

Abstract
The genus Acidihalobacter has three validated species, Acidihalobacter ferrooxydans , Acidihalobacter prosperus and Acidihalobacter aeolinanus, all of which were isolated from Vulcano island, Italy. They are obligately chemolithotrophic, aerobic, acidophilic and halophilic in nature and use either ferrous iron or reduced sulphur as electron donors. Recently, a novel strain was isolated from an acidic, saline drain in the Yilgarn region of Western Australia. Strain F5T has an absolute requirement for sodium chloride (>5 mM) and is osmophilic, growing in elevated concentrations (>1 M) of magnesium sulphate. A defining feature of its physiology is its ability to catalyse the oxidative dissolution of the most abundant copper mineral, chalcopyrite, suggesting a potential role in biomining. Originally categorized as a strain of A. prosperus , 16S rRNA gene phylogeny and multiprotein phylogenies derived from clusters of orthologous proteins (COGS) of ribosomal protein families and universal protein families unambiguously demonstrate that strain F5T forms a well-supported separate branch as a sister clade to A. prosperus and is clearly distinguishable from A. ferrooxydans DSM 14175T and A. aeolinanus DSM14174T. Results of comparisons between strain F5T and the other Acidihalobacter species, using genome-based average nucleotide identity, average amino acid identity, correlation indices of tetra-nucleotide signatures (Tetra) and genome-to-genome distance (digital DNA–DNA hybridization), support the contention that strain F5T represents a novel species of the genus Acidihalobacter . It is proposed that strain F5T should be formally reclassified as Acidihalobacter yilgarnenesis F5T (=DSM 105917T=JCM 32255T).
Keywords: Acidihalobacter, halotolerant, iron and sulfur oxidising, acidophile, Yilgarn Craton, genome-based average nucleotide identity (ANI), average amino acid identity (AAI), genome-to-genome distance (digital DNA-DNA hybridization (dDDH), chalcopyrite bioleaching
Introduction
Bioleaching is a technique where acidophilic micro-organisms are used to catalyse the extraction of metals from mineral ores through the oxidation of metal sulfides, a technology referred to to generically as biomining [1]. As many accessible higher-grade metal ore bodies are now depleted, lower-grade ores are being increasingly exploited, and bioleaching can have both economic and environmental benefits for processing these materials [2]. However, biomining has long faced the challenge of the negative effect of the presence of salt (sodium chloride) in ores and process waters. The ability of bioleaching micro-organisms to tolerate salt varies between genus and species, but most bioleaching micro-organisms cannot tolerate the levels of chloride present in seawater and can be inhibited by concentrations as low as 6.6 g l−1 [3–5]. However, the presence of salt has been shown to enhance the abiotic leaching of the recalcitrant but also most abundant copper-containing mineral in the lithosphere, chalcopyrite (CuFeS2). Therefore, the use of halophilic micro-organisms that are also capable of tolerating low pH while being able to catalyse the oxidative dissolution of chalcopyrite would be of major benefit to the biomining industry [4, 5]. Furthermore, as freshwater resources become increasingly scarce, the mining industry would benefit from using seawater at mining sites to reduce the costs associated with desalination plants [2].
Due to the limited environments that are available for the discovery of the unique micro-organisms that inhabit low pH and highly saline environments and have the ability to oxidize metal sulfide minerals, it is important to isolate and characterize these prokaryotes [5].
The genus Acidihalobacter represents one such group of ferrous iron- and sulfur-oxidizing bacteria that are both extremely acidophilic and halotolerant (tolerating up to 1283 mM NaCl) [6–8]. Three members of the genus Acidihalobacter have been isolated from the Vulcano region Italy. Acidihalobacter prosperus DSM 5130T (previously ‘ Thiobacillus prosperus ’) was isolated from a geothermally heated seafloor at Porto di Levante while the type strains of Acidihalobacter aeolinanus (previously ‘ Acidihalobacter prosperus DSM 14174’) and Acidihalobacter ferrooxydans (previously ‘Acidihalobacter ferrooxidans DSM 14175’) were isolated from a shallow acidic pool by the shore of Baia de Levant [9, 10]. All are members of the family Ectothiorhodospiraceae of the class Gammaproteobacteria in the genus Acidihalobacter , and each has been characterized as the type strain of their respective species [11–14].
More recently, a novel bacterial strain, designated as F5T and belonging to genus Acidihalobacter , was isolated from an acidic saline drain in the Yilgarn region of Western Australia [5]. The isolate was initially considered to be a strain of A. prosperus due to the high sequence similarity (98.7 %) of its 16S rRNA gene to the latter [8]. However, a defining feature of this strain that distinguishes it from other strains of Acidihalobater was its ability to leach chalcopyrite at 508 mM NaCl. This makes it a potentially valuable isolate for the industrial biorecovery of copper through saline water bioleaching [8].
The genome of strain F5T is the only available complete genome of a halotolerant acidophile to date, as well as the first complete genome for a member of the genus Acidihalobacter [8]. The completeness of its genome provides an opportunity for studies of its metabolic capabilities as well as clarification of its taxonomy. Genome-based classification of the other members of the genus Acidihalobacter has recently been completed and has proven to provide a more robust approach for the re-evaluation of taxonomy using bioinformatics-based phylogenomic strategies that are more accurate than 16S rRNA gene phylogeny and morphology alone [14, 15].
Methods
Isolation of strain F5T
Strain F5T was isolated from an enrichment culture obtained from an acidic saline drain in the Yilgarn region in Western Australia (pH 2.1, 463 mM chloride, 25 mM iron (II); GPS coordinates −31.070302° S, 117.43901° E) [5, 8]. The enrichment culture was inoculated onto overlay plates [16] (0.625 % agarose pH 2.5) enriched with (i) FeSO4, (ii) K2S4O6 or (iii) a mixture of both, containing 214 mM NaCl. Single colonies picked from the solid media were resuspended in liquid media containing either 50 mM ferrous sulphate or 5 mM potassium tetrathionate, basal salts (3 mM (NH4)2SO4, 1.6 mM MgSO4 and 2.9 mM KH2PO4) and trace elements (pH 1.8) [7]. DNA extraction and 16S rRNA gene sequences were obtained through Sanger sequencing as described previously [17].
Tolerance to temperature, pH and NaCl
A pure culture of strain F5T was maintained at 30 °C in basal salts containing 50 mM ferrous sulphate and 5 mM potassium tetrathionate at pH 2.5 as described above, and DNA was extracted from these cultures for genome sequencing as described previously [8]. Growth of the isolate was tested at a range of temperatures (17–42.5 °C), pH levels (pH 1– 5) and sodium chloride concentrations (0–1.71 M; data not shown). Bioleaching studies were performed as described elsewhere [8].
For the purpose of this study, further growth tests were performed on various liquid and solid media, including the growth of strain F5T on elemental S, H2 and in K2S4O6-free media containing 10 mM Fe(II) and 200 mM MgSO4. Aerobic growth was tested at different concentrations of MgSO4 (0, 50, 100, 200, 500 and 1000 mM). When the cultures failed to grow, 25 mM NaCl was added in the medium and incubated further for up to 24 days. Tests for optimum concentration of NaCl in cultures containing 200 mM MgSO4 were then performed using 0, 5, 10, 25 and 50 mM NaCl.
Electron microscopy
Electron microscopic studies of strain F5T were performed using the method described previously for the type strain of A. prosperus [18].
Selection of members for phylogenetic assignment
Members for inclusion in the study were identified from the 30 closest phylogenetic neighbours as given by ab initio comparisons of glimmer3 gene candidates with a set of universal proteins and up to 200 unduplicated proteins in the seed and Rapid Annotation of Microbial genomes using Subsystems Technology (rast) [19, 20]. These were verified by comparison to the sequences previously used for the reclassification of the type strain of A. prosperus [15], as well as by comparison with nucleotide databases after running a blastn-based script using an E-value threshold of 1e-5 and the databases greengenes, RDP and silva [21–23].
A total of 15 genomes, including the four members of the genus Acidihalobacter , were selected for inclusion into the following phylogenetic tree reconstructions. Halothiobacillus neapolitanus ATCC 23641 was used as an outgroup (Table S1, available in the online version of this article).
Closest phylogenetic neighbours of the genus Acidihalobacter were selected based on ab initio comparisons of glimmer3 and rast [19, 20]. A total of 14 organisms of the order Chromatiales , including the three validated members of the genus Acidihalobacter together with strain F5T, were selected for inclusion in phylogenetic tree reconstructions.
Phylogenetic tree reconstruction
16S rRNA gene phylogeny
16S rRNA genes of Acidihalobacter species were identified by comparison of genomic sequences against 16S rRNA databases greengenes [21], RDP [22] and silva [23] by blastn [24] using an E-value threshold of 1e-5. Sequences of the taxonomically related genomes from the order Chromatiales were selected from NCBI databases to be included in the 16S rRNA gene phylogenetic tree. All 16S rRNA gene sequences were aligned in mafft version 7 with the L-INS-i iterative refinement [25, 26]. The phylogenetic tree was reconstructed with iqtree, using 1000 replications as bootstrap support [27, 28] with best model fit by iqtree (TN+F+I+G according to the Bayesian information criterion) [29].
Multi-locus sequence analysis (MLSA)
A set of 30 ribosomal proteins associated with COG markers (Table S2) were obtained from the DOE Joint Genome Institute – Integrated Microbial Genomes and Microbiome Samples website (https://img.jgi.doe.gov/cgi-bin/m/main.cgi) for each micro-organism in the study [30, 31]. A multi-locus phylogenomic tree was reconstructed by aligning a concatenated set of the 30 COGs sequences with L-INS-i iterative refinement in mafft version 7 and removal of unreliable regions with gblocks [32, 33]. A maximum-likelihood tree with 1000 replicates was reconstructed with best-fit model LG+F+I+G according to the Bayesian information criterion using iqtree [27, 28].
Nine conserved housekeeping genes (argS, dnaQ, dnaN, era, gltA, gyrB, ppnK, rpoB and rpoD [34–36]) were used to build a multi-gene species tree using a concatenated alignment from members of the order Chromatiales as described previously [14]. The contatenated alignment was reconstructed using the L-INS-i iterative refinement in mafft version 7 [25, 26], which were masked to remove unreliable regions with gblocks [32, 33]. The maximum-likelihood tree was reconstructed with iqtree using the bootstrap method with 1000 replicates [37] and the best-suited substitution model GTR+F+I+G selected by iqtree .
Sequence-based methods for species circumscription
Calculation of average nucleotide identity was based on blast (ANIb) [24, 38, 39] and the correlation indexes of tetra-nucleotide signatures (Tetra) were conducted using Jspecies [39] and JspeciesWS (http://jspecies.ribohost.com/jspeciesws/#Analyse) [40]. The Genome-to-Genome Distance Calculator (GGDC) web tool (http://ggdc.dsmz.de/distcalc2.php) was used to calculate the digital DNA–DNA hybridization (dDDH) values [41, 42]. Average amino acid identity (AAI) [43] values were calculated with the CompareM tool (https://github.com/dparks1134/CompareM).
Gene prediction
Genes potentially encoding terminal oxidases and those involved in ferrous iron and reduced sulphur oxidation were predicted using a bidirectional blastp of the NR databases as described previously [14] and were visualized using Artemis [44].
Results and discussion
The genomes of the different Acidihalobacter isolates included in this study were previously obtained from pure cultures grown in acidified basal salts/trace elements medium supplemented with soluble iron and sulphur sources, and sodium chloride [8, 11–13]. However, key differences can be seen in the pH, temperature and optimum NaCl concentrations required for growth on soluble iron and sulphur sources as well as on the mineral sulfide ore pyrite (Table 1). While the type strain of A. prosperus has been shown to grow on sphalerite, chalcopyrite, arsenopyrite and galena as well as on H2S, no leaching data is available for growth on these substrates [9]. Meanwhile, the type strains of A. aeolianus and A. ferrooxydans have previously been shown to oxidize chalcopyrite when in mixed culture; however, growth of pure isolates has not been tested [45, 46]. Furthermore, growth of the type strains of A. aeolianus and A. ferrooxydans is yet to be tested on other mineral ores. Strain F5T is the only isolate that has been shown to successfully leach the mineral ore pentlandite (at up to 1283 mM NaCl at pH 2.5 [8]). More importantly, it the only known isolate to leach the recalcitrant mineral chalcopyrite at up to 513 mM NaCl (pH 2.5), thereby suggesting its suitability to leach base metals from different sulfide ores at chloride ion concentrations of sea water or above (564 mM NaCl [6]).
Table 1.
Comparison of genomic and phenotypic features of the four members of the genus Acidihalobacter
|
Feature |
Genome |
|||
|---|---|---|---|---|
|
Acidihalobacter strain F5T |
Acidihalobacter prosperus DSM5130T |
Acidihalobacter aeolianus DSM 14174T |
Acidihalobacter ferrooxydans DSM 14175T |
|
|
Genome size (Mbp) |
3.57 |
3.36 |
3.36 |
3.45 |
|
G+C content (mol%) |
59.9 |
64.5 |
62.2 |
61.6 |
|
Predicted coding DNA sequence (CDS) |
3233 |
3088 |
3194 |
3089 |
|
Plasmid |
– |
– |
162 484 bp (pABPV6) |
– |
|
tRNA genes |
47 |
48 |
46 |
45 |
|
Sulphur oxygenase reductase (EC 1.13.11.55) |
– |
+ |
– |
+ |
|
Temperature range for growth (°C) |
24–33 |
20–45 [9] |
26–42 [60] |
26–43 [60] |
|
Optimum temperature for growth (°C) |
30 |
33 [9] |
36 [60] |
36 [60] |
|
pH range for growth |
2.0–4.0 |
1.0–4.5 [9] |
1.5–3.0 [14] |
1.0–3.0 [14] |
|
Optimum pH for growth |
2.5 |
2.0 [9] |
1.8 [14] |
1.8 [14] |
|
NaCl range for growth (mM) |
5–1283 |
70–1030 [18] |
60–1283 [7] |
60–856 [7] |
|
Optimum NaCl (mM) for growth on FeSO4 and K2S4O6 |
428 |
340 [9] |
||
|
Optimum NaCl (mM) for growth on pyrite |
513 |
n/a |
256 [7] |
856 [7] |
|
Optimum NaCl (mM) for growth on chalcopyrite |
254 |
na |
na |
na |
+, Present; −, absent; na, not available.
Growth characteristics of strain F5T
The growth tests on strain F5T performed in this study showed that it can grow on both elemental sulphur and the reduced sulphur oxy-anion, tetrathionate. Growth was also observed when Fe(II) was provided as the sole electron donor and 200 mM MgSO4 as the osmolyte, though no growth was seen when hydrogen was provided as the sole electron donor. Furthermore, the results of the tests using 0, 50, 100, 200, 500 and 1000 mM MgSO4 as the osmotic stressor showed that no growth in the absence of salt. The addition of 25 mM NaCl resulted in good oxidation for cultures containing 50 and 500 mM but not 0 or 1 M MgSO4. This shows that strain F5T has an absolute requirement for NaCl, as has previously been shown for the other members of the genus Acidihalobacter . When NaCl was added in increments (0, 5, 10, 25 and 50 mM) to cultures containing 200 mM MgSO4 iron oxidation was evident in the cultures containing 10 and 25 mM NaCl within 3 days. After 4 days, some oxidation was seen in the 5 mM NaCl containing cultures, while after 12 days the 50 mM NaCl cultures were well oxidized. The salt-free cultures showed very little oxidation even after 12 days. While MgSO4 can meet its requirement for a relatively high external osmotic potential, a minimum of 5 mM NaCl is required for iron oxidation, with 10–25 mM being the optimum NaCl requirement in the presence of 200 mM MgSO4. This NaCl requirement is lower than has been previously shown for the type strains of the three validated Acidihalobacter species (≥60 mM), although these values were determined with NaCl acting as the only significant osmolyte. In total, the results of the growth studies and absolute requirement of strain F5T for NaCl, confirms its obligately osmophilic nature.
Microscopy
Electron microscopic studies revealed that cells of strain F5T were 1–2 µm long straight rods (Fig. 1). Endopsores were not detected.
Fig. 1.
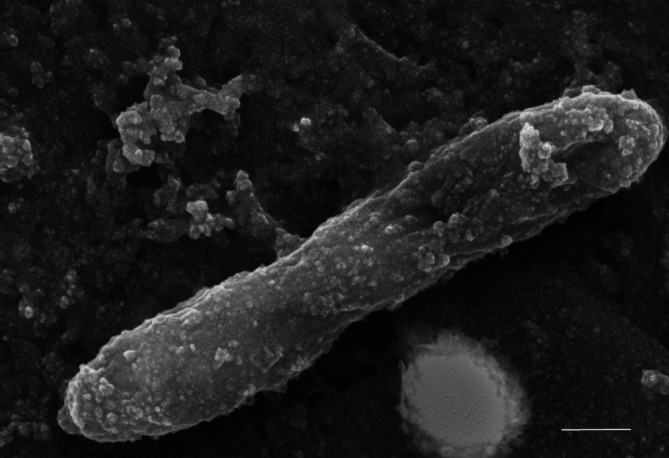
Electron microscopy image of strain F5T grown in the presence of 214 mM NaCl. The scale bar is 200 nm.
Genome and gene information
Members of the family Ectothiorhodospiraceae are known to have a DNA G+C content within the range 50.5–69.7 mol% [47]. The bioinformatically inferred G+C content for the genome of strain F5T was previously found to be 59.9 mol%, which is lower than that of the other members in the genus, but is still within the range of the family Ectothiorhodospiraceae . The genome of strain F5T is 3.57 Mbp and is predicted to have 3233 coding sequences with 47 tRNA genes [8]. Bioinformatically predicted terminal oxidases from the genomes of F5T were as for DSM14174T and DSM14175T and included aa3 (EC 1.9.3.1), bo3 (EC 1.10.3.10), bd-I (EC 1.10.3.14) and fumarate reductase (quinol, EC 1.3.5.1–1.3.5.4). Respiratory quinones predicted from the genomes include ubiquinone ubiABDEGIHJX (EC 1.14.13.-, 1.14.12.240, 2.1.1.222, 2.1.1.64, 2.1.1.163, 2.1.1.201, 2.5.1.39, 2.5.1.129, 4.1.1.98). Phenotypic and genomic features of the four species of the genus Acidihalobacter are compared in Table 1. The genome is predicted to encode a rusticyanin gene cluster thought to be involved in Fe2+ oxidation [8]. The accession number of the genome sequence of strain F5T is CP017415.1.
Phylogeny based on 16S rRNA gene sequence analysis
A 16S rRNA gene phylogenetic tree of strain F5T and three validated members of the genus Acidihalobacter was reconstructed using ten validated species belonging to the family Ectothiorhodospiraceae of the order Chromatiales of the class Gammaproteobacteria using Halothiobacillus neapolitanus ATCC 23641 as an outgroup (Fig. 2). The tree agrees with a previously published 16S rRNA gene phylogenetic tree in the placement of strain F5T within the genus Acidihalobacter and confirms its taxonomic position within the family Ectothiorhodospiraceae [8]. Strain F5T forms a separate branch as a sister clade to A. prosperus DSM 5130T that is well-supported (95 % bootstrap support) and is clearly distinguishable from A. ferrooxydans DSM 14175T and A. aeolinanus DSM 14174T.
Fig. 2.

Maximum-likelihood phylogenetic tree of 16S rRNA gene sequences of strain F5T (in red) and other phylogenetic relatives as described in the text. Bootstrap percentages (1000 replicates) are labelled at the nodes. Scale bar represents 0.03 nucleotide substitution per site. The genetic distance of Halothiobacillus neapolitanus ATCC 23641 is not to scale as indicated by the break lines //. The full list of NCBI accession numbers is given in Table S1.
Phylogeny based on multiple locus sequence analyses (MLSA)
Additional approaches were used to evaluate the phylogenomic position of strain F5T. Phylogenomic trees were reconstructed based on the sequences of 30 concatenated conserved ribosomal proteins [30, 31] (Fig. 3a) and nine concatenated housekeeping genes (Fig. 3b). These multi-locus sequence alignments were sufficiently long to allow mapping of their phylogenetic relationships [30, 34, 35]. Both trees consistently place strain F5T as a sister clade to A. prosperus DSM 5130T with 100 % bootstrap support and clearly show that strain F5T forms a distinct branch from A. ferrooxydans DSM 14175T and A. aeolianus DSM 14174T agreeing with the 16S rRNA gene phylogenetic tree.
Fig. 3.

Phylogenomic trees of 14 members of the order Chromatiales and Halothiobacillus neapolitanus ATCC 23641 as outgroup, including strain F5T (in red), based on (a) 30 concatenated conserved proteins from proposed 34 ribosomal proteins [30, 31] and (b) nine concatenated housekeeping genes. Statistically supported bootstrap values as percentages of 1000 replicates are labelled at the nodes. Scale bar represents 0.07 amino acid and 0.2 nucleotide changes per site, respectively. The full list of COG families is given in Table S2.
MLSA is a powerful tool for determining phylogenetic relationships but it is not widely used to discriminate species and subspecies because it is difficult to decide the depth of clustering that should be used as a threshold for differentiation [48].
Phylogenetic distance based on percentage similarity of 16S rRNA gene sequences
16S rRNA gene sequence similarity analysis is frequently used to infer phylogenetic distance and is used in microbial classification and species identification [49]. The similarity of the 16S rRNA gene sequence of strain F5T to the three validated Acidihalobacter species is reported as a heat map comparison (Fig. 4).
Fig. 4.

Heat maps of the percent difference of 16S rRNA gene sequences between the three validated Acidihalobacter species and strain F5T. The results are displayed as a cladogram based on the 16S rRNA gene phylogenetic tree shown in Fig. 2, using 97, 98.7 and 98.73 % 16S rRNA gene sequence similarity cutoff values (left to right, respectively).
Strain F5T is located in a sister clade to A. prosperus DSM 5130T but can be distinguished from it at a cutoff of 98.7 % sequence similarity (Fig. 4). A cutoff of 97 % 16S rRNA gene sequence similarity has been used to identify a new species [50]. However, in many instances this was not sufficient for species discrimination and a cutoff of 98.5 % similarity has become the new ‘gold-standard’ [51, 52].
Other phylogenomic approaches for species discrimination (dDDH, ANI, AAI and Tetra)
Today, phylogenomic approaches such as dDDH, ANI (average nucleotide identity), AAI and Tetra (Tetra Nucleotide Signature Correlation Index) are frequently used for microbial classification and often provide better criteria for species discrimination than 16S rRNA gene sequence similarity [53]. The currently accepted cutoff values for delimiting species boundaries are about 70 % for dDDH [41, 42], 95 % for ANI [38, 54–56], 95–96 % for AAI [57, 58] and 0.989 for Tetra [39, 59]. Using these approaches, we report the values for the comparisons between the three validated species of Acidihalobacter and strain F5T (Fig. 5). These values support the previously published species designations for A. ferroxydans DSM 14175T, A. aeolianus DSM 14174T and A. prosperus DSM 5130T [14]. The values for the comparison of strain F5T with A. prosperus DSM 5130T are as follows (Fig. 5): 22.1 % (dDDH); 79.21 % (ANI); 83.93 % (AAI); and 0.92 (Tetra). These results are all well below the accepted cutoff values for species delineation, indicating that strain F5T should be considered as representing a new species of the genus Acidihalobacter .
Fig. 5.

Heat maps indicating the genetic relatedness between the three validated Acidihalobacter species and strain F5T displayed as a cladogram using different non-sequence-based methods. (a) dDDH, digital DNA–DNA hybridization, (b) ANI, average nucleotide identity, (c) AAI, average amino acid identity and (d) Tetra, nucleotide signature correlation index. The cutoffs for each of the four methods represent the values accepted as defining different species.
Description of Acidihalobacter yilgarnensis sp. nov.
Acidihalobacter yilgarnensis (yil.garn.en′sis . N.L. masc. adj. yilgarnensis, referring to its isolation from the Yilgarn region, Western Australia).
Cells are Gram-stain-negative, motile, straight rods (1–2 µm long). Extremely acidophilic, optimum pH for growth is pH 2.5 with a range of pH 2.0–4.0. Halotolerant, can grow at up to 1283 mM NaCl with optimal growth at 428 mM NaCl. Mesophilic, optimal growth occurs at 30 °C, and capable of growth between 24 and 33 °C. Chemolithoautotrophic and aerobic. Able to utilize ferrous iron, elemental sulphur and tetrathionate as electron donors. It is able to leach base metals from the sulfide mineral pyrite (FeS2) at up to 846 mM NaCl, pentlandite (Fe,Ni)9S8) at 1283 mM NaCl and chalcopyrite at 508 mM NaCl. Predicted terminal oxidases from the genome include aa3 (EC 1.9.3.1), bo3 (EC 1.10.3.10), bd-I (EC 1.10.3.14) and fumarate reductase (quinol, EC 1.3.5.1–1.3.5.4). Predicted respiratory quinones from the genome include ubiquinone (EC 1.14.13.-, 2.1.1.64, 2.1.1.63, 2.1.1.201, 2.1.1.222, 2.5.1.39, 2.5.1.129, 4.1.1.98). The genome contains a full compliment of sox genes distributed in two clusters (soxXYZ and soxXA) and separated soxA and soxB. It also includes a gene cluster for the predicted biosynthesis of the osmoprotectant ectoine. The G+C content of the DNA is 59.9 mol%. The genome contains one copy of both the 16S and 23S rRNA genes and contains 3233 coding sequences and 47 tRNA genes. The whole-genome sequence of 3 566 941 bp is available (GenBank accession no. CP017415.1).
The type strain is F5T (=DSM 105917T=JCM 32255T), isolated from an acidic saline drain in the Yilgarn region, Western Australia.
Supplementary Data
Funding information
H.N.K. was the recipient of an Australian Government Research Training Program Scholarship and a CSIRO Mineral Resources Postgraduate Scholarship. D.H. was supported by Fondecyt 1 140 617 and the Programa de Apoyo a Centros con Financiamiento Basal AFB 17004 to the Fundación Ciencia and Vida. C.G. was supported by a post-doctoral fellowship FONDECYT 3190792.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Footnotes
Abbreviations: AAI, average amino acid identity; ANIb, average nucleotide identity based on blast; COGS, clusters of orthologous proteins; dDDH, digital DNA–DNA hybridization; MAFFT, Multiple Alignment using Fast Fourier Transform; MLSA, multiple locus sequence analyses; RAST, Rapid Annotation of Microbial genomes using Subsystems Technology; RDP, Ribosomal Database Project; Tetra, a statistical analysis of tetranucleotide usage patterns in genomic fragments.
Two supplementary tables are available with the online version of this article.
References
- 1.Rohwerder T, Gehrke T, Kinzler K, Sand W. Bioleaching review Part A: progress in bioleaching: fundamentals and mechanisms of bacterial metal sulfide oxidation. Appl Microbiol Biotechnol. 2003;63:239–248. doi: 10.1007/s00253-003-1448-7. [DOI] [PubMed] [Google Scholar]
- 2.Watling H. Microbiological advances in Biohydrometallurgy. Minerals. 2016;6:49. doi: 10.3390/min6020049. [DOI] [Google Scholar]
- 3.Shiers DW, Blight KR, Ralph DE. Sodium sulphate and sodium chloride effects on batch culture of iron oxidising bacteria. Hydrometallurgy. 2005;80:75–82. doi: 10.1016/j.hydromet.2005.07.001. [DOI] [Google Scholar]
- 4.Zammit CM, Mangold S, Jonna V, Mutch LA, Watling HR, et al. Bioleaching in brackish waters--effect of chloride ions on the acidophile population and proteomes of model species. Appl Microbiol Biotechnol. 2012;93:319–329. doi: 10.1007/s00253-011-3731-3. [DOI] [PubMed] [Google Scholar]
- 5.Zammit CM, Mutch LA, Watling HR, Watkin ELJ. The characterization of salt tolerance in biomining microorganisms and the search for novel salt tolerant strains. Adv Mat Res. 2009;71-73:283–286. doi: 10.4028/www.scientific.net/AMR.71-73.283. [DOI] [Google Scholar]
- 6.Kaksonen AH, Boxall NJ, Gumulya Y, Khaleque HN, Morris C, et al. Recent progress in biohydrometallurgy and microbial characterisation. Hydrometallurgy. 2018;180:7–25. doi: 10.1016/j.hydromet.2018.06.018. [DOI] [Google Scholar]
- 7.Khaleque HN, Kaksonen AH, Boxall NJ, Watkin ELJ. Chloride ion tolerance and pyrite bioleaching capabilities of pure and mixed halotolerant, acidophilic iron- and sulfur-oxidizing cultures. Miner Eng. 2018;120:87–93. doi: 10.1016/j.mineng.2018.02.025. [DOI] [Google Scholar]
- 8.Khaleque HN, Corbett MK, Ramsay JP, Kaksonen AH, Boxall NJ, et al. Complete genome sequence of Acidihalobacter prosperus strain F5, an extremely acidophilic, iron- and sulfur-oxidizing halophile with potential industrial applicability in saline water bioleaching of chalcopyrite. J Biotechnol. 2017;262:56–59. doi: 10.1016/j.jbiotec.2017.10.001. [DOI] [PubMed] [Google Scholar]
- 9.Huber H, Stetter KO. Thiobacillus prosperus sp. nov., represents a new group of halotolerant metal-mobilizing bacteria isolated from a marine geothermal field. Arch Microbiol. 1989;151:479–485. doi: 10.1007/BF00454862. [DOI] [Google Scholar]
- 10.Simmons S, Norris R. Acidophiles of saline water at thermal vents of Vulcano, Italy. Extremophiles. 2002;6:201–207. doi: 10.1007/s007920100242. [DOI] [PubMed] [Google Scholar]
- 11.Ossandon FJ, Cárdenas JP, Corbett M, Quatrini R, Holmes DS, et al. Draft genome sequence of the iron-oxidizing, acidophilic, and halotolerant "Thiobacillus prosperus" type strain DSM 5130. Genom Announc. 2014;2:e01042–01014. doi: 10.1128/genomeA.01042-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khaleque HN, Ramsay JP, Murphy RJT, Kaksonen AH, Boxall NJ, et al. Draft Genome Sequence of the Acidophilic, Halotolerant, and Iron/Sulfur-Oxidizing Acidihalobacter prosperus DSM 14174 (Strain V6) Genom Announc. 2017;5:e01469–01416. doi: 10.1128/genomeA.01469-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khaleque HN, Ramsay JP, Murphy RJT, Kaksonen AH, Boxall NJ, et al. Draft Genome Sequence of Acidihalobacter ferrooxidans DSM 14175 (Strain V8), a New Iron- and Sulfur-Oxidizing, Halotolerant, Acidophilic Species. Genom Announc. 2017;5:e00413–00417. doi: 10.1128/genomeA.00413-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khaleque HN, González C, Kaksonen AH, Boxall NJ, Holmes DS, et al. Genome-based classification of two halotolerant extreme acidophiles, Acidihalobacter prosperus V6 (= DSM 14174=JCM 32253) and ‘Acidihalobacter ferrooxidans’ V8 (= DSM 14175=JCM 32254) as two new species, Acidihalobacter aeolianus sp. nov. and Acidihalobacter ferrooxydans sp. nov., respectively. Int J Syst Evol Microbiol. 2019;69:1557–1565. doi: 10.1099/ijsem.0.003313. [DOI] [PubMed] [Google Scholar]
- 15.Pablo Cárdenas J, Ortiz R, Norris PR, Watkin E, Holmes DS. Reclassification of 'Thiobacillus prosperus' Huber and Stetter 1989 as Acidihalobacter prosperus gen. nov., sp. nov., a member of the family Ectothiorhodospiraceae . Int J Syst Evol Microbiol. 2015;65:3641–3644. doi: 10.1099/ijsem.0.000468. [DOI] [PubMed] [Google Scholar]
- 16.Johnson DB, McGinness S. A highly effecient and universal solid medium for growing mesophilic and moderately thermophilic, iron-oxidizing, acidophilic bacteria. J Microbiol Methods. 1991;13:113–122. doi: 10.1016/0167-7012(91)90011-E. [DOI] [Google Scholar]
- 17.Zammit CM, Mutch LA, Watling HR, Watkin EL. The recovery of nucleic acid from biomining and acid mine drainage microorganisms. Hydrometallurgy. 2011;108:87–92. doi: 10.1016/j.hydromet.2011.03.002. [DOI] [Google Scholar]
- 18.Dopson M, Holmes DS, Lazcano M, McCredden TJ, Bryan CG, et al. Multiple Osmotic Stress Responses in Acidihalobacter prosperus Result in Tolerance to Chloride Ions. Front Microbiol. 2016;7:2132. doi: 10.3389/fmicb.2016.02132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, et al. The seed and the rapid annotation of microbial genomes using subsystems technology (RAST) Nucleic Acids Res. 2014;42:D206–D214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with glimmer. Bioinformatics. 2007;23:673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, et al. The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, et al. Gapped blast and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katoh K, Misawa K, Kuma K-ichi, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katoh K, Standley DM. MAFFT: iterative refinement and additional methods. Methods Mol Biol. 2014;1079:131–146. doi: 10.1007/978-1-62703-646-7_8. [DOI] [PubMed] [Google Scholar]
- 27.Brown J. Bootstrap hypothesis tests for evolutionary trees and other dendrograms. Proc Natl Acad Sci U S A. 1994;91:12293–12297. doi: 10.1073/pnas.91.25.12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 29.Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yutin N, Puigbò P, Koonin EV, Wolf YI. Phylogenomics of prokaryotic ribosomal proteins. PLoS One. 2012;7:e36972. doi: 10.1371/journal.pone.0036972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 33.Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- 34.López-Hermoso C, de la Haba RR, Sánchez-Porro C, Papke RT, Ventosa A. Assessment of multilocus sequence analysis as a valuable tool for the classification of the genus Salinivibrio . Front Microbiol. 2017;8:1107. doi: 10.3389/fmicb.2017.01107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yonezuka K, Shimodaira J, Tabata M, Ohji S, Hosoyama A, et al. Phylogenetic analysis reveals the taxonomically diverse distribution of the Pseudomonas putida group. J Gen Appl Microbiol. 2017;63:1–10. doi: 10.2323/jgam.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 36.Gomila M, Peña A, Mulet M, Lalucat J, García-Valdés E. Phylogenomics and systematics in Pseudomonas . Front Microbiol. 2015;6:214. doi: 10.3389/fmicb.2015.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 2018;35:518–522. doi: 10.1093/molbev/msx281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, et al. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 39.Richter M, Rosselló-Móra R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A. 2009;106:19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richter M, Rosselló-Móra R, Oliver Glöckner F, Peplies J. JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics. 2016;32:btv681. doi: 10.1093/bioinformatics/btv681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Auch AF, von Jan M, Klenk H-P, Göker M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci. 2010;2:117–134. doi: 10.4056/sigs.531120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meier-Kolthoff JP, Auch AF, Klenk H-P, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics. 2013;14:60. doi: 10.1186/1471-2105-14-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci U S A. 2005;102:2567–2572. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carver T, Harris SR, Berriman M, Parkhill J, McQuillan JA. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics. 2012;28:464–469. doi: 10.1093/bioinformatics/btr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davis-Belmar CS, Nicolle JLC, Norris PR. Ferrous iron oxidation and leaching of copper ore with halotolerant bacteria in ore columns. Hydrometallurgy. 2008;94:144–147. doi: 10.1016/j.hydromet.2008.05.030. [DOI] [Google Scholar]
- 46.Norris PR, Davis-Belmar CS, Nicolle JLC, Calvo-Bado LA, Angelatou V. Pyrite oxidation and copper sulfide ore leaching by halotolerant, thermotolerant bacteria. Hydrometallurgy. 2010;104:432–436. doi: 10.1016/j.hydromet.2010.03.025. [DOI] [Google Scholar]
- 47.Imhoff JF. Family II. Ectothiorhodospiraceae Imhoff 1984b, 339VP. Bergey’s Manual of Systematic Bacteriology: Volume 2: The Proteobacteria, Part B: The Gammaproteobacteria. 2007;2:41. p. [Google Scholar]
- 48.Gevers D, Cohan FM, Lawrence JG, Spratt BG, Coenye T, et al. Opinion: Re-evaluating prokaryotic species. Nat Rev Microbiol. 2005;3:733–739. doi: 10.1038/nrmicro1236. [DOI] [PubMed] [Google Scholar]
- 49.Lane DJ. 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M, editors. Nucleic Acid Techniques in Bacterial Systematics. New York, NY: John Wiley and Sons; 1991. pp. 115–175. [Google Scholar]
- 50.Stackebrandt E, Goebel BM. Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J Syst Evol Microbiol. 1994;44:846–849. doi: 10.1099/00207713-44-4-846. [DOI] [Google Scholar]
- 51.Yarza P, Yilmaz P, Pruesse E, Glöckner FO, Ludwig W, et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol. 2014;12:635–645. doi: 10.1038/nrmicro3330. [DOI] [PubMed] [Google Scholar]
- 52.Rodriguez-R LM, Castro JC, Kyrpides NC, Cole JR, Tiedje JM, et al. How much do rRNA gene surveys underestimate extant bacterial diversity? Appl Environ Microbiol. 2018;84:e00014–00018. doi: 10.1128/AEM.00014-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chun J, Oren A, Ventosa A, Christensen H, Arahal DR, et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int J Syst Evol Microbiol. 2018;68:461–466. doi: 10.1099/ijsem.0.002516. [DOI] [PubMed] [Google Scholar]
- 54.Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9:1–8. doi: 10.1038/s41467-018-07641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim M, Oh HS, Park S-C, Chun J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol. 2014;64:346–351. doi: 10.1099/ijs.0.059774-0. [DOI] [PubMed] [Google Scholar]
- 56.Rodriguez-R LM, Konstantinidis KT. Bypassing cultivation to identify bacterial species. Microbe. 2014;9:111–118. doi: 10.1128/microbe.9.111.1. [DOI] [Google Scholar]
- 57.Thompson CC, Chimetto L, Edwards RA, Swings J, Stackebrandt E, et al. Microbial genomic taxonomy. BMC Genomics. 2013;14:913. doi: 10.1186/1471-2164-14-913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Konstantinidis KT, Tiedje JM. Towards a genome-based taxonomy for prokaryotes. J Bacteriol. 2005;187:6258–6264. doi: 10.1128/JB.187.18.6258-6264.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Teeling H, Meyerdierks A, Bauer M, Amann R, Glöckner FO. Application of tetranucleotide frequencies for the assignment of genomic fragments. Environ Microbiol. 2004;6:938–947. doi: 10.1111/j.1462-2920.2004.00624.x. [DOI] [PubMed] [Google Scholar]
- 60.Simmons S. PhD thesis. University of Warwick; 2001. The microbial ecology of acidic environments. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.