

Abstract
Protection from immunodeficiency virus challenge in nonhuman primates (NHPs) by a first-generation HIV broadly neutralizing antibody (bnAb) b12 has previously been shown to benefit from interaction between the bnAb and Fcγ receptors (FcγR) on immune cells. To investigate the mechanism of protection for a more potent second-generation bnAb currently in clinical trials, PGT121, we carried out a series of NHP studies. These studies included treating with PGT121 at a concentration at which only half of the animals were protected in order to avoid potential masking of FcγR effector function benefits by dominant neutralization and using a new variant that more completely eliminated all rhesus FcγR binding than earlier variants. In contrast to b12, which required FcγR binding for optimal protection, we concluded that PGT121-mediated protection is not augmented by FcγR interaction. Thus, for HIV passive antibody prophylaxis, these results, together with existing literature, emphasize the importance of neutralization potency for clinical antibodies, with effector function requiring evaluation for individual antibodies.
One Sentence Summary
Effector function is not required for a potent HIV envelope-specific broadly neutralizing antibody to protect nonhuman primates from mucosal SHIV infection.
Introduction
Evidence from nonhuman primate (NHP) models of human immunodeficiency virus (HIV) infection indicate that both virus neutralization and Fc-dependent effector functions can contribute to neutralizing Ab (nAb) protection against virus challenge (1, 2). These Fc-dependent effector functions include antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and activation of the complement system (C’). Protection against high-dose vaginal simian-human immunodeficiency virus (SHIV) in the NHP model by the first-generation HIV broadly neutralizing antibody (bnAb) b12 was most effective if neutralization was augmented by Fcγ receptor (FcγR) functions, as shown by reduced protection in NHPs treated with effector function-crippled variants of b12 (1). A b12 variant lacking complement -activation (KA variant) was as protective as the wild-type antibody, suggesting complement is not involved in b12-mediated protection. In animals treated prophylactically, treatment with wild-type or KA-variant b12 resulted in lower and delayed primary viremia compared to controls. However, a L234A/L235A (LALA) variant of b12, which lacks both complement and FcγR binding, was less protective, leading to more infected animals and higher primary viremia compared to wild-type and KA-breakthrough animals. These results implied that FcγR function contributed to protection. Of note, approximately half the animals in the study were protected by the LALA antibody, which implied that neutralization alone can be sufficient for protection mediated by b12. Further, low-dose repeated vaginal challenge in b12-treated NHPs yielded a comparable result to high-dose SHIV challenge (2).
Further NHP studies using FcγRNIa-binding-enhanced antibodies did not show an advantage relative to wild-type antibodies, suggesting that improved FcγRIIIa binding, often associated with enhanced ADCC, does not necessarily lead to improved protection (3). In humanized mice, FcγR-mediated functions, under certain conditions, increased anti-viral activities of bnAbs (4). Interestingly, the authors showed that no FcγR benefits are apparent at high bnAb concentration, suggesting that FcγR contribution might be of particular importance specifically when virus neutralization by bnAbs is not overwhelming. Of note, the humanized mice studies involve infection of cells before antibody is introduced and so are not strictly investigating prophylaxis. Very recently, two studies assessed the ability of variant bnAbs to impact established infection in NHPs. Modest effects of eliminating antibody effector function on viral clearance were observed, and enhanced FcγR binding led to decreased, rather than increased, viral clearance (5, 6).
PGT121 (7) is a second-generation highly potent bnAb, that is currently being evaluated for HIV prophylaxis clinical trials (8). PGT121 was also recently evaluated in the SHIV macaque model (7). In contrast to b12-LALA, PGT121-LALA afforded sterilizing immunity against cell-associated intravenous SHIVSF162P3 challenge, which was similar to wild-type PGT121, suggesting antibody effector functions may be redundant for PGT121-mediated protection (9). A number of differences exist between the conditions used for the published b12 and PGT121 studies, including (1) the former studies used free virus and mucosal challenge whereas the latter used cell-associated virus and intravenous challenge (2) PGT121 protection was measured at high bnAb titers (10), that from mouse studies (4) might mask benefits of bnAb interaction with FcγR (4). Finally, It has previously been found that LALA variants retain considerable binding to human FcγRI (11) that may have influenced the results observed should the finding hold for macaque FcRI.
Here, we carried out protection studies with PGT121 and its LALA variant at high bnAb dose as in Parsons et al. (9) and then at moderate bnAb concentration that may provide deeper insight into any FcγR requirements for protection, as suggested by murine studies (4). We also performed a protection study using a PGT121-LALAPG variant that yielded more complete abrogation of all FcγR binding activity.
Results
Effector functions do not contribute to PGT121 protection even at sub-protective doses.
To determine the effects of reduced effector function on the protective activity of the potent second-generation bnAb, PGT121, a passive immunization experiment was conducted by infusion of 1 mg/kg of each of the Abs PGT121, PGT121-LALA, or anti-Dengue control (DEN3) into antibiotic-treated, menstrual cycle-synchronized female Indian rhesus macaques. In comparison to b12 administered at 25 mg/kg, PGT121 was given at a dose of 1 mg/kg (9,10) to account for its lower neutralization IC50 (0.04 vs 0.005 μg/ml respectively). Following single high-dose vaginal challenge with SHIVSF162P3 (300 tissue culture infectious doses 50 [TCID50]), animals receiving the DEN3 control antibody readily became infected while groups of 5 animals given either PGT121 or PGT121-LALA were fully protected (Fig. 1).
Figure 1. Macaques are equally protected against single high-dose vaginal challenge with SHIVSF162P3 by transfer of 1mg/kg of PGT121 or PGT121-LALA.
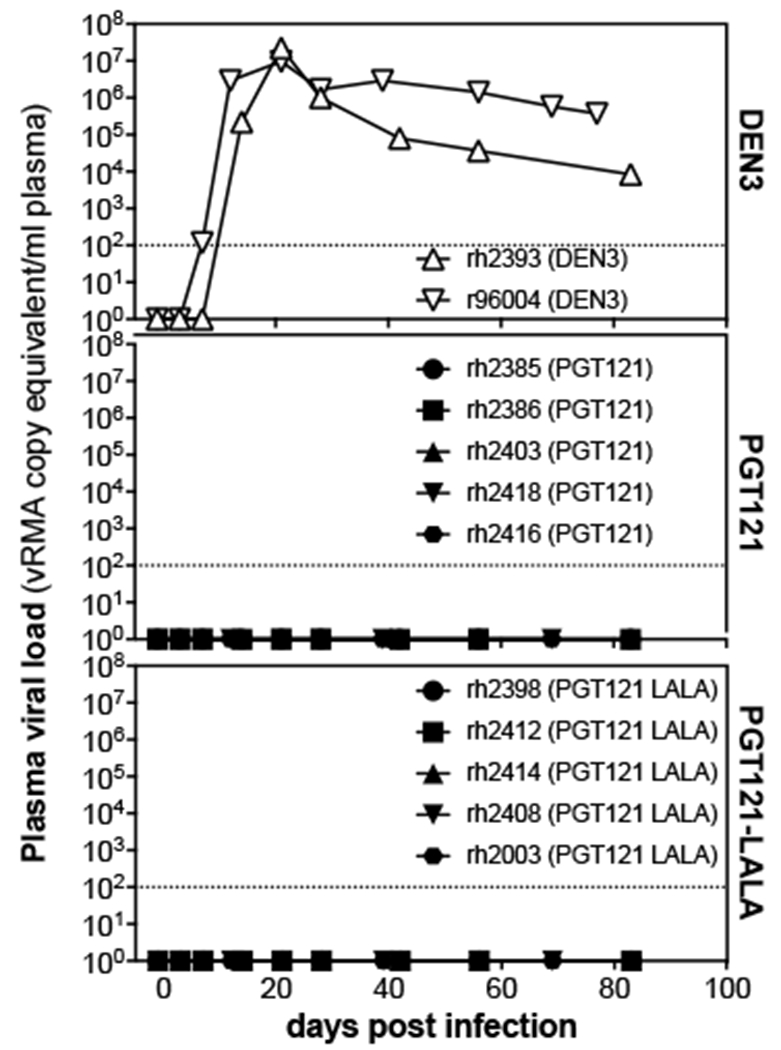
Viral loads after SHIVSF162P3 challenge following passive transfer of PGT121, PGT121-LALA, or a control antibody, DEN3. Animal groups were balanced for weight, FcγR and MHC alleles, and their menstrual cycles were synchronized by administration of DepoProvera 30 days before infection. Daily Benadryl antibiotic application courses were initiated on day −7 to treat latent vaginal infections or inflammation. Indicated antibodies were then infused intravenously one day before intravaginal administration of 300 TCID50 of SHIVSF162P3. Animal identifications are listed in the symbol legend, open symbols represent infected animals, closed symbols denote protected animals. Dashed lines indicate the assay limit of detection.
To investigate whether neutralization at 1mg/kg of PGT121 masked possible protection mediated by Fc effector function (fig. S1), a sub-protective dose of antibody (0.2 mg/kg) was administered in a second set of passive immunization experiments (IS-35, 49, 82, Fig. 2), as differences in protection between Fc-competent and Fc-crippled versions of PGT121 may be more prominent at this semi-protective concentration (10). However, again, there was no significant difference in protection conferred by Fc-competent and Fc-crippled PGT121 (p=0.15, Fig. 2); 7 out of 15 PGT121 and 11 out of 15 PGT121-LALA-immunized animals were protected, suggesting that the lack of Fc-effector function benefits in the previous experiment was not due to the high dose of the antibody.
Figure 2. Macaques are equivalently protected against single high-dose vaginal challenge with SHIVSF162P3 following administration of 0.2mg/kg of PGT121 or PGT121-LALA.
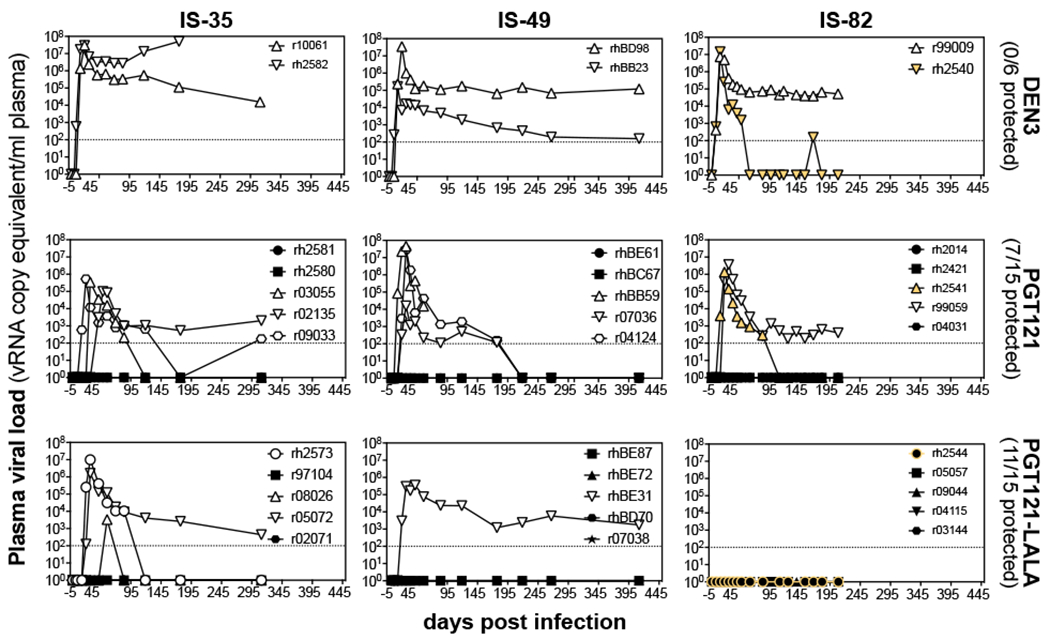
Animals were prepared as in Fig. 1. Low doses of antibodies (0.2mg/kg) were infused intravenously one day before intravaginal administration of 300 TCID50 of SHIVSF162P3. Viral titers are shown for each of the three experiments (IS-35, IS-49 and IS-82; columns). Animal identifications are listed in the symbol legend, open symbols represent infected animals, closed symbols denote protected animals. Symbols in yellow indicate three IS-82 animals that were found to be Chinese/Indian hybrid animals during major histocompatibility complex genotyping (rh2540, rh2541, rh2544).
Wild-type and KA variant b12 antibodies not only reduced the number of infections compared to an isotype control antibody, but also lowered primary viremia and delayed breakthrough in animals (1). For b12-LALA, primary viremia, summarized by the area under the curve (AUC), followed that of control animals more closely, while the AUCs of b12-KA and b12 breakthrough animals were more similar to each other. In this study, we detected no significant difference between PGT121 and PGT121-LALA in terms of primary viremia.
Unexpectedly, day 0 serum concentrations of PGT121 were 25-30% lower than PGT121 LALA (fig. S2) despite identical doses of antibodies being infused in both groups. This difference may be caused by altered serum or tissue distributions of both antibodies. If the serum half-life was calculated including day 0, the apparent serum half-life differed between PGT121 (1.6 days) and PGT121-LALA (2.1 days, fig. S2). However, excluding day 0, both serum half-lives were comparable at 2.9 and 2.5 days for PGT121 and PGT121-LALA, respectively. This suggested the LALA variant diffused into tissue slower than wild-type antibody, yet offered comparable or better protection.
Similar to previous experiments performed with the bnAb 10-1074 (12), another V3-glycan specific antibody, a potential effect arising from PGT121 administration was observed in that 5 animals receiving either PGT121 or PGT121-LALA spontaneously controlled viremia after 90 days, while we did not observe spontaneous control in DEN3 animals. Of note, Chinese/Indian hybrid animals (rh2540, rh2541, rh2544), purchased as Indian rhesus macaques but identified as hybrids during major histocompatibility complex (MHC) and FcγR genotyping, also spontaneously controlled infection after 45 days, even after receiving DEN3.
To determine whether protection across the studies was considered sterile, and to determine the mechanism of the spontaneous control, two controller animals (r04124 and r07036), three protected PGT121-LALA animals (r07038, rhBD70, rhBE72), and two protected PGT121 recipient animals (rhBC67 and rhBE61) were depleted of CD8+ lymphocytes via antibody administration 482 days post infection. No viremia was detected in protected animals. Also, no signs of T cell priming were found in protected animals by tetramer or intracellular cytokine staining of peripheral blood mononuclear cells (PBMCs) from IS-35. In contrast, T cell priming was found in all infected animals evaluated (table S1). Spontaneous controller PGT121-treated animals, r04124 and r07036, became viremic within 3 days following CD8+ lymphocyte depletion (fig. S3). Animal r04124 spontaneously lost viral control around the time of CD8+ lymphocyte depletion yet displayed exacerbated viremia as a result of the depletion. Although this experiment only employed a very limited number of animals, it provides evidence that virus persisted in both animals for over 200 days and indicates that the virus may have been held in check by CD8+ effector cells.
More complete ablation of rhesus FcγR-binding does not affect protection afforded by PGT121.
It was reported that LALA variant IgGs can retain binding to human FcγRI (11). In order to investigate PGT121-LALA binding to rhesus FcγRs, the respective soluble proteins were recombinantly expressed and antibody binding monitored by ELISA (Fig. 3A). PGT121-LALA retained considerable binding to rhesus FcγRI, and weak binding to rhesus FcγRIIa. The latter was sufficient to trigger uptake of opsonized beads by THP-1 cells but was not sufficient to mediate ADCC (Fig. 3B–D). A variant of PGT121 containing a P329G substitution in addition to the L234A/L235A modification was prepared (LALAPG) and was found to abolish all binding to macaque FcγRs, including FcγRI and II (Fig. 3A), and abolished residual bead-phagocytosis (Fig. 3B). This variant retained all other properties of the antibody, including its serum half-life (11, 13).
Figure 3. Differences in effector function activities of PGT121 and b12 fail to provide an explanation for their differing in vivo behaviour.
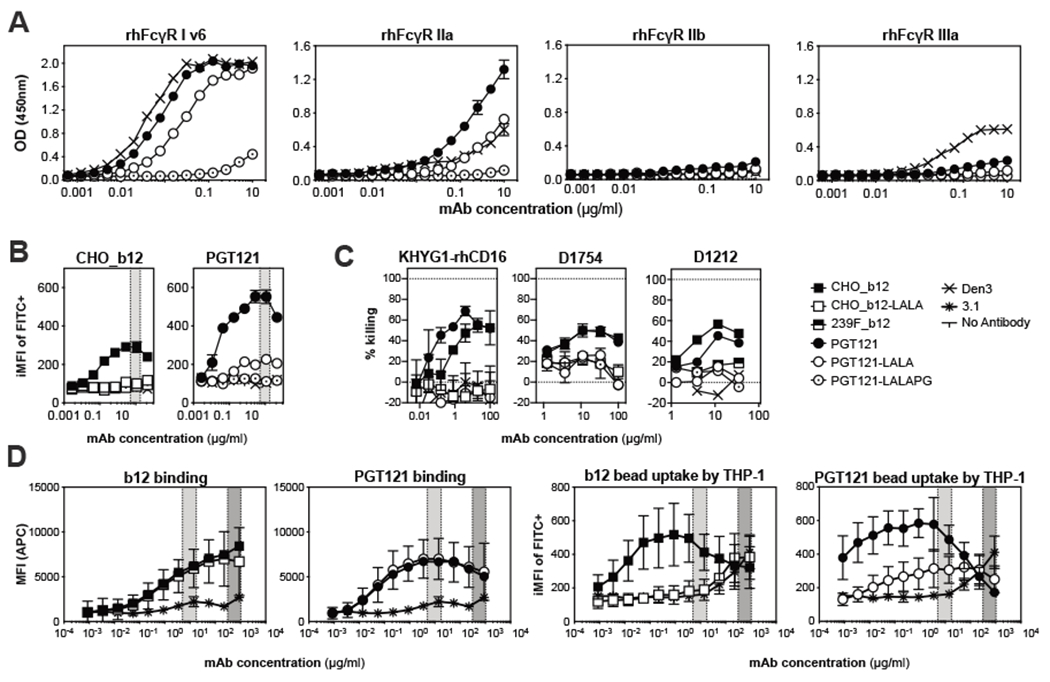
(A) Graphs indicate binding of Fc-variants of PGT121 to recombinantly expressed rhesus FcγRs. Plates were coated with recombinant soluble FcγR at 5μg/ml before titrated amounts of the indicated antibodies were allowed to bind. Bound antibodies were then detected by peroxidase labelled F(ab′)2 anti-human F(ab′)2. Mean and SD from two technical replicates are displayed. OD: optical density (B) THP-1 cell phagocytosis of HIVJR-FL envelope coated beads was compared across PGT121 and b12 and mutants. Titrated amounts of both the wild-type and effector-crippled version of the indicated ultracentifuged bnAbs were used to opsonize HIVJR-FL NFL TD CC+ trimer-coated 1 μm fluorescent beads. THP-1 cells were then added to antibody and bead mixtures and incubated overnight at 37°C. Opsonization was evaluated by flow cytometry and the integrated mean fluorescence intensity (iMFI) was calculated. Data represents mean and SD from two replicates, gray areas indicate the PGT121 serum concentration range in the NHP protection experiments. (C) NK cell-mediated ADCC activities of b12 and PGT121 were compared in vitro. SHIVSF162P3 -infected CEM.NKR.CCR5+Luc+.1c12 cells were incubated with the indicated antibodies and then used as target for KHYG-rhCD16 cells (left) or primary human NK cells (right). The donor ID for the human NK cells is indicated above the panel. Data represents mean and SD from two replicates. (D) THP-1 cell binding and phagocytosis of HIVJR-FL NFL coated beads was evaluated at concentrations reflective of in vivo challenge studies. Data represent mean and standard deviation from 4 independent experiments. Anti-influenza HA mAb 3.1 (29) was used as a negative control and data represent two replicates. Light and dark gray areas indicate the serum concentration range of the respective PGT121 and b12 antibodies in the relevant NHP protection experiments.
Protection afforded by PGT121-LALAPG was then assessed in 15 animals receiving 0.2mg/kg PGT121-LALAPG, 3 animals receiving PGT121, and 5 animals receiving control DEN3 (Fig. 3C). The LALAPG-variant was found at similar serum titers at the day of infection and had a serum half-life of 2.3 days, similar to PGT121. Further, PGT121-LALAPG protected 12 out of 15 animals from a single high-dose vaginal SHIVSF162P3 infection (Fig. 4A). Comparing these data with the data above, the results suggest that the LALAPG variant performs comparably to wild-type PGT121 and that effector function is not contributing to antibody protection.
Figure 4. PGT121-LALAPG variant is equivalently protective as PGT121 or PGT121-LALA.
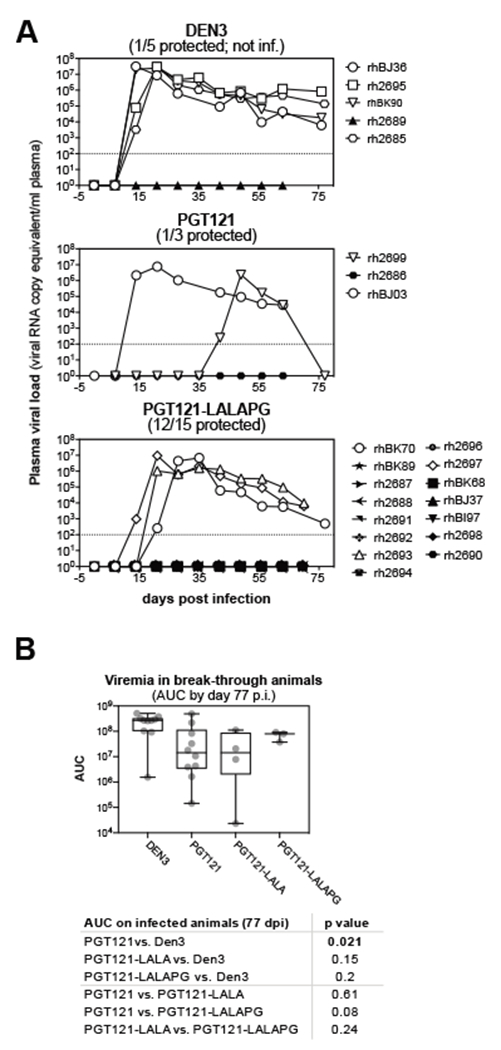
(A) Viral loads after single high-dose SHIVSF162P3 vaginal challenge following transfer of 0.2mg/kg of the indicated antibody are plotted (experiment IS-108/109). Antibodies (0.2mg/kg) were infused intravenously one day before intravaginal administration of 300 TCID50 of SHIVSF162P3. Animal identifications are listed in the symbol legend, open symbols represent infected animals, closed symbols denote protected animals. Dashed lines indicate the limit of detection of the assay. (B) Meta-analysis of viremia in break-through animals. Area under the curve values were integrated for viremic animals from day 0 though day 77 post infection. Data is displayed as individual values (gray dots), mean (central line) interquartile range (box) and minimum to maximum range (whiskers). Indicated p values were determined by Welch two sample t-tests.
A meta-analysis across all individual experiments based on day 0 antibody concentration and neutralizing data binned by outcome revealed that protected animals had significantly higher PGT121 variant concentrations (using Welch two sample t-tests, p=0.000603) or neutralizing titers (p= 0.0008) compared to infected animals (fig. S2D). However, differences in day 0 neutralizing titers were not different for all individual experiments (fig. S2C). When viremia in the breakthrough animals was analyzed, it was found that PGT121-treated animals had significantly lower viremia (p=0.021, Fig. 4B) compared to DEN3-treated animals. In contrast, viremia in PGT121-LALA treated animals was not significantly different as compared to DEN3-treated animals (p=0.15, Fig. 4B). Similarly, PGT121-LALAPG-treated animals did not significantly differ in AUC compared to DEN3-treated control animals (p=0.2), and viremia in these animals was also not different from PGT121 (p=0.08) or PGT121-LALA (p=0.24)-treated animals in Welch two sample t-tests (Fig. 4B). However, it should be noted that the number of break-through animals reported here is small, thus limiting the power of these analyses.
Differences in effector function activities of PGT121 and b12 fail to provide an explanation for their differing in vivo behavior.
To explore potential reasons for the observed differences between b12 and PGT121 in terms of the contribution of effector function towards protection, we investigated effector activities of the two bnAbs. We recognized that it would be difficult to reproduce the conditions in terms of effector and target cells present in the exposed animals, but we considered it worthwhile to investigate a number of assays in vitro. One key factor that we took into account in this study was that typical serum concentrations used in b12 experiments at challenge (310-1029 μg/ml) were much higher than those used for PGT121 (5-17.6 μg/ml).
We therefore investigated the ability of the human NK cell line KHYG-1 expressing rhesus CD16 (14) to kill SHIVSF162P3-infected CEM.NKR CCR5+Luc+.1c12 target cells, a subclone of the widely used CEM.NKR CCR5+Luc+ target cell line (14). As shown in Fig. 3C, PGT121 was somewhat more effective than b12. However, making allowance for the differing antibody concentrations present in vivo, the two bnAbs might have been predicted to be approximately equally effective in our experiments. The LALAPG antibodies were ineffective at promoting ADCC. Therefore, this ADCC assay did not correlate with the differences in protection described between b12 and PGT121 in NHP studies.
We also performed ADCC assays using SHIVSF162P3-infected CEM.NKR CCR5+Luc+.1c12 target cells with human primary NK effector cells. b12 and PGT121 displayed comparable NK cell-mediated ADCC activities at typical assay concentrations (<35μg/ml, Fig. 3C). At high antibody concentrations present in b12 animals at the day of infection (>300μg/ml), there was an in vitro prozone effect observable for all antibodies tested (fig. S4A). Overall, the results from assays using human primary NK cells were similar to those for the rhesus CD16 human KHYG-rhCD16 NK cell line.
We then assessed uptake of HIVJR-FL NFL TD CC+ trimer-coated beads by THP-1 cells, a human monocyte cell line, as a measure of ADCP (Fig. 3B and D). HIVJR-FL was chosen instead of SHIVSF162P3 as there is not currently a structurally stable soluble SHIVSF162P3 envelope trimer and the inhibitory concentrations of both bnAbs are comparable for HIVJR-FL. These in vitro studies revealed that PGT121 and b12 were equally effective at binding and inducing ADCP (Fig. 3D).
We hypothesized that there may be differences in glycosylation of b12 and PGT121 because b12 and b12-LALA were produced in Chinese Hamster Ovary cells (ATCC CCL-61) while all PGT121 variants were produced in human embryonic kidney cells (FreeStyle 293-F Cells; fig. S5). Using mass spectrometry, we found higher frequencies of galactosylation in CHO-produced b12. However, we observed similar frequencies of fucosylation across the antibodies and no difference in ADCP in the trimer-coated beads assay between b12 produced in CHO or 293-F cells. These data suggest that alternative glycosylation may not contribute to differences observed in ADCP. However, we observed that 293F-produced b12 displayed lower NK cell killing compared to its CHO-produced counterpart (Fig. 3C and fig S6), suggesting that this slight difference in glycosylation could contribute to differing ADCC activities (15).
Discussion
It has been long known that antibodies can make use of effector functions mediated through Fc receptors as well as neutralization in protection against viruses (16). For example, FcγR effector functions have been shown to be important for protection against alphavirus infection (17) and protection by heterosubtypic stem-specific antibodies in the context of influenza A virus infection (18). For HIV, studies with a first generation bnAb in NHPs implied that effector functions could contribute to protection, both in terms of sterilizing immunity and in delaying and reducing the magnitude of primary viremia and morbidity (1, 2). Humanized mouse model studies (4) lent support to this view, although the model did not strictly describe prophylactic activity. However, a recent study using a second generation potent bnAb, PGT121, found no evidence for a contribution of effector function to protection from cell-associated virus (19). There were notable differences between that study and the earlier ones, including the route of challenge and form of the challenge virus. In addition, the study was carried out under conditions of high bnAb concentration that may mask contributions of effector functions to protection (4). Concerns have also been expressed that the antibody variant (LALA) typically used to assess the role of effector functions may not completely eliminate all such functions (11). Accordingly, we carried out an extensive study of PGT121 protection to address some of these questions.
Our central finding is that effector function does not contribute to PGT121 protection against single high-dose vaginal SHIV challenge, even at semi-protective concentrations when effector functions might have the greatest opportunity to contribute. The finding with a high Ab dose was in agreement with that of Parsons et al (19), despite aforementioned differences, and is supported by correlation of protection with day 0 serum neutralizing titers. Further, these findings were confirmed using a variant of PGT121 (LALAPG) in which any residual binding to rhesus FcγRs had been eliminated.
We investigated possible in vitro correlates of the different mechanisms of protection mediated by bnAbs b12 and PGT21, despite limitations associated with in vitro assays. These assays provided no clear indication of a difference between b12 and PGT121 that would explain their distinct protection profiles. It is notable that, in previous studies, b12 was used at much higher doses than PGT121, producing much higher serum Ab concentrations in vivo. Yet, when the ability of PGT121 and b12 to induce ADCC or ADCP were compared in vitro, we found no difference between the bnAbs.
Our study does have some limitations. In order to compare PGT121 with the earlier extensive data on b12 protection, we endeavored to keep similar conditions. The time period between administration of the antibodies and virus challenge was maintained at one day although more recent data indicates that a longer time period may lead to better tissue penetrance of antibody. We also used high-dose virus challenge, again for reasons of consistency, although low-dose repeated challenge may have given better statistical power. However, the low-dose repeated challenge model may be associated with greater macaque anti-human antibody problems. A further limitation, referred to above, is the artificiality of many of the best available effector function assays in attempts to explain differences between antibodies. For instance, antibody phagocytosis of protein-coated beads in vitro may be a very poor predictor of phagocytosis of virions in vivo.
The results described to date suggest it will be difficult to generalize confidently with respect to the contributions of antibody effector function in the context of HIV bnAb-mediated protection. It may well be that the more potent second generation bnAbs are less dependent on effector function than the first generation. Alternatively, the site of vulnerability recognized by the bnAb might be critical. A further possibility is that an Fc-dependent effector activity that is not well modeled by the current in vitro assays is essential for bnAb function. Investigation of protection with more bnAbs is required to definitively distinguish these possibilities. Given current information, it is prudent that clinical passive antibody and HIV vaccine efforts emphasize neutralizing potency while more data is gathered on effector functions.
Methods
Study design
The research aim of this study was to test whether a second generation, highly potent bnAb had the same dependence on effector function for protection as a first generation, less potent bnAb. Another hypothesis was whether a contribution of effector function could be masked by high concentrations of bnAb in vivo. To this end, the impacts of antibody dose or functional knockout mutations (L234A, L235A, with or without P329G) on passive immunization protection of NHPs from SHIV challenge infection were assessed. NHP group sizes were determined in consultation with statisticians to maximize statistical power of group comparisons with the minimum number of animals, resulting in group sizes between 3 (DEN3) and 15 animals (PGT121-LALAPG). For logistic reasons the animal experiments were split into two or three repetitions (IS-35, IS-49, IS-82; IS-108, IS-109). Experiments were terminated after 78 days to ensure delayed infection was appropriately ascertained. Data collection for IS-35, IS-49, and IS-82 were extended for a longer period of time to assess potential vaccinal effects. In addition, animals were euthanized if they showed signs of distress.
Animals
70 adult, female, captive-bred rhesus macaques (Macaca mulatta) of Indian origin were obtained from the colonies of Wisconsin National Primate Research Center (WNPRC), or California National Primate Research Center (CNPRC). The median age was 10.3 years (range of 4-21.8 years) with median body weight of 7.66 kg (range of 4.67-14.42 kg). Animals were distributed so MHC-I and FCGR alleles (determined by the Genetics Services unit of the WNPRC) were represented as evenly as possible in the experimental groups.
All animals were cared for at the WNPRC according to Weatherall report guidelines under a protocol approved by the University of Wisconsin - Madison Animal Care and Use Committee and managed according to the research animal husbandry program of the WNPRC. A macaque nutritional plan was closely monitored by animal research technicians and followed the recommendations of the National Research Council (2050 Teklad Global 20% Protein Primate Diet twice daily). The diet was supplemented with a variety of fruits, vegetables, and dairy products as part of the environmental enrichment program.
SHIV-challenged animals were single-housed and twice-daily evaluated for signs of pain, illness, and stress by observing appetite, stool, typical behavior, and physical condition by the staff of the Animal Services Unit. Animals exhibiting distressed behaviors were reported to the Behavioral Management staff and managed accordingly. If any of the above parameters were found to be unacceptable, a member of the WNPRC veterinary staff was notified and their recommendations followed. All efforts were made to minimize suffering.
Intravenous antibody infusion, intravaginal SHIV challenge, and routine blood draws were performed under anesthesia using up to 7 mg/kg ketamine intramuscularly, and up to 0.03 mg/kg dexmedetomidine intramuscularly. Anesthesia was reversed at the conclusion of a procedure by up to 0.3 mg/kg atipamezole intravenously.
Euthanasia was performed at the end of the study or if an animal experienced conditions deemed distressful by one of the veterinarians at the WNPRC. Euthanasia was performed according to the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association (>50 mg/kg of sodium pentobarbital intravenously, preceded by > 15 mg/kg body weight ketamine intramuscularly).
SHIVSF162P3 challenge
Macaque menstrual cycles were synchronized with delivery of 30 mg medroxyprogesterone acetate intramuscularly to each animal 28 days before viral challenge to reduce the thickness of the vaginal epithelium for approximately 28-33 days. Lower reproductive tract inflammation may decrease the threshold for virus infection, thus animals received a seven-day course of broad-spectrum antibiotics prior to viral challenge (2.5mg/kg enrofloxacin intramuscularly, twice-daily). Anesthetized animals received 300 TCID50 of SHIVSF162P3 in the vaginal lumen atraumatically to initiate viral challenge.
Anti-HIV Env monoclonal Antibodies
The HIV bnAbs PGT121 (7) and b12 (20), and variants thereof, were expressed in Freestyle 293-F cells (PGT121, Thermo Fisher Scientific), or a stable CHO cell line (b12 variants, (1)). Supernatant was harvested after 6-7 days, clarified by centrifugation and filtration through 0.22 μm Rapid-Flow filter units (Nalgene). Antibodies were recovered using Protein G Sepharose Fast Flow (GE Healthcare) in columns, washed with Dulbecco’s phosphate buffered saline (DPBS), and eluted with 0.1 M glycine pH 2.5. Antibodies were concentrated and buffer exchanged into DPBS by ultrafiltration. LALA Fc mutations were introduced by site-directed mutagenesis using the Quikchange II mutagenesis kit (Agilent cat. 200523) and the P329G mutation was introduced with the Q5 Site-Directed Mutagenesis Kit (New England BioLabs cat. E0554S) according to the manufacturers’ instructions.
Soluble Env trimer production
HIV envelope trimers BG505 N332 SOSIP.664 v5.2 (21) and JRFL NFL TD CC+ (22) containing a C-terminal Avi tag were expressed in Freestlye 293-F cells by co-transfection with furin and purified using PGT145-affinity columns (23) and size exclusion chromatography (SEC). Intact trimer fractions were pooled, concentrated to 1–2 mg/ml and sterile filtered prior to aliquoting and freezing at −80°C.
Soluble rhesus FcγR production
The ectodomain genes of Fc gamma receptors from Rhesus Macaques (Macaca mulatta) were synthesized fused to a thrombin site, an avi- and a hexa-histidine tags (GeneArt) and cloned into the pcDNA3.4 mammalian expression vector. Proteins were expressed in Freestyle 293-F cells, purified by nickel nitrilotriacetic acid (Ni-NTA) Sepharose and SEC.
Biotinylation of soluble HIV envelope trimers and FcγRs
Avi-tagged recombinant proteins were enzymatically biotinylated using the BirA biotin-protein ligase standard reaction kit (Avidity) according to the manufacturer’s instructions.
Pseudovirus and replication competent virus production
SHIVSF162P3 envelope pseudotyped virus supernatant from transfected cells was sterile filtered 48-60 hours post transfection, ultra-centrifuged at 100,000 × g for 1 hour, resuspended in DMEM containing 10% fetal bovine serum (FBS) and frozen in aliquots at −80°C (24). Pseudovirus was titrated on TZMbl cells using luciferase expression as readout. Replication competent viruses used in ADCC assays were generated as above using a single plasmid expressing the complete SHIVSF162P3 genome transfected in HEK293T cells. Replication competent virus was titrated on CEM.NKR CCR5+Luc+.1c12 cells to determine optimal infection and viability for subsequent ADCC assays.
Detection of Env-specific antibodies in serum by ELISA
BG505.SOSIP.N332T-HIS (3μg/ml) in phosphate buffered saline (PBS)/1% bovine serum albumin (BSA)/0.05% Tween 20 (PBS/BT) was bound to ELISA plates pre-coated with anti-His mAb (2μg/ml in PBS) and pre-blocked with PBS/3% BSA, and washed. Sera were heat-inactivated (30 min, 56°C), serially diluted in PBS/BT and allowed to bind to Env-coated plates for 1 hour. After washing, bound serum antibodies were detected using alkaline phosphatase labelled goat antihuman IgG (1:2000 in PBS/BT, 1 hour) and 4-Nitrophenyl phosphate disodium salt substrate with PGT121 as a standard. Non-linear four parameter Hill Curve regression was performed using 450nm optical density (OD) data, Prism 7 and 8 software (Graphpad Software), and logarithmized x-values. Serum dilution giving half-maximal ODs at 450nm (EC50) was determined, and original antibody concentrations were calculated from PGT121 standards. In cases of incomplete titration curves, curve fitting was constrained with top values determined for the PGT121 standards to avoid curve fitting artifacts.
Evaluation of binding of Ig-variants to rhesus FcγRs by ELISA
Antibodies were serially diluted in PBS/1% BSA and allowed to bind to rhesus FcγR (5μg/ml in PBS) pre-coated and blocked (PBS/3% BSA) clear ELISA plates at room temperature for 1 hour. After washing, bound Ig was detected using horseradish peroxidase-labelled F(ab’)2 goat-anti human F(ab’)2 antibody (Jackson ImmunoResearch, 1:10,000 in PBS/1% BSA, 1 hour) and pre-mixed 3,3′,5,5′-Tetramethylbenzidine (TMB)-substrate (Thermo Fisher Scientific). Color reactions were stopped by the addition of 2N sulfuric acid and ODs were determined at 450nm.
Antibody Dependent Bead Phagocytosis (ADBP)
ADBP was performed as described (25). 1μm fluorescein isothiocyanate (FITC) containing neutravidin coated beads (Invitrogen) were incubated with 25 μg/ml enzymatically biotinylated soluble HIV envelope trimer (HIVJR-FL NFL TD CC+) in PBS/0.1% BSA. Antibody aggregates were sedimented immediately prior to the assay by ultracentifugation of endotoxin-free mAbs at 50,000 revolutions per minute (rpm) and 4°C for 15 min in TLA 100.3 rotor (Beckman Coulter). The top 10% of the supernatant volume was then transferred to a new container, and the bnAb concentration was determined by OD280 measurement. Beads were washed and incubated in 96 well round bottom plates (Corning) with serial bnAb (b12 or PGT121 and mutants, control DEN3) dilutions in DPBS/0.1% BSA. After 2 hours, 50,000 THP-1 cells were added to the beads and incubated at 37°C, 5% CO2, 80% humidity for 16-18 hours overnight. Cells were fixed by the addition of an equal volume of 4% formaldehyde, washed with DPBS containing 2% FCS and 1 mM EDTA, and analyzed on a BD FACS Lyric flow cytometer with a universal plate loader. Statistics (FITC mean fluorescence intensity [MFI], % FITC-positive cells) were calculated with FlowJo v10.4.0. The integrated MFI was calculated by multiplying the (% FITC+) x (FITC MFI) and graphed using Graphpad Prism v7.
Antibody Dependent Cellular Cytotoxicity (ADCC)
The CEM.NKR CCR5+Luc+ cell line (14) was subcloned by limiting dilution and screened for low spontaneous killing by primary human NK cells and high luciferase expression following infection. Subclone 1C12 was used for all subsequent experiments. CEM.NKR CCR5+Luc+.1c12 cells were infected with replication competent HIVJRFL or SHIVSF162P3. Three days later, target cells were opsonized with serial dilutions of HIV bnAbs or control Abs for 1 hour at 37°C. For NK ADCC assays, thawed human PBMCs from leukapheresis (Stemcell) were allowed to rest overnight in RPMI-1640 (Corning) plus 10% FBS (Omega Scientific), penicillin/streptomycin (Corning), and L-glutamine (Corning) (R10 media) before NK cells were isolated using human NK cell enrichment kits (Stemcell). Purity was confirmed by flow cytometry using fluorescent antibodies to CD14, CD16, and CD56 (BD BioSciences) according to the manufacturer’s instructions. 10,800 NK cells at 90 to 95% purity were added to 10,200 infected and opsonized CEM.NKR CCR5+Luc+.1c12 cells at a rate of about 20-40% infected. This provided an effector:infected target ratio of 2.4:1 – 5:1. Cells were incubated overnight. KHYG-1 killer assays were performed as described in (14). Uninfected CEM.NKR CCR5+Luc+.1c12 cells were used to determine baseline luciferase activity, which was subtracted from all values. Antibody-mediated specific killing of HIV-infected target cells was evaluated by measuring luciferase activity and normalizing to non-specific killing in control wells containing infected target cells co-cultured with effector cells but no HIV-specific Abs. The percent specific killing was calculated as percentage of max luciferase signal in wells with no antibody.
Glycoanalyses by mass spectrometry
Glycoanalyses were performed as described in (26). In brief, 100-150 mg of indicated antibody variants were buffer exchanged using Vivaspin 100 kDa columns, denatured, reduced, and alkylated by sequential 1 hour incubations at room temperature. The proteins were then buffer exchanged into 50 mM Tris/HCl, pH 8.0 using Vivaspin 3 kDa columns and aliquots were digested with trypsin or chymotrypsin (Mass Spectrometry Grade, Promega) at a ratio of 1:30 (w/w) for 16 hours at 37°C. The reactions were dried and peptides or glycopeptides were extracted using C18 Zip-tip (Merck Millipore) following the manufacturer’s protocol. The reaction mixture was loaded onto the tip and eluted with 50% acetonitrile, 0.1% trifluoracetic acid. Eluted peptides and glycopeptides were dried again and re-suspended in 0.1% formic acid prior to mass spectrometry analysis. An aliquot of peptides and glycopeptides was analyzed by liquid chromatography–mass spectrometry (LC-MS) with an Easy-nLC 1200 system coupled to an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific) using higher energy collisional dissociation (HCD) fragmentation. Peptides and glycopeptides were separated using an EasySpray PepMap RSLC C18 column (75 mm x 75 cm) with a 275 min. linear gradient consisting of 0%–32% acetonitrile in 0.1% formic acid over 240 minutes followed by 35 minutes of 80% acetonitrile in 0.1% formic acid. The flow rate was set to 200 nL/min. The spray voltage was set to 2.8 kV and the temperature of the heated capillary was set to 275°C. HCD collision energy was set to 50%. Byonic (Version 2.7) and Byologic software (Version 2.3; Protein Metrics Inc.) extracted data evaluated manually and each peptide was scored as true-positive when the correct b and y fragment ions were observed along with oxonium ions corresponding to the glycan identified. The relative abundance of unoccupied versus glycosylated sites was calculated using the extracted ion chromatograms for the true-positive peptides.
SHIVSF162P3 viral load quantitation
Viral RNA was isolated from plasma samples using the Maxwell 16 Viral Total Nucleic Acid Purification kit on the Maxwell 48RSC instrument (Promega) and quantified using a highly sensitive QRT-PCR assay (limit of detection 100 copies/ml) as previously described (27). Primer and probe sequences included: forward primer: 5’- GTCTGCGTCATCTGGTGCATTC, reverse primer: 5’-CACTAGCTGTCTCTGCACTATGTGTTTTG-3′ and probe: 5′-6-carboxyfluorescein-CTTCCTCAGTGTGTTTCACTTTCTCTTCTGCG-BHQ1-3′. Reactions were cycled at 37°C for 15 min, 50°C for 30 min, 95°C for 2 min followed by 50 cycles of 95°C for 15 seconds and 62°C for 1 min.
Neutralization assays
NHP serum collected at various timepoints after passive-immunization was heat inactivated (30 min, 56°C) and incubated for 1 hour at 37°C with pseudovirus, then transferred onto TZM-bl cells (10,000/well) in 96 well plates, in complete DMEM supplemented with 10% FBS (Omega Scientific), penicillin/streptomycin (Corning), L-glutamate (Corning), and 20 μg/mL Diethylaminoethyl (DEAE)-dextran. Supernatant was removed after a 48 hour incubation at 37°C, 5% CO2, 80% humidity and the cells lysed with lysis buffer (25mM Glycylglycine, 15mM MgSO4, 4mM EGTA tetrasodium salt, 5 ml/L Triton X) for 5 min at room temperature. Luciferase activity was measured by the addition of Bright-Glo luciferase-substrate (Promega), and the luminescence signal read using a BioTek Synergy 2 plate reader. Uninfected cells were used to correct for background luciferase expression.
CD8+ lymphocyte depletion
CD8α-expressing lymphocytes were transiently depleted in vivo by intravenous injection of the anti-CD8α monoclonal antibody (clone M-T807R1) in a single dose of 50 mg/kg of body weight. The antibody was provided by the National Institutes of Health Nonhuman Primate Reagent Resource, lot number LH16-12. The efficacy of CD8α depletion was monitored by flow cytometry.
Rhesus macaque MHC-I tetramer staining
Mamu-A*02, Mamu-B*08, and Mamu-B*17 MHC-I tetramers labelled with phycoerythrin (PE), allophycocyanin (APC), or Brilliant Violet (BV)-421 and bound to indicated SIV peptides used to quantify SIV-specific CD8+ T cells as described previously (28). All macaque MHC-I tetramers utilized in this study were obtained from the NIH Tetramer Core Facility. Approximately 800,000 PBMC were incubated in R10 medium with titrated amounts of each tetramer at room temperature for 45 min. Before adding the Mamu-B*17 tetramers to the respective tubes, the cells were pre-incubated with 50 nM of the protein kinase inhibitor Dasatinib for 30 min in order to improve tetramer staining. Following the 45 min tetramer incubation, cells were stained with fluorochrome-labeled mAbs directed against lineage markers CD3 (clone SP34-2; PerCP Cy5.5), CD8α (clone RPA-T8; BV785), CD14 (clone M5E2; BV510), CD16 (clone 3G8; BV510), and CD20 (clone 2H7; BV510) for 25 min at room temperature. A viability marker (ARD; Live/DEAD Fixable Aqua Dead Cell Stain; Life Technologies) was also included in this step (1:4,000 dilution of master stock prepared according to manufacturer’s instruction). Cells were washed with Wash Buffer (Dulbecco’s PBS with 0.1% BSA and 0.45 g/L NaN3) and fixed with PBS/2% paraformaldehyde (PFA) for 20 min at 4°C. Next, cells were washed and analyzed on a Special-Order BD LSRII flow cytometer equipped with 50-mW 405-nm (violet), 100-mW 488-nm (blue), and 30-mW 635-nm (red) lasers with FACSDIVA v6 software.
Data was gated on diagonally clustered singlets in height-vs-area plots for forward scatter height (FSC-H/FSC-A) and side scatter (SSC-H/SSC-A) in FlowJo. Singlets were further gated using a time gate between the 5th and 90th percentiles. T lymphocytes were selected as “Dump channel negative” (ARD−, CD14−, CD16−, CD20−) and CD3+ cells and the lymphocyte population was delineated based on FSC-A/SSC-A properties. Gates were then created to identify CD8+ cells and tetramer+ cells. All tetramer frequencies shown in this study correspond to percentages of live CD3+ CD8+ tetramer+ lymphocytes.
Intracellular cytokine staining (ICS) assay.
Frozen PBMCs were thawed, transferred to 37°C warm R10 medium, and pelleted at 530 x g for 5 min. The pellet was resuspended in R10 and centrifugation was repeated. Pools of peptides (15mers overlapping by 11 amino acids) spanning the SIVmac239 Gag, Vif, and Nef proteins were used for T cell stimulation. Peptides corresponding to SIV epitopes restricted by Mamu-B*08 were also used for T cell stimulation. The Gag peptides were split into two pools covering amino acids 1-263 or 248-510. The Vif (amino acids 1-214) and Nef (amino acids 1-263) peptides were each grouped into a separate pool. The final assay concentration of each 15mer was 1.0 μM. PBMCs were stimulated with the appropriate pools of SIVmac239 peptides in R10 containing co-stimulatory mAbs against CD28 (clone L293; BD Biosciences) and CD49d (clone 9F10; BD Pharmingen) for 9 hours at 37°C/5.0% CO2. A PE-conjugated mAb against CD107a (clone H4A3; PE; Biolegend) was also included in this step. Brefeldin A (BioLegend) and GolgiStop (BD Biosciences) were added to all tubes after 1 hour to inhibit protein transport. The tubes were refrigerated at 4°C at the end of this 9 hour incubation. The surface staining was performed as described above, using the same ARD and mAbs against CD14, CD16, CD20. The surface staining mAb cocktail also included mAbs against CD4 (clone OKT4; BioLegend) and CD8 (clone RPA-T8; BioLegend). After the cells were fixed in 2% PFA, they were permeabilized by homogenization in “Perm Buffer” (1× BD FACS lysing solution 2 (Beckton Dickinson) and 0.05% Tween-20 [Sigma-Aldrich]) for 10 min and then washed with Wash Buffer. Cells were then incubated with mAbs against CD3 (clone SP34-2), interferon γ (IFN-γ; clone 4S.B3), tumor necrosis factor-α (TNF-α; clone mAb11), and CD69 (clone FN50) for 1 hour in the dark at room temperature. Lastly, the cells were washed and then stored at 4 °C until analysis by flow cytometry.
After gating on live CD14− CD16− CD20− CD3+ lymphocytes as described above, subsets were analyzed based on their exclusive expression of either CD4 or CD8. Responses were considered positive if they co-expressed IFN-γ and TNF-α. Leukocyte activation cocktail (LAC; BD Pharmingen)–stimulated cells stained with fluorochrome-labeled control mAbs of the same isotypes as those against IFN-γ and TNF-α guided identification of positive populations. Calculations were performed in Microsoft Excel and results were presented as percentages of IFN-γ+ TNF-α+ CD4+ or CD8+ T cells.
Cell lines
TZM-bl cells (human female HeLa-derived cancer cell line) and HEK293T cells (human embryonic kidney cancer cell line expressing the SV40 large T antigen) were maintained at 37°C and 5% CO2 in high glucose Dulbecco’s Modified Eagle Medium (DMEM, Corning) containing 1X Penicillin-Streptomycin (Corning), 2 mM L-Glutamine (Corning), and 10% heat-inactivated FBS Omega Scientific. FreeStyle 293F cells (Thermo Fisher Scientific) were maintained at 37°C, 80% humidity, and 8% CO2 in shaking incubators set to 135 revolutions per minute in serum and antibiotic free FreeStyle 293 Expression Medium (Thermo Fisher Scientific). CEM.NKR CCR5+Luc+ cell line derivatives were propagated in RPMI-1640 medium supplemented heat-inactivated 10% fetal bovine serum (FBS, Omega Scientific), 25 mM HEPES (Invitrogen), 2 mM L-glutamine (Invitrogen) and 0.1 mg/ml Primocin (InvivoGen). KGHY-1 rhCD16 cells were cultivated the same way as CEM.NKR.CCR5+Luc+ cells with the addition of 10U/ml interleukin-2 (AIDS Research and Reference Reagent Program) and cyclosporine A (Sigma) at 1 μg per ml.
Statistical Analysis
Data was plotted using Prism 7 and 8 (GraphPad Software), or using custom Python scripts based on the Pandas or Seaborn data analysis packages. Statistical analyses were performed in R and included two sample t tests to assess significance of differences between animal groups in terms of protection, logarithmized AUC, day 0 serum ELISA and neutralization titers. Data and scripts used for these analyses have been deposited online (doi:10.5281/zenodo.4474217).
Supplementary Material
Fig. S1. Hypothesis for possible effector function augmentation in relation to antibody concentration.
Fig. S2. PGT121 serum antibody titers.
Fig. S3. CD8 depletion to probe presence of virus in antibody-treated macaques.
Fig. S4. Additional in vitro ADCC assay.
Fig. S5. Glycoanalyses of CHO-produced b12 and 293F cell-produced PGT121 variants.
Fig. S6. Effect of producer cell line on ADCC activity.
Table S1. Assessment of T cell priming in protected animals.
Acknowledgements
We thank Javier Guenaga for providing the JRFL NFL TD CC+ construct, and Matteo Bianchi for the BG505 N332 SOSIP v5.2 construct. We are grateful to the WNPRC Genetics Services, Roger Wiseman and his team, for performing all haplotyping. We thank Diane Kubrik from the Center for Antibody Development and Production at The Scripps Research Institute for producing all antibodies used in vivo and David Evans for providing the CEM.NKR-CCR5-Luc and KHYG cell lines used in this study.
Funding
This work was funded by NIH grants 5R37 A1055332 and UM1AI100663 to DRB and UM1AI144462 to DRB and LH. The Swiss National Science Foundation Fellowship P2ZHP3_174834 supported DB. This nonhuman primate study was performed at the Wisconsin National Primate Research Center supported in part by NCRR grant P51OD011106. MAM was supported by K01 0D023032 from the Office of the Director, NIH.
Footnotes
Competing interests
DRB is a joint holder of a patent on PGT121 (WO2012030904A2 - Human immunodeficiency virus (HIV)-neutralizing antibodies). Dr. Martins has a consulting financial interest in Emmune, Inc., a company that is developing HIV immunotherapies based on the immunoadhesin eCD4-Ig. All other authors have no other conflicting interests to declare.
Data Availability
All data associated with this study are in the paper or deposited on Zenodo (doi:10.5281/zenodo.4474217).
References
- 1.Hessell AJ, Hangartner L, Hunter M, Havenith CEG, Beurskens FJ, Bakker JM, Lanigan CMS, Landucci G, Forthal DN, Parren PWHI, Marx PA, Burton DR, Fc receptor but not complement binding is important in antibody protection against HIV, Nature 449, 101–104 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Hessell AJ, Poignard P, Hunter M, Hangartner L, Tehrani DM, Bleeker WK, Parren PWHI, Marx PA, Burton DR, Effective, low-titer antibody protection against low-dose repeated mucosal SHIV challenge in macaques, Nat. Med 15, 951–954 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moldt B, Schultz N, Dunlop DC, Alpert MD, Harvey JD, Evans DT, Poignard P, Hessell AJ, Burton DR, A panel of IgG1 b12 variants with selectively diminished or enhanced affinity for Fcγ receptors to define the role of effector functions in protection against HIV, J. Virol 85, 10572–10581 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bournazos S, Klein F, Pietzsch J, Seaman MS, Nussenzweig MC, Ravetch JV, Broadly neutralizing anti-HIV-1 antibodies require Fc effector functions for in vivo activity, Cell 158, 1243–1253 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asokan M, Rudicell RS, Louder M, McKee K, apos Dell SO, Stewart-Jones G, Wang K, Xu L, Chen X, Choe M, Chuang G, Georgiev IS, Joyce MG, Kirys T, Ko S, Pegu A, Shi W, Todd JP, Yang Z, Bailer RT, Rao S, Kwong PD, Nabel GJ, Mascola JR, Silvestri G, Ed. Bispecific Antibodies Targeting Different Epitopes on the HIV-1 Envelope Exhibit Broad and Potent Neutralization, J. Virol 89, 12501–12512 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang P, Gajjar MR, Yu J, Padte NN, Gettie A, Blanchard JL, Russell-Lodrigue K, Liao LE, Perelson AS, Huang Y, Ho DD, Quantifying the contribution of Fc-mediated effector functions to the antiviral activity of anti-HIV-1 IgG1 antibodies in vivo, Proc. Natl. Acad. Sci. U.S.A 117, 18002–18009 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien J-P, Wang S-K, Ramos A, Chan-Hui P-Y, Moyle M, Mitcham JL, Hammond PW, Olsen OA, Phung P, Fling S, Wong C-H, Wrin T, Simek MD, Investigators PGP, Koff WC, Wilson IA, Burton DR, Poignard P, Broad neutralization coverage of HIV by multiple highly potent antibodies, Nature 477, 466–470 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahomed S, Garrett N, Capparelli E, Baxter C, Zuma NY, Gengiah T, Archary D, Moore P, Samsunder N, Barouch DH, Mascola J, Ledgerwood J, Morris L, Abdool Karim S, Assessing the safety and pharmacokinetics of the monoclonal antibodies, VRC07-523LS and PGT121 in HIV negative women in South Africa: study protocol for the CAPRISA 012A randomised controlled phase I trial, BMJ Open 9, e030283 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parsons MS, Lloyd SB, Lee WS, Kristensen AB, Amarasena T, Center, Rob J, Keele BF, Lifson JD, LaBranche CC, Montefiori D, Wines BD, Hogarth PM, Swiderek KM, Venturi V, Davenport MP, Kent SJ, Partial efficacy of a broadly neutralizing antibody against cell-associated SHIV infection, Sci. Transl. Med 9, eaaf1483 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Moldt B, Rakasz EG, Schultz N, Chan-Hui P-Y, Swiderek K, Weisgrau KL, Piaskowski SM, Bergman Z, Watkins DI, Poignard P, Burton DR, Highly potent HIV-specific antibody neutralization in vitro translates into effective protection against mucosal SHIV challenge in vivo, Proc. Natl. Acad. Sci. U.S.A 109, 18921–18925 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, Kubbies M, Klein C, Umaña P, Mössner E, Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions, Protein Engineering Design and Selection 29, 457–466 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Nishimura Y, Gautam R, Chun TW, Sadjadpour R, Foulds KE, Shingai M, Klein F, Gazumyan A, Golijanin J, Donaldson M, Donau OK, Plishka RJ, Buckler-White A, Seaman MS, Lifson JD, Koup RA, Fauci AS, Nussenzweig MC, Martin MA, Early antibody therapy can induce long-lasting immunity to SHIV, Nature 543, 559–563 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lo M, Kim HS, Tong RK, Bainbridge TW, Vernes J-M, Zhang Y, Lin YL, Chung S, Dennis MS, Zuchero YJY, Watts RJ, Couch JA, Meng YG, Atwal JK, Brezski RJ, Spiess C, Ernst JA, Effector-attenuating Substitutions That Maintain Antibody Stability and Reduce Toxicity in Mice, J. Biol. Chem 292, 3900–3908 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alpert MD, Heyer LN, Williams DEJ, Harvey JD, Greenough T, Allhorn M, Evans DT, A novel assay for antibody-dependent cell-mediated cytotoxicity against HIV-1- or SIV-infected cells reveals incomplete overlap with antibodies measured by neutralization and binding assays, J. Virol 86, 12039–12052 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomann M, Reckermann K, Reusch D, Prasser J, Tejada ML, Fc-galactosylation modulates antibody-dependent cellular cytotoxicity of therapeutic antibodies, Mol. Immunol 73, 69–75 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Parren PW, Burton DR, The antiviral activity of antibodies in vitro and in vivo, Adv. Immunol 77, 195–262 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Earnest JT, Basore K, Roy V, Bailey AL, Wang D, Alter G, Fremont DH, Diamond MS, Neutralizing antibodies against Mayaro virus require Fc effector functions for protective activity, J. Exp. Med 216, 2282–2301 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DiLillo DJ, Tan GS, Palese P, Broadly neutralizing hemagglutinin stalk-specific antibodies require FcγR interactions for protection against influenza virus in vivo, Nat. Med 20, 143–151 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parsons MS, Lee WS, Kristensen AB, Amarasena T, Khoury G, Wheatley AK, Reynaldi A, Wines BD, Hogarth PM, Davenport MP, Kent SJ, Fc-dependent functions are redundant to efficacy of anti-HIV antibody PGT121 in macaques, J. Clin. Invest 129, 182–191 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roben P, Moore JP, Thali M, Sodroski J, Barbas CF, Burton DR, Recognition properties of a panel of human recombinant Fab fragments to the CD4 binding site of gp120 that show differing abilities to neutralize human immunodeficiency virus type 1, J. Virol 68, 4821–4828 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Torrents de la Peña A, Julien J-P, de Taeye SW, Garces F, Guttman M, Ozorowski G, Pritchard LK, Behrens A-J, Go EP, Burger JA, Schermer EE, Sliepen K, Ketas TJ, Pugach P, Yasmeen A, Cottrell CA, Torres JL, Vavourakis CD, van Gils MJ, LaBranche C, Montefiori DC, Desaire H, Crispin M, Klasse P-J, Lee KK, Moore JP, Ward AB, Wilson IA, Sanders RW , Improving the Immunogenicity of Native-like HIV-1 Envelope Trimers by Hyperstabilization, Cell Rep 20, 1805–1817 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guenaga J, Garces F, de Val N, Stanfield RL, Dubrovskaya V, Higgins B, Carrette B, Ward AB, Wilson IA, Wyatt RT, Glycine Substitution at Helix-to-Coil Transitions Facilitates the Structural Determination of a Stabilized Subtype C HIV Envelope Glycoprotein, Immunity 46, 792–803.e3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pugach P, Ozorowski G, Cupo A, Ringe R, Yasmeen A, de Val N, Derking R, Kim HJ, Korzun J, Golabek M, de Los Reyes K, Ketas TJ, Julien J-P, Burton DR, Wilson IA, Sanders RW, Klasse PJ, Ward AB, Moore JP, Doms RW, Ed. A native-like SOSIP.664 trimer based on an HIV-1 subtype B env gene, J. Virol 89, 3380–3395 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pauthner M, Havenar-Daughton C, Sok D, Nkolola JP, Bastidas R, Boopathy AV, Carnathan DG, Chandrashekar A, Cirelli KM, Cottrell CA, Eroshkin AM, Guenaga J, Kaushik K, Kulp DW, Liu J, McCoy LE, Oom AL, Ozorowski G, Post KW, Sharma SK, Steichen JM, de Taeye SW, Tokatlian T, Torrents de la Peña A, Butera ST, LaBranche CC, Montefiori DC, Silvestri G, Wilson IA, Irvine DJ, Sanders RW, Schief WR, Ward AB, Wyatt RT, Barouch DH, Crotty S, Burton DR, Elicitation of Robust Tier 2 Neutralizing Antibody Responses in Nonhuman Primates by HIV Envelope Trimer Immunization Using Optimized Approaches, Immunity 46, 1073–1088.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McAndrew EG, Dugast A-S, Licht AF, Eusebio JR, Alter G, Ackerman ME, Determining the phagocytic activity of clinical antibody samples, J Vis Exp, e3588 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seabright GE, Cottrell CA, van Gils MJ, D’addabbo A, Harvey DJ, Behrens A-J, Allen JD, Watanabe Y, Scaringi N, Polveroni TM, Maker A, Vasiljevic S, de Val N, Sanders RW, Ward AB, Crispin M, Networks of HIV-1 Envelope Glycans Maintain Antibody Epitopes in the Face of Glycan Additions and Deletions, Structure/Folding and Design, 1–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cline AN, Bess JW, Piatak M, Lifson JD, Highly sensitive SIV plasma viral load assay: practical considerations, realistic performance expectations, and application to reverse engineering of vaccines for AIDS, Journal of Medical Primatology 34, 303–312 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Gonzalez-Nieto L, Domingues A, Ricciardi M, Gutman MJ, Maxwell HS, Pedreño-Lopez N, Bailey V, Magnani DM, Martins MA, Analysis of Simian Immunodeficiency Virus-specific CD8+ T-cells in Rhesus Macaques by Peptide-MHC-I Tetramer Staining, J Vis Exp, e54881 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wyrzucki A, Dreyfus C, Kohler I, Steck M, Wilson IA, Hangartner L, Alternative recognition of the conserved stem epitope in influenza A virus hemagglutinin by a VH3-30-encoded heterosubtypic antibody, J. Virol 88, 7083–7092 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Hypothesis for possible effector function augmentation in relation to antibody concentration.
Fig. S2. PGT121 serum antibody titers.
Fig. S3. CD8 depletion to probe presence of virus in antibody-treated macaques.
Fig. S4. Additional in vitro ADCC assay.
Fig. S5. Glycoanalyses of CHO-produced b12 and 293F cell-produced PGT121 variants.
Fig. S6. Effect of producer cell line on ADCC activity.
Table S1. Assessment of T cell priming in protected animals.