

Abstract
While innate lymphoid cells (ILCs) play fundamental roles in mucosal barrier functionality and tissue homeostasis, ILC-related mechanisms underlying intestinal barrier function, homeostatic regulation, and graft rejection in intestinal transplantation (ITx) patients have yet to be thoroughly defined. We found protective type 3 NKp44+ILCs (ILC3s) to be significantly diminished in newly transplanted allografts, compared to allografts at 6 months, while proinflammatory type 1 NKp44−ILCs (ILC1s) were higher. Moreover, serial immunomonitoring revealed that in healthy allografts, protective ILC3s repopulate by 2–4 weeks postoperatively, but in rejecting allografts they remain diminished. Intracellular cytokine staining confirmed that NKp44+ILC3 produced protective IL-22, while ILC1s produced proinflammatory IFN-γ and TNF-α. Our findings about the paucity of protective ILC3s immediately following transplant and their repopulation in healthy allografts during the first month following transplant were confirmed by RNAseq analyses of serial ITx biopsies. Overall, our findings show that ILCs may play a key role in regulating ITx graft homeostasis and could serve as sentinels for early recognition of allograft rejection and be targets for future therapies.
1. Introduction
The intestine is a major reservoir of lymphoid tissue and involved in many complex pathways critical to gut homeostasis and mucosal barrier functions. The intestinal epithelial barrier is responsible for the compartmentalization of gut microbiota and host immune cells, and diminished function may contribute to intestinal diseases, including inflammatory bowel disease (IBD), ischemia-reperfusion injury (IRI), and metabolic disorders(1, 2).
Intestinal transplantation (ITx) can be a life-saving intervention for patients with irreversible intestinal failure and short bowel syndrome(3–5). Healthy intestinal barrier function and gut homeostasis are critical for the success of ITx. Recent advances in the outcome of ITx have been enabled by improvements in surgical management and immunosuppressive therapies. However, recipients continue to experience lethal and severe complications, including infections, rejection, post-transplant lymphoproliferative disease (PTLD), and graft versus host disease (GVHD).
The high rate of immunologic complications is intimately tied to dysregulation of immunological pathways, seemingly critical to epithelial barrier integrity and intestinal homeostasis. In this context, our group recently demonstrated that intestinal allograft recipients with Crohn’s disease (CD) associated NOD2 mutations show higher rates of severe ITx rejection and allograft failure than do matched control recipients without these genetic alterations(6). Mechanistically, NOD2 mutant ITx recipients exhibit impaired morphology and function of lamina propria (LP) antigen presenting cells (APCs) leading to reduced levels of antimicrobial peptides (AMPs) and subsequent epithelial barrier breakdown, highlighting the importance of intestinal epithelial barrier integrity for ITx viability(6, 7).
The role of key immunologic regulators of intestinal barrier integrity and homeostasis in ITx, such as the recently described innate lymphoid cells (ILCs), is unknown. In contrast to T- and B-cells, ILCs lack lineage markers (Lin-) and somatic rearrangement of antigen specific receptors(8), but do show subunits of cytokine receptors including IL-7 receptor α (IL7Rα; CD127). Different subsets of ILCs have been identified based on distinct expression of transcription factors, cell-surface markers, and cytokines(9, 10). ILC3s have recently been shown to play fundamental roles in mucosal barrier immunity, tissue homeostasis, and immune regulation through the activation of host-derived cytokine expression(8, 11, 12). ILC3s can be characterized by expression of RORγt and, in response to IL-23 and IL-1β(8, 12, 13), secrete IL-22 and IL-17, both of which have been shown to regulate intestinal barrier integrity, mucosal defense, and tissue homeostasis by interacting with IL-17 and IL-22 receptors on intestinal epithelia cells and stem cells(14, 15). Interestingly, ILC3s consist of two distinct subsets based on cell surface expression of natural cytotoxicity receptors (NCR), such as NKp44 in humans and NKp46 in mice(16). NKp44+ ILC3s produce mainly IL-22, whereas NKp44−ILC3s produce IL-17 and limited amounts of IL-22(17).
A number of studies have reported changes in the ILC composition and function in human gut inflammation(18, 19). Recently, it has been reported that intraepithelial CD3− lymphocytes in biopsies from intestinal grafts exhibited phenotypes of ILCs after ITx(20). Subsequently, Weiner et al. (21) showed that donor ILCs can persist for more than 8 years in the gut after ITx. However, the precise composition of mucosal ILCs in ITx, especially in their role as key regulators of ITx homeostasis, mucosal defense, and epithelial barrier viability, has yet to be defined. In this study, we address the following questions: What are the phenotypes and relative composition of mucosal ILC subsets in ITx? Does the composition change over time? Do ILCs play a role in post-transplant outcomes? If so, what are the cellular mechanisms at the protein and genetic levels?
We found that the relative composition of ILC subsets indeed varies over time and between healthy and rejecting allografts. Moreover, we found that a reduced level of protective ILC3s was associated with diminished expression of key genes involved in epithelial barrier integrity and homeostasis pathways.
2. Materials and Methods
2.1. Ethics statement
Written informed consent was obtained from all individuals. This study received IRB approval (IRB studies #2004-008 and #2017-0365).
2.2. Patients and sample collection
Our study cohort was comprised of 38 transplant recipients (Table 1). Pre-operative, surgical, and post-operative care followed standard procedures(6, 7). Samples were collected from the distal ileum as previously described(6) when patients presented for endoscopic procedures for longitudinal immunomonitoring (day 0, week 2–4, month 2–4, and 6 months or beyond post-transplantation). Evaluated biopsies were clinically indicated per our standard of care protocol and read clinically by a pathologist. All biopsies were immediately placed in Allprotect Tissue Reagent (Qiagen, Valencia, CA, USA) and stored according to manufacturer’s instructions, or collected in RPMI 1640 (Gibco). Samples from healthy patients were obtained during the first year of transplantation or subsequent follow-up visits from protocol biopsies of healthy allografts without any evidence of rejection. Samples from rejecting patients were obtained during protocol or for cause biopsies at the time of diagnosis of rejection before initiation of treatment for rejection. Samples were treated as a single cohort to ensure maximal consistency. The exact number of allograft samples analyzed (n) is specified in corresponding figure legends.
Table 1.
Demographic data from intestinal transplant patients followed longitudinally since time of transplant. The first column shows the demographic distribution of the total study population. The second column shows the distribution for those patients who had no allograft rejection on serial endoscopies at 2–4 weeks, 2–4 months, and > 6 months post-transplantation (“healthy controls”). The third column shows patients who had rejection at one or more of the aforementioned time points. P values were tested by chi-square test.
| Total Population (n= 38) |
Healthy (Control) (n= 28) |
Rejection (n= 10) |
P value | |
|---|---|---|---|---|
| Age at Transplantation | ||||
| Pediatric | 13 | 11 | 2 | |
| Sex (M/F) | 17/21 | 14/14 | 3/7 | 0.275 |
| Race/Ethnicity | ||||
| Black/African American | 10 | 8 | 2 | 0.875 |
| Other | 6 | 4 | 2 | |
| Organs Transplanted | ||||
| Combined Liver or Multi-Visceral Transplant | 13 | 12 | 1 | 0.06 |
| Etiology | ||||
| Motility / Malabsorption | 8 | 7 | 1 | |
| Tumor | 2 | 2 | 0 | |
| Other | 3 | 2 | 1 | |
2.3. Lymphocyte isolation from tissue specimens
Isolation of ILCs from endoscopic biopsies was performed based on previously described protocols(22). In brief, biopsy samples were mechanically minced and incubated in RPMI 1640 (Gibco) supplemented with Pen/Strep containing 1mM DTT (Dithiothreitol; Sigma- Aldrich) at room temperature for up to 10 minutes. Following centrifugation (450g for 5 min) supernatant was collected and tissue specimens were enzymatically digested with a cocktail containing DNAse (1 mg ml−1, StemCell Technologies), collagenase (1 mg ml−1, StemCell Technologies), and 1M CaCl2 solution (Sigma-Aldrich) in RPMI 1640 for 30 minutes at 37 °C. The pellet from biopsy samples was stained using a live dead marker Zombie NIR™ dye (BioLegend), reconstituted in PBS for dead cell exclusion, and then blocked with Human TruStain FcX™ (BioLegend) for 15 minutes prior to surface antibody staining.
2.4. FACS analyses
Phenotypic analyses of cells were performed using a BD FACSAria III Cytometer (BD Biosciences) at our Flow Cytometry Core Facility and the following human antibodies (all from Biolegend) were used: lineage negative (PE-conjugated anti-CD19, CD3, CD1a, CD11b, CD34, FcεRIα, CD14, CD11c, CD94), Brilliant Violet 650™ -conjugated anti-CD45, FITC-conjugated anti-CD56, Alexa Fluor® 647-conjuated anti-NKp44, PE/Cy7-conjuated anti-CD117, PerCP/Cyanine5.5 anti-CD294 (CRTH2) and Brilliant Violet 421™ -conjugated anti-CD127. Cells were then washed with cell staining buffer (Biolegend) for removing excess antibody and resuspended in 4% paraformaldehyde solution and incubated at room temperature for 20 minutes. Data were analyzed using FlowJo software (Tree Star, Ashland, OR, USA). The gating strategy utilized appropriate fluorescent minus one (FMO) negative controls. Any samples with viability of 30% or lower (as determined by staining with live dead marker Zombie NIR™, BioLegend) were excluded from all analyses. Any samples with lower than 25% CD45+ were excluded from all analyses.
2.5. Cytokine stimulation and intracellular cytokine staining
Intracellular staining for the detection of cytokines was performed from post transplantation ileostomy reversal tissues. Approximately 1×106cells/ml RPMI supplemented with 10% FBS, 1% Penicillin Streptomycin, and 0.5% Gentamycin were cultured for 48 hours at 37°C in a cell culture flask. Recombinant mouse cytokine concentrations used were 100ng/ml IL-23, 10ng/ml IL1β, and 50ng/ml IL-15 (R&D Systems). Cells were stimulated for 4 hours with Cell Activation Cocktail (phorbol-12-myristat-13-acetat (PMA, 50 ng/ml) and ionomycin, Biolegend) with brefeldin A. Following stimulation, the cells were stained for surface markers as aforementioned. Cells were fixed (IC Fixation buffer, Invitrogen) and permeabilized (Permeabilzation buffer, Invitrogen) according to manufacturer’s instructions. The following antibodies were used for the detection of cytokines: PE/Dazzle™ 594-conjuated anti-IL-17A (Biolegend), PE-CY7-conjuated anti-IL-22 (Invitrogen), Brilliant Violet 650™ -conjugated anti-IFN-γ (Biolegend), and Brilliant Violet 510™-conjugated anti-TNF-α (Biolegend).
2.6. cDNA synthesis and RNA-sequencing
Total RNA was isolated from frozen tissue using the Direct-zol RNA Miniprep kit (Zymo). Library preparation and RNA-sequencing were performed at our Genomics and Epigenomics Shared Resource. Paired-end, dual-indexed libraries for RNA-seq were constructed from 20 ng total RNA using the TruSeq RNA Exome Kit (Illumina) according to manufacturer’s instructions. The resulting sequencing libraries were assessed using the BioAnalyzer 2100 with a DNA 1000 kit (Agilent), and quantified via fluorometry using the Qubit 4.0 (ThermoFisher). Libraries were sequenced on the NextSeq 550 System (Illumina) using the High Output Kit v2.5 (300 Cycles) with paired-end 150 bp read mode to an average depth of >50 M reads PF per sample.
2.7. Transcriptomic analysis
The raw sequences in the RNA-seq data were in the form of fastq files and first trimmed based on length and quality using FqTrim tool (https://ccb.jhu.edu/software/fqtrim/)(23). Then they were aligned to the human reference genome version hg38 using RSEM package(24). The RSEM tool was used to performed quantification of the transcriptome at the isoform and gene level. This analysis was performed on the Cancer Genomics Cloud (CGC), powered by Seven Bridges (https://cgc.sbgenomics.com). The group comparison analysis was performed using the Bioconductor package EdgeR (http://bioconductor.org/packages/edgeR/)(25) on the R statistical platform (https://www.r-project.org/)(26). For this analysis, Exact Tests were performed to compare the expression of genes across the groups of interest. The results obtained from the group comparison analysis were sorted based on False Discovery Rate (FDR) and the top 2,000 results were shortlisted, gene names obtained. This gene list was input into R package EnrichR (https://cran.r-project.org/web/packages/enrichR/index.html), a tool for analyzing gene sets that returns any enrichment of common annotated biological functions (http://amp.pharm.mssm.edu/Enrichr/)(27).
2.8. Histology
Presence or absence of allograft rejection in routine follow up or for cause ITx biopsies was determined by a board-certified transplant pathologist. The results of the pathologic classification were obtained from medical records. For representative histologies, intestinal ileal biopsy samples fixed in 4% paraformaldehyde were processed and stained with H&E by our Histopathology Tissue Shared Resource facility.
2.9. Statistical analysis
Each symbol on scatter plots and bar graphs (Prism, GraphPad Software, Inc.) represents the value for a single scope sample from a single subject. Statistical significance was determined with Mann-Whitney test, paired student t-test, chi-square test, and Wilcoxon singed-rank test. For linear regression test, R software (https://www.r-project.org) was used. Heatmap and volcano plots were made using the seaborn and matplotlib package in Python(28).
3. Results
3.1. Day 0 allografts exhibit a markedly different ILC1 and ILC3 subset composition compared to healthy allografts >6 months post ITx
Given the crucial role of ILCs in mediating intestinal health, we began by investigating their existence and phenotypes in ITx allografts. Using polychromatic flow cytometry, we compared the ILC subset composition in biopsy samples from healthy, histologically normal allografts at >6 months after ITx (the timepoint at which an ITx is considered to be stable(7)) to samples obtained from day-of ITx allografts after reperfusion (d0). As shown in Fig. 1A, all ILC subsets express CD45 but are negative for lineage-specific surface markers and can be divided into NKp44+ and NKp44− subsets based on their expression of NKp44 and CD56. ILC1s and ILC3s were gated based on expression of CD117 (c-Kit) and CD127 (IL7Rα). Type 2 ILCs were barely detectable in the intestinal allografts (data not shown).
Figure 1.
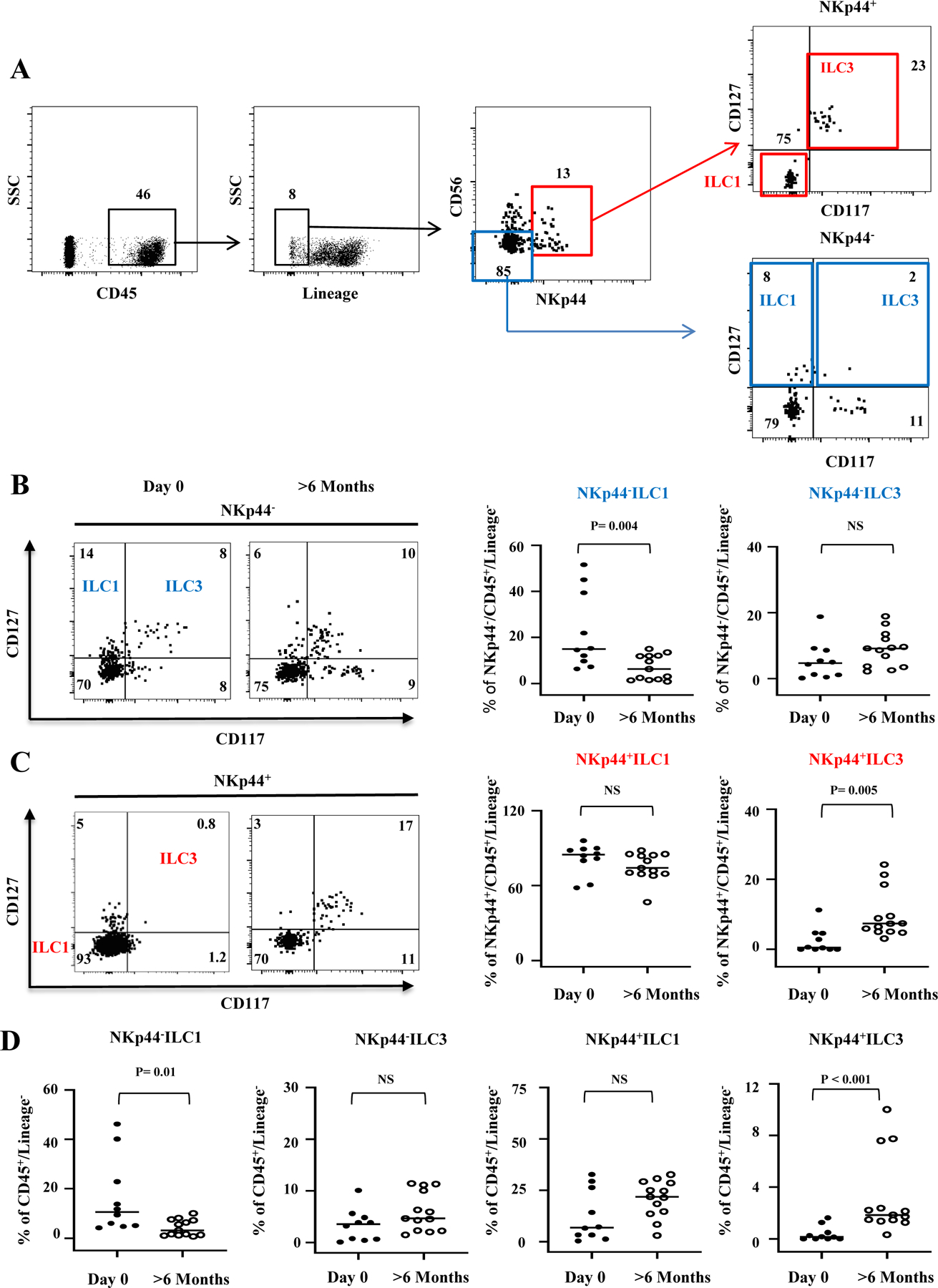
ILC1 and ILC3 subset composition on the day of ITx and >6 months post ITx. (A) ILC gating strategy. LP cells were isolated, and the following phenotypic definition was used for ILCs: lineage negative cells expressing CD45 were identified as type 1 and 3 ILCs by expression of CD56, NKp44, CD117(c-Kit), and CD127 (IL7Rα). Four distinct subsets were further defined as NKp44−ILC1 (CD56−CD127+CD117−), NKp44−ILC3 (CD56−CD127+CD117+), NKp44+ILC1 (CD56 ±CD127−CD117−), and NKp44+ILC3 (CD56±CD127+CD117+). NKp44− ILCs are shown by the blue box and NKp44+ILCs are shown by the red box. (B) Representative flow cytometric gating and frequencies of NKp44− ILC subsets as a % of NKp44+/CD45+/Lineage- at day 0 or 6 month after transplantation in healthy controls. (C) Representative flow cytometric gating and frequencies of NKp44+ILC subsets as a % of NKp44+/CD45+/Lineage- at day 0 or 6 month after transplantation in healthy controls. (D) All ILC frequencies are shown as % of CD45+/Lineage−. Sample size is Day 0 n=10, >6 months n=13. The frequencies of each subset were quantified and compared by Mann-Whitney test. NS = not significant.
Surprisingly, we found that the frequencies of potentially protective NKp44+ILC3s were significantly diminished in d0 allografts compared to healthy allografts >6 months (Fig. 1C and D), whereas proinflammatory NKp44−ILC1s were significantly higher in d0 allografts (Fig. 1B and D). In addition, we found similar frequencies of NKp44−ILC3 (Fig. 1B and 1D) and NKp44+ILC1 (Fig. 1C and 1D) in both d0 and healthy ITx allografts >6 months. Of note, we were able to identify higher frequencies of NKp44+ILC3s in a subset of ITx allograft samples obtained during back-table preparation before ITx was performed (Supplemental Fig. 1), suggesting that potentially protective donor-derived ILC3s are present in the allograft before ITx in at least a fraction of cases.
Together, these findings indicate that the relative composition of potentially protective ILC3 and proinflammatory ILC1 subsets in allografts at d0 is markedly different than in healthy allografts >6 months out (which we define for the purpose of this paper as “healthy ILC subset composition”). The harmful effects of ILC subset composition changes have also been described for other diseases(29).
3.2. A healthy ILC subset composition reemerges in healthy allografts within 4 weeks post-ITx
Having established that a healthy ILC1/ILC3 subset composition appears in healthy, histologically normal allografts >6 months post-transplant, we next explored the reconstitution kinetics via a longitudinal phenotype analysis from histologically normal allograft samples serially obtained from healthy ITx recipients at various time points (2–4 weeks, 2–4 months, and >6 months) during routine surveillance endoscopic follow-up evaluations. We observed that a substantial NKp44+ILC3 population was reached by 2–4 weeks post transplantation in healthy allografts (Fig. 2C), indicating the potential importance of the first 30 postoperative days following ITx. By contrast, the frequencies of proinflammatory NKp44−ILC1 significantly declined by 2–4 weeks, 2–4 months, and >6 months (Fig. 2B), indicating that the unhealthy ILC1/ILC3 composition observed in allografts at d0 adjusts in healthy allografts over time. The observed ILC composition changes were likely unrelated to changes in immunosuppression levels over time, as suggested by absence of significant correlations between ILC subset frequencies and tacrolimus trough level concentrations (Supplemental Fig. 2).
Figure 2.
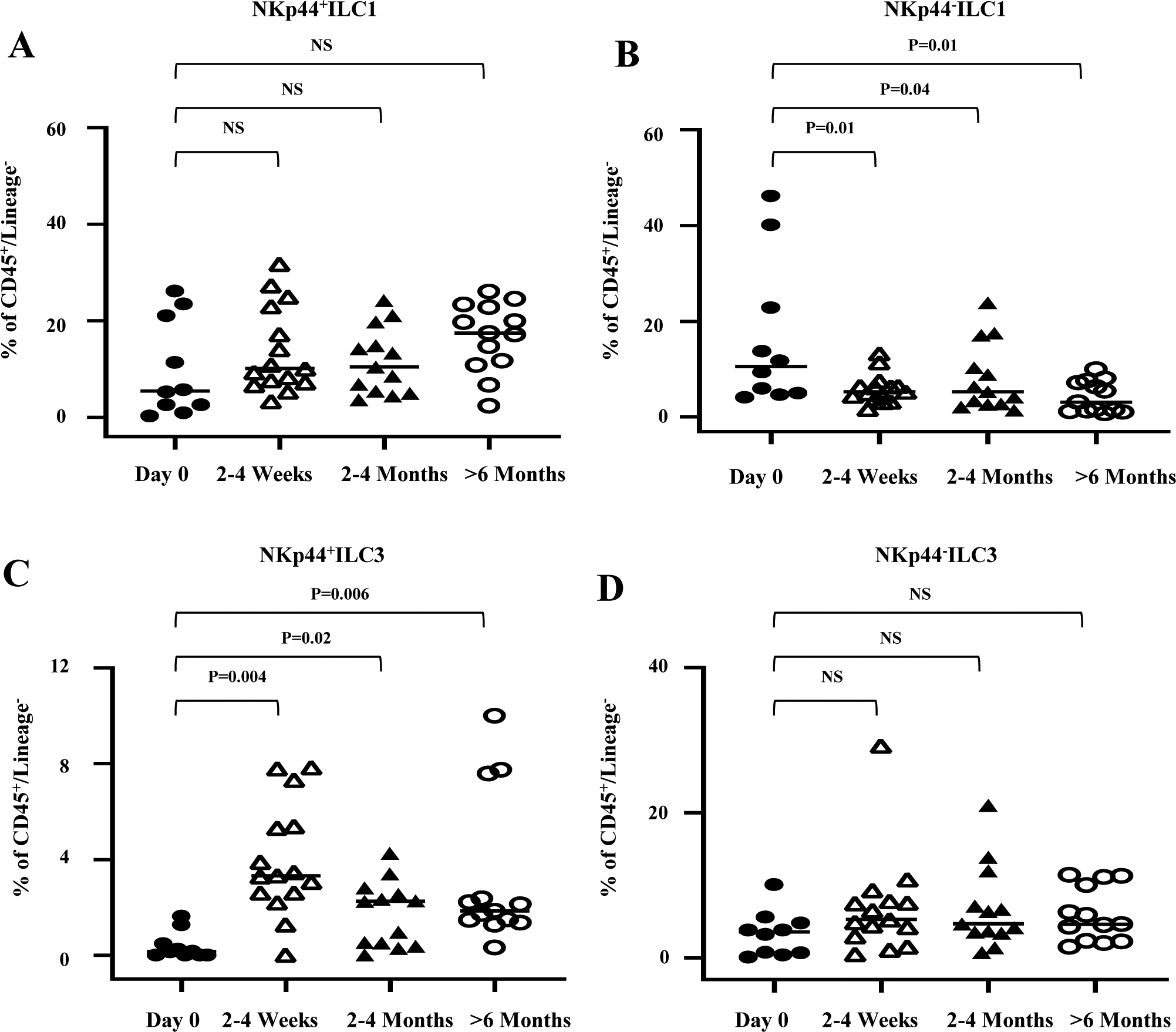
Longitudinal study of change in the frequency of ILC subsets in LP after transplantation. (A-D) Statistical assessment was analyzed by Wilcoxon test for each related sample; NS = not significant. All ILC frequencies are shown as % of CD45+/Lineage−. Sample size is day 0 (n=10), 2–4 weeks (n=15), 2–4 months (n=13) and >6 months (n=13). Each symbol on scatter plots represents the value for a single scope sample from a single subject.
3.3. A healthy ILC subset composition is not observed in rejecting ITx allografts
About half of intestinal transplants are plagued by complications, including infections and rejection(3). Having demonstrated that d0 ITx allografts start out with an unhealthy composition of ILC1 and ILC3 subsets that reconstitutes in healthy, histologically normal allografts over time, we next hypothesized that this reconstitution does not occur in rejecting allografts (i.e. those with a diagnosis of biopsy proven rejection). This hypothesis was supported by prior studies suggesting similar changes in dysregulated ILC compositions have been associated with local and systemic inflammation in other disease models(8, 30). To examine this, we identified ITx allograft biopsies obtained at the aforementioned time points from which both flow cytometry data on the ILC subset composition and clinical pathology results were available, and based on pathology findings classified them into two groups: (1) Histologically normal, healthy allograft samples or (2) rejection allograft samples.
Fig. 3 shows representative endoscopic and histologic images of allograft rejection. Endoscopically, rejecting allografts showed evidence of rejection such as loss of mucosal architecture, ulcers, hyperemia, and spontaneous bleeding (Fig. 3A). Histologically, biopsy specimens obtained from rejection allografts demonstrated an influx of neutrophils and mononuclear cells with effacement of the mucosa, and loss of mucin production in the glands (Fig. 3A).
Figure 3.
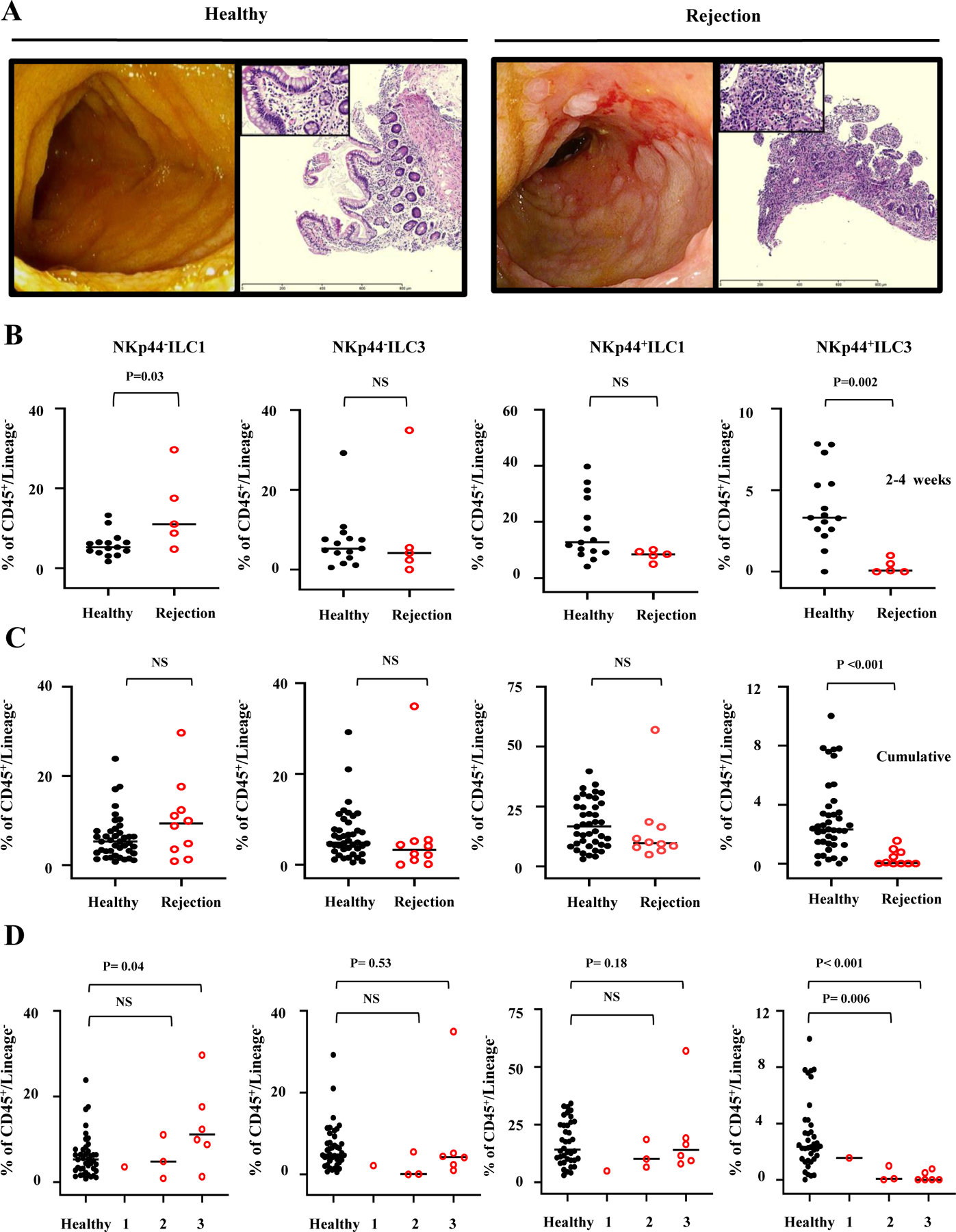
ILC subset composition in healthy versus rejecting ITx allografts. (A) Representative endoscopy and histology of ITx patients with healthy and rejecting allografts. H&E, ×4 (inset, ×40). The slides were visualized on an Olympus BX41 light microscope. (B-D) The rejection biopsies were obtained at the time of diagnosis of rejection and prior to administration of any treatment for rejection. The frequencies of each subset were quantified and compared by Mann-Whitney test (NS = not significant) between healthy controls and rejection patients for (B) 2–4 weeks and (C and D) all cumulative time points. For 2–4 weeks, sample sizes are for healthy allografts n=15 (15 patients) and for rejecting allografts n=5 (5 patients). For all cumulative time points, sample sizes are for healthy allografts n=41 (38 patients) and for rejecting allografts n=10 (10 patients). (D) Scoring system defined as 1 =grade 1 rejection, 2=grade 2 rejection, 3=grade 3 rejection as per pathology consensus guidelines.
Importantly, we found that at the 2–4 week-time point the frequency of potentially proinflammatory NKp44−ILC1s was significantly greater in rejection versus control allografts (Fig. 3B), whereas the frequency of potentially protective NKp44+ILC3 cells was significantly lower (Fig. 3B). Next, we compared the ILC phenotypes between rejection and healthy allografts across all postoperative time points. Critically, the protective NKp44+ ILC3 population was also decreased in the rejection allografts compared to healthy controls (Fig. 3C), whereas the proinflammatory NKp44−ILC1 population was not significantly higher (Fig. 3C).
To correlate the effects of ILC subset composition on the severity of ITx rejection, we assessed the histopathological grade of graft rejection using a scoring system as per pathology consensus guidelines (31). Critically, we found that the frequencies of protective NKp44+ILC3s, when compared to healthy allografts, were significantly diminished in allografts with moderate to severe rejection (both grade 2 and 3, Fig. 3 D), whereas proinflammatory NKp44−ILC1s were significantly higher in allografts with severe grade 3 rejection (Fig. 3D). Moreover, we further evaluated dynamic changes in ILC subset composition in ITx recipients with rejection over time by comparing ILC subset phenotypes in histologically normal, pre-rejection allograft samples to matched rejection allograft samples in the same recipients. Importantly, we found in all cases that protective NKp44+ ILC3s were significantly higher in allografts pre-rejection than at the time of diagnosis of rejection (Supplemental Fig. 3).
Together, these findings clearly suggest that an unhealthy ILC1/ILC3 subset composition predominantly characterized by a significant decrease of protective NKp44+ ILC3s was associated with rejection in ITx allografts.
3.4. Distinct ILCs isolated from ITx allografts produce different protective and proinflammatory cytokine profiles
Having demonstrated that ILC1 and ILC3 subset compositions are associated with allograft health and rejection we next sought to understand their effector functions in ITx. ILC1s have been identified as producers of proinflammatory IFN-γ, and TNF-α(32), whereas ILC3s have been identified as potent sources of IL-17 and IL-22(33, 34), potentially protective cytokines in the gastrointestinal tract in terms of tissue regeneration, mucosal defense, and intestinal homeostasis(34, 35). To determine the cytokine production capacity of ILCs in ITx recipients, we cultured LP lymphocytes freshly isolated from ileostomy takedown specimens in the presence of IL-23, IL-1β, and IL-15 followed by re-stimulation with PMA and ionomycin. Distinct cytokine production profiles (IL-22, IL-17, IFN-γ, and TNF-α) were detected by intracellular staining (Fig. 4A and B). We confirmed that NKp44+ILC3s were the main producers of IL-22, while NKp44−ILC3s were the main source of IL-17. However, ILC1s did not show detectable amount of IL-22 and IL-17 expression but can release proinflammatory IFN-γ and TNF-α, consistent with previous reports(17). Our data clearly show that ILC3s alone can secrete IL-17 and IL-22, which are important for epithelial barrier integrity and mucosal defense through stimulation of AMP production by epithelial cells.
Figure 4.

Intracellular cytokine production in the intestinal ILC population. Fresh isolated cell suspensions from post-transplantation ileostomy reversal specimens (n=13) were stimulated with PMA/ionomycin for 4 hours in the presence of IL-23, IL1β, and IL-15 for 48 hours. (A) IL-17A, IL-22, IFN-γ, and TNF-α expression in each ILC subset are shown. Data in A are representative of seven independent experiments with healthy recipients each. (B) The percentage of each ILC subset was quantified and compared by Mann-Whitney test.
3.5. RNA-seq identified altered genes associated with mucosal defense and intestinal homeostasis in day 0 versus healthy ITx allografts
Finally, we evaluated whether changes in ILC1/ILC3 subset composition are associated with transcriptomic differences of genes associated with intestinal barrier integrity and homeostasis pathways. To identify differentially expressed genes (DEGs) and pathways, RNA sequencing (RNA-seq) experiments were performed on serial ITx biopsy samples from healthy ITx patients. Specifically, we studied the transcript abundance in d0 allografts versus healthy 2–4 weeks and healthy >6 months allografts as shown in Fig. 5A.
Figure 5.

Gene expression analysis by RNA-seq in day 0 versus healthy ITx allografts. (A) Volcano plots for expressed genes at day 0 vs. 6 months (left) and day 0 vs. 2–4 weeks. Y-axis represents log10 (p-value, FDR). All blue dots correspond to genes with significant differences (FDR< 0.05). (B) Top 20 pathways from Gene Ontology (GO) enrichment. (C) Genes of interest (FDR <0.05) on the heatmap were manually selected based on Elsevier Pathway Studio® and STRING database (https://string-db.org). Each test includes at least three patients’ samples to compare 6 months and 2–4 weeks relative to baseline (day 0 post-transplantation). (D) STRING-based network analysis of all 21 genes. The line thickness indicates the strength of data support between each gene.
We found 5,085 DEGs (false discovery rate, FDR<0.05) at d0 versus >6 months and 2,895 DEGs at d0 versus 2–4 weeks, respectively. The data show that more DEGs (3,779 of 5,085, or 74%) were lower at d0 vs. >6 months compared with d0 vs. 2–4 weeks (1,986 of 2,895, or 69%), suggesting changes in gene expression in ITx allografts over time. Gene ontology (GO) analysis revealed that the most significantly changed pathways in ITx allografts were involved in inflammatory responses (Fig. 5B). Among the higher DEGs, we identified numerous genes associated with IRI (Supplemental Fig.4).
Importantly, the top 60 uniquely less abundant genes at d0 (Supplemental Table 1) included S100A8 and S100A9, both known to play key roles in IL22-dependent intestinal antimicrobial barrier maintenance(36, 37). A heatmap was created (Fig. 5C) showing expression differences in selected ILC3-related IL-22 and IL-17 target genes, which play critical roles in intestinal homeostasis, mucosal defense, and epithelial barrier integrity. Critically, we found a markedly lower transcript abundance of important ILC3-related genes including IL-22, IL-17A, IL-23, NOD2, CXCL8, IL1B, S100A8, and S100A9 (Fig. 5C) in d0 allografts in comparison to healthy allografts, suggesting that absence of protective ILC3s in d0 allografts correlated with lower expression of critical genes involved in intestinal barrier viability and homeostasis pathways. To corroborate this, we performed an interaction analysis of all 21 genes included in the heatmap using the STRING(38) database, which confirmed a dense gene association network of all 21 genes involved in ILC-related epithelial barrier integrity, mucosal defense, and intestinal homeostasis pathways (Fig 5d).
4. Discussion
In recent years, ILCs have received increasing attention as novel effector cells that play important roles in regulating intestinal homeostasis, mucosal defense, and barrier integrity through the production of cytokines such as IL-17 and IL-22(15, 35, 37). However, little is known about the phenotype and function of ILC s in ITx, a knowledge gap we sought to address.
Our first key finding is that protective IL-22 producing NK44+ILC3s were diminished in d0 allografts at the time of ITx compared to healthy allografts >6 months post-ITx. In order to draw diagnostic or therapeutic conclusions from this finding, it is important to understand why these protective cell populations are lower at the time of ITx.
Thus, one potential explanation for the absence of NKp44+ILC3s in d0 allografts after ITx could be that they undergo IRI-related cell death, which occurs immediately after reperfusion of the ITx allograft(39, 40). This hypothesis is supported by our RNAseq data showing overexpression of genes involved in ischemia related pathways such as Casp6 in d0 allografts that could induce programmed cell death in ILC3s after graft reperfusion (Supplemental Fig. 4). Interestingly, other conventional apoptotic pathway genes including caspases-1, −2, −3 and −9 and bcl-2 were not significantly different (FDR <0.05) between d0 and healthy allografts over time (data not shown). Relatedly, we cannot rule out the possibility that the lower frequencies of NKp44+ILC3 in d0 allografts could be due to their recruitment to different lymphoid compartments such as mesenteric or systemic lymph nodes.
The unhealthy composition of ILC1 and ILC3 subsets in d0 allografts is not only characterized by diminished NKp44+ILC3 frequencies but also by increased NKp44−ILC1 numbers. These changes could also be explained by the phenotypic and functional plasticity of ILCs. Recent evidence indicates that proinflammatory ILC1s can differentiate into protective ILC3 in the presence of IL-2, IL-23, and IL-1β and that the differentiation of ILC1 and ILC3 is reversible and bidirectional(41). This indicates that NKp44+ILC3 could differentiate into NKp44−ILC1 in allografts at d0. This explanation is supported by our RNAseq data showing that the expression of IL-1β and IL-23 was significantly lower in allografts at d0 vs healthy allografts 2–4 weeks and >6 months post-ITx (Fig. 5C). This could be regulated upstream by NOD2, given we observed less expression of NOD2 in d0 allografts (Fig. 5C), which could lead to reduced secretion of IL-23 and L-1β by NOD2-expressing myeloid APCs, critical regulators of ITx allograft homeostasis(7). In contrast, the gene expression of important ILC1 activation stimuli such as IL15 and IL18 was not significantly altered in d0 allografts (data not shown), suggesting that the cytokine milieu in d0 allografts after reperfusion is predominated by type 1 cytokines that favor ILC1 over ILC3 differentiation, thereby potentially contributing to the unhealthy subset composition. Further studies focused on the impact of IRI, cold preservation of the intestine, and other factors could be important to identify potential targets.
Our second key finding is that NKp44+ILC3s repopulated as early as 2–4 weeks after ITx. We see two possible explanations for this. The first possibility is that the observed resurgence is due to the migration of recipient-derived lymphocytes from other host tissues into ITx allografts. This could be regulated by expression of distinct gut-homing receptors on ILC3s such as α4β7 and CCR9(42, 43). In this context it has been shown that ILC1 and ILC3 subsets can undergo a homing receptor switch and express CCR9 and α4β7, which is regulated in a retinoic acid-mediated manner(44). The second possibility is that the observed resurgence is due to repopulation of donor-derived ILC3s from donor precursors in the allograft itself. In line with this, it has been recently reported that donor-derived ILCs, particularly lymphoid tissue inducer (LTi) cells, have the potential to persist in ITx allografts for more than 8 years after transplantation(21, 45). Moreover, a recent study found evidence that allografts from ITx recipients contain both donor- and recipient-derived hematopoietic stem and progenitor cells, which contribute to circulating blood mixed chimerism in the recipient(46). Together, these findings imply that the resurged NKp44+ILC3 populations in healthy ITx allografts could contain a mix of both donor- and recipient derived ILC3s that can be detected as early as 2–4 weeks postoperatively. The exact repopulation kinetics of host- versus donor-derived ILC3s may also differ at earlier (2–4 weeks) versus later (>6 months) post-transplantation timepoints. Further studies are necessary to understand the dynamics of donor-derived progenitor cell replacement by the recipient within the intestinal allograft.
Our third key finding is that an unhealthy composition of ILC1 and ILC3 subsets, specifically higher proinflammatory NKp44−ILC1s and lower protective NKp44+ILC3s, is associated with ITx allograft rejection. In this context, a number of studies have reported changes in the ILC composition and function in human intestinal inflammation and transplantation. Recent studies showed a decrease of IL-22-producing NKp44+ILC3 in intestinal transplant rejection(47) and CD, in which they also found an increase of IFN-γ–producing NKp46+ cells(19). Bernink et al. identified a frequency of IFN-γ-producing CD127+ILC1 subsets in humans that was found to be accumulated in inflamed intestines of CD patients at the expense of NKp44+ILC3(18). Similarly, a deficiency of ILC3s makes mice highly susceptible for development of dextran sulfate sodium-induced colitis(13). We also found that ILC1s produce proinflammatory IFN-γ and TNF-α, whereas ILC3s secrete potentially protective IL-17 and IL-22. In light of this, we speculate that the unhealthy composition of ILC1/ILC3 subsets in d0 and rejecting allografts may contribute to allograft rejection through disturbed ILC-derived mucosal cytokine profiles, resulting in enhancement of IFN-γ and TNF-α mediated alloreactive inflammation as well as impairment of protective IL-17 and IL-22 dependent mucosal defense and epithelial barrier pathways. This is supported by our RNA-seq data revealing a significantly reduced expression of IL-22 and IL-17 and related genes, including the AMPs S100A8 and S100A9 in d0 allografts. In line with this, expression of S100A8 and S100A9 was found to be significantly lower in Il22−/− mice during Salmonella infections(37). Thus, we speculate that the absence of NKp44+ILC3s and subsequent lower IL-22 expression in d0 allografts may result in lack of S100A8/S100A9-AMP-dependent epithelial barrier protection.
In conclusion, we characterize variations in numbers and phenotypes of ILCs in human intestinal transplant allografts, their critical differences in both allograft health and rejection and describe potential mechanisms for these roles at the genetic level. Our findings can provide insight into the mechanisms underlying rejection in ITx patients, and thus suggest a novel direction to enable prevention, early recognition, and detection of intestinal complications.
Supplementary Material
Supplemental Figure 1 NKp44+ILC3s of ITx allograft samples obtained during backtable preparation before ITx were divided into two groups based on absence (A) or presence (B) of NKp44+ILC3; ILC subsets (NKp44−ILC3, NKp44+ILC1 and NKp44−ILC3) for each group were quantified and compared by Mann-Whitney test (NS = not significant).
Supplemental Figure 2 Correlation between allograft ILC subset frequencies and tacrolimus trough concentrations in intestinal transplant recipients during the first 6 postoperative months. 72 allograft biopsies and corresponding tacrolimus concentrations from 38 transplant patients were analyzed. Pearson correlation coefficients (r) are shown.
Supplemental Figure 3 Flow cytometric analysis of ILC subset frequencies in histologically normal, pre-rejection and rejection allograft biopsies (n=4). Only matched allograft biopsies from the same recipients were used. Rejection biopsies were collected before treatment for rejection was initiated. Pre-rejection samples were collected from normal biopsies without histological evidence of rejection based on pathology reports. The paired t-test was used to measure significant differences within groups.
Supplemental Figure 4 Over-expressed ischemia related genes. Significant genes were selected based on statistical significance (FDR < 0.05).
Supplemental Table 1 Top 60 differently expressed genes at day 0 vs. 6 month and day 0 vs. 2–4 week. Significant genes were selected based on statistical significance (FDR < 0.05). Each test includes at least three patients’ samples to compare 6 months and 2–4 weeks relative to baseline (day 0 post-transplantation).
Acknowledgements
A.K., S.C.R., M.Z., and T.M.F. acknowledge funding support from the National Institute of Allergy and Infectious Diseases (R01AI132389). The authors thank Valerie Bockstette for her critical review of the manuscript.
Abbreviations:
- AMP
antimicrobial peptide
- APCs
antigen presenting cells
- CD
Crohn’s Disease
- DEGs
differentially expressed genes
- FDR
False Discovery Rate
- IBD
inflammatory bowel disease
- ILC
innate lymphoid cell
- IRI
ischemia-reperfusion injury
- ITx
intestinal transplantation
- LP
lamina propria
- NCR
natural cytotoxicity receptor
- NOD2
nucleotide oligomerization biding domain-2
- RNA-seq
RNA sequencing
Footnotes
Disclosures
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
References
- 1.De Mey JR, Freund JN. Understanding epithelial homeostasis in the intestine: An old battlefield of ideas, recent breakthroughs and remaining controversies. Tissue barriers 2013;1(2):e24965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeMeo MT, Mutlu EA, Keshavarzian A, et al. Intestinal permeation and gastrointestinal disease. Journal of clinical gastroenterology 2002;34(4):385–396. [DOI] [PubMed] [Google Scholar]
- 3.Fishbein TM. Intestinal transplantation. The New England journal of medicine 2009;361(10):998–1008. [DOI] [PubMed] [Google Scholar]
- 4.Fishbein TM, Matsumoto CS. Intestinal replacement therapy: timing and indications for referral of patients to an intestinal rehabilitation and transplant program. Gastroenterology 2006;130(2 Suppl 1):S147–151. [DOI] [PubMed] [Google Scholar]
- 5.Kroemer A, Cosentino C, Kaiser J, et al. Intestinal Transplant Inflammation: the Third Inflammatory Bowel Disease. Current gastroenterology reports 2016;18(11):56. [DOI] [PubMed] [Google Scholar]
- 6.Fishbein T, Novitskiy G, Mishra L, et al. NOD2-expressing bone marrow-derived cells appear to regulate epithelial innate immunity of the transplanted human small intestine. Gut 2008;57(3):323–330. [DOI] [PubMed] [Google Scholar]
- 7.Lough D, Abdo J, Guerra-Castro JF, et al. Abnormal CX3CR1(+) lamina propria myeloid cells from intestinal transplant recipients with NOD2 mutations. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2012;12(4):992–1003. [DOI] [PubMed] [Google Scholar]
- 8.Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nature immunology 2011;12(5):383–390. [DOI] [PubMed] [Google Scholar]
- 9.Spits H, Cupedo T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annual review of immunology 2012;30:647–675. [DOI] [PubMed] [Google Scholar]
- 10.Yagi R, Zhong C, Northrup DL, et al. The transcription factor GATA3 is critical for the development of all IL-7Ralpha-expressing innate lymphoid cells. Immunity 2014;40(3):378–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanos SL, Vonarbourg C, Mortha A, et al. Control of epithelial cell function by interleukin-22-producing RORgammat+ innate lymphoid cells. Immunology 2011;132(4):453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 2008;29(6):958–970. [DOI] [PubMed] [Google Scholar]
- 13.Sawa S, Lochner M, Satoh-Takayama N, et al. RORgammat+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nature immunology 2011;12(4):320–326. [DOI] [PubMed] [Google Scholar]
- 14.Hanash AM, Dudakov JA, Hua G, et al. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity 2012;37(2):339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar P, Monin L, Castillo P, et al. Intestinal Interleukin-17 Receptor Signaling Mediates Reciprocal Control of the Gut Microbiota and Autoimmune Inflammation. Immunity 2016;44(3):659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kruse PH, Matta J, Ugolini S, et al. Natural cytotoxicity receptors and their ligands. Immunology and cell biology 2014;92(3):221–229. [DOI] [PubMed] [Google Scholar]
- 17.Mjosberg J, Spits H. Human innate lymphoid cells. The Journal of allergy and clinical immunology 2016;138(5):1265–1276. [DOI] [PubMed] [Google Scholar]
- 18.Bernink JH, Peters CP, Munneke M, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nature immunology 2013;14(3):221–229. [DOI] [PubMed] [Google Scholar]
- 19.Takayama T, Kamada N, Chinen H, et al. Imbalance of NKp44(+)NKp46(−) and NKp44(−)NKp46(+) natural killer cells in the intestinal mucosa of patients with Crohn’s disease. Gastroenterology 2010;139(3):882–892, 892.e881–883. [DOI] [PubMed] [Google Scholar]
- 20.Talayero P, Mancebo E, Calvo-Pulido J, et al. Innate Lymphoid Cells Groups 1 and 3 in the Epithelial Compartment of Functional Human Intestinal Allografts. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2016;16(1):72–82. [DOI] [PubMed] [Google Scholar]
- 21.Weiner J, Zuber J, Shonts B, et al. Long-term Persistence of Innate Lymphoid Cells in the Gut After Intestinal Transplantation. Transplantation 2017;101(10):2449–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kramer B, Goeser F, Lutz P, et al. Compartment-specific distribution of human intestinal innate lymphoid cells is altered in HIV patients under effective therapy. PLoS pathogens 2017;13(5):e1006373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pertea G gpertea/fqtrim: fqtrim release v0.9.7 (Version v0.9.7) Zenodo. 10.5281/zenodo.1185412. In., 2018. [DOI] [Google Scholar]
- 24.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics (Oxford, England) 2010;26(1):139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Team RC. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2013. [Google Scholar]
- 27.Kuleshov MV, Jones MR, Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic acids research 2016;44(W1):W90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hunter J Matplotlib: a 2D graphics environment. Comput Sci Eng 2007;9:99–104. [Google Scholar]
- 29.Soare A, Weber S, Maul L, et al. Cutting Edge: Homeostasis of Innate Lymphoid Cells Is Imbalanced in Psoriatic Arthritis. Journal of immunology (Baltimore, Md : 1950) 2018;200(4):1249–1254. [DOI] [PubMed] [Google Scholar]
- 30.Forkel M, Mjosberg J. Dysregulation of Group 3 Innate Lymphoid Cells in the Pathogenesis of Inflammatory Bowel Disease. Current allergy and asthma reports 2016;16(10):73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruiz P, Bagni A, Brown R, et al. Histological criteria for the identification of acute cellular rejection in human small bowel allografts: results of the pathology workshop at the VIII International Small Bowel Transplant Symposium. Transplant Proc 2004;36(2):335–337. [DOI] [PubMed] [Google Scholar]
- 32.Klose CSN, Flach M, Mohle L, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 2014;157(2):340–356. [DOI] [PubMed] [Google Scholar]
- 33.Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 2009;457(7230):722–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teng MW, Bowman EP, McElwee JJ, et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nature medicine 2015;21(7):719–729. [DOI] [PubMed] [Google Scholar]
- 35.Song X, He X, Li X, Qian Y. The roles and functional mechanisms of interleukin-17 family cytokines in mucosal immunity. Cellular & molecular immunology 2016;13(4):418–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aujla SJ, Chan YR, Zheng M, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nature medicine 2008;14(3):275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Behnsen J, Jellbauer S, Wong CP, et al. The cytokine IL-22 promotes pathogen colonization by suppressing related commensal bacteria. Immunity 2014;40(2):262–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szklarczyk D, Jensen LJ. Protein-protein interaction databases. Methods in molecular biology (Clifton, NJ) 2015;1278:39–56. [DOI] [PubMed] [Google Scholar]
- 39.Kalogeris T, Baines CP, Krenz M, et al. Cell biology of ischemia/reperfusion injury. International review of cell and molecular biology 2012;298:229–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gonzalez LM, Moeser AJ, Blikslager AT. Animal models of ischemia-reperfusion-induced intestinal injury: progress and promise for translational research. American journal of physiology Gastrointestinal and liver physiology 2015;308(2):G63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bernink JH, Krabbendam L, Germar K, et al. Interleukin-12 and −23 Control Plasticity of CD127(+) Group 1 and Group 3 Innate Lymphoid Cells in the Intestinal Lamina Propria. Immunity 2015;43(1):146–160. [DOI] [PubMed] [Google Scholar]
- 42.Johansson-Lindbom B, Svensson M, Wurbel MA, et al. Selective generation of gut tropic T cells in gut-associated lymphoid tissue (GALT): requirement for GALT dendritic cells and adjuvant. The Journal of experimental medicine 2003;198(6):963–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mora JR, Bono MR, Manjunath N, et al. Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature 2003;424(6944):88–93. [DOI] [PubMed] [Google Scholar]
- 44.Kim MH, Taparowsky EJ, Kim CH. Retinoic Acid Differentially Regulates the Migration of Innate Lymphoid Cell Subsets to the Gut. Immunity 2015;43(1):107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gómez-Massa E, Lasa-Lázaro M, Gil-Etayo FJ, et al. Donor helper innate lymphoid cells are replaced earlier than lineage positive cells and persist long-term in human intestinal grafts - a descriptive study. Transpl Int 2020. [DOI] [PubMed] [Google Scholar]
- 46.Fu J, Zuber J, Martinez M, et al. Human Intestinal Allografts Contain Functional Hematopoietic Stem and Progenitor Cells that Are Maintained by a Circulating Pool. Cell stem cell 2019;24(2):227–239.e228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pucci Molineris M, González Polo V, Rumbo C, et al. Acute cellular rejection in small-bowel transplantation impairs NCR(+) innate lymphoid cell subpopulation 3/interleukin 22 axis. Transpl Immunol 2020;60:101288. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1 NKp44+ILC3s of ITx allograft samples obtained during backtable preparation before ITx were divided into two groups based on absence (A) or presence (B) of NKp44+ILC3; ILC subsets (NKp44−ILC3, NKp44+ILC1 and NKp44−ILC3) for each group were quantified and compared by Mann-Whitney test (NS = not significant).
Supplemental Figure 2 Correlation between allograft ILC subset frequencies and tacrolimus trough concentrations in intestinal transplant recipients during the first 6 postoperative months. 72 allograft biopsies and corresponding tacrolimus concentrations from 38 transplant patients were analyzed. Pearson correlation coefficients (r) are shown.
Supplemental Figure 3 Flow cytometric analysis of ILC subset frequencies in histologically normal, pre-rejection and rejection allograft biopsies (n=4). Only matched allograft biopsies from the same recipients were used. Rejection biopsies were collected before treatment for rejection was initiated. Pre-rejection samples were collected from normal biopsies without histological evidence of rejection based on pathology reports. The paired t-test was used to measure significant differences within groups.
Supplemental Figure 4 Over-expressed ischemia related genes. Significant genes were selected based on statistical significance (FDR < 0.05).
Supplemental Table 1 Top 60 differently expressed genes at day 0 vs. 6 month and day 0 vs. 2–4 week. Significant genes were selected based on statistical significance (FDR < 0.05). Each test includes at least three patients’ samples to compare 6 months and 2–4 weeks relative to baseline (day 0 post-transplantation).