

Abstract
Farnesyl diphosphate synthase (FPPS) is an isoprenoid chain elongation enzyme that catalyzes the sequential condensation of dimethylallyl diphosphate (DMAPP; C5) with isopentenyl diphosphate (IPP; C5) and the resulting geranyl diphosphate (GPP; C10) with another molecule of IPP, eventually producing farnesyl diphosphate (FPP; C15), which is a precursor for the biosynthesis of a vast majority of isoprenoids. Previous studies on FPPS have highlighted the importance of the structure around the hydrophobic chain elongation path in determining the product specificity. To investigate what structural features define the final chain length of the product in FPPS from Leishmania major, we designed and expressed six mutants of LmFPPS by replacing small amino acids around the binding pocket with bulky residues. Using enzymatic assays, binding kinetics and crystallographic studies, we analyzed the effects of these mutations on the activity and product specificity of FPPS. Our results revealed that replacement of Thr-164 with tryptophan and phenylalanine completely abolished the activity of FPPS. Intriguingly, T164Y substitution displayed dual product specificity and produced a mixture GPP and FPP as final products, with lower activity for FPP synthesis than that of the wild-type enzyme. These data indicate that Thr-164 is a potential regulator of product specificity.
INTRODUCTION
Farnesyl diphosphate synthase (FPPS) is a branch-point enzyme of the isoprenoid biosynthetic pathway, a widespread metabolic pathway present from prokaryotes to eukaryotes. It is responsible for synthesizing essential isoprenoids such as carotenoids, chlorophylls, ubiquinones, dolichols, sterols, and also prenylated proteins 1–3. About 30,000 individual isoprenoids have been identified to date making these compounds the most diverse class of naturally occurring compounds. They serve numerous biochemical functions: as structural membrane components (prenyl-lipids) in archaea; as photosynthetic pigments (carotenoids and chlorophylls) in algae, cyanobacteria and plants; as plant growth-regulating hormones (gibberellins and abscisic acid); as plant defense compounds (mono-, di- and sesqui-terpenes); in electron-transport chains (quinones) and in subcellular targeting of signaling proteins (prenylation of proteins) 2, 4.
FPPS (EC 2.5.1.10) catalyzes a sequential chain elongation reaction by the head-to-tail condensation of two five-carbon compounds (C5), dimethylallyl diphosphate (DMAPP) and its isomer isopentenyl diphosphate (IPP) to form a ten carbon compound (C10), geranyl diphosphate (GPP), which then condenses with a second molecule of IPP to form a fifteen carbon compound (C15), farnesyl diphosphate (FPP) as the final product 5, 6 (Scheme 1). FPP then serves as a precursor for a number of downstream reactions in the pathway resulting in isoprenoid biosynthesis, for example, it is a substrate of squalene synthase (in sterol synthesis) and undecaprenyl diphosphate synthase (in bacterial cell wall synthesis) 7, 8. In addition, FPP undergoes further condensation reaction to produce a higher chain length diphosphate precursor, geranylgeranyl diphosphate (GGPP; C20), and both FPP and GGPP are important for the prenylation of GTPase signaling proteins such as Ras and Rho 9.
Scheme 1. Chain elongation reaction of FPPS.
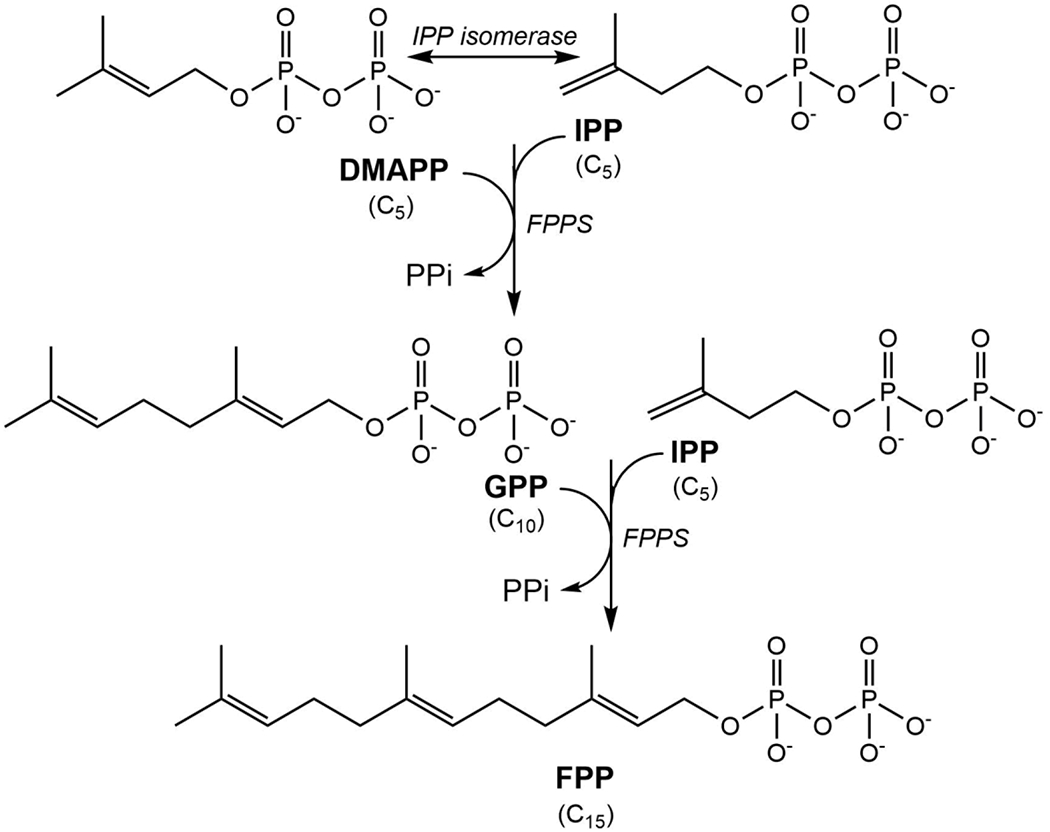
FPPS catalyzes the consecutive condensation of dimethylallyl diphosphate (DMAPP) with isopentenyl diphosphate (IPP) and the resulting geranyl diphosphate (GPP) with another molecule of IPP to form farnesyl diphosphate (FPP).
Structural studies on FPPSs have revealed that FPPS is a homodimer. Each monomer contains two binding pockets in the active site: the allylic binding site to which DMAPP or GPP binds, and the homoallylic binding site to which IPP binds. In the structures of FPPS in complex with bisphosphonate inhibitors (substrate analogs), the bisphosphonates bind to the allylic site and the phosphates of the bisphosphonates are coordinated by three divalent cations (Mg2+ or Ca2+) that bridge the compound to the protein 10–15. All FPPSs contain two highly conserved aspartate-rich motifs that are located on opposite walls of the active site cavity facing each other. The first aspartate-rich motif (FARM) has the sequence DDXX(XX)D, where D is aspartate and X is any amino acid. There are two residues between the aspartates of FARM in eukaryotic FPPS whereas the prokaryotic FPPS has four residues between the aspartates. The second aspartate-rich motif (SARM) has the sequence DDXXD. Both these motifs are essential for catalytic activity and the amino acid residues close to FARM have been reported as the chain length determining residues 16–18. In most eukaryotic FPPSs, two aromatic amino acids at the fourth and fifth positions upstream of the FARM solely determine the chain length, while the product specificity in prokaryotic FPPS is determined by one aromatic amino acid at the fifth position upstream of the FARM, two amino acids inserted in this motif and other modifications 18, 19.
Mutagenesis coupled with kinetic studies on FPPS from Bacillus stearothermophilus have reported that Tyr-81 at the fifth position before the FARM blocks the chain elongation longer than FPP 20, 21. Similarly, Phe-77 of Sulfolobus acidocaldarius GGPPS, the equivalent of Tyr-81 of the BsFPPS, when mutated to serine catalyzes the chain elongation longer than GGPP (C20) 22. Furthermore, replacement of Ser-82 at the fourth position upstream of the FARM with phenylalanine resulted in a BsFPPS that produced GPP as the final product 23. In another important study, Tarshis et al. reported that avian FPPS can be transformed to produce higher chain length products, geranylgeranyl diphosphate (C20) by F112A substitution and geranylfarnesyl diphosphate (C25) by F113S mutation 24. Taken together, these studies have characterized the product specificity of FPPSs from different species clearly demonstrating that the final chain length of the product is governed by the structure around the hydrophobic path through which the product elongates in the active site cavity. However, there is no such study so far on the corresponding FPPS enzyme from Leishmania major, a human pathogen that causes cutaneous leishmaniasis. Moreover, FPPS is of special interest in L. major given the essentiality of this enzyme for parasite survival and its validation as a potential target of bisphosphonates for antiparasitic drug development 25 26, 27.
In the present study, we investigate the product specificity of LmFPPS to identify the amino acids responsible for determining the final chain length of the product. Based on sequence and structural alignments of LmFPPS with FPPSs from other eukaryotic species, His-93 at the fifth position and Phe-94 at the fourth position upstream of FARM (residues 98-102) are thought to be important in determining the product specificity by preventing the chain elongation beyond FPP (Figure S1). Instead of exploring these obvious candidates, we chose to focus our studies on identifying other unknown chain length determining residues that would restrict the condensation beyond GPP. On the basis of an analysis of the structure of wild-type (WT) LmFPPS (PDB ID: 4JZX) previously determined by our lab 15, we focused on amino acids with small and flexible side chains at the floor of the binding pocket, which when replaced with bulky residues with aromatic rings, may shorten the length of the binding pocket and force the chain elongation reaction to stop at the intermediate product GPP. We selected two such residues, Glu-97 and Thr-164 (Figure 1A&B), and constructed and analyzed six mutants of LmFPPS, E97F, E97Y, E97W, T164F, T164Y and T164W to investigate if the synthesis of the final product FPP was indeed dictated by the size of the binding pocket. Here we report biochemical, biophysical and structural studies of the active site mutants that provide insights into the regulation of chain elongation reaction by FPPS.
Figure 1. Structural basis of site-directed mutations in LmFPPS.
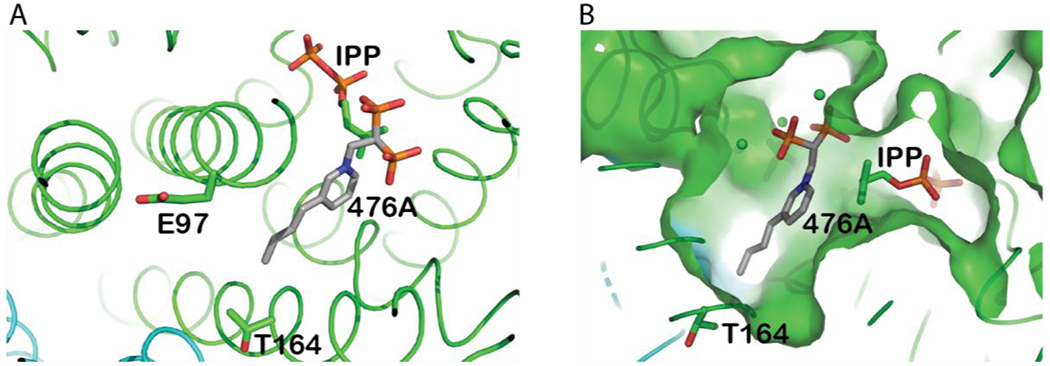
(A) Active site of WT-LmFPPS (PDB ID: 4JZX)in complex with bisphosphonate inhibitor 476A (grey chain; colored by atoms) and substrate IPP(colored by atoms). The amino acid residues mutated in this study, E97 and T164 are illustrated as sticks. (B) Surface representation of the binding pocket of LmFPPS showing 476A and IPP buried in the allylic and homoallylic sites respectively.
MATERIALS AND METHODS
Cloning and expression of LmFPPS.
LmFPPS was cloned into pET-28a vector (Novagen) (encoding an N-terminal His tag) and the recombinant plasmid was used to transform Escherichia coli BL21 (DE3) cells 28. E. coli cells were grown at 37 °C in LB medium (Thermo Fisher Scientific) supplemented with 30 μg/ml kanamycin (GoldBio). Once the OD600 reached 0.6-0.8, protein expression was induced by the addition of 0.1 mM isopropyl β-D-thiogalactoside (GoldBio) and the culture was incubated at 37 °C for 3 hours. Cells were harvested by centrifugation at 6000 g for 10 min at 4 °C and stored at −80 °C. LmFPPS mutants, E97F, E97Y, E97W, T164F, T164Y and T164W were generated using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). The forward and reverse primers designed to introduce these mutations are depicted in Table S1. All LmFPPS mutants were expressed in the same way as the WT protein.
Purification of WT LmFPPS and mutants.
Cell pellets were resuspended in a buffer containing 50 mM sodium phosphate pH 8, 300 mM NaCl, 10 mM imidazole, 1mM TCEP and lysed by microfluidizer. The cell lysate was centrifuged at 12000 rpm for 30 min at 4 °C and the soluble supernatant was loaded onto a nickel affinity HisTrap HP column (GE Healthcare). LmFPPS was eluted with 100 mM imidazole. The eluates were pooled, dialyzed against buffer containing 20 mM Tris pH 8, 50 mM NaCl, 1 mM TCEP and digested with thrombin to cleave the 6X His tag. The protein was further purified by anion-exchange chromatography using a Source Q column (GE Healthcare) by eluting with a linear gradient of 0.05-1 M NaCl. The protein was repurified by nickel affinity chromatography using HisTrap HP column and the flowthrough fractions containing pure LmFPPS were collected. The protein was then dialyzed against a buffer containing 20 mM Tris pH 8, 150 mM NaCl, 1 mM TCEP and concentrated to 10-15 mg/ml 15. All LmFPPS mutants used in this study were purified by the same protocol.
Differential Scanning Fluorimetry analysis.
To examine the thermal stability of WT-LmFPPS and mutants, we used a differential scanning fluorimetry (DSF) assay. It monitors the fluorescence of a dye which binds to hydrophobic regions of the protein that become exposed upon temperature-induced denaturation 29, 30. Proteins were screened against different buffers with pH ranging from 2.5-11. 25 μl reactions were set up in white low-profile 96-well unskirted PCR plates (Bio-Rad) by mixing 10 μl of purified LmFPPS at 1 mg/ml (10 μg final protein amount) with 2.5 μl of 50x SYPRO orange dye (Invitrogen) (5X final SYPRO concentration) and 12.5 μl of each pH of the buffers. Plates were sealed with an optical transparent film and centrifuged at 2500 g for 30 sec. Thermal scanning was performed from 25 to 99 °C (1 °C/min temperature gradient) using a CFX9 Connect real-time polymerase chain reaction (RT-PCR) instrument (Bio-Rad). Melt curves were constructed with the fluorescence intensity from each well versus temperature and a protein unfolding/melting temperature ‘Tm’ was calculated from the maximum value of the negative first derivative of the melt curve using CFX Manager software (Bio-Rad).
Radioactive enzyme assay for LmFPPS.
We used a radioactive-based enzyme assay to characterize the activity of FPPS 19, 31. The reaction mixture contained 50 mM Tris pH 8, 10 mM MgCl2, 1 mM TCEP, 50 μM DMAPP (Sigma-Aldrich), 100 μM 14C-IPP (PerkinElmer) and 140 μg of LmFPPS in a total volume of 500 μl. The reaction was incubated at 37 °C for 3 hours followed with the addition of 1 μl of bacterial alkaline phosphatase (150 U/μl) and overnight incubation at 37 °C to hydrolyze prenyl diphosphates into corresponding alcohols. The alcohols were then extracted with 2 ml of hexane and the volumes were concentrated to 100 μl. The concentrated extracts were analyzed using thin layer chromatography (TLC) by blotting them onto a TLC silica-gel plate (Fisher Scientific) along with the product standards, geraniol and farnesol. Radiolabeled substrates and products were separated by benzene:ethyl acetate (4:1) as the developing solvent. The positions of the standards were revealed with iodine vapor and the radiolabeled solution was blotted onto these spots to further visualize these spots by autoradiography. The plate was then exposed to storage phosphor screen (Molecular Dynamics) overnight and subsequently scanned on a phosphorimager (Amersham).
Surface Plasmon Resonance.
The binding affinities of LmFPPS for FPP and GPP were measured using surface plasmon resonance (SPR) with a Biacore T200 instrument (Cytiva) at 25°C. WT-LmFPPS was diluted to 10 μg/ml in a buffer consisting of 25 mM HEPES pH 7.5, 5 mM MgCl2, 300 mM NaCl, 1 mM DTT and 0.05% Tween 20. The WT protein was then immobilized onto flow cell 2 (Fc2) of a Biacore CM5 (carboxymethyl-dextran) sensor chip via amine coupling to approximately 5000 resonance units (RU). Binding of analytes FPP and GPP were measured with multi-cycle kinetics. FPP was injected over immobilized LmFPPS at four increasing concentrations: 1.5, 6.2, 25 and 100 μM. Similarly, GPP was separately injected at varying concentrations ranging from 0.1 to 25 μM. The mutants were analyzed using the same protocol as the WT-LmFPPS. Reference subtracted and blank subtracted binding curves were fitted into a 1:1 interaction model and the steady-state affinity ‘KD’ (equilibrium dissociation constant) was determined from steady-state affinity analysis using Biacore T200 Evaluation Software v3.2 (Cytiva).
Crystallization of LmFPPS mutants.
Crystals of LmFPPS mutants, E97Y, T164Y and T164W were produced using the hanging drop vapor diffusion technique at 18 °C. Proteins were co-crystallized with inhibitor 476A and substrate IPP in the presence of MgCl2 26. Conditions for crystallization of mutants were slightly varied from those of WT-LmFPPS 15, with PEG 8000 as precipitant and different concentrations of ligands as stated below. The drops were prepared in 1:1 ratio by mixing 1 μl of protein-ligand mixture (12 mg/ml LmFPPS, 0.4 mM IPP, 0.4 mM 476A and 1mM MgCl2) with 1 μl of reservoir buffer (0.1 M MES sodium salt pH 6.5, 0.1-0.2 M calcium acetate and 15-25 % PEG 8000).
Data collection and structure determination.
X-ray diffraction data for E97Y-LmFPPS, T164Y-LmFPPS and T164W-LmFPPS crystals were collected in-house using a Rigaku FR-E Superbright copper rotating anode x-ray generator as the source, with a Rigaku Saturn 944 CCD detector for E97Y-LmFPPS and R-AXIS IV image plate detector for T164Y-LmFPPS and T164W-LmFPPS. Data were indexed, integrated and scaled using HKL-2000 suite 32. The structures of E97Y-LmFPPS and T164Y-LmFPPS were determined by direct refinement using LmFPPS structure (PDB ID: 4JZX). T164W-LmFPPS structure was determined by molecular replacement with the program AMoRe 33 using LmFPPS structure (PDB ID: 4JZX) as the search model. Each of the models was refined in alternate cycles of restrained refinement with REFMAC5 in the CCP4 suite and rebuilding by Coot 34–36. The divalent calcium ions were assigned on the basis of anomalous scattering data of WT-LmFPPS-476A complex structure (PDB ID: 4JZX). The final models were validated using Coot and ‘wwPDB Validation Server’ (https://validate.wwpdb.org). RMSD was calculated between the structures using ‘SSM Superpose’ function in Coot. Figures for the structures were prepared using PyMOL 37, 38.
RESULTS AND DISCUSSION
Design of LmFPPS mutants.
The residues that determine the product chain length in the FPPS enzyme have been reported previously for archaeal, bacterial and avian species 18, 20–22, 24, but there has been no such report on the equivalent FPPS in Leishmania species. We sought to characterize the product specificity of LmFPPS in order to identify the residues that regulate the chain elongation reaction and in particular limit the product length at GPP. Using previously published LmFPPS structure (PDB ID: 4JZX) as a template, we selected two residues, Glu-97 and Thr-164, around the binding pocket of LmFPPS for further studies (Figure 1A&B). We mutated each of these residues independently to bulkier aromatic amino acids by site-directed mutagenesis. In total, six mutants were designed, E97F, E97Y, E97W, T164F, T164Y and T164W. All the recombinant proteins including the WT and the six mutants were expressed in bacteria and purified to homogeneity by a combination of two rounds of nickel affinity chromatography and one round of anion-exchange chromatography. The purity of the resulting proteins was greater than 98% and the yield was ~4-5 mg per liter of cell culture (Figure 2).
Figure 2. Purified recombinant WT-LmFPPS and mutants.
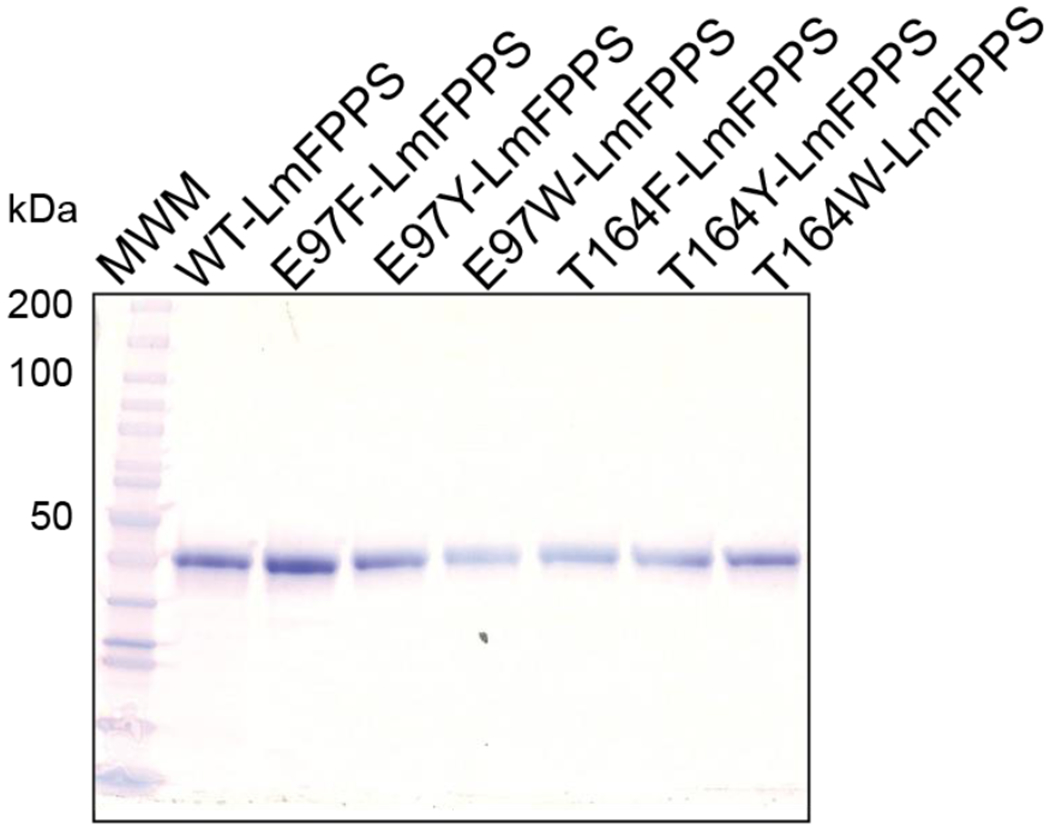
SDS-PAGE analysis of purified LmFPPS proteins. WT-LmFPPS and six mutants are shown(~40 kDa).
Thermal shift assay for WT-LmFPPS and mutants.
We carried out differential scanning fluorimetry (DSF) assay to probe the stability of the WT and mutant proteins. DSF measures the melting temperature ‘Tm’ of a protein which is reported by increased fluorescence of the SYPRO orange dye when bound to hydrophobic protein surfaces exposed upon thermal denaturation. The change in Tm is referred to as thermal shift which indicates the change in protein stability. All the proteins were screened over a wide range of pH from 2.5 to 11 and were observed to be most stable from pH 7.5-8.0, with a very high Tm (>94 °C). We were particularly interested in comparing the stability of WT and mutant LmFPPS proteins in the Tris buffer at pH 8 which is the optimal pH for LmFPPS purification as well as enzymatic assays. At pH 8, the Tm was 95 °C for WT LmFPPS, T164F-LmFPPS and T164W-LmFPPS, and 97 °C for T164Y-LmFPPS (Figure 3A&B). Thus, there was no significant thermal shift between the WT enzyme and the mutants, indicating that the single-point mutations do not disrupt the folding and the stability of the enzymes, and any observed differences in the enzyme activities of these mutants will be more likely due to the disruption of the size of the binding pocket.
Figure 3. DSF analyses of WT-LmFPPS and the mutants.
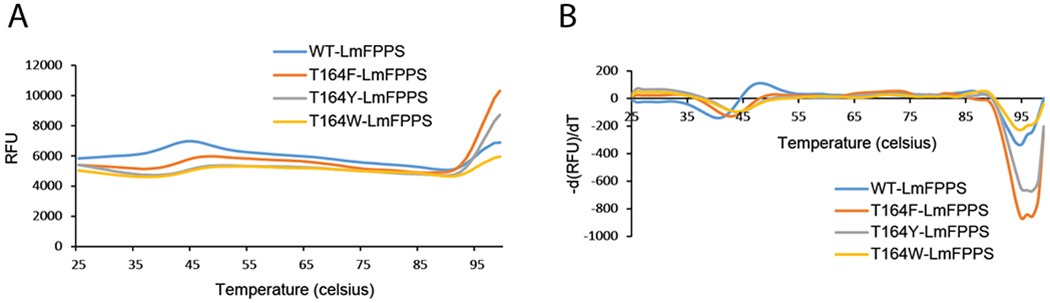
(A) Melt curves for the WT enzyme (shown in blue) and mutants, T164F-LmFPPS, T164Y-LmFPPS and T164W-LmFPPS (shown in orange, grey and yellow respectively) in Tris buffer, pH 8. Data are represented in relative fluorescence units (RFU) versus temperature. (B) The negative derivative of RFU versus temperature. The melting temperature ‘Tm’ corresponds to the peak or maximum of the first negative derivative of the curve.
Enzymatic activity of WT-LmFPPS and the mutants.
To assess and compare the activity of WT enzyme and the six mutants, we used a radioactivity based enzyme assay. After carrying out the FPPS reaction with 100 μM 14C labeled IPP and 50 μM DMAPP, the products were hydrolyzed to corresponding alcohols using alkaline phosphatase and the resulting alcohols were analyzed by thin layer chromatography followed by autoradiography. As expected, WT-LmFPPS displayed a prominent signal corresponding to farnesol (C15), clearly indicating that the WT enzyme is active and synthesizes FPP as the final product (Figure 4). All the Glu-97 mutants, E97F-LmFPPS, E97Y-LmFPPS and E97W-LmFPPS also showed equally intense spots corresponding to farnesol, suggesting that they are as active as the WT enzyme with FPP as the final product (Figure 4). These observations indicate that substitution of Glu-97 to bulky amino acids does not affect the product specificity of FPPS.
Figure 4. T164Y-LmFPPS produces both GPP and FPP as revealed by radioactive assay.
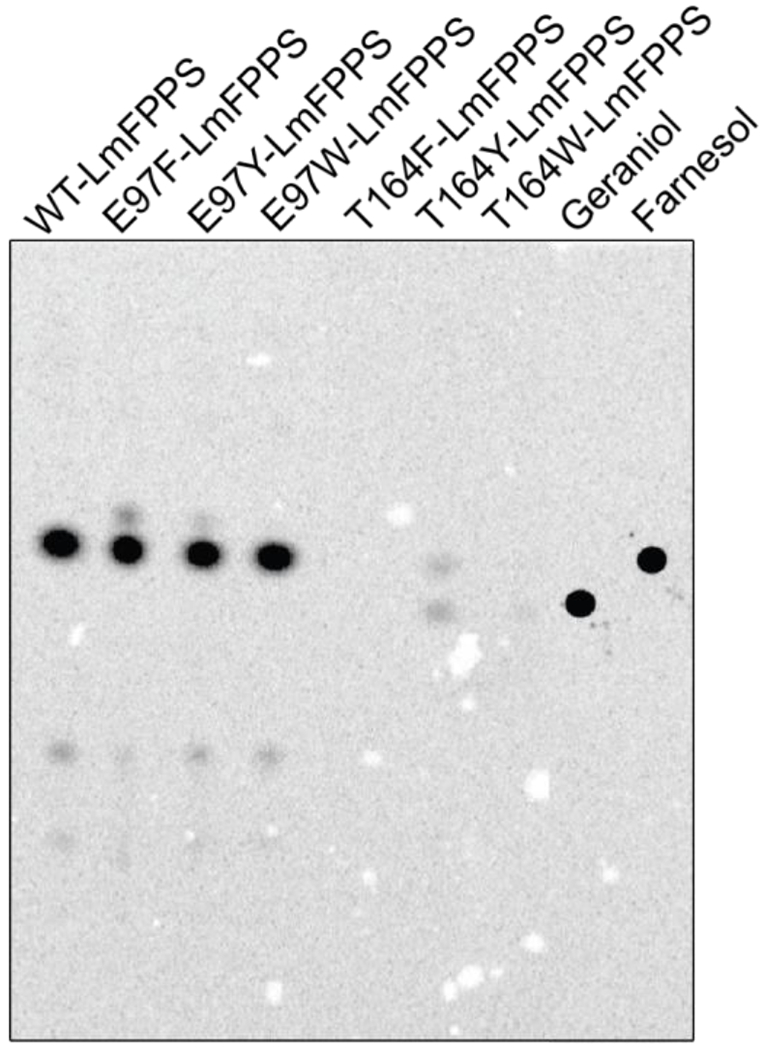
TLC autoradiochromatogram showing the activity of WT and mutant LmFPPSs. The spots for standard alcohols, geraniol and farnesol are shown in the last two lanes.
On the other hand, T164F-LmFPPS and T164W-LmFPPS did not show any spot for either farnesol (C15) or geraniol (C10), suggesting that these two mutants are inactive (Figure 4). Interestingly, T164Y-LmFPPS showed two less intense but distinct spots corresponding to geraniol and farnesol, implying that T164Y-LmFPPS can produce a mixture of both GPP and FPP (Figure 4). These results clearly demonstrate that out of all the three Thr-164 mutants, only T164Y-LmFPPS is active and able to synthesize GPP and FPP as final products, with weak activity for FPP synthesis compared to that of the WT enzyme as can be seen by the intensity of the spots corresponding to farnesol (Figure 4). Though replacement of the side chain of Thr-164 with any of the bulky groups did not completely alter the chain selectivity of FPPS, the dual product specificity of the T164Y mutant suggests its chain length regulating potential in partially preventing the condensation reaction beyond GPP.
Binding kinetics of WT-LmFPPS and mutants.
We used surface plasmon resonance (SPR) experiments to evaluate the binding kinetics and steady-state affinities of the WT enzyme and the mutants, E97Y-LmFPPS, T164Y-LmFPPS and T164W-LmFPPS for FPP as well as GPP. For these experiments, LmFPPS and the mutant proteins were immobilized on different flow cells of a CM5 sensor chip. FPP and GPP were separately injected over immobilized LmFPPS proteins at increasing concentrations ranging from 1.5 to 100 μM and 0.1 to 25 μM respectively. Binding was analyzed by multi-cycle kinetics and the resulting binding curves were fitted to a 1:1 interaction model. Steady-state affinity ‘KD’ was determined by fitting the dose response curve of binding response at steady-state versus analyte concentration to a steady-state affinity model. The resulting analysis revealed that FPP was bound to the WT enzyme with a KD of 36.6 μM, and showed fast association and fast dissociation binding kinetics (Figure 5A, Table 1). Of the three mutants, only E97Y-LmFPPS showed a KD for FPP similar to that of the WT enzyme (Figure 5B, Table 1). In contrast, T164Y-LmFPPS and T164W-LmFPPS showed a binding response that was not saturated up to the concentration of 100 μM FPP used in the assay, indicating a higher KD for FPP than that of the WT enzyme (Figure S2 A&B). These results are in agreement with the enzyme activity assays which showed that E97Y-LmFPPS was as active as the WT enzyme, T164Y-LmFPPS was less active than the WT for FPP synthesis, and T164W-LmFPPS was catalytically inactive.
Figure 5. Binding of FPP and GPP to WT and mutant LmFPPSs determined by SPR.
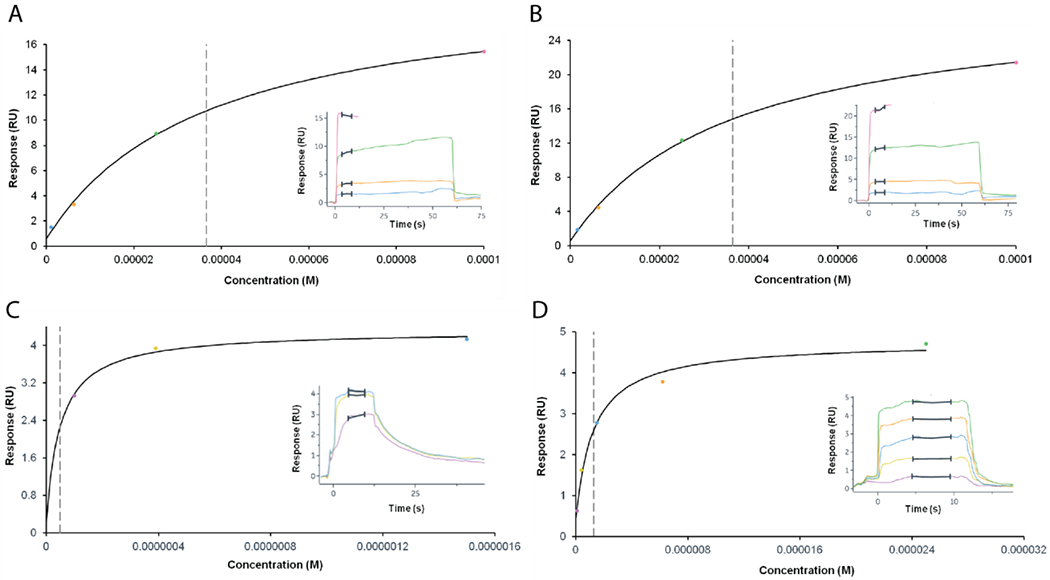
Steady-state dose response curves for binding of FPP to (A) WT-LmFPPS and (B) E97Y-LmFPPS. FPP was loaded in increasing concentrations at 1.5, 6.2, 25 and 100 μM. Dotted vertical grey lines indicate the values of steady-state affinity ‘KD’. Sensorgrams for multi-cycle kinetics are depicted as inserts. Binding of GPP to (C) WT-LmFPPS and (D) E97Y-LmFPPS. GPP was loaded in increasing concentrations at 0.1, 0.39 and 1.5 μM (C) and at 0.1, 0.39, 1.5, 6.2 and 25 μM (D).All SPR experiments were performed two times.
Table 1. Comparative steady-state affinity analysis of WT-LmFPPS and mutants for FPP and GPP using SPR.
Steady-state affinity ‘KD’ and the maximum response “Rmax’ were estimated from non-linear regression by fitting the equilibrium binding data to steady-state affinity equation. ND- not determined.
| Enzyme | Steady-state affinity ‘KD’ (¼M) | Rmax (RU) | ||
|---|---|---|---|---|
| Enzyme | FPP | GPP | FPP | GPP |
| WT-LmFPPS | 36.6 | 0.049 | 20.3 | 4.1 |
| E97Y-LmFPPS | 36.4 | 1.27 | 28.5 | 4.3 |
| T164Y-LmFPPS | ND | ND | ND | ND |
| T164W-LmFPPS | ND | ND | ND | ND |
On the other hand, GPP showed a KD to WT-LmFPPS and E97Y-LmFPPS in the nanomolar to low micromolar range, with fast association but relatively slower dissociation kinetics compared to that of FPP with WT enzyme (Figure 5C&D, Table 1). It is not surprising though that GPP binds more tightly to the enzyme than FPP as previous structural and mechanistic studies have proposed that the final product FPP might be released from the active site of the enzyme (in the open conformation) without the release of intermediate product GPP 12. Unlike the WT enzyme and the E97Y mutant, T164Y-LmFPPS and T164W-LmFPPS displayed a non-saturable steady-state dose response for GPP under standard assay conditions, suggesting lower affinities of the T164 mutants for GPP compared to that of the WT enzyme (Table 1).
Structures of LmFPPS mutants.
We used x-ray crystallography to investigate how the mutations around the binding pocket affect the chain length of the product in the active site. Of the six mutants originally constructed and produced in this study, we were successful in obtaining crystals of three mutants, E97Y-LmFPPS, T164Y-LmFPPS and T164W-LmFPPS. The conditions of crystallization for these mutants were adapted from those of the previously reported WT-LmFPPS protein with minor modifications 15. The crystals were formed in the mother liquor containing 0.1 M MES sodium salt pH 6.5, 15-25% PEG 8000 and 0.1-0.2 M calcium acetate. Like WT-LmFPPS, we co-crystallized the mutant proteins with bisphosphonate inhibitor 476A (allylic substrate analog) and homoallylic substrate IPP. Though the structures of the WT-LmFPPS protein were determined with three different bisphosphonate inhibitors, 476A, 300B and 46I, we chose to crystallize the mutant proteins in complex with 476A over other inhibitors in this study 13, 15, 26, 39. This choice was based on the fact that 476A consists of a relatively longer and flexible alkyl chain, and thus can possibly mimic the carbon tail of allylic substrates and have closer interaction with the mutated residues in the active site.
The structures of E97Y-LmFPPS, T164Y-LmFPPS and T164W-LmFPPS were refined to 2.0 Å, 1.8 Å and 2.1 Å respectively. Full data collection and refinement statistics are shown in Table 2. The structures of the three mutants with bound ligands were highly similar to that of the WT-LmFPPS (PDB ID: 4JZX) reported previously, with the exception of the mutated residues. E97Y-LmFPPS and T164Y-LmFPPS mutants crystallized in the same space group (primitive orthorhombic, P212121) as the WT-LmFPPS, with similar cell dimensions and RMSD of 0.20 Å and 0.17 Å respectively for all 362 Cα atoms. However, crystals of the T164W-LmFPPS mutant had a different space group (primitive monoclinic, P21) and cell dimensions from those of the WT-LmFPPS, with RMSD of 0.22 Å for all 362 Cα atoms. Like the FPPS from other species, all the LmFPPS mutants were homodimers, with each momomer having a large central cavity (active site) formed by ten core α-helices 10–12, 14, 15. In addition, the electron densities for the bisphosphonate inhibitor 476A and substrate IPP were observed at the allylic and homoallylic sites respectively in the active site cavity (Figures 6A–D). In all the mutant structures, the phosphates of the bisphosphonate 476A were coordinated by three Ca2+ ions, that were in turn bound to the side chains of the aspartates of the two highly conserved aspartate-rich motifs (DDXXD), FARM (residues 98-102) and SARM (residues 250-254).
Table 2.
Data collection and refinement statistics for LmFPPS mutants.
| E97Y-LmFPPS-476A-IPP-Ca2+ PDB ID: 6WW1 |
T164W-LmFPPS-476A-IPP-Ca2+ PDB ID: 6W7I |
T164Y-LmFPPS-476A-IPP-Ca2+ PDB ID: 6VJC |
|
|---|---|---|---|
| Data collection | |||
| Space group | P212121 | P21 | P212121 |
| Cell dimensions a,b,c (Å) α,β,γ (Å) |
80.70, 86.08, 107.42 90.00, 90.00, 90.00 |
58.93,79.97, 81.17 90.00, 106.39, 90.00 |
80.52, 85.88, 107.20 90.00, 90.00, 90.00 |
| X-ray source | FR-E Superbright | FR-E Superbright | FR-E Superbright |
| Detector | Saturn 944 CCD | R-AXIS IV | R-AXIS IV |
| Wavelength (Å) | 1.54 | 1.54 | 1.54 |
| Resolution (Å) | 67.17-2.05 (2.09-2.05) |
50.00-2.10 (2.18-2.10) |
50.00-1.80 (1.86-1.80) |
| Completeness (%) | 90.6 (78.7) | 92.1 (57.8) | 99.2 (92.8) |
| Redundancy | 4.7 (2.7) | 3.2 (1.9) | 6.2 (2.6) |
| Rmerge (%) | 7.7 (24.0) | 7.2 (21.7) | 8.1 (68.8) |
| I/σI | 19.91 (4.40) | 26.07 (5.28) | 30.09 (1.4) |
| Measured reflections | 204177 | 123691 | 422649 |
| Unique reflections | 43443 | 38828 | 67996 |
| Refinement statistics | |||
| Rwork/Rfree | 0.21/0.29 (0.22/0.30) | 0.18/0.23 (0.21/0.31) | 0.15/0.19 (0.49/0.48) |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.016 | 0.009 | 0.010 |
| Bond Angles (°) | 1.81 | 1.16 | 1.65 |
| B-factors (Å2) | |||
| Protein | 17.9 | 19.0 | 24.6 |
| Ligand | 14.0 | 14.7 | 19.6 |
| Water | 24.9 | 26.6 | 24.3 |
| Monomers in asymmetric unit | 2 | 2 | 2 |
| Number of protein atoms | 5763 | 5856 | 5806 |
| Number of water molecules | 600 | 471 | 698 |
| Ligands | 476A, IPP, Ca2+ | 476A, IPP, Ca2+ | 476A, IPP, Ca2+ |
| Ramachandran plot statistics | |||
| Preferred regions | 96.25 % | 96.55 % | 97.75 % |
| Outliers | 0.56 % | 0.43 % | 0.28 % |
Values in parentheses are for the highest resolution shells.
Figure 6. Structural insights into LmFPPS mutants.
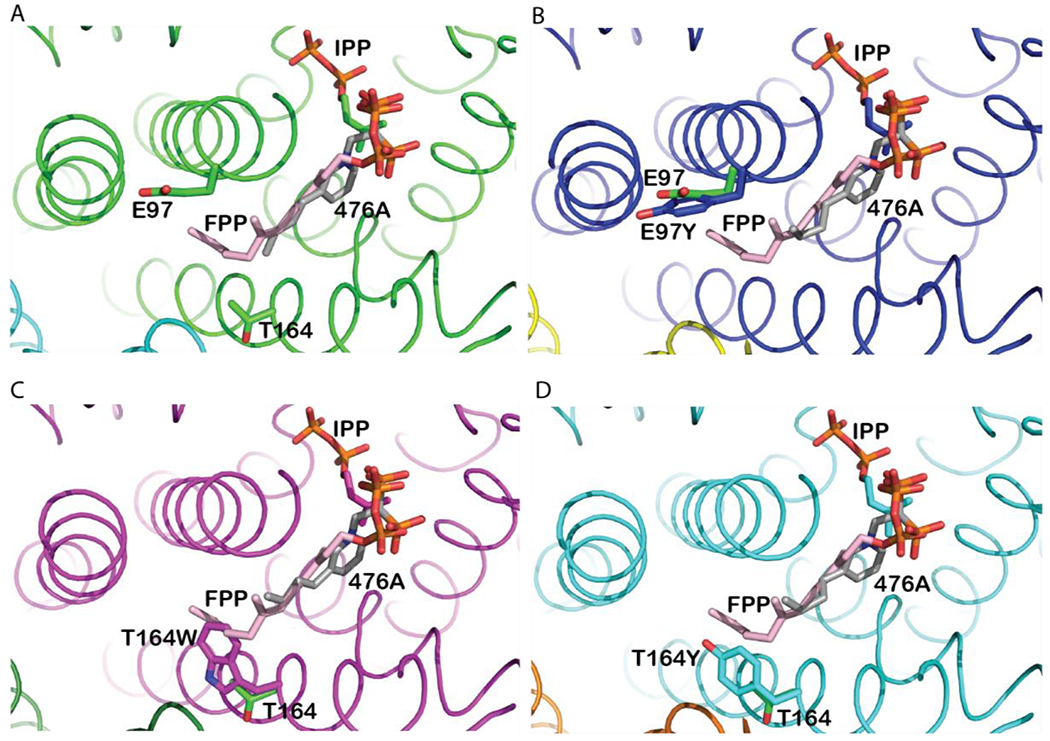
Active sites of (A) WT-LmFPPS (PDB ID: 4JZX) (shown in green). (B) E97Y-LmFPPS (shown in blue), (C) T164W-LmFPPS (shown in magenta) and (D) T164Y-LmFPPS (shown in cyan) structures. The mutant structures are aligned with the WT-LmFPPS structure. Bound ligands, inhibitor 476A (grey chain; colored by atoms) and substrate IPP (colored by atoms) are shown in all the structures. Product FPP (pink chain; colored by atoms) was modeled from avian FPPS structure (PDB ID: 1UBX). The WT residues (E97 and T164) and the mutations (E97Y, T164W and T164Y) are illustrated as sticks.
To compare the orientation of each of the three mutated residues in the binding pocket with respect to the corresponding residues in WT-LmFPPS structure, we aligned the structures of each of these mutants independently with the previously determined WT-LmFPPS structure (PDB ID: 4JZX). In each of the structures, we modeled FPP from avian (Gallus gallus) FPPS structure (PDB ID: 1UBX) to further investigate the interaction of each of the mutated residues with the FPP 24. On aligning the structures of WT-LmFPPS and mutants with that of the avian FPPS, the hydrocarbon tails of the allylic substrate analog 476A and modeled FPP were observed as oriented in the same confirmation, running down the hydrophobic pocket towards the mutated sites (Figure 6A–D).
In E97Y-LmFPPS structure, Tyr-97 faces away from the active site similar to Glu-97 of WT-LmFPPS (Figure 6B). This was against the expectation that a bulky aromatic amino acid would position its aromatic ring towards the active site to shorten the size of the binding pocket and prevent the condensation of GPP to FPP. This structural observation is consistent with our activity assay results which showed that E97Y-LmFPPS as well as the other two bulky mutants of Glu-97 (E97F-LmFPPS and E97W-LmFPPS) are equally active as the WT enzyme producing FPP as the final product. In addition, the steady-state affinity ‘KD’ of E97Y-LmFPPS for FPP is comparable with that of the WT enzyme. Collectively, the structural conformation of Tyr-97 and activity and affinity studies suggest that Glu-97 is not essential for regulating the chain length of the product.
Unlike the tyrosine in E97Y-LmFPPS, the aromatic amino acid Trp-164 in the T164W-LmFPPS structure is oriented towards the active site. Most importantly, the aromatic ring of tryptophan clearly overlaps with the hydrocarbon tail of the modeled FPP, forming a cap near the floor of the binding pocket (Figure 6C). Activity assay and binding kinetics showed that the T164W-LmFPPS mutant is catalytically inactive and has a weak affinity for FPP and GPP compared to the WT enzyme. Though the structural analysis explains the impaired activity of T164W-LmFPPS in synthesizing FPP, however its inability to produce GPP as product is not fully understood from this structure. It is possible that Trp-164 decreases the depth of the hydrophobic binding pocket to an extent where it can no longer accommodate GPP, though not evident from the present structure. Alternatively, the aromatic rings of Trp-164 might present steric hindrance strong enough to completely inhibit the chain elongation reaction in the active site. Regardless of the mechanism, the catalytic inactivity of T164W-LmFPPS reported in this study points towards a role of Thr-164 in facilitating the chain elongation reaction.
On the other hand, the side chain of Tyr-164 in T164Y-LmFPPS mutant, being relatively shorter than that of the tryptophan with only one aromatic ring, does not overlap with the isoprene tail of modeled FPP (Figure 6D). In addition, Tyr-164 is at H-bonding distance with NE2 of His-93 and shows a pi-pi interaction with the phenyl ring of Phe-94. Given the structural orientation of Tyr-164, it is not surprising that T164Y-LmFPPS is catalytically active and can form FPP in contrast to T164W-LmFPPS. However, T164Y-LmFPPS shows weak activity for the synthesis of FPP compared to that of the WT enzyme. Most importantly, this mutant exhibits dual product specificity and is capable of producing both GPP and FPP as products. To further investigate what structural change in T164Y mutant restricts the complete condensation of GPP, we analyzed the size of the binding pocket. To our observation, the bulky aromatic ring of Tyr-164 decreased the depth of the binding pocket by ~2.1 Å compared with that of the WT enzyme. This alteration in the size of the binding pocket could partially interfere with ~5.1 Å increase in chain length from GPP (C10) to FPP (C15) which explains the dual product specificity of T164Y-LmFPPS in synthesizing FPP as well as GPP, and a weak activity and affinity of this mutant for FPP.
CONCLUSIONS
Taken together, the crystal structures of the mutants, E97Y-LmFPPS, T164W-LmFPPS and T164Y-LmFPPS bound with 476A, IPP and modeled FPP provide a strong evidence that these mutations produced the observed effects by altering the size of the binding pocket for the growing isoprenoid chain in the active site of the enzyme. Though none of the Thr-164 substitutions with bulky amino acids could force the chain elongation reaction to stop at the formation of GPP as may have been expected, the present evidence still weighs in the favor of Thr-164 as a potential chain length regulating residue for its ability to partially restrict the chain elongation beyond GPP. In summary, our data on biochemical and biophysical characterization together with structural analysis of the active site mutants suggest that threonine at position 164 in LmFPPS is important for determining the product specificity of the enzyme and is a promising element for the development of double mutants that will stop the reaction at GPP.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Eric Oldfield for the gift of 476A bisphosphonate inhibitor and for his critical reading of the manuscript. This research was funded in part by the US Department of Defense, DOD CDMRP BC151831 (S.B.G.) and NIH NHLBI (HL128743) (L.M.A).
ABBREVIATIONS
- FPPS
farnesyl diphosphate synthase
- FPP
farnesyl diphosphate
- GPP
geranyl diphosphate
- IPP
isopentenyl diphosphate
- DMAPP
dimethylallyl diphosphate
- GGPP
geranylgeranyl diphosphate
- FARM
first aspartate rich motif
- SARM
second aspartate rich motif
- WT
wild-type
- DSF
differential scanning fluorimetry
- SPR
surface plasmon resonance
Footnotes
ASSOCIATED CONTENT
Supporting information. Table S1, Figures S1 and S2 (PDF)
Accession Codes. The atomic coordinates and structure factors of E97Y-LmFPPS, T164W-LmFPPS and T164Y-LmFPPS crystal structures in complex with inhibitor 476A and substrate IPP have been deposited in the Protein Data Bank with accession codes, 6WW1, 6W7I, 6VJC and respectively. Uniprot entry for LmFPPS is Q4QBL1.
Notes. The authors declare no competing financial interests.
REFERENCES
- [1].Chappell J (1995) The Biochemistry and Molecular Biology of Isoprenoid Metabolism, Plant Physiol 107, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lange BM, Rujan T, Martin W, and Croteau R (2000) Isoprenoid biosynthesis: the evolution of two ancient and distinct pathways across genomes, Proc Natl Acad Sci U S A 97, 13172–13177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hunter WN (2007) The non-mevalonate pathway of isoprenoid precursor biosynthesis, J Biol Chem 282, 21573–21577. [DOI] [PubMed] [Google Scholar]
- [4].Dhar MK, Koul A, and Kaul S (2013) Farnesyl pyrophosphate synthase: a key enzyme in isoprenoid biosynthetic pathway and potential molecular target for drug development, N Biotechnol 30, 114–123. [DOI] [PubMed] [Google Scholar]
- [5].Kellogg BA, and Poulter CD (1997) Chain elongation in the isoprenoid biosynthetic pathway, Curr Opin Chem Biol 1, 570–578. [DOI] [PubMed] [Google Scholar]
- [6].Thulasiram HV, and Poulter CD (2006) Farnesyl diphosphate synthase: the art of compromise between substrate selectivity and stereoselectivity, J Am Chem Soc 128, 15819–15823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Krishna G, Whitlock HW Jr., Feldbruegge DH, and Porter JW (1966) Enzymic conversion of farnesyl pyrophosphate to squalene, Arch Biochem Biophys 114, 200–215. [DOI] [PubMed] [Google Scholar]
- [8].Teng KH, and Liang PH (2012) Undecaprenyl diphosphate synthase, a cis-prenyltransferase synthesizing lipid carrier for bacterial cell wall biosynthesis, Mol Membr Biol 29, 267–273. [DOI] [PubMed] [Google Scholar]
- [9].McTaggart SJ (2006) Isoprenylated proteins, Cell Mol Life Sci 63, 255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tarshis LC, Yan M, Poulter CD, and Sacchettini JC (1994) Crystal structure of recombinant farnesyl diphosphate synthase at 2.6-A resolution, Biochemistry 33, 10871–10877. [DOI] [PubMed] [Google Scholar]
- [11].Rondeau JM, Bitsch F, Bourgier E, Geiser M, Hemmig R, Kroemer M, Lehmann S, Ramage P, Rieffel S, Strauss A, Green JR, and Jahnke W (2006) Structural basis for the exceptional in vivo efficacy of bisphosphonate drugs, ChemMedChem 1, 267–273. [DOI] [PubMed] [Google Scholar]
- [12].Gabelli SB, McLellan JS, Montalvetti A, Oldfield E, Docampo R, and Amzel LM (2006) Structure and mechanism of the farnesyl diphosphate synthase from Trypanosoma cruzi: implications for drug design, Proteins 62, 80–88. [DOI] [PubMed] [Google Scholar]
- [13].Huang CH, Gabelli SB, Oldfield E, and Amzel LM (2010) Binding of nitrogen-containing bisphosphonates (N-BPs) to the Trypanosoma cruzi farnesyl diphosphate synthase homodimer, Proteins 78, 888–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Aripirala S, Szajnman SH, Jakoncic J, Rodriguez JB, Docampo R, Gabelli SB, and Amzel LM (2012) Design, synthesis, calorimetry, and crystallographic analysis of 2-alkylaminoethyl-1,1-bisphosphonates as inhibitors of Trypanosoma cruzi farnesyl diphosphate synthase, J Med Chem 55, 6445–6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Aripirala S, Gonzalez-Pacanowska D, Oldfield E, Kaiser M, Amzel LM, and Gabelli SB (2014) Structural and thermodynamic basis of the inhibition of Leishmania major farnesyl diphosphate synthase by nitrogen-containing bisphosphonates, Acta Crystallogr D Biol Crystallogr 70, 802–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Joly A, and Edwards PA (1993) Effect of site-directed mutagenesis of conserved aspartate and arginine residues upon farnesyl diphosphate synthase activity, J Biol Chem 268, 26983–26989. [PubMed] [Google Scholar]
- [17].Song L, and Poulter CD (1994) Yeast farnesyl-diphosphate synthase: site-directed mutagenesis of residues in highly conserved prenyltransferase domains I and II, Proc Natl Acad Sci U S A 91, 3044–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ohnuma S, Hirooka K, Ohto C, and Nishino T (1997) Conversion from archaeal geranylgeranyl diphosphate synthase to farnesyl diphosphate synthase. Two amino acids before the first aspartate-rich motif solely determine eukaryotic farnesyl diphosphate synthase activity, J Biol Chem 272, 5192–5198. [DOI] [PubMed] [Google Scholar]
- [19].Koyama T, Obata S, Osabe M, Takeshita A, Yokoyama K, Uchida M, Nishino T, and Ogura K (1993) Thermostable farnesyl diphosphate synthase of Bacillus stearothermophilus: molecular cloning, sequence determination, overproduction, and purification, J Biochem 113, 355–363. [DOI] [PubMed] [Google Scholar]
- [20].Ohnuma S, Nakazawa T, Hemmi H, Hallberg AM, Koyama T, Ogura K, and Nishino T (1996) Conversion from farnesyl diphosphate synthase to geranylgeranyl diphosphate synthase by random chemical mutagenesis, J Biol Chem 271, 10087–10095. [DOI] [PubMed] [Google Scholar]
- [21].Ohnuma S, Narita K, Nakazawa T, Ishida C, Takeuchi Y, Ohto C, and Nishino T (1996) A role of the amino acid residue located on the fifth position before the first aspartate-rich motif of farnesyl diphosphate synthase on determination of the final product, J Biol Chem 271, 30748–30754. [DOI] [PubMed] [Google Scholar]
- [22].Ohnuma S, Hirooka K, Hemmi H, Ishida C, Ohto C, and Nishino T (1996) Conversion of product specificity of archaebacterial geranylgeranyl-diphosphate synthase. Identification of essential amino acid residues for chain length determination of prenyltransferase reaction, J Biol Chem 271, 18831–18837. [DOI] [PubMed] [Google Scholar]
- [23].Narita K, Ohnuma S, and Nishino T (1999) Protein design of geranyl diphosphate synthase. Structural features that define the product specificities of prenyltransferases, J Biochem 126, 566–571. [DOI] [PubMed] [Google Scholar]
- [24].Tarshis LC, Proteau PJ, Kellogg BA, Sacchettini JC, and Poulter CD (1996) Regulation of product chain length by isoprenyl diphosphate synthases, Proc Natl Acad Sci U S A 93, 15018–15023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mukherjee S, Basu S, and Zhang K (2019) Farnesyl pyrophosphate synthase is essential for the promastigote and amastigote stages in Leishmania major, Mol Biochem Parasitol 230, 8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sanders JM, Song Y, Chan JM, Zhang Y, Jennings S, Kosztowski T, Odeh S, Flessner R, Schwerdtfeger C, Kotsikorou E, Meints GA, Gomez AO, Gonzalez-Pacanowska D, Raker AM, Wang H, van Beek ER, Papapoulos SE, Morita CT, and Oldfield E (2005) Pyridinium-1-yl bisphosphonates are potent inhibitors of farnesyl diphosphate synthase and bone resorption, J Med Chem 48, 2957–2963. [DOI] [PubMed] [Google Scholar]
- [27].Branco Santos JC, de Melo JA, Maheshwari S, de Medeiros W, de Freitas Oliveira JW, Moreno CJ, Mario Amzel L, Gabelli SB, and Sousa Silva M (2020) Bisphosphonate-Based Molecules as Potential New Antiparasitic Drugs, Molecules 25, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ortiz-Gomez A, Jimenez C, Estevez AM, Carrero-Lerida J, Ruiz-Perez LM, and Gonzalez-Pacanowska D (2006) Farnesyl diphosphate synthase is a cytosolic enzyme in Leishmania major promastigotes and its overexpression confers resistance to risedronate, Eukaryot Cell 5, 1057–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Pantoliano MW, Petrella EC, Kwasnoski JD, Lobanov VS, Myslik J, Graf E, Carver T, Asel E, Springer BA, Lane P, and Salemme FR (2001) High-density miniaturized thermal shift assays as a general strategy for drug discovery, J Biomol Screen 6, 429–440. [DOI] [PubMed] [Google Scholar]
- [30].Lo MC, Aulabaugh A, Jin G, Cowling R, Bard J, Malamas M, and Ellestad G (2004) Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery, Anal Biochem 332, 153–159. [DOI] [PubMed] [Google Scholar]
- [31].Fujii H, Koyama T, and Ogura K (1982) Efficient enzymatic hydrolysis of polyprenyl pyrophosphates, Biochim Biophys Acta 712, 716–718. [PubMed] [Google Scholar]
- [32].Otwinowski Z, and Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode, Macromolecular Crystallography, Pt A 276, 307–326. [DOI] [PubMed] [Google Scholar]
- [33].Navaza J (1994) Amore - an Automated Package for Molecular Replacement, Acta Crystallogr A 50, 157–163. [Google Scholar]
- [34].Winn MD, Isupov MN, and Murshudov GN (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement, Acta Crystallogr D 57, 122–133. [DOI] [PubMed] [Google Scholar]
- [35].Winn MD, Ashton AW, Briggs PJ, Ballard CC, and Patel P (2002) Ongoing developments in CCP4 for high-throughput structure determination, Acta Crystallogr D Biol Crystallogr 58, 1929–1936. [DOI] [PubMed] [Google Scholar]
- [36].Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot, Acta Crystallogr D 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Delano WL (2002) The PyMOL Molecular Graphics System. [Google Scholar]
- [38].Schrodinger LLC (2015) The PyMOL Molecular Graphics System, V.1.8. [Google Scholar]
- [39].Yang G, Zhu W, Kim K, Byun SY, Choi G, Wang K, Cha JS, Cho HS, Oldfield E, and No JH (2015) In Vitro and In Vivo Investigation of the Inhibition of Trypanosoma brucei Cell Growth by Lipophilic Bisphosphonates, Antimicrob Agents Chemother 59, 7530–7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.