

Abstract
To better understand primary and recall T cell responses during coronavirus disease 2019 (COVID-19), it is important to examine unmanipulated severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-specific T cells. By using peptide-human leukocyte antigen (HLA) tetramers for direct ex vivo analysis, we characterized CD8+ T cells specific for SARS-CoV-2 epitopes in COVID-19 patients and unexposed individuals. Unlike CD8+ T cells directed toward subdominant epitopes (B7/N257, A2/S269, and A24/S1,208) CD8+ T cells specific for the immunodominant B7/N105 epitope were detected at high frequencies in pre-pandemic samples and at increased frequencies during acute COVID-19 and convalescence. SARS-CoV-2-specific CD8+ T cells in pre-pandemic samples from children, adults, and elderly individuals predominantly displayed a naive phenotype, indicating a lack of previous cross-reactive exposures. T cell receptor (TCR) analyses revealed diverse TCRαβ repertoires and promiscuous αβ-TCR pairing within B7/N105+CD8+ T cells. Our study demonstrates high naive precursor frequency and TCRαβ diversity within immunodominant B7/N105-specific CD8+ T cells and provides insight into SARS-CoV-2-specific T cell origins and subsequent responses.
Keywords: COVID-19, SARS-CoV-2-specific CD8+, T cells, TCR, immunodominant
Graphical abstract
Examining unmanipulated SARS-CoV-2-specific T cells is important for understanding primary and recall responses during COVID-19. Nguyen et al. analyze ex vivo CD8+ T cells specific for SARS-CoV-2 epitopes and find that immunodominant B7/N105-specific CD8+ T cells are present at high frequencies in blood samples from unexposed, acute COVID-19, and convalescence individuals, which is underpinned by diverse TCR repertoires.
Introduction
As global research efforts are moving at record speed to develop and evaluate severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) vaccines, concurrent efforts are needed to provide an understanding of the optimal immune responses in coronavirus disease 2019 (COVID-19), thus enabling the rational design of long-lasting and broadly protective vaccines and immunotherapies. Virus-specific CD8+ T cells can greatly ameliorate recovery from respiratory diseases such as avian influenza, SARS, and Middle East respiratory syndrome (MERS) (Channappanavar et al., 2014; Wang et al., 2015; Wang et al., 2018; Zhao et al., 2017) and persist long-term as cross-reactive memory pools (Koutsakos et al., 2019). Therefore, understanding human epitope-specific CD8+ T cell responses to SARS-CoV-2 directly ex vivo is needed to better define the role of CD8+ T cells in primary SARS-CoV-2 infection, long-term memory persistence, and their subsequent recall after re-infection and/or vaccination.
SARS-CoV-2-specific CD8+ T cells can be elicited during primary COVID-19 and survive into short-term convalescence (Dan et al., 2021). Activated CD8+ T cells, displaying a broad range of cytotoxic molecules, appear in patients’ blood prior to recovery (Koutsakos et al., 2021; Thevarajan et al., 2020), suggesting involvement of CD8+ T cells in the resolution of COVID-19. Activated T cells directed toward spike (S), membrane (M), nucleocapsid (N) and other open reading frames can be detected in ∼70% of acute and convalescent COVID-19 patients by utilizing human leukocyte antigen (HLA) class I and II predicted peptide “megapools” to stimulate peripheral blood mononuclear cells (PBMCs) (Grifoni et al., 2020; Weiskopf et al., 2020). Stimulation of PBMCs from COVID-19 patients with SARS-CoV-2 overlapping peptides also leads to interferon (IFN)-γ production and clonal expansion of CD8+ T cells in vitro. Thus, both ex vivo and in vitro studies indicate that CD8+ T cells get activated during primary SARS-CoV-2 infection.
SARS-CoV-2 CD8+ T cell epitopes (peptides + major histocompatibility complex [MHC]-I) are identified for dominant HLA class I alleles, including HLA-A∗01:01, A∗02:01, A∗03:01, A∗11:01, HLA-B∗07:02, B∗27:05, B∗40:01, and B∗44:03, by using both peptide stimulations and peptide-MHC class I tetramers (Ferretti et al., 2020; Habel et al., 2020; Peng et al., 2020; Schulien et al., 2021). Identification of SARS-CoV-2 CD8+ T cell epitopes allows accurate determination of the magnitude and phenotype of SARS-CoV-2-specific CD8+ T cells directly ex vivo in COVID-19 patients and in pre-pandemic PBMCs. It also allows us to precisely define the persistence of long-term memory CD8+ T cells and their recall after SARS-CoV-2 re-infection and/or vaccination. Ex vivo frequencies of SARS-CoV-2-specific CD8+ T cells appear to be present mainly in the range of ∼1 × 10−5 to 5 × 10−5 in the CD8+ T cell population, which is ∼2- to 10-fold lower than the frequency of long-term memory CD8+ T cells specific for influenza or Epstein-Barr virus (EBV). HLA-A∗02:01-restricted SARS-CoV-2 epitopes appear to exhibit the lowest frequency of ∼1 × 10−5 in the CD8+ T cell population (Habel et al., 2020; Peng et al., 2020). Direct ex vivo phenotypic analysis of A2/S269 +CD8+ T cells from COVID-19 convalescent individuals revealed that A2/S269 +CD8+ T cells were suboptimally stimulated and composed of naive, stem cell memory, and central memory T cells rather than effector memory populations. In contrast, the HLA-B∗07:02-restricted N105-113 epitope (B7/N105) appears to be the most dominant SARS-CoV-2 CD8+ T cell specificity known to date (Ferretti et al., 2020; Peng et al., 2020; Schulien et al., 2021), applicable to 1 in 12 people globally (Gonzalez-Galarza et al., 2020). The question remains, however, whether these immunodominant B7/N105 +CD8+ T cell responses arise from pre-existing memory pools established after infection with a cross-reactive seasonal human coronavirus or other pathogens, or whether they represent a high frequency of naive pools found across HLA-B∗07:02-expressing individuals.
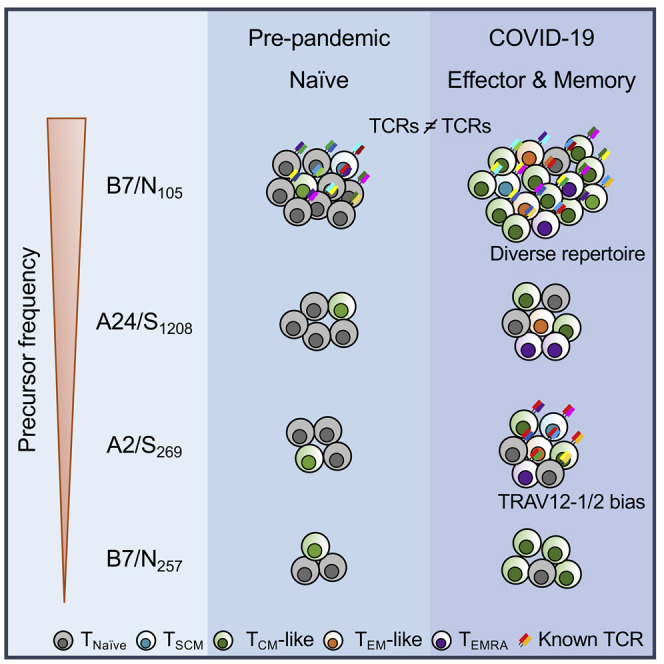
To provide an in-depth understanding of CD8+ T cell responses directed at the immunodominant B7/N105 epitope, we performed direct ex vivo analyses in PBMCs from COVID-19 patients as well as in pre-pandemic PBMCs, tonsils, lungs, and spleens. With ex vivo tetramer enrichment, single-cell reverse transcriptase (RT)-PCR, and in vitro IFN-γ/tumor necrosis factor (TNF) intracellular cytokine secretion (ICS) assay, we determined the magnitude, naive/effector/memory phenotype, and molecular T cell receptor (TCR)αβ signatures. Overall, we assessed CD8+ T cells directed at 4 SARS-CoV-2 epitopes: B7/N105, B7/N257, A2/S269, and A24/S1208. In contrast to the 3 subdominant SARS-CoV-2-specific CD8+ T cell populations, B7/N105-specific CD8+ T cell pools were numerically immunodominant by 4.89- to 38.03-fold in both COVID-19 patients and pre-pandemic donors. Tetramer-specific CD8+ T cell populations directed at all 4 SARS-CoV-2 epitopes were predominantly of a naive phenotype in 22 of the 27 pre-pandemic donors. Together with the TCR data, the findings support our conclusion that high precursor frequency and promiscuity in αβ TCR pairing could underpin CD8+ T cell responses to an immunodominant SARS-CoV-2 nucleocapsid epitope.
Results
COVID-19 patient cohort and pre-pandemic controls
We recruited a total of 21 COVID-19 subjects, including 8 acute hospitalized patients and 13 convalescent COVID-19 patients (Figure 1 A; Table S1). Of the 8 patients hospitalized, 1 ICU and 3 ward patients required oxygen support. Ten of the convalescents were community SARS-CoV-2 infections. All COVID-19 patients seroconverted for SARS-CoV-2 antibodies. The median age of COVID-19 patients was 54 years, and 33% of patients were females. As controls, we analyzed pre-pandemic PBMC and tonsil samples from 31 subjects across 3 age groups: 4 children (median age 6 years, range 3–15), 20 adults (median age 46 years, range 24–63) and 7 elderly (median age 72 years, range 65–76), 37.6% were females. Additionally, we tested pre-existing B7/CD8+ T cell populations in lung and spleen tissues from 9 HLA-B7 individuals (median age 46 years, range 22–63).
Figure 1.

CD4+ and CD8+ T cell responses to SARS-CoV-2 overlapping peptide pools and individual SARS-CoV-2 HLA-B∗07:02-restricted peptides
(A) Overview of cohort and samples collected.
(B) CD4+ and CD8+ T cell responses to SARS-CoV-2 S, N, and M peptide pools in COVID-19 cases and suspected cases. Representative fluorescence-activated cell sorting (FACS) plots showing IFN-γ and TNF staining of CD4+ or CD8+ T cell populations; background staining values are shown in brackets. Frequencies of IFN-γ+, TNF+, MIP-1β+, or CD107a+ within the CD4+ or CD8+ T cells, with background staining subtracted in COVID-19 cases (n = 3, 2 time points each) and suspected cases (n = 4); data are shown as mean with SD. Statistical significance was determined with Mann-Whitney test.
(C) FACS plots showing IFN-γ staining of CD4+ or CD8+ T cell populations from COVID-19 case CA2 V1 and V2 after stimulation with S, N, and M peptide pools; the number of IFN-γ+ cells are shown. Paired frequencies of IFN-γ+CD4+ and CD8+ T cells for S, N, and M peptide pools (right).
(D) FACS plots of CD8+ IFN-γ/TNF staining after stimulation with individual N-derived SARS-CoV-2 peptides (N66, N105, N257, N219, and N222) and FACS plots showing CD8+ T cell staining with B7/SARS-CoV-2 tetramers (B7/N66, B7/N105, B7/N257) on expanded cells. Frequency of IFN-γ+CD8+ or tetramer+CD8+ T cells are shown.
Within this study, a traveler cohort included 3 male SARS-CoV-2 cases (a 19-year-old CA3 index case with moderate COVID-19, 1 21-year-old CA2 individual with mild symptoms, and 1 24-year-old CA1 individual who remained asymptomatic; Table S1). Four asymptomatic suspected cases (SU1, SU2, SU3, SU4; all 20 years old) were also investigated. All were returned travelers. Receptor binding domain (RBD) and spike immunoglobulin (Ig)G antibody levels were significantly (p < 0.05) higher in cases (log10 median titer 3.195 RBD IgG; 3.237 spike IgG) than in suspected cases (log10 median titer 1.762 RBD IgG; 1.994 spike IgG) and in healthy unexposed individuals (log10 median titer 2.024 RBD IgG; 2.239 spike IgG) (Figure S1A). IgM titers in COVID-19 cases were significantly elevated in comparison with those found in healthy unexposed individuals (Figure S1A). RBD- and spike-reactive B cell responses were concordant with the antibody titers and showed significantly increased frequencies of RBD- and spike-specific B cells in COVID-19 cases when compared to the suspected cases (Figures S1B–S1D). Minimal immune cell activation across a broad range of cell populations (CD56+ NK cells, CD3+ γδ T cells, CD8+ T cells, CD4+ T cells, antibody-secreting cells [ASCs], T follicular helper cells [Tfhs], and monocytes), low granzyme/perforin levels within CD8+ and CD4+ T cells, and normal inflammatory milieu (Figure S2) verified lack of acute COVID-19 in the traveler cohort.
CD4+ and CD8+ T cell responses react to SARS-CoV-2 overlapping peptide pools
Probing SARS-CoV-2-specific CD4+ and CD8+ T cells in the traveler cohort was performed by using overlapping SARS-CoV-2 peptide pools spanning the entire N and M proteins and the immunogenic regions of S protein, followed by an ICS assay (Figure 1B). PBMCs were stimulated with 1 peptide pool, expanded for 10 days before the analysis of SARS-CoV-2-reactive T cells by ICS for intracellular IFN-γ, TNF, MIP-1β, and CD107a (Figure 1B; Figure S3A). In agreement with the antibody and B cell data, SARS-CoV-2-reactive CD4+ and CD8+ T cells were detected in COVID-19 cases but not in the suspected cases, with CD4+ T cells generally dominating over CD8+ T cell populations, as previously reported (Habel et al., 2020). No differences were observed between the responses against S, N, and M proteins for both CD4+ and CD8+ T cells (Figure 1B, right panel). The exception was CA2 with highly prominent CD8+ T cell responses directed toward the N peptide pool (IFN-γ production up to ∼25% of CD8+ T cells), markedly above IFN-γ-producing CD4+ T cells (range 0.24%–1.15%) (Figure 1C). Because CA2 was HLA-B∗07:02+ and HLA-A∗02:01+ (Table S1), we further dissected the CD8+ T cell response to 5 previously reported immunogenic peptides derived from N and restricted by both HLA-B∗07:02 and HLA-A∗02:01 (B7/N66, B7/N105, B7/N257, A2/N219, and A2/N222) (Ferretti et al., 2020; Schulien et al., 2021). In CA2, immunodominant CD8+ T cell responses were directed toward B7/N105, whereas subdominant CD8+ T cell responses were against B7/N257, as shown by both IFN-γ production toward those peptides and B7/N105, B7/N257, and B7/N66 tetramer staining (Figure 1D; Figure S3B).
Immunodominant B7/N105+CD8+ T cells possess a high precursor frequency
To determine the magnitude of SARS-CoV-2-specific CD8+ T cell populations in 19 COVID-19 patients and 23 pre-pandemic unexposed individuals expressing HLA-B∗07:02, HLA-A∗02:01, and/or HLA-A∗24:02, we used tetramer-associated magnetic enrichment (TAME) directly ex vivo (Nguyen et al., 2017b; Valkenburg et al., 2016) for the immunodominant B7/N105 and subdominant B7/N257, A2/S269 (Habel et al., 2020), and A24/S1208 (Ferretti et al., 2020). After validating tetramers in peptide-expanded T cell lines (as shown in Figure 1D), further ex vivo tetramer validation experiments were performed with direct ex vivo dual-tetramer staining of the immunodominant B7/N105 tetramers conjugated to phycoerythrin (PE) and allophycocyanin (APC) fluorophores, as well as dual TAME with B7/N105 tetramer PE and an irrelevant B7/EBV tetramer (EBNA-3379-387, RPPIFIRRL) conjugated to APC (Figures S4A and S4B). Following TAME (Figure 2 ), CD8+ T cells specific for the immunodominant B7/N105 epitope could be readily detected ex vivo in all COVID-19 patients (Figure 2A; Figure S4C) at a mean frequency of 6.88 × 10−4 (n = 11; Figure 2D), with the B7/N105 +CD8+ T cells being easily detected without enrichment (Figure 2A). B7/N105-specific CD8+ T cell pools in COVID-19 patients were numerically immunodominant when compared to 3 subdominant SARS-CoV-2-specific CD8+ T cell populations directed at B7/N257, A2/S269, and A24/S1208 epitopes (by 38.03-, 21.54-, and 8.92-fold respectively; p < 0.05; Figure 2D).
Figure 2.

High precursor frequency for immunodominant B7/N105-specific CD8+ but not subdominant B7/N257-, A2/S269-, and A24/S1208-specific CD8+ T cells
(A and B) B7/N105-, B7/N257-, A2/S269-, and A24/S1208-specific CD8+ T cells were identified directly ex vivo from COVID-19 PBMCs, as well as from healthy pre-pandemic PBMCs and tonsils by tetramer magnetic enrichment. Representative FACS plots of enriched and unenriched B7/N105-, B7/N257-, A2/S269-, and A24/S1208-specific CD8+ T cells from (A) acute and convalescent COVID-19 donors and (B) pre-pandemic children (PBMCs and tonsils), adult, and elderly donors. Pre-pandemic A2/S269 donors from Habel et al. [2020].
(C) B7/N105-specific CD8+ T cell precursor frequencies were calculated for matched children’s PBMC and tonsil samples, with statistical significance analyzed by Wilcoxin test.
(D) CD8+ precursor frequencies were calculated for B7/N105-, B7/N257-, A2/S269-, and A24/S1208-specific CD8+ T cells enriched from PBMCs and tonsils. Dots represent individual donors; data are shown as mean. Donors with undetectable precursor frequencies are included on the graph to show the number of donors tested; these donors were not included in statistical analyses. Statistical significance was determined with Dunn’s multiple comparison test unless indicated otherwise.
See also Figure S4.
The frequency of B7/N105-specific CD8+ T cells in COVID-19 patients was significantly higher than that in adult (mean 3.00 × 10−5; p = 0.0017) and elderly (mean 6.76 × 10−5; p = 0.0023) pre-pandemic PBMC samples (Figures 2B and 2D), suggesting clonal expansion after SARS-CoV-2 infection. The values for pre-pandemic children’s tonsil samples (mean 2.76 × 10−4) and elderly PBMCs (mean 6.76 × 10−5) were variable, with some elderly individuals showing a decline in the magnitude of B7/N105-specific CD8+ T cells, most likely reflecting a loss of tetramer-positive CD8+ T cells with age (Nguyen et al., 2018). Moreover, the magnitude of the immunodominant B7/N105 in combined pre-pandemic adult and elderly PBMCs (mean 4.64 × 10−5) was higher than that for the subdominant B7/N257 (mean 1.52 × 10−6; p < 0.0001), the previously described A2/S269 frequencies (Habel et al., 2020) (mean 1.65 × 10−6; p = 0.0022), and A24/S1208, although not statistically significant (mean 9.5 × 10−6; p = 0.2484 Mann-U) (Figure 2D), suggesting that the immunodominance of B7/N105-specific CD8+ T cell responses in COVID-19 reflects higher precursor frequencies in unexposed individuals. Although the frequency of SARS-CoV-2-specific B7/N105 tetramer-positive CD8+ T cells in children’s pre-pandemic tonsils (mean 2.76 × 10−4) appeared higher than that found in matched PBMCs (mean 2.5 × 10−5) (Figure 2C), this difference was not statistically significant (p = 0.125). Analysis of spleen and lung samples did not show any robust distinct populations of B7/N105-specific CD8+ T cells in pre-pandemic tissues (Figure S4D). It thus seems that the immunodominant B7/N105-specific CD8+ T cells are present in high precursor frequencies in pre-pandemic adult PBMCs and increase by 14.82-fold during SARS-CoV-2 infection in the peripheral blood.
SARS-CoV-2 tetramer-positive CD8+ T cells in pre-pandemic individuals show predominantly naive phenotype
The activation profiles of B7/N105-specific CD8+ T cells tested by tetramer staining directly ex vivo from COVID-19 patients and pre-pandemic donors across different ages were assessed by CD27, CD45RA, and CD95 staining to determine the prevalence of the naive (TNaive) (CD27+CD45RA+CD95−), stem cell memory (TSCM) (CD27+CD45RA+CD95+), central memory (TCM)-like (CD27+CD45RA−), effector memory (TEM)-like (CD27−CD45RA−), and effector memory CD45RA (TEMRA) (CD27−CD45RA+) subsets (Figure 3 ). As expected, COVID-19 donors displayed the lowest proportion of TNaive B7/N105 +CD8+ T cells (mean of 10%) and instead had the highest proportion of TCM-like B7/N105 +CD8+ T cells (mean of 61%). Conversely, pre-pandemic children and adult B7/N105 +CD8+ T cells were predominantly of the TNaive phenotype (p < 0.0001 for both age groups in comparison with COVID-19 samples) and had minimal activation profiles across all the T cell subsets tested (Figure 3C), indicating that these B7/N105 +CD8+ T cells constitute a naive precursor pool rather than a pre-existing memory population from exposure to other coronaviruses or pathogens. Although cross-reactive memory T cell responses between human coronaviruses and SARS-CoV-2 have been described after stimulation of pre-pandemic PBMCs with peptide megapools and readouts by IFN-γ production, these T cells usually express distinct memory markers (Le Bert et al., 2020; Peng et al., 2006), indicating that SARS-CoV-2 tetramer-reactive T cells in our pre-pandemic samples are truly naive. Both TNaive and TCM-like subsets varied across different elderly individuals, suggesting that B7/N105 +CD8+ T cells in some elderly individuals were either previously activated by cross-reactive peptides or underwent age-related dysregulated homeostatic proliferation, as commonly observed with age (LeMaoult et al., 2000; Messaoudi et al., 2006; Valkenburg et al., 2012).
Figure 3.

Ex vivo activation profiles of SARS-CoV-2-specific CD8+ T cells in COVID-19 and pre-pandemic donors
(A and B) Representative FACS plots of B7/N105-, B7/N257-, A2/S269-, and A24/S1208-specific CD8+ T cells from (A) acute and convalescent COVID-19 donors and (B) pre-pandemic children (PBMCs and tonsils), adult, and elderly donors showing TNaive (CD27+CD45RA+CD95−), TSCM (CD27+CD45RA+CD95+), TCM-like (CD27+CD45RA−), TEM-like (CD27−CD45RA−), and TEMRA (CD27−CD45RA+) subsets. To account for donor variability in the phenotype markers and day-to-day variations in flow cytometry settings (e.g., laser power, compensation, calibration), phenotype gates were first selected on the larger parent CD8+ T cell population for each individual before applying the gates to the smaller tetramer+ population. The same gates were used across time points from the same individual if they were acquired on the same day.
(C) Stacked plots display the proportion of each phenotype subset within the B7/N105-, B7/N257-, A2/S269-, and A24/S1208-specific CD8+ T cells. Only donors above the detection limit (≥10 tetramer-positive-enriched events) were included for analysis; mean with SD is shown, and statistical significance was determined with Tukey’s multiple comparisons test.
Naive profiles in pre-pandemic subdominant B7/N257-, A2/S269-, and A24/S1208-specific CD8+ T cells largely reflected those found for the immunodominant pre-pandemic B7/N105-specific CD8+ T cells (Figure 3C). In COVID-19 patients, activation profiles of subdominant B7/N257-specific CD8+ T cells were similar to those in immunodominant B7/N105-specific CD8+ T cells and consisted largely of TCM phenotypes. In contrast, A2/S269-specific CD8+ and A24/S1208-specific CD8+ T cells in COVID-19 were more skewed toward TNaive, TSCM, and/or TEMRA phenotypes.
Frequencies and phenotypes of SARS-CoV-2-specific B7/N105+CD8+ T cells remain largely stable in convalescent COVID-19 individuals
To assess whether the high numbers and TCM phenotype of B7/N105-specific CD8+ T cells changed or remained stable over time, we longitudinally sampled HLA-B∗07:02-expressing COVID-19 individuals from acute to convalescent time points for up to 270 days post-disease onset (n = 9 COVID-19 patients, 22 samples). The longitudinal frequencies of B7/N105-specific CD8+ T cells remained stable and relatively high, whereas the very low numbers of subdominant B7/N257-specific CD8+ T frequencies, many below the detection limit (<10 tetramer-positive-enriched events), did not expand over time (Figures 4A and 4C). Individual phenotypes were then tracked over time and across age (Figures 4B and 4C), which generally showed very stable populations of TCM-like B7/N105-specific CD8+ T cells in most COVID-19 individuals, as well as in 1 individual with longitudinal B7/N257-specific CD8+ T cell responses (Figure 4B). However, TEMRA-like populations increased over time in 4 individuals (CA6, 10, 11, and 13), 3 out of 4 being elderly donors (65 years and over), whereas TSCM populations either increased or decreased in certain individuals. Overall, apart from some fluctuations in TSCM- and TEMRA-like populations, both the frequencies and TCM-like phenotypes remained stable over time in convalescent individuals up to 270 days post-disease onset.
Figure 4.

Tracking longitudinal COVID-19 B7/CD8+ T cell responses up to day 270 post disease onset
(A) TAME-enriched precursor frequencies of B7/N105-specific CD8+ and B7/N257-specific CD8+ T cells from each individual with multiple time points.
(B) Stacked bar graphs of each individual phenotype profiles of each donor across age.
(C) Representative FACS plots of donors’ longitudinal B7/N105-specific CD8+ T cell responses in terms of TAME-enriched (i) tetramer and (ii) phenotype profiles.
CD8+ T cells specific for B7/N105 display diverse TCRαβ repertoire and promiscuous TCRα-TCRβ pairing
The nature of the TCR repertoire can affect CD8+ T cell immunodominance, functionality, and protection (Messaoudi et al., 2002; Ndhlovu et al., 2015; Price et al., 2009). We therefore defined the molecular signatures underpinning the immunodominant B7/N105 +CD8+ T cell response. We dissected TCRαβ clonal composition and diversity with direct ex vivo tetramer staining and human single-cell TCRαβ multiplex RT-PCR, as described previously (Nguyen et al., 2017a; Valkenburg et al., 2016). Here, we showed ex vivo dissection of TCRαβ repertoires for immunodominant B7/N105-specific CD8+ T cells in PBMCs from 4 HLA-B∗07:02-expressing COVID-19 patients and 4 pre-pandemic PBMC samples. We examined a total of 264 B7/N105-specific CD8+ T cells TCRαβ pairs in total (Figure 5 ; Table S2).
Figure 5.

Diverse TCRαβ repertoire and promiscuous TCRα-TCRβ pairing within B7/N105-specific CD8+ T cells
(A and B) B7/N105-specific CD8+ T cells were enriched by TAME and then single-cell sorted for TCRαβ analysis. Pie charts of TRBV and TRAV gene usage in B7/N105+CD8+ T cells in (A) COVID-19 (n = 4) and (B) pre-pandemic donors (n = 4). All COVID-19 patients were from convalescent samples with one exception, where 5 out of 36 TCR clonotypes for Donor CA12 were from the acute time point. Segments shown by the same color represent TCRαβ clonotypes with the same V segment usage but different CDR3 sequences.
(C) Circos plots of TRBV and TRAV clonotype pairings; left arch and segment color indicate TRBV usage, and right outer arch color depicts TRAV usage.
(D) Bubble plot showing the distribution (number of donors and frequency) of TRBV/TRAV gene usage in COVID-19 patients.
(E) Dominant clonotypes identified in HLA-B∗07:02 donors specific to B7/N105+CD8+ T cells. ND, not determined.
See also Table S2.
TCRαβ sequences were first analyzed for the overall TCRαβ diversity on a per-individual basis (Figure 5A). Segments shown by the same color represent TCRαβ clonotypes with the same V segment usage but different CDR3 sequences. The B7/N105-specific CD8+ TCRαβ repertoire of each COVID-19 patient was generally diverse, with the presence of 1 to 3 largely expanded TCRαβ clonotypes and a number (mean of 19 per donor) of smaller TCRαβ clonotypes. Clone B (TRBV6-6/TRAV17) was detected in 2 COVID-19 patients (Figures 5A and 5E), thus representing a shared TCRαβ clonotype for the B7/N105 +CD8+ repertoire. Apart from clone B, each donor had distinct usage of TRAV, TRBV, TRAJ, and TRBJ gene segments, with no common gene features found across the COVID-19 patients and no common motifs within CDR3α and CDR3β sequences (Figure 5E; Table S2). Similarly, the pre-pandemic B7/N105-specific CD8+ repertoires were highly diverse between donors, with no predominant TCRαβ V-J gene sharing between them (Figure 5B).
Lack of common TCRαβ segment features was even more evident when we analyzed the TCRαβ repertoire on a per-individual basis with circos plots (Figure 5C), with the color of the segment indicating TRBV gene usage (on the left), and the pairing with a specific TRAV segment depicted by the color of the outer arch (on the right). Again, our circos analysis revealed a remarkable level of diversity in TRAV and TRBV usage within the individual COVID-19 patients and across the donors for both COVID-19 and pre-pandemic, suggesting the plasticity of TCRα-TCRβ pairings. Across all COVID-19 and pre-pandemic TCRs, only 1 COVID-19 patient (CA4, 2 clonotypes) shared the same TRAV-TRBV pairing (TRBV29-1/TRAV13-1) with 1 pre-pandemic individual (PP24, 8 clonotypes), with very similar CDR3αβ regions (Figure 5E). To further investigate TCRα-TCRβ pairings, we represented TRAV versus TRBV gene usage in a bubble plot for COVID-19 patients. We found 26 different TRBVs capable of pairing across 31 distinct TRAVs gene segments (Figure 5D). This shows that the TCRαβ diversity in B7/N105-specific CD8+ T cells arises from distinct TCRαβ gene segment usage and TRAV-TRBV pairings both across and within the donors and not from being associated with any CDR3α or CDR3β motifs across different donors. These data suggest that promiscuity in TCRα-TCRβ pairing and CDR3α-CDR3β sequences (rather than stringent requirements for TCRαβ common features) underlie high naive precursor frequency within immunodominant B7/N105-specific CD8+ T cells, capable of expansion after SARS-CoV-2 infection.
A2/S269+CD8+ TCRαβ repertoire contains common TRAV/TRAJ motifs
Because CD8+ T cells directed at SARS-CoV-2-specific A2/S269 represent subdominant and suboptimal T cell specificity in COVID-19 (Habel et al., 2020), we also investigated ex vivo TCRαβ repertoires within A2/S269-specific CD8+ T cells and how it differs to that of immunodominant B7/N105-specific CD8+ T cells. As we and others previously reported (Habel et al., 2020; Peng et al., 2020), A2/S269-specific CD8+ T cells detected ex vivo with tetramers represented a small but distinct population in COVID-19 patients (n = 5; Figure 3A). The TCRαβ repertoire displayed a considerable level of diversity by both circos and bubble plots (Figures 6 A–6C), although common TRBV (43% of total 175 TCRs: TRBV2 at 11%, TRBV7-9 at 13%, TRBV20-1 at 20%), TRBJ (68% of total TCRs: TRBJ2-2 at 59%, TRBJ2-7 at 9%), TRAV (32% of total TCRs: TRAV12-1 at 23%, TRAV12-2 at 3%, TRAV14/DV4 at 6%), and TRAJ (24% of total TCRs: TRAJ43 at 18%, TRAJ30 at 6%) gene segments were found across different COVID-19 patients (Figures 6A and 6B; Table S3). More importantly, 2 key TCRα motifs within the CDR3α loop were found across the COVID-19 patients (Figure 6D). These included (1) TRAV12-1/TRAJ43 CVVNXXXDMRF motif (where X denotes any amino) found across all COVID-19 patients (16% of total TCR repertoire), paired with different prominent TRBV segments, and (2) TRAV12-2/TRAJ30 CAVNXDDKIIF pairing with TRBV7-9 across 4 donors (except CA6) (3.4% of TCR repertoire), as also recently reported by Shomuradova and colleagues (Shomuradova et al., 2020). These findings suggest that, in contrast to the B7/N105-specific CD8+ repertoire with no common motif and lack of shared TRBV, TRBJ, TRAV, or TRAJ gene segment usage, TCRαβ clonotypes within the subdominant A2/S269-specific CD8+ T cells had dominant TRBV, TRBJ, TRAV, or TRAJ gene segment usage across the donors and well-defined CDR3α motifs, suggesting more rigid requirements for TCRαβ clones capable of recognizing A2/S269 epitopes. Such lack of TCRαβ plasticity most likely results in such low naive precursor frequency (Habel et al., 2020).
Figure 6.

Common TRAV/TRAJ motifs within suboptimal A2/S269-specific CD8+ TCRαβ repertoire
A2/S269+CD8+ T cells from COVID-19 PBMCs were identified ex vivo and enriched by TAME before single-cell sorting for TCRαβ analysis.
(A) Pie charts of TRBV and TRAV gene usage in A2/S269+CD8+ T cells in acute and convalescent COVID-19 donors. Segments shown by the same color represent TCRαβ clonotypes with the same V segment usage but different CDR3 sequences.
(B) Circos plots of TRBV and TRAV clonotype pairings; left arch and segment color indicates TRBV usage, and right outer arch color depicts TRAV usage. Ac, acute; Fup, follow-up convalescent sample.
(C) Bubble plot showing the distribution (number of donors and frequency) of TRBV/TRAV gene usage.
(D) Dominant clonotypes identified in HLA-A∗02:01 donors specific to A2/S269+CD8+ T cells. X denotes any amino acid, and (n) denotes any number of additional amino acids.
See also Table S3.
CD8+ T cells specific for B7/N105 display high TCR diversity within pre-pandemic PBMCs
To determine the extent of TCR diversity between pre-pandemic B7/N105, COVID-19 B7/N105, and A2/S269 TCR repertoires, an independent analysis was performed with the TCRdist framework (Dash et al., 2017) across equivalent numbers of TCRαβ pairs per group to calculate diversity scores for single alpha and beta chains and paired TCRαβ clonotypes (Figure 7 A; Table S4). As observed in our earlier analyses, the A2/S269 TCR repertoire (paired TCRdiv = 147.9) was less diverse than both pre-pandemic and COVID-19 B7/N105 repertoires, in which diversity was mainly driven by the beta chain. Pre-pandemic B7/N105-specific TCRs were extremely diverse (paired TCRdiv = 730.4) and then narrowed after primary COVID-19 infection with different individuals (paired TCRdiv = 299.9), although the latter were still considered highly diverse. To contextualize the COVID-19 TCR diversity with other well-established acute (influenza A) and chronic viral infections (EBV and human cytomegalovirus [CMV]), the TCR diversity against the B7/N105 epitope was reminiscent of the relatively diverse TCR repertoires against the chronic and immunodominant A2/CMV-pp65495-503 epitope. Independent analysis of the common CDR3 motifs was also conducted with TCRdist (Dash et al., 2017) to generate highly significant alpha and beta amino acid motifs for A2/S269 and B7/N105 TCRs (Figure 7B; Figure S5). Similar to our initial analysis (Figure 6D), COVID-19 A2/S269 TCR repertoire generated 2 dominant alpha motifs (TRAV12-2/12-1-TRAJ43/30), where the gene pairing of TRAV12-1 with TRAJ43 was highly significant (p < 2.3E−07, Figure S6). In comparison, COVID-19 B7/N105 TCR repertoire encompassed several alpha and beta chain motifs which, surprisingly, did not overlap with the pre-pandemic B7/N105 motifs.
Figure 7.

TCR repertoire diversity and distinct single-chain motifs within COVID-19 and pre-pandemic individuals
(A) Single alpha, beta, and paired TCRdiv diversity values between COVID-19 A2/S269, B7/N105 and pre-pandemic (PP) B7/N105 repertoires. COVID-19 A2/S269 (43 out of 75) and B7/N105 TCRs (42 out of 82) were randomly down-sampled to obtain equivalent TCR pairs for comparison with the pre-pandemic B7/N105 group. The higher value indicates higher diversity.
(B) Enriched alpha and beta amino acid motifs in the CDR3 region were generated by TCRdist for all TCR pairs. Each TCR chain motif depicts the variable (left side) and joining (right side) gene frequencies, CDR3 amino acid sequences (middle), and inferred rearrangement structure (bottom bars colored by source region: light gray, V-region; dark gray, J-region; black, diversity [D]-region; red, insertions). Chi-square values greater than 50 were considered highly significant; values below 50 were borderline significant. The full set of motifs are shown in Figure S5.
(C) Probabilities of generation (Pgen; log10 transformed), and number of nucleotide insertions and deletions for all single alpha and beta chains, were generated with the TCRdist pipeline and contextualized with publicly available data from A2/EBV-BMLF1280-288, A2/M158-66 (influenza A), and A2/CMV-pp65495-503 TCR repertoires, which were not included in the statistical analysis. Statistical analysis between COVID-19 A2/S269, B7/N105, and B7/N105 pre-pandemic (PP) repertoires for variations in Pgen, insertions, and deletions are further described in the STAR Methods with linear mixed models. p values were adjusted (padj) for multiple testings with the Benjamini and Hochberg FDR method. Box plots represent the median (middle bar), 75% quantile (upper hinge), and 25% quantile (lower hinge), with whiskers extending 1.5 times the inter-quartile range.
The probability of generating (Pgen) TCR alpha and beta chains were then calculated with TCRdist, which correlated with the number of insertions and deletions within the CDR3 region (Figure 7C). Within the TCR alpha chain, Pgen values were similar for COVID-19 and well-established IAV, EBV, and CMV epitopes. The probability of generating beta chains in the B7/N105 groups was comparable between pre-pandemic and COVID-19, but both COVID-19 and pre-pandemic B7/N105’s Pgen were significantly lower than those of A2/S269 (p adj = 0.0012 and 0.0435, respectively) but similar to the A2/CMV repertoire. The lower beta Pgen values for B7/N105 were supported by both B7/N105 groups having significantly more beta N-insertions than A2/S269; however, lower numbers of N deletions were only significantly observed for the COVID-19 B7/N105 TCR repertoire, but not for pre-pandemic B7/N105 or A2/S269. Taken together, the lower probability of generating B7/N105 TCRs, by way of more insertions, reflects the extreme nature in diversity for both pre-pandemic and COVID-19 B7/N105 TCR repertoires.
Discussion
Our study deciphered the most immunodominant CD8+ T cell response known to date in COVID-19, the B7/N105 +CD8+ T cell population. Our direct ex vivo approach, without any in vitro manipulations, revealed high precursor frequencies of B7/N105-specific CD8+ T cells in COVID-19 patients (∼6.88 × 10−4), comparable to the magnitude of a well-established influenza-specific A2/M158-specific CD8+ T cell memory population (3.04 × 10−4). High precursor frequencies were also detected in pre-pandemic adult and elderly PBMCs (3.00 × 10−5 and 6.76 × 10−5, respectively), much higher than the precursor frequencies previously observed for naive CD8+ T cells directed toward HLA-A∗02:01-restricted NY-ESO-11157-1165, WT1126-134, HIV Gag p1777-85, HCV Core132-140, and CMV pp65 antigens (between 0.6 × 10−6 to 5.3 × 10−6) in cancer-free and seronegative individuals (Alanio et al., 2010; Nguyen et al., 2017b). It was thus quite surprising that these B7/N105-specific CD8+ T cells in pre-pandemic samples mainly displayed a naive phenotype, indicating a lack of previous cross-reactive exposures. Because the SARS-CoV-2-derived N105-113 peptide SPRWYFYYL displays a close amino acid (aa) homology with the corresponding peptide LPRWYFYYL derived from N in the globally distributed coronavirus strains HCoV-OC43 and HCoV-HKU1, there is potential for cross-reactive CD8+ T cell responses, which was notably observed in all 4 donors tested by Ferretti et al.’s study via genome-wide epitope screening technology and the secretion of granzyme B (Ferretti et al., 2020; Schulien et al., 2021). However, the overall conclusion was that the COVID-19 CD8+ T cell response was not significantly shaped by pre-existing immunity to endemic coronaviruses. However, because 100% of the pediatric and 81.3% of adult pre-pandemic donors in our study had a prototypical naive (and not even TSCM) B7/N105-specific CD8+ T cell phenotype directly ex vivo, this suggests that either the HCoV-OC43/HKU1 peptide is not presented on the infected cell surface (for example due to different processing of the peptide within the cell), the SARS-CoV-2-derived N105–113 peptide is not cross-reactive with the corresponding N-derived peptides originating from other human coronaviruses, or our pre-pandemic adult and pediatric donors were not exposed to those circulating coronaviruses. Although we cannot exclude that these CD8+ T cells are naive antigen-experienced T cells that express a naive CD45RA+CD27−CD95− phenotype, our previous study indicated that the SARS-CoV2 A2/S269-specific CD8+ T cells with naive CD45RA+CD27+CD95− phenotype could not respond to the peptide stimulation (Habel et al., 2020). These data are in line with previous phenotypical analysis of human coronavirus-specific T cells which do not express a naive phenotype (Le Bert et al., 2020; Peng et al., 2006), indicating that infection with human coronaviruses might not induce SARS-CoV-2 B7/N105–113-specific T cells. We have not found effector and/or memory SARS-CoV-2 tetramer-positive CD8+ T cells in lungs or spleens from pre-pandemic donors, in contrast to readily detected memory influenza tetramer-positive CD8+ T cells across different epitopes found at these anatomical sites in our previous studies (Koutsakos et al., 2019; Sant et al., 2018).
Both the magnitude and phenotype of B7/N105-specific CD8+ T cells were highly variable in the pre-pandemic elderly. Reduced magnitude of B7/N105-specific CD8+ T cells in elderly individuals corresponds with published reports providing clear evidence for a loss of tetramer-positive CD8+ T cells as well as naive CD8+ T cell precursors with aging (Nguyen et al., 2018; Valkenburg et al., 2012). Although B7/N105-specific CD8+ T cells within 4 elderly donors mainly had a naive phenotype, 3 donors had a high prevalence of TEMRA (75%) and TCM-like (37%–73%) subsets. This could be due to either priming with cross-reactive peptides derived from circulating coronaviruses occurring sequentially with age or SARS-CoV-2 CD8+ T cells undergoing age-related dysregulated homeostatic proliferation, as commonly observed with age and CMV seropositivity (LeMaoult et al., 2000; Messaoudi et al., 2006; Valkenburg et al., 2012; Wertheimer et al., 2014). The high frequency of TEMRA-phenotype T cells observed in PP27 could be a result of age-related CD45RA upregulation observed previously in CMV-specific CD8+ T cells (Griffiths et al., 2013). It is, however, unclear whether this upregulation also occurs in antigen-specific CD8+ T cells in acute infection in contrast to constant exposure to antigens derived from persistent infections such as CMV, given that persistent infections can induce different memory phenotypes (Appay et al., 2002).
Because our TCRαβ analysis of the B7/N105-specific CD8+ T cells obtained from the pre-pandemic elderly individual (PP27) with the high-frequency TEMRA population showed a highly clonal TCRαβ repertoire, atypical of what we observed in other COVID-19 and pre-pandemic individuals, this suggested that the clonal B7/N105-specific CD8+ TEMRA population in this elderly individual represented a highly proliferating non-COVID-19-specific CD8+ T cell population. Whether or not the frequencies of SARS-CoV-2 tetramer-positive CD8+ T cells within adult tonsils are higher than those in adult blood will require future investigation. From our previous study, we found higher levels of influenza-specific CD8+ T cells in healthy adult tonsils than in unmatched healthy adult blood (Koutsakos et al., 2019).
Our analyses of longitudinal COVID-19 samples for B7/N105 +CD8+ T cell populations demonstrated the maintenance of predominant TCM-like phenotypes into the long-term memory (up to d270 for some patients) after disease onset, similar to what we observed in patients hospitalized with H7N9 in Shanghai (Wang et al., 2015). Our findings are in agreement with studies showing that SARS-CoV-specific memory T cells persist into long-term memory for at least 6 to 11 years (Peng et al., 2006; Tang et al., 2011). However, it should be noted that distinct memory populations can be established after infection with different viruses (Appay et al., 2002) or even across different epitope-specific CD8+ T cells, as exemplified by our recent study (Habel et al., 2020) showing mainly TSCM/naive phenotypes within A2/S269 +CD8+ T cells during convalescence.
Ex vivo TCRαβ analyses for the remaining COVID-19 patients and pre-pandemic samples across all ages revealed that a diverse TCRαβ repertoire and promiscuity in TCRα-TCRβ pairing could underlie such high naive precursor frequencies of B7/N105-specific CD8+ T cells. The importance of TCRαβ repertoire diversity on disease outcome was previously reported (LeMaoult et al., 2000) in mice, macaques, and humans (Messaoudi et al., 2002; Chen et al., 2012; Price et al., 2009). Generally, a diverse TCRαβ repertoire provides a greater scope for selection of TCRαβ clonotypes with high peptide-MHC-I avidity. CD8+ T cells with diverse TCR repertoires can generate broadly protective responses and are often capable of recognizing both the wild-type virus and newly emerging mutants. Our single-cell ex vivo TCRαβ analysis, including independent analysis with the TCRdist framework, demonstrated higher TCRαβ diversity within immunodominant B7/N105-specific CD8+ T cells with variable usage of TRAV, TRBV, TRAJ, and TRBJ gene segments. Furthermore, no common gene features and no common motifs within CDR3α and CDR3β sequences were observed across the donors. Such TCRαβ repertoire diversity, combined with the promiscuity of TCRα-TCRβ pairing, might reflect a high degree of TCRαβ plasticity to recognize the immunodominant B7/N105 epitope. Consequently, this plasticity could lead to the generation of a large and diverse pool of naive TCRαβ precursors and, hence, high precursor frequency of B7/N105-specific CD8+ T cells. When it comes to immunodominance hierarchies, both naive precursor frequency and antigen dose play a key role (La Gruta et al., 2006). Thus, a high naive precursor frequency in pre-pandemic samples could underlie highly immunodominant B7/N105 +CD8+ T cell responses in COVID-19 patients during acute disease and convalescence. Future crystallography studies of ternary N105 peptide in the context of HLA-B∗07:02 and TCRαβs are needed to provide key insights into promiscuous TCRαβ recognition of the B7/N105 epitope, as well as the inclusion of more nucleocapsid-derived epitopes and tetramers for further TCR repertoire studies. The TCRαβ repertoire within the dominant B7/N105-specific CD8+ T cells contrasted those of subdominant SARS-CoV-2-specific A2/S269-specific CD8+ T cells containing clear CDR3α motifs and TRAV usage (as shown by our study and the recent report by Shomuradova et al., [2020]). The latter reflected numerous previously analyzed TCRαβ repertoires of viral-specific CD8+ T cell populations in mice and humans, characterized by biased usage of TCR gene segments (Kedzierska and Koutsakos, 2020).
Overall, our study suggests that high precursor frequency and plasticity of TCRα-TCRβ pairing underpin the immunodominance of SARS-CoV-2-specific B7/N105 +CD8+ T cell responses. As both the immunodominant B7/N105 and subdominant B7/N257 SARS-CoV-2-specific epitopes encompass nucleocapsid-derived peptides, infection with SARS-CoV-2 clearly leads to robust CD8+ T cell immunity in HLA-B∗07:02 individuals. Better understanding of T cells directed toward immunodominant epitopes and their origins provides important insights into the rational design of next-generation vaccine strategies to optimize long-lasting CD8+ T cell immunity.
Limitations of the study
We recognize that our pre-pandemic and COVID-19 donors originate from 1 country and thus do not represent the global population. This might be especially relevant when CD8+ T cell responses toward the circulating human coronavirus strains are assessed. Cross-reactivity of CD8+ T cell responses against different SARS-CoV-2 variants was not addressed in the current manuscript and is of interest for future studies. The number of pre-pandemic children’s samples was relatively small to perform in-depth analyses. Further ex vivo examination of tetramer-positive CD8+ T cells in SARS-CoV-2-infected children would be of a great interest to understand SARS-CoV-2-specific CD8+ T cells responses in acute COVID-19 and convalescence.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD71 M-A712 BV421 | BD Biosciences | Cat#562995; RRID: AB_2737939 |
| CD4 SK3 BV650 | BD Biosciences | Cat#563875; RRID: AB_2744425 |
| CD27 L128 BV711 | BD Biosciences | Cat#563167; RRID: AB_2738042 |
| CD38 HIT2 BV786 | BD Biosciences | Cat#563964; RRID: AB_2738515 |
| CCR7 150503 AF700 | BD Biosciences | Cat#561143; RRID: AB_10562031 |
| CD14 MΦP9 APC-H7 | BD Biosciences | Cat#560180; RRID: AB_1645464 |
| CD19 SJ25C1 APC-H7 | BD Biosciences | Cat#560177; RRID: AB_1645470 |
| CD45RA HI100 FITC | BD Biosciences | Cat#555488; RRID: AB_395879 |
| CD8a SK1 PerCP-Cy5.5 | BD PharMingen | Cat#565310; RRID: AB_2687497 |
| CD95 DX2 PE-CF594 | BD Biosciences | Cat#562395; RRID: AB_11153666 |
| PD-1 EH12.1 PE-Cy7 | BD Biosciences | Cat#561272; RRID: AB_10611585 |
| CD3 OKT3 BV510 | BioLegend | Cat#317332; RRID: AB_2561943 |
| HLA-DR L243 BV605 | BioLegend | Cat#307640; RRID: AB_2561913 |
| CD69 FN50 BV421 | BioLegend | Cat#310930; RRID: AB_2561909 |
| CD103 Ber-ACT8 FITC | BioLegend | Cat#350203; RRID: AB_10642828 |
| CD45RO UCHL1 PE-Cy7 | Thermo Fisher Scientific | Cat#25-0457-41; RRID: AB_10717670 |
| CD56 MEM-188 APC | BioLegend | Cat#304610; RRID: AB_314452 |
| CD16 3G8 AF700 | BioLegend | Cat#302026; RRID: AB_2278418 |
| CD19 HIB19 BV570 | BioLegend | Cat#302236; RRID: AB_2563606 |
| CD3 UCHT1 PECF594 | BD Biosciences | Cat#562280; RRID: AB_11153674 |
| CD8a RPA-T8 BV421 | BioLegend | Cat#301036; RRID: AB_10960142 |
| Granzyme B GB11 AF700 | BD Biosciences | Cat#560213; RRID: AB_1645453 |
| Granzyme K G3H69 PerCP-eFluor710 | Thermo Fisher Scientific | Cat#46-8897-42; RRID: AB_2573854 |
| Granzyme M 4B2G4 eFluor660 | Thermo Fisher Scientific | Cat#50-9774-42; RRID: AB_2574374 |
| Perforin B-D48 Pe-Cy7 | BioLegend | Cat#353316; RRID: AB_2571973 |
| CD19 J4.119 ECD | Beckman Coulter | Cat#IM2708U; RRID: AB_130854 |
| IgM G20-127 BUV395 | BD Biosciences | Cat#563903; RRID: AB_2721269 |
| CD21 B-ly4 BUV737 | BD Biosciences | Cat#564437; RRID: AB_2738807 |
| IgD IA6-2 PE-Cy7 | BD Biosciences | Cat#561314; RRID: AB_10642457 |
| IgG G18-145 BV786 | BD Biosciences | Cat#564230; RRID: AB_2738684 |
| Streptavidin BV510 | BD Biosciences | Cat#563261; RRID: AB_2869477 |
| CD20 2H7 APC-Cy7 | BioLegend | Cat#302314; RRID: AB_314262 |
| CD14 M5E2 BV510 | BioLegend | Cat#301841; RRID: AB_2561379 |
| CD8a RPA-T8 BV510 | BioLegend | Cat#301048; RRID: AB_2561942 |
| CD16 3G8 BV510 | BioLegend | Cat#302048; RRID: AB_2562085 |
| CD10 HI10a BV510 | BioLegend | Cat#312220; RRID: AB_2563835 |
| CD27 O323 BV605 | BioLegend | Cat#302829; RRID: AB_11204431 |
| IFN-γ RUO V450 | BD Bioscience | Cat#560371; RRID: AB_1645594 |
| TNF Mab11 AF700 | BD Bioscience | Cat#557996; RRID: AB_396978 |
| MIP-1b D21-1351 APC | BD Bioscience | Cat#560686; RRID: AB_1727565 |
| CD107a eBioH4A3 ΩAF48 | Thermo Fisher Scientific | Cat#53-1079-42; RRID: AB_2016657 |
| Streptavidin PE | BD Biosciences | Cat#349023, RRID: AB_2868860 |
| Streptavidin APC | BD Biosciences | Cat#349024, RRID: AB_2868861 |
| Streptavidin PE | Thermo Fisher Scientific | Cat#S866 |
| APC Conjugation Lightning-Link Kit | Abcam | Cat#ab201807 |
| Peroxidase AffiniPure goat anti-human IgG, Fcγ fragment specific | Jackson ImmunoResearch | Cat#109-035-098; RRID: AB_2337586 |
| Rat anti-human IgA mAb MT20, alkaline phosphate-conjugated | MabTech | Cat#3860-9A; RRID: AB_10736550 |
| anti-human IgM mAb MT22, biotinylated | MabTech | Cat#3880-6-250 |
| Bacterial and Virus Strains | ||
| SARS-CoV-2 isolate CoV/Australia/VIC01/2020 | Caly et al., 2020 | N/A |
| Biological Samples | ||
| Blood samples (peripheral blood mononuclear cells (PBMCs), serum and plasma samples) from COVID-19 patients and healthy control individuals | Alfred Hospital, Austin Health, The University of Melbourne, James Cook University, Australian Red Cross LifeBlood and Launceston General Hospital (Australia) | N/A |
| Tissue samples (tonsil, spleen and lung samples) from healthy control individuals | Launceston General Hospital, Alfred Hospital’s Lung Tissue Biobank and DonateLife Victoria (Australia) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| AccuCheck Counting Beads | Thermo Fisher Scientific | Cat#PCB100 |
| 3,3¢,5,5¢-Tetramethylbenzidine (TMB) Liquid Substrate System for ELISA, peroxidase substrate | Sigma | Cat#T0440-1L |
| Alkaline phosphatase yellow (pNPP) liquid substrate for ELISA | Sigma | Cat#P7998-100ML |
| Pierce High Sensitivity Streptavidin-HRP | Thermo Fisher Scientific | Cat#21130 |
| SARS-CoV-2 RBD protein | Amanat et al., 2020 | N/A |
| SARS-CoV-2 RBD protein | Juno et al., 2020 | N/A |
| SARS-CoV-2 Spike protein | Juno et al., 2020 | N/A |
| PepTivator® SARS-CoV-2 Prot_S | Miltenyi Biotec | Cat#130-126-700 |
| PepTivator® SARS-CoV-2 Prot_N | Miltenyi Biotec | Cat#130-126-698 |
| PepTivator® SARS-CoV-2 Prot_M | Miltenyi Biotec | Cat#130-126-702 |
| SARS-CoV-2 peptides - B7/N66-74 FPRGQGVPI; B7/N105-113 SPRWYFYYL; B7/N257-265 KPRQKRTAT; A2/N219-227 LALLLLDRL; A2/N222-230 LLLDRLNQL; A24/S1208 QYIKWPWYI | GenScript | N/A |
| HLA-B∗07:02 and HLA-A∗24:02 monomers with N105, N257, N66 (for HLA-B∗07:02) or S1208 (for HLA-A∗24:02) peptide | This paper | N/A |
| A2/S269 monomer (SARS-CoV-2, S269, YLQPRTFLL) | Habel et al., 2020 | N/A |
| B7/EBV monomer (EBNA-3379-387, RPPIFIRRL) | Rowntree et al., 2020 | N/A |
| Critical Commercial Assays | ||
| eBioscience™ Foxp3/Transcription Factor Staining Buffer Set | Thermo Fisher Scientific | Cat#00-5521-00 |
| LEGENDplex™ Human Inflammation Panel 1 kit | BioLegend | Cat#740809 |
| Experimental Models: Cell Lines | ||
| Vero C1008, African Green monkey kidney cells | ATCC | Cat#CRL-1586; Lot#3338237; RRID: CVCL_0574 |
| Software and Algorithms | ||
| R v3.6.2 | The Comprehensive R Archive Network | https://cran.r-project.org |
| Circlize R package | Gu et al., 2014 | https://cran.r-project.org/package=circlize |
| ggplot R package | Wickham, 2016 | https://ggplot2.tidyverse.org |
| TCRdist pipeline | Dash et al., 2017 | https://github.com/phbradley/tcr-dist |
| lme4 R package | Bates et al., 2014 | https://www.jstatsoft.org/article/view/v067i01/ |
| FlowJo v10.5.3 | FlowJo | https://www.flowjo.com |
| Prism v8.3.1 or v9.1.0 | GraphPad | https://www.graphpad.com |
| BD FACS Diva v8.0.1 | BD Biosciences | https://www.bdbiosciences.com/en-us/instruments/research-instruments/research-software/flow-cytometry-acquisition/facsdiva-software |
| Other | ||
| Anti-PE MicroBeads | Miltenyi Biotec | Cat# 130-048-801, RRID: AB_244373 |
| Anti-APC MicroBeads | Miltenyi Biotec | Cat# 130-090-855, RRID: AB_244367 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Katherine Kedzierska (kkedz@unimelb.edu.au).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The published article includes all datasets generated or analyzed during the study.
Experimental model and subject details
A total of 21 COVID-19 cases (CA) and 40 pre-pandemic (PP) subjects were recruited into this study (Table S1). The traveler cohort included 3 COVID-19 cases (CA1-3) at convalescence and 4 suspected cases (SU1-4). Acute and convalescent COVID-19 patients were recruited via the Alfred Hospital, Austin Hospital, University of Melbourne or James Cook University. Eight of the donors were admitted to hospital during their active infection (Table S1). Healthy pre-pandemic blood donors were recruited via the University of Melbourne or buffy packs obtained from the Australian Red Cross LifeBlood (West Melbourne, Australia) (Table S1). Healthy COVID-19-unexposed and pre-pandemic tonsils, spleens and lungs were also obtained. Matched tonsils and PBMCs were obtained from healthy individuals undergoing tonsillectomy (Launceston General Hospital, Tasmania, Australia), lung samples via the Alfred Hospital’s Lung Tissue Biobank, while spleens were provided by DonateLife Victoria. Donors were HLA typed by VTIS (Melbourne, Australia). Peripheral blood was collected in heparinized tubes and peripheral blood monocular cells (PBMCs) were isolated via Ficoll-Paque separation. Single cell suspensions were isolated from tissues as previously described (Habel et al., 2020; Koutsakos et al., 2018).
Experiments conformed to the Declaration of Helsinki Principles and the Australian National Health and Medical Research Council Code of Practice. Written informed consents were obtained from all blood donors prior to the study. Lung and spleen tissues were obtained from deceased organ donors after written informed consents from the next of kin. Written informed consents were obtained from participants’ parental or guardians for underage tonsil tissue donors. The study was approved by the Alfred Hospital (#280/14), Austin Health (HREC/63201/Austin-2020); the University of Melbourne (#2057366.1, #2056901.1, #2056689, #2056761, #1442952, #1955465, and #1443389), the Australian Red Cross Lifeblood (ID 2015#8), the Tasmanian Health and Medical (ID H0017479) and the James Cook University (H7886) Human Research Ethics Committees.
Method details
Peptides and peptide-HLA class I tetramers
Overlapping synthetic peptides spanning the SARS-CoV-2 Nucleocapsid (N) and Membrane (M) proteins, and the immunogenic regions of Spike (S) were purchased from Miltenyi Biotec and reconstituted in 80% DMSO. SARS-CoV-2 peptides shown to bind HLA-B∗07:02, HLA-A∗02:01 and HLA-A∗24:02 (B7/N66-74 FPRGQGVPI; B7/N105-113 SPRWYFYYL; B7/N257-265 KPRQKRTAT; A2/N219-227 LALLLLDRL; A2/N222-230 LLLDRLNQL and A24/S1208 QYIKWPWYI) were purchased from GenScript and reconstituted in DMSO. Tetramers were generated from soluble, biotinylated HLA-B∗07:02 or HLA-A∗24:02 monomers. Briefly, HLA α-heavy chain with C-terminal BirA biotinylation motif and β2-microglobulin were expressed and purified as inclusion bodies in E. coli, solubilized in 6M Guanidine HCl and refolded with either N105, N257, N66 (for HLA-B∗07:02) or S1208 (for HLA-A∗24:02) peptide, in buffer containing 50mM Tris pH8, 3M urea, 0.4M Arginine, 2mM oxidised Glutathione, 20mM Glutathione, 2mM EDTA, 10mM PMSF and cOmplete protease inhibitor (Roche). Following dialysis in 10mM Tris, HLA monomers were purified via DEAE and HiTrapQ ion exchange chromatography, and biotinylated with BirA ligase in 50mM Bicine pH 8.3, 10mM ATP, 10mM magnesium acetate and 100μm d-biotin. Following S200 gel permeation chromatography fully biotinylated HLA monomers were stored at −80°C and conjugated to fluorescently-labeled streptavidin (SA), PE-SA or APC-SA (BD Biosciences) at an 8:1 monomer to SA molar ratio to form pMHC-I tetramers. The A2/S269 (SARS-CoV-2, S269, YLQPRTFLL) and B7/EBV (EBNA-3379-387, RPPIFIRRL) tetramers were generated as previously described (Habel et al., 2020) (Rowntree et al., 2020).
Ex vivo tetramer enrichment
PBMCs, tonsils and spleens (7-50x106) were stained with B7/N105-PE, B7/N257-APC, A2/S269-PE or A24/S1208-APC tetramers at room temperature for 1 h in MACS buffer (PBS with 0.5% BSA and 2 mM EDTA). Cells were then incubated with anti-PE and anti-APC microbeads (Miltenyi) and tetramer+ cells were enriched using magnetic separation (Nguyen et al., 2017b; Valkenburg et al., 2016). Lymphocytes were stained with anti-CD71-BV421 (#562995), anti-CD4-BV650 (#563875), anti-CD27-BV711 (#563167), anti-CD38-BV786 (#563964), anti-CCR7-AF700 (#561143), anti-CD14-APC-H7 (#560180), anti-CD19-APC-H7 (#560177), anti-CD45RA-FITC (#555488), anti-CD8-PerCP-Cy5.5 (#565310), anti-CD95-PE-CF594 (#562395), anti-PD1-PE-Cy7 (#561272) (BD Biosciences), anti-CD3-BV510 (#317332), anti-HLA-DR-BV605 (#307640) (BioLegend, San Diego, CA, USA) and Live/Dead near-infrared (#L10119, Invitrogen) stain for 30 min, washed, resuspended in MACS buffer and analyzed by flow cytometry. Lung cells were stained with tetramer for 1 h then cell surface stained using a modified panel to include anti-CD69-BV421 (#310930), anti-CD103-FITC (#350203) (BioLegend) and anti-CD45RO-PE-Cy7 (#25-0457-41, Thermo Fisher Scientific). In some experiments, cells were fixed with 1% PFA and washed before acquiring on a LSRII Fortessa (BD) or single cell sorted using the FACSAriaIII (BD) for TCR analyses. FCS files were analyzed using FlowJo v10 software.
TCRαβ repertoire analysis
Tetramer+ CD8+ T cells were single-cell sorted into empty 96-well Twin.tec PCR plates (Eppendorf, Hamburg, Germany), centrifuged then stored at −80°C. Multiplex-nested RT-PCR amplification of paired CDR3a and CDR3b regions were performed as described (Nguyen et al., 2017b; Valkenburg et al., 2016). TCR sequences were analyzed using IMGT/V-QUEST. Data visualization was performed in R using packages for circular layout (Gu et al., 2014) and graphics generation (Wickham, 2016).
Phenotypic whole blood immune analyses
Fresh whole blood was used to analyze immune cell populations using three human antibody panels for enumerating immune cell activation (monocytes and T/B/NK/γδ cells), TFH and ASC cell activation, and cytotoxicity profiles of T cell’s expressing intracellular granzymes (A, B, K and M) and perforin, as previously described (Thevarajan et al., 2020). PBMCs were stained, red blood cell lysed, then fixed in 1% PFA, or intracellularly stained using the eBioscience™ Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher Scientific, Carlsbad, CA, USA) (Thevarajan et al., 2020). Samples were acquired on a BD LSRII Fortessa and analyzed using FlowJo v10 software.
Detection of SARS-CoV-2 RBD- and Spike-reactive B cells
PBMCs were stained with SARS-CoV-2 Spike and RBD probes, as previously described (Juno et al., 2020). Probes were generated by sequential addition of streptavidin-phycoerythrin (PE) (Thermo Fisher Scientific) to trimeric S protein biotinylated using recombinant Bir-A (Avidity), while SARS-CoV-2 RBD was labeled to APC using an APC Conjugation Lightning-Link Kit (Abcam). PBMCs were surface stained with Aqua viability dye (Thermo Fisher) and monoclonal antibodies against CD19-ECD (#IM2708U, Beckman Coulter), IgM BUV395 (#563903), CD21 BUV737 (#564437), IgD PE-Cy7 (#561314), IgG BV786 (#564230), streptavidin-BV510 (#563261) (BD Biosciences), CD20 APC-Cy7 (#302314), CD14 BV510 (#301841), CD3 BV510 (#317332), CD8a BV510 (#301048), CD16 BV510 (#302048), CD10 BV510 (#312220) and CD27 BV605 (#302829) (BioLegend). Cells were washed, fixed with 1% formaldehyde and acquired on a BD LSRII Fortessa.
Intracellular cytokine staining (ICS)
PBMCs were stimulated with 0.6 nmol of overlapping SARS-CoV-2 peptides for 10 days in RF-10 medium (+10U/mL IL-2) (Koutsakos et al., 2019). On d10, cells were stimulated with peptides for 5-6 h in the presence of GolgiPlug and GolgiStop (BD Biosciences) plus 10U/mL IL-2, and SARS-CoV-2-reactive T cells were quantified using anti-IFN-γ-V450 (#560371), anti-TNF-AF700 (#557996), anti-MIP-1b-APC (#560686) (BD Biosciences), and anti-CD107a-AF488 (#53-1079-42, Thermo Fisher Scientific) in an ICS assay as previously described (Clemens et al., 2016).
SARS-CoV-2 RBD and Spike ELISA
RBD- and Spike-specific ELISA for detection of IgM, IgG and IgA antibodies was performed as described (Amanat et al., 2020; Stadlbauer et al., 2020), with some modifications: Nunc MaxiSorp flat bottom 96-well plates (Thermo Fisher Scientific) were used for antigen coating, blocking with PBS (containing w/v 1% BSA) and serial dilutions performed in PBS (containing v/v 0.05% Tween and w/v 0.5% BSA). For detection of IgG and IgA, peroxidase-conjugated goat anti-human IgG (Fcγ fragment specific; Jackson ImmunoResearch) or alkaline phosphate-conjugated rat anti-human IgA (mAb MT20; MabTech) was used and developed with TMB (Sigma) substrate for IgG or pNPP (Sigma) for IgA. For IgM, biotinylated mAb MT22 and peroxidase-conjugated streptavidin (Pierce; Thermo Fisher Scientific) was used. Peroxidase reactions were stopped using 1M H3PO4 and plates read on a Multiskan plate reader (Labsystems). Inter- and intra-experimental measurements were normalized using a positive control plasma from a COVID-19 patient run on each plate. Endpoint titers were determined by interpolation from a sigmodial curve fit (all R-squared values > 0.95; GraphPad Prism 8) as the reciprocal dilution of plasma that produced > 15% (for IgA and IgG) or > 30% (for IgM) absorbance of the positive control at a 1:31.6 (IgG and IgM) or 1:10 dilution (IgA). Seroconvertion was defined when titers were above the mean titer (plus 2 standard deviations) of healthy non-COVID-19 donors.
Microneutralisation assay
Microneutralisation activity of serum samples was assessed as previously described (Juno et al., 2020). SARS-CoV-2 isolate CoV/Australia/VIC01/2020 (Caly et al., 2020) was propagated in Vero cells and stored at −80°C. Sera were heat-inactivated at 56°C for 30 min and serially diluted. Residual virus infectivity in the serum/virus mixtures was assessed in quadruplicate wells of Vero cells incubated in serum-free media containing 1 μg/mL of TPCK trypsin at 37°C and 5% CO2. Viral cytopathic effect was read on day 5. The neutralising antibody titer was calculated using the Reed–Muench method, as described (Juno et al., 2020).
Cytokine analysis
Patients’ plasma was diluted 1:2 for measuring IL-1β, IFN-α2, IFNγ, TNFα, MCP-1 (CCL2), IL-6, IL-8 (CXCL8), IL-10, IL-12p70, IL-17A, IL-18, IL-23 and IL-33 using the LEGENDplex Human Inflammation Panel 1 kit, as per manufacturer’s instructions (BioLegend).
Quantification and statistical analysis
TCR statistical analysis
Single-chain alpha and beta TCR sequences were independently parsed and analyzed using the TCRdist pipeline to detect statistically significant amino acid motifs and to model CDR3 insertions, deletions, and probabilities of generation (Pgen) (Dash et al., 2017). To test for variations in Pgen, insertions, and deletions across epitope specificities, we used linear mixed models (Bates et al., 2014) with the number of sequences per subject per epitope included as a covariate and subject included as a random effect in order to control for unintentional differences in sequencing effort and non-independence of the data across subjects, respectively. Log10 of Pgen was modeled using a Gaussian distribution, whereas insertions and deletions were analyzed using a generalized model for the negative binomial distribution. P values were adjusted (p adj) for multiple testing using the Benjamini & Hochberg FDR method. We also used the TCRdist framework on a subset of the data to characterize repertoire diversity using TCRdiv, which was contextualized using publicly available data from A2/EBV-BMLF1280-288, A2/M158-66 (influenza A), and A2/CMV-pp65495-503 repertoires. For this analysis, we only considered cells that had functional, paired alpha and beta sequences, and we randomly down-sampled repertoires such that each were derived from an equivalent number of donors and had an equivalent number of TCRs for comparison (i.e., A2/S269, B7/N105 COVID-19, A2/EBV, A2/M1 and A2/CMV). The subsampled and full repertoires are detailed in Table S4.
Statistical analysis
Statistical significance of nonparametric datasets (two-tailed) were determined using GraphPad Prism v9 software. Mann-Whitney (unpaired) and Wilcoxin (paired) tests were used for comparisons between two groups. Kruskal-Wallis (unmatched) test with Dunn’s multiple comparisons was used to compare more than two groups. Tukey’s multiple comparison test compared row means between more than two groups.
Acknowledgments
We thank all the participants involved in the study, as well as Robyn Esterbauer, Hannah Kelly, Jane Batten, Helen Kent, and Kanta Subbarao for support with the cohorts and assays. We thank Jill Garlick, Janine Roney, Anne Paterson, and the research nurses at the Alfred Hospital. We acknowledge all DRASTIC (The use of cytokines as a preDictoR of disease Severity in criTically Ill Covid-patients) investigators from Austin Health and thank the participants involved. We thank Ana Copaescu for laboratory work and study coordination for the DRASTIC study. We thank Thomas Loudovaris and Stuart I. Mannering from St Vincent’s Institute of Medical Research for access to spleen samples. This work was supported by the Clifford Craig Foundation to K.L.F. and K.K.; an NHMRC Leadership Investigator Grant to K.K. (1173871); an NHMRC Emerging Leadership Level 1 Investigator Grant to THON (1194036); an NHMRC program grant to D.L.D. (1132975); the Research Grants Council of the Hong Kong Special Administrative Region, China (T11-712/19-N) to K.K.; the Victorian government to S.J.K. and A.K.W.; an MRFF award (2002073) to S.J.K. and A.K.W.; an MRFF award (1202445) to K.K.; an MRFF award (2005544) to K.K., S.J.K., J.A.J., and A.K.W.; an NHMRC program grant (1149990) to S.J.K.; an NHMRC project grant (1162760) to A.K.W.; and NIH contract CIVR-HRP (HHS-NIH-NIAID-BAA2018) to P.G.T. and K.K. A.K.W. is supported by an Emerging Leadership 1 Investigator Grant (1173433), J.A.J. by an NHMRC Early Career Fellowship (ECF) (1123673), D.L.D. by an NHMRC Principal Research Fellowship (1137285), and S.J.K. by an NHMRC Senior Principal Research Fellowship (1136322). C.E.S. has received funding from the European Union’s Horizon 2020 research, innovation program under the Marie Skłodowska-Curie grant agreement (792532) and the Doherty Collaborative Seed Grant. J.R. is supported by an ARC Laureate fellowship. J.R.H. and W.Z. are supported by the Melbourne Research Scholarship from the University of Melbourne. L.H. is supported by the Melbourne International Research Scholarship (MIRS) and the Melbourne International Fee Remission Scholarship (MIFRS) from The University of Melbourne. J.C.C. and P.G.T. are supported by NIH NIAID (R01 AI136514-03) and ALSAC at St. Jude.
Author contributions
K.K. led the study. K.K., J.T., and J.R. supervised the study. K.K., J.T., J.R., T.H.O.N., L.C.R., J.P., B.Y.C., and L.K. designed the experiments. T.H.O.N., L.C.R., J.P., B.Y.C., L.H., L.K., P.C., H.T., J.R.H., W.Z., L.A., L.E., K.Y.M., J.A.J., K.W., F.L.M., and A.K.W. performed and analyzed experiments. F.A., F.K., N.A.M., and A.K.W. provided reagents. D.L.D., K.L.F., S.S., J.K., L.M.W., G.P.W., F.J., E.M., C.L.G., N.E.H., O.C.S., J.A.T., A.C.C., P.H., P.C., and S.J.K. recruited the patient cohorts. T.H.O.N., L.C.R., J.P., J.R., J.T., and K.K. provided intellectual input into the study design and data interpretation. T.H.O.N., L.C.R., C.E.S., J.C.C., and P.G.T. analyzed TCR sequences. T.H.O.N., L.C.R., and K.K. wrote the manuscript. All authors reviewed and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: April 15, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.immuni.2021.04.009.
Supplemental information
References
- Alanio C., Lemaitre F., Law H.K., Hasan M., Albert M.L. Enumeration of human antigen-specific naive CD8+ T cells reveals conserved precursor frequencies. Blood. 2010;115:3718–3725. doi: 10.1182/blood-2009-10-251124. [DOI] [PubMed] [Google Scholar]
- Amanat F., Stadlbauer D., Strohmeier S., Nguyen T.H.O., Chromikova V., McMahon M., Jiang K., Arunkumar G.A., Jurczyszak D., Polanco J. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat. Med. 2020;26:1033–1036. doi: 10.1038/s41591-020-0913-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appay V., Dunbar P.R., Callan M., Klenerman P., Gillespie G.M., Papagno L., Ogg G.S., King A., Lechner F., Spina C.A. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat. Med. 2002;8:379–385. doi: 10.1038/nm0402-379. [DOI] [PubMed] [Google Scholar]
- Bates D., Mächler M., Bolker B., Walker S. Fitting Linear Mixed-Effects Models Usinglme4. J. Stat. Softw. 2014;61 doi: 10.18637/jss.v067.i01. [DOI] [Google Scholar]
- Caly L., Druce J., Roberts J., Bond K., Tran T., Kostecki R., Yoga Y., Naughton W., Taiaroa G., Seemann T. Isolation and rapid sharing of the 2019 novel coronavirus (SARS-CoV-2) from the first patient diagnosed with COVID-19 in Australia. Med. J. Aust. 2020;212:459–462. doi: 10.5694/mja2.50569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Channappanavar R., Fett C., Zhao J., Meyerholz D.K., Perlman S. Virus-specific memory CD8 T cells provide substantial protection from lethal severe acute respiratory syndrome coronavirus infection. J. Virol. 2014;88:11034–11044. doi: 10.1128/JVI.01505-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Ndhlovu Z.M., Liu D., Porter L.C., Fang J.W., Darko S., Brockman M.A., Miura T., Brumme Z.L., Schneidewind A. TCR clonotypes modulate the protective effect of HLA class I molecules in HIV-1 infection. Nat. Immunol. 2012;13:691–700. doi: 10.1038/ni.2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens E.B., Grant E.J., Wang Z., Gras S., Tipping P., Rossjohn J., Miller A., Tong S.Y., Kedzierska K. Towards identification of immune and genetic correlates of severe influenza disease in Indigenous Australians. Immunol. Cell Biol. 2016;94:367–377. doi: 10.1038/icb.2015.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan J.M., Mateus J., Kato Y., Hastie K.M., Yu E.D., Faliti C.E., Grifoni A., Ramirez S.I., Haupt S., Frazier A. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science. 2021;371:eabf4063. doi: 10.1126/science.abf4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash P., Fiore-Gartland A.J., Hertz T., Wang G.C., Sharma S., Souquette A., Crawford J.C., Clemens E.B., Nguyen T.H.O., Kedzierska K. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature. 2017;547:89–93. doi: 10.1038/nature22383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti A.P., Kula T., Wang Y., Nguyen D.M.V., Weinheimer A., Dunlap G.S., Xu Q., Nabilsi N., Perullo C.R., Cristofaro A.W. Unbiased Screens Show CD8+ T Cells of COVID-19 Patients Recognize Shared Epitopes in SARS-CoV-2 that Largely Reside outside the Spike Protein. Immunity. 2020;53:1095–1107.e3. doi: 10.1016/j.immuni.2020.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Galarza F.F., McCabe A., Santos E.J.M.D., Jones J., Takeshita L., Ortega-Rivera N.D., Cid-Pavon G.M.D., Ramsbottom K., Ghattaoraya G., Alfirevic A. Allele frequency net database (AFND) 2020 update: gold-standard data classification, open access genotype data and new query tools. Nucleic Acids Res. 2020;48(D1):D783–D788. doi: 10.1093/nar/gkz1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths S.J., Riddell N.E., Masters J., Libri V., Henson S.M., Wertheimer A., Wallace D., Sims S., Rivino L., Larbi A. Age-associated increase of low-avidity cytomegalovirus-specific CD8+ T cells that re-express CD45RA. J. Immunol. 2013;190:5363–5372. doi: 10.4049/jimmunol.1203267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifoni A., Weiskopf D., Ramirez S.I., Mateus J., Dan J.M., Moderbacher C.R., Rawlings S.A., Sutherland A., Premkumar L., Jadi R.S. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell. 2020;181:1489–1501.e15, e1415. doi: 10.1016/j.cell.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z., Gu L., Eils R., Schlesner M., Brors B. circlize Implements and enhances circular visualization in R. Bioinformatics. 2014;30:2811–2812. doi: 10.1093/bioinformatics/btu393. [DOI] [PubMed] [Google Scholar]
- Habel J.R., Nguyen T.H.O., van de Sandt C.E., Juno J.A., Chaurasia P., Wragg K., Koutsakos M., Hensen L., Jia X., Chua B. Suboptimal SARS-CoV-2-specific CD8+ T cell response associated with the prominent HLA-A∗02:01 phenotype. Proc. Natl. Acad. Sci. USA. 2020;117:24384–24391. doi: 10.1073/pnas.2015486117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juno J.A., Tan H.-X., Lee W.S., Reynaldi A., Kelly H.G., Wragg K., Esterbauer R., Kent H.E., Batten C.J., Mordant F.L. Humoral and circulating follicular helper T cell responses in recovered patients with COVID-19. Nat. Med. 2020;26:1428–1434. doi: 10.1038/s41591-020-0995-0. [DOI] [PubMed] [Google Scholar]
- Kedzierska K., Koutsakos M. The ABC of Major Histocompatibility Complexes and T Cell Receptors in Health and Disease. Viral Immunol. 2020;33:160–178. doi: 10.1089/vim.2019.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsakos M., Wheatley A.K., Loh L., Clemens E.B., Sant S., Nüssing S., Fox A., Chung A.W., Laurie K.L., Hurt A.C. Circulating TFH cells, serological memory, and tissue compartmentalization shape human influenza-specific B cell immunity. Sci. Transl. Med. 2018;10:eaan8405. doi: 10.1126/scitranslmed.aan8405. [DOI] [PubMed] [Google Scholar]
- Koutsakos M., Illing P.T., Nguyen T.H.O., Mifsud N.A., Crawford J.C., Rizzetto S., Eltahla A.A., Clemens E.B., Sant S., Chua B.Y. Human CD8+ T cell cross-reactivity across influenza A, B and C viruses. Nat. Immunol. 2019;20:613–625. doi: 10.1038/s41590-019-0320-6. [DOI] [PubMed] [Google Scholar]
- Koutsakos M., Rowntree L.C., Hensen L., Chua B.Y., van de Sandt C.E., Habel J.R., Zhang W., Jia X., Kedzierski L., Ashhurst T.M. Integrated immune dynamics define correlates of COVID-19 severity and antibody responses. Cell Rep Med. 2021;2:100208. doi: 10.1016/j.xcrm.2021.100208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Gruta N.L., Kedzierska K., Pang K., Webby R., Davenport M., Chen W., Turner S.J., Doherty P.C. A virus-specific CD8+ T cell immunodominance hierarchy determined by antigen dose and precursor frequencies. Proc. Natl. Acad. Sci. USA. 2006;103:994–999. doi: 10.1073/pnas.0510429103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bert N., Tan A.T., Kunasegaran K., Tham C.Y.L., Hafezi M., Chia A., Chng M.H.Y., Lin M., Tan N., Linster M., Chia W.N., Chen M.I.C., Wang L-F, Ooi E.E., Kalimuddin S., Tambyah P.A., Low J.G-H., Tan Y-J., Bertoletti A. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature. 2020;584:457–462. doi: 10.1038/s41586-020-2550-z. [DOI] [PubMed] [Google Scholar]
- LeMaoult J., Messaoudi I., Manavalan J.S., Potvin H., Nikolich-Zugich D., Dyall R., Szabo P., Weksler M.E., Nikolich-Zugich J. Age-related dysregulation in CD8 T cell homeostasis: kinetics of a diversity loss. J. Immunol. 2000;165:2367–2373. doi: 10.4049/jimmunol.165.5.2367. [DOI] [PubMed] [Google Scholar]
- Messaoudi I., Guevara Patiño J.A., Dyall R., LeMaoult J., Nikolich-Zugich J. Direct link between mhc polymorphism, T cell avidity, and diversity in immune defense. Science. 2002;298:1797–1800. doi: 10.1126/science.1076064. [DOI] [PubMed] [Google Scholar]
- Messaoudi I., Warner J., Nikolich-Zugich D., Fischer M., Nikolich-Zugich J. Molecular, cellular, and antigen requirements for development of age-associated T cell clonal expansions in vivo. J. Immunol. 2006;176:301–308. doi: 10.4049/jimmunol.176.1.301. [DOI] [PubMed] [Google Scholar]
- Ndhlovu Z.M., Kamya P., Mewalal N., Kløverpris H.N., Nkosi T., Pretorius K., Laher F., Ogunshola F., Chopera D., Shekhar K. Magnitude and Kinetics of CD8+ T Cell Activation during Hyperacute HIV Infection Impact Viral Set Point. Immunity. 2015;43:591–604. doi: 10.1016/j.immuni.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T.H., Bird N.L., Grant E.J., Miles J.J., Thomas P.G., Kotsimbos T.C., Mifsud N.A., Kedzierska K. Maintenance of the EBV-specific CD8+ TCRαβ repertoire in immunosuppressed lung transplant recipients. Immunol. Cell Biol. 2017;95:77–86. doi: 10.1038/icb.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T.H., Tan A.C., Xiang S.D., Goubier A., Harland K.L., Clemens E.B., Plebanski M., Kedzierska K. Understanding CD8+ T-cell responses toward the native and alternate HLA-A∗02:01-restricted WT1 epitope. Clin. Transl. Immunology. 2017;6:e134. doi: 10.1038/cti.2017.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T.H.O., Sant S., Bird N.L., Grant E.J., Clemens E.B., Koutsakos M., Valkenburg S.A., Gras S., Lappas M., Jaworowski A. Perturbed CD8+ T cell immunity across universal influenza epitopes in the elderly. J. Leukoc. Biol. 2018;103:321–339. doi: 10.1189/jlb.5MA0517-207R. [DOI] [PubMed] [Google Scholar]
- Peng H., Yang L.T., Wang L.Y., Li J., Huang J., Lu Z.Q., Koup R.A., Bailer R.T., Wu C.Y. Long-lived memory T lymphocyte responses against SARS coronavirus nucleocapsid protein in SARS-recovered patients. Virology. 2006;351:466–475. doi: 10.1016/j.virol.2006.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y., Mentzer A.J., Liu G., Yao X., Yin Z., Dong D., Dejnirattisai W., Rostron T., Supasa P., Liu C., Oxford Immunology Network Covid-19 Response T cell Consortium. ISARIC4C Investigators Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020;21:1336–1345. doi: 10.1038/s41590-020-0782-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price D.A., Asher T.E., Wilson N.A., Nason M.C., Brenchley J.M., Metzler I.S., Venturi V., Gostick E., Chattopadhyay P.K., Roederer M. Public clonotype usage identifies protective Gag-specific CD8+ T cell responses in SIV infection. J. Exp. Med. 2009;206:923–936. doi: 10.1084/jem.20081127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowntree L.C., Nguyen T.H.O., Farenc C., Halim H., Hensen L., Rossjohn J., Kotsimbos T.C., Purcell A.W., Kedzierska K., Gras S., Mifsud N.A. A Shared TCR Bias toward an Immunogenic EBV Epitope Dominates in HLA-B∗07:02-Expressing Individuals. J. Immunol. 2020;205:1524–1534. doi: 10.4049/jimmunol.2000249. [DOI] [PubMed] [Google Scholar]
- Sant S., Grzelak L., Wang Z., Pizzolla A., Koutsakos M., Crowe J., Loudovaris T., Mannering S.I., Westall G.P., Wakim L.M. Single-Cell Approach to Influenza-Specific CD8+ T Cell Receptor Repertoires Across Different Age Groups, Tissues, and Following Influenza Virus Infection. Front. Immunol. 2018;9:1453. doi: 10.3389/fimmu.2018.01453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulien I., Kemming J., Oberhardt V., Wild K., Seidel L.M., Killmer S., Sagar, Daul F., Salvat Lago M., Decker A. Characterization of pre-existing and induced SARS-CoV-2-specific CD8+ T cells. Nat. Med. 2021;27:78–85. doi: 10.1038/s41591-020-01143-2. [DOI] [PubMed] [Google Scholar]
- Shomuradova A.S., Vagida M.S., Sheetikov S.A., Zornikova K.V., Kiryukhin D., Titov A., Peshkova I.O., Khmelevskaya A., Dianov D.V., Malasheva M. SARS-CoV-2 epitopes are recognized by a public and diverse repertoire of human T cell receptors. Immunity. 2020;53:1245–1257.e5. doi: 10.1016/j.immuni.2020.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadlbauer D., Amanat F., Chromikova V., Jiang K., Strohmeier S., Arunkumar G.A., Tan J., Bhavsar D., Capuano C., Kirkpatrick E. SARS-CoV-2 Seroconversion in Humans: A Detailed Protocol for a Serological Assay, Antigen Production, and Test Setup. Curr. Protoc. Microbiol. 2020;57:e100. doi: 10.1002/cpmc.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F., Quan Y., Xin Z.T., Wrammert J., Ma M.J., Lv H., Wang T.B., Yang H., Richardus J.H., Liu W., Cao W.C. Lack of peripheral memory B cell responses in recovered patients with severe acute respiratory syndrome: a six-year follow-up study. J. Immunol. 2011;186:7264–7268. doi: 10.4049/jimmunol.0903490. [DOI] [PubMed] [Google Scholar]
- Thevarajan I., Nguyen T.H.O., Koutsakos M., Druce J., Caly L., van de Sandt C.E., Jia X., Nicholson S., Catton M., Cowie B. Breadth of concomitant immune responses prior to patient recovery: a case report of non-severe COVID-19. Nat. Med. 2020;26:453–455. doi: 10.1038/s41591-020-0819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valkenburg S.A., Venturi V., Dang T.H., Bird N.L., Doherty P.C., Turner S.J., Davenport M.P., Kedzierska K. Early priming minimizes the age-related immune compromise of CD8+ T cell diversity and function. PLoS Pathog. 2012;8:e1002544. doi: 10.1371/journal.ppat.1002544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valkenburg S.A., Josephs T.M., Clemens E.B., Grant E.J., Nguyen T.H., Wang G.C., Price D.A., Miller A., Tong S.Y., Thomas P.G. Molecular basis for universal HLA-A∗0201-restricted CD8+ T-cell immunity against influenza viruses. Proc. Natl. Acad. Sci. USA. 2016;113:4440–4445. doi: 10.1073/pnas.1603106113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Wan Y., Qiu C., Quiñones-Parra S., Zhu Z., Loh L., Tian D., Ren Y., Hu Y., Zhang X. Recovery from severe H7N9 disease is associated with diverse response mechanisms dominated by CD8+ T cells. Nat. Commun. 2015;6:6833. doi: 10.1038/ncomms7833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Zhu L., Nguyen T.H.O., Wan Y., Sant S., Quiñones-Parra S.M., Crawford J.C., Eltahla A.A., Rizzetto S., Bull R.A. Clonally diverse CD38+HLA-DR+CD8+ T cells persist during fatal H7N9 disease. Nat. Commun. 2018;9:824. doi: 10.1038/s41467-018-03243-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiskopf D., Schmitz K.S., Raadsen M.P., Grifoni A., Okba N.M.A., Endeman H., van den Akker J.P.C., Molenkamp R., Koopmans M.P.G., van Gorp E.C.M. Phenotype and kinetics of SARS-CoV-2-specific T cells in COVID-19 patients with acute respiratory distress syndrome. Sci. Immunol. 2020;5:eabd2071. doi: 10.1126/sciimmunol.abd2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertheimer A.M., Bennett M.S., Park B., Uhrlaub J.L., Martinez C., Pulko V., Currier N.L., Nikolich-Žugich D., Kaye J., Nikolich-Žugich J. Aging and cytomegalovirus infection differentially and jointly affect distinct circulating T cell subsets in humans. J. Immunol. 2014;192:2143–2155. doi: 10.4049/jimmunol.1301721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. Springer-Verlag; 2016. ggplot2: Elegant Graphics for Data Analysis. [Google Scholar]
- Zhao J., Alshukairi A.N., Baharoon S.A., Ahmed W.A., Bokhari A.A., Nehdi A.M., Layqah L.A., Alghamdi M.G., Al Gethamy M.M., Dada A.M. Recovery from the Middle East respiratory syndrome is associated with antibody and T-cell responses. Sci. Immunol. 2017;2:eaan5393. doi: 10.1126/sciimmunol.aan5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during the study.