

Abstract
Although the pathogenesis of steroid-sensitive nephrotic syndrome (SSNS) remains elusive, multiple epidemiologic, clinical, and experimental studies converge on the common theme of immune dysregulation. Initially, T-cell adaptive immunity was solely emphasized; however, the role of humoral immunity in nephrotic syndrome has gained recognition. The study by Colucci and colleagues provides preliminary evidence that production of deglycosylated IgM that is unable to regulate T-cell function in the presence or absence of corticosteroid may be responsible for a steroid-dependence course in SSNS. This study provides invaluable insights into the mechanistic roles of both T-cell and B-cell responses in the pathogenesis and clinical course of SSNS.
Idiopathic nephrotic syndrome (NS) is the most common cause of glomerular disease in the pediatric population worldwide.1 The pathogenesis of nephrotic syndrome is not completely known; however, epidemiological, clinical, experimental animal, and cell culture studies show that the majority of cases are due to dysregulation of the immune system.2 What is not known is the relative contribution of T- and B-cell adaptive immunity to disease pathogenesis. Early studies by Shaloub2 suggest that dysregulation of the T-cell population is probably central to the pathogenesis of NS. Evidence for this suggestion includes (i) alterations in T-cell subsets during relapse of nephrotic syndrome, (ii) induction of remission of NS during active infections with measles and malaria, infectious agents that are known to suppress the T-cell population, (iii) NS manifesting as paraneoplastic syndrome in some lymphoreticular malignancies such as Hodgkin’s lymphoma, and last but not the least, (iv) the fact that many therapeutic agents that are used to induce or maintain remission in NS are modulators of T-cell functions.2 On the other hand, the role of B cells in disease pathogenesis, while less clear-cut, are becoming more relevant. Recent evidence suggests that B cells play a prominent role in disease pathogenesis, and therapies targeting B cells qualitatively and quantitatively may be potent tools in the treatment of NS. In addition, recent genetic studies implicate variants in major histocompatibility complex, class II, DQ alpha 1 (HLA-DQA1), other major histocompatibility class II loci, and phospholipase C gamma 2 (PLCG2) as important risk factors for NS and further support the importance of defective B-cell functions in the pathogenesis of the disease.1,3,4 Corticosteroid is the mainstay of treatment for NS, and the disease is classified as steroid-sensitive nephrotic syndrome (SSNS), frequent relapsing (FR), steroid dependent (SD), and steroid-resistant NS based on response to corticosteroid therapy. The reasons for variability in response to corticosteroid therapy in a disease in which dysregulation of immune cells is central to pathogenesis is largely unknown. The study by Colucci and colleagues5 in this issue provides novel insight into aspects of this disparate immunological response to corticosteroid in NS and possible convergence points for T- and B-cell dysregulation in disease pathogenesis.
In this study, the authors reported for the first time the atypical presence of IgM on the surface of T cells (T-cell IgM) in a cohort of children with NS and showed that this T-cell population is seen more abundantly in children with an SD course compared with children who had an infrequent relapsing SSNS or FR SSNS course. These cell populations were found to be present in children before commencement of therapy, suggesting that they may represent a biomarker for tailoring immunosuppression. The authors observed that this phenomenon was not associated with an abundance of IgM in the circulation; however, they found that in children with an SD clinical course, natural IgM, which was not antigenic specific, was less glycosylated compared with the infrequent relapsing and FR SSNS groups. The reduced glycosylation seems to promote more avid binding of IgM to T cells; reduced internalization of IgM, leading to uncontrolled T-cell activation and proliferation; and hyporesponsiveness to immunomodulatory effects of corticosteroids. Furthermore, a Cox proportional hazard regression model revealed that T-cell IgM intensity was predictive of relapse, treatment with anti-CD20 monoclonal antibody significantly reduced the T-cell IgM level, and repopulation of these T cells was associated with post-treatment relapse. Additionally, CD19+CD5+IgM+ B cells were significantly elevated in SSNS at disease onset, although the correlation with relapse was unclear. This B-cell subset has been known to produce natural IgM and has been found in other autoimmune diseases such as systemic lupus erythematous and IgA nephropathy.6 Thus the data emphasized additional roles for B cells in the pathogenesis of NS and provided evidence that B-cell defects also may be an important prognostic factor in NS. The data also demonstrated for the first time potential explanations about why B-cell modulating agents are useful in the treatment of SDNS. The model based on these data is that for some unknown reasons, B cells in a subset of patients with an NS and SD clinical course are prone to producing natural IgM with reduced glycosylation, reminiscent of what is seen in IgA nephropathy. The poorly glycosylated IgM is unable to regulate T-cell functions and also prevents T cells from responding to corticosteroid, leading to an SD clinical course (Figure 1). We congratulate the authors for this seminal observation to understand the mechanisms by which these unique T-cell populations may affect the pattern of therapy response.
Figure 1 |. Proposed mechanism of atypical IgM-bearing T cells in steroid-dependent (SD) nephrotic syndrome.
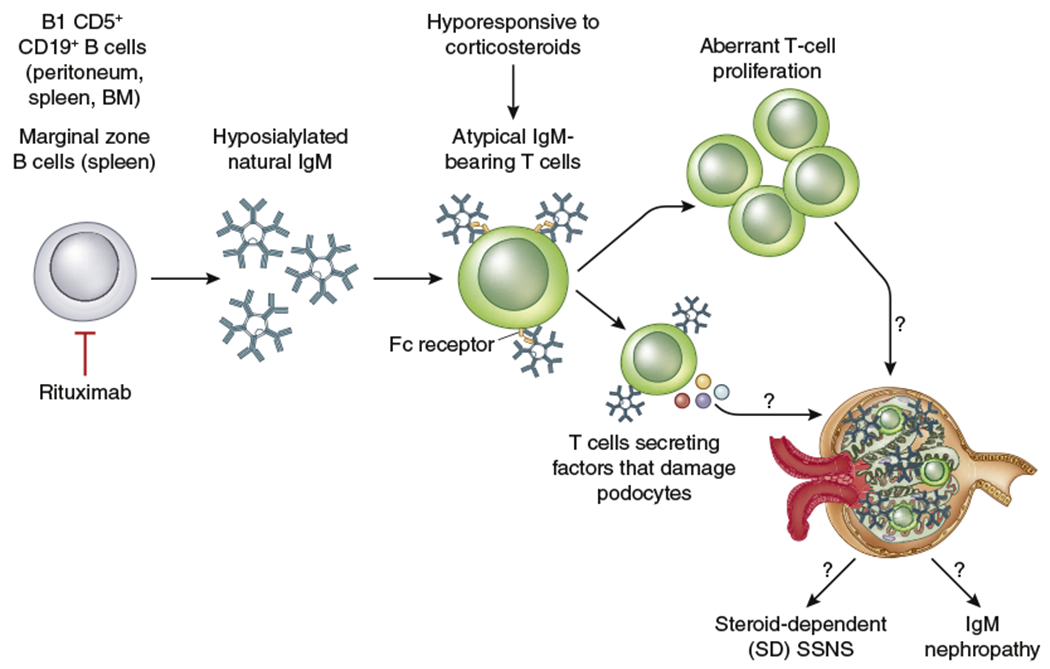
CD5+CD19+ B cells produce hyposialylated natural IgM that binds to T cells via the crystallizable fragment of immunoglobulin molecule (Fc) receptor. Internalization of natural IgM is impaired and remains on the cell surface. As a consequence, this T-cell population proliferates, secretes potential factors that cause podocyte injury, and is hyporesponsive to corticosteroids. In turn, this mechanism leads to a steroid-dependence course in children with steroid-sensitive nephrotic syndrome (SSNS). B-cell depletion with rituximab can reduce CD5+CD19+ B cells, IgM, and atypical IgM-bearing T cells to induce nephrotic syndrome remission. BM, bone marrow.
It will be fascinating to see if IgM-bearing T cells are also associated with the morphologic type of NS that some investigators have described as IgM nephropathy.7 IgM nephropathy is characterized by granular or diffuse deposition of IgM in the mesangium and mesangial proliferation and in some cases accumulation of extracellular mesangial matrix.7 The majority of patients with this histologic picture tend to have an FR and SD course. If this is the case, it is possible that this unique T-cell population with poorly glycosylated IgM could be identified in renal biopsies and therefore could be associated with IgM nephropathy (Figure 1). Indeed, in this study, the authors provided indirect evidence for this outcome because they showed in vitro that T cells incubated with nonglycosylated IgM secrete factors that result in podocyte injury and proteinuria.5
This work provides an exciting catalyst for future investigation. For example, it is unknown why some patients will have B-cell populations that produce poorly glycosylated IgM. Certain individuals may be genetically prone to this phenomenon because it has been described in IgA nephropathy, or defective glycosylation may be the second hit for NS, the first hit being variations in genes encoding for major histocompatibility complex II molecules.8 The differences between FR and SD in the T-cell IgM population are puzzling because clinically both subtypes of NS behave similarly. Lastly, natural IgM through crystallizable fragment of immunoglobulin molecule (Fc) receptor binding also modulates B-cell function and homeostasis.6,9 Similar to what the authors found with T cells, nonglycosylated IgM also could dysregulate B cells, which deserves further examination. From these insights, there is a need for a comprehensive, prospective study to look at the role of defective IgM glycosylation, as well as IgM-bearing B- and T-cell subsets, in relation to the pattern of corticosteroid response in NS and other glomerular diseases. Furthermore, we also can use the same approach reported in this study to determine the biological basis for variable response to other immunosuppressive agents used in the treatment of NS. In conclusion, if reproduced in larger studies, IgM-bearing T-cell populations, CD19+CD5+IgM+ B cells, and/or glycosylation state of IgM in patients with NS may represent clinically relevant biomarkers or potential therapeutic targets for personalized monitoring and immunosuppressive strategies.
ACKNOWLEDGMENTS
Funding was provided by NIH NIDDK grants 5R01DK098135 and 5R01DK094987 and by the Doris Duke Clinical Scientist Development Award to RG and the Duke Health Scholars Award to ETC.
Footnotes
DISCLOSURE
All the authors declared no conflicts of interest.
REFERENCES
- 1.Gbadegesin RA, Adeyemo A, Webb NJ, et al. HLA-DQA1 and PLCG2 are candidate risk loci for childhood-onset steroid-sensitive nephrotic syndrome. J Am Soc Nephrol. 2015;26:1701–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shalhoub RJ. Pathogenesis of lipoid nephrosis: a disorder of T-cell function. Lancet. 1974;2:556–560. [DOI] [PubMed] [Google Scholar]
- 3.Debiec H, Dossier C, Letouzé E, et al. Transethnic, genome-wide analysis reveals immune-related risk alleles and phenotypic correlates in pediatric steroid-sensitive nephrotic syndrome. J Am Soc Nephrol. 2018;29:2000–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jia X, Horinouchi T, Hitomi Y, et al. Strong association of the HLA-DR/DQ locus with childhood steroid-sensitive nephrotic syndrome in the Japanese population. J Am Soc Nephrol. 2018;29:2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colucci M, Carsetti R, Rosado MM, et al. Atypical IgM on T cells predict relapse and steroid dependence in idiopathic nephrotic syndrome. Kidney Int. 2019;96:971–982. [DOI] [PubMed] [Google Scholar]
- 6.Wang H, Coligan JE, Morse HC. Emerging functions of natural IgM and its Fc receptor FCMR in immune homeostasis. Front Immunol. 2016;7:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen AH, Border WA, Glassock RJ. Nephrotic syndrome with glomerular mesangial IgM deposits. Lab Invest. 1978;38:610–619. [PubMed] [Google Scholar]
- 8.Kiryluk K, Li Y, Moldoveanu Z, et al. GWAS for serum galactose-deficient IgA1 implicates critical genes of the O-glycosylation pathway. PLoS Genet. 2017;13:e1006609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakamura T, Kubagawa H, Ohno T, Cooper MD. Characterization of an IgM Fc-binding receptor on human T cells. J Immunol. 1993;151:6933–6941. [PubMed] [Google Scholar]