

Abstract
[URE3] is an amyloid-based prion of Ure2p, a negative regulator of poor nitrogen source catabolism in Saccharomyces cerevisiae. Overproduced Btn2p or its paralog Cur1p, in processes requiring Hsp42, cure the [URE3] prion. Btn2p cures by collecting Ure2p amyloid filaments at one place in the cell. We find that rpl4aΔ, rpl21aΔ, rpl21bΔ, rpl11bΔ, and rpl16bΔ (large ribosomal subunit proteins) or ubr2Δ (ubiquitin ligase targeting Rpn4p, an activator of proteasome genes) reduce curing by overproduced Btn2p or Cur1p. Impaired curing in ubr2Δ or rpl21bΔ is restored by an rpn4Δ mutation. No effect of rps14aΔ or rps30bΔ on curing was observed, indicating that 60S subunit deficiency specifically impairs curing. Levels of Hsp42p, Sis1p, or Btn3p are unchanged in rpl4aΔ, rpl21bΔ, or ubr2Δ mutants. Overproduction of Cur1p or Btn2p was enhanced in rpn4Δ and hsp42Δ mutants, lower in ubr2Δ strains, and restored to above wild-type levels in rpn4Δ ubr2Δ strains. As in the wild-type, Ure2N-GFP colocalizes with Btn2-RFP in rpl4aΔ, rpl21bΔ, or ubr2Δ strains, but not in hsp42Δ. Btn2p/Cur1p overproduction cures [URE3] variants with low seed number, but seed number is not increased in rpl4aΔ, rpl21bΔ or ubr2Δ mutants. Knockouts of genes required for the protein sorting function of Btn2p did not affect curing of [URE3], nor did inactivation of the Hsp104 prion-curing activity. Overactivity of the ubiquitin/proteasome system, resulting from 60S subunit deficiency or ubr2Δ, may impair Cur1p and Btn2p curing of [URE3] by degrading Cur1p, Btn2p or another component of these curing systems.
Keywords: UBR2, RPL4A, RPL21B, RPN4
Introduction
Most of the prions (infectious proteins) of Saccharomyces cerevisiae are self-propagating amyloids, each a linear polymer of a single protein with an in-register parallel folded β-sheet architecture (reviewed in Liebman and Chernoff 2012; Wickner et al. 2015). [URE3] is an amyloid-based prion of the Ure2 protein (Wickner 1994; Masison and Wickner 1995; Edskes et al. 1999; Brachmann et al. 2005), a repressor of genes for utilization of poor nitrogen sources (Cooper 2002). [PSI+] is an amyloid-based prion of Sup35p (Wickner 1994; Glover et al. 1997; King et al. 1997; Paushkin et al. 1997; King and Diaz-Avalos 2004; Tanaka et al. 2004), a subunit of the translation termination factor (Stansfield et al. 1995; Zhouravleva et al. 1995). [PIN+], a prion of Rnq1p (function unknown), is detected by its ability to rarely prime [PSI+] formation (Derkatch et al. 1997, 2001; Sondheimer and Lindquist 2000). Most variants of [PSI+] and [URE3] prions are either lethal or cause severely slowed growth (Mcglinchey et al. 2011), but even the mild variants of each (such as those used in most lab experiments) are rare in wild strains (Nakayashiki et al. 2005), demonstrating that even these are detrimental (Kelly et al. 2012). In fact, there have not yet been reproducible benefits of any variant of either prion (reviewed in Wickner et al. 2015). Thus, it is not surprising that, through the action of a series of anti-prion systems, normal cells are found to eliminate most variants of [URE3] and of [PSI+] as they arise(reviewed by Wickner et al. 2018; Son and Wickner 2020).
Among these anti-prion systems, normal levels of Btn2p and Cur1p cure a large majority of [URE3] variants arising in their absence (Kryndushkin et al. 2008; Wickner et al. 2014). In general, those [URE3] variants with low-propagation/seed numbers are cured by normal levels of Btn2p or Cur1p, while [URE3] variants with higher seed number, such as [URE3-1], require overproduction of Btn2p or Cur1p to eliminate the prion (Kryndushkin et al. 2008; Wickner et al. 2014). Normal levels of Btn2p and Cur1p measurably lower the propagon number of [URE3-1] although they cannot cure this variant (Kryndushkin et al. 2008). Overproduced Btn2p collects Ure2p amyloid filaments at one location in the cell, co-incident with Btn2p itself (Kryndushkin et al. 2008; Kanneganti et al. 2011). It is proposed that this sequestration of prion filaments explains the frequent loss of the prion as cells divide (Kryndushkin et al. 2008). Sites in Ure2p outside the prion domain affecting curing of [URE3] by overproduced Btn2p may identify regions of interaction with Btn2p (Shailesh et al. 2020). Btn2p also binds to and collects other amyloids and other non-amyloid aggregates, but does not cure the variants of [PSI+] or [PIN+] that have been tested (Kryndushkin et al. 2008, 2012; Malinovska et al. 2012; Barbitoff et al. 2017). Btn2p also cures an artificial prion based on a domain of Nrp1p (Malinovska et al. 2012). Cur1p, although paralogous to Btn2p, does not co-localize with the amyloid filaments during curing and may act by a different mechanism (Kryndushkin et al. 2008; Barbitoff et al. 2017).
Studying an artificial prion, in cells grown at 37°C, in the presence of the proteasome inhibitor MG132, Malinovska et al. (2012) found that overexpression of Sis1p prevented prion curing by overexpressed Btn2p or overexpressed Cur1p. They proposed that Btn2p and Cur1p cure prions by sequestration in the nucleus of Sis1p, an Hsp40 necessary for the propagation of the [URE3], [PSI+], and [PIN+] prions (Higurashi et al. 2008). However, this proposal explains neither Btn2p’s co-localization with Ure2p prion amyloid (Kryndushkin et al. 2008), nor the retention of [URE3]-curing activity after deletion of the Btn2p nuclear localization sequence (Kryndushkin et al. 2008), nor the curing of 90% of [URE3] variants by the normal levels of Btn2p or Cur1p (Wickner et al. 2014), which are 20- and 400-fold (respectively) below that of Sis1p (https://pax-db.org, Wang et al. 2015). Moreover, although Sis1p is necessary for the propagation of the [URE3], [PSI+] and [PIN+] prions (Higurashi et al. 2008), neither [PSI+] nor [PIN+] was cured by Btn2p or Cur1p overproduction (Kryndushkin et al. 2008). Perhaps the overproduced Sis1p binds to and prevents the curing action of Btn2p or Cur1p, the reverse of the hypothesized mechanism. Nonetheless, overproduced Cur1p may cure by the suggested Sis1p removal mechanism (Barbitoff et al. 2017).
Btn2p has another, apparently unrelated, role as a late endosome mediator of protein sorting (Chattopadhyay et al. 2003; Kama et al. 2007, 2011; Kanneganti et al. 2011). BTN2 was first identified as a gene overexpressed in strains deleted for BTN1, the yeast homolog of the Batten’s disease gene (CLN3) (Chattopadhyay et al. 2000). In btn2Δ strains, Yif1p is mislocalized to the vacuole and Kex2p to the late endosome (Chattopadhyay et al. 2003; Kama et al. 2007). Btn2p co-localizes with Snx4p and Vps27p, two endosome markers, and immunoprecipitates with endocytic SNARE proteins Snc1p, Snc2p, Tlg1p, Tlg2p, and Vti1p (Kama et al. 2007). Kama et al. (2007) have shown that the protein sorting function of Btn2p requires at least Snc1p, Tlg2p, Vps26p, Snx4p, Ypt6p, Chs4p, and Rhb1p, but whether the same components are involved in the prion-curing activity of Btn2p is unknown. In a two-hybrid screen with Btn2p as bait, Btn3p was detected (Kanneganti et al. 2011). Overproduced Btn3p is evenly distributed in the cytoplasm, but is concentrated in discrete loci with overproduced Btn2p, binds directly to Btn2p, and inhibits both the protein-sorting and prion-curing activities of Btn2p (Kanneganti et al. 2011). Btn3p is necessary for normal endosomal sorting of ubiquitylated cargos and endosomal recycling of the Snc1 SNARE, even without Btn2p overproduction (Morvan et al. 2015).
Here, we report a screen for co-factors whose mutation prevents curing of [URE3] by overproduced Btn2p or Cur1p. Our screen utilizes the HERMES transposon, originally from house flies, but adapted for use in S. cerevisiae (Gangadharan et al. 2010; Guo et al. 2013), and utilized for the discovery of genes preventing [URE3] prion pathology (Edskes et al. 2018). We find that mutation of 60S ribosomal subunit protein genes or UBR2, encoding an E3 ubiquitin protein ligase, substantially impair curing activities of Btn2p and Cur1p. We examine the mechanism of these effects. We also tested seven of the proteins involved in the protein sorting functions of Btn2p, and, remarkably, none were necessary for the prion curing activity of either Btn2p or Cur1p.
Materials and methods
Strains and media
Strains of S. cerevisiae are listed in Table 1. Media are described by Sherman (1991). The reduced—adenine medium ½ YPD is 5 g/l yeast extract, 20 g/l peptone, 20 g/l dextrose, and 20 g/l agar and is used for scoring the [URE3] phenotype. DAL5 transcription is strongly repressed by Ure2p in the presence of ammonia or other good nitrogen source (Cooper 2002), and we use a DAL5-promoted ADE2 to measure Ure2p activity, and thereby, the presence or absence of [URE3] (Schlumpberger et al. 2001; Brachmann et al. 2005). [URE3] strains are white or pink, while [ure-o] strains are red on ½ YPD. Synthetic complete and dropout medium contained 6.67 g/l Yeast Nitrogen Base without amino acids (Difco), glucose (2%, dex) or galactose (3%) and raffinose (2%) (gal/raf), and an appropriate dropout mix from Sunrise Science (e.g., 1.92 g/L of SC-W). All incubations were at 30°C unless noted otherwise.
Table 1.
Strains of Saccharomyces cerevisiae
| Strain | Parent | Genotype | Source |
|---|---|---|---|
| 1735 | MA116-8A | MATα his- ura2-60 [URE3-1] | (Lacroute 1971; Aigle and Lacroute 1975) |
| BEE1027 | 74D-694 | MAT a ura3 leu2 trp1 his3 ade1-14 [PIN+] [psi-] | (Chernoff et al. 1995) |
| BEE1021 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 [URE3-1] | (Brachmann et al. 2005) |
| 4591 | MATα his3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 [ure-o] | (Brachmann et al. 2005) | |
| BEE1190 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 hsp42::KanMX [URE3-1] | (Wickner et al. 2014) |
| BEE1192 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 ubr2::KanMX [URE3-1] | This study |
| BEE1251 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 rpl21b::KanMX [URE3-1] | This study |
| BEE1272 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 rpl4a::KanMX [URE3-1] | This study |
| BEE1318 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 ubr2::KanMX rpn4::URA3 [URE3-1] | This study |
| BEE1363 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 rpl16b::KanMX [URE3-1] | This study |
| BEE1364 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 rpl11b::KanMX [URE3-1] | This study |
| BEE1372 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 rps14a::KanMX [URE3-1] | This study |
| BEE1378 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 rps30b::KanMX [URE3-1] | This study |
| BEE1086 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 snc1::KanMX [URE3-1] | This study |
| BEE1096 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 snc2::KanMX [URE3-1] | This study |
| BEE1132 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 tlg2::KanMX [URE3-1] | This study |
| BEE1125 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 vps27::KanMX [URE3-1] | This study |
| BEE1219 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 vps35::KanMX [URE3-1] | This study |
| BEE1088 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 vps26::KanMX [URE3-1] | This study |
| BEE1012 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 btn2::TRP1 cur1::kanMX [URE3-1] | (Kryndushkin et al. 2008) |
| BEE1182 | BY245 | MATα his3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 [ure-o] | This study |
| BEE1200 | 5169-3D | MATα trp1 kar1 DAL5p-ADE2 leu2 ura3 Hsp104T160M—URA3 [ure-o] | This study |
| BEE1201 | 5169-7B | MATα trp1 kar1 DAL5p-ADE2 leu2 ura3 [ure-o] | This study |
| BEE1202 | 5168-9A | MATα trp1 kar1 DAL5p-ADE2 leu2 ura3 Hsp104T160M—URA3 [ure-o] | This study |
| BEE1203 | 5169-5A | MATα trp1 kar1 DAL5p-ADE2 leu2 ura3 [ure-o] | This study |
| BEE1407 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 rpl21a::KanMX [URE3-1] | This study |
| BEE1319 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 rpn4::KanMX [URE3-1] | Emily Stroobant and Herman Edskes |
| BEE1103 | 779-6A | MATα kar1 ade2.1 SUQ5 his3Δ202 leu2Δ1 trp1Δ63 ura3.52 [PSI+ strong] | |
| BEE1104 | 779-6A | MATα kar1 ade2.1 SUQ5 his3Δ202 leu2Δ1 trp1Δ63 ura3.52 [PSI+ Sc4] | Mike Reidy |
| BEE1105 | 779-6A | MATα kar1 ade2.1 SUQ5 his3Δ202 leu2Δ1 trp1Δ63 ura3.52 [PSI+ Sc37] | Mike Reidy |
| BEE1106 | 779-6A | MAT a kar1 ade2.1 SUQ5 his3Δ202 leu2Δ1 trp1Δ63 ura3.52 [PSI+ LiebW] | Mike Reidy |
| BEE1107 | 779-6A | MAT a kar1 ade2.1 SUQ5 his3Δ202 leu2Δ1 trp1Δ63 ura3.52 [PSI+ LiebS] | Mike Reidy |
| BEE1386 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-CAN1: CAN1 kar1 arg1::TRP1 lys2::LEU2 [URE3-1] | Morgan DeWilde and Herman Edskes |
| BEE1387 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-CAN1: CAN1 kar1 arg1::TRP1 lys2::LEU2 hsp42::KanMX [URE3-1] | This study |
| BEE1392 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-CAN1: CAN1 kar1 arg1::TRP1 lys2::LEU2 rpl4a::KanMX [URE3-1] | This study |
| BEE1395 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-CAN1: CAN1 kar1 arg1::TRP1 lys2::LEU2 rpl21b::KanMX [URE3-1] | This study |
| BEE1401 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-CAN1: CAN1 kar1 arg1::TRP1 lys2::LEU2 rps30b::KanMX [URE3-1] | This study |
| BEE1271 | BY241 | MAT a ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 rpl4a::KanMX [ure-0] | This study |
| BEE1368 | BY241 | MATα ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 rpl4a::KanMX [ure-0] | This study |
| BEE1370 | BY241 | MATα ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 [ure-0] | This study |
| BEE1417 | BY241 | MATa ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 rpl21b::KanMX rpn4::URA3 [URE3-1] | This study |
| 5991 | BY241 | MATa ura3 leu2 trp1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 tma10::kanMX | This study |
| 74-D694 | MATa kar1 ade1-14 his3 leu2 trp1 ura3 [psi-][PIN+] | (Chernoff et al. 1995) | |
| 6010 | BY241 | MATα ura3 leu2 trp1 his3::TRP1 P-DAL5: ADE2 P-DAL5: CAN1 kar1 | This study |
Strain 1735 (MA116-8A) (Lacroute 1971; Aigle and Lacroute 1975) was used as cytoduction donor to BY241 BY241 (Brachmann et al. 2005) forming BEE1021. Segregants of crosses 5168 and 5169 were produced by crossing a strain carrying hsp104-T160M-URA3 (Hung and Masison 2006) twice with BY241 producing strains BEE1200 to BEE1203.
Plasmid construction
Plasmids used are listed in Table 2.
Table 2.
Plasmids
| Plasmid | Promoter | Gene | Marker | Copy number | Source |
|---|---|---|---|---|---|
| pBEE34 | CUP1 | BTN2 | TRP1 | CEN | This study |
| pBEE42 | CUP1 | CUR1 | TRP1 | CEN | This study |
| pBEE95 | HSP42 | HSP42-HA | LEU2 | CEN | This study |
| pBEE92 | BTN3 | BTN3-HA | LEU2 | CEN | This study |
| pSG36 | GAL1 | Hermes transposase & Hermes-natMX6 transposon | URA3 | CEN | (Gangadharan et al. 2010) |
| pBEE28 | GAL1 | BTN2-RFP, pYES52-BTN2-RFP | URA3 | 2 μm | (Kryndushkin et al. 2008) |
| pBEE29 | URE2 | URE2N-GFP, pVTG12 | LEU2 | CEN | (Edskes et al. 1999) |
| pBEE64 | GAL1 | RPL4A | LEU2 | CEN | This study |
| pSL1066 | CUP1 | SUP35NM-GFP | URA3 | CEN | (Zhou et al. 2001) |
| pRS314 | none | empty vector | TRP1 | CEN | (Sikorski and Hieter 1989) |
| pH316 | GAL1 | empty vector | LEU2 | CEN | (Edskes and Wickner 2002) |
| pRS315 | none | empty vector | LEU2 | CEN | (Sikorski and Hieter 1989) |
| pBEE1 | GAL1 | BTN2 | LEU2 | CEN | (Zhao et al. 2018) |
| pBEE2 | GAL1 | CUR1 | LEU2 | CEN | (Zhao et al. 2018) |
| pBEE103 | CUP1 | SUP35NM-GFP | TRP1 | CEN | This study |
| pET28-BTN2-HIS | T7 | BTN2-HIS | Kanamycin | Shailesh Kumar, NIDDK | |
| pET28-CUR1-HIS | T7 | CUR1-HIS | Kanamycin | Shailesh Kumar, NIDDK | |
| pH381 | GAL1 | Ure2 1-89 | LEU2 | CEN | (Edskes and Wickner, 2000) |
| p1289 | BTN2 | BTN2 | URA3 | 2 μm | (Kryndushkin et al. 2008) |
| p1488 | CUR1 | CUR1 | URA3 | 2 μm | (Wickner et al. 2014) |
| pRS426 | none | empty vector | URA3 | 2 μm | (Christianson et al. 1992) |
| P1678 | GAL1 | TMA10 | HIS3 | CEN | This study |
All plasmids are E. coli—S. cerevisiae shuttle plasmids except for pET28-BTN2-HIS and pET28-CUR1-HIS.
pBEE103 (CUP1 promoter—SUP35NM CEN TRP1) was constructed from pSL1066 (CUP1 promoter—SUP35NM CEN URA3) by homologous recombination in yeast. StuI-treated pSL1066 (cutting inside URA3) and a PCR product containing TRP1 and regions of homology upstream and downstream of the URA3 marker in pSL1066 were transformed into yeast selecting for Trp+. The TRP1 PCR product was obtained using primers BEE3065 (5’CGCATCTGTGCGGTATTTCACACCGCATAGaacgacattactatatatat3’) and BEE3066 (5’GCAGATTGTACTGAGAGTGCACCATACCACcaggcaagtgcacaaacaat3’) and pRS314 as template. The resulting pBEE103 containing TRP1 instead of URA3 was used to make pBEE34 (CUP1 promoter—BTN2 CEN TRP1) and pBEE42 (CUP1 promoter—CUR1 CEN TRP1) by homologous recombination of pBEE103 digested by MscI (cutting inside SUP35NM) with PCR products containing the BTN2 or CUR1 ORFs flanked by sequences in pBEE103 just upstream and downstream of the SUP35NM ORF. The PCR product for creation of pBEE34 was obtained using primers BEE3067 (5’GTACAATCAATCAATCAATCAGGATCCACAATGTTTTCCA TATTCAATTC3’) and BEE3068 (5’TACGACTCACTATAGGGCGAATTGGAGCTCTTATATCTCCTCAATAATAG3’) and pBEE1 as template. The PCR product for creation of pBEE42 was obtained using primers BEE3069 (5’GTACAATCAATCAATCAATCAGGATCCACAATGGCTGCCGCATGCATTTG3’) and BEE3070 (5’TACGACTCACTATAGGGCGAATTGGAGCTCTTACCGCCCATTCAATCTTC3’) and pBEE2 as template.
The CEN LEU2 BTN3 plasmid pBEE92 was constructed from PstI, shrimp alkaline phosphatase-treated pRS315 to which was ligated a PstI-treated BTN3-HA construct including 562 bp 5’ of the start codon. The BTN3-HA PCR product was from genomic DNA of BY241 using primers BEE3389 (5’ ccctgcagCCGCAGCCTTGATTCTGCCTCGGG3’) and BEE3390 (5’ ccctgcagTTAAGCGTAATCTGGAACATCGTATGGGTAAGAAAGGATGGTGGCGTCG3’).
To make pBEE95 (CEN LEU2 HSP42 promoter (587 bp) HSP42-HA), a PCR product obtained using primers BEE3391 (5’ ccctgcagCACTTACAATTTAGGCCGCGCC3’) and BEE3392 (5’ ccctgcagTCAAGCGTAATCTGGAACATCGTATGGGTAATTTTCTACCGTAGGGTTGGG3’) was digested with PstI and ligated to PstI and shrimp alkaline phosphatase—treated pRS315.pBEE1 (GAL1-promoted BTN2 CEN LEU2) was constructed by inserting BamHI-PstI-treated PCR product with the BTN2 coding sequence into BamHI-PstI-treated plasmid pH316 (LEU2 CEN GAL1 promoter, Edskes and Wickner 2002). The PCR product with BTN2 was made from p1475 (Kryndushkin et al. 2008) using primers: ATAGGATCCATGTTTTCCATATTCAATTC (BTN2BamHIF) and ATACTGCAGTTATATCTCCTCAATAATAG (BTN2PstIR) (Zhao et al. 2018). pBEE2 was constructed by inserting a BamHI-HindIII-treated PCR product with the CUR1 coding sequence into BamHI-HindIII-treated plasmid pH316 (Edskes and Wickner 2002). The PCR product with CUR1 was obtained using p1488 as template and primers: ATAGGATCCATGGCTGCCGCATGCATTTG (CUR1BamHIF) and GCGAAGCTTTTACCGCCCATTCAATCTTC (CUR1HindIIIR).pBEE64 (GAL1 promoter-RPL4A CEN LEU2) was constructed by in vivo recombination in yeast substituting the RPL4A ORF for the BTN2 ORF in pBEE1 using a PCR product carrying RPL4A flanked by pBEE1 sequences upstream and downstream of BTN2. This PCR product was obtained using DNA from strain BEE1021 as template and primers BEE3334 (5’GTTAATATACCTCTATACTTTAACGTCAAGGAGAAAAAACCCCGGATCCATGTCCCGTCCACAAGTTAC3’) and BEE3335 (5’CTTGATTGGAGACTTGACCAAACCTCTGGCGAAGAAGTCCAAAGCTTCAGCTGCTGCAGTTAATCGTGTTTCAAAGTTT3’).
Antibodies used
See Table 3.
Table 3.
Antibodies used
| Specificity | Animal | Titer | Source |
|---|---|---|---|
| Anti-Btn2 polyclonal | Rabbit | 1:5,000 | custom generated by COVANCE |
| Anti-Cur1 polyclonal | Rabbit | 1:5,000 | custom generated by COVANCE |
| Anti-Sis1 | Rabbit | 1:5,000 | kind gift from Daniel Masison laboratory at NIDDK NIH |
| Anti-HA tag polyclonal | Rabbit | 1:1,000 | Abcam ab 184643 |
| Anti-Pgk1 monoclonal | Mouse | 1:5,000 | Abcam ab 113687 |
| Donkey anti-rabbit IgG-AP | Donkey | 1:5,000 | Santa Cruz Biotechnology sc-2057 used in Figure 2 |
| Anti-rabbit IgG(Fc), AP Conjugate | 1:7,500 | Promega AP conjugate S3738 used in Figure 3 |
Anti-Btn2 and anti-Cur1 antibodies were generated by COVANCE using affinity purified (Ni-NTA agarose, Qiagen) proteins Btn2-His6 and Cur1-His6 produced from pET28-BTN2-HIS and pET28-CUR1-HIS.×
Western blots
Yeast cells in log phase from 50 ml of synthetic complete media were collected, washed 3 times in 1 ml of water and resuspended in 600 μl of lysis buffer containing 8 M urea, 1 mM DTT, 1 mM EDTA, 1 mM PMSF, 1 mM Pefabloc SC, 1x complete EDTA-free protease inhibitor tablet (Pierce), 5% glycerol, 1xPBS and placed in 2 ml tubes with screw caps. Glass beads (0.5 mm) were added to fill the tubes and samples were homogenized at 4 C for 3 min using a Bead Beater (BioSpec) at maximum shaking parameters. Using a hot needle, a hole was made at the bottom of the 2 ml tube which was placed then inside a 5 ml tube with screw cap containing a hole in the cap which fits the 2 ml tube. Samples were centrifuged for 5 minutes at 3000 g at 4°C. The supernatant was placed in 1.5 ml tubes and centrifuged at maximum speed in a microcentrifuge. The supernatant was transferred to a fresh 1.5 ml tube, protein measured by BCA (Pierce) and stored at −80°C. Protein concentrations of samples for acrylamide gel electrophoresis were equalized, and samples were made 1x LDS sample buffer (ThermoFisher Scientific), and 175 mM DTT. After 5 min incubation at 100 °C and 5 min centrifugation, samples were loaded on precasted 4%−12% NuPAGE Bis-Tris gels (ThermoFischer Scientific) and electrophoresed for 35 min at 200 V in 1X NuPAGe MES SDS running buffer (ThermoFischer Scientific). Western blotting was performed on isopropanol activated PVDF membrane for 1 h 30 min in 1x NuPAGE transfer buffer (ThermoFischer Scientific) at 30 V. For detection of Btn2p, Sis1p and HA tag PVDF membranes were stained using SNAP i.d. 2.0 (Millipore) according to the recommendations of the manufacturer with 1X TBS (20 mM Tris Cl pH 7.4, 136 mM NaCl) with 0.1% Tween 20 as washing solution and 1X TBS, 0.1% Tween 20, 1% BSA as blocking solution (see Table 3 for antibodies). Before addition of secondary antibodies, membranes were washed once with blocking solution.
Construction of mutant strains
Disruption cassettes carrying the kanMX cassette were obtained by amplifying yeast genomic DNA of corresponding strains from the S. cerevisiae knockout collection (Winzeler et al. 1999) using a forward primer specific to the region 200 base pairs upstream of the start codon of the gene to be knocked out and a reverse primer specific to the region 200 base pairs downstream of the stop codon. Disrupted mutants were then obtained by transforming the resulting PCR fragment into yeast and selecting for G418-resistant colonies at a final concentration of 1 g/l of G418. In each case, disruption was confirmed by PCR.
Curing of [URE3] by overexpressed Btn2p or Cur1p
Strains carrying [URE3-1] were transformed with pBEE34 (CUP1 promoter-BTN2) or pBEE42 (CUP1 promoter-CUR1), and transformants were grown in 2 ml liquid SD media without adenine or tryptophan to stationary phase (OD600 ∼3–5). The OD600 of the liquid cultures was measured and cells were transferred to 2 ml of dex -Trp liquid media containing 0.004% adenine and 0.250 mM copper sulfate starting at OD600 = 0.001. Cells were grown at 250 rpm in 14 ml tubes until they reached stationary phase, and dilutions were plated on ½ YPD plates. The percentage of red colored colonies was used as a measurement of [URE3] curing efficiency by Btn2 or Cur1 overproduction.
Hermes transposon mutagenesis to select Btn2-incurable mutants
The [URE3] strain BY241 [URE3] was transformed with pBEE34 (CUP1promoter-BTN2) and pSG36 [HERMES transposon with natMX4 producing resistance to nourseothricin and GAL1- promoted HERMES transposase (Gangadharan et al. 2010)]. Cells were then grown for 2 days in filter-sterilized gal/raf liquid media (-Trp) starting from OD600 = 0.1. The transposase excises the HERMES transposon and inserts it into the genome (after which the plasmid is not functional). Cells were collected by centrifugation and placed in the same volume of the same liquid media containing additionally 1 g/L of 5-FOA and 0.004% uracil (5-FOA selects cells which have lost the URA3-carrying pSG36) and were grown overnight. Cells were collected by centrifugation and placed in the same medium containing additionally 0.1 g/L of nourseothricin sulfate (GoldBio) and grown overnight at 30°C. The culture was then diluted 300-fold in liquid media containing 6.67 g/L of yeast nitrogen base, 3% glucose, 1.92 g/L of SC-W dropout mix, 0.01% adenine, 0.002% uracil, 1 g/L 5-FOA, 0.1 g/L of nourseothricin sulfate and 0.25 mM copper sulfate and cells were grown for two days to induce Btn2 overexpression. Finally, cells were spread on 1/2YPD plates. Clones which were not cured of the prion (still white) were selected for further analysis. The nat-resistant mutants were subjected to repeated copper induction of Btn2 to verify that [URE3-1] was not cured by Btn2p overproduction. Genomic DNA was purified (YeaStar Genomic DNA Kit, Zymo Research) from clones which showed Btn2-incurability but were curable by growth on 5 mM guanidine. The genomic DNA was digested overnight with MseI, cutting a site inside of the Hermes transposon and in the genomic DNA, and then treated with T4 DNA ligase overnight at 4°C. The region of genomic DNA containing part of the self-ligated MseI-digested Hermes transposon and the adjacent yeast genomic DNA was PCR amplified using primers BEE3054 (5’AGCACAATTGTACTCATAAG3’) and BEE3055 (5’TCATTGATTCATCGACACTC3’): 4 ul of ligase mixture was used as a template for a 25 ul PCR reaction. PCR products were purified by QIAquick PCR purification kit (Qiagen) and were used as templates for a second round of PCR with primers BEE3049 (5’GCAAGTGGCGCATAAGTATCAAAATAAGCCACTTGTTG3’) and BEE3056 (5’GTTTTGTCGTGTCGTTCTGCG3’. The resulting PCR products were purified by QIAquick PCR purification kit and sequenced using primers BEE3049 and BEE3056. If transposon insertions in the same gene were identified two or more times, this gene was knocked out in strain BY241 carrying [URE3-1] using the KanMX cassette from the knockout collection (G418-resistance). Btn2 overexpression curability of [URE3-1] was then tested in the newly generated mutant.
[URE3] seed number measurements
Cells were grown to single colonies on YPAD plates with 3 mM or 5 mM guanidine HCl. Single colonies were picked up in their entirety, suspended in 200 μL of water and each spread on an SC—Ade plate. The number of Ade+ colonies was counted and used as a representation of the prion seed number in the founding cell of the colony (Cox et al. 2003). Ten colonies were examined for each strain in each experiment.
Cytoduction
Cytoplasmic transfer (cytoduction) was performed using the kar1-1 mutation (Conde and Fink 1976) that largely blocks nuclear fusion after mating. Heterokaryons are formed by mating which, on cell division, produce haploids with the parental nuclei and a mixture of cytoplasms of the parents.
Microscopy
A Zeiss LSM 780 confocal scanhead (Carl Zeiss Microscopy, LLC, Thornwood, NY) mounted on a Zeiss AxioObserver.Z1 microscope, running ZEN 2.3 software, was used to collect 2 D images of red/green fluorescence and DIC images. The objective lens used was the Zeiss Plan-Apo 100x/1.4 Oil M27. For Ure2N-GFP fluorescence: excited by the 488 nm laser, emission was filtered by spectral detector band pass 420–475 and 500–610 nm. For Btn2-RFP fluorescence: excited by the 555 nm laser; emission was filtered by spectral detector long pass 560 nm. Differential Interference Contrast (DIC) images were acquired simultaneously with the Ure2N-GFP channel.
Preparation of samples for SILAC
Media (for 1 liter): Tyr 50 mg, Ade 80 mg, Asp 80 mg, His 20 mg, Ile 50 mg, Leu 100 mg, Met 20 mg, Phe 50 mg, Thr 100 mg, Trp 50 mg, Ura 20 mg, Val 140 mg, Pro 200 mg, YNB 6.7 g, and Glucose 20 g. For preparation of unlabeled media unlabeled L-Arg (20 mg/L) and L-Lys (30 mg/L) were added. For preparation of labeled media 20 mg/L of L-Arg-2HCl (13C6 99%; 15N4 99%) and 30 mg/L of L-Lys-2HCl (13C6 99%; 15N2 99%) were added. A pre-culture of cells carrying [URE3] and originating from a single colony (strains BEE1386, BEE1387, BEE1392, BEE1395, and BEE1401) was grown to stationary phase in 2 ml of 1X Synthetic Complete—Ade (Sunrise Inc.) liquid media with 0.67% YNB (Difco) and 2% glucose in a 14 ml cultivation tube. 12 µl of the pre-culture was inoculated in 60 ml of unlabeled or labeled media in a 250 ml flask and was grown to OD600 in the range 0.6–0.9. Then 50 ml of culture was placed into a 50 ml conical tube and spun for 3 min in a cold room (3000 g) and the pellet was transferred with cold water to a 1.5 ml microfuge tube. The tube was spun for 20 s in microcentrifuge at 4°C, supernatant was removed and the pellet was washed 3 times with cold water. The pellet was resuspended in 500 ul of cold water, frozen in ethanol with dry ice and placed at –80°C for storage until all samples were collected. Unlabeled wild-type samples were then mixed with equal amounts of labeled mutant samples (based on cell pellet weight). All procedures were made on ice. There were three repeats of each mix of samples. Mixed samples were pelleted in a microcentrifuge, the supernatant was removed and cells were resuspended in 600 ul of lysis buffer (1% sodium dodecanoate, 8 M urea, 1 mM dithiothreitol, 1 mM EDTA, 100 mM PMSF, 1x complete EDTA free protein inhibitor, 5% glycerol, 1 M Tris-HCl pH 7.4) in a 2 ml tube with screw cap. Glass beads were added almost to the top of the tube and cells were homogenized in the cold room for 3 minutes. Using a hot needle, a hole was poked in the bottom of the 2 ml tube, it was placed into a 5 ml tube and subjected to centrifugation at 4000 g for 5 min at 4 C. The supernatant was placed in a 1.5 ml microfuge tube, centrifuged for 5 min at 4 C in a microcentrifuge and the supernatant was placed to a new microfuge tube. An aliquot of the supernatant was taken for BCA assay and the rest of the supernatant was stored at −80°C before trypsinolysis, purification and mass spectrometry. After further reduction and alkylation of thiols samples were digested with trypsin, subjected to off-line high pH concatenated separation, and subjected to LC/MS/MS with data analysis using MaxQuant (Rappsilber et al. 2007; Cox and Mann 2008; Wang et al. 2011) (details in Edskes et al., submitted).
Statistics
The curing data follows the binomial distribution, because each data point expresses two alternative results, loss of [URE3] or its retention. The number of observations (N), the probability of curing (p) and of not curing (q) are used in calculations. The results should be approximately normally distributed as long as Npq ≫ 1, which was true for all our data. We want to calculate the probability that two sets of data (e.g., w.t. vs mutant) could be samples from the same population. If pw, qw, Nw vs pm, qm, Nm are the parameters for w.t. and mutant, respectively, then, combining populations, the overall chance of prion loss is p = (pw•Nw+pm•Nm)/(Nw+Nm), expected loss number in the mutant clones is p•Nm with standard deviation S = ((Nw+Nm)p(1-p))1/2. The difference p•Nm—pm•Nm between expected and observed number of mutant clones that lost the prion, is divided by the standard deviation S, and the probability of this difference or greater from the expected result is obtained from a normal distribution table. Data in Supplementary Figure S2 was analyzed using the Mann-Whitney U-test.
Data availability
Strains and plasmids are available upon request. The authors affirm that all data necessary for confirming the conclusions of the article are present within the article, figures, tables, and Supplementary material which are available at figshare: https://doi.org/10.25386/genetics.13549292.
Results
Isolation of mutants with impaired Btn2p or Cur1p curing of [URE3-1]
In order to find which genes are involved in the process of Btn2p overproduction curing of [URE3], strain BY241 was transformed with pBEE34 carrying BTN2 under the CUP1 promoter and by pSG36 carrying a HERMES transposon containing NatMX (nourseothricin-resistance) and its transposase under the GAL1 promoter. Transposition was induced in galactose, then cells lacking the transposon-carrying plasmid were selected. Among these cells, those resistant to nourseothricin, and thus presumably carrying an insertion of HERMES in the genome, were selected. Then Btn2p overproduction was induced by adding copper (Supplementary Figure S1) and, after a period of growth under this condition, cells were plated on ½ YPD to screen for those retaining [URE3] by their white colony color (Ade+). [URE3] cells have much reduced Ure2p repressor activity, consequently increased DAL5-promoted ADE2 activity and are Ade+ (white). [ure-o] cells have normal Ure2p repression of the DAL5 promoter, little transcription of the DAL5: ADE2 reporter, and accumulation of an adenine pathway intermediate that forms a red pigment. DNA was isolated from these mutants resistant to Btn2-curing of [URE3] and the site of the HERMES insertion was identified by PCR. Their resistance to curing was confirmed by repeated Btn2p overproduction in copper-containing media, and their sensitivity to guanidine-curing [by inhibition of Hsp104, needed for prion propagation (Ferreira et al. 2001; Jung and Masison 2001; Jung et al. 2002)] confirmed that the Ade+ phenotype was still due to the [URE3] prion. Strains with insertions in UBR2 and RPL4A and upstream of RPL21B showed greatly reduced curing of [URE3] by overproduced Btn2. Replacing the Btn2p over-expression plasmid did not improve the curing ruling out a plasmid mutation as the basis of the lack of curing. To confirm that the transposon insertion in these strains was producing the reduction in curing, we made knockouts in the corresponding genes. These knockout strains showed almost no loss of [URE3] on overproduction of Btn2p (Table 4, Figure 1). RPL4A and RPL21B are duplicated genes encoding 60S ribosomal proteins, and each is the major source of the respective protein, with comparable slowing of growth and depletion of 60S subunits in rpl4aΔ and rpl21b strains (Steffen et al. 2008; Pillet et al. 2015). Ubr2p is an ubiquitin ligase that has as one of its targets Rpn4, a transcription factor stimulating expression of proteasomal genes (Wang et al. 2004).
Table 4.
Btn2p and Cur1p overproduction (under the CUP1-promoter) curing of [URE3] in mutants and wild-type
| Overproduction |
||||
|---|---|---|---|---|
| Host | Btn2p |
Cur1p |
||
| # | % Curing | # | % Curing | |
| WT | 732 | 60 | 209 | 90 |
| hsp42Δ | 614 | 0.5* | 112 | 12* |
| ubr2Δ | 708 | 1.6* | 112 | 9* |
| ubr2Δ rpn4Δ | 354 | 59†† | 110 | 21 |
| rpl21bΔ | 1152 | 1.1* | 122 | 8* |
| rpl4aΔ | 1063 | 0.7* | 150 | 46* |
| WT | — | — | 99 | 29 |
| ubr2Δ | — | — | 85 | 3* |
| ubr2Δ rpn4Δ | — | — | 84 | 31** |
| WT | 361 | 94 | 290 | 76 |
| rpl21bΔ | 251 | 7* | 272 | 6* |
| rpn4Δ#1 | 580 | 57 | 393 | 54 |
| rpn4Δ#2 | 207 | 70 | 423 | 65 |
| rpl21bΔ rpn4Δ | 246 | 67† | 162 | 55† |
Cells were grown with inducing concentrations of copper for 12 generations before plating. The stability of [URE3] in the wild-type and mutant strains was tested before introduction of the CUR1/BTN2 overexpression plasmids as well as a 0 mM copper control for the WT (using “copper-free” YNB). Loss of [URE3] was <1% in all of those cases.
= differs from w.t. with p < 2 × 10−4,
= differs from ubr2Δ with p < 2 × 10−3,
= differs from rpl21bΔ with p < 10−4,
= differs from ubr2Δ with p < 10−4.
Figure 1.

Loss of Btn2-overproduction curing of [URE3] in mutants. [URE3-1] strains carrying pBEE34 (CUP1 promoter—BTN2) were inoculated in synthetic complete minus tryptophan media containing 0.25 mM copper sulfate, starting at OD600 = 0.001, grown for 12 generations and dilutions were plated on ½ YPD to score loss of [URE3]. Red color development by ubr2Δ rpn4Δ cells was less than other strains and the Ade- phenotype and loss of [URE3] was confirmed by testing on synthetic complete−Ade plates and mating with [ure-o] cells (Supplementary Figure S7). The stability of [URE3] in the wild-type and mutant strains was tested before introduction of the CUR1/BTN2 overexpression plasmids and loss of [URE3] was <1% in all of those cases.
Subclones of the deletion mutants that had lost the Btn2 overproduction plasmid were obtained and pBEE42 carrying CUP1-promoted CUR1 was introduced. Both ubr2Δ and rpl21bΔ strains were resistant to curing by Cur1 overproduction (Table 4, Supplementary Figure S2).
Has an incurable [URE3] variant been selected?
Curability by normal levels of Btn2p and Cur1p was often an unstable property of newly isolated [URE3] variants, with relatively curing-resistant derivatives arising frequently even in a btn2Δ cur1Δ host (Wickner et al. 2014). [URE3] from Btn2p-incurable mutants hsp42Δ, ubr2Δ, rpl4aΔ, rpl21bΔ or 1021 (wild-type) was cytoduced into wild-type recipient 4591, five cytoductants in each case were transformed by a plasmid with BTN2 or CUR1 under the CUP1 promoter and transformants were subjected to copper induction. All cytoductants analyzed were curable upon Btn2p/Cur1p overproduction (Table 5, Supplementary Figure S3), indicating that the [URE3-1] variant used had not changed to an incurable form. Other attempts to select a variant of [URE3] incurable by Btn2p- or Cur1p- overproduction were unsuccessful.
Table 5.
Cytoduction of [URE3] from Btn2-incurable mutants into wild-type recipient and subsequent Btn2p or Cur1p overproduction
|
Cytoductants into WT
from |
% cured upon
Btn2↑ |
% cured upon
Cur1↑ |
|---|---|---|
| Wild-type | 28–65 | 15–61 |
| hsp42Δ | 24–50 | 27–53 |
| ubr2Δ | 44–58 | 20–44 |
| rpl21bΔ | 8–62 | 22–49 |
| rpl4aΔ | 32–60 | 43–65 |
To test whether the [URE3] prion in incurable mutants was the incurable property, cytoplasm was transferred from the curable w.t. (BY241 [URE3-1]) or the isogenic incurable mutants to the w.t. [ure-o] strain 4,591. Five cytoductants for each case were transformed with pBEE34 (CUP1-BTN2) or pBEE42 (CUP1-CUR1), grown 13–14 generations with Cu and plated on ½ YPD. The stability of [URE3] in these strains was tested before introduction of the CUR1/BTN2 overexpression plasmids and loss of [URE3] was <1% in each cases. Dot plot of data is shown in Supplementary Figure S3. The Mann-Whitney U-test shows there is no statistical difference between the wild-type and any of the mutants.
Western blot analysis of lysates of Btn2p-incurable mutants
Lower levels of Btnp2 or Cur1p overproduction (Kryndushkin et al. 2008), overproduction of Sis1p (Malinovska et al. 2012; Wickner et al. 2014), decreased Hsp42 (Wickner et al. 2014) or elevated Btn3 (Kanneganti et al. 2011) could explain the failure of [URE3] curing in these mutants. We examined levels of Btn2p, Cur1p, Sis1p, Hsp42-HA, and Btn3-HA in mutants and wild-type lysates by western blot (Figures 2 and 3). Levels of Sis1p and Hsp42-HA did not change in the Btn2-incurable mutants. The level of overproduced Btn2p was lower in the ubr2Δ mutant than in wild-type, but rpl4aΔ and rpl21bΔ did not show this effect (Figure 3, Supplementary Figure S4). Levels of Btn3-HA did not increase in the mutants, except in the hsp42Δ mutant (Figure 2B). Because elevated Btn3p prevents curing of [URE3] by elevated Btn2p (Kanneganti et al. 2011), it is possible that our observed increase in Btn3p in the hsp42Δ mutant is part of the explanation for failure of [URE3] curing by Btn2p overproduction in this strain (Wickner et al. 2014).
Figure 2.
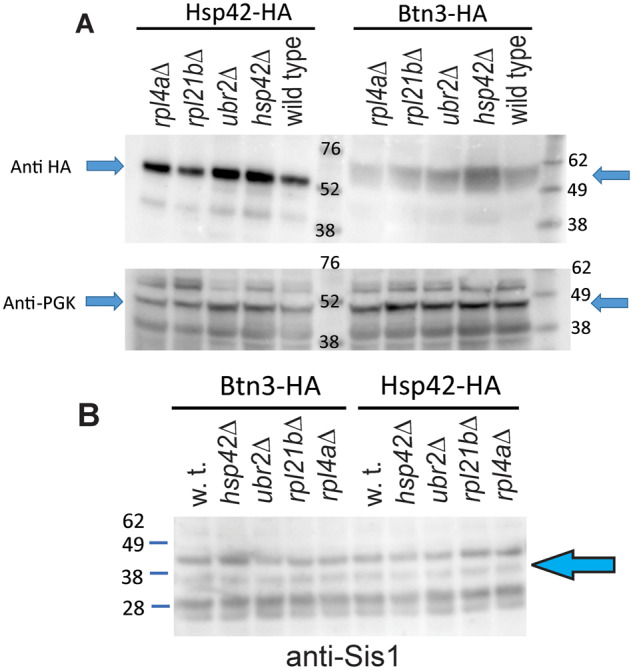
Western blot analysis of Hsp42, Btn3 and Sis1 levels in incurable mutants. (A) Hsp42-HA and Btn3-HA were expressed from their own promoters on CEN plasmids in log phase cells and were visualized by staining with anti-HA tag antibodies. Anti PGK staining was used as a loading control. Btn3 is 57.6 kDa; Hsp42 42.8 kDa; PGK1 44.7 kDa, and the HA tag adds 1.1 kDa. For Western blots 10 μg of protein per lane was loaded. (B) Anti-Sis1 staining of lysates of wild-type and Btn2-incurable mutants. Strains carried CEN plasmids expressing Btn2-HA and Hsp42-HA from their own promoters.
Figure 3.

Btn2p and Cur1p levels in incurable mutants. After pregrowth on 3 mM guanidine HCl to remove [URE3], cells were grown in SC-Trp with 250 μM copper sulfate. Extracts (10 μg per lane) were loaded on a 12% MOPS polyacrylamide gel. The primary antibody was anti-Btn2p or affinity-purified rabbit anti-Cur1p, and the secondary antibody was Promega anti-rabbit IgG(Fc) AP conjugate S3738. Cur1p has a predicted molecular mass of 29.2 kDal and Btn2p is 47.1 kDa. Lane 1: BEE1012 btn2Δ cur1Δ; Lane 2: BY241 wild-type; Lane 3: BEE1319 rpn4Δ; Lane 4: BEE1190 hsp42Δ; Lane 5: BE1192 ubr2Δ; Lane 6: BEE1272 rpl4aΔ; Lane 7: BEE1251 rpl21bΔ; Lane 8: BEE1417 rpl21bΔ rpn4Δ; Lane 9: BEE1318 ubr2Δ rpn4Δ.
The levels of overproduced Cur1p were lower in the ubr2Δ strain, explaining the incurability of [URE3] in this strain. Cur1p is substantially higher in the hsp42Δ host (Figure 3, Supplementary Figure S4). Cur1p was not lower in the rpl4aΔ or rpl21bΔ strains, although both strains were resistant to curing by overproduced Cur1p (Table 4). The reduced levels of Cur1p in ubr2Δ strains are restored to above-normal levels by including the rpn4Δ mutation that prevents overactivity of the proteasome system (Figure 3, Supplementary Figure S4).
Colocalization of Btn2p with Ure2p in mutants
Btn2-RFP and Ure2N-GFP were expressed from different plasmids in wild-type and Btn2-incurable mutants. Living cells then were subjected to microscopy. It was found that Btn2-RFP foci still largely colocalized with Ure2N-GFP aggregates in ubr2Δ, rpl4aΔ, and rpl21bΔ as well as in wild-type, but did not colocalize in hsp42Δ (Figure 4, Supplementary Table S1).
Figure 4.
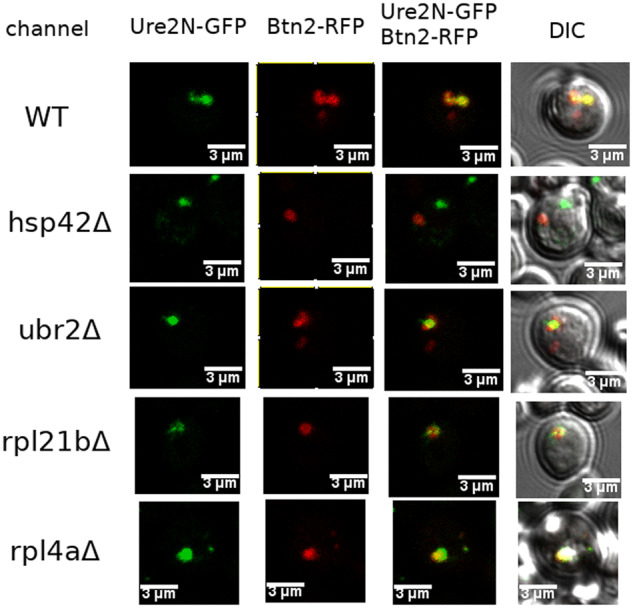
Co-localization of Btn2-RFP and Ure2-GFP in wild-type, ubr2Δ, rpl4aΔ, and rpl21bΔ, but not in hsp42Δ incurable mutants. Btn2-RFP was expressed from pBEE28 under a GAL1 promoter, and Ure2N-GFP was expressed from pBEE29 under a constitutive promoter. Wild-type and mutant cells were transformed by both pBEE28 and pBEE29 plasmids. Cells grown to stationary phase in liquid synthetic media with 3% raffinose were diluted to OD600 = 0.5 in liquid synthetic media with 2% raffinose and 3% galactose, were grown overnight and then subjected to microscopy.
Seed number of [URE3] in Btn2p-incurable mutants
At normal levels, Btn2p and Cur1p cure [URE3] prion variants with particularly low seed numbers, but not those with higher seed numbers, which can only be cured by overproducing the curing proteins (Wickner et al. 2014). Seed number of [URE3-1] in ubr2Δ, rpl4aΔ, and rpl21bΔ was determined using the guanidine method of Cox et al. (2003) (see Material and Methods section) and showed no consistent increase in these mutants in comparison with wild-type in three experiments (Figure 5). Paradoxically, hsp42Δ strains showed a modest tendency toward lower seed numbers than wild-type, even though they were resistant to curing (Figure 5).
Figure 5.

Propagon/seed number of [URE3-1] in wild-type and Btn2-incurable mutants. Strains with [URE3-1] were plated on YPAD containing guanidine (3 mM in the top and bottom panels, 5 mM in the middle panel) to inhibit Hsp104 (Jung et al. 2002) and thus block prion propagation by preventing filament cleavage (Ness et al. 2002). Each of 10 colonies was resuspended in water and plated on SC-Ade, and the number of colonies forming is a measure of propagon number (Cox et al. 2003). Separate experiments with the same mutant are shown as separate “whisker” diagrams.
Btn2p or Cur1p overproduction curing of [URE3] in Hsp104 T160M mutants
Overproduction of Hsp104 is best known for its curing of [PSI+] (Chernoff et al. 1995), but overproduced Hsp104 can also, less efficiently, cure [URE3] (Kryndushkin et al. 2008; Matveenko et al. 2018). Mutants in the N-terminal domain of Hsp104 eliminate its curing ability while not affecting its ability to promote prion propagation (Hung and Masison 2006). We overproduced Btn2p or Cur1p in one such mutant, Hsp104 T160M, and found that curing proceeded efficiently, indicating that the Hsp104 curing activity is not required for the curing of [URE3] by Btn2p or Cur1p overproduction (Table 6, Supplementary Figure S5). In addition, co-overproduction of Hsp104 with Btn2p did not affect curing of [URE3].
Table 6.
Btn2 or Cur1 overproduction curing of [URE3] in Hsp104 T160M mutants
| Strain | Prion curing (%) |
|---|---|
| WT BTN2↑ | 94–98 |
| T160M BTN2↑ | 83–94 |
| WT CUR1↑ | 77–95 |
| T160M CUR1↑ | 52–83 |
Wild-type (strains BEE1201, BEE1203) and hsp104(T160M) mutants (strains BEE1200 and BEE1202) carrying [URE3-1] were transformed with pBEE34 (CUP1-BTN2) or pBEE42 (CUP1-CUR1), grown with CuSO4 to induce Btn2/Cur1 production and plated on ½ YPD to measure curing. A total of >1,500 colonies were counted in each case and the range of curing frequency is shown. The stability of [URE3] in the wild-type and mutant strains was tested before introduction of the CUR1/BTN2 overexpression plasmids as well as a 0 mM copper control for the WT (using “copper-free” YNB). Loss of [URE3] was <1% in all of these cases. See also Supplementary Figure S5.
Rpl4Ap overproduction does not cure [URE3]
As deletion of RPL4A stabilizes [URE3] against challenge by Btn2p or Cur1p, it was possible that Rpl4Ap has an anti-prion action independent of Btn2p/Cur1p overproduction. However, we find that GAL1—promoted Rpl4Ap overproduction does not cure the [URE3-1] prion in strain BY241 (Supplementary Figure S6). We also examined 96 [URE3] variants generated in an rpl4aΔ host, and find that none were destabilized on restoration of RPL4A by mating with a wild-type host. Thus, there are no (or very few) [URE3] variants for which Rpl4Ap by itself has an anti-prion action.
Deficiency of 60S ribosomal subunit genes prevents Btn2/Cur1 overproduction curing of [URE3]
To determine whether the effects on prion curing of rpl21b is paralog—specific, we tested an rpl21aΔ mutant, and found that it too prevented [URE3] curing by overproduced Btn2p or Cur1p (Table 7). Both rpl21aΔ and rpl21bΔ noticeably slow cell growth (Steffen et al. 2008; Bhattacharya et al. 2010; Yoshikawa et al. 2011) indicating that each contributes substantially to the pool of the Rpl21 ribosomal protein. Indeed, we find that Btn2p and Cur1p overproduction curing of [URE3] was inhibited in rpl11bΔ and rpl16bΔ mutants which decrease 60S ribosomal subunits levels, but curing was still occurring in rps14aΔ and rps30bΔ strains which decrease 40S ribosomal subunits levels (Figure 6). Thus, 60S ribosomal subunit deficiency, rather than slowing of translation per se, seems to be the factor preventing prion curing. Note that rps14aΔ and rps30bΔ each limit translation enough to slow growth as much as rpl21bΔ or rpl11bΔ (Steffen et al. 2008; Bhattacharya et al. 2010; Yoshikawa et al. 2011).
Table 7.
Btn2 and Cur1 overproduction curing of [URE3] prion is impaired in rpl21aΔ mutant (RPL21A is a paralog of RPL21B)
| Sample plated on ½ YPD | Total | % Curing |
|---|---|---|
| Wild-type Btn2↑ | 612 | 71 |
| rpl21aΔ#1 Btn2↑ | 580 | 3* |
| rpl21aΔ#2 Btn2↑ | 460 | 2* |
| Wild-type Cur1↑ | 846 | 41 |
| rpl21aΔ#1 Cur1↑ | 564 | 4* |
| rpl21aΔ#2 Cur1↑ | 592 | 4* |
Wild-type strain BEE1021 and two verified rpl21aΔ knockout isolates (#1, #2) were transformed with pBEE34 (CUP1-BTN2) or pBEE42 (CUP1-CUR1) and Btn2p or Cur1p overproduction was induced by growth with CuSO4 as described in Methods. Aliquots of the indicated dilutions of each culture were plated on ½ YPD and red/white colonies were scored. The stability of [URE3] in the wild-type and mutant strains was tested before introduction of the CUR1/BTN2 overexpression plasmids as well as a 0 mM copper control for the WT (using “copper-free” YNB). Loss of [URE3] was <1% in all of these cases.
= differs from w.t. with p < 10−4.
Figure 6.

60S ribosomal subunit deficiency prevents [URE3-1] curing. Mutants in genes encoding 60S ribosomal subunit proteins carrying [URE3-1] and overproducing Btn2p or Cur1p were examined as described in section Methods. The stability of [URE3] in the wild-type and mutant strains was tested before introduction of the CUR1/BTN2 overexpression plasmids and loss of [URE3] was <1% in all cases.
Restoration of Btn2/Cur1 overproduction curing of [URE3] in ubr2Δ rpn4Δ and rpl21bΔ rpn4Δ double mutants
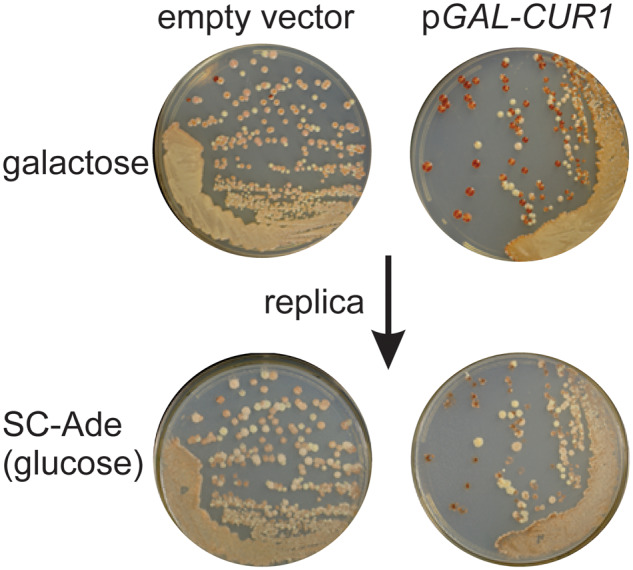
Ubr2 is an E3 ubiquitin ligase whose target is Rpn4, a transcription factor promoting expression of proteasome components (Wang et al. 2004). If the abrogation of Btn2/Cur1-curing of [URE3] is because of this action of Ubr2p, we expected that in a ubr2Δ rpn4Δ double mutant Btn2p or Cur1p overproduction and prion curing would be restored in comparison with a single ubr2Δ mutant. Indeed this is the case (Table 4, Figures 1 and 3). The ubr2Δ rpn4Δ double mutant colonies cured of [URE3] were pink on ½ YPD (instead of red as in wild-type), but were Ade- (Supplementary Figure S7A), and curing was confirmed by mating of the pink mutant colonies with wild-type [ure-0] cells and replica plating them on a media without adenine (Supplementary Figure S7B). Moreover, the reduced levels of Cur1p found in ubr2Δ strains were restored in the ubr2Δ rpn4Δ double mutant (Figure 3).
Because mutations in 60S ribosomal subunit proteins (but not in 40S proteins) result in elevated proteasome components (Cheng et al. 2019), we examined whether rpn4Δ would reverse the effects of rpl21bΔ mutations on curing of [URE3] by Btn2p or Cur1p. Indeed, we found that in a rpn4Δ rpl21bΔ strain, either Btn2p or Cur1p cured [URE3] with efficiencies similar to what is found in a wild-type host (Table 4).
Btn2p overproduction curing of [URE3] in mutants of Btn2p-interactors
In its protein—trafficking role, Btn2p interacts with SNARE and endosome proteins (Kama et al. 2007). Btn2p overproduction was performed in knockouts of genes shown to interact with Btn2p (Kama et al. 2007) but there was little influence on Btn2p overproduction curing in these mutants in comparison with wild-type (Table 8).
Table 8.
Btn2 overproduction (from copper promoter) curing in mutants of genes shown to interact with Btn2 (Kama et al. 2007)
| Strain | Btn2 overproduction |
Cur1 overproduction |
||
|---|---|---|---|---|
| Colonies | % Curing | Colonies | % Curing | |
| Wild-type | 637 | 69 | 408 | 59 |
| snc1Δ | 538 | 68 | 349 | 70 |
| snc2Δ | 417 | 87 | 230 | 73 |
| pep8Δ | 301 | 88 | 233 | 81 |
| vps27Δ | 401 | 79 | 559 | 66 |
| tlg2Δ | 224 | 34 | 248 | 50 |
| vps35Δ | 419 | 92 | 433 | 84 |
Strain BY241 (w.t.) and isogenic knockout strains were tested for curing as described in Methods. The stability of [URE3] in the wild-type and mutant strains was tested before introduction of the CUR1/BTN2 overexpression plasmids as well as a 0 mM copper control for the WT (using “copper-free” YNB). Loss of [URE3] was <1% in all of these cases. In all mutants, robust curing was observed by overproduction of either Btn2 or Cur1. While there were statistically significant differences (e.g., the tlg2Δ strain was slightly more slowly for curing by Btn2p, while the pep8 Δ and vps35Δ were slightly more rapidly cured by Btn2 or Cur1, vps27Δ by Cur1 and snc2Δ by Btn2), all were well cured.
Generation of [PSI+] variants curable by Cur1p overproduction
None of the three [PSI+] variants previously tested were cured by Btn2p or Cur1p overexpression (Kryndushkin et al. 2008; Barbitoff et al. 2017). We examined five different [PSI+] variants (strong [PSI+], Sc4, Sc37, Lieb W, and Lieb S), transforming each with 2 µ plasmids carrying BTN2 (p1289) or CUR1 (p1488) under their own promoters and no curing was observed.
In strain 74-D694 carrying BTN2 (pBEE1) or CUR1 (pBEE2) under a GAL1 promoter on a CEN plasmid and pSL1066 with CUP1-promoted SUP35-NM-GFP, we generated [PSI+] variants by growth with CuSO4. Sixty BTN2- and 60 CUR1- plasmid containing [PSI+] isolates were streaked on minimal dextrose or galactose plates with limited adenine (10 mg/l). Nine clones for CUR1- and 4 clones for BTN2-overexpression showed more red or darker pink color on galactose than on dextrose plates. In the next step, candidates were streaked on YPAD plates to remove plasmids and retransformed with an empty vector or a plasmid with BTN2 or CUR1. Three subclones of each Btn2/Cur1-sensitive [PSI+] variant were analyzed. The effect of curing was reproduced only for 1 subclone (out of 3 subclones) for 2 CUR1-sensitive [PSI+] clones (Figure 7). Thus, these variants are unusual and quite unstable.
Figure 7.
Rare [PSI+] variant sensitive to curing by Cur1p overproduction. In strain 74D-694 carrying pBEE2 (CEN LEU2 GAL1promoter-CUR1) and pSL1066 (CEN URA3 CUP1promoter-SUP35NM-GFP) [PSI+] variants were copper-induced. Cur1-sensitive colonies were found by subcloning on galactose media and picking those which became [psi-]. The original [PSI+] Cur1-sensitive variants were streaked on YPAD plates to allow loss of pBEE2 and pSL1066, were retransformed with pBEE2 or empty vector control (pH316) and tested for Cur1-sensitivity again by streaking cells on selective plates containing 2% galactose and 10 mg/l adenine. The galactose plates were replica plated to dextrose plates lacking adenine (below).
SILAC of Btn2-incurable mutants
To detect proteins whose elevated or depressed levels may be affecting Btn2 curing, we measured the ratio of over 4600 proteins from isogenic mutant and wild-type cells using SILAC (see Methods section), labeling one with L-Arginine-13C6,15N4 hydrochloride and L-Lysine-13C6,15N2 hydrochloride, and the other with 12C, 14N amino acids. Cells were mixed, extracted, and proteins were digested with trypsin, so that each peptide had one amino acid differently labeled between mutant and wild-type. Digested extract mixtures were analyzed by mass spectrometry (ONG et al. 2002).
Several proteins showed modest changes in curing-resistant strains compared to the isogenic wild-type (Supplementary Tables S2 and S3), some with evident connection to the auxotrophies of these strains. None of these seemed clearly related to the curing process. However, Tma10p was dramatically elevated in hsp42Δ, rpl4aΔ, and rpl21bΔ strains 28-, 22-, and 39-fold, respectively, but only slightly elevated in rps30bΔ, which does not affect Btn2p curing of [URE3]. It was possible that elevated levels of Tma10p would antagonize Btn2p’s curing activity, or that Tma10p is necessary for [URE3] propagation. Overexpressing Tma10p from a GAL promoter (p1678) in parallel with GAL-promoted Btn2p (pBEE1), did not, however, diminish the curing efficiency in a wild-type host (Supplementary Table S3). Likewise, a BY241 tma10::kanMX host (5991) maintained [URE3-1] as stably as an isogenic wild-type.
Changes in Hsp42, Hsp104, Sis1p, Ydj1p, Swa2p, Cpr7, Btn3p, or Ure2p itself could affect loss of the prion (Aigle and Lacroute 1975; Moriyama et al. 2000; Higurashi et al. 2008; Kanneganti et al. 2011; Wickner et al. 2014; Kumar et al. 2015; Troisi et al. 2015). None of these proteins were changed significantly more in rpl4a and rpl21b than in rps30a (Table 9).
Table 9.
SILAC levels of some proteins affecting [URE3]
|
Mutant=>
Protein |
rpl4aΔ/w.t. | rpl21bΔ/w.t. | rps30bΔ/w.t. |
|---|---|---|---|
| Hsp42 | 1.34 ± 0.07 | 1.27 ± 0.27 | 1.27 ± 0.16 |
| Btn2 | NSD | NSD | NSD |
| Cur1 | NSD | NSD | NSD |
| Hsp82 | 0.84 ± 0.25 | 0.82 ± 0.24 | 0.80 ± 0.37 |
| Hsp104 | 0.95 ± 0.14 | 0.99 ± 0.22 | 0.94 ± 0.34 |
| Rpn4 | NSD | NSD | NSD |
| Ubr2 | NSD | NSD | NSD |
| Ubi4 | 0.90 ± 0.02 | 0.93 ± 0.07 | 0.94 ± 0.13 |
| Sis1 | 0.86 ± 0.06 | 0.86 ± 0.05 | 0.91 ± 0.04 |
| Ydj1 | 0.74 ± 0.04 | 0.82 ± 0.06 | 0.83 ± 0.04 |
| Hsp26 | 1.50 ± 1.1 | 1.50 ± 1.9 | 1.4 ± 1.9 |
| Swa2 | 0.78 ± 0.03 | 0.86 ± 0.07 | 0.82 ± 0.2 |
| Ure2 | 0.95 ± 0.11 | 0.99 ± 0.06 | 0.99 ± 0.25 |
| Btn3 | 0.76 ± 0.15 | 0.81 ± 0.12 | 0.83 ± 0.17 |
| Cpr7 | 0.86 ± 0.02 | 0.94 ± 0.05 | 0.90 ± 0.13 |
Each ratio was the result of three independent determinations (mean ± standard deviation). NSD = not sufficient data.
Discussion
We used two approaches to find genes affecting Btn2p and Cur1p curing of the [URE3] prion. We examined, as candidates, genes known to be involved in Btn2p’s action in protein sorting. Btn2 interacts in two-hybrid assays with Snc1 and Snc2, and binds in extracts to Tlg1, Tlg2 and Vti1, as well as Snc1 and Snc2, all endocytic SNARE components mediating membrane fusion of vesicles with endosomes (Kama et al. 2007). Btn2p also co-localizes with Vps27p and co-IPs with Vps26p and Vps35p. We find that snc1Δ, snc2Δ, tlg2Δ, vps27Δ, vps26Δ, and vps35Δ (TLG1 and VTI1 are essential) have no substantial effect on the Btn2p overproduction curing of [URE3], suggesting that Btn2p has two separate functions. Of course, Btn3p, shown to bind to Btn2p, inhibits both activities when overproduced (Kanneganti et al. 2011).
We also used HERMES transposon mutagenesis in an unbiased screen for genes whose mutation would interfere with curing of [URE3] by Btn2p or Cur1p. Our screen identified rpl4a, rpl21b, and ubr2 mutants with this property. Deletion mutants of these genes had the same effect. Curing was also diminished by rpl21a, rpl11b, or rpl16b, but not by rps14a or rps30b, indicating that 60S subunit deficiency was specifically involved, but not a particular 60S ribosomal subunit protein.
Consequences of 60S subunit deficiency
Mutation of ribosomal protein genes has the potential to produce specific phenotypes by a variety of mechanisms. Fifty-nine ribosomal proteins are encoded by paralogous pairs of genes, which can differ in encoded sequence, but in the most carefully studied cases, differences in paralog expression levels have explained the observed phenotype differences (e.g., Palumbo et al. 2017). The extensive modifications of rRNAs and ribosomal proteins could produce a large array of distinct ribosomes, but there is little evidence that this produces functional varieties (reviewed by Dinman 2016). Extraribosomal functions of ribosomal proteins have been documented in a few cases (Blumenthal and Carmichael 1979), reviewed in Warner and McIntosh (2009), but since all 60S ribosomal protein gene deletions tested suppressed curing, it is not likely an extraribosomal effect here. An overall deficiency of active ribosomes might be expected to retard translation of some mRNAs more than others, but should be produced equally by either 40S or 60S deficiency. However, several cases of defects selective for 60S subunits rather than 40S have been described.
Mutations in any of 30 chromosomal genes (MAK for maintenance of killer) lose the M1 dsRNA satellite dsRNA of the L-A virus and have lowered copy number of L-A itself (Wickner 2013). Eighteen of the 30 MAK genes were deficient in 60S ribosomal subunits, many encoding 60S subunit proteins (Ohtake and Wickner 1995). None had 40S subunit protein defects. It has been suggested that impaired expression of the viral nonpolyA mRNA relative to cellular polyA+ mRNAs (Edskes et al. 1998) combined with the preferential use of the viral coat proteins by the L-A helper virus, can explain the preferential loss of the satellite segment. Except for viral mRNAs, there are no known nonpolyA mRNAs in S. cerevisiae.
60S subunit deficiency also prolongs the replicative life span of S. cerevisiae, the number of daughter cells that can be made by a single mother cell (Steffen et al. 2008). This phenomenon requires the action of Gcn4p (Steffen et al. 2008), best known for its role in the general control of amino acid biosynthesis (Hinnebusch 2005). Gcn4p expression requires the read-through of several small upstream open reading frames, and 60S subunit deficiency is known to facilitate this readthrough (Martín-Marcos et al. 2007). While rpl21bΔ showed life span extension, rpl4aΔ, rpl11bΔ, rpl16bΔ, and rpl21aΔ did not consistently do so (Steffen et al. 2008), suggesting that the prion curing suppression effect was not due to elevated Gcn4p. Moreover, our SILAC data did not show significant changes in Gcn4p. However, there are other yeast genes known to have small upstream ORFs which may be involved (Lawless et al. 2009).
Recently, Tye et al. showed that limiting 60S subunit biogenesis by lowering levels of certain rRNA processing RNAses, results in elevated Hsf1p, a transcription factor promoting heat shock protein and other proteostasis factor transcription (Tye et al. 2019). Chaperones sequestered by the aggregated excess ribosomal subunit were believed to induce Hsf1p and consequent transcription of BTN2, HSP42, and HSP82, among other genes. While there is a slight increase of Hsp42 in our incurable mutants, there is no change of Hsp82 (Table 9) unlike the dramatic increase seen by Tye et al. (2019). Moreover, the effect observed by Tye et al. would be expected to facilitate [URE3] curing, rather than impair it. We also looked for other proteins involved in [URE3] propagation that showed substantial change in rpl4aΔ and rpl21bΔ, but not in rps30aΔ. The SILAC data does not show differential depression or elevation of any such proteins, including several that are Hsf1p-controlled (Table 9). However, it remains possible that Hsp42 capture by aggregates of ribosomal components plays a part in limiting the action of Btn2p or Cur1p.
A proteomic study of mutants deficient in a series of 60S or 40S subunit proteins found that no specific class of proteins was differentially affected in 40S subunit protein mutants, but that the ubiquitin/proteasome system (UPS) components were overproduced specifically in 60S subunit protein mutants (Cheng et al. 2019). Ubr2p is part of the cytoplasmic Mub1-Ubr2-Rad6 E3 ubiquitin ligase complex that targets Rpn4p, a transcription factor that promotes expression of genes for components of the UPS (Wang et al. 2004). Thus, ubr2Δ and rplxΔ strains should also have a hyperactive UPS system, and this effect should be blocked in ubr2Δ rpn4Δ and rplxΔ rpn4Δ strains, as we have observed. Hyperactivity of the UPS system could thus explain the effects of both rplxΔ and ubr2Δ mutants.
Btn2p and Cur1p are normally present at very low levels, but are elevated on inhibition of the UPS system (Malinovska et al. 2012; H. K. Edskes et al., unpublished). Our western blots of Btn2p and Cur1p showed that overproduction of these proteins was diminished in ubr2Δ strains, with levels restored by an rpn4Δ mutation, indicating that effects on curing by Btn2p and Cur1p is mediated by the effects of ubr2Δ on the ubiquitin-proteasome system. The levels of overexpressed Btn2p and Cur1p were not significantly reduced in rpl21bΔ or rpl4aΔ. However, the effect of the rplxΔ mutations on curing was eliminated by rpn4Δ, again implicating the proteasome. It is possible that another factor involved in Btn2p and Cur1p overproduction curing is affected by proteasome activity. We also examined the levels of Btn3p, Sis1p, and Hsp42, all known to affect prion curing by Btn2p or Cur1p, but none were substantially altered in rplxΔ or ubr2Δ strains.
Our proteomic analysis of rpl4aΔ and rpl21bΔ strains by SILAC mass spectrometry revealed a specific dramatic elevation of Tma10p in these strains, but not in an rps30bΔ strain. However, co-overexpression of Tma10p from the very strong GAL1 promoter did not impair curing of [URE3] by Btn2p. This implies that Tma10p elevation is not sufficient for the diminished curing, but does not rule out some role for this protein. We also found that Tma10p is not necessary for [URE3] propagation. While several modestly over- or under-produced proteins were detected by the SILAC experiments (Supplementary Table S2), none had obvious connection to the curing process. The unamplified Btn2p levels in these strains were too low to be reliably detected by this method. It is possible that the protein(s) mediating the mutant effects on curing of [URE3] are not detected by the SILAC experiment.
Roles of Btn3 and Hsp42
Hsp42 overproduction can cure [URE3] and is necessary for overproduction curing by Btn2p or Cur1p (Wickner et al. 2014). Btn3p is an inhibitor of Btn2p for both its protein sorting and [URE3]-curing activities (Kanneganti et al. 2011). Btn3p directly binds to Btn2p and changes its localization, explaining the other effects (Kanneganti et al. 2011). We find that Btn3 is substantially elevated in hsp42Δ, a finding that could explain, in part, the lack of [URE3] curing by elevated Btn2p in hsp42Δ strains (Wickner et al. 2014). We also noted that Btn2-RFP is not co-localized with Ure2-GFP in [URE3-1] hsp42Δ strains, consistent with Btn3p blocking Btn2p action in this strain. Alternatively, Hsp42, which is known to bind to aggregated proteins (Specht et al. 2011), may be part of a Btn2-Hsp42-amyloid complex in which Btn2 [human HOOK1 homolog (Kama et al. 2007)] attaches to the cytoskeleton or microtubules to direct the complex to its special site.
Overall, our results suggest that large ribosomal protein gene mutations or ubr2 mutation, through their known activation of proteasomes, increase the degradation of Btn2p or Cur1p or their cofactors, resulting in failure of curing of the [URE3] prion. Current work on a variety of human amyloidoses is revealing that they have many close similarities to the classical PrP-based prion diseases. S. cerevisiae has a wide array of anti-prion systems, blocking prion generation, curing most prion variants as they arise and reducing the toxicity of those that do arise, in each case without overexpression or deficiency of any components (reviewed in Wickner et al. 2018). These systems are analogous to the innate immunity systems of mammals, active against virus or bacterial infection. It is hoped that detailed understanding of these systems in yeast will facilitate the discovery and utilization of analogous or homologous systems in humans active against amyloid diseases. A region of Btn2p is homologous to human HOOK1 (Kama et al. 2007), a microtubule-binding protein involved in clathrin-independent endocytosis (Maldonado-Baez et al. 2013) and associated with tau aggregates in Alzheimer’s disease (Herrmann et al. 2015). The association of the E3 ubiquitin ligases, HECTD2 and PARKIN with susceptibility to PrP-based prion disease and Parkinson’s disease (Shimura et al. 2000; Lloyd et al. 2009), respectively, is an interesting parallel with our findings.
Conflicts of interest
None declared.
Acknowledgments
We thank the NIDDK Advanced Light Microscopy and Image Analysis Core (ALMIAC) for use of their equipment, David Young and Nick Guydosh for ribosomal advice, and Eric Anderson for the skilled performance of the SILAC analyses (NIDDK Mass Spec Facility). The authors are grateful to John Woolford (Carnegie Mellon University), and Mike Reidy, Shailesh Kumar, Dan Masison, Emily Stroobant, and Morgan DeWilde (NIDDK) for strains and plasmids used in this work.
Funding
This study was supported by the Intramural Program of the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health.
Literature cited
- Aigle M, Lacroute F.. 1975. Genetical aspects of [URE3], a non-Mendelian, cytoplasmically inherited mutation in yeast. Mol Gen Genet. 136:327–335. [DOI] [PubMed] [Google Scholar]
- Barbitoff YA, Matveenko AG, Moskalenko SE, Zemlyanko OM, Newnam GP, et al. 2017. To CURe or not to CURe? Differential effects of the chaperone sorting factor Cur1 on yeast prions are mediated by the chaperone Sis1. Mol Microbiol. 105:242–257. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A, McIntosh KB, Willis IM, Warner JR.. 2010. Why Dom34 stimulates growth of cells with defects of 40S ribosomal subunit biosynthesis. Mol Cell Biol. 30:5562–5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal T, Carmichael GG.. 1979. RNA replication: function and structure of Qbeta-replicase. Annu Rev Biochem. 48:525–548. [DOI] [PubMed] [Google Scholar]
- Brachmann A, Baxa U, Wickner RB.. 2005. Prion generation in vitro: amyloid of Ure2p is infectious. Embo J. 24:3082–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay S, Muzaffar NE, Sherman F, Pearce DA.. 2000. The yeast model for batten disease: mutations in BTN1, BTN2, and HSP30 alter pH homeostasis. J Bacteriol. 182:6418–6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay S, Roberts PM, Pearce DA.. 2003. The yeast model for Batten disease: a role for Btn2p in the trafficking of the Golgi-associated vesicular targeting protein, Yif1p. Biochem Biophys Res Commun. 302:534–538. [DOI] [PubMed] [Google Scholar]
- Cheng Z, Mugler CF, Keskin A, Hodapp S, Chan LY-L, et al. 2019. Small and large ribosomal subunit deficiencies lead to distinct gene expression signatures that reflect cellular growth rate. Mol Cell 73:36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernoff YO, Lindquist SL, Ono B-I, Inge-Vechtomov SG, Liebman SW.. 1995. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi]. Science 268:880–884. [DOI] [PubMed] [Google Scholar]
- Christianson TW, Sikorski RS, Dante M, Shero JH, Hieter P.. 1992. Multifunctional yeast high-copy-number shuttle vectors. Gene 110:119–122. [DOI] [PubMed] [Google Scholar]
- Conde J, Fink GR.. 1976. A mutant of Saccharomyces cerevisiae defective for nuclear fusion. Proc Natl Acad Sci USA. 73:3651–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper TG. 2002. Transmitting the signal of excess nitrogen in Saccharomyces cerevisiae from the Tor proteins to the GATA factors: connecting the dots. FEMS Microbiol Rev. 26:223–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox BS, Ness F, Tuite MF.. 2003. Analysis of the generation and segregation of propagons: entities that propagate the [PSI+] prion in yeast. Genetics 165:23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M.. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b. - range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 26:1367–1372. [DOI] [PubMed] [Google Scholar]
- Derkatch IL, Bradley ME, Hong JY, Liebman SW.. 2001. Prions affect the appearance of other prions: the story of [PIN(+)]. Cell 106:171–182. [DOI] [PubMed] [Google Scholar]
- Derkatch IL, Bradley ME, Zhou P, Chernoff YO, Liebman SW.. 1997. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics 147:507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinman JD. 2016. Pathways to specialized ribosomes: the Brussels lecture. J Mol Biol. 428:2186–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edskes H K, , Wickner R B. 2000. A protein required for prion generation: [URE3] induction requires the Ras-regulated Mks1 protein. Proceedings of the National Academy of Sciences. 97:6625–6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edskes HE, Mukhamedova M, Edskes BK, Wickner RB.. 2018. Hermes transposon mutagenesis shows [URE3] prion pathology prevented by a ubiquitin-targeting protein: evidence for carbon/nitrogen assimilation cross-talk and a second function for Ure2p. Genetics 209:789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edskes HK, Gray VT, Wickner RB.. 1999. The [URE3] prion is an aggregated form of Ure2p that can be cured by overexpression of Ure2p fragments. Proc Natl Acad Sci USA. 96:1498–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edskes HK, Ohtake Y, Wickner RB.. 1998. Mak21p of Saccharomyces cerevisiae, a homolog of human CAATT-binding protein, is essential for 60S ribosomal subunit biogenesis. J Biol Chem. 273:28912–28920. [DOI] [PubMed] [Google Scholar]
- Edskes HK, Wickner RB.. 2002. Conservation of a portion of the S. cerevisiae Ure2p prion domain that interacts with the full - length protein. Proc Natl Acad Sci USA. 99:16384–16391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira PC, Ness F, Edwards SR, Cox BS, Tuite MF.. 2001. The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol Microbiol. 40:1357–1369. [DOI] [PubMed] [Google Scholar]
- Gangadharan S, Mularoni L, Fain-Thornton J, Wheelan SJ, Craig NL.. 2010. DNA transposon Hermes inserts into DNA in nucleosome-free regions in vivo. Proc Natl Acad Sci USA. 107:21966–21972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover JR, Kowal AS, Schirmer EC, Patino MM, Liu J-J, et al. 1997. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell 89:811–819. [DOI] [PubMed] [Google Scholar]
- Guo Y, Park JM, Cui B, Humes E, Gangadharan S, et al. 2013. Integration profiling of gene function with dense maps of transposon integration. Genetics 195:599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann L, Wiegmann C, Arsalan-Werner A, Hilbrich I, Jager C, et al. 2015. Hook proteins: association with Alzheimer pathology and regulatory role of hook3 in amyloid beta generation. PLoS One 10:e0119423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higurashi T, Hines JK, Sahi C, Aron R, Craig EA.. 2008. Specificity of the J-protein Sis1 in the propagation of 3 yeast prions. Proc Natl Acad Sci USA. 105:16596–16601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG. 2005. Translational regulation of GCN4 and the general amino acid control in yeast. Annu Rev Microbiol. 59:407–450. [DOI] [PubMed] [Google Scholar]
- Hung GC, Masison DC.. 2006. N-terminal domain of yeast Hsp104 chaperone is dispensable for thermotolerance and prion propagation but necessary for curing prions by Hsp104 overexpression. Genetics 173:611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung G, Jones G, Masison DC.. 2002. Amino acid residue 184 of yeast Hsp104 chaperone is critical for prion-curing by guanidine, prion propagation, and thermotolerance. Proc Natl Acad Sci USA. 99:9936–9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung G, Masison DC.. 2001. Guanidine hydrochloride inhibits Hsp104 activity in vivo: a possible explanation for its effect in curing yeast prions. Curr Microbiol. 43:7–10. [DOI] [PubMed] [Google Scholar]
- Kama R, Kanneganti V, Ungermann C, Gerst JE.. 2011. The yeast Batten disease ortholog Btn1 controls endosome-Golgi retrograde transport via SNARE assembly. J Cell Biol. 195:203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kama R, Robinson M, Gerst JE.. 2007. Btn2, a Hook1 ortholog and potential Batten disease-related protein, mediates late endosome-Golgi protein sorting in yeast. Mol Cell Biol. 27:605–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanneganti V, Kama R, Gerst JE.. 2011. Btn3 is a negative regulator of Btn2-mediated endosomal protein trafficking and prion curing in yeast. Mol Biol Cell 22:1648–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly AC, Shewmaker FP, Kryndushkin D, Wickner RB.. 2012. Sex, prions and plasmids in yeast. Proc Natl Acad Sci USA. 109:E2683–E2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C-Y, Diaz-Avalos R.. 2004. Protein-only transmission of three yeast prion strains. Nature 428:319–323. [DOI] [PubMed] [Google Scholar]
- King C-Y, Tittmann P, Gross H, Gebert R, Aebi M, et al. 1997. Prion-inducing domain 2-114 of yeast Sup35 protein transforms in vitro into amyloid-like filaments. Proc Natl Acad Sci USA. 94:6618–6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryndushkin D, Ihrke G, Piermartiri TC, Shewmaker F.. 2012. A yeast model of optineurin proteinopathy reveals a unique aggregation pattern associated with cellular toxicity. Mol. Microbiol. 86:1531–1547. [DOI] [PubMed] [Google Scholar]
- Kryndushkin D, Shewmaker F, Wickner RB.. 2008. Curing of the [URE3] prion by Btn2p, a Batten disease-related protein. Embo J. 27:2725–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N, Gaur D, Gupta A, Puri A, Sharma D.. 2015. Hsp90-associated immunophilin homolog Cpr7 is required for the mitotic stability of [URE3] prion in Saccharomyces cerevisiae. PLoS Genet. 11:e1005567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroute F. 1971. Non-Mendelian mutation allowing ureidosuccinic acid uptake in yeast. J Bacteriol. 106:519–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawless C, Pearson RD, Selley JN, Smirnova JB, Grant CM, et al. 2009. Upstream sequence elements direct post-transcriptioinal regulation of gene expression under stress conditions in yeast. BMC Genomics 10:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebman SW, Chernoff YO.. 2012. Prions in yeast. Genetics. 191:1041–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd S, Maytham EG, Pota H, Grizenkova J, Molou E, et al. 2009. HECTD2 is associated with susceptibility to mouse and human prion disease. PLoS Genet. 5:e1000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado-Baez L, Cole NB, Kramer H, Donaldson JG.. 2013. Microtubule-dependent endosomal sorting of clathrin-independent cargo by Hook1. J Cell Biol. 201:233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinovska L, Kroschwald S, Munder MC, Richter D, Alberti S.. 2012. Molecular chaperones and stress-inducible protein-sorting factors coordinate the spaciotemporal distribution of protein aggregates. Mol Biol Cell 23:3041–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín-Marcos P, Hinnebusch AG, Tamame M.. 2007. Ribosomal protein L33 is required for ribossome biogenesis, subunit joining, and repression of GCN4 translation. Mol Cell Biol. 27:5968–5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masison DC, Wickner RB.. 1995. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells. Science 270:93–95. [DOI] [PubMed] [Google Scholar]
- Matveenko AG, Barbitoff YA, Jay-Garcia LM, Chernoff YO, Zhouravleva GA.. 2018. Differential effects of chaperones on yeast prions: CURrent views. Curr Genet. 64:317–325. [DOI] [PubMed] [Google Scholar]
- McGlinchey R, Kryndushkin D, Wickner RB.. 2011. Suicidal [PSI+] is a lethal yeast prion. Proc Natl Acad Sci USA. 108:5337–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama H, Edskes HK, Wickner RB.. 2000. URE3] prion propagation in Saccharomyces cerevisiae: requirement for chaperone Hsp104 and curing by overexpressed chaperone Ydj1p. Mol Cell Biol. 20:8916–8922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morvan J, de Craene J-O, Rinaldi B, Addis V, Misslin C, et al. 2015. Btn3 regulates the endosomal sorting function of the yeast Ent3 epsin, an adaptor for SNARE proteins. J Cell Sci. 128:706–716. [DOI] [PubMed] [Google Scholar]
- Nakayashiki T, Kurtzman CP, Edskes HK, Wickner RB.. 2005. Yeast prions [URE3] and [PSI+] are diseases. Proc Natl Acad Sci USA. 102:10575–10580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness F, Ferreira P, Cox BS, Tuite MF.. 2002. Guanidine hydrochloride inhibits the generation of prion "seeds" but not prion protein aggregation in yeast. Mol Cell Biol. 22:5593–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtake Y, Wickner RB.. 1995. Yeast virus propagation depends critically on free 60S ribosomal subunit concentration. Mol Cell Biol. 15:2772–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong S-E, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, et al. 2002. Stable isotope labeling by amion acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 1:376–386. [DOI] [PubMed] [Google Scholar]
- Palumbo RJ, Fuchs G, Lutz S, Curcio MJ.. 2017. Paralog-specific functions of RPL7A and RPL7B mediated by ribosomal protein or snlRNA dosage in Saccharomyces cerevisiae. G3 (Bethesda). 7:591–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD.. 1997. In vitro propagation of the prion-like state of yeast Sup35 protein. Science 277:381–383. [DOI] [PubMed] [Google Scholar]
- Pillet B, Garcia-Gomez JJ, Pausch P, Lalquet L, Bange G, et al. 2015. The dedicated chaperone Acl4 escorts ribosomal protein Rpl4 to its nuclear pre-assembly site. PLoS Genet. 11:e1005565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, Ishihama Y.. 2007. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2:1896–1906. [DOI] [PubMed] [Google Scholar]
- Schlumpberger M, Prusiner SB, Herskowitz I.. 2001. Induction of distinct [URE3] yeast prion strains. Mol Cell Biol. 21:7035–7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shailesh K, Dine EA, Paddock E, Masison DC.. 2020. Mutations outside the Ure2 amyloid-forming region disrupt [URE3] prion propagation and alter interactions with protein quality control factors. Mol Cell Biol. 40:e00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F. 1991. Getting started with yeast. In: Guthrie Cand Fink GR editors. Guide to Yeast Genetics and Molecular Biology. San Diego: Academic Press. p. 3–21. [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, et al. 2000. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 25:302–305. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P.. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son M, Wickner RB.. 2020. Normal levels of ribosome-associated chaperones cure two groups of [PSI+] variants. Proc Natl Acad Sci USA. 117:26298–26306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondheimer N, Lindquist S.. 2000. Rnq1: An epigenetic modifier of protein function in yeast. Mol Cell 5:163–172. [DOI] [PubMed] [Google Scholar]
- Specht S, Miller SBM, Mogk A, Bukau B.. 2011. Hsp42 is required for sequestration of protein aggregates into deposition sites in Saccharomyces cerevisiae. J Cell Biol. 195:617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfield I, Jones KM, Kushnirov VV, Dagkesamanskaya AR, Poznyakovski AI, et al. 1995. The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. Embo J. 14:4365–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen KK, MacKay VL, Kerr EO, Tsuchiya M, Hu D, et al. 2008. Yeast life span extension by depletion of 60S ribosomal subunits is mediated by Gcn4. Cell 133:292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Chien P, Naber N, Cooke R, Weissman JS.. 2004. Conformational variations in an infectious protein determine prion strain differences. Nature. 428:323–328. [DOI] [PubMed] [Google Scholar]
- Troisi EM, Rockman ME, Nguyen PP, Oliver EE, Hines JK.. 2015. Swa2, the yeast homolog of mammalian auxilin, is specifically required for the propagation of the prion variant [URE3-1]. Mol Microbiol. 97:926–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tye BW, Commins N, Ryazanova LV, Wuhr M, Springer M, et al. 2019. Proteotoxicity from aberrant ribosome biogenesis compromises cell fitness. Elife 8:e43002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Mao X, Ju D, Xie Y.. 2004. Rpn4 is a physiological substrate of the Ubr2 ubiquitin ligase. J Biol Chem. 279:55218–55223. [DOI] [PubMed] [Google Scholar]
- Wang M, Herrmann CJ, Simonovic M, Szklarczyk D, Mering C.. 2015. Version 4.0 of PaxDb: protein abundance data, integrated across model organisms, tissues, and cell-lines. Proteomics 15:3163–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yang F, Gritsenko MA, Wang Y, Clauss T, et al. 2011. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 11:2019–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner JD, McIntosh KB.. 2009. How common are extraribosomal functions of ribosomal proteins? Mol. Cell. 34:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner RB. 1994. [ URE3] as an altered URE2 protein: evidence for a prion analog in S. cerevisiae. Science 264:566–569. [ [DOI] [PubMed] [Google Scholar]
- Wickner RB. 2013. Viruses and prions of yeasts, fungi and unicellular eukaryotes. In: Knipe DMand Howley PM editors. Fields Virology, 6th ed. Philadelphia: Wolters Kluwer/Lippincott Williams & Wilkins. p. 2355–2383. [Google Scholar]
- Wickner RB, Bezsonov E, Bateman DA.. 2014. Normal levels of the antiprion proteins Btn2 and Cur1 cure most newly formed [URE3] prion variants. Proc Natl Acad Sci USA. 111:E2711–E2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner RB, Bezsonov EE, Son M, Ducatez M, DeWilde M, et al. 2018. Anti-prion systems in yeast and inositol polyphosphates. Biochemistry. 57:1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner RB, Shewmaker F, Bateman DA, Edskes HE, Gorkovskiy A, et al. 2015. Yeast prions: structure, biology and prion-handling systems. Microbiol Mol Biol Rev. 79:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, et.al .1999. Functional Characterization of the S. cerevisiae Genome by Gene Deletion and Parallel Analysis. Science. 285:901–906. [DOI] [PubMed] [Google Scholar]
- Yoshikawa K, Tanaka T, Ida Y, Furusawa C, Hirasawa T, et al. 2011. Comprehensive phenotypic analysis of single-gene deletion and overexpression strains of Saccharomyces cerevisiae. Yeast 28:349–361. [DOI] [PubMed] [Google Scholar]
- Zhao X, Lanz J, Steinberg D, Pease T, Ahearn JM, et al. 2018. Real-time imaging of yeast cells reveals several distinct mechanisms of curing of the [URE3] prion. J Biol Chem. 293:3104–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Derkatch IL, Liebman SW.. 2001. The relationship between visible intracellular aggregates that appear after overexpression of Sup35 and the yeast prion-like elements [PSI+] and [PIN+]. Mol Microbiol. 39:37–46. [DOI] [PubMed] [Google Scholar]
- Zhouravleva G, Frolova L, Le Goff X, Le Guellec R, Inge-Vechtomov S, et al. 1995. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. Embo J. 14:4065–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Strains and plasmids are available upon request. The authors affirm that all data necessary for confirming the conclusions of the article are present within the article, figures, tables, and Supplementary material which are available at figshare: https://doi.org/10.25386/genetics.13549292.