

Abstract
The past decade has produced an explosion in the number and variety of genetic tools available to neuroscientists, resulting in an unprecedented ability to precisely manipulate the genome and epigenome in behaving animals. However, no single resource exists that describes all of the tools available to neuroscientists. Here, we review the genetic, transgenic, and viral techniques that are currently available to probe the complex relationship between genes and cognition. Topics covered include types of traditional transgenic mouse models (knockout, knock-in, reporter lines), inducible systems (Cre-loxP, Tet-On, Tet-Off) and cell- and circuit-specific systems (TetTag, TRAP, DIO-DREADD). Additionally, we provide details on virus-mediated and siRNA/shRNA approaches, as well as a comprehensive discussion of the myriad manipulations that can be made using the CRISPR-Cas9 system, including single base pair editing and spatially- and temporally-regulated gene-specific transcriptional control. Collectively, this review will serve as a guide to assist neuroscientists in identifying and choosing the appropriate genetic tools available to study the complex relationship between the brain and behavior.
Keywords: Transgenic mice, knockouts, Cre, DREADDs, CRISPR, brain, behavior
The development of transgenic mice dates back to the early twenty century when it was first discovered that homologous genes could cross over and recombine (Morgan, 1911). More than 60 years later, it became known that during meiosis eukaryotes recruit similar machinery to exchange segments of DNA between homologous chromosomes. Subsequent work by Richard Palmiter, Gali Martin and Nobel lauretes Richard Axel (2004), Mario Capecchi, Oliver Smithies, and Martin Evans (2007) led to the development of procedures for inactivating a targeted gene in the mouse genome using ES cells (Brinster et al., 1981; Brinster et al., 1982; Capecchi, 1989; Martin, 1981; Thomas and Capecchi, 1987; Thomas et al., 1986; Wigler et al., 1977). These efforts ultimately led a number of labs to independently generate the first gene knockout mice (Joyner et al., 1989; Koller et al., 1989; Koller et al., 1990; Schwartzberg et al., 1989; Zijlstra et al., 1989). This “knockout” technology has let scientists study the function of specific genes in physiology, development and pathology. The knockout mice generated using this technology became an essential tool that helped fuel many of the initial discoveries in neuroscience and, while their use has declined due to more advanced methodology, continue to be a useful tool today (Rocha-Martins et al., 2015).
After the establishment of early knockout mice, several important developments and modifications have widened its use in different fields of study. For example, today neuroscientists can choose from global knockout lines, gene knock-ins and a variety of reporter lines. Additionally, site-specific recombinases (SSR) have been developed to modify the DNA with temporal and cell type specificity. The use of these systems and others have enabled researchers to conditionally induce or suppress the expression of a gene of interest in a temporally-controlled and/or cell type-specific manner (Bouabe and Okkenhaug, 2013). In this review, we will discuss the different transgenic mouse lines available to neuroscientists, citing specific examples of how these diverse tools have been used in ways that have significantly advanced our current understanding of the nervous system. The emphasis here is on the lines available as opposed to methods on how to develop transgenic mice, which has been discussed in detail previously (Doyle et al., 2012). Additionally, we will discuss other genetic tools available to neuroscientists, such as siRNAs, viral-mediate approaches, and variations of the CRISPR-Cas9 system.
Section 1: Knockouts, Knock-ins and Reporter Lines
Knockouts
Generation of knockout mice is a common procedure in neuroscience research. An engineered DNA sequence is introduced into ES cells that are isolated from a mouse blastocyst. This plasmid contains mutations in the target DNA sequence, resulting in a partial or complete loss of the coding gene. ES cells that incorporated the plasmid or knock-out gene are then isolated and inserted into a mouse blastocyst, which is then implanted into the uterus of a female mouse. The offspring will be chimeras, which are then crossbred with other wildtype mice to produce a heterozygous line (F1). Mice obtained in the F1 line are then interbred to result in an F2 line in which some of the offspring will be homozygous for the knockout gene. Importantly, wild-type littermates from the F2 generation can then be used as the proper control group for the homozygous knockouts.
Global knockouts are the most basic type of genetically modified mice and, while they have a number of important limitations (Eisener-Dorman et al., 2009), have been critical in initially evaluating the importance of a specific gene to nervous system function. For example, knockout mouse lines were the first to implicate a number of transcription factors and immediate early genes in synaptic plasticity and memory formation in the brain (Ahn et al., 2008; Bourtchuladze et al., 1994; Gupta et al., 2010; Jarome et al., 2015; Kogan et al., 1997; Migaud et al., 1998; Plath et al., 2006; Ramamoorthi et al., 2011; Selcher et al., 2001; Silva et al., 1992; Zhou et al., 2016). However, due to the limitations of these knockouts, many conflicting results have sometimes been obtained using the same transgenic line, as in the study of the Nuclear Factor Kappa B protein p50 in synaptic plasticity and spatial memory (Denis-Donini et al., 2008; Kassed et al., 2002; Lehmann et al., 2010; Oikawa et al., 2012). Some studies have shown that p50 deletion results in deficits in synaptic plasticity and memory formation, while others have reported that the same deletion enhances learning and memory. These conflicting results are likely due to compensation effects triggered by the permanent gene deletion, an effect that is especially likely in cases in which a number of different proteins have redundant functions within a specific cellular signaling pathway or molecular process. Additionally, in some instances, global knockouts are lethal or lead to gross abnormalities in physiology and brain development (Gangloff et al., 2004; Li et al., 1992; Yamashita et al., 2005), preventing the characterization of a specific gene in a given neurological process. While global knockouts serve an initial purpose as a “screener” of potential gene candidates for a given biological process, conditional mutations that better control the regional, temporal and cell-type characteristics of the knockout are usually needed to fully understand the importance of the identified candidate gene to nervous system function.
Knock-ins
While the development of a knock-out mice line can be a useful means of determining the role of a particular gene in a given biological process, in other cases it is best to leave the DNA sequence present in the genome but instead alter the function of the coding gene. Knock-in mice provide a way of doing this by altering the function of a particular gene through the replacement of the original DNA sequence with a modified version. This technique has enabled researchers to introduce a mutated gene to a host mouse genome and investigate the roles of that mutation in a variety of biological processes (Harper, 2010). Knock-in mice have been used to examine a wide range of hypotheses in neuroscience by generating loss-of-function and gain-of-function mice for numerous gene targets. For example, dominant-negative mutations in which a catalytically impaired mutant out-competes the endogenous gene, ultimately impairing that gene’s function, have been used to assess the importance of protein function for a variety of genes during synaptic plasticity and memory formation (Gao et al., 2015; Kang et al., 2001; Kwapis et al., 2018; Malleret et al., 2001; Vogel-Ciernia et al., 2013; White et al., 2016). Additionally, knock-in approaches can be used to create constitutively active forms of a protein (Bach et al., 1995; Mayford et al., 1995; Serita et al., 2017; Suzuki et al., 2011). Other knock-in lines consist of point mutations that can prevent DNA binding (Oitzl et al., 2001) or in which a single codon is substituted, resulting in a change in the translated amino acid. Such a method is particularly useful when trying to assess the function of a specific protein phosphorylation site in a given biological function (Briand et al., 2015; Giese et al., 1998; Lee et al., 2003). However, if the knock-in mutations are not spatially and temporally controlled, then the transgenic mouse lines developed using this approach can suffer from many of the same limitations as the knockout mouse.
Another widely used form of knock-in includes “humanized” mutations in which a mouse has a human gene inserted into its genome. This approach has been very useful in studying neurodevelopmental and neurodegenerative disorders. For example, one well-known transgenic mouse line contains a knock-in of the mutated human beta-amyloid precursor protein (APP), which has been widely used in the study of Alzheimer’s disease (Hsiao et al., 1996; Sasaguri et al., 2017), while another carries a Shank3 mutation associated with human autism spectrum disorders (Yoo et al., 2019). Additionally, humanized knock-in mouse lines have also been used extensively in other preclinical models, such as understanding tumor growth, infectous disease and immune system function (Walsh et al., 2017). Consequently, while dominant-negative and other loss- or gain-of-function knock-in mice may be useful in determining the role of a specific gene in a given biological process, humanized knock-in lines are better suited for translational research relevant to human disease.
Reporter lines
While knockout and knock-in mice provide a means of controlling the expression and function of a particular gene, in some cases it may be more useful to track the subcellular localization of a specific gene or protein or monitor cellular activity. In these cases, reporter mouse lines are ideal. One of the most robust tools to visualize and trace the cells and their behaviors in living animals is using fluorescent proteins. Initially, beta-galactosidase of Escherichia coli (LacZ) was the tool often used as a reporter in fixed samples (Abe and Fujimori, 2013). However, GFP, the gene encoding green fluorescent protein, was cloned (Prasher et al., 1992) and subsequently used as a fluorescent label in vivo (Chalfie et al., 1994). This led to the generation of the first mouse line that expresses green fluorescent protein (Okabe et al., 1997), which is now widely used to study the biological characteristics of living cells, including the localization of subcellular structures, gene transcription and translation, and cell cycle progression (Abe and Fujimori, 2013). To date, a wide variety of transgenic reporter lines are available, including expression patterns that are native (EGFP, mCherry, etc.) or localized to the nucleus (H2B-GFP, H2B-mCherry), membrane (m-tdTomato/m-EGFP), microtubules (Tau-EGFP), Golgi apparatus (Golgi-GFP), mitochondria (Mito-EGFP) or actin cytoskeleton (Venus-Actin) (Abe et al., 2011; Kawamoto et al., 2000; Kurotaki et al., 2007; Muzumdar et al., 2007; Pratt et al., 2000). While these are just a few examples, we refer the reader to an excellent review by Abe and Fujimori (2013) that provides an exhaustive list of available reporter mouse lines.
Linkage of fluorescent reporters to specific genes has allowed tracking of receptor insertion and immediate-early gene expression in the brain during memory formation (Mitsushima et al., 2011; Xie et al., 2014), though the major advantage in these reporter lines has been mapping circuits and systems involved in specific neurological processes (Barth, 2007). GFP or LacZ under control of immediate-early gene (IEG) promoters such as fos and arc have allowed neuroscientists to track neuronal activity in a specific population of cells in response to a variety of stimulations or events in vivo (Barth et al., 2004; Clem and Barth, 2006; Wang et al., 2006). These IEG-based reporter lines recently have been integrated with inducible technology, allowing unprecedented mapping of network-wide neuronal activity and will be discussed in more detail in a later section.
In addition to fluorescent reporters, neuronal activity and protein expression can also be monitored in transgenic mice using luciferase-based reporters (Ishimoto et al., 2015). Luciferases are a class of oxidative enzymes that use a chemical reaction to convert energy into light. Promoter regions or whole transgenes can be inserted upstream of the luciferase gene to allow the researcher to accurately measure enzymatic activity by quantitating the emission of light. Thus, in a luciferase-based reporter mouse line, the gene of interest (or its promoter region) is fused to a bioluminescent reporter, allowing for quantification of the resulting luciferase expression. The luciferase gene can be under the control of almost any promoter and, unlike fluorescent proteins, it is not subject to high background from tissue autofluorescence (Contag and Bachmann, 2002; Serganova and Blasberg, 2005), resulting in a superior signal-to-noise ratio. However, if the goal is to visually track the expression of a gene or protein, then luciferase is not a good option as it is strictly quantitative and fluorescent reporters should be used.
Section 2: Spatial and Temporal Specific Promoter Lines
As mentioned above, a major limitation to the use of global gene knockouts or knock-ins is their lack of specificity in both time and space. One way to overcome these issues is to put the targeted mutation under the control of a cell-type specific promoter. Using a promoter to drive expression can provide some spatial control by targeting selective cell types that populate distinct brain regions. Some promoters can also provide a degree of temporal control, restricting transgene expression to a specific developmental time window. The AllenBrain Institute (http://connectivity.brain-map.org/transgenic/) has some great resources for identifying appropriate cell-type-specific promoters for neurons and other brain cell types to help researchers choose the correct promoter. In this section we will focus on two of the most widely used promoters in neuroscience (CaMKIIα and EMX1), but will also list others that have been developed and used with some frequency. However, it should be noted that this is not meant to be an exhaustive list of the available transgene promoters.
CaMKIIα
One of the most widely used transgenic promoter lines is the 1.3 Kb promoter derived from the calcium/calmodulin-dependent kinase II alpha (CaMKIIα) gene, which allows the mutation to be restricted to excitatory neurons in the neocortex and hippocampus with an expression pattern that starts during early development. This promoter has been widely used as a method to spatially control gene deletions and dominant negative and constitutively active mutations in a neuron-specific manner during complex physiological states (Hasegawa et al., 2009; Mansuy et al., 1998; Mayford et al., 1996; Winder et al., 1998; Zhou et al., 2016). Typically, CaMKIIα is used when the researcher aims to manipulate the gene of interest in forebrain excitatory neurons after development. For example, some researchers have used this promoter to overexpress a truncated form of the protein phosphatase calcineurin, which allowed localization of the transgene to the forebrain and hippocampus, and found significant effects on long-term potentiation (LTP) and memory formation (Mansuy et al., 1998; Winder et al., 1998).
EMX1
EMX1 is a second promoter that enables improved spatial and temporal control of the intended manipulation. EMX1 is a mouse homologue of the Drosophila homeobox gene empty spiracles that is expressed throughout the developing and adult telencephalon, including the cerebral cortex, olfactory bulbs, and hippocampus, and begins around embryonic day 9.5 (E9.5). As expression of cortical Emx1 is primarily restricted to projection neurons and has not been detected in glia, it is often used to drive neuron-specific transgene expression. This approach has been particularly useful in identifying the role of specific genes in hippocampus-dependent memory formation, including CaMKII and Intraflagellar Transport 88 (Achterberg et al., 2014; Berbari et al., 2014; Haettig et al., 2013).
Examples of other neuron- and region-specific promoters
CaMKIIα and EMX1 are not the only promoters that specifically target neurons and provide a degree of spatial and temporal control over transgene expression. Other neuron-specific promoters include the human synapsin 1 (hSyn) (Kugler et al., 2003) promoter, the neuron-specific enolase (NSE) promoter, and the platelet-derived growth factor beta chain promoter. Of these, hSyn has the highest specificity for neuronal expression (Hioki et al., 2007) and has been used for successful transgene expression and altered memory formation (Jaitner et al., 2016). To further improve specificity, distinct populations of neurons can be targeted using promoters that selectively target distinct cell types. For example, the choline acetyltransferase (ChAT) promoter can be used to express a transgene in cholinergic neurons (Bloem et al., 2014; Lopez et al., 2019). Similarly, glutamate acid decarboxylase 67 (GAD67) and glutamate receptor 1 (GluR1) promoters can selectively target GABAergic neurons. To target dopaminergic neurons, a researcher can use either the dopamine receptor D1a (Drd1a) or the preprotachykinin 1 (Tac1) promoter (Delzor et al., 2012). Some promoters even allow for region-specific control; gonadotropin-releasing hormone (mGnRH) promoter, for example, specifically targets the hypothalamus (Kim et al., 2002). Conversely, gene mutations can be targeted to astrocytes using the glial fibrillary acidic protein (GFAP) promoter, which has been used to limit LacZ reporter gene expression to only astrocytes throughout the central nervous system (Brenner et al., 1994).
It is important to note that the right promoter depends on the experimental conditions, including the known expression pattern of the gene of interest, the intended cell- and region-specific targets, and the desired onset of the transgene (e.g. immediately versus later in development). For instance, CaMKIIα start to express around postnatal day 1 (P1; Kool et al., 2016), while EMX1 mRNA is detected at E9.5 in mouse brains (Chan et al., 2001). Syn1 expression in rats starts at E13 (Ye and Marth, 2004), ChAT at E11 in mouse neurons (Huber and Ernsberger, 2006) and GAD67 at E17 in rats (Popp et al., 2009). In many cases there may be more than one promoter which can achieve the desired transgene expression profile, so pilot studies may be needed to determine which promoter works best under your specific experimental conditions.
Limitations
Although promoter lines can improve temporal and spatial specificity, they also have limitations. Many of these promoters (including both the EMX and CaMKII promoters) express early in development although their use is often intended to manipulate gene expression in adult animals. This could lead to gross changes in neuroanatomy or cellular physiology. Further, the persistent nature of these manipulations might trigger compensatory mechanisms which could confound the effects of the gene mutations. Finally, while these promoters can be effective at limiting the number and type of affected cells, some leakiness may occur, affecting unintended cell types, albeit at lower levels. As a result, while these transgenic lines are widely used and are largely an effective means of determining a specific gene’s function in the brain, often they need to be combined with more sophisticated approaches to improve specificity.
Section 3: Viruses
In Sections 4–7 we will discuss several genetic and post-transcriptional manipulations that that have a high level of temporal and spatial specificity. Because most cell types in the brain are difficult to transfect, the foreign material (DNA) designed to induce these manipulations is often packaged into a virus for efficient delivery into cells. There are several different types of viral vectors that can be used for this purpose, each with its own advantages and disadvantages (Table 1). In this section, we will provide details on the different viral approaches that can be used for transgene delivery.
Table 1:
Viruses
| Vector | Type | Capacity | Onset | Duration | Advantages | Disadvantages |
|---|---|---|---|---|---|---|
| Adeno | dsDNA | ~8kb | Days | Weeks | High packaging capacity Transient Integrates into genome |
High immune response BSL2 |
| AAV | ssDNA | ~5kb | Weeks | Years | Safe (BS1) Easy production Long lasting Low immune response Good penetration Anterograde or retrograde delivery |
Limited packaging compacity Does not integrate into genome |
| Lentivirus | RNA | ~8kb | Weeks | Years | High packaging capacity Integrates into genome Easy production Long lasting Low immune response |
BSL2 Expression is spatially limited |
| Rabies Virus | RNA | ~5kb | Days | Weeks | Rapid expression | BSL2 Cytotoxicity by 2 weeks |
| HSV-1 | dsDNA | ~30–50kb | Hours | 8–10 days | Largest packaging capacity Neurotropic Transient |
BSL2 More difficult to produce |
| LT-HSV-1 | dsDNA | ~30–50kb | Weeks | Indefinite | Largest packaging capacity Retrograde transfer properties Neurotropic Long lasting |
BSL2 More difficult to produce |
AAV: Adenoassociated Virus; HSV-1: Herpes Simplex Virus
Adenoviruses
Adenoviruses infect a wide range of cells, including dividing and non-dividing cells, with a high DNA packaging capacity (~8kb), but the strong immunogenicity of adenoviruses poses a significant limitation to their use (Robbins and Ghivizzani, 1998). Recent improvements in adenoviral vectors have drastically reduced this adversive immune response, however, making them more attractive as delivery vehicles for transgenic DNA or shRNA (discussed in the next section). As adenoviruses express in a short-term, episomal manner, they are particularly effective for conditional transgene expression as the manipulation will be transient (Lundstrom, 2018), though this would not be case for gene editing studies since DNA recombination would have already occurred. Importantly, this limited expression pattern can be a limitation if long-term transgene expression is desired. Adenoviral vectors have many reported uses including carrying reporter genes to the target cells (Hermens et al., 1997), CRISPR-Cas9 gene editing (Cheng et al., 2014), local delivery of Cre recombinase and cancer gene therapy (Wold and Toth, 2013). An important consideration when using adenoviruses is that they pose a risk to humans, requiring operation with biosafety level 2 (BSL2), which can also be a limitation depending on the facilities available.
Adeno-associated viruses (AAV)
Adeno-associated virus (AAV) is a non-human pathogen and a member of the parvovirus family that is the most widely used viral delivery system in neuroscience. AAV can infect both dividing and non-dividing cells, albeit with a limited DNA capacity (<5 kb) and without integration into the genome. (Deyle and Russell, 2009; Robbins and Ghivizzani, 1998). A major advantage of AAVs are that they produce a low immune response and provide long-term transgene expression, lasting years in some animals (Lundstrom, 2018; Wojno et al., 2013). This relatively safe viral vector (BSL1) has both retrograde (like AAV6) and anterograde (like AAV2) serotypes, and could therefore be used to as circuit-specific questions. Recently, a group of researchers developed a series of AAVs that can be injected into the tail vein and cross the blood-brain barrier to infect most areas of the brain (Deverman et al., 2016), allowing the researcher to avoid using invasive and technical stereotaxic surgeries to deliver the virus. Other uses of AAV include, but are not limited to, injecting AAV vectors containing Cre recombinase into brain areas of flox mice (discussed in Section 5), faster and more efficient gene knock out animals (Wang et al., 2018b), optogenetics (Parr-Brownlie et al., 2015), and gene therapy, such as Rett syndrome preclinical research (Sinnett and Gray, 2017) and phase I/II clinical trials for Parkinson’s disease (Sun and Schaffer, 2018).
Lentiviruses
Lentiviruses belong to the ssRNA family of retroviruses that are usually based on HIV-1. The lentiviral particle has the reverse transcriptase, integrase, and proteinase needed for the replication, allowing lentiviruses to infect both dividing and non-dividing cells (Escors and Breckpot, 2010). They can infect a broad range of hosts with low cytotoxicity, making them appropriate for a wide range of model organisms. Further, like AAVs (Lundstrom, 2018), lentiviruses produce persistent expression, in part because they permanently integrate into the host genome and can even be transduced to the next generation of mitotic cells (Chen and Gonçalves, 2016). Through pseudotyping (expressing glycoproteins originating from a different virus), lentiviruses can be used to achieve a high level of spatial specificity with limited spread (Parr-Brownlie et al., 2015). These vectors are used in zinc-finger nucleases (Lombardo et al., 2007), CRISPR-mediated multiple gene editing (Zetsche et al., 2016), delivery of shRNAs (Li et al., 2014) or “decoys” (Dias et al., 2014) and have been used in clinical trails (Escors and Breckpot, 2010). As with adenoviruses, lentiviruses need to be handled at BSL2, as there is a small risk to humans.
Herpes simplex virus (HSV)
Herpes simplex virus (HSV) is a large enveloped dsDNA with a relatively wide host range that also requires handling at BSL2. HSVs are naturally neurotropic, making them ideal for specifically targeting neurons without the additional requirement of a neuron-specific promoter. Further, they do not integrate into the host DNA but rather stay episomal as a circular molecule, minimizing off-target effects that might occur during viral integration into the host genome. As HSV is also a retrograde virus, it will be transferred to the CNS even after injection into the peripheral nervous system (Artusi et al., 2018; Lundstrom, 2018). Newer generations of replication-deficient HSV vectors cause a low immune response with an extremely large insert capacity (>30kb) (Lundstrom, 2018). Short-term HSVs are expressed rapidly (peak at ~3d post-injection) and transiently (gone by ~10d) at the site of injection (Neve et al., 2005). Modified long-term retrograde HSV vectors, in comparsion, are retroactively transported from the axon terminal to the cell body and are indefinitely expressed following injection, making it possible to do circuit-specific manipulations (Fenno et al., 2014; Sarno and Robison, 2018). Many researchers use HSV in gene therapy, CRISPR technology (Wang et al., 2018a) or optogenetic methods (Li et al., 2019).
Limitations
Viral manipulations of gene expression have many benefits over breeding-based approaches, including the ability to spatially and temporally restrict the manipulation through stereotaxic delivery of the viruses. Further, as many viruses are intended to manipulate gene expression in wild type animals, it is unnecessary to maintain a genetic mouse line for these manipulations, avoiding expensive and time-consuming colony maintenance. Nonetheless, viruses also have limitations. For example, although stereotaxically delivering the viruses can produce spatial specificity, stereotaxic surgery can have adverse affects and requires specialized equipment and training not necessary for breeding-based manipulations. Further, as mentioned above, viral vectors require safety precautions and handling under BSL1 or BSL2 conditions (Collins et al., 2017). Packaging size can also be a limiting factor for the use of some viruses, such as AAV, and an adverse immune response can limit the utility of other viral vectors, especially adenoviruses. Finally, the genetic manipulation is affected by the targeting and spread of the virus, which can change across batches, making viral manipulations inherently more variable than traditional breeding methods. This concern, however, can be mitigated by normalizing the titer before injection and verifying viral expression and spread in each animal to ensure that only animals with the desired manipulation are used in analyses. Viral approaches therefore have unique advantages and disadvantages and the most appropriate virus will be dictated by experimental factors, including the size of the desired transgene, the spatial precision necessary, and the temporal requirements of the task.
Section 4: Spatially controlled manipulations via siRNA and shRNAs
Knockout and knock-in approaches aim to alter gene expression by inserting DNA to either delete/modify, overexpress or outcompete endogenous genes. Another strategy to manipulate gene expression while gaining some temporal and spatial control is post-transcriptional gene silencing through the use of RNA interference (RNAi) techniques in the cytoplasm (Langlois et al., 2005) and nucleus (Robb et al., 2005). RNAi technology has a number of potential uses in neuroscience (Miller et al., 2005) and has been used to study the role of specific genes in synaptic plasticity, memory formation and disease. In this section we will discuss RNAi technology, focusing on siRNA, shRNA and recently developed Accell siRNA.
siRNAs
In RNAi, a complementary RNA molecule binds to a target mRNA transcript to silence its expression. This evolutionary conserved process is believed to be a mechanism that host cells use to protect against invading pathogens. This defense system has been repurposed to control gene expression using small interfering RNAs (siRNA) to silence expression of the target gene (Lovett-Racke et al., 2005). siRNAs get incorporated into the RNA-induced silencing complex (RISC), resulting in siRNA strand separation and binding of one strand of siRNA to the target mRNA. This binding allows RISC to cleave the mRNA and degrade it (Hannon, 2002). This method is transient, as the targeted gene continues to be transcribed as the siRNA molecules are degraded, ultimately resulting in a loss gene silencing (Morris, 2011). Despite this, a large number of studies have successfully used siRNA technology to investigate the role(s) of specific genes in memory formation, including transcription factors, protein phosphatases and epigenetic modifiers (Kremer et al., 2018; Peters et al., 2009).
shRNAs
To address this transient expression limitation of siRNAs, researchers have designed a variety of vectors that are capable of integrating into DNA by encoding a long inverted repeat, which when transcribed in vivo forms a short hairpin RNA (shRNA). The difference here is that while siRNAs are delivered directly to the cytosol, shRNA integrates into the host cell DNA and gets transcribed via RNA polymerase II or III. Subsecuently, it is transferred into the cytoplasm, cleaved by Dicer, and processed into siRNA (Jacob, 2006; Moore et al., 2010). Similar to siRNAs, a number of studies have successfully manipulated a variety pf biological processes and memory formation through brain-region specific expression of shRNAs (i.e., Li et al., 2014; Neuner et al., 2015). The choice between shRNA and siRNA depends on factors such as time demands and the need for a more or less stable knock-down of the gene of interest. There are also limitations to siRNAs, including toxicity that can occur as a result of the transfection reagents needed to get siRNA into cells in vivo, the possibility of off-target effects, and a reduction in siRNA concentration following cell division. siRNA may require multiple injections to maintain an optimal level of gene silencing whereas shRNA is continuously transcribed, even in daughter cells, and significantly increases the reproducibility of results (Moore et al., 2010).
Accell siRNAs
The simplest way to deliver RNAi is through cytosolic introduction of si/shRNA plasmids. This method, however, is primarily used for transient in vitro studies and cell types amenable to transfection. As transfection is difficult to achieve in brain cells in vivo, other methods have been developed to introduce RNAi into neurons in animals, including the use of viral vectors (e.g. adenovirus or lentivirus) to deliver siRNA or shRNA into the brain. However, this can be an expensive and time-consuming process. Recently, the development of novel siRNA technology has circumvented this problem. Accell siRNA is a functionally validated commercial product in which a chemical modification allows for siRNA delivery into difficult-to-transfect cells without the need for viral vectors or other transfection reagents (Nakajima et al., 2012). These Accell siRNAs are neuron-preferring and have a rapid induction profile in which expression typically peaks in less than 48 hours post-injection. We have used these siRNAs extensively in the rodent brain and have consistently achieved successful, rapid gene knockdown for a variety of different targets in both rats and mice (Alaghband et al., 2018; Jarome et al., 2015; Jarome et al., 2018; Kwapis et al., 2018; Webb et al., 2017). As a result, Accell siRNA can be especially advantegous if the researcher desires rapid, transient knockdown with no biosafety concern. Compared to viral approaches, however, knockdowns made with Accell siRNA are relatively difficult to verify following a behavioral experiment, as the knockdown is transient and the siRNA does not contain an easily visualized epitope tag. As with other manipulations, the specific siRNA delivery method (virus vs Accell) should be chosen based on the specific requirements of the experiment.
Section 5: Spatially- and temporally-specific conditional genetic manipulations
Although much information has been learned using knockout, knock-in, or specific promoter-based approaches to manipulate genetic information, these methods lack both the spatial and temporal specificity needed to answer basic neuroscience questions. Promoter lines are not typically capable of restricting the genetic modification to a specific brain structure, such as the basolateral amygdala or the dorsal hippocampus. Further, many promoter lines used to temporally restrict genetic manipulations to the post-development period, such as CaMKIIα, still express relatively early in development (~3 weeks postnatally), making it difficult to determine whether effects observed in the adult or aged mouse are due to the long-term manipulation of this gene throughout the animal’s lifespan (Kojima et al., 1997). Furthermore, while siRNAs and shRNAs are a powerful mechanism of controlling post-transcriptional processing, they are often transient, can only target a small number of cells and a single brain region at a time and are limited only to gene silencing. To address these limitations, conditional knockout approaches were developed to improve both spatial- and temporal-specificity. There are a number of methods used to create conditional genetic manipulations, including the Cre/lox system, the FLP/FRT system, and the Dre/rox system. More recently, these recombination systems have been modified to be ligand-sensitive, responsive to either tetracycline (e.g. Tet-On and Tet-Off systems) or tamoxifen (e.g. CreERT2) to enable precise temporal control over when the recombination event (and thus the desired genetic manipulation) occurs. All of these approaches can selectively alter gene expression in a tissue-specific manner to control when and where the genetic manipulation occurs, and each system has benefits and limitations that should be considered when designing an experiment, as discussed below.
Recombination-based approaches: Cre, FLP, and Dre
The development of site-specific in vivo DNA recombination systems in the early 1990s allowed neuroscientists to ask more precise questions about where individual genes function in the brain and when these genes are needed across the lifespan. The first system to be developed was the Cre/lox recombination system (Orban et al., 1992), which remains the most widely-used conditional approach to date. In the Cre/lox system, Cre recombinase recognizes loxP sequences and drives their recombination, removing any genetic material between loxP sites that share the same orientation (Sauer, 1998). Typically, conditional knockout mice are bred with loxP sites flanking the gene of interest, so that exposure to Cre recombinase drives the selective removal of that gene only in tissues where Cre is expressed (Figure 1A). For example, Tsien and colleagues crossed NR1 floxed mice (the NR1 subunit of the NMDA receptor is flanked with loxP sites) with mice expressing Cre under the CaMKIIα or KA1 promoter (Tsien et al., 1996). The resulting mice had selective deletions of the NR1 subunit of the NMDA receptor in either area CA1 (CaMKII promoter) or CA3 (KA1 promoter) of the dorsal hippocampus, allowing the researchers to determine that NR1-containing NMDARs are critical for spatial learning in area CA1 and for pattern completion in CA3 (Cui et al., 2004).
Figure 1. Cre-loxP system.
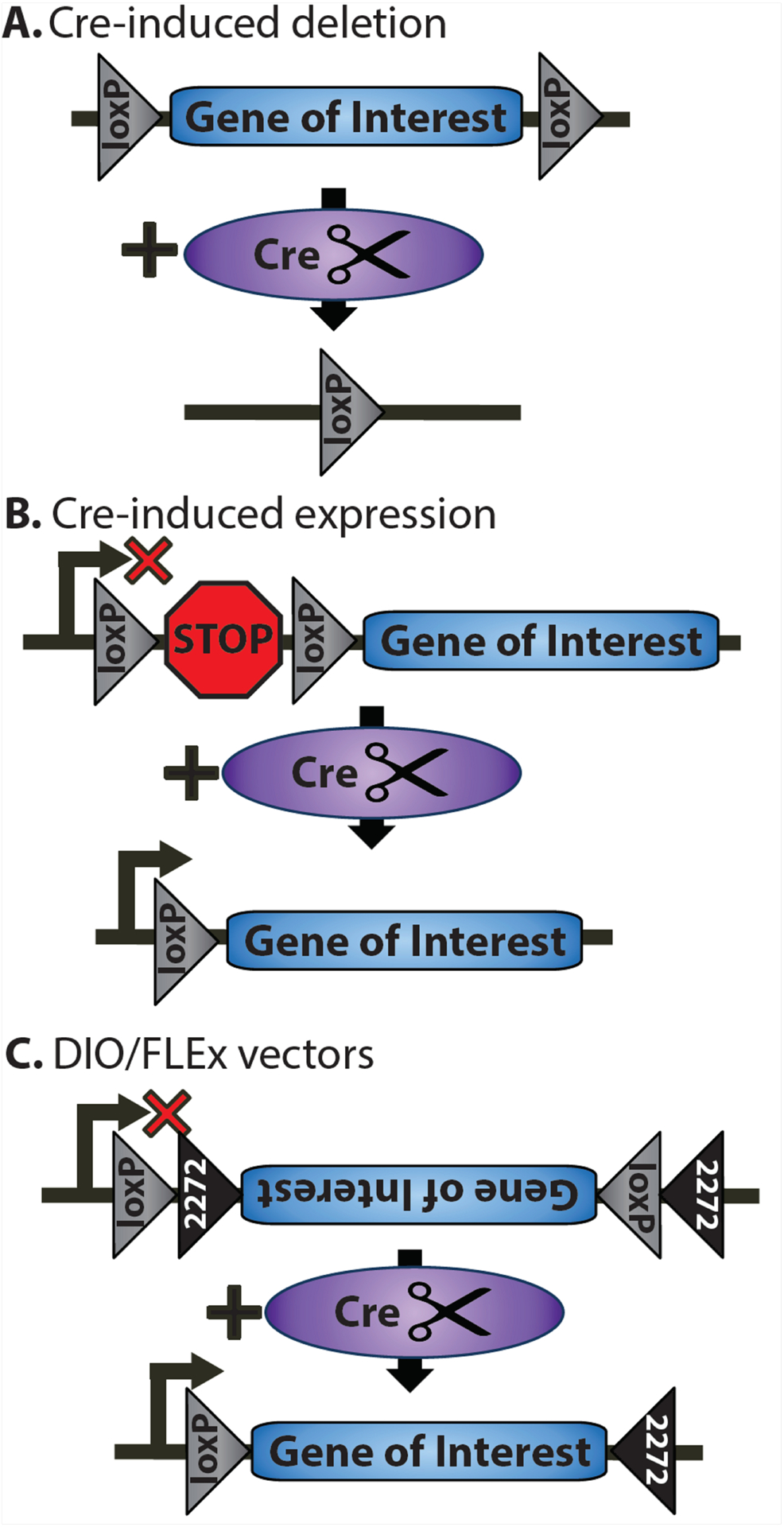
Cre-induced recombination can be used to induce either deletion or expression of a target gene. (A) Schematic depicting Cre-induced deletion. Cre cuts at LoxP sites flanking the gene of interest, removing the gene in any region expressing Cre. (B) Schematic depicting a typical Cre-induced expression system. LoxP sites flank a stop sequence that prevents expression of the gene. Exposure to Cre excises the stop sequence, allowing the target gene to be expressed. (C) Schematic of the DIO (Double Inverted Orientation) system (also called the Flip-Excision (FLEx) system). The vector expresses the gene of interest in an antisense orientation flanked by two unique lox sites (loxP and lox2272). The addition of Cre catalyzes recombination of these two sites that permantly flips the gene into the sense orientation, allowing for expression.
The Cre/lox system can also be used to drive expression of a target gene (such as a fluorescent reporter or a dominant-negative transgene) by removing a floxed transcription termination sequence (or “stop” sequence) inserted between the promoter and the transgene (Madisen et al., 2010). In this case, a mutant mouse carrying the LoxP-flanked “stop” sequence is bred to a promoter line that expresses Cre recombinase in a site- and time-specific manner, leading to selective expression of the gene of interest wherever (and whenever) Cre is expressed (Figure 1B).
The Cre/lox system has been used to create a wide range of genetic deletions using a variety of promoter lines that selectively express Cre within specific cell types (e.g. excitatory forebrain neurons) during specific points in development (e.g. early postnatal development). For a comprehensive review of the different Cre promoter lines available to drive tissue- and time-specific deletion of LoxP-flanked genes, the reader is referred to Kim et al., (2018) as well as The Jackson Laboratory (the JAX Cre repository: https://www.jax.org/research-and-faculty/resources/cre-repository). Thus, in this Cre/lox system, the spatial and temporal precision of the genetic deletion is limited by the specificity of the promoter line used to express Cre.
In addition to the widely-used Cre/lox system, other recombination-based systems have been developed to manipulate gene expression in a site- and time-specific manner, including the FLP/FRT system and the Dre/rox system. In the FLP/FRT system, the yeast-derived flippase (FLP) recombinase recognizes FRT (Flippase Recognition Target) sites and drives their recombination, removing any genetic material between FRT sites. Although the FLP/FRT system was first identified around the same time as the Cre/lox system, it was initially less efficient than Cre (Buchholz et al., 1998), leading to a slower adoption in the field. Since its initial discovery, FLP has been modified to become more efficient at mammalian body temperatures and the latest version of FLP, FLPo, has recombination efficiency similar to that of Cre (Kranz et al., 2010). Despite its improved efficiency, FLP/FRT is not as commonly used as Cre/loxP, which has a wider array of promoter lines and other tools available that do not yet exist for FLP/FRT.
The Dre/rox system is a second, more recently described alternative to the Cre/lox approach. As with Cre/lox, the Dre/rox system consists of a recombinase (DreO) that recognizes and recombines two identical DNA sequences (called rox sites), deleting any genomic DNA in between. Like Cre/Lox, Dre/rox is extremely efficient in driving recombination in specific cell types (Anastassiadis et al., 2009). Although functionally identical to the Cre/lox system, Dre/rox relies on a unique recombinase and distinct recombination sites, making it a complementary system that can be used to complement and extend Cre/lox-based manipulations.
As most conditional knockout approaches rely on the Cre/lox system, the number of promoter lines and other tools available for this system far outnumber those available for either the FLP/FRT or DRE/rox systems. The strength in these redundant approaches is twofold. First, the FLP/FRT and DRE/rox systems can be used to verify the results of a Cre/lox manipulation with an independent recombinase system. Second, as each system uses a unique recombinase and recognition site combination, these systems are complementary to each other and can be multiplexed in vivo to create relatively complex gene programs across development (Fenno et al., 2014; Schonhuber et al., 2014). By combining the three systems, researchers can begin to address questions about how different genes interact in specific brain regions during development or during complex behaviors.
Narrowing the temporal window: Using viruses to drive site-specific recombination.
Conditional genetic manpulations typically rely on promoter lines to express Cre (or FLP or Dre) in a site- and time-specific manner by placing recombinase expression under the control of a promoter or enhancer element that expresses in a specific cell type. Thus, the manipulation specificity is achieved by breeding mice carrying the recombination recognition (e.g. LoxP) sites in all cells with mice that only express the recombinase (e.g. Cre) in certain cell types. Although this is a powerful method of controlling specific subtypes of neurons in broad regions of the brain, it is limited by the availability and accuracy of the promoter line used to express Cre. If no promoter line exists for a particular cell type or brain region, for example, it may not be possible to achieve site-specific recombination through this method. Further, most promoter lines turn on relatively early in development, altering the gene early in the lifespan, potentially impacting both development and the aging process. Finally, other organisms used in neuroscience have relatively few promoter lines compared to mice, limiting the scope and specificity of the manipulations that can be achieved through breeding-based methods.
One way to avoid these issues is to use viruses to express Cre and other transgenes in a site-specific manner in the adult brain. (Jarome et al., 2015; Kwapis et al., 2018; McQuown et al., 2011; Shu et al., 2018). Cre can be delivered through a modified virus (Table 1, see Section 3 for an in-depth comparison of viral delivery systems) directly into the brain structure of interest to provide tight spatial and temporal control of the genetic manipulation. As Cre is only expressed where the virus is delivered, this method can be used to achieve site-specific manipulation of the target gene. Within the injected region, cell-type specificity can be achieved through both the viral capsid protein (e.g., AAV 2/1 is packaged in the capsid from serotype 1, which preferentially infects neurons (Burger et al., 2004)) and the promoter driving Cre expression (e.g., the CaMKIIα promoter primarily drives expression in forebrain excitatory neurons (Mayford et al., 1996)). Finally, as the floxed animal expresses the target gene normally until delivery of the virus encoding Cre, it develops and ages normally until the moment of virus-mediated Cre delivery. This avoids the spatial and temporal limitations of promoter-line approaches to delivering Cre recombinase. For example, in a recent paper, we injected AAV-CaMKII-Cre into the dorsal hippocampus of 18-month-old HDAC3flox/flox mice to show that hippocampus-specific deletion of HDAC3 improved long-term memory in aging mice (Kwapis et al., 2018). Importantly, this virus-based strategy allowed the animals to develop and age with the target gene intact before the virus was injected at 18 months of age.
As with transgenic mouse lines, viral vectors can be used to express a wide range of genetic manipulations beyond encoding Cre recombinase. For example, the virus can simply code for a gene of interest to test the effects of overexpression in the injected brain region (e.g., Kaas et al., 2013). Alternatively, a virus can express a mutant version of the gene of interest that it is catalytically dead (Alaghband et al., 2017; Kwapis et al., 2017) or encodes for a phosphomimic or phosphomutant protein of interest (Han et al., 2007; Vogel Ciernia et al., 2017). Expression of the transgene can also be placed under the control of Cre using either a floxed “stop” cassette, as described above (see Fig. 1B) or using the DIO (Double Inverted Orientation) system (also called the Flip-Excision, or FLEx system; Fig. 1C). In the DIO/FLEx system, the viral vector expresses the transgene in an antisense orientation between two unique lox sites (loxP and lox2272) so that no functional gene product is expressed in the absence of Cre. The lox sites are organized in oppositing orientations (compared to being in the same orientation, see Fig. 1A, 1B), allowing Cre to invert the genetic material between compatible lox sites. This allows two sequential recombination events to occur: 1) Cre inverts the transgene between the loxP sites into the sense orientation and 2) the genetic material between the lox2272 sites (now in the same orientation) is removed, excising one of the loxP sites. The resulting product has two incompatible lox sequences, preventing further recombination. Thus, the transgene is flipped into the correct orientation and expresses in any Cre-positive cells. This enables both cell type-specific expression of the gene of interest and enables circuit-specific expression through the use of a combination of retrograde and anterograde viruses, as described in the next section.
Inducible systems: Tet-On, Tet-Off and CreER
Although the Cre/lox system allows the experimenter to induce the manipulation at a specific time, the genetic manipulation typically has a gradual onset (as it requires expression on the promoter or virus to drive the manipulation) and is permanent once recombination is complete. These limitations can be overcome with the use of ligand-sensitive manipulations that are rapidly induced and reversible, providing more precise temporal specificity over when the genetic information is expressed. There are two major inducible systems capable of this tight temporal control: tetracycline-based and taxmoxifen-based.
In the basic Tet expression system, a transcriptional activator induces expression of the transgene only in the presence (or absence) of tetracycline or its derivative, doxycycline (Dox) (Figure 2). Both the “Tet-Off” and the “Tet-On” systems contain two core components: 1) the doxycycline-sensitive transcriptional activator and 2) the target gene under the control of a tetracycline-responsive promoter element (TRE), consisting of the tetO binding site fused to a short promoter sequence. In the Tet-Off system, the tetracycline-controlled transactivator protein (tTa) binds to the TRE only in the absence of Dox, allowing the experimenter to prevent expression of the transgene by maintaining the animal on Dox, typically provided in the diet. Thus, the investigator can remove Dox from the diet at a specfic timepoint in the experiment to restrict transgene expression to a discrete experimental window, such as a training session. Administration of Dox after this timepoint (e.g. immeciately after training) will close the window, preventing further expression of the transgene. In an early demonstration of this system, Mayford and colleagues (Mayford et al., 1996) used the Tet-Off system to conditionally express a constitutively active mutant CaMKIIα transgene in adult mice. Removal of dox before LTP or behavioral training led to expression of this calcium-independent CaMKIIα and impaired both hippocampal LTP and spatial memory. This was a reversible effect, as placing the animal back on Dox suppressed expression of the mutant CaMKII transgene and ameliorated these impairments in LTP and memory. Therefore, the authors were able to demonstrate for the first time that CaMKIIα was critical for LTP and memory formation in the adult brain, independent of its role in development, which was not affected by their temporally-restricted manipulation.
Figure 2. Tet-inducible systems.
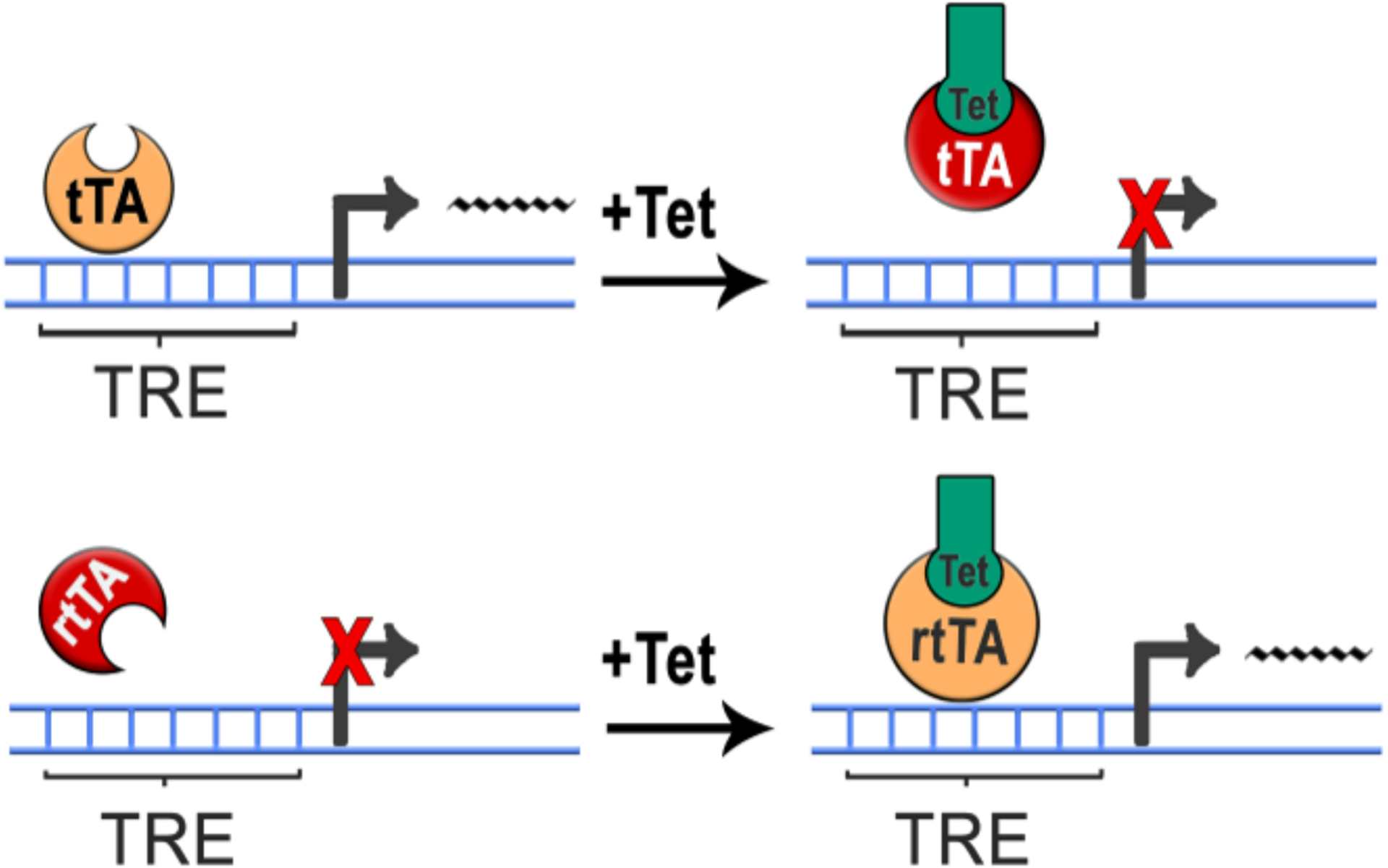
The target gene is under control of the tetracycline-responsive promoter element (TRE), which consists of a TetO binding site fused to a short promoter sequence. Transcriptional control of target gene is achieved by presence or absence of doxycycline (Tet). (A) In the Tet-Off system, the tetracycline-controlled transactivator protein (tTA) binds to the TRE only in the absence of Tet, which allows active transcription of the target gene (left). When Tet is applied, tTA is unable to bind TRE and transcription of the transgene is repressed (right). (B) In the Tet-On system, the reverse tetracycline-controlled transactivator protein (rtTA) only binds to the TRE in the presence of Tet, which allows active transcription of the target gene (right). When Tet is not present, tTA is unable to bind TRE and transcription of the transgene is repressed (left).
To prevent expression of the transgene outside of the desired temporal window, the Tet-Off system requires long-term, continuous administration of Dox, which may cause off-target effects (Sultan et al., 2013). Further, expression of the target gene in the Tet-Off system depends on the removal of Dox from the appropriate tissues, which limits how rapidly the transgene can be induced (Das et al., 2016). To avoid these limitations, the Tet-On system was developed, in which a reverse tetracycline-controlled transactivator (rtTA) only recognizes the TRE in the presence of doxycycline. Therefore, in the Tet-On system, the transgene remains off until Dox is administered, allowing the researcher to rapidly drive expression by acutely administering Dox. As with the Tet-Off system, this expression is transient and reversible, allowing the investigator to control a tight window of transgene expression.
As with the Cre/lox system, the Tet-On and Tet-Off systems can gain spatial specificity through the use of different promoters to drive expression of the tTA or rtTA transactivator in specific cell types. Thus, the basic tetracycline system is limited by the specificity of the chosen promoters. As with the Cre/lox system, viral methods can be used to provide additional spatial specificity by restricting one of the two components (usually the tTA) to a specific region of the brain. Notably, these tetracycline systems have been elegantly redesigned to allow cells active during a discrete window of time (e.g. during a memory acquisition session) to be tagged for future manipulation, as discussed in the next session.
A more recent addition to the conditional manipulation toolbox is the development of the CreER system, which combines the powerful Cre-based approach with ligand-dependent activation to gain temporal specificity over the genetic manipulation (Denny et al., 2014; Guenthner et al., 2013). The CreER system consists of two components: 1) the target transgene floxed by loxP sites and 2) a tamoxifen-sensitive Cre recombinase, called CreERT2. To make the tamoxifen-dependent CreERT2, Cre is fused to a mutated human estrogen receptor that responds to tamoxifen and 4-hydroxytamoxifen (4-OHT) rather than endogenous estradiol. In the absence of tamoxifen, CreERT2 is sequestered in the cytoplasm, but application of tamoxifen allows CreERT2 to translocate to the nucleus, where it drives recombination of loxP sites flanking the transgene. The CreER system not only improves the temporal resolution of the Cre/loxP system (recombination occurs within a few days of tamoxifen exposure (Brocard et al., 1997)), it can also improve the spatial specificity of the deletion, as tamoxifen can be locally injected into the appropriate brain region. Combining this site- and temporal precision with promoter-specific expression of CreERT2 and/or the target transgene allows the experimenter to precisely control which cells express the manipulation at a specific point in time.
The advent of tamoxifen- and tetracycline-inducibule genetic manipulations has greatly expanded the questions that can be addressed with conditional knockouts, as the fine-tuned spatial resolution can more carefully control when the manipulation is induced. Nonetheless, there are important drawbacks to using these systems that should be carefully considered. First, leakiness has been reported for both systems, so that some transgene expression has been observed in the “off” state, even before the removal of Dox (tetracycline-based system) or addition of 4-OHT (tamoxifen-based system) (Forster et al., 1999; Heffner et al., 2012; Kristianto et al., 2017; Schmidt et al., 2000; Vooijs et al., 2001; Zhu et al., 2001). It is therefore critically important to include control mice (e.g. mice with the transgene but maintained on Dox or not given 4-OHT) both when characterizing transgene expression and when studying behavior. Second, although these ligand-inducible systems avoid many of the off-target effects that can occur with long-term Cre exposure (Loonstra et al., 2001; Pfeifer et al., 2001), injections of tamoxifen/4-OHT or tetracycline/doxycycline can also cause unintended side effects, such as diarrhea, colitis, and apoptosis of gastric parietal cells (Huh et al., 2012; Riond and Riviere, 1988). As these issues can alter behavior, it is again important to include appropriate control groups that receive tamoxifen/doxycycline without the inducible transgene. Finally, there are limits to the administration protocol itself, including differences in the time window of effectiveness and differences in recombination efficiency across both strain and sex that can introduce variability (Abram et al., 2014; Sandlesh et al., 2018; Valny et al., 2016). Therefore, if tight spatial control over the genetic manipulation is not necessary for a particular experimental question, a standard, non-inducible manipulation may be more appropriate to avoid some of these issues.
Section 6: Cell- and circuit-specific expression
The conditional genetic systems described above have not only allowed researchers to restrict genetic manipulations in both time and space but have also laid the foundation for the ultra-precise manipulations that have been developed in the past decade. Recent advances in this field have made it possible for neuronal activity and even genetic information to be altered within specific circuits during defined time windows of activity. These technical improvements have allowed researchers to identify the roles of discrete neuronal networks in specific phases of behavior. Further, it is now possible to “tag” a population of neurons active during a discrete time window and manipulate the activity of those cells at a later timepoint. This unprecedented precision has fundamentally changed how researchers approach questions about the role of genetics in behavior as well as the function of active cells in memory formation.
Identification of active cells: TetTag, CreERT2
To identify and access the subset of neurons engaged by a particular task, the Mayford lab developed the TetTag mouse, which persistently tags neurons activated during a specific time window controlled by the investigator (Reijmers and Mayford, 2009; Reijmers et al., 2007). This mouse line takes advantage of the dynamic, activity-responsive immediate early gene (IEG) cFos, which is rapidly (~5 minutes) upregulated in active neurons after a learning event (Strekalova et al., 2003; Webb et al., 2017) or other stimulation event (Kaczmarek, 1992). The tTA element is placed under the control of the cFos promoter, so that tTA expression is limited to cells activated by a sufficiently salient event (Reijmers et al., 2007). In the second part of the system, the tetO promoter drives bidirectional transcription of a reporter gene (LacZ) as well as a dox-insensitive tTA mutant (tTA*) capable of maintaining its own expression through a transcriptional feedback loop (Figure 3A). Thus, the removal of Dox opens a window during which cFos drives persistent expression of tTA* and the LacZ reporter gene selectively in active neurons. Placing the mice back on Dox prevents tagging of new neurons without affecting the Dox-insensitive tTA*-mediated transcription, which supports continued LacZ expression from the tagged neurons, maintaining the tag that was induced when those cells became active.
Figure 3. TetTag and TRAP systems allow neurons active during specific time windows to be persistently tagged.
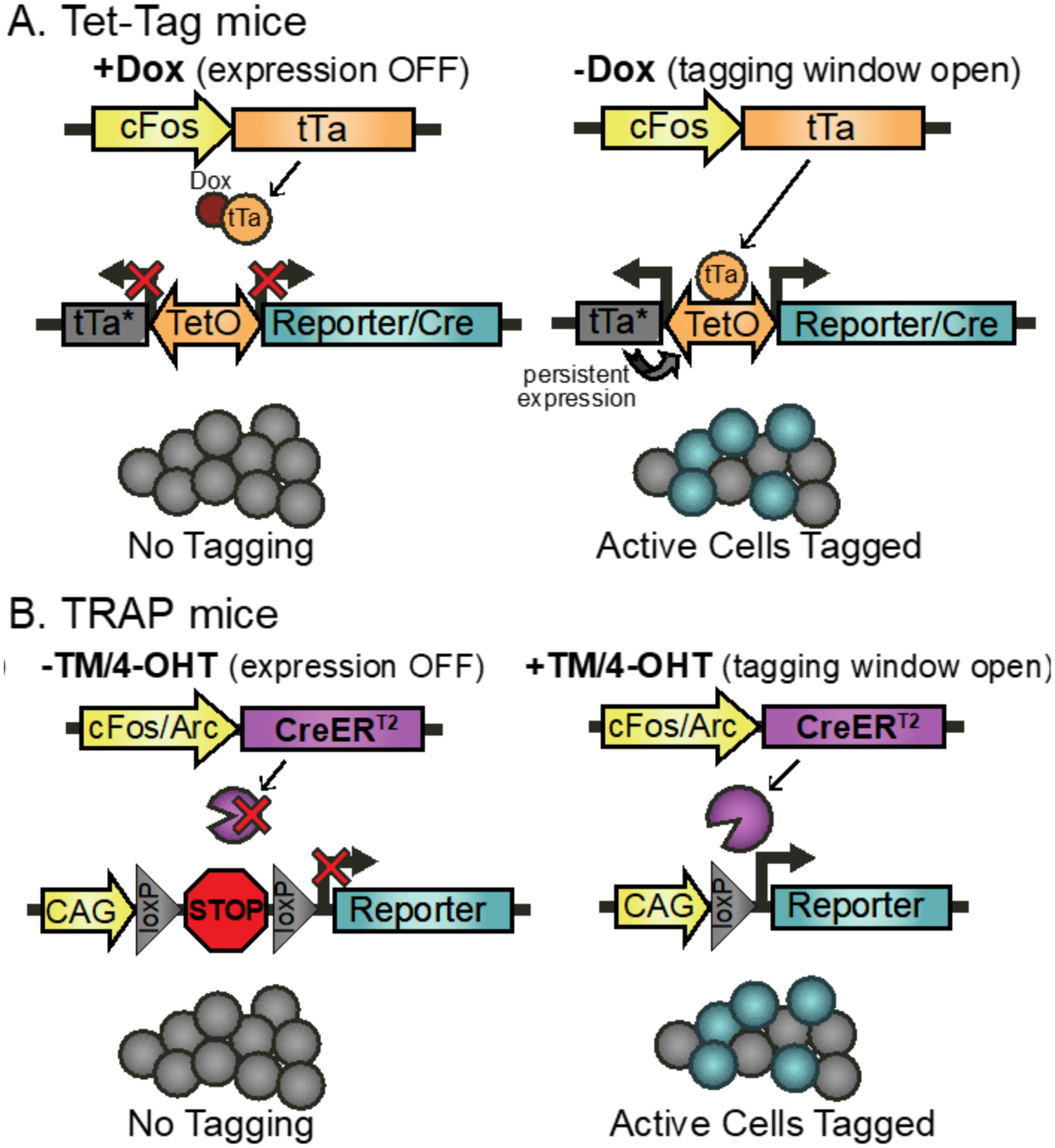
(A) Tet-tag system. Two transgenes control tagging of active neurons: 1) tTA under the control of the activity-dependent cFos promoter and a TetO promoter that drives both the gene of interest (e.g. a reporter gene) and the dox-insensitive tTA* mutant that drives persistent expression. The presence of Doxycyline (Dox) at rest prevents tTa-TetO-mediated expression (left). Removing Dox (right) opens a temporary tagging window during which tTa binds the TetO promoter and drives expression of the desired gene selectively in cells activated during that window. (B) The TRAP system also requires two transgenes: 1) The tamoxifen-dependent CreERT2 under the control of an activity-sensitive promoter like cFos or Arc and 2) a transgene that expresses the gene of interest (e.g. a reporter gene) in a Cre-dependent manner. In the absence of tamoxifen (TM) or its metabolite 4-OHT (left), Cre is sequestered to the cytoplasm, preventing expression of the target gene. Injecting TM or 4-OHT (right) opens a tagging window in which CreERT2 recombination occurs selectively in activated cells.
In a similar manner, the ArcCreERT2 and TRAP (Targeted Recombination in Active Populations; Figure 3) mouse lines both exploit the activity-dependent expression pattern of the IEGs Arc and cFos to selectively express the tamoxifen-dependent CreERT2 in active neural populations (Denny et al., 2014; Guenthner et al., 2013). Co-expressing the IEG-dependent CreERT2 with a cre-dependent fluorescent reporter allows active neurons to drive permanent recombination of the reporter gene only when tamoxifen is present. A researcher can therefore open the tagging window before a stimulation event, such as a training session, by injecting tamoxifen or 4-OHT (the major active metabolite of tamoxifen), to allow active neurons to persistently express the fluorescent tag for future visualization. This window automatically closes as tamoxifen/4-OHT is metabolized, within approximately 12h. This CreERT2-based system has a few advantages over the TetTag system, including a more precise temporal window for tagging (hours compared to days), permanent recombination-based modifications, a wide array of available Cre-based genetic tools, and the ability to delete endogenous genes (Guenthner et al., 2013; Reijmers and Mayford, 2009).
Manipulation of active cells: Optogenetics, DREADDs, DIO/FLEx
The TetTag and CreERT2 systems were initially used to observe the pattern of cells active during memory acquisition or sensory stimulation to demonstrate that cells active during fear memory acquisition are preferentially reactivated during the retrieval of that memory (Denny et al., 2014; Reijmers et al., 2007; Tayler et al., 2013). Although their initial use was purely observational, the TetTag and CreERT2 systems have since been modified to express different regulators of neural activity that allow bidirectional control over tagged neurons, to test the role of these cells in behavior. For example, the Hen lab bred ArcCreERT2 mice with mice expressing a STOP-floxed inhibitory optogenetic receptor Archaerhodopsin-3 (Arch). By injecting these mice with 4-OHT before training, they were able to express Arch selectively in cells active during context fear acquisition. Optogenetic inhibition of these tagged cells at a later timepoint revealed that this population of cells is critical for successful memory retrieval (Denny et al., 2014). The ArcCreERT2 mouse line has also been used with the excitatory optogenetic receptor channel rhodopsin (ChR2) to allow the tagged population of neurons to be stimulated at a later timepoint, allowing for bidirectional control (Lacagnina et al., 2019). The TetTag and CreERT2 systems can also be combined with DREADDs (Designer Receptors Exclusively Activated by Designer Drugs) to allow for chemogenetic control over the tagged population of cells (Garner et al., 2012). Together, these methods have been used to interrogate the memory “engram;” by stimulating or inhibiting these cell populations, researchers have forced or prevented memory recall (Cowansage et al., 2014; Denny et al., 2014; Tanaka et al., 2014), changed the information encoded in an endogenous memory (Garner et al., 2012), improved extinction memory (Denny et al., 2014; Lacagnina et al., 2019), and even created a false memory (Ramirez et al., 2013).
The use of viruses with anterograde and retrograde properties (see Section 3) has also made it possible to manipulate neural circuits with incredible precision. It is now possible to selectively stimulate or inhibit populations of cells that connect two brain regions. The observation that optogenetic receptors get transported from the soma to the axon terminal made it relatively simple for researchers to test the role of specific neural circuits in behavior. By injecting an anterograde virus into region A and implanting an optic fiber in region B, the cells making efferent projections from region A to B can be selectively stimulated or inhibited at the axon terminal (Fig. 4A) (Carreno et al., 2016; Carter and de Lecea, 2011). Similarly, retrograde viruses can also be used to determine the role of specific projection pathways; injection of a retrograde virus that enters through axon terminals in combination with optical fiber implantation in an upstream structure allows for selective activation or silencing of cells connecting these regions (Carter and de Lecea, 2011).
Figure 4. Circuit-specific use of optogenetics and DREADDs.
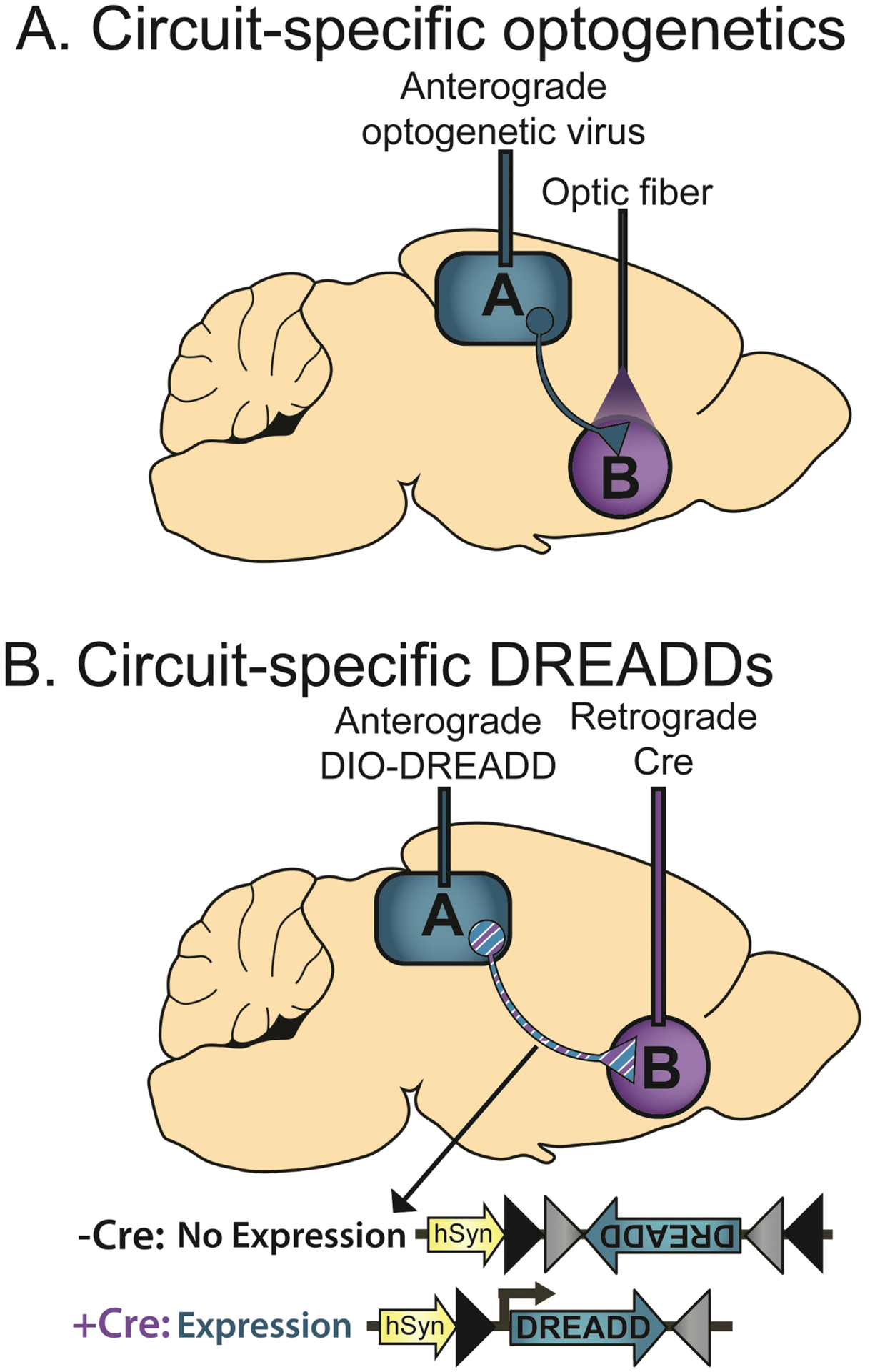
(A) An example of the use of optogenetics to selectively interrogate neurons projecting in a specific circuit. Here, an anterograde optogenetic virus (e.g. AAV1-ChR2) is injected into brain region “A” and the terminals are optically stimulated with an optical fiber implanted into efferent region “B” to selectively activate neurons projecting from A to B. (B) An example of the use of DIO-DREADDs to selectively interrogate a neural circuit. An anterograde DIO-DREADD is injected into region A and a retrograde Cre virus is injected into efferent region B. As the DIO-DREADD is inactive in the absence of Cre, only cells that receive both viruses will express DREADDS and will respond to systemic injection of CNO.
The ability to manipulate projection-specific neural pathways has been extended to DREADD systems (Figure 4B) as well (Smith et al., 2016). Although they lack the precise temporal resolution of optogenetic manipulations, DREADDs allow for remote activation or inhibition of the target circuit with a systemic injection of the designer drug CNO (Clozapine-n-oxide), rather than requiring an invasive optical implant to allow light to be delivered to a specific brain region. In order to target a specific population of neurons with DREADDs, Cre-based recombination in a specific cell type (using a transgenic Cre promoter line or a virus expressing Cre under a cell type-specific promoter) can be combined with a virus encoding the DREADD gene in a flipped DIO/FLEx configuration (see Section 5). The DIO/FLEx system allows the DREADD virus to remain dormant until Cre application, which flips the gene into the sense orientation and allows its expression. Thus, in a promoter line, the locally-injected DIO-DREADD is only expressed in a functional orientation in cell types expressing Cre. To achieve pathway-specific control, a retrograde virus (e.g. CAV-2) expressing Cre can be injected into target region B and an anterograde virus encoding the DIO-DREADD can be expressed in upstream region A (Fig. 4B). Only cells projecting from A to B will receive both viruses, allowing for DREADD expression specifically in this group of cells (Boender et al., 2014). Thus, exposure to a systemic injection of CNO will selectively activate or repress this pathway-specific group of cells.
Section 7: The CRISPR revolution
The combination of IEG-based promoters, Cre recombination, and methods of modulating neural activity (optogenetics and DREADDs) have allowed researchers to manipulate the activity of cells at specific timepoints in distinct circuits. Understanding the roles of individual genes within these neural circuits has been relatively difficult until very recently, however. The recent advent of genome editing technology, such as zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs) and CRISPR/Cas9, has made it possible to bidirectionally interrogate single genes in a circuit-specific manner. Due to the many advantages of CRISPR/Cas9, such as versatility, cost and ease of use, we will focus specifically on it as opposed to ZFNs and TALENS. Although this work is at a very early stage, especially in in vivo studies of brain function, this simple and powerful technique has already greatly expanded our ability to drive precise genetic manipulations within specific cell types.
CRISPR as a tool to make site-specific genetic and epigenetic modifications
The CRISPR/Cas9 system is a flexible and powerful way to edit an organism’s genome or bidirectionally alter the expression of endogenous target genes (Wang et al., 2016). CRISPR/Cas9 has two core components: 1) the CRISPR-associated endonuclease (Cas) protein, which cuts the target DNA and 2) the single guide RNA (sgRNA) which directs Cas9 to the correct genomic site, although there are restrictions to where the sgRNA can target (e.g. the sequence must be unique and contain a protospacer adjacent motif, or PAM (Walters et al., 2015)). In the simplest iteration of this system, CRISPR/Cas9 is used to create a gene knockout (Figure 5A). To this end, the sgRNA, which is programmed to target the gene of interest, forms a complex with Cas9 and directs it to the target gene, where it creates a double-strand break (DSB) of the DNA at that locus. Repairing that break with the non-homologous end joining (NHEJ) pathway typically results in insertion or deletion errors at the site, causing frameshift mutations that disrupt expression of the target gene. In instances in which the NHEJ pathway corrects the DSB without mutating the sequence, Cas9 cuts the site again to increase the likelihood of mutation. Thus, in theory, the system can be targeted to delete any gene of interest simply by designing the sgRNA to direct the Cas9 to the appropriate locus.
Figure 5. Basic CRISPR-Cas9 systems.
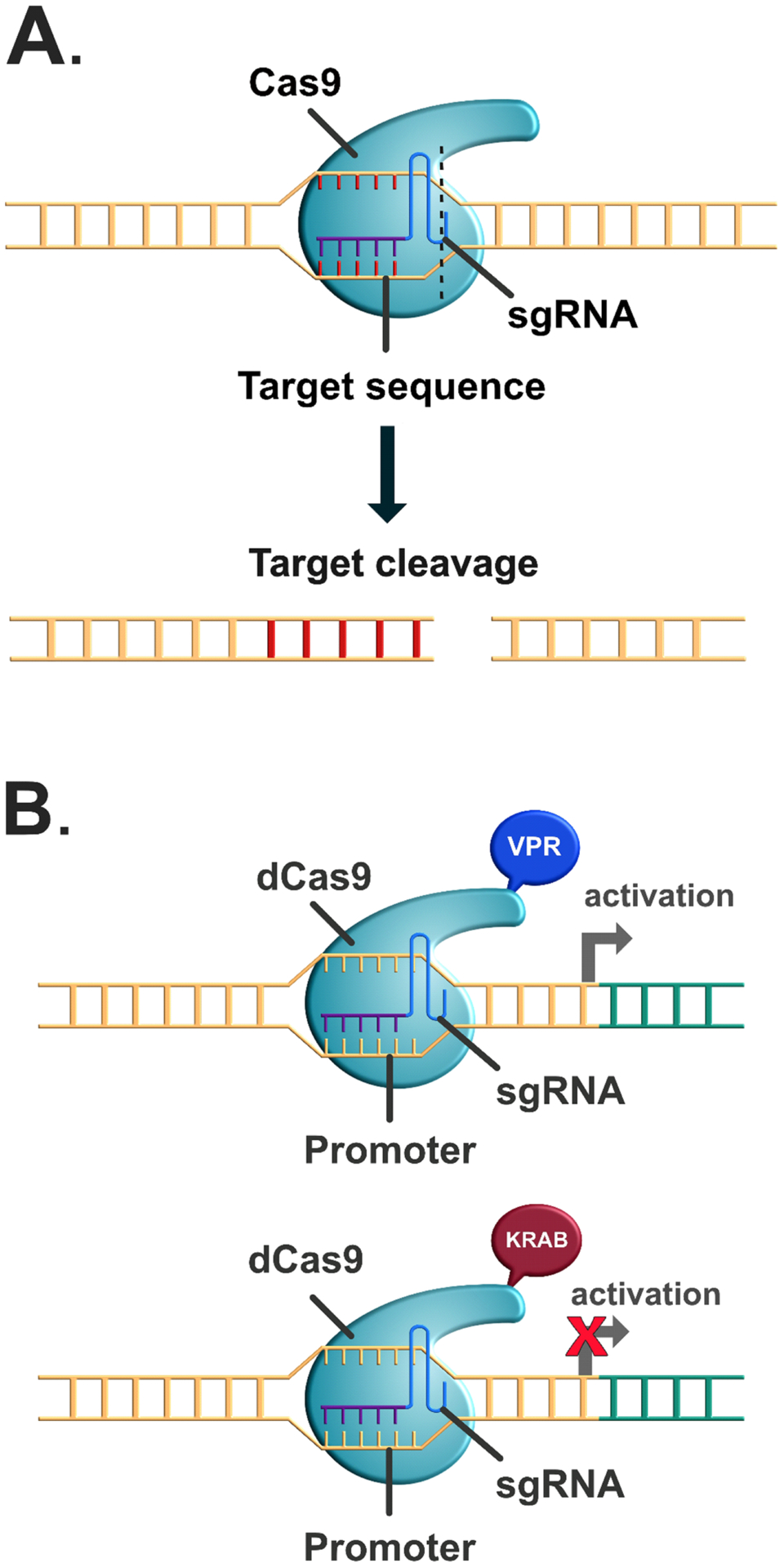
The CRISPR-Cas9 system can be used to edit a gene out or to control gene-specific transcriptional activation or silencing. (A) In the traditional CRISPR-Cas9 system, the catalytically active Cas9 complex is recruited to a target DNA region via a synthetic guide RNA (sgRNA; top). The Cas9 will “cut” the DNA, resulting in a double-stranded break (bottom). (B) In the CRISPR-dCas9 system, the Cas9 is catalytically inactive, so cannot cut DNA, but can still bind DNA as directed by the sgRNA. The dCas9 is fused to a transcriptional activator such as p65 (top) or repressor such as KRAB (bottom), which allows endogenous transcriptional control without editing the genome.
The CRISPR/Cas9 system can also be used to alter genomic DNA, such as making a single nucleotide substitution (to make a catalytically inactive point mutant, for example) or adding in an epitope tag for visualization of the gene (Ran et al., 2013). To drive these knock-in changes in the gene sequence, the researcher must provide a repair template that serves to “fix” the DSB with homology directed repair. Although this system can produce precise modifications to endogenous genes, it is relatively inefficient and produces a heterogenous population of cells that are unedited or mutated with the NHEJ pathway in addition to cells containing the desired genetic edit. Further, as these knock-in mutations rely on the homology-dependent repair pathway, which is not typically present in neurons (Ran et al., 2013), efforts to modify genetic information with this system in the brain have not yet been successful (Walters et al., 2015). Another method of generating mutations in vivo includes the use of Cas9 nickases (nCas9) or enzymatically-inactive “dead” Cas9 (dCas9) to generate single base edits. Cas9 nickases are modified to cut only one strand of DNA, rather than creating a double strand break like a traditional Cas9 enzyme. Thus, two nCas9 must be combined to generate a double-strand break to produce a traditional CRISPR-based knockout, which improves precision. To generate a single base mutation, nCas9 or dCas9 can be fused with a DNA deaminase to modify a single nucleotide in a target gene in vivo (Eid et al., 2018; Molla and Yang, 2019). Although this technique can be used relatively efficiently to produce C→T and A→G mutations, there are a number of limitations that restrict its usefulness (Eid et al., 2018). For example, these deaminases cannot make other types of point mutations besides the C→T and A→G conversions. Further this system lacks precision in the face of a string of Cs or As, so that all of the cytadine or adenine bases within the editing window of the CRISPR system will be modified. Finally, as with homology-driven knock-ins, the efficacy of single base editing in the brain has yet to be determined. Therefore, despite its promise as a simple mechanism to modify genetic information in vivo, there are a number of technical issues that need to be resolved before CRISPR/Cas9 or dCas9/nCas9-based nucleotide edits can be used to modify genomic DNA in the brain of a behaving animal.
In addition to deletions and knock-ins, the CRISPR/Cas9 system has been modified to transcriptionally activate or transcriptionally repress an endogenous gene without affecting the underlying sequence (Figure 5B). In these systems, the Cas9 enzyme is inactivated with point mutations, making it unable to cut the DNA at the targeted site. Rather, the dead Cas9 serves as a scaffold for transcriptional activators, repressors, or even chromatin modifiers to alter the expression of a target gene (Adli, 2018). A simple form of this system consists of the dCas9 fused to either a transcriptional activator (e.g. VP64) or repressor (e.g. KRAB) to drive or inhibit transcription of the target gene, respectively. To further enhance expression of the target gene, slight modifications in the CRISPR activation system can recruit additional transcriptional activators to augment expression of the target gene. For example, the dCas9-VPR activator system contains dCas9 fused to three different transcriptional activators: VP64, p65, and Rta that together increase expression of the target gene more than a single activator alone (Chavez et al., 2015; Chavez et al., 2016; Gilbert et al., 2013; Mali et al., 2013). Similarly, the CRISPR-SAM (Synergistic Activation Mediator) system uses a modified sgRNA containing MS2 aptamers to recruit additional activators (p65 and HSF1) fused to an MS2 coat protein to the target gene (Konermann et al., 2015). The modified sgRNA recruits both MS2-p65-HSF1 and the dCas9-VP64 fusion protein to the target gene to synergistically drive transcription. The CRISPR-SAM system has been successfully used in vivo to drive expression of the circadian gene Per1 in the dorsal hippocampus to improve long-term memory formation in aging mice (Kwapis et al., 2018). Finally, more by fusing chromatin modifiers to dCas9 (or driving the recruitment of a chromatin modifier through the use of MS2), researchers can theoretically modify the chromatin at a precise genomic locus. For example, targeting a dCas9-HAT (e.g. p300) to a specific gene should promote acetylation at that site, promoting an open chromatin formation that is permissive to gene expression without directly activating transcription. Finally, CRISPR can also be used to target long non-coding RNA (lncRNA) sequences to a specific genomic locus with the dCas9-based CRISPR Display (CRISPR-Disp) system, in which the long RNA sequence is incorporated into the sgRNA as an “aptameric accessory” domain (Perez-Pinera et al., 2015; Shechner et al., 2015). Although this work is in an early phase and much is unknown, the CRISPR/Cas9 system has already fundamentally changed how researchers can study gene function in the brain.
As CRISPR systems get more sophisticated, increasingly precise and complex manipulations are becoming feasible. Entire gene programs can theoretically be coordinated by multiplexing different sgRNAs with transcriptional activators and repressors. Introducing multiple sgRNAs to target dCas9-VPR to different regions along the promoter of a single gene can drive a larger increase in transcription than one sgRNA alone (Savell et al., 2019), indicating that dCas9-based activation scaffolds can be targeted along a promoter to synergistically increase transcription. Further, multiplexing sgRNAs targeting three different genes can increase the expression of each of the three target genes in neuronal culture (Savell et al., 2019), indicating that the CRISPR/dCas9 system can coordinately increase the expression of multiple genes at the same time. Researchers can gain an extra layer of control by also shaping the chromatin landscape across the genome using multiple sgRNAs to target chromatin modifying enzymes to specific genes, promoters, or enhancer elements. This ability to simultaneously manipulate multiple genetic and epigenetic events is critical for understanding how any individual gene contributes to behavior within the millieu of reactions happening within each neuron. Although there is much research on the effects of individual genetic manipulations in vivo, very little is understood about how these genes function relative to other genetic variants, epigenetic manipulations, or compensatory mechanisms, in part due to the lack of appropriate tools. With the ability to mimic a relatively complex and nuanced genetic program, CRISPR tools have the potential to broaden our understanding of the interactions between these mechaisms.
How to express CRISPR systems in the brain
CRISPR/Cas9 can be expressed in the central nervous system via three different methods: a transgenic mouse line, with viruses or using a nanoparticle delivery system. One popular approach is to use a transgenic mouse expressing Cas9 at the ROSA26 locus under a CAG promoter, which produces constitutive, brain-wide expression of cas9 (Platt et al., 2014). To create a mutation, a researcher simply needs to introduce a sgRNA (or multiple sgRNAs) to target Cas9 to the appropriate gene(s) using a virus. As Cas9 is a large protein that requires its own AAV due to packaging limitations, this transgenic line avoids the need to introduce multiple viruses; the researcher needs only to express the sgRNA (or even multiple sgRNAs), which readily fit into a typical AAV cassette. Additionally, a Cre-sensitive version of this knock-in mouse is also available, allowing for cell- and circuit-specific expression of Cas9.
Transgenic lines encoding dCas9 are also becoming available to allow for CRISPR-based gene activation or repression. For example, Cre-inducible versions of the gene-activating SunTag system are now available from The Jackson Laboratory. In the SunTag system, dCas9 is fused to a repeating polypeptide sequence that is recognized by the GCN4 antibody. Fusing GCN4 antibodies to transcriptional activators like VP64, p65, and HSF1, allows multiple copies of these transcriptional activators to be scaffolded to the gene locus targeted by the sgRNA (Tanenbaum et al., 2014). A researcher can therefore drive expression of a target gene in a site-specific manner by introducing both Cre and an appropriate sgRNA in the target brain region. Although there are very few dCas9-based transgenic lines currently available, as CRISPR-based technologies advance, it is likely that the number of available dCas9 transgenic mouse lines will continue to improve, as well.
A second method to express CRISPR/Cas9 in the brain is through the use of viruses. Virus-mediated expression of the full CRISPR/Cas9 system has the major advantage of improving flexibility; the researcher does not need to maintain a transgenic mouse line and can apply the system to a wide range of animal models at a desired age with a localized intracranial virus injection. The major disadvantage, as discussed above, is that the CRISPR system is too large to be packaged in a single AAV. In order to express Cas9/dCas9, the sgRNA, and any other required elements, multiple AAVs must be used. Although a basic Cas9 and sgRNA with short promoter sequences could be expressed in a single lentivirus, which has a larger packaging limit than AAV, a dCas9 system including transcriptional activators or repressors and a fluorescent protein for localization easily exceeds the ~9.7kB limit. Multiple viruses (three AAVs or two lentiviruses) expressing different dCas9-based activation systems have been successfully used to drive gene expression in different brain regions in vivo, including the hippocampus, nucleus accumbens, and prefrontal cortex (Kwapis et al., 2018; Savell et al., 2019). Using multiple viruses is problematic, however, as only the cells infected with all of the viruses get a functional CRISPR system. An alternative solution to this problem is to use HSVs, which are neurotropic and have a much larger packaging limit (>30kB) (Sarno and Robison, 2018) that can accommodate the entire CRISPR/dCas9 system, avoiding the requirement for multiple viruses. HSV-mediated expression of a target gene is more transient than expression with AAVs or lentiviruses; expression peaks around 3–5d and eliminated by 8–10d after infection (Sarno and Robison, 2018). Thus, if the behavioral task and the research question are amenable to this shorter timecourse, the use of HSVs are an effective delivery method for large CRISPR/dCas9 systems into the brain (Lorsch et al., 2019; Walters et al., 2017).
New nanoparticle-based delivery systems like CRISPR-Gold are currently being developed to avoid the use of viruses altogether, which have limitations that restrict its use in a clinical setting. CRISPR-Gold uses a gold nanoparticle complex to package the Cas9 molecule and sgRNA inside of a polymer that allows the package to cross the cell membrane before allowing the Cas9 and sgRNA to be released (Lee et al., 2017). Thus, like viruses, CRISPR-Gold allows for site-specific expression of CRISPR but has the additional advantage of packaging the entire CRISPR system in a single nanoparticle. Further, CRISPR-Gold avoids both the toxicity and the immune response induced by viral CRISPR infusions (Lee et al., 2017) and its transient expression prevents the off-target effects observed following persistent viral-induced Cas9 expression. Since its initial development, CRISPR-Gold has been used to edit genes in the adult mouse brain, most notably the metabotropic glutamate receptor 5 (mGluR5) gene, deletion of which reduced the excessive repetitive behaviors observed in a mouse model of fragile X syndrome (Lee et al., 2018). As nanoparticle and viral delivery methods continue to improve, it is becoming increasingly likely that CRISPR/Cas9 will be a viable therapeutic method for gene editing in patients.
Finally, CRISPR/Cas9 plasmids can also be delivered into the brain using the PolyPlus transfection reagent in vivo Jet-PEI. This polymer-based reagent acts by condensing nucleic acids into stable nanoparticle-like structures small enough to diffuse into tissue, which occurs via endocytosis. This transfection reagent has a number of advantages over virus-mediated systems, including no biohazard concerns and low costs. However, disadvantages include the proprietary nature of the product, which limits our understanding of exactly how it works to deliver foreign material into the cell, and the lack of cell type specificity, though this could be overcome through the use of specifc promoters in the CRISPR constructs. Similar to CRISPR-Gold, few studies have used this CRISPR delivery system to date. However, one recent study was able to express CRISPR-dCas9 constructs in the mouse hippocampus using in vivo Jet-PEI and achieved transcriptional control over their target gene and altered memory formation (Butler et al., 2019).
Using CRISPR to direct circuit-specific genetic modifications
In addition to improving the ease of precise genetic manipulations, CRISPR/Cas9 can also be used to interrogate the roles of specific genes within defined neural circuits in the brain. Although it is possible to overexpress a gene or knock down a gene in a specific neural circuit by combining promoter-line Cre expression with a Cre-dependent construct, CRISPR/Cas9 makes it possible to create knockouts or overexpression from endogenous genes in a population of cells with a specific projection target. Creating a circuit-specific manipulation using CRISPR/Cas9 or CRISPR/dCas9 can be accomplished with either a Cre-based CRISPR system or by using retrograde virus expression to express a portion of the functional CRISPR system. Transgenic models encoding Cre-sensitive versions of both Cas9 and dCas9 systems are available from The Jackson Laboratory (www.jax.org) that allow the researcher to limit expression of the CRISPR system to cell populations expressing Cre. Therefore, by crossing these mice with a promoter line expressing Cre in a specific population of neurons or by locally injecting a virus encoding Cre under a cell type-specific promoter, expression of the system can be restricted to a subset of neurons with similar properties. Viruses with retrograde properties can also be used to restrict CRISPR-based manipulations to a specific neural circuit. By splitting up the components of a functional CRISPR system into two components (the sgRNA and the dCas9) and delivering the dCas9 component via a retrograde HSV into one brain region (“B”) and the sgRNA via local viral expression in a brain region that projects to this site (“A”), CRISPR could be used to selectively delete or modify the expression of a single gene in neurons projecting from brain region A to region B (Sarno and Robison, 2018).
CRISPR has also been incorporated into inducible systems to restrict when the gene editing occurs during behavioral experiments. Using these systems, it is possible to induce a CRISPR-based manipulation during a specific phase of memory formation, for example. There are two primary methods for temporally restricting Cas9: incorporation into a Tet-On (or Tet-Off) system or using light-sensitive effectors. In the Tet-On system, a transgene coding for Cas9/dCas9 and any necessary effector domains are placed under the control of doxycycline, so that exposure to doxycycline (or removal of Dox in the Tet-Off system) drives transcription of the CRISPR system (Cao et al., 2016). As with other Tet-based systems (see Figure 2), activity-regulated promoters like Arc or cFos can be used to express the tTA or rtTA gene, limiting expression of Cas9/dCas9 to cells activated by stimulation during the “active” window. Thus, the CRISPR system can be used to delete, inhibit, or activate a gene selectively in cells activated by a specific event, such as a training session.
A second type of inducible CRISPR uses a light-sensitive effector domain that can be activated with light stimulation, allowing for even more fine temporal control over gene expression. In the most popular of these systems, light-activated CRISPR effector (LACE), the blue light-sensitive cryptochrome 2 (CRY2) protein is fused to an effector domain (VP64 or p65) that can promote transcription upon binding to CIB1, which is fused to the dCas9. In the absence of light, the CRY2-effector fusion is unable to bind CIB1. When exposed to blue light, however, CRY2 undergoes a reversible conformational change that allows it to bind CIB1, scaffolding the effector domain to the target gene to induce its transcription (Nihongaki et al., 2015; Polstein and Gersbach, 2015). LACE therefore allows rapid and reversible activation of a gene during behavior.
As CRISPR technology continues to improve, the list of questions that can be experimentally addressed also continues to expand. For example, until very recently it was not possible to determine whether epigenetic modifications at a specific gene during memory formation affected its expression and ultimate effects on memory (Heller et al., 2014). Clever systems now combine dCas9 with epigenetic modifiers, allowing the researcher to direct site-specific chromatin modifications during behavior by scaffolding a chromatin-modifying enzyme to a specific gene promoter. In theory, this method could be used to “open” or “close” chromatin at a specific locus, allowing the endogenous transcriptional machinery to produce physiologically relevant expression of a specific gene under either permissive or restrictive chromatin conditions, respectively. Whether this system can be successfully used to modify chromatin in vivo during behavior and, if so, whether these precise chromatin modifications are sufficient to improve or impair memory remains to be seen. CRISPR systems will continue to grow more sophisticated as Cas9 gets incorporated into existing systems of “tagging” active neurons, such as the TRAP system (Fig. 3B) and new tools, such as light-sensitive CRISPR-based interference, are developed. The addition of CRISPR into the neuroscientist’s toolkit has given us the ability to make precise manipulations that were once inaccessible, opening the door to answering incredibly detailed questions about the causal role of genetic and epigenetic modifications in altering behavior in vivo.
Conclusions
As genetic manipulations become more common and more sophisticated, neuroscientists can answer increasingly detailed questions about the role of different genetic and epigenetic mechanisms in brain function. In addition to time-tested techniques like gene knockouts and knock-ins, more recent tools, including CRISPR-Cas9, are now available to manipulate specific genes in spatially- and temporally-specific systems. Neuroscientists can now probe the function of a specific gene within a desired circuit at a specific point in time, such as during a learning event. With this powerful, ever-expanding toolkit available to us, neuroscientists are poised to drastically advance our understanding of brain function.
Acknowledgements
This work was supported in part by startup funds from the College of Agricultural and Life Sciences and the School of Neuroscience at Virginia Tech (TJJ). It was also supported by startup funds from the Eberly College of Science and Department of Biology at Pennsylvania State University and the National Institute on Aging (NIA) grant K99/R00 AG056596 (JLK).
Footnotes
Declaration of Interest
Declarations of interest: None
References
- Abe T, Fujimori T, 2013. Reporter mouse lines for fluorescence imaging. Development, growth & differentiation 55, 390–405. [DOI] [PubMed] [Google Scholar]
- Abe T, Kiyonari H, Shioi G, Inoue K, Nakao K, Aizawa S, Fujimori T, 2011. Establishment of conditional reporter mouse lines at ROSA26 locus for live cell imaging. Genesis 49, 579–590. [DOI] [PubMed] [Google Scholar]
- Abram CL, Roberge GL, Hu Y, Lowell CA, 2014. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J. Immunol. Methods 408, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achterberg KG, Buitendijk GH, Kool MJ, Goorden SM, Post L, Slump DE, Silva AJ, van Woerden GM, Kushner SA, Elgersma Y, 2014. Temporal and region-specific requirements of alphaCaMKII in spatial and contextual learning. J Neurosci 34, 11180–11187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adli M, 2018. The CRISPR tool kit for genome editing and beyond. Nat Commun 9, 1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn HJ, Hernandez CM, Levenson JM, Lubin FD, Liou HC, Sweatt JD, 2008. c-Rel, an NF-kappaB family transcription factor, is required for hippocampal long-term synaptic plasticity and memory formation. Learn Mem 15, 539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alaghband Y, Kramar E, Kwapis JL, Kim ES, Hemstedt TJ, Lopez AJ, White AO, Al-Kachak A, Aimiuwu OV, Bodinayake KK, Oparaugo NC, Han J, Lattal KM, Wood MA, 2018. CREST in the Nucleus Accumbens Core Regulates Cocaine Conditioned Place Preference, Cocaine-Seeking Behavior, and Synaptic Plasticity. J Neurosci 38, 9514–9526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alaghband Y, Kwapis JL, Lopez AJ, White AO, Aimiuwu OV, Al-Kachak A, Bodinayake KK, Oparaugo NC, Dang R, Astarabadi M, Matheos DP, Wood MA, 2017. Distinct roles for the deacetylase domain of HDAC3 in the hippocampus and medial prefrontal cortex in the formation and extinction of memory. Neurobiol. Learn. Mem 145, 94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastassiadis K, Fu J, Patsch C, Hu S, Weidlich S, Duerschke K, Buchholz F, Edenhofer F, Stewart AF, 2009. Dre recombinase, like Cre, is a highly efficient site-specific recombinase in E. coli, mammalian cells and mice. Dis. Model. Mech 2, 508–515. [DOI] [PubMed] [Google Scholar]
- Artusi S, Miyagawa Y, Goins WF, Cohen JB, Glorioso JC, 2018. Herpes Simplex Virus Vectors for Gene Transfer to the Central Nervous System. Diseases 6, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach ME, Hawkins RD, Osman M, Kandel ER, Mayford M, 1995. Impairment of spatial but not contextual memory in CaMKII mutant mice with a selective loss of hippocampal LTP in the range of the theta frequency. Cell 81, 905–915. [DOI] [PubMed] [Google Scholar]
- Barth AL, 2007. Visualizing circuits and systems using transgenic reporters of neural activity. Curr Opin Neurobiol 17, 567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth AL, Gerkin RC, Dean KL, 2004. Alteration of neuronal firing properties after in vivo experience in a FosGFP transgenic mouse. J Neurosci 24, 6466–6475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berbari NF, Malarkey EB, Yazdi SM, McNair AD, Kippe JM, Croyle MJ, Kraft TW, Yoder BK, 2014. Hippocampal and cortical primary cilia are required for aversive memory in mice. PLoS One 9, e106576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloem B, Schoppink L, Rotaru DC, Faiz A, Hendriks P, Mansvelder HD, van de Berg WD, Wouterlood FG, 2014. Topographic mapping between basal forebrain cholinergic neurons and the medial prefrontal cortex in mice. J Neurosci 34, 16234–16246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boender AJ, de Jong JW, Boekhoudt L, Luijendijk MC, van der Plasse G, Adan RA, 2014. Combined use of the canine adenovirus-2 and DREADD-technology to activate specific neural pathways in vivo. PLoS One 9, e95392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouabe H, Okkenhaug K, 2013. Gene targeting in mice: a review, Virus-Host Interactions. Springer, pp. 315–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ, 1994. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 79, 59–68. [DOI] [PubMed] [Google Scholar]
- Brenner M, Kisseberth WC, Su Y, Besnard F, Messing A, 1994. GFAP promoter directs astrocyte-specific expression in transgenic mice. J Neurosci 14, 1030–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briand LA, Lee BG, Lelay J, Kaestner KH, Blendy JA, 2015. Serine 133 phosphorylation is not required for hippocampal CREB-mediated transcription and behavior. Learn Mem 22, 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster RL, Chen HY, Trumbauer M, Senear AW, Warren R, Palmiter RD, 1981. Somatic expression of herpes thymidine kinase in mice following injection of a fusion gene into eggs. Cell 27, 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster RL, Chen HY, Warren R, Sarthy A, Palmiter RD, 1982. Regulation of metallothionein–thymidine kinase fusion plasmids injected into mouse eggs. Nature 296, 39. [DOI] [PubMed] [Google Scholar]
- Brocard J, Warot X, Wendling O, Messaddeq N, Vonesch JL, Chambon P, Metzger D, 1997. Spatio-temporally controlled site-specific somatic mutagenesis in the mouse. Proceedings of the National Academy of Sciences of the United States of America 94, 14559–14563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz F, Angrand PO, Stewart AF, 1998. Improved properties of FLP recombinase evolved by cycling mutagenesis. Nat. Biotechnol 16, 657–662. [DOI] [PubMed] [Google Scholar]
- Burger C, Gorbatyuk OS, Velardo MJ, Peden CS, Williams P, Zolotukhin S, Reier PJ, Mandel RJ, Muzyczka N, 2004. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol Ther 10, 302–317. [DOI] [PubMed] [Google Scholar]
- Butler AA, Johnston DR, Kaur S, Lubin FD, 2019. Long noncoding RNA NEAT1 mediates neuronal histone methylation and age-related memory impairment. Sci Signal 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Wu L, Zhang SM, Lu M, Cheung WK, Cai W, Gale M, Xu Q, Yan Q, 2016. An easy and efficient inducible CRISPR/Cas9 platform with improved specificity for multiple gene targeting. Nucleic Acids Res 44, e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capecchi MR, 1989. Altering the genome by homologous recombination. Science 244, 1288–1292. [DOI] [PubMed] [Google Scholar]
- Carreno FR, Donegan JJ, Boley AM, Shah A, DeGuzman M, Frazer A, Lodge DJ, 2016. Activation of a ventral hippocampus-medial prefrontal cortex pathway is both necessary and sufficient for an antidepressant response to ketamine. Mol Psychiatry 21, 1298–1308. [DOI] [PubMed] [Google Scholar]
- Carter ME, de Lecea L, 2011. Optogenetic investigation of neural circuits in vivo. Trends Mol Med 17, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC, 1994. Green fluorescent protein as a marker for gene expression. Science 263, 802–805. [DOI] [PubMed] [Google Scholar]
- Chan CH, Godinho LN, Thomaidou D, Tan SS, Gulisano M, Parnavelas JG, 2001. Emx1 is a Marker for Pyramidal Neurons of the Cerebral Cortex. Cerebral Cortex 11, 1191–1198. [DOI] [PubMed] [Google Scholar]
- Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, E PRI, Lin S, Kiani S, Guzman CD, Wiegand DJ, Ter-Ovanesyan D, Braff JL, Davidsohn N, Housden BE, Perrimon N, Weiss R, Aach J, Collins JJ, Church GM, 2015. Highly efficient Cas9-mediated transcriptional programming. Nat Methods 12, 326–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez A, Tuttle M, Pruitt BW, Ewen-Campen B, Chari R, Ter-Ovanesyan D, Haque SJ, Cecchi RJ, Kowal EJK, Buchthal J, Housden BE, Perrimon N, Collins JJ, Church G, 2016. Comparison of Cas9 activators in multiple species. Nat Methods 13, 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Gonçalves MAFV, 2016. Engineered Viruses as Genome Editing Devices. Mol Ther 24, 447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng R, Peng J, Yan Y, Cao P, Wang J, Qiu C, Tang L, Liu D, Tang L, Jin J.J.F.l., 2014. Efficient gene editing in adult mouse livers via adenoviral delivery of CRISPR/Cas9. 588, 3954–3958. [DOI] [PubMed] [Google Scholar]
- Clem RL, Barth A, 2006. Pathway-specific trafficking of native AMPARs by in vivo experience. Neuron 49, 663–670. [DOI] [PubMed] [Google Scholar]
- Collins DE, Reuter JD, Rush HG, Villano JS, 2017. Viral Vector Biosafety in Laboratory Animal Research. Comp. Med 67, 215–221. [PMC free article] [PubMed] [Google Scholar]
- Contag CH, Bachmann MH, 2002. Advances in in vivo bioluminescence imaging of gene expression. Annu Rev Biomed Eng 4, 235–260. [DOI] [PubMed] [Google Scholar]
- Cowansage KK, Shuman T, Dillingham BC, Chang A, Golshani P, Mayford M, 2014. Direct reactivation of a coherent neocortical memory of context. Neuron 84, 432–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z, Wang H, Tan Y, Zaia KA, Zhang S, Tsien JZ, 2004. Inducible and reversible NR1 knockout reveals crucial role of the NMDA receptor in preserving remote memories in the brain. Neuron 41, 781–793. [DOI] [PubMed] [Google Scholar]
- Das AT, Tenenbaum L, Berkhout B, 2016. Tet-On Systems For Doxycycline-inducible Gene Expression. Curr Gene Ther 16, 156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delzor A, Dufour N, Petit F, Guillermier M, Houitte D, Auregan G, Brouillet E, Hantraye P, Deglon N, 2012. Restricted transgene expression in the brain with cell-type specific neuronal promoters. Hum Gene Ther Methods 23, 242–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis-Donini S, Dellarole A, Crociara P, Francese MT, Bortolotto V, Quadrato G, Canonico PL, Orsetti M, Ghi P, Memo M, Bonini SA, Ferrari-Toninelli G, Grilli M, 2008. Impaired adult neurogenesis associated with short-term memory defects in NF-kappaB p50-deficient mice. J Neurosci 28, 3911–3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny CA, Kheirbek MA, Alba EL, Tanaka KF, Brachman RA, Laughman KB, Tomm NK, Turi GF, Losonczy A, Hen R, 2014. Hippocampal memory traces are differentially modulated by experience, time, and adult neurogenesis. Neuron 83, 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deverman BE, Pravdo PL, Simpson BP, Kumar SR, Chan KY, Banerjee A, Wu W-L, Yang B, Huber N, Pasca SP, Gradinaru V, 2016. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol 34, 204–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deyle DR, Russell DW, 2009. Adeno-associated virus vector integration. Curr Opin Mol Ther 11, 442–447. [PMC free article] [PubMed] [Google Scholar]
- Dias BG, Goodman JV, Ahluwalia R, Easton AE, Andero R, Ressler KJ, 2014. Amygdala-dependent fear memory consolidation via miR-34a and Notch signaling. Neuron 83, 906–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle A, McGarry MP, Lee NA, Lee JJ, 2012. The construction of transgenic and gene knockout/knockin mouse models of human disease. Transgenic Res 21, 327–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid A, Alshareef S, Mahfouz MM, 2018. CRISPR base editors: genome editing without double-stranded breaks. Biochem J 475, 1955–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisener-Dorman AF, Lawrence DA, Bolivar VJ, 2009. Cautionary insights on knockout mouse studies: the gene or not the gene? Brain Behav Immun 23, 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escors D, Breckpot K, 2010. Lentiviral vectors in gene therapy: their current status and future potential. Arch Immunol Ther Exp (Warsz) 58, 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenno LE, Mattis J, Ramakrishnan C, Hyun M, Lee SY, He M, Tucciarone J, Selimbeyoglu A, Berndt A, Grosenick L, Zalocusky KA, Bernstein H, Swanson H, Perry C, Diester I, Boyce FM, Bass CE, Neve R, Huang ZJ, Deisseroth K, 2014. Targeting cells with single vectors using multiple-feature Boolean logic. Nat Methods 11, 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster K, Helbl V, Lederer T, Urlinger S, Wittenburg N, Hillen W, 1999. Tetracycline-inducible expression systems with reduced basal activity in mammalian cells. Nucleic Acids Res. 27, 708–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff YG, Mueller M, Dann SG, Svoboda P, Sticker M, Spetz JF, Um SH, Brown EJ, Cereghini S, Thomas G, Kozma SC, 2004. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol Cell Biol 24, 9508–9516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q, Yao W, Wang J, Yang T, Liu C, Tao Y, Chen Y, Liu X, Ma L, 2015. Post-training activation of Rac1 in the basolateral amygdala is required for the formation of both short-term and long-term auditory fear memory. Front Mol Neurosci 8, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner AR, Rowland DC, Hwang SY, Baumgaertel K, Roth BL, Kentros C, Mayford M, 2012. Generation of a synthetic memory trace. Science 335, 1513–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese KP, Fedorov NB, Filipkowski RK, Silva AJ, 1998. Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science 279, 870–873. [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, Lim WA, Weissman JS, Qi LS, 2013. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenthner CJ, Miyamichi K, Yang HH, Heller HC, Luo L, 2013. Permanent genetic access to transiently active neurons via TRAP: targeted recombination in active populations. Neuron 78, 773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Kim SY, Artis S, Molfese DL, Schumacher A, Sweatt JD, Paylor RE, Lubin FD, 2010. Histone methylation regulates memory formation. J Neurosci 30, 3589–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haettig J, Sun Y, Wood MA, Xu X, 2013. Cell-type specific inactivation of hippocampal CA1 disrupts location-dependent object recognition in the mouse. Learn Mem 20, 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JH, Kushner SA, Yiu AP, Cole CJ, Matynia A, Brown RA, Neve RL, Guzowski JF, Silva AJ, Josselyn SA, 2007. Neuronal competition and selection during memory formation. Science 316, 457–460. [DOI] [PubMed] [Google Scholar]
- Hannon GJ, 2002. RNA interference. Nature 418, 244–251. [DOI] [PubMed] [Google Scholar]
- Harper A, 2010. Mouse models of neurological disorders—A comparison of heritable and acquired traits. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1802, 785–795. [DOI] [PubMed] [Google Scholar]
- Hasegawa S, Furuichi T, Yoshida T, Endoh K, Kato K, Sado M, Maeda R, Kitamoto A, Miyao T, Suzuki R, Homma S, Masushige S, Kajii Y, Kida S, 2009. Transgenic up-regulation of alpha-CaMKII in forebrain leads to increased anxiety-like behaviors and aggression. Mol Brain 2, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffner CS, Herbert Pratt C, Babiuk RP, Sharma Y, Rockwood SF, Donahue LR, Eppig JT, Murray SA, 2012. Supporting conditional mouse mutagenesis with a comprehensive cre characterization resource. Nat Commun 3, 1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller EA, Cates HM, Pena CJ, Sun H, Shao N, Feng J, Golden SA, Herman JP, Walsh JJ, Mazei-Robison M, Ferguson D, Knight S, Gerber MA, Nievera C, Han MH, Russo SJ, Tamminga CS, Neve RL, Shen L, Zhang HS, Zhang F, Nestler EJ, 2014. Locus-specific epigenetic remodeling controls addiction- and depression-related behaviors. Nature neuroscience 17, 1720–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermens WT, Giger RJ, Holtmaat AJ, Dijkhuizen PA, Houweling DA, Verhaagen J.J.J.o.n.m., 1997. Transient gene transfer to neurons and glia: analysis of adenoviral vector performance in the CNS and PNS. 71, 85–98. [DOI] [PubMed] [Google Scholar]
- Hioki H, Kameda H, Nakamura H, Okunomiya T, Ohira K, Nakamura K, Kuroda M, Furuta T, Kaneko T, 2007. Efficient gene transduction of neurons by lentivirus with enhanced neuron-specific promoters. Gene Ther 14, 872–882. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G, 1996. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274, 99–102. [DOI] [PubMed] [Google Scholar]
- Huber K, Ernsberger U, 2006. Cholinergic differentiation occurs early in mouse sympathetic neurons and requires Phox2b. Gene Expr 13, 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh WJ, Khurana SS, Geahlen JH, Kohli K, Waller RA, Mills JC, 2012. Tamoxifen induces rapid, reversible atrophy, and metaplasia in mouse stomach. Gastroenterology 142, 21–24.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimoto T, Mano H, Mori H, 2015. In vivo imaging of CREB phosphorylation in awake-mouse brain. Sci Rep 5, 9757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob T.C.J.T.D.S.M.M.i.I.R.B., 2006. 10 RNAi and Applications in Neurobiology. 177. [PubMed] [Google Scholar]
- Jaitner C, Reddy C, Abentung A, Whittle N, Rieder D, Delekate A, Korte M, Jain G, Fischer A, Sananbenesi F, Cera I, Singewald N, Dechant G, Apostolova G, 2016. Satb2 determines miRNA expression and long-term memory in the adult central nervous system. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarome TJ, Butler AA, Nichols JN, Pacheco NL, Lubin FD, 2015. NF-kappaB mediates Gadd45beta expression and DNA demethylation in the hippocampus during fear memory formation. Front Mol Neurosci 8, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarome TJ, Perez GA, Hauser RM, Hatch KM, Lubin FD, 2018. EZH2 Methyltransferase Activity Controls Pten Expression and mTOR Signaling during Fear Memory Reconsolidation. J Neurosci 38, 7635–7648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyner AL, Skarnes WC, Rossant J, 1989. Production of a mutation in mouse En-2 gene by homologous recombination in embryonic stem cells. Nature 338, 153. [DOI] [PubMed] [Google Scholar]
- Kaas GA, Zhong C, Eason DE, Ross DL, Vachhani RV, Ming GL, King JR, Song H, Sweatt JD, 2013. TET1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron 79, 1086–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarek L, 1992. Expression of c-fos and other genes encoding transcription factors in long-term potentiation. Behav Neural Biol 57, 263–266. [DOI] [PubMed] [Google Scholar]
- Kang H, Sun LD, Atkins CM, Soderling TR, Wilson MA, Tonegawa S, 2001. An important role of neural activity-dependent CaMKIV signaling in the consolidation of long-term memory. Cell 106, 771–783. [DOI] [PubMed] [Google Scholar]
- Kassed CA, Willing AE, Garbuzova-Davis S, Sanberg PR, Pennypacker KR, 2002. Lack of NF-kappaB p50 exacerbates degeneration of hippocampal neurons after chemical exposure and impairs learning. Exp Neurol 176, 277–288. [DOI] [PubMed] [Google Scholar]
- Kawamoto S, Niwa H, Tashiro F, Sano S, Kondoh G, Takeda J, Tabayashi K, Miyazaki J, 2000. A novel reporter mouse strain that expresses enhanced green fluorescent protein upon Cre-mediated recombination. FEBS Lett 470, 263–268. [DOI] [PubMed] [Google Scholar]
- Kim H, Kim M, Im SK, Fang S, 2018. Mouse Cre-LoxP system: general principles to determine tissue-specific roles of target genes. Lab Anim Res 34, 147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HH, Wolfe A, Smith GR, Tobet SA, Radovick S, 2002. Promoter sequences targeting tissue-specific gene expression of hypothalamic and ovarian gonadotropin-releasing hormone in vivo. J Biol Chem 277, 5194–5202. [DOI] [PubMed] [Google Scholar]
- Kogan JH, Frankland PW, Blendy JA, Coblentz J, Marowitz Z, Schutz G, Silva AJ, 1997. Spaced training induces normal long-term memory in CREB mutant mice. Curr Biol 7, 1–11. [DOI] [PubMed] [Google Scholar]
- Kojima N, Wang J, Mansuy IM, Grant SG, Mayford M, Kandel ER, 1997. Rescuing impairment of long-term potentiation in fyn-deficient mice by introducing Fyn transgene. Proceedings of the National Academy of Sciences of the United States of America 94, 4761–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koller BH, Hagemann LJ, Doetschman T, Hagaman JR, Huang S, Williams PJ, First NL, Maeda N, Smithies O, 1989. Germ-line transmission of a planned alteration made in a hypoxanthine phosphoribosyltransferase gene by homologous recombination in embryonic stem cells. Proceedings of the National Academy of Sciences 86, 8927–8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koller BH, Marrack P, Kappler JW, Smithies O, 1990. Normal development of mice deficient in beta 2M, MHC class I proteins, and CD8+ T cells. Science 248, 1227–1230. [DOI] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, Nureki O, Zhang F, 2015. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool MJ, van de Bree JE, Bodde HE, Elgersma Y, van Woerden GM, 2016. The molecular, temporal and region-specific requirements of the beta isoform of Calcium/Calmodulin-dependent protein kinase type 2 (CAMK2B) in mouse locomotion. Scientific Reports 6, 26989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranz A, Fu J, Duerschke K, Weidlich S, Naumann R, Stewart AF, Anastassiadis K, 2010. An improved Flp deleter mouse in C57Bl/6 based on Flpo recombinase. Genesis 48, 512–520. [DOI] [PubMed] [Google Scholar]
- Kremer EA, Gaur N, Lee MA, Engmann O, Bohacek J, Mansuy IM, 2018. Interplay between TETs and microRNAs in the adult brain for memory formation. Sci Rep 8, 1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristianto J, Johnson MG, Zastrow RK, Radcliff AB, Blank RD, 2017. Spontaneous recombinase activity of Cre-ERT2 in vivo. Transgenic Res. 26, 411–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugler S, Kilic E, Bahr M, 2003. Human synapsin 1 gene promoter confers highly neuron-specific long-term transgene expression from an adenoviral vector in the adult rat brain depending on the transduced area. Gene Ther 10, 337–347. [DOI] [PubMed] [Google Scholar]
- Kurotaki Y, Hatta K, Nakao K, Nabeshima Y, Fujimori T, 2007. Blastocyst axis is specified independently of early cell lineage but aligns with the ZP shape. Science 316, 719–723. [DOI] [PubMed] [Google Scholar]
- Kwapis JL, Alaghband Y, Kramar EA, Lopez AJ, Vogel Ciernia A, White AO, Shu G, Rhee D, Michael CM, Montellier E, Liu Y, Magnan CN, Chen S, Sassone-Corsi P, Baldi P, Matheos DP, Wood MA, 2018. Epigenetic regulation of the circadian gene Per1 contributes to age-related changes in hippocampal memory. Nat Commun 9, 3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwapis JL, Alaghband Y, Lopez AJ, White AO, Campbell RR, Dang RT, Rhee D, Tran AV, Carl AE, Matheos DP, Wood MA, 2017. Context and Auditory Fear are Differentially Regulated by HDAC3 Activity in the Lateral and Basal Subnuclei of the Amygdala. Neuropsychopharmacology 42, 1284–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacagnina AF, Brockway ET, Crovetti CR, Shue F, McCarty MJ, Sattler KP, Lim SC, Santos SL, Denny CA, Drew MR, 2019. Distinct hippocampal engrams control extinction and relapse of fear memory. Nature neuroscience 22, 753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois M-A, Boniface C, Wang G, Alluin J, Salvaterra PM, Puymirat J, Rossi JJ, Lee N.S.J.J.o.B.C., 2005. Cytoplasmic and nuclear retained DMPK mRNAs are targets for RNA interference in myotonic dystrophy cells. 280, 16949–16954. [DOI] [PubMed] [Google Scholar]
- Lee B, Lee K, Panda S, Gonzales-Rojas R, Chong A, Bugay V, Park HM, Brenner R, Murthy N, Lee HY, 2018. Nanoparticle delivery of CRISPR into the brain rescues a mouse model of fragile X syndrome from exaggerated repetitive behaviours. Nat Biomed Eng 2, 497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, Wenthold RJ, Gallagher M, Huganir RL, 2003. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 112, 631–643. [DOI] [PubMed] [Google Scholar]
- Lee K, Conboy M, Park HM, Jiang F, Kim HJ, Dewitt MA, Mackley VA, Chang K, Rao A, Skinner C, Shobha T, Mehdipour M, Liu H, Huang WC, Lan F, Bray NL, Li S, Corn JE, Kataoka K, Doudna JA, Conboy I, Murthy N, 2017. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat Biomed Eng 1, 889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann ML, Brachman RA, Listwak SJ, Herkenham M, 2010. NF-kappaB activity affects learning in aversive tasks: possible actions via modulation of the stress axis. Brain Behav Immun 24, 1008–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R, 1992. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69, 915–926. [DOI] [PubMed] [Google Scholar]
- Li F, Lu Z, Wu W, Qian N, Wang F, Chen T, 2019. Optogenetic gene editing in regional skin. Cell Research 29, 862–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wei W, Zhao QY, Widagdo J, Baker-Andresen D, Flavell CR, D’Alessio A, Zhang Y, Bredy TW, 2014. Neocortical Tet3-mediated accumulation of 5-hydroxymethylcytosine promotes rapid behavioral adaptation. Proceedings of the National Academy of Sciences of the United States of America 111, 7120–7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee Y-L, Kim KA, Ando D, Urnov FD, Galli C, Gregory PD, Holmes MC, Naldini L, 2007. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol 25, 1298–1306. [DOI] [PubMed] [Google Scholar]
- Loonstra A, Vooijs M, Beverloo HB, Allak BA, van Drunen E, Kanaar R, Berns A, Jonkers J, 2001. Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Proc. Natl. Acad. Sci. U. S. A 98, 9209–9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez AJ, Jia Y, White AO, Kwapis JL, Espinoza M, Hwang P, Campbell R, Alaghband Y, Chitnis O, Matheos DP, Lynch G, Wood MA, 2019. Medial habenula cholinergic signaling regulates cocaine-associated relapse-like behavior. Addict Biol 24, 403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorsch ZS, Hamilton PJ, Ramakrishnan A, Parise EM, Salery M, Wright WJ, Lepack AE, Mews P, Issler O, McKenzie A, Zhou X, Parise LF, Pirpinias ST, Ortiz Torres I, Kronman HG, Montgomery SE, Loh YE, Labonte B, Conkey A, Symonds AE, Neve RL, Turecki G, Maze I, Dong Y, Zhang B, Shen L, Bagot RC, Nestler EJ, 2019. Stress resilience is promoted by a Zfp189-driven transcriptional network in prefrontal cortex. Nat. Neurosci 22, 1413–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett-Racke AE, Cravens PD, Gocke AR, Racke MK, Stüve O, 2005. Therapeutic Potential of Small Interfering RNA for Central Nervous System Diseases. JAMA Neurology 62, 1810–1813. [DOI] [PubMed] [Google Scholar]
- Lundstrom K, 2018. Viral Vectors in Gene Therapy. Diseases 6, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H, 2010. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nature neuroscience 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, Yang L, Church GM, 2013. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol 31, 833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malleret G, Haditsch U, Genoux D, Jones MW, Bliss TV, Vanhoose AM, Weitlauf C, Kandel ER, Winder DG, Mansuy IM, 2001. Inducible and reversible enhancement of learning, memory, and long-term potentiation by genetic inhibition of calcineurin. Cell 104, 675–686. [DOI] [PubMed] [Google Scholar]
- Mansuy IM, Mayford M, Jacob B, Kandel ER, Bach ME, 1998. Restricted and regulated overexpression reveals calcineurin as a key component in the transition from short-term to long-term memory. Cell 92, 39–49. [DOI] [PubMed] [Google Scholar]
- Martin GR, 1981. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proceedings of the National Academy of Sciences 78, 7634–7638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER, 1996. Control of memory formation through regulated expression of a CaMKII transgene. Science 274, 1678–1683. [DOI] [PubMed] [Google Scholar]
- Mayford M, Wang J, Kandel ER, O’Dell TJ, 1995. CaMKII regulates the frequency-response function of hippocampal synapses for the production of both LTD and LTP. Cell 81, 891–904. [DOI] [PubMed] [Google Scholar]
- McQuown SC, Barrett RM, Matheos DP, Post RJ, Rogge GA, Alenghat T, Mullican SE, Jones S, Rusche JR, Lazar MA, Wood MA, 2011. HDAC3 is a critical negative regulator of long-term memory formation. J Neurosci 31, 764–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O’Dell TJ, Grant SG, 1998. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature 396, 433–439. [DOI] [PubMed] [Google Scholar]
- Miller VM, Paulson HL, Gonzalez-Alegre P, 2005. RNA interference in neuroscience: progress and challenges. Cell Mol Neurobiol 25, 1195–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsushima D, Ishihara K, Sano A, Kessels HW, Takahashi T, 2011. Contextual learning requires synaptic AMPA receptor delivery in the hippocampus. Proceedings of the National Academy of Sciences of the United States of America 108, 12503–12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molla KA, Yang Y, 2019. CRISPR/Cas-Mediated Base Editing: Technical Considerations and Practical Applications. Trends Biotechnol. [DOI] [PubMed] [Google Scholar]
- Moore CB, Guthrie EH, Huang MT-H, Taxman DJ, 2010. Short hairpin RNA (shRNA): design, delivery, and assessment of gene knockdown. Methods Mol Biol 629, 141–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan TH, 1911. Random segregation versus coupling in Mendelian inheritance. Science 34, 384–384. [DOI] [PubMed] [Google Scholar]
- Morris KV, 2011. The Use of Small Noncoding RNAs to Silence Transcription in Human Cells, in: Harper SQ (Ed.), RNA Interference Techniques. Humana Press, Totowa, NJ, pp. 39–57. [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L, 2007. A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605. [DOI] [PubMed] [Google Scholar]
- Nakajima H, Kubo T, Semi Y, Itakura M, Kuwamura M, Izawa T, Azuma YT, Takeuchi T, 2012. A rapid, targeted, neuron-selective, in vivo knockdown following a single intracerebroventricular injection of a novel chemically modified siRNA in the adult rat brain. J Biotechnol 157, 326–333. [DOI] [PubMed] [Google Scholar]
- Neuner SM, Wilmott LA, Hope KA, Hoffmann B, Chong JA, Abramowitz J, Birnbaumer L, O’Connell KM, Tryba AK, Greene AS, Savio Chan C, Kaczorowski CC, 2015. TRPC3 channels critically regulate hippocampal excitability and contextual fear memory. Behav Brain Res 281, 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve RL, Neve KA, Nestler EJ, Carlezon WA Jr., 2005. Use of herpes virus amplicon vectors to study brain disorders. Biotechniques 39, 381–391. [DOI] [PubMed] [Google Scholar]
- Nihongaki Y, Yamamoto S, Kawano F, Suzuki H, Sato M, 2015. CRISPR-Cas9-based photoactivatable transcription system. Chem Biol 22, 169–174. [DOI] [PubMed] [Google Scholar]
- Oikawa K, Odero GL, Platt E, Neuendorff M, Hatherell A, Bernstein MJ, Albensi BC, 2012. NF-kappaB p50 subunit knockout impairs late LTP and alters long term memory in the mouse hippocampus. BMC Neurosci 13, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oitzl MS, Reichardt HM, Joels M, de Kloet ER, 2001. Point mutation in the mouse glucocorticoid receptor preventing DNA binding impairs spatial memory. Proceedings of the National Academy of Sciences of the United States of America 98, 12790–12795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y, 1997. ‘Green mice’as a source of ubiquitous green cells. FEBS letters 407, 313–319. [DOI] [PubMed] [Google Scholar]
- Orban PC, Chui D, Marth JD, 1992. Tissue- and site-specific DNA recombination in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America 89, 6861–6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parr-Brownlie LC, Bosch-Bouju C, Schoderboeck L, Sizemore RJ, Abraham WC, Hughes SM, 2015. Lentiviral vectors as tools to understand central nervous system biology in mammalian model organisms. Front Mol Neurosci 8, 14–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Pinera P, Jones MF, Lal A, Lu TK, 2015. Putting Non-coding RNA on Display with CRISPR. Mol Cell 59, 146–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters M, Bletsch M, Catapano R, Zhang X, Tully T, Bourtchouladze R, 2009. RNA interference in hippocampus demonstrates opposing roles for CREB and PP1alpha in contextual and temporal long-term memory. Genes Brain Behav 8, 320–329. [DOI] [PubMed] [Google Scholar]
- Pfeifer A, Brandon EP, Kootstra N, Gage FH, Verma IM, 2001. Delivery of the Cre recombinase by a self-deleting lentiviral vector: efficient gene targeting in vivo. Proc. Natl. Acad. Sci. U. S. A 98, 11450–11455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plath N, Ohana O, Dammermann B, Errington ML, Schmitz D, Gross C, Mao X, Engelsberg A, Mahlke C, Welzl H, Kobalz U, Stawrakakis A, Fernandez E, Waltereit R, Bick-Sander A, Therstappen E, Cooke SF, Blanquet V, Wurst W, Salmen B, Bosl MR, Lipp HP, Grant SG, Bliss TV, Wolfer DP, Kuhl D, 2006. Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron 52, 437–444. [DOI] [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, Graham DB, Jhunjhunwala S, Heidenreich M, Xavier RJ, Langer R, Anderson DG, Hacohen N, Regev A, Feng G, Sharp PA, Zhang F, 2014. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polstein LR, Gersbach CA, 2015. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat Chem Biol 11, 198–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp A, Urbach A, Witte OW, Frahm C, 2009. Adult and embryonic GAD transcripts are spatiotemporally regulated during postnatal development in the rat brain. PLoS One 4, e4371–e4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasher DC, Eckenrode VK, Ward WW, Prendergast FG, Cormier MJ, 1992. Primary structure of the Aequorea victoria green-fluorescent protein. Gene 111, 229–233. [DOI] [PubMed] [Google Scholar]
- Pratt T, Sharp L, Nichols J, Price DJ, Mason JO, 2000. Embryonic stem cells and transgenic mice ubiquitously expressing a tau-tagged green fluorescent protein. Dev Biol 228, 19–28. [DOI] [PubMed] [Google Scholar]
- Ramamoorthi K, Fropf R, Belfort GM, Fitzmaurice HL, McKinney RM, Neve RL, Otto T, Lin Y, 2011. Npas4 regulates a transcriptional program in CA3 required for contextual memory formation. Science 334, 1669–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez S, Liu X, Lin PA, Suh J, Pignatelli M, Redondo RL, Ryan TJ, Tonegawa S, 2013. Creating a false memory in the hippocampus. Science 341, 387–391. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F, 2013. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijmers L, Mayford M, 2009. Genetic control of active neural circuits. Front Mol Neurosci 2, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijmers LG, Perkins BL, Matsuo N, Mayford M, 2007. Localization of a stable neural correlate of associative memory. Science 317, 1230–1233. [DOI] [PubMed] [Google Scholar]
- Riond JL, Riviere JE, 1988. Pharmacology and toxicology of doxycycline. Vet. Hum. Toxicol 30, 431–443. [PubMed] [Google Scholar]
- Robb GB, Brown KM, Khurana J, Rana T.M.J.N.s., biology, m., 2005. Specific and potent RNAi in the nucleus of human cells. 12, 133. [DOI] [PubMed] [Google Scholar]
- Robbins PD, Ghivizzani SC, 1998. Viral Vectors for Gene Therapy. Pharmacology & Therapeutics 80, 35–47. [PubMed] [Google Scholar]
- Rocha-Martins M, Cavalheiro GR, Matos-Rodrigues GE, Martins RA, 2015. From gene targeting to genome editing: transgenic animals applications and beyond. Anais da Academia Brasileira de Ciências 87, 1323–1348. [DOI] [PubMed] [Google Scholar]
- Sandlesh P, Juang T, Safina A, Higgins MJ, Gurova KV, 2018. Uncovering the fine print of the CreERT2-LoxP system while generating a conditional knockout mouse model of Ssrp1 gene. PLoS One 13, e0199785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarno E, Robison AJ, 2018. Emerging role of viral vectors for circuit-specific gene interrogation and manipulation in rodent brain. Pharmacology, biochemistry, and behavior 174, 2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaguri H, Nilsson P, Hashimoto S, Nagata K, Saito T, De Strooper B, Hardy J, Vassar R, Winblad B, Saido TC, 2017. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J 36, 2473–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer B, 1998. Inducible gene targeting in mice using the Cre/lox system. Methods 14, 381–392. [DOI] [PubMed] [Google Scholar]
- Savell KE, Bach SV, Zipperly ME, Revanna JS, Goska NA, Tuscher JJ, Duke CG, Sultan FA, Burke JN, Williams D, Ianov L, Day JJ, 2019. A Neuron-Optimized CRISPR/dCas9 Activation System for Robust and Specific Gene Regulation. eNeuro 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EE, Taylor DS, Prigge JR, Barnett S, Capecchi MR, 2000. Illegitimate Cre-dependent chromosome rearrangements in transgenic mouse spermatids. Proc. Natl. Acad. Sci. U. S. A 97, 13702–13707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonhuber N, Seidler B, Schuck K, Veltkamp C, Schachtler C, Zukowska M, Eser S, Feyerabend TB, Paul MC, Eser P, Klein S, Lowy AM, Banerjee R, Yang F, Lee CL, Moding EJ, Kirsch DG, Scheideler A, Alessi DR, Varela I, Bradley A, Kind A, Schnieke AE, Rodewald HR, Rad R, Schmid RM, Schneider G, Saur D, 2014. A next-generation dual-recombinase system for time- and host-specific targeting of pancreatic cancer. Nat. Med 20, 1340–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzberg PL, Goff SP, Robertson EJ, 1989. Germ-line transmission of a c-abl mutation produced by targeted gene disruption in ES cells. Science 246, 799–803. [DOI] [PubMed] [Google Scholar]
- Selcher JC, Nekrasova T, Paylor R, Landreth GE, Sweatt JD, 2001. Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learning. Learn Mem 8, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serganova I, Blasberg R, 2005. Reporter gene imaging: potential impact on therapy. Nucl Med Biol 32, 763–780. [DOI] [PubMed] [Google Scholar]
- Serita T, Fukushima H, Kida S, 2017. Constitutive activation of CREB in mice enhances temporal association learning and increases hippocampal CA1 neuronal spine density and complexity. Sci Rep 7, 42528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechner DM, Hacisuleyman E, Younger ST, Rinn JL, 2015. Multiplexable, locus-specific targeting of long RNAs with CRISPR-Display. Nat Methods 12, 664–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu G, Kramar EA, Lopez AJ, Huynh G, Wood MA, Kwapis JL, 2018. Deleting HDAC3 rescues long-term memory impairments induced by disruption of the neuron-specific chromatin remodeling subunit BAF53b. Learn Mem 25, 109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Paylor R, Wehner JM, Tonegawa S, 1992. Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science 257, 206–211. [DOI] [PubMed] [Google Scholar]
- Sinnett SE, Gray S.J.J.D.m., 2017. Recent endeavors in MECP2 gene transfer for gene therapy of Rett syndrome. 24, 153–159. [PubMed] [Google Scholar]
- Smith KS, Bucci DJ, Luikart BW, Mahler SV, 2016. DREADDS: Use and application in behavioral neuroscience. Behav Neurosci 130, 137–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strekalova T, Zorner B, Zacher C, Sadovska G, Herdegen T, Gass P, 2003. Memory retrieval after contextual fear conditioning induces c-Fos and JunB expression in CA1 hippocampus. Genes Brain Behav 2, 3–10. [DOI] [PubMed] [Google Scholar]
- Sultan S, Gebara E, Toni N, 2013. Doxycycline increases neurogenesis and reduces microglia in the adult hippocampus. Front Neurosci 7, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Schaffer DV, 2018. Engineered viral vectors for functional interrogation, deconvolution, and manipulation of neural circuits. Curr Opin Neurobiol 50, 163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Fukushima H, Mukawa T, Toyoda H, Wu LJ, Zhao MG, Xu H, Shang Y, Endoh K, Iwamoto T, Mamiya N, Okano E, Hasegawa S, Mercaldo V, Zhang Y, Maeda R, Ohta M, Josselyn SA, Zhuo M, Kida S, 2011. Upregulation of CREB-mediated transcription enhances both short- and long-term memory. J Neurosci 31, 8786–8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka KZ, Pevzner A, Hamidi AB, Nakazawa Y, Graham J, Wiltgen BJ, 2014. Cortical representations are reinstated by the hippocampus during memory retrieval. Neuron 84, 347–354. [DOI] [PubMed] [Google Scholar]
- Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD, 2014. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 159, 635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayler KK, Tanaka KZ, Reijmers LG, Wiltgen BJ, 2013. Reactivation of neural ensembles during the retrieval of recent and remote memory. Curr Biol 23, 99–106. [DOI] [PubMed] [Google Scholar]
- Thomas KR, Capecchi MR, 1987. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 51, 503–512. [DOI] [PubMed] [Google Scholar]
- Thomas KR, Folger KR, Capecchi MR, 1986. High frequency targeting of genes to specific sites in the mammalian genome. Cell 44, 419–428. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S, 1996. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 87, 1327–1338. [DOI] [PubMed] [Google Scholar]
- Valny M, Honsa P, Kirdajova D, Kamenik Z, Anderova M, 2016. Tamoxifen in the Mouse Brain: Implications for Fate-Mapping Studies Using the Tamoxifen-Inducible Cre-loxP System. Front. Cell. Neurosci 10, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel-Ciernia A, Matheos DP, Barrett RM, Kramar EA, Azzawi S, Chen Y, Magnan CN, Zeller M, Sylvain A, Haettig J, Jia Y, Tran A, Dang R, Post RJ, Chabrier M, Babayan AH, Wu JI, Crabtree GR, Baldi P, Baram TZ, Lynch G, Wood MA, 2013. The neuron-specific chromatin regulatory subunit BAF53b is necessary for synaptic plasticity and memory. Nature neuroscience 16, 552–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel Ciernia A, Kramar EA, Matheos DP, Havekes R, Hemstedt TJ, Magnan CN, Sakata K, Tran A, Azzawi S, Lopez A, Dang R, Wang W, Trieu B, Tong J, Barrett RM, Post RJ, Baldi P, Abel T, Lynch G, Wood MA, 2017. Mutation of neuron-specific chromatin remodeling subunit BAF53b: rescue of plasticity and memory by manipulating actin remodeling. Learn. Mem 24, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vooijs M, Jonkers J, Berns A, 2001. A highly efficient ligand-regulated Cre recombinase mouse line shows that LoxP recombination is position dependent. EMBO Rep 2, 292–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh NC, Kenney LL, Jangalwe S, Aryee KE, Greiner DL, Brehm MA, Shultz LD, 2017. Humanized Mouse Models of Clinical Disease. Annu Rev Pathol 12, 187–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters BJ, Azam AB, Gillon CJ, Josselyn SA, Zovkic IB, 2015. Advanced In vivo Use of CRISPR/Cas9 and Anti-sense DNA Inhibition for Gene Manipulation in the Brain. Front Genet 6, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters BJ, Mercaldo V, Gillon CJ, Yip M, Neve RL, Boyce FM, Frankland PW, Josselyn SA, 2017. The Role of The RNA Demethylase FTO (Fat Mass and Obesity-Associated) and mRNA Methylation in Hippocampal Memory Formation. Neuropsychopharmacology 42, 1502–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Wang X-W, Peng X-C, Xiang Y, Song S-B, Wang Y-Y, Chen L, Xin VW, Lyu Y-N, Ji J, Ma Z-W, Li C-B, Xin H-W, 2018a. CRISPR/Cas9 genome editing technology significantly accelerated herpes simplex virus research. Cancer Gene Therapy 25, 93–105. [DOI] [PubMed] [Google Scholar]
- Wang H, La Russa M, Qi LS, 2016. CRISPR/Cas9 in Genome Editing and Beyond. Annu Rev Biochem 85, 227–264. [DOI] [PubMed] [Google Scholar]
- Wang KH, Majewska A, Schummers J, Farley B, Hu C, Sur M, Tonegawa S, 2006. In vivo two-photon imaging reveals a role of arc in enhancing orientation specificity in visual cortex. Cell 126, 389–402. [DOI] [PubMed] [Google Scholar]
- Wang Y, Hu Z, Ju P, Yin S, Wang F, Pan O, Chen J, 2018b. Viral vectors as a novel tool for clinical and neuropsychiatric research applications. General Psychiatry 31, e000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb WM, Sanchez RG, Perez G, Butler AA, Hauser RM, Rich MC, O’Bierne AL, Jarome TJ, Lubin FD, 2017. Dynamic association of epigenetic H3K4me3 and DNA 5hmC marks in the dorsal hippocampus and anterior cingulate cortex following reactivation of a fear memory. Neurobiol Learn Mem 142, 66–78. [DOI] [PubMed] [Google Scholar]
- White AO, Kramar EA, Lopez AJ, Kwapis JL, Doan J, Saldana D, Davatolhagh MF, Alaghband Y, Blurton-Jones M, Matheos DP, Wood MA, 2016. BDNF rescues BAF53b-dependent synaptic plasticity and cocaine-associated memory in the nucleus accumbens. Nat Commun 7, 11725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigler M, Silverstein S, Lee L-S, Pellicer A, Cheng Y. c., Axel R, 1977. Transfer of purified herpes virus thymidine kinase gene to cultured mouse cells. Cell 11, 223–232. [DOI] [PubMed] [Google Scholar]
- Winder DG, Mansuy IM, Osman M, Moallem TM, Kandel ER, 1998. Genetic and pharmacological evidence for a novel, intermediate phase of long-term potentiation suppressed by calcineurin. Cell 92, 25–37. [DOI] [PubMed] [Google Scholar]
- Wojno AP, Pierce EA, Bennett J, 2013. Seeing the light. Sci. Transl. Med 5, 175fs178. [DOI] [PubMed] [Google Scholar]
- Wold WSM, Toth K, 2013. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr Gene Ther 13, 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Liu Y, Zhu Y, Ding X, Yang Y, Guan JS, 2014. In vivo imaging of immediate early gene expression reveals layer-specific memory traces in the mammalian brain. Proceedings of the National Academy of Sciences of the United States of America 111, 2788–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Ying SX, Zhang GM, Li C, Cheng SY, Deng CX, Zhang YE, 2005. Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell 121, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Z, Marth JD, 2004. N-glycan branching requirement in neuronal and postnatal viability. Glycobiology 14, 547–558. [DOI] [PubMed] [Google Scholar]
- Yoo YE, Yoo T, Lee S, Lee J, Kim D, Han HM, Bae YC, Kim E, 2019. Shank3 Mice Carrying the Human Q321R Mutation Display Enhanced Self-Grooming, Abnormal Electroencephalogram Patterns, and Suppressed Neuronal Excitability and Seizure Susceptibility. Front Mol Neurosci 12, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B, Heidenreich M, Mohanraju P, Fedorova I, Kneppers J, DeGennaro EM, Winblad N, Choudhury SR, Abudayyeh OO, Gootenberg JS, Wu WY, Scott DA, Severinov K, van der Oost J, Zhang F, 2016. Multiplex gene editing by CRISPR–Cpf1 using a single crRNA array. Nat Biotechnol 35, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Greenhill S, Huang S, Silva TK, Sano Y, Wu S, Cai Y, Nagaoka Y, Sehgal M, Cai DJ, Lee YS, Fox K, Silva AJ, 2016. CCR5 is a suppressor for cortical plasticity and hippocampal learning and memory. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Ma B, Homer RJ, Zheng T, Elias JA, 2001. Use of the tetracycline-controlled transcriptional silencer (tTS) to eliminate transgene leak in inducible overexpression transgenic mice. J. Biol. Chem 276, 25222–25229. [DOI] [PubMed] [Google Scholar]
- Zijlstra M, Li E, Sajjadi F, Subramani S, Jaenisch R, 1989. Germ-line transmission of a disrupted β2microglobulin gene produced by homologous recombination in embryonic stem cells. Nature 342, 435. [DOI] [PubMed] [Google Scholar]