re-evaluated by ‘back-filling’ the analysis to give a more consistent set of data. As much as possible, data for these methods should also be obtained for therapeutically relevant cell types, such as hematopoietic stem cells. A recently created National Institute of Standards and Technology genome editing consortium (Box 1) will develop measurements and standards to increase confidence in the use of these technologies.
Improved machine learning
Two main factors have facilitated the development of ML-based algorithms for off-target prediction. The evolution of NGS made it affordable for researchers to screen larger numbers of potential off-target sites with much greater sensitivity, resulting in datasets sufficient for model training, whereas in-depth research into the mechanism of CRISPR–Cas9 editing provided more potential features affecting cutting efficiency and specificity for consideration during model development. With further increases in the amount of off-target data and the rapid progress of basic research in ML, it is expected that ML-based off-target scoring algorithms will aid both off-target prediction and the gRNA design process.
Personalized off-target analysis
One major limitation of most existing off-target analysis tools concerns mapping of sequencing reads. This is currently done using the reference human genome, which is mostly comprised of a single individual with 70% of the reference derived from donor RP11 (ref. 126). A recent study of 910 African genomes revealed 300 million bases of new DNA spread across 120,000 contigs not found in the reference genome, with 40% of this new DNA shared with Korean and Chinese genomes127. This large variability across genomes raises the possibility that distinct human populations or individuals might harbor novel off-target sites and events that will go undetected by in silico tools that search the reference genome and by experimental assays, because sequence reads are filtered out when mapping to the reference genome. This is especially important considering that clinical trials are underway for patients of African descent with SCD. Future studies of off-target effects in gene editing using programmable nucleases for therapeutic application should take into account the genome of the patient, reflecting a truly personalized medicine approach.
Data availability
The data sources are available at https://github.com/baolabrice/OT-review. Supplementary Tables 3, 4 and 5 contain the data used in the computational techniques performance assessment.
Code availability
The processing scripts are available at https://github.com/baolab-rice/OT-review.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (UG3HL151545, R01HL152314 and OT2HL154977 to G.B.) and the Cancer Prevention and Research Institute of Texas (RR140081 to G.B.).
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41596-020-00431-y.
Peer review information Nature Protocols thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kim YG, Cha J & Chandrasegaran S Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl Acad. Sci. USA 93, 1156–1160 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Porteus MH & Baltimore D Chimeric nucleases stimulate gene targeting in human cells. Science 300, 763 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Urnov FD et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435, 646–651 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Cermak T et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 39, 7879–7879 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller JC et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 29, 143–148 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Mussolino C et al. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 39, 9283–9293 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gasiunas G, Barrangou R, Horvath P & Siksnys V Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl Acad. Sci. USA 109, E2579–E2586 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jinek M et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cobb RE, Wang YJ & Zhao HM High-efficiency multiplex genome editing of streptomyces species using an engineered CRISPR/Cas system. ACS Synth. Biol. 4, 723–728 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaudelli NM et al. Programmable base editing of A.T to G.C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anzalone AV et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rees HA & Liu DR Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 19, 770–788 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anzalone AV, Koblan LW & Liu DR Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 38, 824–844 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Sander JD & Joung JK CRISPR–Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 32, 347–355 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carroll D Genome engineering with zinc-finger nucleases. Genetics 188, 773–782 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joung JK & Sander JD TALENs: a widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 14, 49–55 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cox DB, Platt RJ & Zhang F Therapeutic genome editing: prospects and challenges. Nat. Med. 21, 121–131 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wagner DL et al. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 25, 242–248 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Charlesworth CT et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 25, 249–254 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simhadri VL et al. Prevalence of pre-existing antibodies to CRISPR-associated nuclease Cas9 in the USA population. Mol. Ther. Methods Clin. Dev. 10, 105–112 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li A et al. AAV-CRISPR gene editing is negated by pre-existing immunity to Cas9. Mol. Ther. 28, 1432–1441 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tong S, Moyo B, Lee CM, K. L & Bao G Engineered materials for in vivo delivery of genome-editing machinery. Nat. Rev. Mater. 4, 726–737 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsu PD et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu Y et al. High-frequency off-target mutagenesis induced by CRISPR–Cas nucleases in human cells. Nat. Biotechnol. 31, 822–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cradick TJ, Fine EJ, Antico CJ & Bao G CRISPR/Cas9 systems targeting beta-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 41, 9584–9592 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frock RL et al. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 33, 179–186 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsai SQ et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR–Cas nucleases. Nat. Biotechnol. 33, 187–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiu P et al. Mutation detection using Surveyor nuclease. Biotechniques 36, 702–707 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Kim HJ, Lee HJ, Kim H, Cho SW & Kim JS Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res. 19, 1279–1288 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brinkman EK, Chen T, Amendola M & van Steensel B Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsiau T et al. Inference of CRISPR edits from Sanger trace data. Preprint at https://www.biorxiv.org/content/10.1101/251082v3 (2019). [DOI] [PubMed]
- 32.Potapov V & Ong JL Examining sources of error in PCR by single-molecule sequencing. PLoS ONE 12, e0169774 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jarrett KE et al. Somatic genome editing with CRISPR/Cas9 generates and corrects a metabolic disease. Sci. Rep. 7, 44624 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zykovich A, Korf I & Segal DJ Bind-n-Seq: high-throughput analysis of in vitro protein–DNA interactions using massively parallel sequencing. Nucleic Acids Res. 37, e151 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perez EE et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 26, 808–816 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Segal DJ et al. Evaluation of a modular strategy for the construction of novel polydactyl zinc finger DNA-binding proteins. Biochemistry 42, 2137–2148 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Gupta A, Meng X, Zhu LJ, Lawson ND & Wolfe SA Zinc finger protein-dependent and -independent contributions to the in vivo off-target activity of zinc finger nucleases. Nucleic Acids Res. 39, 381–392 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Segal DJ, Dreier B, Beerli RR & Barbas CF 3rd Toward controlling gene expression at will: selection and design of zinc finger domains recognizing each of the 5′-GNN-3′ DNA target sequences. Proc. Natl Acad. Sci. USA 96, 2758–2763 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bulyk ML, Huang X, Choo Y & Church GM Exploring the DNA-binding specificities of zinc fingers with DNA microarrays. Proc. Natl Acad. Sci. USA 98, 7158–7163 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang L et al. Systematic in vitro profiling of off-target affinity, cleavage and efficiency for CRISPR enzymes. Nucleic Acids Res. 48, 5037–5053 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuscu C, Arslan S, Singh R, Thorpe J & Adli M Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 32, 677–683 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Wu X et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 32, 670–676 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pattanayak V, Ramirez CL, Joung JK & Liu DR Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat. Methods 8, 765–770 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pattanayak V et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 31, 839–843 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fu BX, St Onge RP, Fire AZ & Smith JD Distinct patterns of Cas9 mismatch tolerance in vitro and in vivo. Nucleic Acids Res. 44, 5365–5377 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huston NC et al. Identification of guide-intrinsic determinants of Cas9 specificity. CRISPR J 2, 172–185 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim D et al. Digenome-seq: genome-wide profiling of CRISPR–Cas9 off-target effects in human cells. Nat. Methods 12, 237–243 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Cameron P et al. Mapping the genomic landscape of CRISPR–Cas9 cleavage. Nat. Methods 14, 600–606 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Tsai SQ et al. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR–Cas9 nuclease off-targets. Nat. Methods 14, 607–614 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lazzarotto CR et al. CHANGE-seq reveals genetic and epigenetic effects on CRISPR–Cas9 genome-wide activity. Nat Biotechnol. 38, 1317–1327 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim D & Kim JS DIG-seq: a genome-wide CRISPR off-target profiling method using chromatin DNA. Genome Res. 28, 1894–1900 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crosetto N et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat. Methods 10, 361–365 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ran FA et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yan WX et al. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat. Commun. 8, 15058 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wienert B et al. Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science 364, 286–289 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang X et al. Unbiased detection of off-target cleavage by CRISPR–Cas9 and TALENs using integrase-defective lentiviral vectors. Nat. Biotechnol. 33, 175–178 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Gabriel R et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat. Biotechnol. 29, 816–823 (2011). [DOI] [PubMed] [Google Scholar]
- 58.Hu J et al. Detecting DNA double-stranded breaks in mammalian genomes by linear amplification-mediated high-throughput genome-wide translocation sequencing. Nat. Protoc. 11, 853–871 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hruscha A et al. Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development 140, 4982–4987 (2013). [DOI] [PubMed] [Google Scholar]
- 60.Zhu X et al. An efficient genotyping method for genome-modified animals and human cells generated with CRISPR/Cas9 system. Sci. Rep. 4, 6420 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Langmead B, Trapnell C, Pop M & Salzberg SL Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Montague TG, Cruz JM, Gagnon JA, Church GM & Valen E CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 42, W401–W407 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O’Brien A & Bailey TL GT-Scan: identifying unique genomic targets. Bioinformatics 30, 2673–2675 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Langdon WB Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. BioData Min. 8, 1 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heigwer F, Kerr G & Boutros M E-CRISP: fast CRISPR target site identification. Nat. Methods 11, 122–123 (2014). [DOI] [PubMed] [Google Scholar]
- 66.Moreno-Mateos MA et al. CRISPRscan: designing highly efficient sgRNAs for CRISPR–Cas9 targeting in vivo. Nat. Methods 12, 982–988 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cancellieri S, Canver MC, Bombieri N, Giugno R & Pinello L CRISPRitz: rapid, high-throughput, and variant-aware in silico off-target site identification for CRISPR genome editing. Bioinformatics 36, 2001–2008 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zschemisch NH et al. Zinc-finger nuclease mediated disruption of Rag1 in the LEW/Ztm rat. BMC Immunol. 13, 60 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Watanabe T et al. Non-transgenic genome modifications in a hemimetabolous insect using zinc-finger and TAL effector nucleases. Nat. Commun. 3, 1017 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sebastiano V et al. In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells 29, 1717–1726 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang P et al. Heritable gene targeting in zebrafish using customized TALENs. Nat. Biotechnol. 29, 699–700 (2011). [DOI] [PubMed] [Google Scholar]
- 72.Lei Y et al. Efficient targeted gene disruption in Xenopus embryos using engineered transcription activator-like effector nucleases (TALENs). Proc. Natl Acad. Sci. USA 109, 17484–17489 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Heigwer F et al. E-TALEN: a web tool to design TALENs for genome engineering. Nucleic Acids Res. 41, e190 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim Y et al. A library of TAL effector nucleases spanning the human genome. Nat. Biotechnol. 31, 251–258 (2013). [DOI] [PubMed] [Google Scholar]
- 75.Doyle EL et al. TAL effector-nucleotide targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res. 40, W117–W122 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grau J, Boch J & Posch S TALENoffer: genome-wide TALEN off-target prediction. Bioinformatics 29, 2931–2932 (2013). [DOI] [PubMed] [Google Scholar]
- 77.Streubel J, Blücher C, Landgraf A & Boch J TAL effector RVD specificities and efficiencies. Nat. Biotechnol. 30, 593–595 (2012). [DOI] [PubMed] [Google Scholar]
- 78.Fine EJ, Cradick TJ, Zhao CL, Lin Y & Bao G An online bioinformatics tool predicts zinc finger and TALE nuclease off-target cleavage. Nucleic Acids Res. 42, e42 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Meckler JF et al. Quantitative analysis of TALE–DNA interactions suggests polarity effects. Nucleic Acids Res. 41, 4118–4128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Haeussler M et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 17, 148 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bae S, Park J & Kim JS Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jacquin ALS, Odom DT & Lukk M Crisflash: open-source software to generate CRISPR guide RNAs against genomes annotated with individual variation. Bioinformatics 35, 3146–3147 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xiao A et al. CasOT: a genome-wide Cas9/gRNA off-target searching tool. Bioinformatics 30, 1180–1182 (2014). [DOI] [PubMed] [Google Scholar]
- 84.Stemmer M, Thumberger T, Del Sol Keyer M, Wittbrodt J & Mateo JL CCTop: an intuitive, flexible and reliable CRISPR/Cas9 target prediction tool. PLoS ONE 10, e0124633 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sternberg SH, Redding S, Jinek M, Greene EC & Doudna JA DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Singh R, Kuscu C, Quinlan A, Qi Y & Adli M Cas9-chromatin binding information enables more accurate CRISPR off-target prediction. Nucleic Acids Res. 43, e118 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Thurman RE et al. The accessible chromatin landscape of the human genome. Nature 489, 75–82 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Doench JG et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR–Cas9. Nat. Biotechnol. 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Abadi S, Yan WX, Amar D & Mayrose I A machine learning approach for predicting CRISPR–Cas9 cleavage efficiencies and patterns underlying its mechanism of action. PLoS Comput. Biol. 13, e1005807 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peng H, Zheng Y, Zhao Z, Liu T & Li J Recognition of CRISPR/Cas9 off-target sites through ensemble learning of uneven mismatch distributions. Bioinformatics 34, i757–i765 (2018). [DOI] [PubMed] [Google Scholar]
- 91.Listgarten J et al. Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nat. Biomed. Eng. 2, 38–47 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lin J & Wong KC Off-target predictions in CRISPR–Cas9 gene editing using deep learning. Bioinformatics 34, i656–i663 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chuai G et al. DeepCRISPR: optimized CRISPR guide RNA design by deep learning. Genome Biol. 19, 80 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang SX, Li XT, Lin QZ & Wong KC Synergizing CRISPR/Cas9 off-target predictions for ensemble insights and practical applications. Bioinformatics 35, 1108–1115 (2019). [DOI] [PubMed] [Google Scholar]
- 95.Luo JS, Chen W, Xue L & Tang B Prediction of activity and specificity of CRISPR-Cpf1 using convolutional deep learning neural networks. BMC Bioinformatics 20, 332 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kim D et al. Genome-wide target specificities of CRISPR RNA-guided programmable deaminases. Nat. Biotechnol. 35, 797–797 (2017). [DOI] [PubMed] [Google Scholar]
- 97.Rees HA & Liu DR Publisher Correction: Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 19, 801 (2018). [DOI] [PubMed] [Google Scholar]
- 98.Zuo EW et al. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science 364, 289–292 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jin S et al. Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science 364, 292–295 (2019). [DOI] [PubMed] [Google Scholar]
- 100.Grunewald J et al. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 569, 433–437 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rees HA, Wilson C, Doman JL & Liu DR Analysis and minimization of cellular RNA editing by DNA adenine base editors. Sci. Adv. 5, eaax5717 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhou CY et al. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature 571, 275–278 (2019). [DOI] [PubMed] [Google Scholar]
- 103.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nishida K et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 353, aaf8729 (2016). [DOI] [PubMed] [Google Scholar]
- 105.Kim D, Kim DE, Lee G, Cho SI & Kim JS Genome-wide target specificity of CRISPR RNA-guided adenine base editors. Nat. Biotechnol. 37, 430–435 (2019). [DOI] [PubMed] [Google Scholar]
- 106.Liang PP et al. Genome-wide profiling of adenine base editor specificity by EndoV-seq. Nat. Commun. 10, 67 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yu Y et al. Cytosine base editors with minimized unguided DNA and RNA off-target events and high on-target activity. Nat. Commun. 11, 2052 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Doman JL, Raguram A, Newby GA & Liu DR Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nat. Biotechnol. 38, 620–628 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kim D, Luk K, Wolfe SA & Kim JS Evaluating and enhancing target specificity of gene-editing nucleases and deaminases. Annu. Rev. Biochem. 88, 191–220 (2019). [DOI] [PubMed] [Google Scholar]
- 111.Clement K et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cho SW et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 24, 132–141 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kim D, Kim S, Kim S, Park J & Kim JS Genome-wide target specificities of CRISPR–Cas9 nucleases revealed by multiplex Digenome-seq. Genome Res. 26, 406–415 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Park SH et al. Highly efficient editing of the beta-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res. 47, 7955–7972 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gomez-Ospina N et al. Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat. Commun. 10, 4045 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pavel-Dinu M et al. Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat. Commun. 10, 1634 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vaidyanathan S et al. High-efficiency, selection-free gene repair in airway stem cells from cystic fibrosis patients rescues CFTR function in differentiated epithelia. Cell Stem Cell 26, 161–171 e164 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pedregosa E et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011). [Google Scholar]
- 119.Sledzinski P, Nowaczyk M & Olejniczak M Computational tools and resources supporting CRISPR–Cas experiments. Cells 9, 1288 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Park J, Lim K, Kim JS & Bae S Cas-analyzer: an online tool for assessing genome editing results using NGS data. Bioinformatics 33, 286–288 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Guell M, Yang L & Church GM Genome editing assessment using CRISPR Genome Analyzer (CRISPR-GA). Bioinformatics 30, 2968–2970 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.DiGiusto DL et al. Preclinical development and qualification of ZFN-mediated CCR5 disruption in human hematopoietic stem/progenitor cells. Mol. Ther. Methods Clin. Dev. 3, 16067 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fraietta JA et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 558, 307–312 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hacein-Bey-Abina S et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 302, 415–419 (2003). [DOI] [PubMed] [Google Scholar]
- 125.Miller JC et al. Enhancing gene editing specificity by attenuating DNA cleavage kinetics. Nat. Biotechnol. 37, 945–952 (2019). [DOI] [PubMed] [Google Scholar]
- 126.Schneider VA et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 27, 849–864 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sherman RM et al. Assembly of a pan-genome from deep sequencing of 910 humans of African descent. Nat. Genet. 51, 30–35 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cradick TJ, Ambrosini G, Iseli C, Bucher P & McCaffrey AP ZFN-site searches genomes for zinc finger nuclease target sites and off-target sites. BMC Bioinformatics 12, 152 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Iseli C, Ambrosini G, Bucher P & Jongeneel CV Indexing strategies for rapid searches of short words in genome sequences. PLoS ONE 2, e579 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mandell JG & Barbas CF 3rd Zinc finger tools: custom DNA-binding domains for transcription factors and nucleases. Nucleic Acids Res. 34, W516–W523 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Altschul SF, Gish W, Miller W, Myers EW & Lipman DJ Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- 132.Liu G, Zhang Y & Zhang T Computational approaches for effective CRISPR guide RNA design and evaluation. Comput. Struct. Biotechnol. J 18, 35–44 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.McKenna A & Shendure J FlashFry: a fast and flexible tool for large-scale CRISPR target design. BMC Biol. 16, 74 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sources are available at https://github.com/baolabrice/OT-review. Supplementary Tables 3, 4 and 5 contain the data used in the computational techniques performance assessment.