

Abstract
Multiple sclerosis is a chronic inflammatory disease of the CNS1. Astrocytes contribute to the pathogenesis of multiple sclerosis2, but little is known about the heterogeneity of astrocytes and its regulation. Here we report the analysis of astrocytes in multiple sclerosis and its preclinical model experimental autoimmune encephalomyelitis (EAE) by single-cell RNA sequencing in combination with cell-specific Ribotag RNA profiling, assay for transposase-accessible chromatin with sequencing (ATAC–seq), chromatin immunoprecipitation with sequencing (ChIP–seq), genome-wide analysis of DNA methylation and in vivo CRISPR–Cas9-based genetic perturbations. We identified astrocytes in EAE and multiple sclerosis that were characterized by decreased expression of NRF2 and increased expression of MAFG, which cooperates with MAT2α to promote DNA methylation and represses antioxidant and anti-inflammatory transcriptional programs. Granulocyte–macrophage colony-stimulating factor (GM-CSF) signalling in astrocytes drives the expression of MAFG and MAT2α and pro-inflammatory transcriptional modules, contributing to CNS pathology in EAE and, potentially, multiple sclerosis. Our results identify candidate therapeutic targets in multiple sclerosis.
Astrocytes are abundant CNS-resident cells that have important functions in health and disease3,4 and are thought to contribute to the pathogenesis of multiple sclerosis (MS)5–9. In MS and other neurological disorders, astrocyte function is controlled by factors provided by microglia10,11, the microbiome12 and the environment13. An understanding of the pathogenesis of MS requires the identification of the cell populations involved and the mechanisms that control them, but little is known about astrocyte heterogeneity and its control in MS. Single-cell RNA sequencing (scRNA-seq) enables the unbiased investigation of cells in complex tissues in order to identify novel populations and regulatory gene circuits in development and disease14–16. Recently, scRNA-seq has been used to study the heterogeneity of oligodendrocytes, neurons, microglia and other myeloid cells in MS and EAE17–21. The functional and phenotypic diversity of other CNS glial cells17–19,22 suggests that multiple astrocyte populations are likely to contribute to the pathogenesis of MS.
scRNA-seq analysis of astrocytes in EAE
Bulk studies have investigated the transcriptional profile of astrocytes in cluster 4 astrocytes. In addition, our computational analysis idenin EAE11–13,23,24, but an alternative to marker-based analysis of tissues tified five genes as candidate transcriptional regulators of cluster is unsupervised sampling and scRNA-seq. Thus, we induced EAE in wild-type B6 mice by immunization with the myelin oligodendrocyte glycoprotein epitope MOG35–55 in complete Freund’s adjuvant (CFA), followed by injection of pertussis toxin, and analysed 24,275 CNS cells using droplet-based scRNA-seq (Drop-seq), which enables the analysis of gene expression in thousands of individual cells25 (Fig. 1a). These scRNA-seq studies identified multiple cell populations in mice with EAE that differed from those found in control mice (Extended Data Fig. 1a–f, Supplementary Table 1). By focusing on astrocytes expressing S100b, Gja1, Aldh1l1, Gfap and Aqp4, we identified several subpopulations of astrocytes (Fig. 1b–d, Supplementary Table 2, Extended Data Fig. 1g–i), suggesting that astrocytes adopt multiple transcriptional states during EAE.
Fig. 1 |. scRNA-seq analysis of astrocytes in EAE.
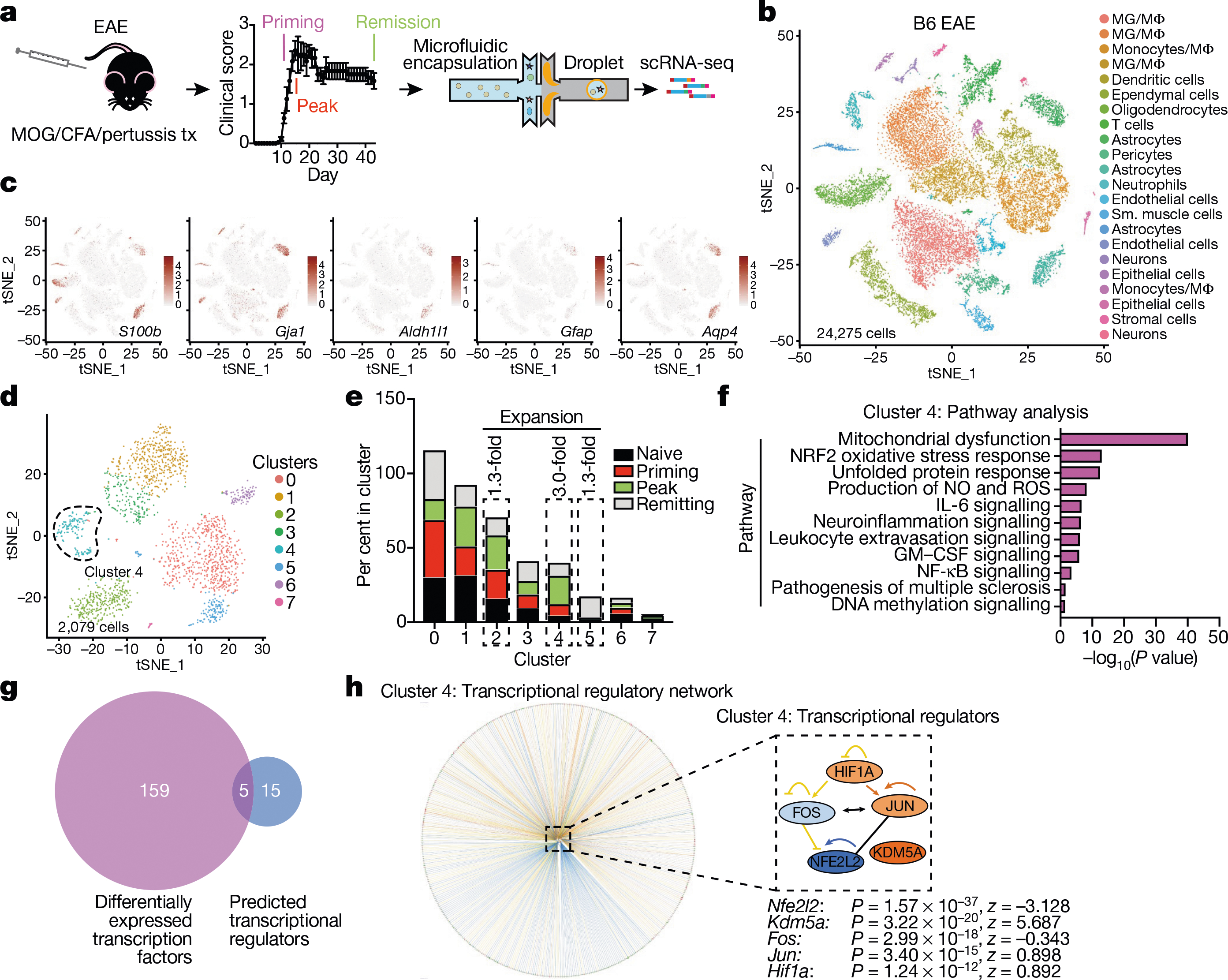
a, scRNA-seq analysis of CNS samples. Mice per group are n = 6 naive, n = 4 priming, n = 6 peak, n = 6 remission, n = 3 CFA. b, Unsupervised clustering t-distributed stochastic neighbour embedding (tSNE) plot of CNS cells from mice with EAE. MG, microglia; MΦ, macrophages. c, Expression scatterplots of astrocyte markers in population from b. n = 6 naive, n = 4 priming, n = 6 peak, n = 6 remission, n = 3 CFA. d, Unsupervised clustering tSNE plot of astrocytes. e, Cluster analysis of astrocytes based on per cent composition in EAE. f, Ingenuity pathway analysis (IPA) of cluster 4 astrocytes (n = 2,079 cells). g, Overlap between differentially expressed cluster 4 transcription factors and IPA predicted cluster 4 transcriptional regulators. h, Regulatory network of the intersection shown in g in cluster 4 astrocytes (n = 2,079 cells). Right-tailed Fisher’s exact test. Data shown as mean ± s.e.m.
We identified cluster 4 astrocytes as the most expanded subpopulation during EAE (Fig. 1e); this population was characterized by pro-inflammatory and neurotoxic pathways linked to astrocyte pathogenic activities in MS and EAE, such as the unfolded protein response (UPR)13 and the activation of NF-κB and inducible nitric oxide synthase (iNOS) pathways10,13 (Fig. 1f). We also detected increased GM-CSF signalling in cluster 4 astrocytes. In addition, our computational analysis identified five genes as candidate transcriptional regulators of cluster 4 astrocytes: Nfe2l2, Kdm5a, Hif1a, Fos and Jun (Fig. 1g–h). Notably, expression of Nfe2l2 was lower than that of Kdm5a, Hif1a, Fos and Jun (Fig. 1h, Extended Data Fig. 1j). Nfe2l2 encodes the transcription factor NRF2, which controls cellular responses that limit oxidative stress and inflammation26; this suggests that NRF2 is a negative regulator of pro-inflammatory and neurotoxic pathways in cluster 4 astrocytes.
NRF2 in astrocytes suppresses EAE
To generate an scRNA-seq dataset enriched for Gfap+ astrocytes, we performed scRNA-seq on 24,963 TdTomato+ cells isolated from TdTomatoGfap mice (which express the TdTomato reporter under the control of a Cre recombinase driven by the Gfap promoter) (Fig. 2a, Extended Data Figs. 2, 3, Supplementary Table 3). Consistent with our previous results, we found a subpopulation of astrocytes emerging in peak EAE (cluster 5) that were characterized by decreased expression of NRF2 target genes (Fig. 2b–d, Extended Data Fig. 3c). TdTomatoGfap cluster 5 astrocytes showed increased activation of pathways associated with pathogenic activities in EAE and MS, including UPR signalling13, sphingolipid metabolism27, and NF-κB10,11 and pro-inflammatory signalling12 (Fig. 2c, d). In addition, a pseudotime analysis of gene expression in TdTomatoGfap cluster 5 astrocytes indicated that decreased NRF2 activation precedes increased DNA methyltransferase activity and enhanced biosynthesis of S-adenosyl l-methionine (SAM), which acts as a substrate for DNA methylation28 (Fig. 2e). These data suggest that decreased NRF2 signalling facilitates the emergence of astrocyte functions that promote CNS inflammation.
Fig. 2 |. NRF2 suppresses pro-inflammatory astrocyte responses in EAE.
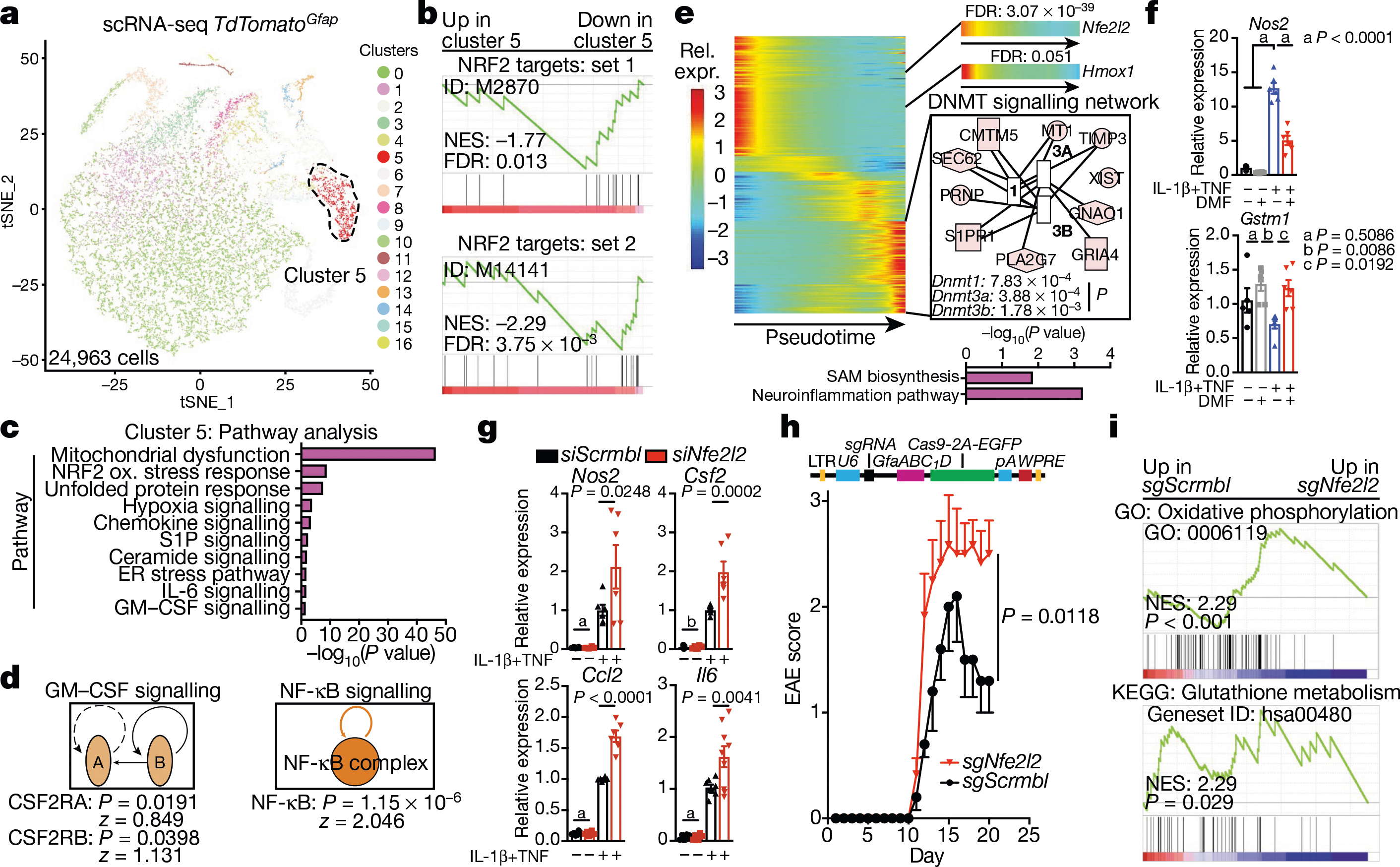
a, Unsupervised clustering tSNE plot of TdTomatoGfap astrocytes. n = 4 naive, n = 4 peak, n = 4 remission. b, Pre-ranked gene set enrichment analysis (GSEA) of NRF2 pathways in cluster 5 astrocytes. FDR, false discovery rate. NES, normalized enrichment score. y-axis, enrichment score; x-axis, rank in ordered dataset. n = 804 cells. c, d, IPA analysis of cluster 5 astrocytes showing differentially modulated pathways (c) and predicted regulators (d). n = 804 cells. e, Pseudotime and IPA analysis of TdTomatoGfap astrocytes. Benjamini–Hochberg test. n = 24,963 cells. f, Quantitative PCR (qPCR) analysis of Nos2 and Gstm1 expression in activated astrocytes with or without DMF. n = 6 biologically independent samples per condition, n = 5 for vehicle, experiment repeated twice. One-way ANOVA, Tukey post-test. g, qPCR analysis of proinflammatory gene expression in astrocytes after short interfering RNA (siRNA)-mediated knockdown of Nfe2l2 (siNfe2l2). siScrmbl, scrambled control siRNA. n = 6 biologically independent samples per condition, n = 5 siScrmbl for vehicle Csf2, n = 8 siScrmbl for cytokines Il6, n = 9 for siNfe2l2 cytokines. Experiment repeated twice. Two-way ANOVA, Sidak post-test. a, P > 0.9999; b, P = 0.9992. h, EAE development in mice transduced with sgScrmbl (n = 19) or sgNfe2l2 (n = 10) single guide RNAs. Top, schematic of lentiviral vector. Experiment repeated twice. Two-way repeated measures ANOVA. i, GSEA of RNA-seq data for astrocytes from mice transfected with sgScrmbl or sgNfe2l2. n = 3 mice per group. GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes. Benjamini–Hochberg test. y-axis, enrichment score; x-axis, rank in ordered dataset. Data shown as mean ± s.e.m.
To evaluate the role of NRF2 in astrocytes, we used primary cultures of mouse astrocytes activated with IL-1β and TNF, which have been linked to MS pathology29,30 and induce transcriptional responses similar to those detected in EAE (Extended Data Fig. 4a). Notably, IL-1β and TNF activate NF-κB, which can suppress NRF2 signalling through multiple mechanisms31. Activation of astrocytes by IL-1β and TNF boosted the expression of Nos2 and decreased NRF2-driven expression of Gstm1; these effects were reversed by activation of NRF2 using dimethyl fumarate (DMF)26 (Fig. 2f). Similarly, knockdown of Nfe2l2 in primary mouse astrocytes increased pro-inflammatory gene expression induced by IL-1β and TNF (Fig. 2g, Extended Data Fig. 4b).
To investigate the role of NRF2 in astrocytes, we inactivated Nfe2l2 using a lentiviral vector that co-expresses Gfap-driven CRISPR–Cas9 and a targeting single guide RNA (sgRNA)13 (Extended Data Fig. 4c). Inactivation of Nfe2l2 worsened EAE and increased the activation of pro-inflammatory pathways in astrocytes, as determined by RNA-seq (Fig. 2h, i, Extended Data Fig. 4d, e). Collectively, these data suggest that NRF2 signalling limits the transcriptional responses in astrocytes that promote the pathogenesis of EAE.
MAFG drives pathogenic astrocytes
To identify additional astrocyte regulators that are active during EAE we used RibotagGfap mice, which express a haemagglutinin-tagged ribosome subunit under the control of a Cre recombinase driven by the Gfap promoter (Extended Data Fig. 5a, b, Supplementary Table 4). The transcriptional analysis of RibotagGfap mice detected decreased expression of NRF2-driven genes during EAE (Extended Data Fig. 5c). The projection of the EAE transcriptional response of RibotagGfap astrocytes (Supplementary Table 4) onto the one detected in TdTomatoGfap cluster 5 astrocytes suggested that increased levels of the small MAF protein MAFG have a functional role in astrocytes during peak EAE, concomitant with decreased NRF2 expression (Fig. 3a).
Fig. 3 |. MAFG in astrocytes promotes CNS inflammation.
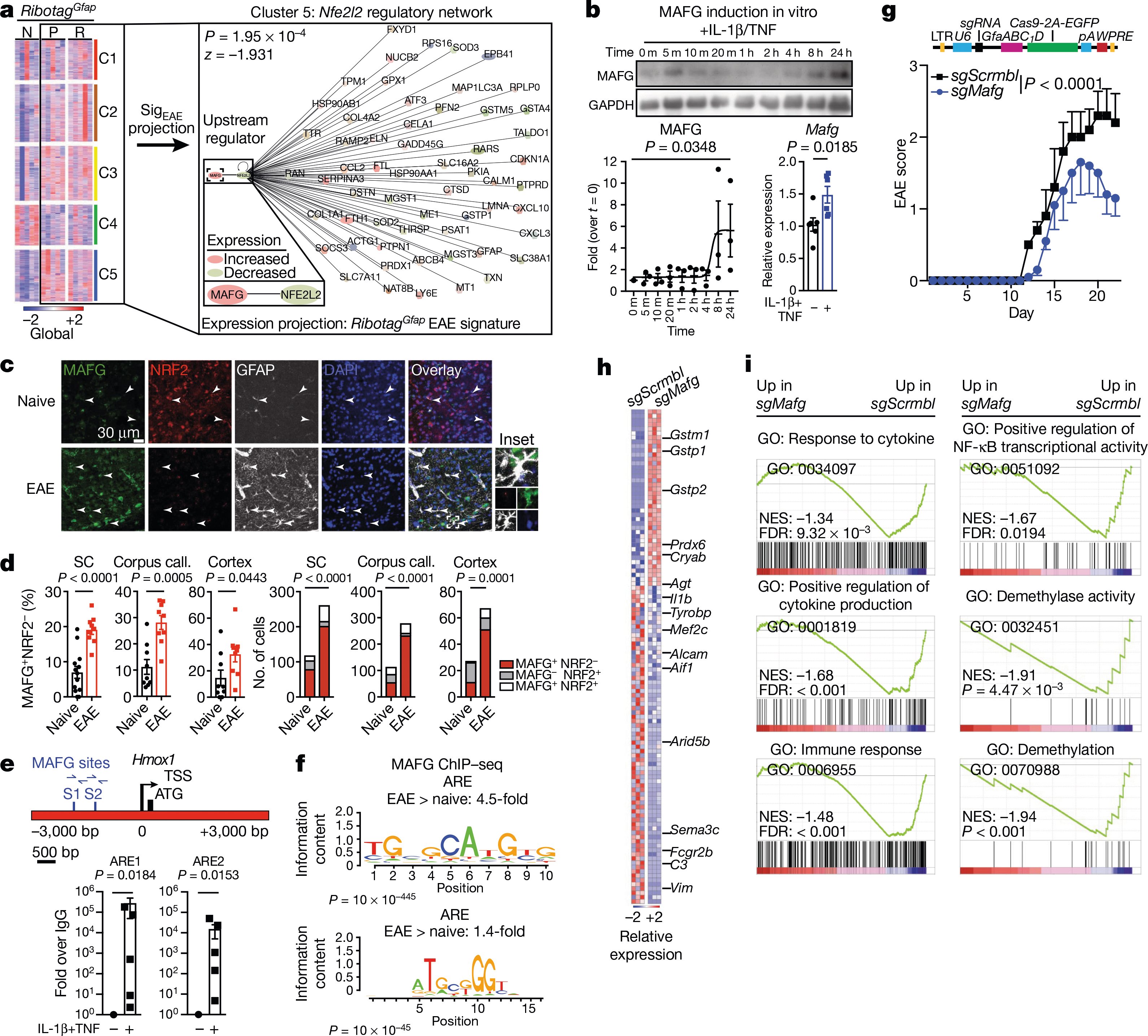
a, K-means clustering by region of expression data from RibotagGfap astrocytes projected onto cluster 5 (C5) TdTomatoGfap Nfe2l2 IPA network. Naive, n = 3; peak: n = 3 for fourth and fifth columns, n = 2 for first, second and third columns; remission: n = 3 for second, fourth and fifth columns, n = 2 for first and third columns. First column, cerebellum; second, cortex; third, cranial nerves; fourth, parenchyma; fifth, spinal cord. N, naive; P, peak; R, remission. b, Western blot (top) and quantification of induction (bottom left) of MAFG expression in activated astrocytes. Kruskal–Wallis test, uncorrected Dunn’s test. qPCR analysis (bottom right). n = 6 independent biological samples per condition, unpaired two-tailed t-test. c, d, Immunostaining (c) and quantification (d) of MAFG+NRF2− astrocytes in mice with and without EAE (naive). Left, per cent MAFG+NRF2− astrocytes. Spinal cord (SC), n = 117 cells from naive mice, n = 260 from mice with EAE; corpus callosum, n = 112 naive, n = 277 EAE; cortex, n = 27 naive, n = 67 EAE; χ2 test. Right, number of MAFG+NRF2− astrocytes. n = 13 sections for spinal cord naive, n = 9 sections otherwise, from n = 3 mice; unpaired two-tailed t-test. e, ChIP–qPCR analysis of MAFG recruitment to the Hmox1 promoter, n = 5 per group. One-sample t-test on log-normalized data. f, Antioxidant response element (ARE) motif analysis by ChIP–seq (day 23 after induction of EAE). Panels denote ARE motif sequences. n = 3 mice per group. g, EAE progression in mice transduced with sgScrmbl (n = 25) or sgMafg (n = 15). Top: schematic of lentiviral vector. Experiment repeated three times. Two-way repeated measures ANOVA. h, i, RNA-seq analysis (h) and GSEA (i) of gene expression in astrocytes from mice transduced with sgScrmbl or SgMafg. n = 3 mice per group (day 22). Data shown as mean ± s.e.m.
Small MAF proteins (sMAFs) lack a transactivation domain but can associate with other proteins to regulate gene expression; sMAFs also form homodimers that repress the expression of target genes32. MAFG is enriched in the nervous system33 and is detectable by scRNA-seq (Extended Data Figs. 3f, 5e), but its role in astrocytes is unknown.
Stimulation of primary mouse astrocytes in culture by IL-1β and TNF increased the expression of MAFG (Fig. 3b). Moreover, the number of MAFG+NRF2− astrocytes was increased during EAE in the spinal cord, corpus callosum, and cortex (Fig. 3c, d, Extended Data Fig. 6a, b) consistent both with reports of astrocyte regional signatures in EAE23 and with the upregulation of MAFG expression in astrocytes during CNS inflammation.
MAFG interacts with other proteins to regulate gene expression32. MAFG–NRF2 heterodimers participate in NRF2-driven gene expression, but MAFG homodimers compete for MAFG–NRF2-responsive elements and thereby suppress target gene expression32. ChIP–seq showed that recruitment of MAFG to NRF2-responsive antioxidant responsive elements34 was increased in astrocytes during EAE and after cytokine stimulation in vitro, suggesting that in the context of decreased NRF2 levels, MAFG suppresses NRF2-driven gene expression (Fig. 3e, f).
To study the effects of MAFG in astrocytes on EAE, we co-delivered Gfap-driven CRISPR–Cas9 and Mafg-targeting or control sgRNA using a lentiviral vector13. Inactivation of Mafg in astrocytes ameliorated EAE, increased the expression of the NRF2 target genes Gstm1, Gstp1, Gstp2 and Prdx6 (detected by RNA-seq) and decreased pro-inflammatory gene expression (Fig. 3g–i, Extended Data Fig. 6b, c, Supplementary Table 5). These data suggest that decreased NRF2 signalling in astrocytes concomitant with increased MAFG expression promotes CNS inflammation during EAE.
MAFG and MAT2α control astrocytes in EAE
MAFG has been reported to cooperate with DNA methyltransferase 3B (DNMT3B) to limit gene expression35. Indeed, we detected the activation of DNA methylation programs in astrocytes from mice with EAE (Fig. 2e). Inactivation of Mafg decreased the expression of pathways associated with DNA methylation in astrocytes during EAE, and ATAC–seq showed that it increased chromatin accessibility in DNMT3B-controlled genes (Figs. 3i, 4a, Supplementary Table 5). To further evaluate the control of DNA methylation in astrocytes by MAFG, we performed whole-genome bisulfite sequencing (WGBS). In astrocytes from mice with EAE, there was increased promoter and exon DNA methylation, including at NRF2 target genes (Fig. 4b, c); inactivation of Mafg reduced methylation of NRF2 target genes (Fig. 4d, e).
Fig. 4 |. MAFG and MAT2α cooperate to promote pathogenic activity of astrocytes.
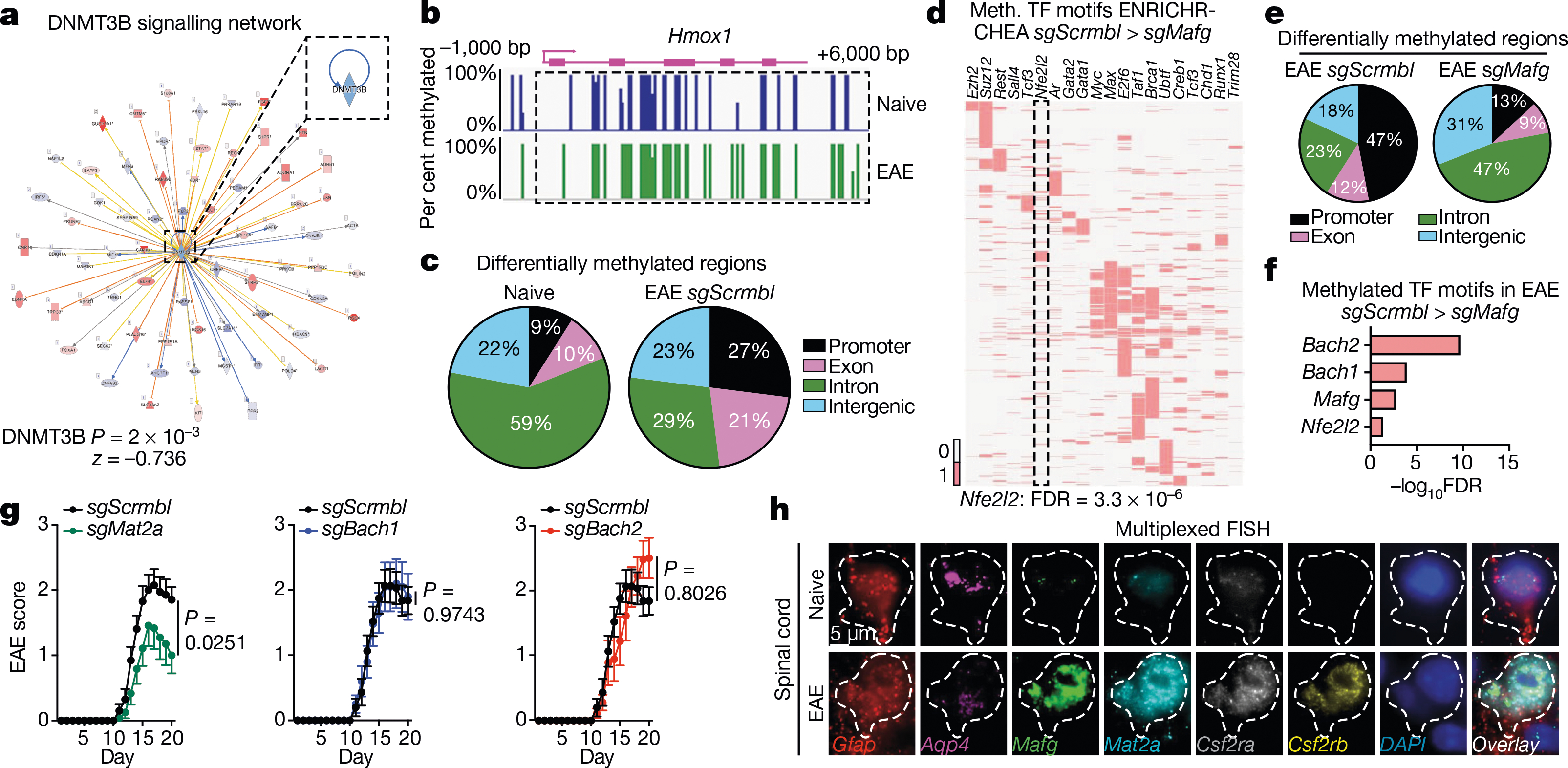
a, ATAC–seq analysis of DNMT3B network in sgMafg compared to sgScrmbl EAE mice (EAE day 22). b, WGBS of Hmox1 promoter. n = 3 mice per group. c, DNA methylation pattern in astrocytes during EAE. n = 3 mice per group. d, Transcription factor (TF) motif analysis of astrocytes from sgScrmbl compared to sgMafg EAE mice using CHEA TF motif analysis in ENRICHR. n = 3 mice per group. e, DNA methylation in astrocytes following Mafg inactivation. n = 3 mice per group. f, Methylated transcription factor motif analysis by WGBS (day 22). n = 3 mice per group. g, EAE in mice transduced with sgMat2a (n = 12), sgBach1 (n = 10), sgBach2 (n = 9) or sgScrmbl (n = 20) lentiviruses. Two-way repeated measures ANOVA. h, Multiplexed FISH showing gene coexpression in astrocytes from EAE and naive mice. Representative images from three independent experiments, n = 3 mice per group.
Several cofactors cooperate with sMAFs to modulate gene expression, including methionine adenosyltransferase IIα (MAT2α) which generates the DNA methylation substrate SAM28, and the transcription repressors BACH1 and BACH232. Our scRNA-seq (Fig. 2e) and WGBS (Fig. 4f) studies identified potential roles for MAT2α, BACH1, and BACH2 in controlling NRF2 signalling by DNA methylation. Therefore, we used lentivirus-delivered CRISPR–Cas9 and targeting sgRNA to inactivate these cofactors in astrocytes13 and evaluate their role in the control of the transcriptional response by MAFG. The inactivation of Mat2a, but not of Bach1 or Bach2, ameliorated EAE (Fig. 4g, Extended Data Fig. 7a), suggesting that although interactions between MAFG and BACH1 or BACH2 interfere with NRF2 signalling32, MAT2α-dependent DNA methylation has a dominant role in the control of astrocytes by MAFG during EAE. Indeed, in multiplexed fluorescent in situ hybridization (FISH) studies of EAE CNS samples, we detected co-expression of Mat2a, Mafg, Gfap and Aqp4 in astrocytes that also expressed the GM-CSF receptor subunits Csf2ra and Csf2rb (Fig. 4h). These findings suggest that in the context of reduced NRF2 expression during EAE, MAFG-driven DNA methylation controls anti-oxidant and pro-inflammatory transcriptional modules in astrocytes.
GM-CSF boosts pathogenic astrocytes
We detected increased GM-CSF signalling in cluster 4 and TdTomatoGfap cluster 5 astrocytes during EAE (Figs. 1f, 2c, d, 4h). GM-CSF secreted by encephalitogenic T cells36,37 is thought to act on microglia38 and recruited monocytes39 to promote CNS pathology, but its effects on astrocytes are unknown. To investigate the role of GM-CSF signalling, we generated Csf2rbAldh1l1-creERT2 knockout mice, which lack the GM-CSF receptor in astrocytes. Deletion of Csf2rb ameliorated EAE and decreased the expression of Mafg, Mat2a and pro-inflammatory pathways in astrocytes (Fig. 5a–d, Extended Data Fig. 7b, c, Supplementary Table 6), suggesting that GM-CSF signalling promotes the pathogenic activities of astrocytes during CNS inflammation.
Fig. 5 |. GM-CSF signalling promotes pathogenic activity in astrocytes.
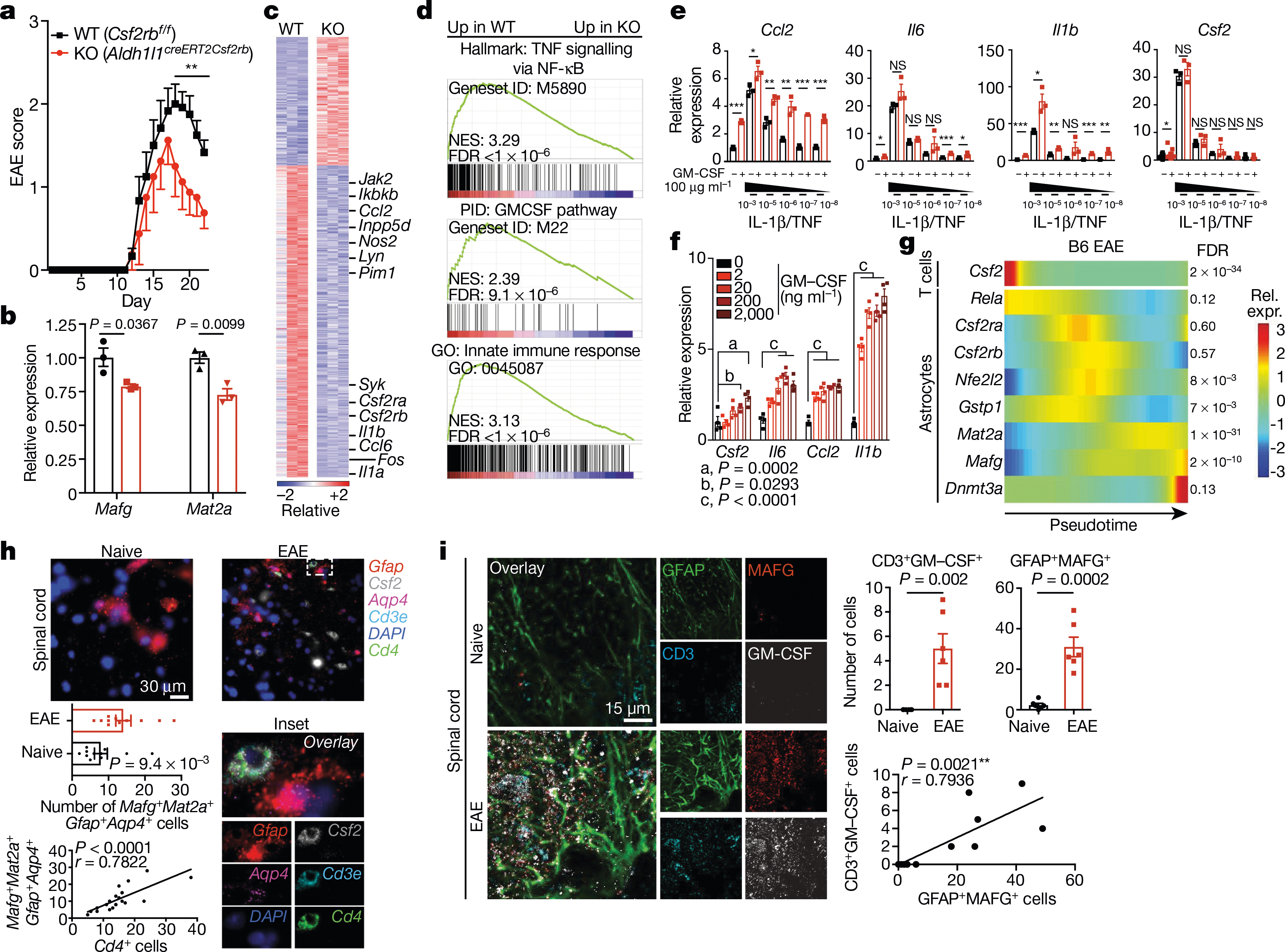
a, EAE progression in wild-type (WT) mice and mice lacking Csf2rb (KO). n = 12 (WT), n = 8 (KO). Experiment repeated twice. Two-way repeated measures ANOVA. Statistical values by day (d) are: d18 (**P = 0.0078), d19 (**P = 0.0015), d20 (**P = 0.0009), d21 (**P = 0.0078), d22 (**P = 0.0097). b, qPCR analysis of expression of Mafg and Mat2a in astrocytes from WT and KO mice (EAE day 22). n = 3 per group; unpaired two-tailed t-test per gene. c, RNA-seq analysis of differential gene expression between astrocytes from WT and KO mice (EAE day 22). n = 3 per group. d, GSEA for genes differentially expressed in WT versus KO mice. n = 3 mice per condition. e, qPCR analysis of gene expression in primary astrocytes with or without different doses of GM-CSF or escalating doses of IL-1β/TNF. n = 3 per condition; n = 18 for Csf2–vehicle. Unpaired two-tailed t-test per grouping, Csf2 data log-normalized. f, qPCR of GM-CSF dose response in primary astrocytes. n = 4 biologically independent samples per condition. Experiment repeated twice. Two-way ANOVA, Dunnett post-test. g, Pseudotime analysis of scRNA-seq data from B6 mice with EAE. n = 9,629 cells. h, Multiplexed FISH analysis of gene co-expression in astrocytes from mice with or without EAE. n = 11 per group from n = 3 mice. Unpaired two-tailed Mann–Whitney test (top), Pearson’s correlation (bottom). i, Immunostaining (left) and quantification (right) of MAFG+ astrocytes in close proximity to GM-CSF+ T cells in spinal cord from mice with or without EAE. n = 6 images per group from n = 3 mice. Unpaired two-tailed t-test (top), Pearson’s correlation (bottom). ***P < 0.001, **P < 0.01, *P < 0.05, NS P > 0.05 (not significant). Data shown as mean ± s.e.m.
To further investigate the effects of GM-CSF signalling, we activated primary mouse astrocytes in vitro with escalating doses of IL-1β and TNF in the presence or absence of GM-CSF. We found that GM-CSF boosted the expression of pro-inflammatory genes in a dose-dependent manner (Fig. 5e, f). Moreover, in a pseudotime trajectory analysis, Csf2 expression in CNS-infiltrating T cells preceded the downregulation of Nfe2l2 and Gstp1 in astrocytes and the upregulation of Rela, Csf2ra, Csf2rb, Mafg, Mat2a and Dnmt3a during EAE (Fig. 5g). Consistent with GM-CSF-driven activation of astrocytes during EAE, multiplexed FISH and immunostaining identified MAFG+ astrocytes in close proximity to GM-CSF+ T cells (Fig. 5h, i, Extended Data Fig. 7d). These data suggest that GM-CSF boosts the pathogenic activity of astrocytes in EAE.
MAFG-driven astrocytes in MS
To study the transcriptional signature of astrocytes in MS, we used scRNA-seq to analyse CNS samples from four patients with MS who underwent euthanasia followed by rapid autopsy, and five surgically resected samples from individuals without MS (Extended Data Fig. 8, Supplementary Table 7). Following batch correction, the analysis of 43,670 cells identified cell clusters that expressed astrocyte markers (Extended Data Fig. 8, Supplementary Table 8). To expand our dataset, we identified astrocytes in data from scRNA-seq analysis of cortical and cerebellar samples from patients with MS and control individuals17,20,40 (Extended Data Fig. 9, Supplementary Tables 9–11). This approach generated an integrated scRNA-seq dataset containing 9,673 cortical and cerebellar astrocytes derived from 20 patients with MS and 28 control individuals (Fig. 6a, Extended Data Fig. 10a–e, Supplementary Table 12). In this combined dataset we identified an astrocyte population that was expanded 25.2-fold in samples from patients with MS compared to control samples; this astrocyte population was detected in 12 out of 20 patients with MS from all three MS patient cohorts (Fig. 6b) and was characterized by decreased NRF2 activation and increased MAFG activation, DNA methylation, GM-CSF signalling and pro-inflammatory pathway activity (Fig. 6c–e, Extended Data Fig. 10f). The re-analysis of subsets of region-matched cortical and cerebellar astrocyte samples17,20,40 identified a similar MS-associated astrocyte population, ruling out the possibility that this population results from differences in sampled CNS regions or quality (Extended Data Fig. 9, Supplementary Tables 9–11).
Fig. 6 |. MAFG activation characterizes an astrocyte population associated with MS.
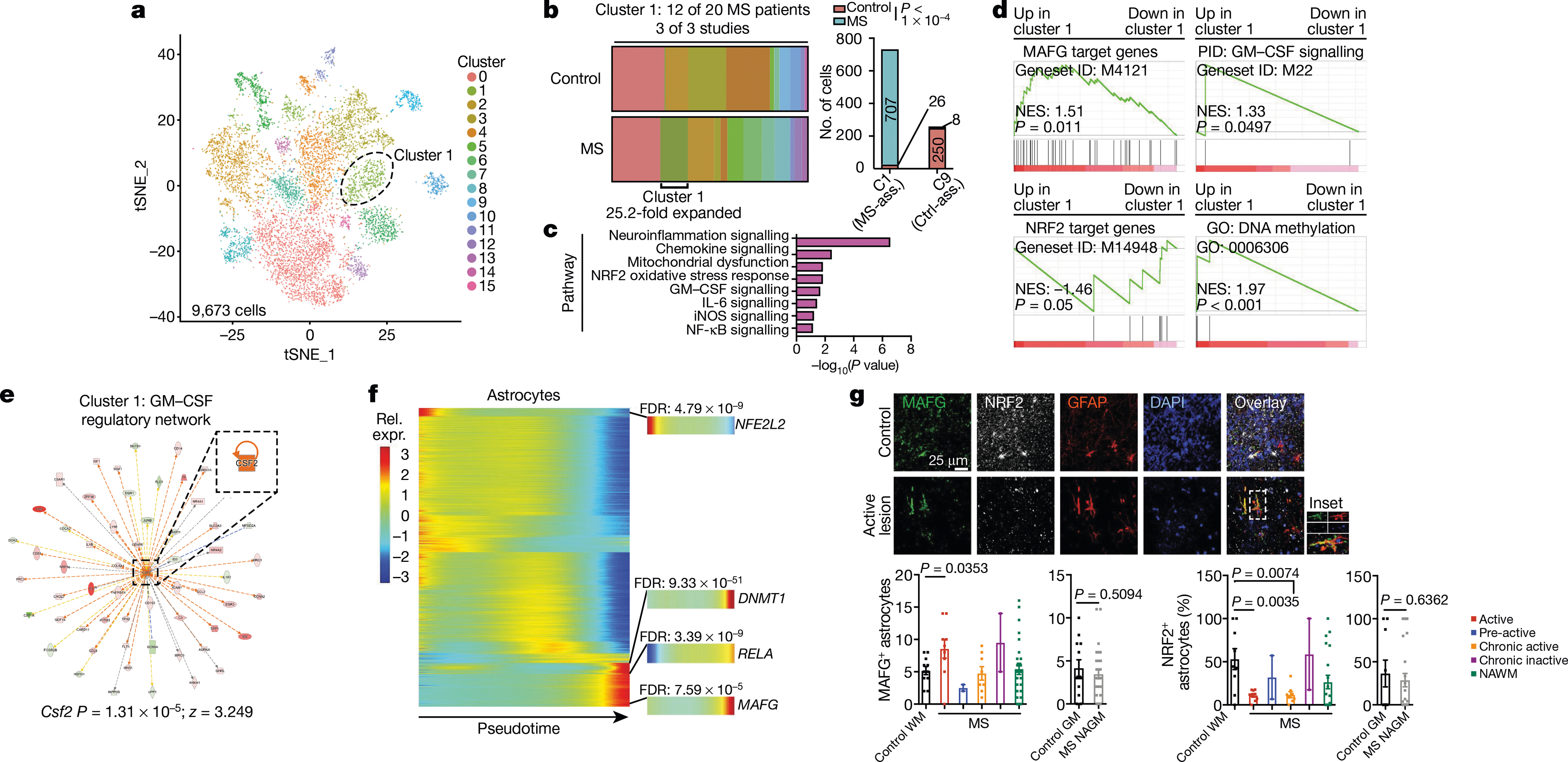
a, tSNE plot by cluster for astrocytes from patients with MS and control individuals from Schirmer et al.20, Jäkel et al.17, Lake et al.40, and this study (n = 9,673 cells). b, Fraction of cells per cluster overall (left) and by patient (right). MS-ass., MS-associated cluster; Ctrl-ass., control-associated cluster. Fisher’s exact test. c, IPA pathway chart of n = 733 cluster 1 astrocytes. d, Pre-ranked GSEA in n = 733 cluster 1 astrocytes. e, GM-CSF regulatory network in cluster 1 astrocytes. f, Pseudotime analysis of gene expression in human astrocytes (n = 9,673 cells). n = 20 MS patients and n = 28 controls by scRNA-seq. g, Immunostaining (top) and quantification (bottom) of MAFG+ astrocytes and NRF2+ astrocytes in tissue samples from patients with MS (n = 5) and control individuals (n = 4). WM, white matter; GM, grey matter; NAWM, normally appearing white matter; NAGM, normally appearing grey matter. Sample sizes from left to right: MAFG analysis: n = 11 sections, n = 9, n = 2, n = 8, n = 2, n = 27, n = 12, n = 30. NRF2 analysis: n = 9, n = 9, n = 2, n = 8, n = 2, n = 20, n = 9, n = 28. Unpaired two-tailed t-test. Data shown as mean ± s.e.m.
Similarly, a pseudotime analysis of differentially expressed genes detected upregulation of MAFG, DNMT1 and RELA concomitant with decreased NFE2L2 expression (Fig. 6f). Notably the integration of human and mouse scRNA-seq data identified common transcriptional modules in astrocytes from patients with MS and mice with EAE, including decreased NRF2 signalling regulated by MAFG activation and GM-CSF (Extended Data Fig. 10g).
Finally, to validate our findings, we performed immunostaining on additional samples from patients with MS and control individuals. In agreement with our scRNA-seq-based identification of a MAFG-driven astrocyte population that is associated with active lesions in MS (Extended Data Fig. 9a, b), our immunostaining studies detected increased astrocyte expression of MAFG in active lesions in white matter from patients with MS (Fig. 6g). In addition, astrocytes in active and chronically active white matter lesions showed decreased NRF2 expression compared to controls (Fig. 6g). Together, these data (Fig. 6a–g, Extended Data Figs. 8–10) identify a disease-associated astrocyte population characterized by decreased NRF2-driven gene expression and the activation of transcriptional modules associated with MAFG and GM-CSF signalling, DNA methylation and the promotion of CNS pathology.
Discussion
The analysis of oligodendrocyte, neuron and myeloid cell heterogeneity has provided important insights into MS pathogenesis17–22. Although a population of neurotoxic astrocytes has been identified in several neurological diseases41, little is known about astrocyte heterogeneity and its regulation in MS. We have identified astrocytes characterized by reduced NRF2-driven gene expression and increased MAFG and MAT2α signalling that promote CNS inflammation in EAE and potentially contribute to the pathogenesis of MS.
Large MAF proteins such as c-Maf and MafB participate in the transcriptional control of the development and functions of T cells, macrophages and microglia42–47. However, less is known about the immunoregulatory roles of sMAFs. Whereas c-Maf and MafB contain transactivation domains, MAFG lacks these domains and interacts with other proteins, such as NRF2, to control gene expression32. Our data suggest that, in the context of reduced NRF2 expression during EAE and potentially MS, MAFG outcompetes MAFG–NRF2 at antioxidant responsive elements and cooperates with MAT2α to establish DNA methylation marks that control disease-promoting activities in a population of astrocytes. Notably, previous analyses of tissue from patients with MS detected altered DNA methylation in CNS-resident cells48 but not peripheral immune cells49, supporting the notion that epigenetic changes in the CNS contribute to the pathogenesis of MS. These data are reminiscent of recent reports of trained immunity in microglia in Alzheimer’s disease50,51, highlighting common mechanisms that regulate the pathogenic activity of glial cells in MS and other neurological diseases. In addition, our findings suggest that GM-CSF, produced by inflammatory T cells recruited to the CNS in MS and EAE, amplifies MAFG/MAT2α-driven pro-inflammatory genomic programs in astrocytes. These findings define previously undescribed mechanisms of disease pathogenesis and identify epigenetic modifiers as candidate targets to suppress the pathogenic activity of astrocytes in MS.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586–020-1999–0.
Methods
Mice
Adult male and female mice and postnatal pups were used on a C57Bl/6J background (#000664, The Jackson Laboratory). B6.Cg-Tg(Gfap-cre)73.12Mvs/J mice52 (The Jackson Laboratory, #012886) mice were crossed with B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato) Hze/J mice53 (The Jackson Laboratory, #007909) to generate Gfapcre/+ TdTomatof/+ (TdTomatoGfap) mice. B6N.129-Rpl22tm1.1Psam/J mice54 (The Jackson Laboratory, #011029) were bred to Gfapcre mice to generate Gfapcre/+Ribotagf/f (RibotagGfap) mice. Aldh1l1creERT2 mice55 (The Jackson Laboratory, #029655) were bred to Csf2rbf/f mice39 to generate Aldh1l1creERT2 Csf2rbf/f mice. Conditional deletion of Csf2rb was induced at 5 weeks of age with tamoxifen (225 mg/kg; Sigma-Aldrich, #T5648) diluted in corn oil (Sigma-Aldrich, #C8267); EAE was induced 3 weeks later. Mice were kept in a pathogen-free facility at the Hale Building for Transformative Medicine at Brigham and Women’s Hospital in accordance with the IACUC guidelines. Eight-to-twelve-week-old mice were used for stereotactic injection and EAE induction. Pups were killed between postnatal day 0 (P0) and P3 for collection and culture of astrocytes. All procedures were reviewed and approved under the IACUC guidelines at Brigham and Women’s Hospital.
Ribotag immunoprecipitation
Ribotag immunoprecipitations were performed as described previously54,56, with some modifications. All surfaces were cleaned with 70% ethanol and wiped down with RNaseZap (Thermo Fisher, #AM9782). Dounce homogenizers were washed with double-distilled water (ddH2O) and cleaned with RNaseZap 3× before being placed on ice. Neural tissue regions were added to supplemented homogenization buffer at 5% (w/v) (HB-S) containing 1% NP-40 (Sigma-Aldrich, #11332473001), 100 mM KCl (Santa Cruz, #sc-301585), 100 mM Tris-HCl pH 7.4 (Sigma-Aldrich, #T2663–1L), and 12 mM MgCl2 (Sigma-Aldrich, #M1028–100ML), which was supplemented with 100 μg/ml cycloheximide (Sigma-Aldrich, #C7698–5G), protease inhibitor cocktail (Sigma-Aldrich, #P8340–5ML, 1:100 dilution), 1 mg/ml heparin (Sigma-Aldrich, #H3393–100KU), RNase inhibitors (Promega, #N2115), and 1 mM dithiothreitol (DTT) (Thermo Fisher, #P2325). Tissue homogenization was performed on ice and 1,200 μl of tissue homogenate was centrifuged at 10,000 rpm at 4 °C for 10 min. Approximately 750 μl of supernatant was used for immunoprecipitation (IP). Each sample was incubated with mouse anti-HA antibody (Sigma-Aldrich, #H9658-.2ML, 1:150 dilution) for 4 h at 4 °C with inversion. Next, before IP, Pierce Protein A/G Magnetic Beads (Thermo Fisher, #88803) were equilibrated in HB-S for 30 min at 4 °C with agitation. Equilibrated beads were added at a concentration of 25 μl beads per 100 μl supernatant sample. Bead–antibody–sample complexes were incubated overnight at 4 °C with inversion. After 16–18 h, samples were pulse centrifuged and magnetic beads were separated from the supernatant. Beads were washed three times with high salt buffer containing 300 mM KCl, 1% NP-40, 50 mM Tris-HCl pH 7.4, 12 mM MgCl2, 100 μg/ml cycloheximide, and 500 μM DTT. Next, ribosome–RNA complexes bound to the beads were lysed in buffer RLT (Qiagen, #79216) containing 1% 2-mercaptoethanol (Sigma-Aldrich, #M3148). Samples were then processed using Qiagen RNeasy Mini Kit (Qiagen, #74106) with on-column DNase digestion (Qiagen, #79254). Purified RNA was enriched for polyadenylated mRNA using the Dynabeads mRNA DIRECT purification kit (Thermo Fisher, #61012). The RNA integrity number (RIN) was determined using the Agilent RNA 6000 Pico Kit (Agilent Technologies, #5067–1513). Libraries for RNA sequencing were created using a derivation of massively parallel single-cell RNA sequencing (MARS-seq)57. The final library concentration was measured with a Qubit fluorometer (Life Technologies) and the mean molecule size was determined using a 2200 TapeStation instrument (Agilent Technologies). Libraries were sequenced using an Illumina NextSeq 500.
Drop-seq
Drop-seq was performed according to the original protocol25. A microfluidic mask was fabricated at 125 μm in height using soft lithography. The curing agent and PDMS prepolymer (Momentive, #RTV615) were mixed at 1:10 and degassed in a vacuum chamber. The PDMS mixture was poured onto the master mould, further degassed, and baked at 65 °C for 4 h. The PDMS replica was punched with a 0.75-mm biopsy punch (Harris Uni-Core) and bonded to a glass slide (75 × 50 × 1.0 mm, Fisher Scientific, 12–550C) using a plasma bonder (Technics Plasma Etcher 500-II). The device was placed on a hot plate at 150 °C for 10 min, baked at 65 °C for 4 h, and treated with Aquapel to render it hydrophobic. A cell suspension was prepared using flow cytometry-sorted cells or a freshly prepared cell suspension from mouse CNS (brain and spinal cord) or human samples. Cells were counted and resuspended at 250,000 cells/ml (final concentration 125 cells/μl) in PBS and 16% Optiprep (Sigma-Aldrich, #D1556–250ML). The cell mixture was loaded into a 3-ml syringe (BD Biosciences, #309657) with a 27 gauge needle (BD Biosciences. #305109) and connected to the microfluidic device using tubing (Scientific Commodities, #BB31695-PE/2). Barcoded beads (Fisher Scientific, #NC0927472) were resuspended in lysis buffer consisting of: 57% Opti-prep (Sigma-Aldrich, #D1556–250ML), 2.4% Ficoll PM-400 (GE Healthcare, #17–0300-10), 0.2% Sarkosyl (Teknova, #S3376), 20 mM EDTA pH 8.0, 200 mM Tris-HCl pH 7.4 (Sigma-Aldrich, #T2663–1L), and 50 mM DTT (Sigma-Aldrich, #646563–10X.5ML) at a concentration of 300,000 beads/ml. Beads resuspended in lysis buffer were loaded into a 3-ml syringe with a magnetic mixing disc (V&P Scientific, #772DP-N42–5-2) and gently stirred during encapsulation using a magnetic mixer (V&P Scientific, #710D2). A third syringe was loaded with oil for droplet generation (Biorad, #186–4006). To perform Drop-seq experiments, pumps were run at 1,500 μl/hour (cell mixture), 1,500 μl/hour (barcoded beads), and 4,500 μl/hour (oil) for approximately 15 min per sample. Droplets were collected, broken with 1H, 1H, 2H, 2H-perfluoro-1-octanol (PFO) (Sigma-Aldrich, #370533) added at a ratio of 1:3 PFO:oil, washed with 10 ml 6× SSC (National Diagnostics, #EC-873), and centrifuged for 1 min at 1,000g. Beads at the interface were removed, oil was eliminated, and beads were washed 3× with 6× SSC. Beads were next washed with Maxima H-minus 1× reverse transcription (RT) buffer (Thermo Fisher Scientific, #EP0753). Beads were resuspended in 50 μl Maxima H-minus 1× RT buffer. The following RT mixture was added to each tube of beads: 40 μl 20% Ficoll PM-400, 30 μl 5× Maxima H-minus RT buffer, 2 μl 100 mM dNTPs (Life Technology, #4368813), 5 μl 100 μM template-switching oligonucleotide (TSO) primer (IDT), 5 μl RNase inhibitor (Lucigen, #30281–2), 58 μl nuclease-free water, and 10 μl Maxima H-minus reverse transcriptase (Thermo Fisher Scientific, #EP0753). Beads suspended in RT mixture were incubated at room temperature for 30 min on an inverter, followed by 90 min at 42 °C on an inverter. Following reverse transcription, beads were washed 1× with 1 ml TE-SDS (10 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0, 0.5% SDS), and 2× with TE-TW (10 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0, 0.01% Tween-20) and pelleted during each step by centrifuging at 1,000g for 1 min. Beads were washed 1× with 10 mM Tris-HCl pH 8.0 and resuspended in exonuclease mix consisting of: 170 μl nuclease-free water, 20 μl 10× exonuclease I buffer, and 10 μl exonuclease I (Thermo Fisher Scientific, #EN0582). Resuspended beads were incubated for 45 min at 37 °C with inversion. After treatment, beads were washed 1× with TE-SDS, 2× with TE-TW, and 2× with nuclease-free water. Beads were counted and resuspended at a concentration of 80 beads/μl in preparation for PCR. Beads were then added to a PCR tube at a concentration of 2,000 beads per tube. PCR mix (25 μl HiFi HotStart ReadyMix (Kapa Biosystems, KK2602); 0.4 μl 100 μM SMART PCR primer (IDT)) was added to each tube and beads were manually mixed before PCR. PCR cycling conditions were: 95 °C (3 min.); 4 cycles of: 98 °C (20 s), 65 °C (45 s), 72 °C (3 min); 9 cycles of: 98 °C (20 s), 67 °C (20 s), 72 °C (3 min); 72 °C (5 min); 4 °C hold. Following PCR, cDNA samples were purified in a 96-well plate (Biorad, #HSP9611) on a magnetic stand (NEB, #S1511S) using Agencourt AMPure XP magnetic beads (Beckman Coulter, #A63881) at a 0.6× ratio according to the standard protocol. The sample from each PCR tube was eluted in 10 μl nuclease-free water, and technical replicates were pooled following elution. cDNA was run on a Bioanalyzer High Sensitivity DNA chip (Agilent Technologies, #5067–4626) on a 2100 Bioanalyzer (Agilent). For cDNA library tagmentation, 600 pg of cDNA in 5 μl was added to 10 μl of Nextera Tagment DNA buffer and 5 μl of Amplicon Tagment Mix (Illumina, #FC-131–1096). cDNA was tagmented at 55 °C for 5 min, followed by addition of 5 μl room temperature-equilibrated Neutralize Tagment buffer (Illumina, #FC-131–1096). Samples sat at room temperature for 5 min followed by addition of 15 μl Nextera PCR Mix (Illumina, #FC-131–1096), 8 μl nuclease-free H2O, 1 μl 10 μM New-P5-SMART PCR hybrid oligo (IDT), and 1 μl of 10 μM Nextera indexing oligonucleotide (Invitrogen). Samples underwent PCR using the following conditions: 95 °C (30 s); 12 cycles of: 95 °C (10 s), 55 °C (30 s), 72 °C (30 s); 72 °C (5 min); 4 °C hold. Tagmented libraries were purified using Agencourt AMPure XP magnetic beads at a 0.6× ratio according to the manufacturer’s protocol and eluted in 10 μl nuclease-free H2O. Samples were then run on a Bioanalyzer 2100 to assess library size. Libraries were quantified by qPCR using a Library Quantification Kit (Kapa Biosystems, #KK4824). Tagmented libraries were pooled at ≥2 nM concentration and sequenced using either a HiSeq 4000 or NovaSeq S2 at the Broad Institute using a Custom Read 1 primer. The sequences of the primers used were: TSO: 5′- AAGCAGTGGTATCAACGCAGAGTGAATrGrGrG-3′; SMART PCR Primer: 5′-AAGCAGTGGTATCAACGCAGAGT-3′; New-P5-SMART PCR hybrid oligo: AATGATACGGCGACCACCGAGATCTACACG CCTGTCCGCGGAAGCAGTGGTATCAACGCAGAGT*A*C; Custom Read 1 primer: 5′-GCCTGTCCGCGGAAGCAGTGGTATCAACGCAGAGTAC-3′; Nextera indexing oligonucleotide: 5′-CAAGCAGAAGACGGCATACGAG ATNNNNNNNNGTCTCGTGGGCTCGG-3′, where NNNNNNNN indicates index sequence, ranging from N701 to N729. The RIN was determined using the Agilent RNA 6000 Pico Kit (Agilent Technologies, #5067–1513). For tissue obtained from patients who underwent euthanasia followed by rapid autopsy, cells from all samples were used for scRNA-seq. At the end of the experiment, RNA was isolated from any remaining cells from a given sample and the RIN was measured. Note that we analysed our data using the mRIN program described in ref. 58 and compared its results with the RIN values obtained by Bioanalyzer from the same samples. mRIN did not accurately predict the RIN values of any of these samples (Bioanalyzer RIN: 6.3 ± 0.8 (mean ± s.e.m.); mRIN: 0). This could be explained by the fact that the mRIN analysis models RNA integrity on the basis of 3′ bias, a problematic feature considering that 3′ bias is inherent to all scRNA-seq techniques because they are based on the capture of mRNAs by their 3′ end59. However, the mean RINs obtained for available samples were comparable to those recently published17,20.
scRNA-seq analysis
Trim Galore (version 0.6.0) was used to trim the adaptor contents and the 5′ primers. Single-cell paired-end reads were then processed using Drop-seq software (version 2.0.0)25. After associating every read with its associated cell barcode and unique molecular identifiers (UMIs), read alignment was performed using STAR software (2.5.2) to human and mouse reference genomes, which were prepared from the ENSEMBL Human Genome (GRCh38.p12) and the ENSEMBL Mouse Genome (GRCm38.p6), respectively. After alignment, .bam files were sorted using Picard software, and bead synthesis errors (indels or substitutions) were detected and fixed by using Drop-seq software. After we fixed bead synthesis errors, gene expression was quantified using Drop-seq software, using the parameter of ‘min_num_genes_per_cell = 500’ for B6 EAE studies and ‘min_num_genes_per_cell = 200’ otherwise. The optimization of scRNA-seq methods such as the Drop-seq method used in this study is likely to increase the number of genes detected per cell, which is likely to lead to improved in-depth transcriptional signatures for the different clusters identified in our studies.
Using Seurat60,61, the expression matrices of samples were log-normalized and doublets removed. Canonical correlation analysis60 was performed to correct for batch effects and to integrate different samples within each dataset. After the integration of different samples within each dataset, principal component analysis was performed to determine the number of principal components (PCs) to use during the dimension reduction and clustering analysis. In all cases, the first 15 PCs were used. To cluster cells, the Louvain algorithm was applied to the integrated datasets with a resolution parameter of 0.5. After clustering, the MAST algorithm62 was used to conduct differential expression analysis for each cluster compared to all other cells. The results of the differential expression analysis were used in further downstream analysis. Visualization was performed using tSNE63,64. To generate pseudotime series analyses, Monocle (v3.9)65–67 was used to define genes that were differentially expressed during pseudotime states. Statistics for pseudotime analyses were determined using the Benjamini–Hochberg test. Following cell type classification, only cortical and cerebellar astrocytes were analysed from dataset in this study. Cells from each experiment were characterized by the following parameters: B6 EAE: 1,839 ± 11 UMIs (mean ± s.e.m.), 891 ± 3.2 genes, 21,282 ± 632 reads per cell; TdTomatoGfap EAE: 918 ± 7.6 UMIs, 521 ± 3.2 genes, 8,198 ± 162 reads per cell; human samples: 1,257 ± 7.6 UMIs, 688 ± 3.0 genes, 10,572 ± 735 reads per cell.
Western blotting
Western blotting was performed largely as described previously13. Protein lysates were prepared by lysing astrocytes with boiling 1× Laemmli buffer (Boston BioProducts, #BP-111R) followed by boiling at 95 °C for 5 min. SDS–PAGE was performed using Bolt 4–12% Bis-Tris Plus gradient gels (Invitrogen, #NW04125BOX). Western blotting was performed by transferring proteins onto a PVDF membrane (Millipore, #IPVH15150) in 1× NuPAGE buffer (Thermo Fisher, #NP00061). Membranes were blocked in 10% milk (Laboratory Scientific, #M0841) in TBS-T (Boston BioProducts, #IBB-180–2L). Primary antibodies used in this study were: rabbit anti-MAFG (Genetex, #GTX114541, 1:1,000), rabbit anti-GAPDH (Cell Signaling Technology, #2118S, 1:1,000), rabbit anti-cyclophilin B (Thermo Fisher Scientific, #PA1027A, 1:1,000), and rabbit anti-MAT2α (Novus Biologicals, #NB110–94158, 1:1,000). The secondary antibody used in this study was anti-rabbit IgG-HRP conjugate (Cell Signaling Technology, #7074S, 1:1,000). HRP-conjugated blots were developed using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, #34095) and CL-XPosure Film (Thermo Fisher Scientific, #34090). Some HRP-conjugated blots were developed using the KwikQuant imaging system (Kindle Biosciences). Film was developed using a M35A X-OMAT Film Processor (Kodak).
Immunostaining
Mice were intracardially perfused with ice cold 1× PBS followed by ice cold 4% PFA. Brains were removed, post-fixed in 4% PFA overnight at 4 °C, and dehydrated in 30% sucrose for 2 days at 4 °C. Brains were then frozen in optimal cutting temperature compound (OCT; Sakura, #4583) and 30-μm sections were obtained by cryostat on SuperFrost Plus slides (Fisher Scientific, #22–037-246). A hydrophobic barrier was drawn (Vector Laboratories, #H-4000) and sections were washed 3× for 5 min with 0.1% Triton X-100 in PBS (PBS-T). Sections were permeabilized with 0.3% PBS-T for 20 min, then washed 3× with 0.1% PBS-T. Sections were blocked with 5% donkey serum (Sigma-Aldrich, #D9663) in 0.1% PBS-T at room temperature for 30 min. Sections were then incubated with primary antibodies diluted in blocking buffer overnight at 4 °C. Following primary antibody incubation, sections were washed 3× with 0.1% PBS-T and incubated with secondary antibodies diluted in blocking buffer for 2 h at room temperature. Following secondary incubation, sections were washed 3× with 0.1% PBS-T and dried, and coverslips were mounted using Fluoromount-G with DAPI (SouthernBiotech, #0100–20) or without DAPI (SouthernBiotech, #00–4958-02). Primary antibodies used in this study were: mouse anti-GFAP (Millipore, 1:500, #MAB360), chicken anti-GFP (Abcam, #ab13970, 1:1,000), rabbit anti-NRF2 (Abcam, #137550, 1:100), rabbit anti-MAFG (Genetex, #GTX114541, 1:100), rabbit anti-CD3ε (Abcam, #ab215212, 1:500), and rabbit anti-GM-CSF (Abcam, #ab9741, 1:100). Secondary antibodies used in this study were: Alexa Fluor 647 donkey anti-mouse (Abcam, #ab150107), donkey anti-rabbit IgG (H+L) Highly Cross-Adsorbed, Alexa Fluor 568 (Life Technologies, #A10042), Rhodamine Red-X-AffiniPure Fab Fragment donkey anti-rabbit IgG (H+L) (Jackson Immunoresearch, #711–297-003), Alexa Fluor 647 AffiniPure Fab Fragment donkey anti-rabbit IgG (H+L) (Jackson Immunoresearch, #711–607-003), goat anti-rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 405 (Thermo Fisher, #A-31556), Alexa Fluor 488-AffiniPure donkey anti-rabbit IgG (H+L) (Jackson Immunoresearch, #711–545-152), and goat anti-chicken IgY (H+L) Alexa Fluor 488 (Life Technologies, #A11039), all at 1:500 working dilution. For CD3ε staining, heat-mediated antigen retrieval was performed using citrate buffer (IHC World, #IW-1100) before staining, as described68. Iterative labelling using rabbit primary antibodies was accomplished by incubating with a single primary antibody on day 1, staining with the anti-rabbit Fab fragment on day 2, washing 6× with PBS-T, and then incubating with primary and secondary antibodies as described above. Cortical and corpus callosum brain regions analysed were studied between bregma +1.32 and −0.82. Spinal cord sections were taken from T11 to L4.
Imaging
Sections were imaged on a Zeiss LSM710 confocal using a 20× objective. For all analyses, regions were randomly chosen on the basis of DAPI fluorescence. Imaging was performed using the LSM710 smart setup parameters with each channel acquired in sequence to minimize crosstalk between channels to essentially zero. Quantification of MAFG staining in brain sections was performed by first quantifying the number of positive cells for the given marker and then counting the number of those cells that were GFAP+ with a visible cell body in the DAPI channel.
Lentivirus production
Lentiviral constructs were generated by modifying the pLenti-U6-sgScramble-Gfap-Cas9–2A-EGFP-WPRE lentiviral backbone, as described previously13. This backbone contains derivatives of the previously described reagents lentiCRISPR v2 (a gift from F. Zhang, Addgene plasmid #5296169), and lentiCas9-EGFP (a gift from P. Sharp and F. Zhang, Addgene plasmid #6359270). The Gfap promoter is the ABC1D gfa2 GFAP promoter71. Substitution of sgRNAs was performed through a PCR-based cloning strategy using Phusion Flash HF 2× Master Mix (Thermo Fisher, #F548L). A three-way cloning strategy was developed to substitute sgRNAs using the following primers: U6-PCR-F 5′-AAAGGCGCGCCGAGGGCCTATTT-3′, U6-PCR-R 5′-TTTTTTGGTCTCCCGGTGTTTCGTCCTTTCCAC-3′, cr-RNA-F 5′-AAAAAAGGTCTCTACCG(N20) GTTTTAGAGCTAGAAATAGCAAGTT-3′ and cr-RNA-R 5′-GTTCCCTGCAGGAAAAAAGCACCGA-3′, where N20 marks the sgRNA substitution site. Amplicons were purified using the QIAquick PCR Purification Kit (Qiagen, #28104) and digested using DpnI (NEB, #R0176S), BsaI-HF (NEB, #R3535/R3733), AscI (for U6 fragment) (NEB, #R0558), or SbfI-HF (for crRNA fragment) (NEB, #R3642). The pLenti backbone was cut with AscI–SbfI-HF and purified using the QIAquick PCR purification kit. Ligations were performed overnight at 16 °C using T4 DNA Ligase Kit (NEB, #M0202L). Ligations were transformed into NEB Stable Cells (NEB, #C3040) at 37 °C, single colonies were picked, and DNA was prepared using QIAprep Spin Miniprep Kit (Qiagen, #27104). Lentiviral plasmids were transfected into HEK293FT cells according to the ViraPower Lentiviral Packaging Mix protocol (Thermo Fisher Scientific, #K497500) and lentiviruses were packaged with pLP1, pLP2, and pseudotyped with pLP/VSVG. Medium was changed the next day and lentivirus was collected 48 h later and concentrated using Lenti-X Concentrator (Clontech. #631231) overnight at 4 °C followed by centrifugation according to the manufacturer’s protocol and resuspension in 1/100–1/500 of the original volume in 1× PBS. Delivery of lentiviruses via intracerebroventricular injection was performed largely as described previously13. In brief, mice were anaesthetized using 1–3% isoflurane mixed with oxygen. Heads were shaved and cleaned using 70% ethanol and Betadine (Thermo Fisher, #19–027132) followed by a medial incision of the skin to expose the skull. The ventricles were targeted bilaterally using the coordinates: ±1.0 (lateral), −0.44 (posterior) and −2.2 (ventral) relative to the bregma. Mice were injected with approximately 107 total IU of lentivirus delivered by two 10-μl injections using a 25-μl Hamilton syringe (Sigma-Aldrich, #20787) on a stereotaxic alignment system (Kopf, #1900), sutured, and permitted to recover in a separate clean cage. Mice were permitted to recover for between 4–7 days before induction of EAE. CRISPR–Cas9 sgRNA sequences were designed using a combination of the Broad Institute’s sgRNA GPP Web Portal (https://portals.broad-institute.org/gpp/public/analysis-tools/sgrna-design) and Synthego (https://design.synthego.com/#/validate), and cross-referenced with activity-optimized sequences contained within the Addgene library #1000000096 (a gift from D. Sabatini and E. Lander)72. sgRNAs used in this study were: sgMafg: 5′-GAGTTGAACCAGCACCTGCG-3′, sgNfe2l2: 5′-TGACTTTAGTCAGCGACAGA-3′, sgBach1: 5′-GCTATGCACAGAGGACTCGT-3′, sgBach2: 5′-GGACTCATATACATACATGG-3′, sgMat2a: 5′-ACGAGGCGTTCATTGAGGAG-3′, and sgScramble: 5′-GCACTACCAGAGCTAACTCA-3′ (sequence from Origene, #GE100003).
EAE
EAE was induced as described13. All mice used were on the C57Bl/6 background. EAE was induced in mice that had been injected with lentivirus 4–7 days post-transduction using 25 μg of MOG35–55 (Genemed Synthesis Inc., #110582) mixed with freshly prepared complete Freund’s adjuvant (using 20 ml incomplete Freund’s adjuvant (BD Biosciences, #BD263910) mixed with 100 mg M. tuberculosis H-37Ra (BD Biosciences, #231141)) at a ratio of 1:1 (v/v at a concentration of 5 mg/ml). For wild-type B6 scRNA-seq studies (Figs. 1, 5g, Extended Data Figs. 1, 4a, 5e, 10g), TdTomatoGfap studies, and RibotagGfap studies (Fig. 2a–e, 3a, Extended Data Figs. 3, 5a, c, d), 150 μg of MOG35–55 was used. For Csf2rbAldh1l1-creERT2 studies, 200 μg of MOG35–55 was used (Fig. 5a–d, Extended Data Fig. 7b, c). All mice received two subcutaneous injections of 100 μl each of the MOG–CFA mix. All mice then received a single intraperitoneal injection of pertussis toxin (List Biological Laboratories, #180) at a concentration of 2 ng/μl in 200 μl PBS. Mice received a second injection of pertussis toxin at the same concentration two days after the initial EAE induction. Mice were monitored and scored daily thereafter. EAE clinical scores were defined as follows: 0, no signs; 1, fully limp tail; 2, hindlimb weakness; 3, hindlimb paralysis; 4, forelimb paralysis; 5, moribund (as described previously)11–13,73. Mice were randomly assigned to treatment groups. All mice were scored blind to genotype. Treatment regimes for genetic perturbations were chosen on the basis of prior experience with lentiviral transduction13 and tamoxifen-induced genetic deletion11.
WGBS
Genomic DNA was isolated from flow cytometry-sorted cells using a DNeasy Blood & Tissue Kit (Qiagen, #69504). Genomic DNA (100 ng) was bisulfite converted and libraries were prepared according to the Pico Methyl-Seq Library Prep Kit (Zymo Research, #D5455). Libraries were analysed on the 2100 Bioanalyzer (Agilent Technologies) and sequenced either by Genewiz using a HiSeq 4000 or at the Harvard Biopolymers Facility on a NextSeq 500 Mid Output.
For data processing, sequences were first assessed using FASTQC (v.0.11.5). Reads were trimmed using Cutadapt (v.1.14) to remove adapters and low quality reads. Trimmed reads were aligned to GRCm38 genome using Bismark (v.0.20.1) and Bowtie2 (v.0.20.1) with default settings. Sequence reads were first transformed into fully bisulfite-converted forward (C>T) and reverse read (G>A conversion of the forward strand) versions, before they were aligned to similarly converted versions of the genome (also C>T and G>A converted). Duplicated reads, which aligned to the same position in the same orientation, were removed. Methylation counts were extracted from the Bismark alignment and data was formatted using awk for downstream analysis, consisting of total reads and methylation reads for each CpG site. Percentages of methylation were visualized using IGV (v.2.5.1), averaging replicates per condition.
Differential methylation sites were determined with DSS (v.2.30.1), using the Wald test against all CpG sites between groups. Regions with many statistically significant CpG sites were identified as differential methylated regions (DMRs). DMRs with P < 0.01 were considered significant, based on differential methylated loci (P < 0.001). Gene and regions for promoter, intron, exon, and intergenic were annotated using the annotatr package (v.1.8.0) and visualized as pie charts with genomation (v.1.4.2).
Primary astrocyte cultures
Procedures were performed largely as described previously13. Brains of mice aged P0–P3 were dissected into PBS on ice. Cortices were discarded and the brain parenchyma were pooled, centrifuged at 500g for 10 min at 4 °C and resuspended in 0.25% Trypsin-EDTA (Thermo Fisher Scientific, #25200–072) at 37 °C for 10 min. DNase I (Thermo Fisher Scientific, #90083) was then added at 1 mg/ml to the solution, and the brains were digested for 10 more minutes at 37 °C. Trypsin was neutralized by adding DMEM/F12 + GlutaMAX (Thermo Fisher Scientific, #10565018) supplemented with 10% FBS (Thermo Fisher Scientific, #10438026) and 1% penicillin/streptomycin (Thermo Fisher Scientific, #15140148), and cells were passed through a 70-μm cell strainer. Cells were centrifuged at 500g for 10 min at 4 C, resuspended in DMEM/F12 + GlutaMAX with 10% FBS/1% penicillin/streptomycin and cultured in T-75 flasks (Falcon, #353136) at 37 °C in a humidified incubator with 5% CO2 for 7–10 days until confluency was reached. Astrocytes were shaken for 30 min at 180 rpm, the supernatant was aspirated and the medium was changed, and then astrocytes were shaken for at least 2 h at 220 rpm and the supernatant was aspirated and the medium was changed again. The medium was replaced every 2–3 days.
RNA isolation from cultured mouse astrocytes
Primary astrocytes were lysed in buffer RLT (Qiagen) and RNA was isolated from cultured astrocytes using the Qiagen RNeasy Mini kit (Qiagen, #74106). cDNA was transcribed using the High-Capacity cDNA Reverse Transcription Kit (Life Technologies, #4368813). Gene expression was then measured by qPCR using Taqman Fast Universal PCR Master Mix (Life Technologies, #4367846). Taqman probes used in this study were: Gapdh (Mm99999915_g1), Nos2 (Mm00440502_m1), Csf2 (Mm01290062_m1), Ccl2 (Mm00441242_m1), Il6 (Mm00446190_m1), Gstm1 (Mm00833915_g1), Mafg (Mm00521961_g1), Nfe2l2 (Mm00477784_m1), and Il1b (Mm00434228_m1). qPCR data were analysed by the ddCt method by normalizing the expression of each gene for each replicate to Gapdh and then to the control group.
Primary astrocyte pharmacological studies
Compound treatment was performed for 18–20 h with compounds diluted in DMEM/F12 + GlutaMAX (Life Technologies, #10565042) that was supplemented with 10% FBS (Life Technologies, #10438026) and 1% penicillin/streptomycin (Life Technologies, #15140122). Compounds used in these studies were: 100 ng/ml IL-1β (R&D Systems, #401-ML-005, 100 μg/ml stock in PBS), 50 ng/ml TNF (R&D Systems, #410-MT-010, 100 μg/ml stock in PBS), 20 μM dimethyl fumarate (Santa Cruz Biotechnology, #sc-239774, 10 mM stock in PBS, diluted at 1:500), and 2–2,000 ng/ml GM-CSF (PeproTech, #315–03, 20 μg/ml stock in PBS). For dilution curves involving IL-1β, TNF, and GM-CSF, stocks of IL-1β/TNF were diluted to achieve the maximal dose, and then subsequently diluted as indicated. GM-CSF was used at 200 ng/ml in dilution curve studies. For siRNA transfections, 1 μl of a 15 μM siRNA pool was mixed with 1 μl Interferin (Polyplus-transfection, #409–10) in 50 μl Opti-MEM (Life Technologies, #31985062). The mix was incubated for 10 min at room temperature and added to a well of a 48-well plate containing 250 μl of astrocyte medium. After 48 h, cells were used for assays. siRNA pools used were siNfe2l2 (Dharmacon, L-040766–00-0005) and siScrmbl (Dharmacon, D-001810–10-20).
In silico promoter analysis
The Mus musculus Mat2a genomic sequence was obtained using Ensembl74. The DNA sequence ~2,000 bp upstream of the protein-coding transcript Mat2a-201 was analysed. p65/RelA DNA binding sites were defined using Mulan75.
Chromatin immunoprecipitation
Approximately >1 million astrocytes were treated for 1 h followed by cell preparation according to the ChIP-IT Express Enzymatic Shearing and ChIP protocol (Active Motif, #53009). In brief, cells were fixed in 1% formaldehyde for 10 min with gentle agitation, washed in 1× PBS, washed for 5 min in 1× glycine Stop-Fix solution in PBS, and scraped in 1× PBS supplemented with 500 μM PMSF. Cells were pelleted, nuclei isolated, and chromatin sheared using the Enzymatic Shearing Cocktail (Active Motif) for 10 min at 37 °C with vortexing every 2 min. Sheared chromatin was immunoprecipitated according to the Active Motif protocol overnight at 4 °C with rotation. The next day, the protein-bound magnetic beads were washed 1× with ChIP buffer 1, 1× with ChIP buffer 2, and 1× with 1× TE. Cross-links were reversed in 100 μl of 0.1% SDS and 300 mM NaCl in 1× TE at 63 °C for 4–5 h, as described13,76. DNA was purified using QIAquick PCR Purification Kit (Qiagen, #28104). qPCR was performed using Fast SYBR Green Master Mix (Thermo Fisher Scientific, #4385612). Anti-IgG immunoprecipitation and input were used as controls. We used rabbit anti-MAFG (Genetex, GTX114541, 1:100), and rabbit IgG polyclonal isotype control (Cell Signaling, 2975S, 1:100). PCR primers were designed with Primer377 to generate 50–150-bp amplicons. Primer sequences used were: MafG-Hmox1-ARE1-F: 5′- CCTCCTGC TTAGGAACACCA-3′, MafG-Hmox1-ARE1-R: 5′- GGCCTTGAGCCTCA TGTTT-3′, MafG-Hmox1-ARE2-F: 5′- AGAGATGGCCTGTGGTTGAC-3′, and MafG-Hmox1-ARER-R: 5′- GGCATTGGATCCCCTAGAAC-3′. Data were analysed by ddCt relative to IgG control.
Isolation of cells from adult mouse CNS
Astrocytes were isolated by flow cytometry as described12,13,27 and by modifying a previously described protocol78. In brief, mice were perfused with 1× PBS and the CNS was isolated into 10 ml of enzyme digestion solution consisting of 75 μl papain suspension (Worthington, #LS003126) diluted in enzyme stock solution (ESS) and equilibrated to 37 °C. ESS consisted of 10 ml 10× EBSS (Sigma-Aldrich, #E7510), 2.4 ml 30% d(+)-glucose (Sigma-Aldrich, #G8769), 5.2 ml 1 M NaHCO3 (VWR, #AAJ62495-AP), 200 μl 500 mM EDTA (Thermo Fisher Scientific, #15575020), and 168.2 ml ddH2O, filter-sterilized through a 0.22-μm filter. Samples were shaken at 80 rpm for 30–40 min at 37 °C. Enzymatic digestion was stopped with 1 ml of 10× hi ovomucoid inhibitor solution and 20 μl 0.4% DNase (Worthington, #LS002007) diluted in 10 ml inhibitor stock solution (ISS). 10× hi ovomucoid inhibitor stock solution contained 300 mg BSA (Sigma-Aldrich, #A8806) and 300 mg ovomucoid trypsin inhibitor (Worthington, #LS003086) diluted in 10 ml 1× PBS and filter sterilized using a 0.22-μm filter. ISS contained 50 ml 10× EBSS (Sigma-Aldrich, #E7510), 6 ml 30% d(+)-glucose (Sigma-Aldrich, #G8769), and 13 ml 1 M NaHCO3 (VWR, #AAJ62495-AP) diluted in 170.4 ml ddH2O and filter-sterilized through a 0.22-μm filter. Tissue was mechanically dissociated using a 5-ml serological pipette and filtered through a 70-μm cell strainer (Fisher Scientific, #22363548) into a fresh 50-ml conical tube. Tissue was centrifuged at 500g for 5 min and resuspended in 10 ml of 30% Percoll solution (9 ml Percoll (GE Healthcare Biosciences, #17–5445-01), 3 ml 10× PBS, 18 ml ddH2O). Percoll suspension was centrifuged at 500g for 25 min with no breaks. Supernatant was discarded and the cell pellet was washed once with 1× PBS, centrifuged at 500g for 5 min and prepared for downstream applications.
Flow cytometry
Cells were stained in the dark on ice for 15 min with flow cytometry antibodies. Cells were then washed once with 1× PBS and resuspended in 1× PBS for sorting as described previously11–13,73. Antibodies used in this study were: PE anti-mouse CD45R/B220 (BD Biosciences, #553089, 1:100), PE anti-mouse TER-119 (Biolegend, #116207, 1:100), PE anti-O4 (R&D Systems, #FAB1326P, 1:100), PE anti-CD105 (eBioscience, #12–1051-82, 1:100), PE anti-CD140a (eBioscience, #12–1401-81, 1:100), PE anti-Ly-6G (Biolegend, #127608, 1:100), PerCP anti-Ly-6C (Biolegend, #128028, 1:100), APC anti-CD45 (eBioscience, #17–0451-83, 1:100), APC-Cy7 anti-CD11c (BD Biosciences, #561241, 1:100), and FITC anti-CD11b (eBioscience, #11–0112-85, 1:100). All cells were gated on the following parameters: CD105negCD140anegO4negTer119negLy-6GnegCD45Rneg. Astrocytes were subsequently gated on: CD11bnegCD45negLy-6CnegCD11cneg. Microglia were subsequently gated on: CD11bhighCD45lowLy-6Clow. Pro-inflammatory monocytes were subsequently gated on: CD11bhighCD45highLy-6Chigh. Compensation was performed on single-stained samples of cells and an unstained control. Cells were sorted on a FACS Aria IIu (BD Biosciences). For sorting of TdTomato+ astrocytes, cells were sorted according to TdTomato fluorescence judged against a wild-type control animal using a yellow-green laser on a FACS Aria IIu.
Fluorescence-activated cell sorting analysis of T cells
T cells were analysed largely as described previously12,13. Single-cell suspensions from the CNS and from the spleen were stimulated using 500 ng/ml PMA (phorbol 12-myristate 13-acetate) (Sigma-Aldrich, #P1585), 500 ng/ml ionomycin (Sigma-Aldrich, #I9657), and GolgiSTOP (BD Biosciences, #554724, 1:1,000) diluted in T-cell culture medium (RPMI (Life Technologies, #11875119) containing 10% FBS, 1% penicillin/streptomycin, 50 μM 2-mercaptoethanol (Sigma-Aldrich, #M6250), and 1% non-essential amino acids (Life Technologies, #11140050)) for 4 h. Following stimulation, T cells were washed with 1× PBS, centrifuged, and incubated with antibodies against surface markers, using a live/dead cell marker (Thermo Fisher Scientific, #L34966) for 15 min on ice. Cells were then washed once with 1× PBS followed by use of a fixation and intracellular antibody labelling kit (eBioscience, #00–5523). Antibodies used were: eFluor 450 anti-CD3 (eBioscience, #48–0032-82, 1:100), FITC anti-CD4 (BioLegend, #100510, 1:50), 405 Aqua LIVE/DEAD cell stain kit (Thermo Fisher Scientific, #L34966, 1:400), APC-Cy7 anti-IFNγ (BD Biosciences, #561479, 1:100), PE anti-IL-17a (eBioscience, #12–7177-81, 1:100), APC anti-IL-10 (BioLegend, #505010, 1:100) and PerCP-Cy5.5 anti-FoxP3 (eBioscience, #45–5773-82, 1:100). Compensation was performed on single-stained samples and an unstained control. Gating of the CNS was performed on 5,000 live CD3+CD4+ cells and gating for the spleen was performed on 10,000 live CD3+CD4+ cells. Fluorescence-activated cell sorting (FACS) was performed on an LSRII (BD Biosciences).
Preparation of MS samples
For control tissue, white and grey matter tissue samples were obtained during elective surgery on 58–62-year-old patients. Sampling was performed in the surgical corridor distant from the expected primary pathology (hippocampal sclerosis in two cases, glioblastoma in one). Use of the tissues was approved by the Montreal Neurological Institute and Hospital (MNI/H) Neurosciences Research Ethics Board under REB approval ANTJ 1988/3. Cells were isolated within 2 h of the surgery using mechanical and trypsin enzymatic digestion as previously described79. An isotonic Percoll gradient was subsequently used to remove myelin. cDNA libraries were created on the same day as surgery using the Chromium v.2.0 (10X Genomics) established protocol80 and were subsequently sequenced using Illumina HiSeq 4000 by paired-end 75-bp sequencing. For two control samples and samples from patients with MS, tissue was transported by submerging in Hibernate (Life Technologies, #A1247601) plus a B-27 supplement (Thermo Fisher Scientific, #17504044) for 8–18 h at 4 °C. Tissue was then isolated in a similar way to mouse brain samples. Dead cells were removed by magnetic bead purification (Miltenyi Biotech, #130–090-101) according to the manufacturer’s protocol before Drop-seq was performed. Each sample was independently processed in the scRNA-seq pipeline rather than pooled. Ethical approval was given before autopsy. Use of the tissues was approved by the Neuroimmunology Research Laboratory, Centre de Recherche du Centre Hospitalier de l’Université de Montréal (CRCHUM) under ethical approval number BH07.001.
Immunostaining of human brain tissue
Human brain tissue was obtained from patients with a clinical and neuropathological diagnosis of MS according to the revised 2010 McDonald’s criteria81. Tissue samples were collected from healthy donors and patients with MS under full ethical approval (BH07.001) and with informed consent as approved by the local ethics committee. Samples removed at autopsy were preserved and lesions classified using Luxol Fast Blue/haematoxylin & eosin staining and Oil Red O staining as previously described82,83. Specifically, active lesions were characterized by immune cell infiltration, lipid debris (Oil Red O staining) and the presence of macrophages and/or microglia throughout the lesion area; pre-active lesions were characterized by immune cell presence in the perivascular space and some microglial activation signified by Luxol Fast Blue staining; chronic active lesions were characterized by a hypocellular lesion centre that is demyelinated and has a hypercellular rim; and chronic inactive lesions are hypocellular and demyelinated with a near absence of macrophages and microglia. Four-micrometre-thick paraffin-embedded brain sections from six patients with MS and four healthy control individuals were deparaffinized, washed in PBS and treated with heat-induced antigen retrieval in citrate buffer pH 6. Endogenous avidin/biotin was blocked using an avidin-biotin blocking kit (Life Technologies, #004303) and non-specific binding was further blocked with 10% goat serum (Sigma, #D9663) in 0.1% PBS-T. Sections were incubated with rabbit anti-NRF2 (Abcam, #ab31163, 1:50) and GFAP-Cy3 (Sigma, #C9205, 1:500) in blocking buffer overnight at 4 °C. On the next day, slides were washed with 1% PBS-T and subsequently incubated with donkey anti-rabbit Fab fragments (Jackson Immunoresearch, #711–007-003, 1:32) and goat anti-donkey biotin (Thermo Fisher, #PA1–28737, 1:500) for 40 min at room temperature. Sections were washed and endogenous peroxidase was blocked using 0.3% H2O2 for 15 min followed by incubation with streptavidin-HRP (BD Biosciences, #BD554066, 1:1,000) for 40 min at room temperature and developed with Tyramide-AF647 (Life Technologies, #B40958). Slides were washed again and incubated with rabbit anti-MAFG (Abcam, #ab154318, 1:50) in blocking buffer overnight at 4 °C. On the next day, slides were washed with 1% PBS-T and incubated with goat anti-rabbit polymer-HRP (Life Technologies, #B40922) for 40 min at room temperature and developed with Tyramide-AF488 (Life Technologies, #B40922). After extensive washing, sections were counterstained with DAPI (Sigma, #D9542, 1:500) and mounted in Mowiol containing prolong gold (Life Technologies, #P36934). As controls, primary antibodies were omitted to control for non-specific binding. Images (z-stacks) were acquired using a Leica SP5 confocal microscope with Leica LAS AF software and processed using Fiji and LAS X. All settings were kept the same. MAFG and NRF2 were quantified per field of view in GFAP+ cells.
Multiplexed FISH
To quantify the expression of astrocyte and T-cell markers, we used a previously published pipeline84 to design 30-nt hybridization regions for FISH probes to Gfap (NM_010277), Aqp4 (NM_009700), Mafg (NM_010756), Mat2a (NM_145569) Csf2ra (NM_009970), Csf2rb (NM_007780), Csf2 (NM_009969), Cd3e (NM_007648), and Cd4 (NM_013488). These target regions were selected to have a narrow melting temperature range (65–75 °C), narrow GC range (40–60%), and few abundant off-targets. The set of target regions for each gene was concatenated to the 5′ end of a unique, previously published84 20-mer ‘readout sequence’ first introduced for multiplexed error-robust FISH (MERFISH) with the following readout sequences assigned to the following genes: Gfap (RS0015), Aqp4 (RS0406), Mafg (RS0095), Mat2a (RS0109), Csf2ra (RS0307), Csf2rb (RS0255), Csf2 (RS0247), Cd3e (RS0175) and Cd4 (RS0237). A total of 96 target regions were designed for each RNA, and each target region was allowed to overlap with other target regions by as much as 20 nt. To design template molecules capable of amplifying these desired probe sequences, a T7 promoter was concatenated to the 5′ end of the target region, and two random 20-mer primers were introduced at each of the 3′ and 5′ ends of these molecules. These template molecules were ordered as a complex oligopool from Genscript, and amplified into single-stranded DNA probes using a previously published protocol84. In brief, an in vitro transcription template set was made via limited cycle PCR from this complex oligopool, RNAs were transcribed from these templates, and then single-stranded DNA probes were made from these RNAs via reverse transcription. Alkaline hydrolysis was then used to remove the RNA template from the final probes. We modified the published protocol slightly. First, we used SPRI beads for all purification steps. Second, we used a reverse transcription primer in which the 5′ nucleobase was an RNA nucleotide rather than a DNA nucleotide. This modification allowed the reverse transcription primer to be efficiently cleaved from the final probes during alkaline hydrolysis. Tissue slices of the spinal cord of three healthy mice and three mice with EAE were prepared as described above. These tissue slices were deposited on silanized coverslips containing orange fiducial beads (used to reregister different images of the same sample region), fixed, permeabilized, and hybridized with the above FISH probes at a total concentration of ~4 μM using published protocols85. To reduce background, the FISH probes were supplemented with a previously published anchor probe85—a 50% LNA dT probe that targets the polyA tail of mRNAs and contains a terminal acrydite moiety—and these samples were embedded in a thin 4% 19:1 polyacrylamide/bis-acrylamide film and then aggressively cleared and washed as described previously85. To image nuclei and the total polyA mRNA content of the samples, each sample was stained with 4 μg/ml DAPI and with a readout probe, complementary to a readout sequence on the anchor probes, conjugated via a disulfide bond to Alexa 488, as described previously85. Individual RNA molecules were imaged using sequential rounds of staining with pairs of readout probes that target the individual readout sequences associated with different probe sets using the buffers and protocols described for MERFISH previously85. Readout probes associated with RNAs were conjugated, via disulfide bonds, to Cy5 or Alexa750. The samples were imaged on a home-built epi-fluorescence microscope using 500 mW of 750-nm illumination (Alexa750), 200 mW of 635-nm illumination (Cy5), 50 mW of 545-nm illumination (orange fiducial beads), 50 mW of 473-nm illumination (Alexa488), and 10 mW of 408-nm illumination (DAPI) provided by a Celesta light engine (Lumencor). Samples were imaged with a 60× Nikon Oil PlanApo objective and a scientific CMOS camera (Hamatasu Orca Flash). As described previously85, pairs of RNAs were measured in individual rounds of imaging by staining with a pair of readout probes, imaging the sample, removing fluorescence by reductively cleaving the disulfide bonds linking fluorophores to readout probes, and then repeating the process with an additional pair of readout probes.
Bulk RNA-seq
Sorted cells were lysed and RNA isolated using the Qiagen RNeasy Micro kit (Qiagen, #74004) with on-column DNase I digestion (Qiagen, #79254). RNA was concentrated with a Savant Speedvac Concentrator (Thermo Fisher Scientific, #DNA120) when needed. RNA was suspended in 10 μl of nuclease-free water at 0.5–1 ng/μl and sequenced using 3′ Digital Gene Expression86 or SMARTSeq287 at the Broad Institute. For 3′ Digital Gene expression, reads were first demultiplexed using Picard (v.2.17.11) IlluminaBaseCallsToSam and then aligned against the GRCm38 mouse genome using STAR (v2.4.2a) with default parameters. Duplicate reads were marked with Picard UmiAwareMarkDuplicates-WithMateCigar to identify duplicate reads based on UMI sequence and alignment start site. Genes were then quantified using RSEM (v1.2.21) with paired-end option and assessed for quality using RNA-SeQC. Processed RNA-seq data were filtered, removing genes with low read counts and using the top-expressed isoform as a proxy for gene expression. Read counts were normalized using TMM normalization and CPM (counts per million) were calculated to create a matrix of normalized expression values. Differential binding analysis was performed using R and DESeq2 (1.20.0). A P value of <0.05 was used to determine differentially expressed genes. Heatmaps were generated with using the GENE-E program of the Broad Institute.
For SMART-seq RNA-seq, reads were first assessed using FASTQC (v.0.11.2). Paired-end reads were then aligned against the GRCM38 mouse genome using STAR (v2.5.3) with parameters –twopassMode–alignIntronMax 1000000–sjdbOverhang 37–alignMatesGapMax 1000000. Duplicates were marked using Picard (v.2.7.1) and reads were then filtered with mapping quality less than 30, unmapped reads, duplicates, and multi-mapped. FeatureCounts from the SubRead (v.1.5.0) package was used to quantify reads mapping to the genes and generated a count matrix for downstream analysis. SMART-Seq differential gene analysis, heatmaps, and pathway analysis were done in a similar manner as 3′ digital gene expression (DGE).
Ribotag RNA-sequencing
Sequencing reads were first quality-tested using FASTQC. In brief, the left sequence read consisted of the barcode and the molecule of origin (UMI barcode). The right reads were trimmed using fastx (v.0.0.13) fastx_trimmer. Ribotag data were aligned against the mm10 genome using STAR (v.2.5.3a) with default parameters and –quantMode geneCounts to identify and quantify the number of reads mapped per gene. DESeq2 and R software were used for differential gene analysis and clustering. For clustering, the z-scores were calculated for significant genes with P < 0.05 and twofold change, using the mean expression of biological replicates per disease stage or region and then subsequently clustered using K-means. A scree plot was used to assess the number of clusters.
ChIP–seq
Approximately 400,000 cells were fixed, nuclei collected, and chromatin isolated using the ChIP-IT Express Enzymatic Shearing Kit (Active Motif, #53009). Chromatin complexes were immunoprecipitated using a rabbit anti-MAFG antibody (Genetex, #GTX114541, 1:100). Cross-links were reversed in buffer (1% SDS, 0.1 M NaHCO3 in H2O) by heating at 65 °C for 8–12 h. DNA was purified using a QIAquick PCR Purification Kit (Qiagen, #28104). DNA was analysed using a 2100 Bioanalyzer (Agilent) and High Sensitivity DNA Kit (Agilent Technologies, #5067–4626). DNA libraries were then prepared for sequencing using the NEBNext Ultra II DNA Library Prep Kit (New England Biolabs, #E7645S) and sequencing adaptors were ligated using NEBNext Multiplex Oligos for Illumina (#E7500S, #E7335S) as described. ChIPed DNA was amplified by using 14 cycles according to the NEBNext protocol. DNA libraries were not size selected but primer dimers were removed via purification using Agencourt AMPure XP Beads (Beckman Coulter, #A63881). DNA libraries consisting of ChIPed DNA and input DNA for each sample were sequenced on an Illumina HiSeq 4000 using 2 × 75 paired-end sequencing.
Paired-end ChIP–seq reads were accessed using FASTQC. Reads were trimmed using cutadapt (v.1.14) to remove adapters and low-quality reads below 30. Trimmed reads were then aligned using BWA mem (v.0.7.8)88 against the GRCm39/mm10 mouse genome assembly with default settings. Duplicated reads were marked using Picard (v.2.5.0). Alignments were filtered with SAMtools (v.1.3)89 to exclude reads with mapping quality <30, not properly paired, duplicated, aligned to mitochondrial genome, and aligned to ENCODE blacklist regions90. For peak calling, MACS2 callpeaks (v.2.1.1)91 were called on merged replicates for ChIP (treatment) and input (control) pairs, using P < 0.01 as threshold. To generate signal tracks, fold enrichment for merged replicates per condition was calculated using MACS2 bdcmp. Regions of interest were then binned into 10-bp windows with Bedtools makeWindows for smoothing and Bedtools map to calculate the coverage for each bin in every replicate. The coverage for both groups (naive and EAE) were pooled by summing the total reads, and density plots were created using IGV (v.2.5.0) to visualize the genomic regions. For differential peaks, merged peaks were mapped to specific gene regions using bedtools intersect and reads were counted using subread featureCounts (v.1.6.2) to produce a count matrix. DESeq2 was used to find differential peaks.
Motif binding was analysed using the SeqPos motif tool (v.1.0.0) in Cistrome (http://cistrome.org/). The top 5,000 peaks by P value significance from MACS2 results were used as input against public databases (Transfac, JASPER, PBM, Y1H, hPDI) with P < 0.05 cut-off and 600 width scanning region. For motif enrichment between EAE and naive, sequences ±300 around the summit for the top 5,000 peaks were extracted from the GRCm38 genome with bedtools (v.2.27.1) getfasta. Meme Suite’s (v.5.0.5) CentriMo was used for motif enrichment. Statistical analysis in SeqPos was performed using Fisher’s exact test.
ATAC–seq
Sequencing libraries were prepared largely as described previously13,92,93. After isolation of nuclei, transposition was performed using the kit (Illumina, #FC-121–1030) according to the Buenrostro protocol. DNA was then amplified using NEBNext High Fidelity 2× PCR Master Mix (New England Biolabs, #M0541S) for five cycles. DNA quantity was then measured using a Viia 7 Real-Time PCR System (Thermo Fisher Scientific) and the number of cycles required to achieve 1/3 of maximal SYBR green fluorescence was determined and libraries amplified accordingly. TruSeq adaptors (universal: Ad1_noMX and barcoded: Ad2.1-Ad2.24) were used according to the Buenrostro protocol. Libraries were purified using MiniElute PCR Purification Kit (Qiagen, #28006) followed by double-sided Agencourt AMPure XP bead purification (Beckman Coulter, #A63881) to remove primer dimers and large DNA fragments. Libraries were analysed on a 2100 Bioanalyzer (Agilent Technologies) and High Sensitivity DNA Kit (Agilent Technologies, #5067–4626). Libraries were sequenced by Genewiz on an Illumina HiSeq 4000 by 2 × 75-bp paired-end sequencing.
Paired-end ATAC–seq reads were first accessed using FASTQC. Reads were trimmed using cutadapt to remove adapters and low quality reads below 30. Paired-end reads were then aligned against the GRCm39/mm10 mouse genome assembly with Bowtie (v.2.3.0)94 in local mode, sensitive settings, and a maximum fragment size of 2000. Duplicated reads were marked using Picard (v.2.5.0). Alignments were filtered with SAMtools (v.1.3) to exclude reads with mapping quality <30, not properly paired, duplicated, aligned to mitochondrial genome, and/or aligned to ENCODE blacklist regions. Alignments with an insertion size of >100 bp were removed to enrich for nucleosome-free reads. ATAC–seq peaks were called for each replicate using MACS2, using –format BAMPE and –keep-dup all. IDR (v.2.0.2) was used to determine consistency of peak detection between individual replicates and peaks with a threshold below 0.10 were merged between replicates for downstream analysis. For differential peaks, merged peaks were mapped to specific genic regions using bedtools intersect and reads were counted using subread featureCounts (v.1.6.2) to produce a count matrix. DESeq2 was then used to find differential peaks. For signal tracks, ATAC–seq was processed in similar manner to ChIP–seq.
Pathway and statistical analysis
GSEA or GSEAPreranked analyses were used to generate enrichment plots for bulk RNA-seq or scRNA-seq data95,96 using MSigDB molecular signatures for canonical pathways: KEGG/Reactome/Biocarta (c2. cp.all), Motif (c3.all), gene ontology (c5.cp.all), and Hallmark (h.all). Alternatively, ENRICHR97,98 was used when a list of differentially expressed genes by group was calculated to identify overrepresented transcriptional motifs or pathways. Protein class identity was classified using PantherDB99. To identify regulators of gene expression networks, IPA software (Qiagen) was used by inputting gene expression datasets with corresponding log(FoldChange) expression levels compared to other groups. ‘Canonical pathways’ and ‘upstream analysis’ metrics were considered significant at P < 0.05. To identify upstream regulatory networks, once a specific ‘upstream regulator’ was identified, the ‘Build>Grow’ function was used to identify molecules upstream of the selected network. NRF2 target genes in Extended Data Fig. 1j were identified using IPA network analysis (Fig. 1h), BIOCARTA_ARENRF2_PATHWAY, ID: M14339, NRF2_Q4, ID: M14141, and NRF2_01, ID: M14948. For IPA analyses of regulatory networks, yellow lines indicate interactions inconsistent with the predicted activation state. The number of inconsistent predictions are: Fig. 1h, 189 out of 603; Fig. 4a, 21 out of 63; Fig. 6e, 12 out of 58; Extended Data Fig. 10g, 6 out of 38. In all cases, statistical analysis using Qiagen IPA was carried out with a right-tailed Fisher’s exact test. In all cases, statistical analysis using GSEA or GSEAPreranked was determined by one-tailed t-test in GSEA. In all cases, ENRICHR FDR values were calculated using the Benjamini–Hochberg test in ENRICHR and P values were calculated using Fisher’s exact test in ENRICHR. All samples were randomly allocated into treatment groups. No statistical methods were used to predetermine sample size. Sample sizes were chosen in accordance with previous studies in the field11–13,27. In all cases, data are shown as mean ± s.e.m. unless otherwise indicated.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Sequencing data have been deposited into the Gene Expression Omnibus (GEO) under the SuperSeries accession number GSE130119. Clinical data for patient samples can be found in Supplementary Table 7. All other data and code that support the findings of this study are available from the corresponding author on reasonable request. Data from Schirmer et al.20 were accessed at PRJNA544731 and https://cells.ucsc.edu/?ds=ms. Data from Jäkel et al.17 were accessed at GSE118257. Data from Lake et al.40 were accessed at GSE97942.
Extended Data
Extended Data Fig. 1 |. Control analyses for scRNA-seq of B6 EAE mice.
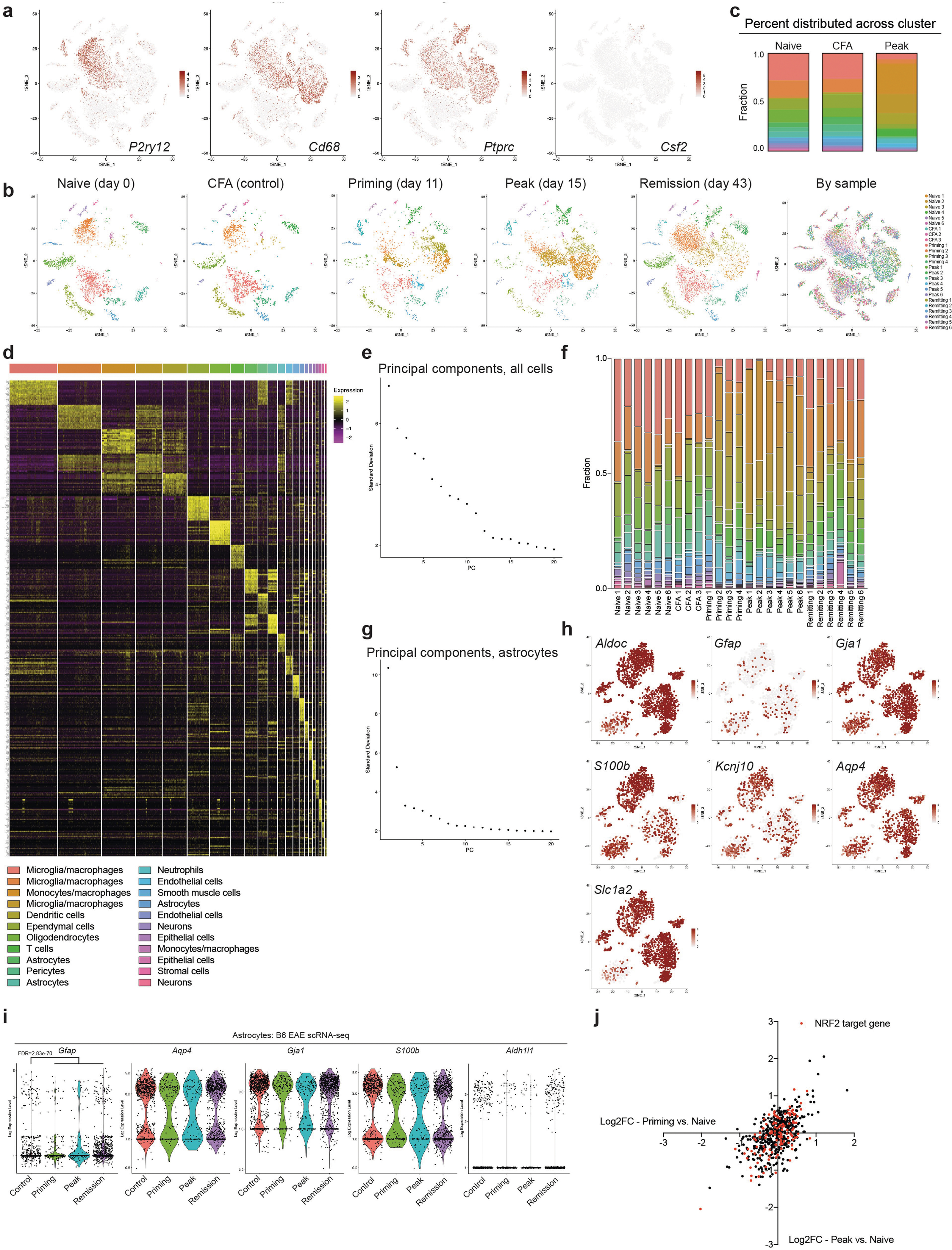
a, Cell type marker expression in mice with EAE. n = 24,275 cells. b, Unsupervised clustering tSNE plots of CNS cells from mice with EAE. n = 6 per group, n = 4 priming, n = 3 CFA. n = 24,275 cells. c, Analysis of cluster occupation by cells across EAE time points. d, Significantly enriched genes by cell type cluster. e, PCs used in study. n = 24,275 cells. f, Cluster distribution by replicates. g, Principal componentsC used in astrocyte subclustering. n = 2,079 cells. h, Gene scatterplots of astrocyte markers in the astrocyte tSNE analysis. n = 2,079 cells. i, Astrocyte marker gene expression by time point during EAE. n = 2,079 cells. j, Correlation of NRF2 target gene expression during priming and peak EAE phases compared to in naive mice. NRF2 target genes are marked in red. In total, 88 out of 123 genes were decreased at at least one time point, whereas 40 out of 123 were decreased at both time points. n = 69 cells from cluster 4.
Extended Data Fig. 2 |. Sorting of TdTomatoGfap astrocytes.
The forward versus side scatter (FSC versus SSC) gating strategy, followed by exclusion of FSC and SSC doublets, and TdTomato fluorescence in the phycoerythrin (PE) channel.
Extended Data Fig. 3 |. Expression of Mafg and Nfe2l2 in astrocytes.
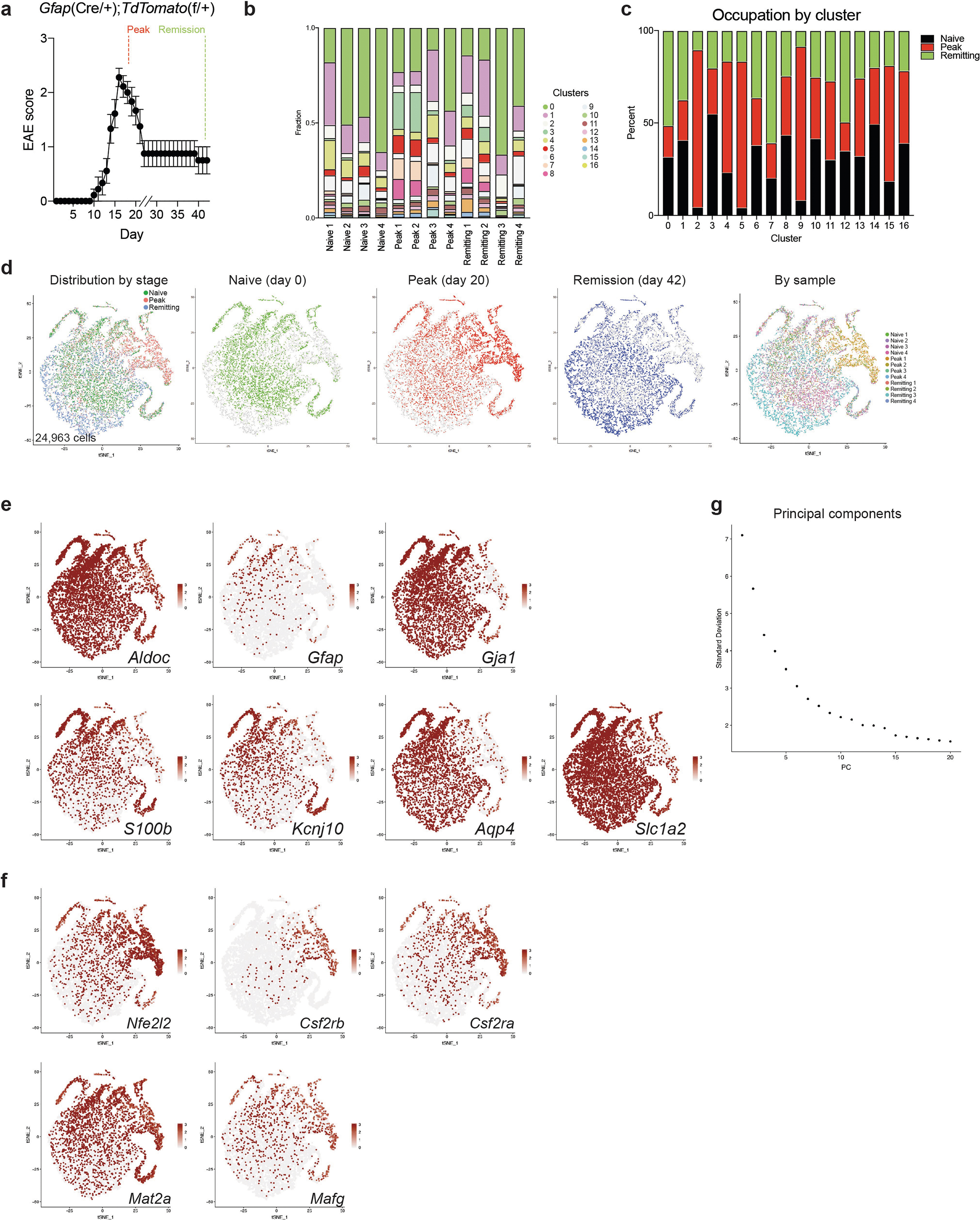
a, EAE in TdTomatoGfap mice used for scRNA-seq. n = 4 mice per time point. b, Cluster composition by replicate. c, Cluster composition by EAE time point. d, Unsupervised clustering tSNE plot of TdTomatoGfap astrocytes from mice with EAE. n = 4 per group (peak, day 20 post-induction; remission, day 42 post-induction). e, Scatterplots of astrocyte markers. f, Scatterplots of genes of interest in this study. g, Principal components used in this analysis. n = 24,963 cells for all scRNA-seq experiments.
Extended Data Fig. 4 |. Nfe2l2 knockdown in astrocytes.
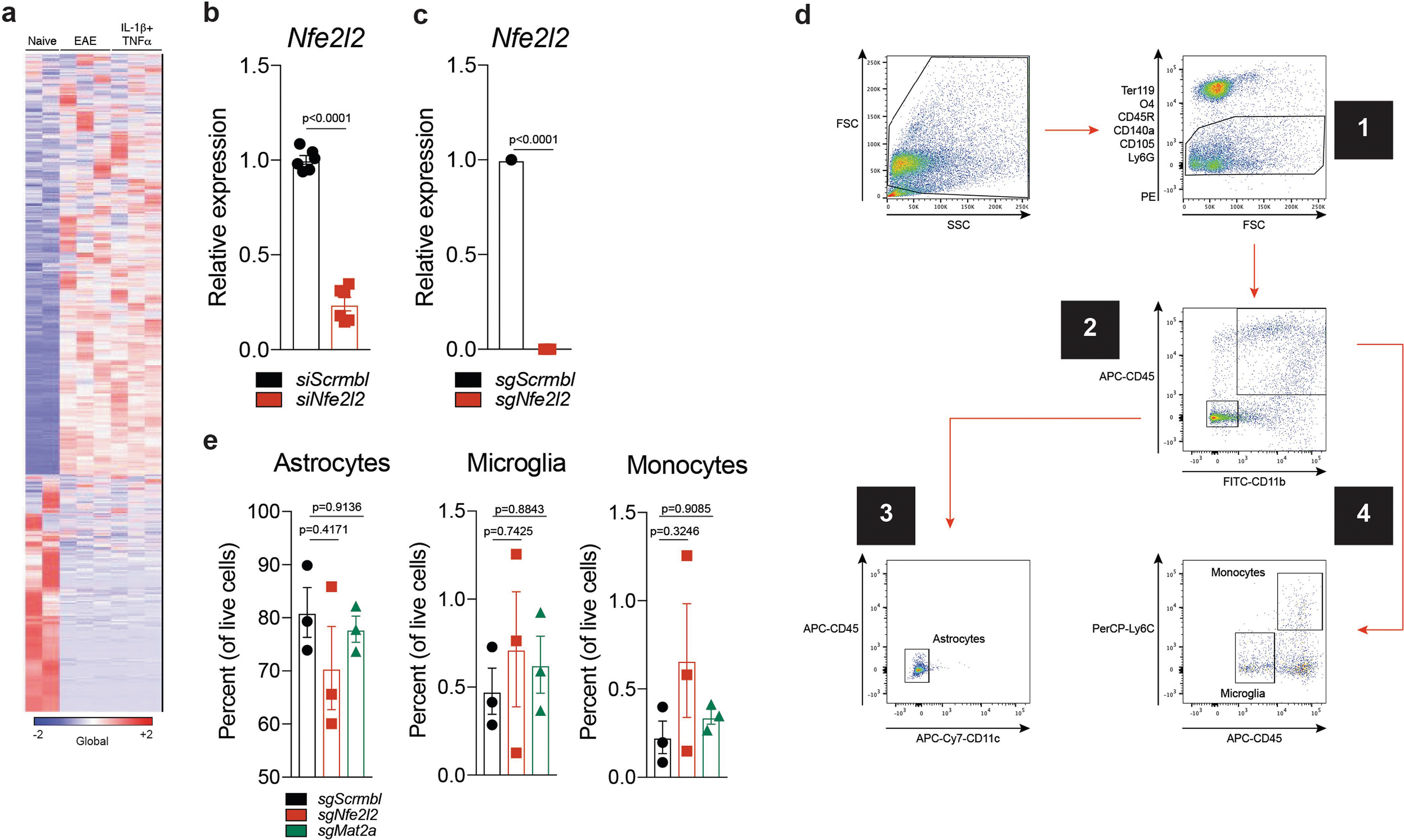
a, RNA-seq analysis of astrocytes following intracerebroventricular injection of IL-1β/TNF, EAE induction, or no treatment. n = 3 per group, n = 2 naive. b, siRNA-based knockdown of Nfe2l2 in primary astrocytes. n = 6 biologically independent samples per condition. Experiment repeated twice. Unpaired two-tailed t-test. c, Nfe2l2 expression determined by qPCR in flow cytometry-sorted astrocytes. n = 3 mice per group. One-sample t-test. d, Flow cytometry sorting strategy for astrocytes, microglia and pro-inflammatory monocytes. e, Quantification of astrocytes, microglia and pro-inflammatory monocytes. n = 3 mice per condition. One-way ANOVA, Tukey post-test. Data shown as mean ± s.e.m. ***P < 0.001, NS (not significant) P > 0.05.
Extended Data Fig. 5 |. Analysis of Nfe2l2–Mafg signature by Ribotag and scRNA-seq.
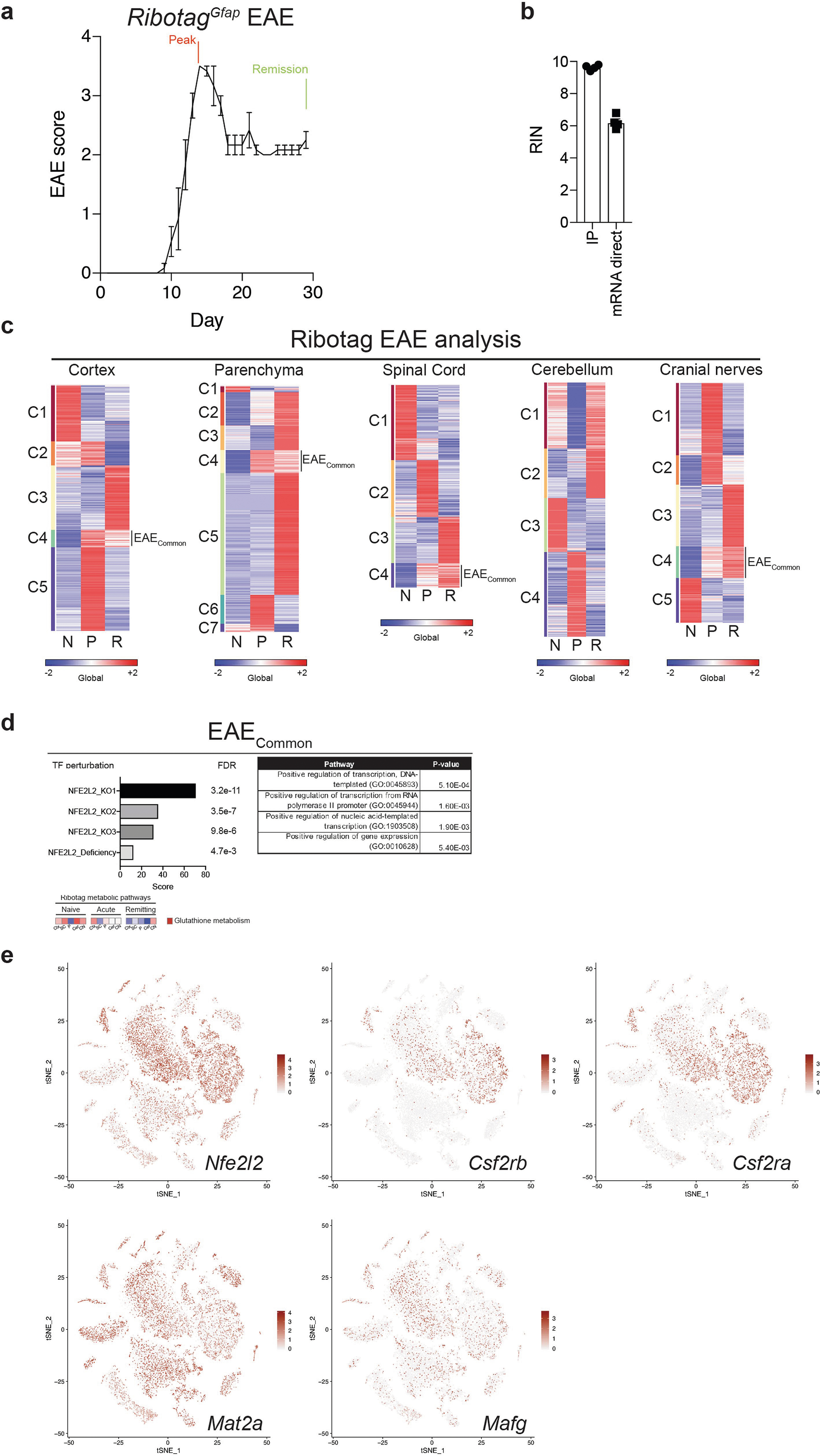
a, EAE in mice used in RibotagGfap studies. n = 3 mice per time point. b, RINs for Ribotag preparation. IP, immunoprecipitated HA-tagged ribosomes. mRNA direct, enrichment of polyadenylated mRNA using mRNA direct kit (Thermo Fisher, #61011). n = 4 biologically independent samples per condition. c, K-means clustering of RibotagGfap RNA-seq data for five CNS regions. d, ENRICHR analysis of upregulated genes in EAE (top). Analysis of gene expression associated with the altered glutathione metabolism KEGG pathway by CNS region (bottom). Number of independent mouse samples studied: n = 7 cortex, n = 8 spinal cord, n = 7 parenchyma, n = 9 cerebellum, n = 8 cranial nerves. e, Gene expression scatterplots of genes of interest in B6 EAE scRNA-seq studies. n = 24,275 cells. Data shown as mean ± s.e.m.
Extended Data Fig. 6 |. Mafg knockdown in astrocytes.
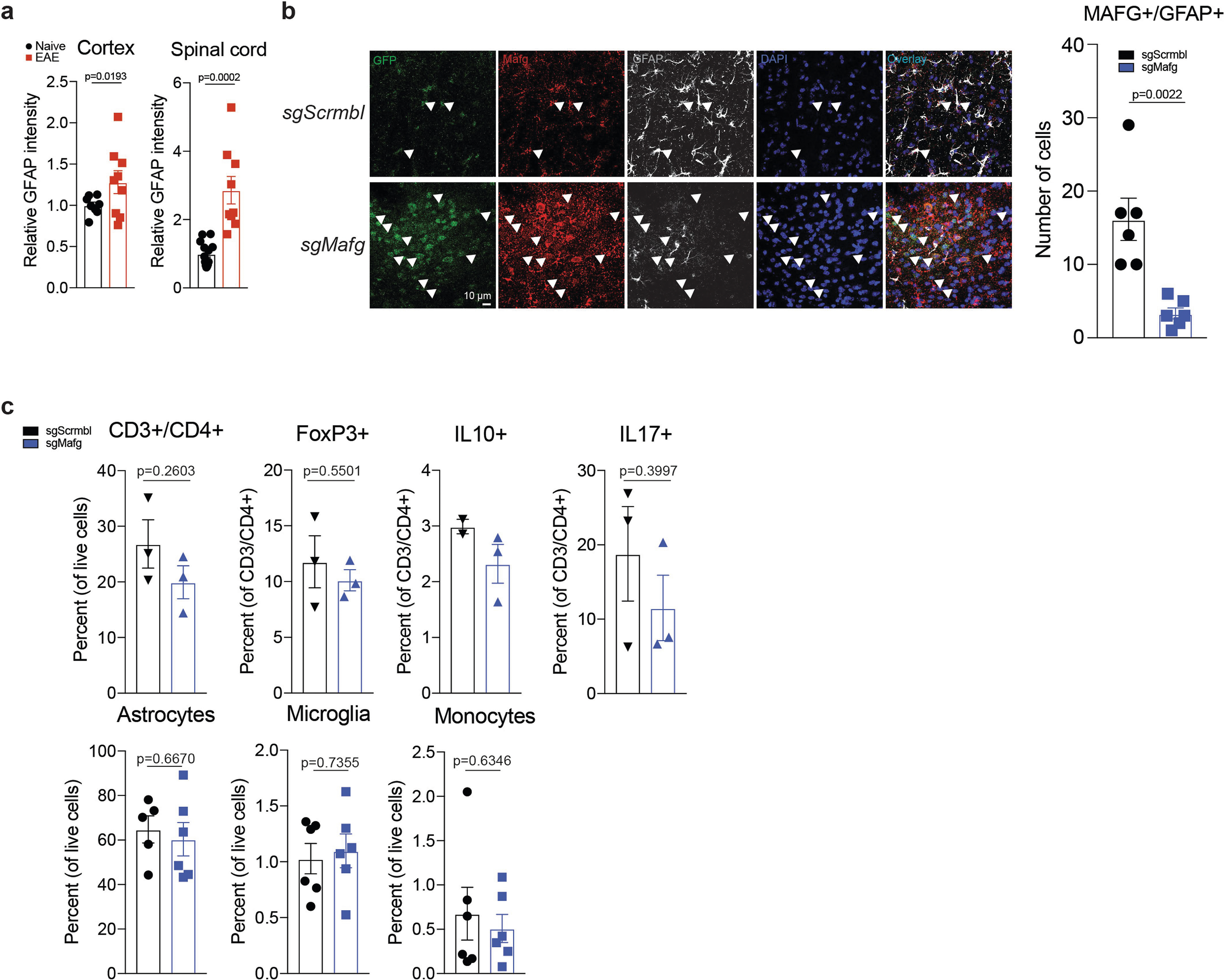
a, Quantification of GFAP immunoreactivity in CNS samples from naive or EAE mice. Cortex: n = 8 naive, n = 9 EAE; spinal cord: n = 13 naive, n = 9 EAE. Kolmogorov–Smirnov test. b, Immunostaining (left) and quantification (right) of MAFG+ GFAP+ astrocytes in mice targeted with sgMafg-delivering or sgScrmbl-delivering lentiviruses. n = 6 images per group from n = 3 mice. Two-tailed Mann–Whitney test. c, T cell subsets, astrocytes, microglia and pro-inflammatory monocytes in sgMafg-targeted versus sgScrmbl-targeted mice. n = 3 per condition for T cells; n = 2 for sgScrmbl IL-10+ group. Unpaired two-tailed t-test. n = 6 per condition for astrocytes, microglia, and monocytes. Experiment repeated twice. Unpaired two-tailed t-test. Data shown as mean ± s.e.m.
Extended Data Fig. 7 |. Mat2a and Csf2rb knockdown in astrocytes.
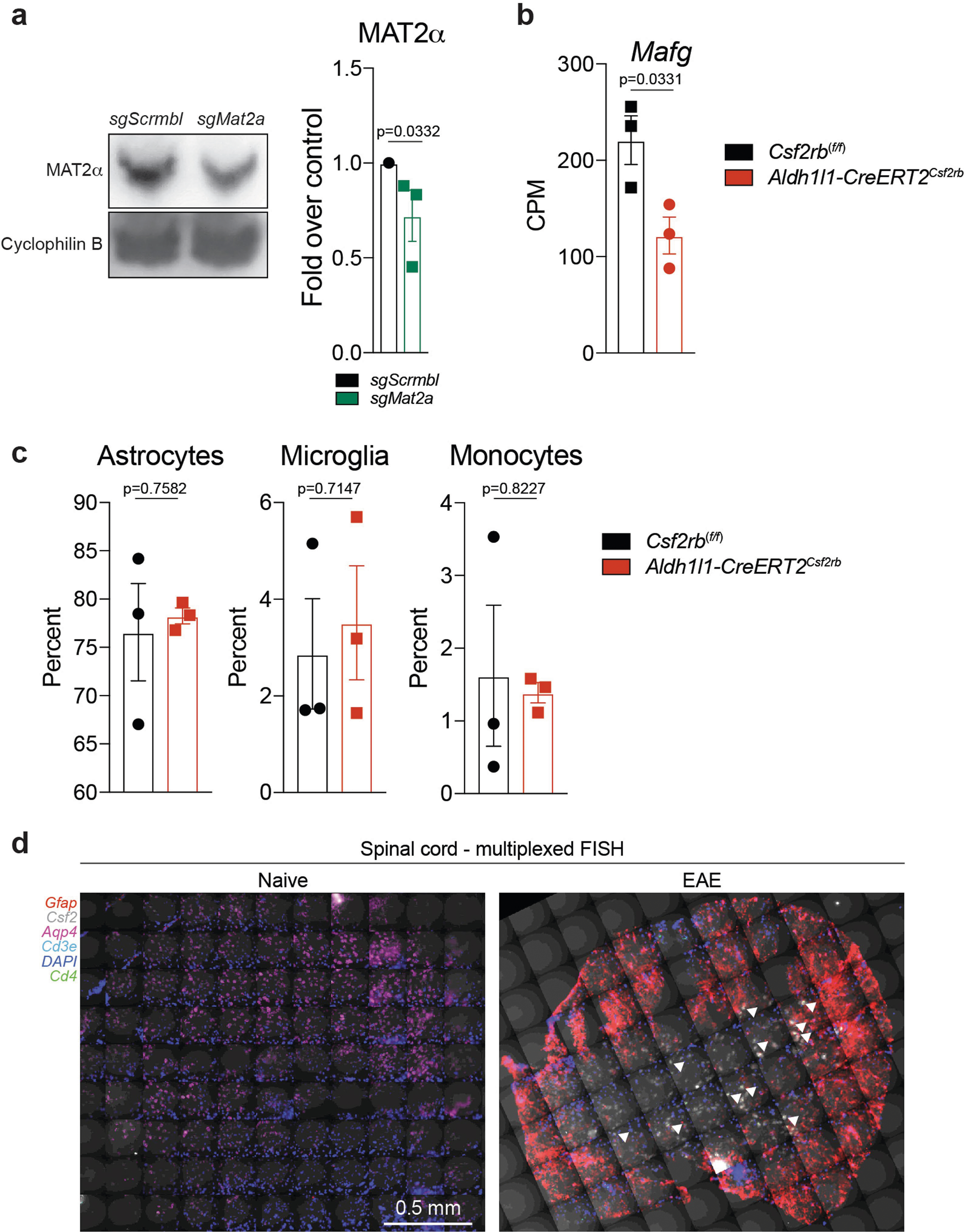
a, Validation of Mat2a knockdown by western blot. n = 3 biologically independent samples per group. Single sample t-test. b, Quantification of Mafg expression by scRNA-seq in Csf2rb conditional knockout mice. n = 3 per condition. Unpaired two-tailed t-test. c, Quantification of cell populations in Csf2rb conditional knockout mice. n = 3 per condition. Unpaired two-tailed t-test. d, Large area scan of down-sampled stitched multiplexed FISH images. Arrowheads indicate T cells. Representative images from three independent experiments with n = 3 mice per group. Data shown as mean ± s.e.m.
Extended Data Fig. 8 |. Control analyses of scRNA-seq on human samples.
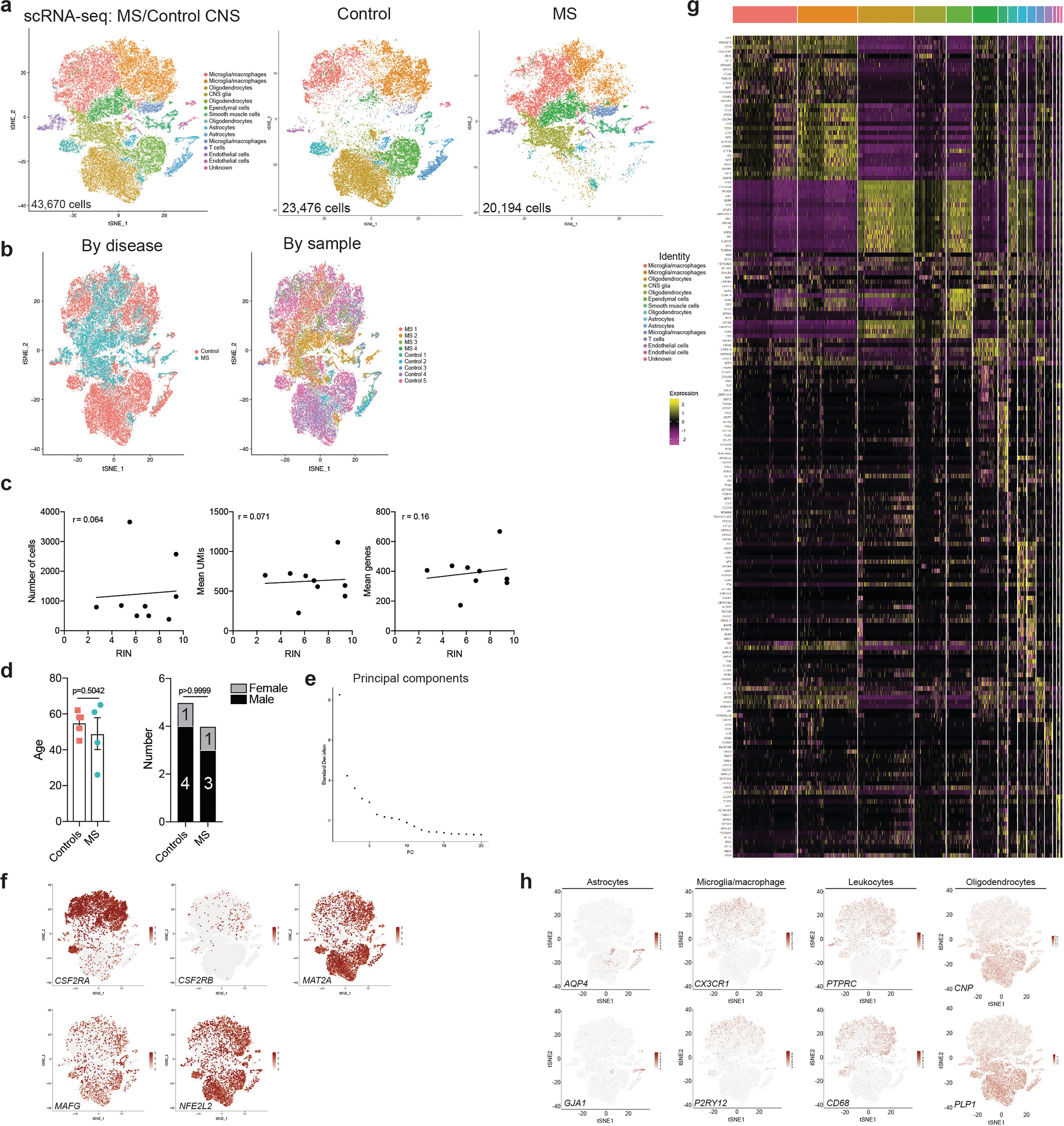
a, tSNE plots of MS and control cells. b, Cluster occupation by disease state and patient. c, RINs and scRNA-seq data quality. n = 9 per analysis. Pearson’s correlation. d, Age and sex corresponding to samples analysed. n = 5 control, n = 4 MS. Unpaired two-tailed t-test (age), Fisher’s exact test (sex). e, Principal components used in this analysis. f, Expression scatterplots of genes of interest. n = 43,670 cells. g, Cell type classification based on significantly enriched genes by cluster. h, Cell type marker scatterplots. n = 43,670 cells. Data shown as mean ± s.e.m.
Extended Data Fig. 9 |. Validation of GM-CSF–MAFG–NRF2 transcriptional signature in multiple human scRNA-seq datasets.
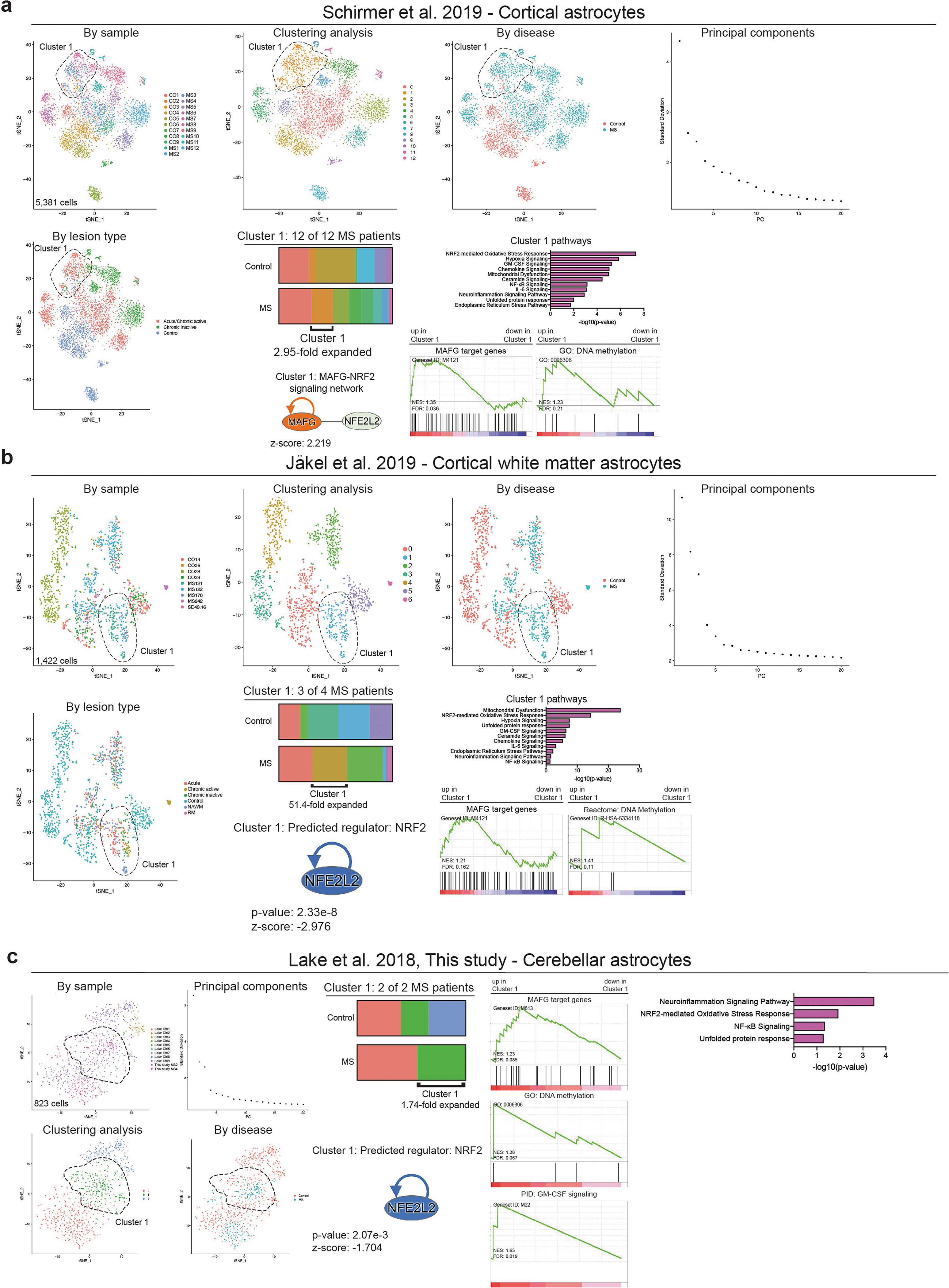
a, Analysis of regionally matched cortical astrocytes derived from patients with MS and control individuals analysed by Schirmer et al.20. n = 9 controls, n = 12 patients. b, Analysis of regionally matched white matter cortical astrocytes derived from patients with MS and control individuals analysed by Jäkel et al.17. n = 5 controls, n = 4 patients. c, Analysis of regionally matched cerebellar astrocytes derived from patients with MS analysed in this study and control individuals analysed by Lake et al.40. n = 9 controls, n = 2 patients.
Extended Data Fig. 10 |. Control analyses of human astrocytes.
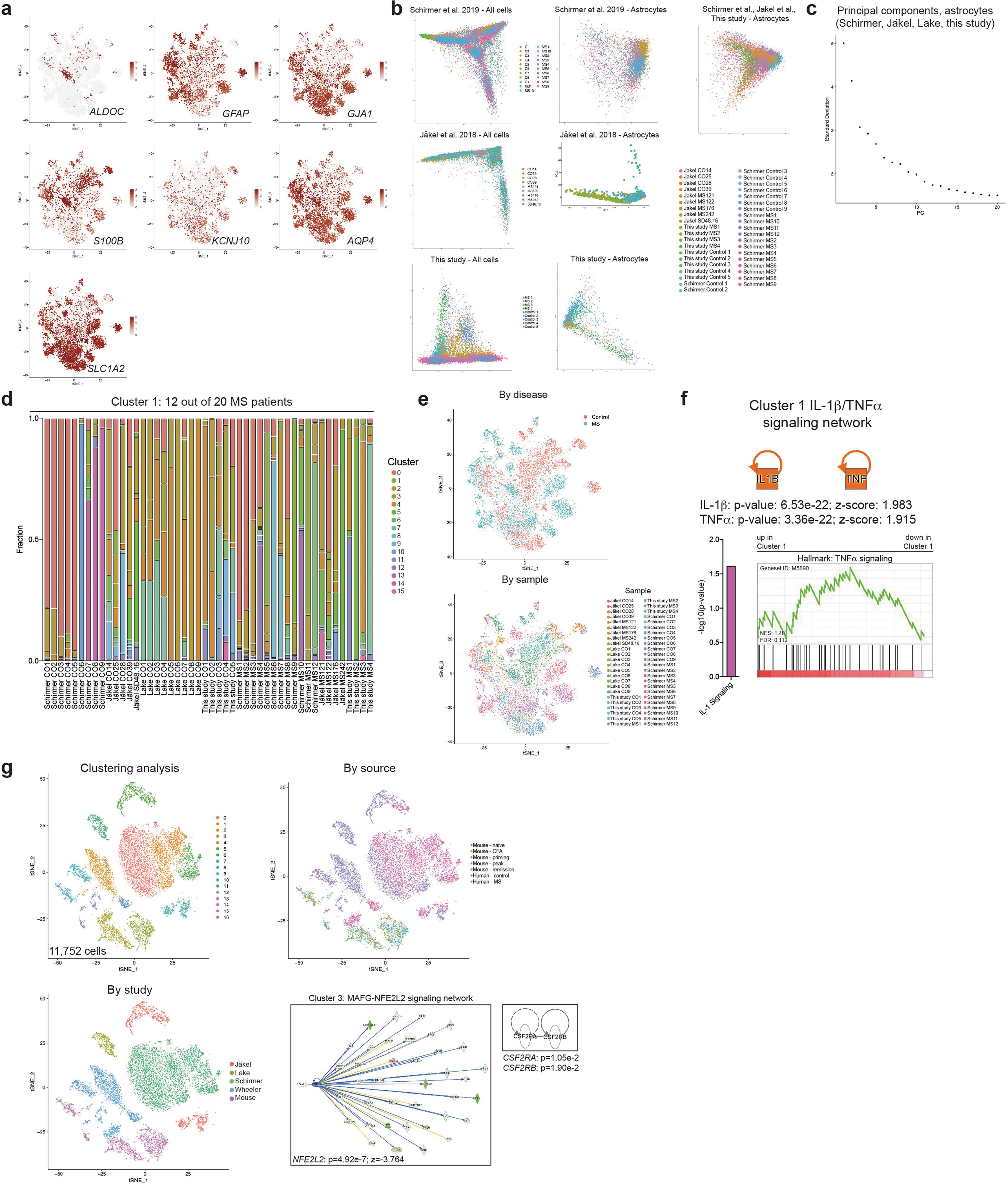
a, Gene scatterplots for astrocyte-specific markers in dataset representing control individuals and patients with MS compiled using data from Schirmer et al.20, Jäkel et al.17, Lake et al.40, and this study. n = 28 controls, n = 20 patients. n = 9,673 cells. b, Principal component analysis of all cells and astrocytes in each study. Number of cells analysed: Schirmer et al.20: 48,919 (all), 5,831 (astrocytes); Jäkel et al.17: 17,799 (all), 1,422 (astrocytes); this study: 43,670 cells (all), 2,332 (astrocytes). c, Principal components used in this study. d, Fraction occupation by cluster by patient. e, tSNE plot by condition and study (n = 9,673 cells). f, Analysis of IL-1β/TNF signalling in cluster 1 astrocytes from n = 28 controls and n = 20 patients with MS from the four studies. g, Canonical correlation analysis of mouse (Fig. 1, B6 EAE) and human (Fig. 6) astrocyte clusters and IPA analysis.
Supplementary Material
Acknowledgements
This work was supported by grants NS102807, NS087867, ES02530, AI126880, and AI093903 from the NIH; RSG-14–198-01-LIB from the American Cancer Society; and RG4111A1 and JF2161-A-5 from the National Multiple Sclerosis Society (to F.J.Q.) and the Canada Foundation for Innovation (CGEn and CFI-LOF 32557 to J.R.). F.J.Q., A.P. and J.P.A. received support from the International Progressive MS Alliance (PA-1604–08459). A.P. holds the T1 Canada Research Chair in Multiple Sclerosis and is funded by the Canada Institute of Health Research, the National Multiple Sclerosis Society, and Foundation of Canada. M.A.W. was supported by a training grant from the NIH and Dana-Farber Cancer Institute (T32CA207201), a postdoctoral fellowship from the NIH (F32NS101790), and a traveling neuroscience fellowship from the Program in Interdisciplinary Neuroscience at Brigham and Women’s Hospital. M.A.W. and I.C.C. received funding from the Women’s Brain Initiative at Brigham and Women’s Hospital. Sanger sequencing was carried out at the DNA Resource Core of Dana-Farber/Harvard Cancer Center (funded in part by NCI Cancer Center support grant 2P30CA006516– 48). We thank S. Jung and L. Chappell-Maor (Weizmann Institute of Science) for help in sequencing the Ribotag dataset; S. Young, T. Mason, and S. Garamszegi for assistance with coordinating deep sequencing; all members of the Quintana laboratory for helpful advice and discussions; P. Hewson for technical assistance; D. Kozoriz, A. Chicoine, and R. Krishnan for technical assistance with flow cytometry studies; Y. C. Wang for human control scRNA-seq library preparation; and the patients and their families that agreed to participate in this study.
Footnotes
Competing interests The authors declare no competing interests.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41586–020-1999–0.
Reprints and permissions information is available at http://www.nature.com/reprints.
References
- 1.Reich DS, Lucchinetti CF & Calabresi PA Multiple sclerosis. N. Engl. J. Med 378, 169–180 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baecher-Allan C, Kaskow BJ & Weiner HL Multiple sclerosis: mechanisms and immunotherapy. Neuron 97, 742–768 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Allen NJ & Lyons DA Glia as architects of central nervous system formation and function. Science 362, 181–185 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sofroniew MV Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci 16, 249–263 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colonna M & Butovsky O Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol 35, 441–468 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fontana A, Fierz W & Wekerle H Astrocytes present myelin basic protein to encephalitogenic T-cell lines. Nature 307, 273–276 (1984). [DOI] [PubMed] [Google Scholar]
- 7.Ousman SS et al. Protective and therapeutic role for αB-crystallin in autoimmune demyelination. Nature 448, 474–479 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Sun D & Wekerle H Ia-restricted encephalitogenic T lymphocytes mediating EAE lyse autoantigen-presenting astrocytes. Nature 320, 70–72 (1986). [DOI] [PubMed] [Google Scholar]
- 9.Wheeler MA & Quintana FJ Regulation of astrocyte functions in multiple sclerosis. Cold Spring Harb. Perspect. Med 9, a029009 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liddelow SA & Barres BA Reactive astrocytes: production, function, and therapeutic potential. Immunity 46, 957–967 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Rothhammer V et al. Microglial control of astrocytes in response to microbial metabolites. Nature 557, 724–728 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rothhammer V et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med 22, 586–597 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wheeler MA et al. Environmental control of astrocyte pathogenic activities in CNS inflammation. Cell 176, 581–596.e18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giladi A & Amit I Single-cell genomics: a stepping stone for future immunology discoveries. Cell 172, 14–21 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Stubbington MJT, Rozenblatt-Rosen O, Regev A & Teichmann SA Single-cell transcriptomics to explore the immune system in health and disease. Science 358, 58–63 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanay A & Regev A Scaling single-cell genomics from phenomenology to mechanism. Nature 541, 331–338 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jäkel S et al. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 566, 543–547 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jordão MJC et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 363, eaat7554 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Masuda T et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 566, 388–392 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Schirmer L et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 573, 75–82 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Falcão AM Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat. Med 24, 1837–1844 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ajami B et al. Single-cell mass cytometry reveals distinct populations of brain myeloid cells in mouse neuroinflammation and neurodegeneration models. Nat. Neurosci 21, 541–551 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Itoh N et al. Cell-specific and region-specific transcriptomics in the multiple sclerosis model: focus on astrocytes. Proc. Natl Acad. Sci. USA 115, E302–E309 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chao C-C et al. Metabolic control of astrocyte pathogenic activity via cPLA2-MAVS. Cell 179, 1483–1498.e22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Macosko EZ et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cuadrado A et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat`. Rev. Drug Discov 18, 295–317 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Mayo L et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat. Med 20, 1147–1156 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katoh Y et al. Methionine adenosyltransferase II serves as a transcriptional corepressor of Maf oncoprotein. Mol. Cell 41, 554–566 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Lin CC & Edelson BT New insights into the role of IL-1β in experimental autoimmune encephalomyelitis and multiple sclerosis. J. Immunol 198, 4553–4560 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valentin-Torres A et al. Sustained TNF production by central nervous system infiltrating macrophages promotes progressive autoimmune encephalomyelitis. J. Neuroinflammation 13, 46 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wardyn JD, Ponsford AH & Sanderson CM Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans 43, 621–626 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katsuoka F & Yamamoto M Small Maf proteins (MafF, MafG, MafK): history, structure and function. Gene 586, 197–205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shavit JA et al. Impaired megakaryopoiesis and behavioral defects in mafG-null mutant mice. Genes Dev. 12, 2164–2174 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raghunath A et al. Antioxidant response elements: discovery, classes, regulation and potential applications. Redox Biol. 17, 297–314 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fang M, Ou J, Hutchinson L & Green MR The BRAF oncoprotein functions through the transcriptional repressor MAFG to mediate the CpG Island Methylator phenotype. Mol. Cell 55, 904–915 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Codarri L et al. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol 12, 560–567 (2011). [DOI] [PubMed] [Google Scholar]
- 37.El-Behi M et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol 12, 568–575 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ponomarev ED et al. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J. Immunol 178, 39–48 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Croxford AL et al. The cytokine GM-CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity 43, 502–514 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Lake BB et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol 36, 70–80 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liddelow SA et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Apetoh L et al. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat. Immunol 11, 854–861 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matcovitch-Natan O et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science 353, aad8670 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Xu M et al. c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature 554, 373–377 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aziz A, Soucie E, Sarrazin S & Sieweke MH MafB/c-Maf deficiency enables selfrenewal of differentiated functional macrophages. Science 326, 867–871 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Soucie EL et al. Lineage-specific enhancers activate self-renewal genes in macrophages and embryonic stem cells. Science 351, aad5510 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gandhi R et al. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3+ regulatory T cells. Nat. Immunol 11, 846–853 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huynh JL et al. Epigenome-wide differences in pathology-free regions of multiple sclerosis-affected brains. Nat. Neurosci 17, 121–130 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baranzini SE et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature 464, 1351–1356 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wendeln AC et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556, 332–338 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ayata P et al. Epigenetic regulation of brain region-specific microglia clearance activity. Nat. Neurosci 21, 1049–1060 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garcia AD, Doan NB, Imura T, Bush TG & Sofroniew MV GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat. Neurosci 7, 1233–1241 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Madisen L et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanz E et al. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc. Natl Acad. Sci. USA 106, 13939–13944 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Srinivasan R et al. New transgenic mouse lines for selectively targeting astrocytes and studying calcium signals in astrocyte processes in situ and in vivo. Neuron 92, 1181–1195 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anderson MA et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jaitin DA et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343, 776–779 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feng H, Zhang X & Zhang C mRIN for direct assessment of genome-wide and gene-specific mRNA integrity from large-scale RNA-sequencing data. Nat. Commun 6, 7816 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang X et al. Comparative analysis of droplet-based ultra-high-throughput single-cell rna-seq systems. Mol. Cell 73, 130–142.e135 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stuart T et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Finak G et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Becht E et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol 37, 38–44 (2018). [DOI] [PubMed] [Google Scholar]
- 64.van der Maaten L & Hinton G Visualizing data using t-SNE. J. Mach. Learn. Res 9, 2579–2605 (2008). [Google Scholar]
- 65.Qiu X et al. Single-cell mRNA quantification and differential analysis with Census. Nat. Methods 14, 309–315 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Qiu X et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Trapnell C et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Takenaka MC et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci 22, 729–740 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sanjana NE, Shalem O & Zhang F Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen S et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 160, 1246–1260 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee Y, Messing A, Su M & Brenner M GFAP promoter elements required for region-specific and astrocyte-specific expression. Glia 56, 481–493 (2008). [DOI] [PubMed] [Google Scholar]
- 72.Wang T et al. Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic Ras. Cell 168, 890–903.e815 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rothhammer V et al. Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proc. Natl Acad. Sci. USA 114, 2012–2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yates A et al. Ensembl 2016. Nucleic Acids Res. 44, D710–D716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ovcharenko I et al. Mulan: multiple-sequence local alignment and visualization for studying function and evolution. Genome Res. 15, 184–194 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.van Galen P et al. A multiplexed system for quantitative comparisons of chromatin landscapes. Mol. Cell 61, 170–180 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Untergasser A et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 40, e115 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Foo LC Purification of rat and mouse astrocytes by immunopanning. Cold Spring Harb. Protoc 2013, 421–432 (2013). [DOI] [PubMed] [Google Scholar]
- 79.Esmonde-White C et al. Distinct function-related molecular profile of adult human A2B5+ pre-oligodendrocytes versus mature oligodendrocytes. J. Neuropathol. Exp. Neurol 78, 468–479 (2019). [DOI] [PubMed] [Google Scholar]
- 80.Zheng GX et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Polman CH et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann. Neurol 69, 292–302 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dhaeze T et al. CD70 defines a subset of proinflammatory and CNS-pathogenic TH1/TH17 lymphocytes and is overexpressed in multiple sclerosis. Cell. Mol. Immunol 16, 652–665 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kuhlmann T et al. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 133, 13–24 (2017). [DOI] [PubMed] [Google Scholar]
- 84.Moffitt JR et al. High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization. Proc. Natl Acad. Sci. USA 113, 11046–11051 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moffitt JR et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362, eaau5324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Soumillon M, Cacchiarelli D, Semrau S, van Oudenaarden A & Mikkelsen TS Characterization of directed differentiation by high-throughput single-cell RNA-Seq. Preprint at 10.1101/003236v1 (2014). [DOI]
- 87.Trombetta JJ et al. Preparation of single-cell RNA-seq libraries for next generation sequencing. Curr. Protoc. Mol. Biol 107, 4.22.1–4.22.17 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li H & Durbin R Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li H et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Buenrostro JD, Wu B, Chang HY & Greenleaf WJ ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol 109, 21.29.1–21.29.9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mootha VK et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273 (2003). [DOI] [PubMed] [Google Scholar]
- 97.Chen EY et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kuleshov MV et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mi H, Muruganujan A, Ebert D, Huang X & Thomas PD PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data have been deposited into the Gene Expression Omnibus (GEO) under the SuperSeries accession number GSE130119. Clinical data for patient samples can be found in Supplementary Table 7. All other data and code that support the findings of this study are available from the corresponding author on reasonable request. Data from Schirmer et al.20 were accessed at PRJNA544731 and https://cells.ucsc.edu/?ds=ms. Data from Jäkel et al.17 were accessed at GSE118257. Data from Lake et al.40 were accessed at GSE97942.