

Abstract
Analogs of nucleosides and nucleotides represent a promising pool of potential therapeutics. This work describes a new synthetic route leading to 2′-deoxy-2′-fluorotetradialdose D-nucleoside phosphonates. Moreover, a new universal synthetic route leading to tetradialdose d-nucleosides bearing purine nucleobases is also described. All new compounds were tested as triphosphate analogs for inhibitory potency against a variety of viral polymerases. The fluorinated nucleosides were transformed to phosphoramidate prodrugs and evaluated in cell cultures against various viruses including influenza and SARS-CoV-2.
Keywords: Nucleoside phosphonate, Triphosphate, Prodrug, Tetradialdose d-nucleoside, 2′-Fluoronucleoside
Graphical abstract

1. Introduction
Synthesis of phosphonate derivatives of nucleosides, followed by the preparation of an appropriate phosphonodiphosphate or a prodrug form, represents a validated approach in the search for novel nucleoside-based antiviral agents [1]. In general, the introduction of a phosphonate group brings several advantages. It directly bypasses the first phosphorylation step in the cascade, which is often the rate-limiting step, leading to a biologically active nucleoside triphosphate. Moreover, unlike nucleoside monophosphates, phosphonates exhibit increased metabolic stability against cleavage by phosphoesterases [1]. To profile their biological activity, the phosphonodiphosphates and lipophilic prodrugs of phosphonate derivatives of nucleosides were prepared. The former can be used directly in enzyme assays, whereas the latter can be screened in cell-based assays due to the improved uptake compared to parent nucleotides.
Continuing our search for novel nucleoside phosphonates [[2], [3], [4]], we turned our attention to a tetradialdose nucleosides bearing O-phosphonomethyl group. Tetradialdose nucleoside 1 was first prepared by Kim et al., in 1991 (Fig. 1 ) [5]. More recently, teams of Herdewijn [6,7] (2, 3) and Cihlář [8] (4) reported synthesis of 3′-fluoro nucleosides and nucleosides derived from L-sugars. Potent anti-HIV activity was reported in the case of compound 4. The potential of 2′-fluoro nucleosides has been demonstrated by the use of sofosbuvir, an approved medication used to treat HCV [9]. Surprisingly, the synthesis of d-2′-fluorotetradialdose nucleoside phosphonates has not been reported so far. Moreover, in the case of d-tetraaldose phosphonates bearing purine nucleobase, only the synthesis of adenine derivative 1 has been published [5]. Therefore, we report here the synthesis of novel 2′-fluorotetradialdose nucleoside phosphonates 5 and phosphonate analogs 6 (Fig. 2 ), and their biochemical evaluation against several types of viruses including influenza and coronavirus.
Fig. 1.

Examples of the reported tetradialdose nucleoside phosphonates.
Fig. 2.

Structures of the prepared tetradialdose nucleoside phosphonates.
2. Results and discussion
2.1. Synthesis of d-2′-fluorotetradialdose nucleoside phosphonates
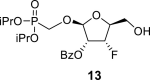
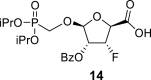
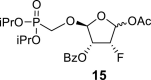
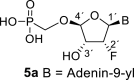
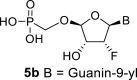
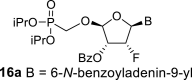
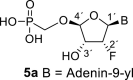
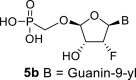
The synthetic strategy leading to 2′-fluoro analogs was developed based on the previously published synthetic route leading to l-dialdose analogs 3 [7]. Starting from l-xylose 7, we prepared methyl glycoside 9 with 5′-protected hydroxyl in two steps (Scheme 1 ). The first key step of the synthetic sequence consisted of the introduction of the fluorine atom. To avoid elimination reactions and the formation of side products during the fluorination step, it was reported that the reaction had to be performed on a precursor bearing unprotected trans diol [10,11]. Therefore, l-xylose glycoside 9 was treated with DAST to afford the fluorinated product in 54% yield. In agreement with the literature [6,7], we observed the regioselective formation of 3-fluoro-l-ribofuranoside 10. Compound 10 was then treated with sulfuric acid in a mixture of acetic anhydride and acetic acid. In the next step, the 1-O-acetyl derivative was converted to phosphonate using the Vorbrüggen reaction in the presence of diisopropyl trimethylsilyloxymethylphosphonate. In our case, the reaction using silylated hydroxymethylphosphonate afforded improved yield of the glycosidation reaction over the reaction performed in the presence of hydroxymethylphosphonate [7]. Afterwards, the acyl groups were removed under basic conditions to afford phosphonate 12. The next goal was to prepare a precursor with free primary hydroxyl. Therefore, we protected the primary hydroxyl with the dimethoxytrityl group and the secondary hydroxyl with benzoyl, and we then deprotected the primary hydroxyl with TFA to obtain the precursor 13. In the next steps, we transformed the hydroxymethyl group to acetate, which was then used for the nucleosidation step [7]. Specifically, the hydroxymethyl group of the compound 13 was oxidized to the carboxylate 14 using (diacetoxyiodo)benzene in the presence of TEMPO, followed by oxidative decarboxylation using lead tetraacetate to yield acetate 15. This precursor could be used for a nucleosidation reaction under the Vorbrüggen conditions. The precursor 15 was treated with silylated 6-N-benzoyladenine in the presence of tin tetrachloride. Because of the absence of the directing 2-O-acyl group, the reaction resulted in a mixture of β and α anomers in a 2:1 ratio, resp. The mixture was separated on a silica gel, and the desired β-anomer 16 a was deprotected to yield the novel d-2′-fluorotetradialdose nucleoside phosphonate 5a product. Using a similar approach with silylated 2-N-acetyl-6-O-diphenylcarbamoylguanine, we also prepared guanine derivative 5b (Scheme 1, Fig. 2).
Scheme 1.

Synthesis of 2′-fluorotetradialdose nucleoside phosphonates. Reagents and conditions: (a) HCl, MeOH, rt, 16 h, quant.; (b) p-Toluoyl chloride, pyridine, 0 °C to rt, 16 h, 48%; (c) DAST, MeCN, 0 °C to rt, 16 h, 54%; (d) H2SO4, Ac2O, AcOH, 0 °C to rt, 2 h, 84%; (e) hexamethyldisilazane, (iPrO)2P(O)CH2OH, saccharine, 100 °C, 8 h; (f) SnCl4, MeCN, 55 °C, 1 h; (g) MeNH2, EtOH, rt, 16 h, 85%; (h) DMTrCl, pyridine, rt, 16 h; (i) BzCl, DMAP, pyridine, rt, 8 h; (j) TFA, DCM, rt, 10 min, 74%; (k) PhI(OAc)2, TEMPO, MeCN, rt, 16 h, 90%; (l) Pb(OAc)4, THF, rt, 16 h, 60%; (m) SnCl4, DCE, BSA, N6-benzoyladenine, rt, 30 min 43%; (n) TMSBr, pyridine, rt, 8 h; (o) MeNH2, EtOH, rt, 73%.
2.2. Synthesis of d-tetradialdose nucleoside phosphonates
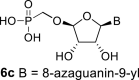
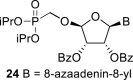
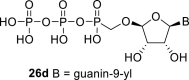
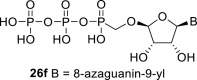
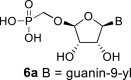
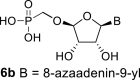
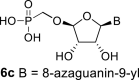
In our experiment, the synthetic sequence for the preparation of 2′-hydroxy compound 1 described by Kim et al. [5] failed to afford the guanine derivative 6a. Therefore, we adapted a synthetic approach similar to the approach used for the 2′-fluoro nucleosides. Starting from l-ribose, we prepared d-nucleoside 1 and the new nucleosides 6a-6c bearing guanine, 8-azaadenine and 8-azaguanine, resp. (Scheme 2 , Fig. 2).
Scheme 2.

Synthesis of tetradialdose nucleoside phosphonates. Reagents and conditions: (a) HCl, MeOH, rt, 16 h; (b) H2SO4, Ac2O, AcOH, 0 °C to rt, 2 h; (c) hexamethyldisilazane, (iPrO)2P(O)CH2OH, saccharine, 100 °C, 8 h; (d) SnCl4, MeCN, 55 °C, 1 h; (e) NH3, MeOH, 90%; (f) TBDPSCl, imidazole, DMF, rt, 24 h; (g) BzCl, Et3N, DMAP, DCM, rt, 2 h; (h) TBAF, THF, rt, 8 h; 78%; (i) PhI(OAc)2,TEMPO, MeCN/H2O, rt, 16 h; (j) Pb(OAc)4, THF, rt, 16 h; 53% (k) SnCl4, MeCN, BSA, N6-benzoyladenine, 50%; (l) TMSBr, pyridine, rt, 8 h; (m) MeNH2, EtOH, rt, 80%.
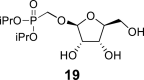
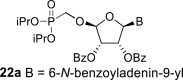
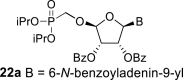
l-Tetraacetylribose 18 was phosphonylated using the Vorbrüggen reaction with diisopropyl trimethylsilyloxymethylphosphonate. Then, the acetyl groups were removed under basic conditions to afford the phosphonate 19. Next, the primary hydroxyl was protected with the TBDPS group, and cis diol was protected with the benzoyl groups. The TBDPS group was subsequently removed using TBAF to obtain the phosphonate 20. The hydroxymethyl group was converted to acetate 21 using the above-mentioned sequence. A nucleosidation reaction using the Vorbrüggen reaction, silylated 6-N-benzoyladenine and 2-N-acetyl-6-O-diphenylcarbamoylguanine afforded compounds 22a and 22b as adenin-9-yl and guanin-9-yl β-anomers in good yields. After deprotection, compounds 1 and 6a were obtained.
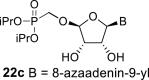
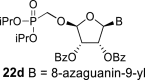
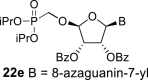
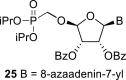
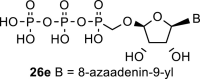
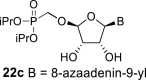
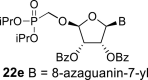
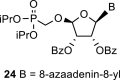
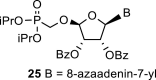
In contrast, in the case of the 8-azaadenine derivative, we observed the formation of 8-azaadenin-7-yl, 8-azaadenin-8-yl and 8-azaadenin-9-yl regioisomers during the Vorbrüggen reaction. Thus, the nucleosidation of acetate 21 with 8-azaadenine afforded a mixture of three regioisomers 23, 24 and 25 in a 2:1:6 ratio (Scheme 2, Fig. 3 ). It is worth noting that the nucleosidation using silylated 8-azaadenine afforded the undesired regioisomer 24 as the only product. Regioisomer 24 was separated easily on a silica gel. Unfortunately, we were not able to separate the mixture of regioisomers 23 and 25 (in ratio 1:3), either on a silica gel or by using reverse phase chromatography. Therefore, we took advantage of the reported difference in the stability of the regioisomers of 8-azaadenosine under acidic conditions. It has been shown that the N7-regioisomer of 2′-deoxy-8-azaadenosine was hydrolyzed 75 times faster than the N9-regioisomer [12]. As an analogy, we exploited this phenomena to depurinate undesired N7-regioisomer 25. First, we treated the mixture of compounds 23 and 25 with ammonia in methanol to remove the benzoyl groups, and then treated it with 80% AcOH until the N7-regioisomer depurinated. The N9-regioisomer 22c was then easily purified on reverse phase, affording the desired 8-azaadenin-9-yl derivative as a β-anomer. After the final cleavage of protecting groups, compound 6b was obtained (Scheme 2).
Fig. 3.

Assignment of the structure of 8-azaadenine derivatives.
We performed NMR study to assign the structures of N9-, N8- and N7-regioisomers 23, 24 and 25 (Fig. 3). These regioisomers could be distinguished on the basis of long-range C,H couplings observed in the 2D-H,C-HMBC spectra. The only C–H proton of 8-azaadenine, proton H-2, has long-range couplings with carbon C-4 and C-6 in all three regioisomers N9, N8 and N7 (see red arrows in 23, 24 and 25 in Fig. 3). However, ribose proton H-1′ showed coupling to carbon C-4 in the N9-regioisomer, and coupling to C-5 in the N7-regioisomer (see blue arrows in 23 and 25 in Fig. 3), while it had no coupling to base carbons in the N8-regioisomer (proton H-1′ is separated by four bonds from both C-4 and C-5).
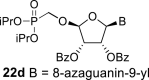
In the case of the 8-azaguanine derivative, we obtained the desired N9-regioisomer using the nucleosidation reaction in the presence of silylated 8-azaguanine (Scheme 2). The nucleosidation of acetate 21 afforded only two regioisomers. The 8-azaguanin-9-yl (22d) and 8-azaguanin-7-yl (22e) were obtained in a 1:5 ratio. The regioisomers were easily separated on a silica gel. Finally, the compound 22d was deprotected to afford nucleotide 6c. The structure of the regioisomers was assigned based on the 2D-H,C-HMBC spectra, as in case of 8-azaadenine derivative 6b.
2.3. Synthesis of nucleoside phosphonodiphosphates
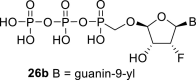
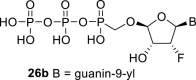
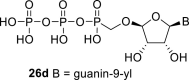
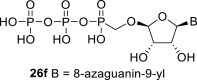
Next, we continued with the preparation of nucleoside phosphonodiphosphates. The tetrabutylammonium salts of the phosphonic acids 5a-5b, 1 and 6a-6c were converted to the corresponding phosphonoimidazolide using PPh3, imidazole and Aldrithiol™ [13]. Afterwards the imidazolides were converted to the corresponding phosphonodiphosphates 26a-f using tributylammonium salt of pyrophosphate, and finalized as sodium salts (Scheme 3 ). The compound 26c was already prepared and tested as a substrate of RNA polymerase in vitro assay by Koh et al. [14].
Scheme 3.

Synthesis of nucleoside phosphonodiphosphates. Reagents and conditions: (a) Imidazole, trioctylamine, PPh3, Aldrithiol™, DMF, 16 h, rt; (b) tributylammonium pyrophosphate, DMF, 16 h, rt; (c) Dowex® 50 (Na+ cycle), 45%.
2.4. Synthesis of phosphoramidate prodrugs
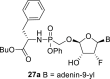
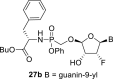
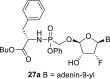
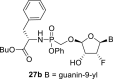
The prodrug forms of the phosphonates 27a-27b were prepared according to combined procedures of Klejch et al. [15] and Mackman et al. [16] (Scheme 4 ). We selected l-phenylalanine butyl ester phosphoramidates as a prodrug candidate based on the positive results of similar prodrug forms reported in the literature [17]. The phosphonic acids 5a-5b were treated with l-phenylalanine butyl ester and phenol in the presence of triphenylphosphine and Aldrithiol™ to afford epimeric mixtures of the phosphoramidates in approximately a 1:1 ratio. Despite all efforts, we were not able to separate the epimeric mixtures, and therefore the phosphoramidate prodrugs were screened as a mixture of both epimers.
Scheme 4.

Synthesis of prodrugs. Reagents and conditions: (a) Phenol, l-phenylalanine butyl ester, Et3N, pyridine, 15 min, 60 °C (b) PPh3, Aldrithiol™, pyridine, 16 h, 60 °C, 56%.
2.5. Biochemical evaluation
The phosphonodiphosphates 26a-26f were tested for the inhibition of RNA template elongation by viral polymerases (RdRp) from Zika virus, HCV and Poliovirus, according to the conditions published previously [18,19]. Radioactivity based competition assay did not show any inhibition of these enzymes at a 20 μM concentration of phosphonodiphosphates 26a-26f.
The potential antiviral effect of the prodrugs 27a-27b was determined by screening against Coxsackie B3, Dengue 2, Influenza A, HIV-1 and Sars-CoV-2 viruses as published previously [20]. Unfortunately, the prodrugs showed low or no inhibition activity of these viruses.
3. Conclusion
We developed a synthetic route leading to the novel 2′-fluoro-d-tetraaldose nucleoside phosphonates and the synthesis of d-tetraaldose nucleoside phosphonates suitable for the preparation of tetraaldose nucleosides bearing purine nucleobases. These compounds were converted to appropriate phosphonodiphosphates and tested against viral polymerases from the Zika virus, HCV and Poliovirus. 2′-Fluoro nucleotides were also converted to prodrugs and screened against coxsackie, dengue, influenza, HIV-1 and Sars-CoV-2 viruses. Unfortunately, no biological activity was found. Nevertheless, the tetraaldose nucleoside phosphonates represent an interesting group of compounds and further exploration of its potential, e.g. synthesis of oligonucleotides bearing isosteric phosphonomethoxy linkages, is in progress.
4. Experimental section
Unless stated otherwise, all solvents used were anhydrous. Mass spectra were recorded on the ZAB-EQ (VG Analytical) instrument using FAB (ionization with Xe, accelerating voltage 8 kV; glycerol and thioglycerol as matrices) and on the LTQ Orbitrap XL (Thermo Fisher Scientific) using ESI ionization. The NMR spectra were measured on a Bruker AVANCE-600 instrument (1H at 600.13 MHz and 13C at 150.9 MHz) with a cryoprobe and a Bruker AVANCE-500 instrument with a cryoprobe (31P at 202.4 MHz) in DMSO‑d 6 and/or D2O at 25 °C. The 1H and 13C spectra in DMSO were referenced to the solvent peak (using δ H(DMSO) = 2.50 ppm; δ C(DMSO) = 39.7 ppm) and spectra in D2O were referenced to internal dioxane (using δ H(dioxane) = 3.75 ppm; δ C(dioxane) = 69.30 ppm). The 31P spectra were referenced to external H3PO4. The structural assignment of the proton and carbon signals was achieved combining 1D-1H and 13C-spectra with homonuclear 2D-H,H–COSY, 2D-H,H-ROESY and heteronuclear 2D-H,C-HSQC and 2D-H,C-HMBC spectra. IR spectra were recorded on a Nicolet 6700 (Thermo Electron Corp.).
4.1. Methyl l-xylofuranoside (8)
A solution of acetyl chloride (5 mL; 70 mmol) and dry MeOH (100 mL) was added dropwise to a suspension of l -xylose 7 (100 g; 0.66 mol) in dry MeOH (1.2 L), and the reaction mixture was vigorously stirred for 16 h at room temperature. When the solution became homogenous, Dowex® 1 × 4 (OH− cycle) was added until its pH became neutral. The resin was filtered, and the reaction mixture was concentrated. Methyl l-xylofuranoside 8 (112 g, quant. yield) was obtained as a colorless viscous oil, and was used in the next step without further purification.
4.2. Methyl 5-O-(4-methylbenzoyl)-l-xylofuranoside (9)
p-Toluoyl chloride (88 mL; 0.66 mol) in 200 mL of dry pyridine was added dropwise to a solution of methyl l-xylofuranoside 8 (112 g; 0.66 mol) in dry pyridine (500 mL) at 0 °C. The reaction mixture was warmed to room temperature, stirred for 16 h, then quenched by the addition of 50 mL of water, evaporated and extracted (1 L EtOAc/500 mL NaCl and 2 × 500 mL NaHCO3). The organic phase was dried over MgSO4, and product 9 was isolated by chromatography on a silica gel (0–100% EtOAc in toluene) affording 90 g (48% over two steps) as a colorless viscous oil. Compound 9 was characterized by low resolution mass spectrometry: (M + H)+ for C14H19O6 calculated: 283.1; measured: 283.1.
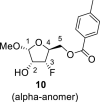
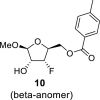
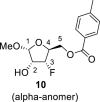
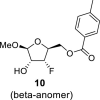
4.3. Methyl 3-deoxy-3-fluoro-5-O-(4-methylbenzoyl)-l-xylofuranoside (10)
DAST (135 mL, 0.96 mol) diluted with 300 mL of ACN was added dropwise to a solution of compound 9 (90 g, 0.32 mol) in ACN (1 L). The reaction mixture was warmed to room temperature and stirred for 16 h. Next, 100 mL of water was added. The reaction mixture was evaporated and extracted (1 L toluene/500 mL sat. NaCl and 2 × 500 mL of sat. NaHCO3). The organic phase was dried over MgSO4, and product 10 was isolated by chromatography on a silica gel (0–60% EtOAc in cyclohexane) as a mixture of both anomers in a yield of 49 g (54%) of colorless viscose oil. Alpha anomer: HRMS (M + Na)+ for C14H17O5FNa calculated: 307.09522; measured: 307.09492; IR (coating, cm−1): 2841, 1720, 1612, 1450, 1380, 1272, 1179, 1074, 1020, 790, 752, 691, 475. For NMR data, see Table Ia, Table Ib b.
Table Ia.
1H NMR data of compounds 10–15 and 19–21.k
| Compound | Solvent | H-1 | H-2 | H-3 | H-4 | H-5a; H-5b |
|---|---|---|---|---|---|---|
 |
DMSO a | 4.86 d 1,2 = 4.7 |
4.045 bdq 2,1 = 4.7 2,F = 26.5 2, OH = 7.5 2,3 = 5.5 |
4.91 ddd 3,F = 56.2 3,2 = 5.5 3,4 = 1.4 |
4.44 dtd 4,F = 26.6 4,5b = 4.4 4,5a = 3.9 4,3 = 1.4 |
4.38 dd 5a, 5b = 11.8; 5a,4 = 3.9 4.35 dd 5b, 5a = 11.8; 5b,4 = 4.4 |
 |
DMSO b | 4.77 t 1,2 = 2.0 1,F = 2.0 |
4.06 um | 5.08 dt 3,F = 53.7 3,2 = 4.5 3,4 = 4.5 |
4.42 m | 4.44 m 4.30 m |
 alpha + beta anomer (56:44) alpha + beta anomer (56:44) |
DMSO c | 6.09 d 1,2 = 2.6 1,F = 1.4 |
5.36 ddd 2,F = 11.0 2,3 = 4.8 2,1 = 2.6 |
5.52 ddd 3,F = 52.0 3,2 = 4.8 3,4 = 3.8 |
4.65 dq 4,F = 20.9 4,5b = 4.2 4,3 = 3.8 4,5a = 3.6 |
4.56 dd 5a, 5b = 12.2; 5a,4 = 3.6 4.39 dd 5b, 5a = 12.2; 5b,4 = 4.2 |
| 6.39 d 1,2 = 4.8 |
5.20 dt 2,F = 25.1 2,3 = 5.2 2,1 = 4.8 |
5.37 ddd 3,F = 55.0 3,2 = 5.2 3,4 = 1.3 |
4.76 dtd 4,F = 26.7 4,5b = 4.2 4,5a = 3.7 4,3 = 1.3 |
4.44 dd 5a, 5b = 12.0; 5a,4 = 3.7 4.42 dd 5b, 5a = 12.0; 5b,4 = 4.2 |
||
 |
DMSO d | 4.905 dd 1,2 = 2.6 |
4.03 dddd 2,1 = 2.6 2,3 = 4.4 2, OH = 5.7 2,F = 11.7 |
4.86 dt 3,2 = 4.4 3.F = 53.9 3,4 = 3.8 |
4.08 dtd 4,3 = 3.8 4,F = 20.9 4,5a = 5.5 4,5b = 5.5 |
3.45 m (2H) |
 |
DMSO e | 5.40 dd 1,2 = 3.1 1,F = 1.4 |
5.31 ddd 2,1 = 3.1 2,3 = 4.9 2,F = 11.7 |
5.30 ddd 3,2 = 4.9 3.F = 53.6 3,4 = 3.1 |
4.29 dtd 4,3 = 3.1 4,F = 22.6 4,5a = 5.4 4,5b = 5.0 |
3.57 m (2H) |
 |
DMSO f | 5.47 dd 1,2 = 2.4 1,F = 1.3 |
5.34 ddd 2,1 = 2.4 2,3 = 4.7 2,F = 9.4 |
5.53 ddd 3,2 = 4.7 3.F = 52.6 3,4 = 3.4 |
4.77 dd 4,3 = 3.4 4,F = 23.2 |
– |
 beta and alpha epimers (83:17) beta and alpha epimers (83:17) |
DMSO g data for major β-epimer only |
5.65 d 1,2 = 3.1 |
5.38 ddd 2,1 = 3.1 2,3 = 4.4 2,F = 17.0 |
5.44 ddd 3,2 = 4.4 3.F = 50.5 3,4 = 1.2 |
6.31 dd 4,3 = 1.2 4,F = 10.8 |
– |
 |
DMSO h | 4.80 s 1,2 = 0 |
3.74 bd 2,1 = 0 2,3 = 4.6 |
3.87 dd 3,2 = 4.6 3,4 = 7.2 |
3.78 ddd 4,3 = 7.2 4,5a = 3.2 4,5b = 6.1 |
3.54 dd 5a, 5b = 11.8; 5a,4 = 3.2 3.34 dd 5b, 5a = 11.8; 5b,4 = 4.4 |
 |
DMSO i | 5.38 d 1,2 = 1.5 |
5.51 dd 2,1 = 1.5 2,3 = 5.0 |
5.54 dd 3,2 = 5.0 3,4 = 5.8 |
4.42 ddd 4,3 = 5.8 4,5a = 4.6 4,5b = 5.1 |
3.68 ddd 5a, 5b = 12.0; 5a,4 = 4.6 5a, OH = 5.8 3.61 ddd 5b, 5a = 12.0; 5b,4 = 5.1 5b, OH = 6.0 |
 beta and alpha epimers (76:24) beta and alpha epimers (76:24) |
DMSO j | 5.65 d 1,2 = 2.1 |
5.60 dd 2,1 = 2.1 2,3 = 5.1 |
5.67 dd 3,2 = 5.1 3,4 = 2.4 |
6.46 d 4,3 = 2.4 |
– |
| 5.64 d 1,2 = 0.9 |
5.59 dd 2,1 = 0.9 2,3 = 5.1 |
5.67 dd 3,2 = 5.1 3,4 = 4.9 |
6.60 d 4,3 = 4.9 |
– |
1-OMe: 3.33 s; 2-OH: 5.12 br; 5-OTol: 7.84 m (2x o-ArH), 7.35 m (2x m-ArH), 2.385 s (CH3).
1-OMe: 3.22 s; 2-OH: 5.70 br; 5-OTol: 7.88 m (2x o-ArH), 7.35 m (2x m-ArH), 2.38 s (CH3).
α-anomer: 1-OAc: 1.88 s; 2-OAc: 2.13 s; 5-OTol: 7.92 m (2x o-ArH), 7.36 m (2x m-ArH), 2.39 s (CH3); β-anomer: 1-OAc: 2.07 s; 2-OAc: 2.12 s; 5-OTol: 7.87 m (2x o-ArH), 7.34 m (2x m-ArH), 2.39 s (CH3).
2-OH: 5.66 bd, J = 5.7 Hz; 5-OH: 4.96 bt, J = 5.5 Hz; O–CH2–P = O(OiPr)2: 3.84 dd, J = 13.8, 9.0 Hz and 3.73 dd, J = 13.8, 9.1 Hz (P–CH2–O), 4.59 m, 2H, 1.235 d, J = 6.2 Hz, 1.237 d, J = 6.2 Hz, 1.244 d, J = 6.2 Hz and 1.246 d, J = 6.2 Hz (2x OiPr).
2-OBz: 8.01 m (2x o-ArH), 7.57 m (2x m-ArH), 7.71 m (p-ArH); 5-OH: 5.14 bt, J = 5.6 Hz; O–CH2–P = O(OiPr)2: 3.95 dd, J = 13.9, 9.0 Hz and 3.87 dd, J = 13.9, 8.9 Hz (P–CH2–O), 4.69 m, 2H, 1.213 d, 3H, J = 6.2 Hz, 1.218 d, 3H, J = 6.2 Hz, 1.223 d, 3H, J = 6.2 Hz and 1.231 d, 3H, J = 6.2 Hz (2x OiPr).
2-OBz: 8.00 m (2x o-ArH), 7.57 m (2x m-ArH), 7.71 m (p-ArH); O–CH2–P = O(OiPr)2: 4.06 dd, J = 13.8, 8.2 Hz and 3.92 dd, J = 13.8, 9.4 Hz (P–CH2–O), 4.59 m, 2H, 1.210 d, 6H, J = 6.2 Hz, 1.221 d, 3H, J = 6.2 Hz, 1.225 d, 3H, J = 6.2 Hz (2x OiPr).
2-OBz: 8.02 m (2x o-ArH), 7.58 m (2x m-ArH), 7.73 m (p-ArH); 4-OAc: 2.10 s; O–CH2–P = O(OiPr)2: 3.92 m, 2H (P–CH2–O), 4.60 m, 2H, 1.205 d, 3H, J = 6.0 Hz, 1.213 d, 3H, J = 6.2 Hz, 1.216 d, 3H, J = 6.0 Hz and 1.231 d, 3H, J = 6.1 Hz (2x OiPr).
O–CH2–P = O(OiPr)2: 3.84 dd, J = 13.8, 8.9 Hz and 3.65 dd, J = 13.8, 8.7 Hz (P–CH2–O), 4.58 m, 2H, 1.245 d, 3H, J = 6.2 Hz, 1.242 d, 3H, J = 6.2 Hz and 1.234 d, 6H, J = 6.6 Hz (2x OiPr).
2-OBz: 7.87 m (2x o-ArH), 7.47 m (2x m-ArH), 7.65 m (p-ArH); 3-OBz: 7.85 m (2x o-ArH), 7.44 m (2x m-ArH), 7.62 m (p-ArH); 5-OH: 5.08 t, J = 5.8 and 6.0 Hz; O–CH2–P = O(OiPr)2: 4.00 dd, J = 13.8, 9.0 Hz and 3.87 dd, J = 13.8, 8.9 Hz (P–CH2–O), 4.64 m, 2H, 1.260 d, 6H, J = 6.2 Hz, 1.264 d, 3H, J = 6.2 Hz and 1.268 d, 3H, J = 6.2 Hz (2x OiPr).
Major epimer: 2-OBz: 7.88 m (2x o-ArH), 7.465 m (2x m-ArH), 7.655 m (p-ArH); 3-OBz: 7.88 m (2x o-ArH), 7.465 m (2x m-ArH), 7.655 m (p-ArH); O–CH2–P = O(OiPr)2: 3.95 dd, J = 13.7, 9.0 Hz and 3.92 dd, J = 13.7, 9.3 Hz (P–CH2–O), 4.64 m, 2H, 1.250 d, 3H, J = 6.2 Hz, 1.260 d, 3H, J = 6.2 Hz, 1.266 d, 3H, J = 6.2 Hz and 1.270 d, 3H, J = 6.2 Hz (2x OiPr); 4-OAc: 2.13 s; minor epimer: 2-OBz: 8.005 m (2x o-ArH), 7.565 m (2x m-ArH), 7.705 m (p-ArH); 3-OBz: 7.76 m (2x o-ArH), 7.42 m (2x m-ArH), 7.62 m (p-ArH); O–CH2–P = O(OiPr)2: 3.99 dd, J = 13.7, 9.0 Hz and 3.96 dd, J = 13.7, 8.7 Hz (P–CH2–O), 4.64 m, 2H, 1.252 d, 6H, J = 6.2 Hz, 1.273 d, 3H, J = 6.2 Hz and 1.277 d, 3H, J = 6.2 Hz (2x OiPr).
Coupling constants are written in italics in a shortened form (e.g. instead J(1‘,2‘) = 8.6 Hz we type simply 1,2 = 8.6).
Table Ib.
13C,31P and19F NMR data of compounds 10–15 and 19–21k
| Compound | Solvent | C-1 | C-2 | C-3 | C-4 | C-5 | 31P | 19F |
|---|---|---|---|---|---|---|---|---|
 |
DMSO a | 102.64 | 71.74 2,F = 16.4 |
90.88 3,F = 185.2 |
80.09 4,F = 24.8 |
64.09 5,F = 10.3 |
– | −189.10 |
 |
DMSO b | 108.19 1,F = 3.2 |
73.35 2,F = 15.0 |
92.11 3,F = 186.6 |
78.47 4,F = 25.0 |
64.03 5,F = 5.9 |
– | −205.56 |
 beta and alpha anomers (56:44) beta and alpha anomers (56:44) |
DMSO c β-epimer |
98.20 1,F = 1.9 |
74.69 2,F = 14.2 |
89.68 3,F = 189.9 |
81.91 4,F = 24.7 |
63.20 5,F = 6.8 |
– | −203.10 |
| α-epimer | 93.30 | 71.00 2,F = 14.7 |
88.84 3,F = 188.1 |
81.09 4,F = 24.8 |
63.63 5,F = 10.1 |
– | −190.71 | |
 |
DMSO d | 107.71 1,P = 12.0 1,F = 2.4 |
73.56 2,F = 15.3 |
82.48 3,F = 185.8 |
82.32 4,F = 22.2 |
61.78 5,F = 6.3 |
20.56 | −203.93 |
 |
DMSO e | 105.30 1,P = 12.2 1,F = 1.4 |
75.56 2,F = 14.2 |
90.81 3,F = 188.1 |
83.40 4,F = 21.9 |
61.29 5,F = 7.3 |
19.95 | −201.10 |
 |
DMSO f | 105.47 1,P = 12.1 |
74.82 2,F = 13.8 |
92.21 3,F = 193.1 |
80.56 4,F = 23.4 |
170.81 5,F = 9.2 |
19.92 | −199.95 |
 beta and alpha epimers (83:17) beta and alpha epimers (83:17) |
DMSO g data for major β-epimer only |
107.10 1,P = 13.4 |
75.26 2,F = 14.3 |
91.96 3,F = 188.8 |
98.26 4,F = 33.1 |
– | 18.98 | −206.24 |
 |
DMSO h | 107.73 1,P = 11.6 |
74.40 | 70.73 | 84.03 | 62.80 | 21.08 | – |
 |
DMSO i | 105.23 1,P = 12.2 |
74.87 | 72.27 | 82.20 | 61.98 | 20.12 | – |
 beta and alpha epimers (76:24) |
DMSO j β-epimer |
106.98 1,P = 13.1 |
74.63 | 75.29 | 98.82 | – | 18.03 | – |
| α-epimer | 105.53 1,P = 12.3 |
72.28 | 70.16 | 93.14 | – | 19.19 | – |
Substituents.
1-OMe: 55.11; 2-OTol: 165.61 (C O), 126.77 (i-ArC), 129.43 (2x o-ArC), 129.61 (2x m-ArC), 144.11 (p-AQrC), 21.38 (CH3).
1-OMe: 55.16; 2-OTol: 165.65 (C O), 126.92 (i-ArC), 129.46 (2x o-ArC), 129.53 (2x m-ArC), 144.03 (p-ArC), 21.37 (CH3).
α-anomer: 1-OAc: 169.19 (C O), 20.74 (CH3); 2-OAc: 169.53 (C O), 20.48 (CH3); 2-OTol: 165.42 (C O), 126.71 (i-ArC), 129.53 (2x o-ArC), 129.58 (2x m-ArC), 144.20 (p-ArC), 21.35 (CH3); β-anomer: 1-OAc: 169.77 (C O), 21.06 (CH3); 2-OAc: 169.73 (C O), 20.40 (CH3); 2-OTol: 165.50 (C O), 126.59 (i-ArC), 129.48 (2x o-ArC), 129.56 (2x m-ArC), 144.20 (p-ArC), 21.34 (CH3).
O–CH2–P = O(OiPr)2: 61.12 d, J = 166.9 Hz (P–CH2–O), 70.70 d, J = 5.0 Hz and 70.57 d, J = 5.0 Hz (2x O–CH<), 23.93 d, J = 4.6 Hz (2x CH3), and 24.06 d, J = 3.4 Hz (2x CH3).
2-OBz: 164.92 (C O), 128.74 (i-ArC), 129.67 (2x o-ArC), 129.22 (2x m-ArC), 134.29 (p-ArC), O–CH2–P = O(OiPr)2: 61.46 d, J = 166.3 Hz (P–CH2–O), 70.81 d, J = 6.3 Hz (2x O–CH<), 23.88 d, J = 4.6 Hz (CH3), 23.87 d, J = 4.6 Hz (CH3), 24.03 d, J = 3.7 Hz (CH3) and 24.03 d, J = 3.7 Hz (CH3).
2-OBz: 164.82 (C O), 128.61 (i-ArC), 129.68 (2x o-ArC), 129.19 (2x m-ArC), 134.30 (p-ArC), O–CH2–P = O(OiPr)2: 60.85 d, J = 165.6 Hz (P–CH2–O), 70.77 d, J = 6.2 Hz and 70.80 d, J = 6.2 Hz (2x O–CH<), 23.82 d, J = 4.7 Hz (CH3), 23.86 d, J = 4.7 Hz (CH3), 23.97 d, J = 3.6 Hz (CH3) and 24.00 d, J = 3.8 Hz (CH3).
2-OBz: 164.86 (C O), 128.41 (i-ArC), 129.81 (2x o-ArC), 129.32 (2x m-ArC), 134.54 (p-ArC), 4-OAc: 169.17 (C O), 21.03 (CH3); O–CH2–P = O(OiPr)2: 62.32 d, J = 166.0 Hz (P–CH2–O), 70.95 d, J = 6.2 Hz and 70.93 d, J = 6.1 Hz (2x O–CH<), 23.87 d, J = 4.4 Hz (2x CH3), 24.03 d, J = 3.5 Hz (2x CH3).
O–CH2–P = O(OiPr)2: 60.47 d, J = 166.9 Hz (P–CH2–O), 70.46 d, J = 6.0 Hz and 70.42 d, J = 6.0 Hz (2x O–CH<), 24.02 d, J = 4.5 Hz (2x CH3), 23.89 d, J = 4.8 Hz (2x CH3).
2-OBz: 164.73 (C O), 128.88 (i-ArC), 129.43 (2x o-ArC), 129.01 (2x m-ArC), 134.09 (p-ArC), 3-OBz: 165.06 (C O), 128.73 (i-ArC), 129.37 (2x o-ArC), 128.92 (2x m-ArC), 133.94 (p-ArC); O–CH2–P = O(OiPr)2: 61.07 d, J = 166.6 Hz (P–CH2–O), 70.71 d, J = 6.3 Hz (2x O–CH<), 23.86 d, J = 4.5 Hz (2x CH3), 24.00 d, J = 3.7 Hz (2x CH3).
Major β-epimer: 2-OBz: 164.67 (C O), 128.42 (i-ArC), 129.56 (2x o-ArC), 129.03 (2x m-ArC), 134.25 (p-ArC), 3-OBz: 164.62 (C O), 128.41 (i-ArC), 129.50 (2x o-ArC), 129.00 (2x m-ArC), 134.21 (p-ArC); O–CH2–P = O(OiPr)2: 61.88 d, J = 166.0 Hz (P–CH2–O), 70.83 d, J = 6.2 Hz and 70.77 d, J = 6.2 Hz (2x O–CH<), 23.81 d, 2C, J = 4.6 Hz (2x CH3), 23.96 d, J = 3.5 Hz and 23.97 d, J = 3.7 Hz (2x CH3); 4-OAc: 169.39 (C O), 20.95 (CH3); minor α-epimer: 2-OBz: 164.70 (C O), 128.83 (i-ArC), 129.54 (2x o-ArC), 129.14 (2x m-ArC), 134.23 (p-ArC), 3-OBz: 164.46 (C O), 128.44 (i-ArC), 129.28 (2x o-ArC), 129.03 (2x m-ArC), 134.15 (p-ArC); O–CH2–P = O(OiPr)2: 61.82 d, J = 166.1 Hz (P–CH2–O), 70.83 d, J = 6.2 Hz and 70.77 d, J = 6.2 Hz (2x O–CH<), 23.98 d, J = 3.7 Hz (2x CH3), 23.86 d, J = 4.6 Hz (CH3) and 23.84 d, J = 4.6 Hz (CH3); 4-OAc: 169.27 (C O), 20.85 (CH3).
Coupling constants are written in italics in a shortened form (e.g. instead J(C2,F) = 16.4 Hz we type simply 2,F = 16.4).
Beta anomer: HRMS (M + Na)+ for C14H17O5FNa calculated: 307.09522; measured: 307.09494; IR (coating, cm−1): 2841, 1720, 1612, 1450, 1380, 1272, 1179, 1074, 1020, 790, 752, 691, 475. For NMR data, see Table Ia, Table Ibb.
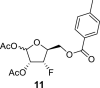
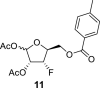
4.4. 1,2-Di-O-acetyl-3-deoxy-3-fluoro-5-O-(4-methylbenzoyl)-l-xylofuranose (11)
H2SO4 (1 mL), diluted with 20 mL of Ac2O, was added dropwise to a solution of compound 10 (49 g; 172 mmol) in AcOH (80 mL) and Ac2O (160 mL). The mixture was stirred at room temperature for 2 h, and then treated with water (200 mL). Then, the reaction mixture was neutralized with 10 g of NaHCO3 and concentrated. The crude mixture was extracted (1 L DCM/500 mL sat. NaCl and 2 × 500 mL of sat. NaHCO3). The organic phase was dried over MgSO4, and product 11 was isolated by chromatography on a silica gel (0–10% EtOAc in toluene) as a mixture of both anomers in a yield of 51 g (84%). HRMS (M + Na)+ for C17H19O7FNa calculated: 377.10070; measured: 377.10126; IR (coating, cm−1): 1751, 1723, 1612, 1454, 1374, 1310, 1274, 1239, 1179, 1109, 1061, 1047, 883, 790, 667, 602, 474. For NMR data, see Table Ia, Table Ibb.
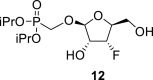
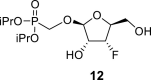
4.5. Diisopropylphosphonomethyl 3-deoxy-3-fluoro-β-l-ribofuranoside (12)
Hexamethyldisilazane (100 mL) and a catalytic amount of saccharin were added to diisopropyl (hydroxymethyl)phosphonate (30.4 g; 190 mmol), and the reaction mixture was stirred for 8 h at 100 °C. Hexamethyldisilazane was evaporated, and the residue was diluted with toluene and co-evaporated (3 × 100 mL). Then the compound 11 (34.4 g; 97 mmol) was added to silylated diisopropyl (trimethylsilyloxymethyl)phosphonate in ACN (1 L). Finally, SnCl4 (24 mL; 200 mmol) was added. The reaction mixture was stirred for 1 h at 55 °C, quenched with 65 mL of pyridine, filtered and concentrated. Next, the crude mixture was dissolved in 800 mL of 33% MeNH2 in EtOH, stirred for 8 h and concentrated. Product 12 was isolated by chromatography on a silica gel (0–10% EtOH in CHCl3) in a yield of 26.9 g (84%). HRMS (M + Na)+ for C12H24O7FNaP calculated: 353.11359; measured: 353.11368; IR (coating, cm-1): 3340, 1468, 1387, 1376, 1238, 1179, 1105, 1054, 999. For NMR data, see Table Ia, Table Ibb.
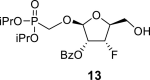
4.6. Diisopropylphosphonomethyl 2-O-benzoyl-3-deoxy-3-fluoro-β-l-ribofuranoside (13)
DMTr-Cl (14.2 g; 40 mmol) was added to a solution of phosphonate 12 (12.7 g; 38 mmol) in 400 mL of pyridine, and the reaction mixture was stirred at room temperature for 8 h. Next, BzCl (6.6 mL; 57 mmol) and DMAP (3 g; 24.6 mmol) were added. The reaction mixture was stirred for 8 h at room temperature, treated with water (100 mL), evaporated and extracted (1 L toluene/500 mL sat. NaCl and 2 × 500 mL of sat. NaHCO3). The organic phase was dried over MgSO4, filtered, concentrated, diluted with 400 mL of dry DCM, and treated with TFA (5 mL) for 10 min at room temperature. The solution was neutralized by the addition of 20 mL of mixture MeOH/Et3N (1:3) and concentrated. Product 13 was isolated by chromatography on a silica gel (0–90% EtOAc in toluene) in a yield of 12.2 g (74%). HRMS (M + Na)+ for C19H28O8FNaP calculated: 457.13980; measured: 457.13949; IR (coating, cm-1): 3372, 2981, 1730, 1602, 1585, 1491, 1452, 1387, 1376, 1316, 1273, 1179, 1142, 1062, 998, 891, 857, 713, 673. For NMR data, see Table Ia, Table Ibb.
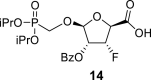
4.7. (2S,3R,4R,5R)-4-(benzoyloxy)-5-((diisopropoxyphosphoryl)methoxy)-3-fluorotetrahydrofuran-2-carboxylic acid (14)
BAIB (18 g; 56 mmol) and a catalytic amount of TEMPO were added to a solution of 13 (12.2 g; 28 mmol) in 50% ACN/water, and the reaction mixture was stirred for 16 h at room temperature. The reaction was quenched by the addition of 100 mL of EtOH, concentrated and adsorbed onto a silica gel. Product 14 was isolated by chromatography on a silica gel (0–100% EtOAc in cyclohexane), and then eluted by 20% MeOH in CHCl3 in a yield of 12.5 g (quantitative). HRMS (M + Na)+ for C19H26O9FNaP calculated: 471.11907; measured: 471.11857; IR (coating, cm−1): 3159, 2591, 1732, 1695, 1602, 1583, 1452, 1416, 1388, 1377, 1316, 1276, 1270, 1179, 1105, 1006, 938, 893, 850, 712, 677. For NMR data, see Table Ia, Table Ibb.
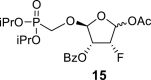
4.8. (2R,3R,4R)-5-acetoxy-2-((diisopropoxyphosphoryl)methoxy)-4-fluorotetrahydrofuran-3-yl benzoate (15)
Pb(OAc)4 (25 g, 56 mmol) was added to a solution of 14 in THF (300 mL), and the reaction mixture was stirred for 8 h at room temperature. The resulting suspension was filtered and concentrated, and the acetate 15 was isolated by chromatography on a silica gel (0–50% EtOAc in toluene) in a yield of 7.8 g (60%). HRMS (M + Na)+ for C20H28O9FNaP calculated: 485.13472; measured: 485.13416; IR (coating, cm−1): 2981, 1755, 1733, 1602, 1585, 1493, 1453, 1386, 1376, 1317, 1272, 1246, 1179, 1106, 1025, 1006, 990, 889, 846, 713, 673, 603. For NMR data, see Table Ia, Table Ibb.
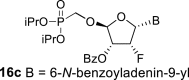
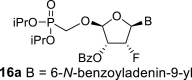
4.9. (2R,3R,4R,5R)-5-(6-benzamido-9H-purin-9-yl)-2-((diisopropoxyphosphoryl)methoxy)-4-fluorotetrahydrofuran-3-yl benzoate (16a)
Bis(trimethylsilyl)acetamide (7.3 mL; 30 mmol) was added to N-benzoyladenine (3.1 g; 13 mmol) in 1,2-dichloroethane (100 mL), and the reaction mixture was stirred for 1 h at 60 °C. The mixture was concentrated, co-evaporated with dry toluene (2 × 50 mL), added to the acetate 15 (3 g, 6.5 mmol) in 1,2-dichloroethane (100 mL) and finally SnCl4 (6 mL; 52 mmol) was added. The reaction was stirred for 1 h at room temperature. Next, it was cooled in ice bath, quenched by the addition 16 mL of pyridine, filtered and concentrated. The reaction afforded mixture of compounds 16a and 16c in a 2:1 ratio. The product 16a was isolated by chromatography on a silica gel (25–35% acetone in toluene) in a yield of 1.8 g (43%) as a faster eluting isomer. HRMS (M + Na)+ for C30H33O8N5FNaP calculated: 664.19430; measured: 664.19346; IR (coating, cm-1): 3068, 3032, 1700, 1603, 1582, 1512, 1490, 1452, 14101335, 1317, 1274, 1247, 1220, 1178, 1119, 1106,1080, 1031, 989, 904, 853, 797, 754, 711, 687, 646, 506. For NMR data, see Table IIa, Table IIb b.
Table IIa.
1H NMR data of compounds 5, 6, 16 and 22–27x
| Compound | Solvent | H-1‘ | H-2‘ | H-3‘ | H-4‘ | Base |
|---|---|---|---|---|---|---|
 |
D2O a | 6.44 dd 1,2 = 4.3 1,F = 14.6 |
5.71 dt 2,F = 50.8 2,1 = 4.3 2,3 = 4.4 |
4.59 ddd 3,2 = 4.4 3,F = 7.1 3,4 = 2.0 |
5.33 dd 4,3 = 2.0 4,F = 1.7 |
H-2: 8.15 s H-8: 8.33 s |
 |
D2O b | 6.31 dd 1,2 = 4.2 1,F = 15.4 |
5.72 dt 2,F = 51.1 2,1 = 4.2 2,3 = 4.4 |
4.62 ddd 3,2 = 4.4 3,F = 8.1 3,4 = 2.3 |
5.32 dd 4,3 = 2.3 4,F = 1.6 |
H8: 8.06 s |
 |
DMSO c | 6.74 dd 1,2 = 3.6 1,F = 17.0 |
6.35 ddd 2,1 = 3.6 2,F = 49.8 2,3 = 4.9 |
5.91 ddd 3,2 = 4.9 3,F = 9.2 3,4 = 2.5 |
5.71 dd 4,3 = 2.5 4,F = 1.0 |
H-2: 8.80 s H-8: 8.65 s |
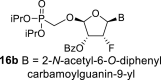 |
DMSO d | 6.59 dd 1,2 = 3.2 1,F = 17.6 |
6.31 ddd 2,1 = 3.2 2,F = 49.9 2,3 = 5.0 |
6.02 ddd 3,2 = 5.0 3,F = 10.3 3,4 = 2.7 |
5.70 dd 4,3 = 2.7 4,F = 0.6 |
H-8: 8.59 s |
 |
DMSO e | 6.995 dd 1,2 = 3.7 1,F = 16.8 |
5.79 ddd 2,1 = 3.7 2,F = 52.2 2,3 = 4.5 |
5.72 ddd 3,2 = 4.5 3,F = 15.3 3,4 = 3.5 |
5.95 dd 4,3 = 3.5 4,F = 1.3 |
H-2: 8.81 s H-8: 8.60 d 8,F = 2.7 |
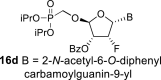 |
DMSO f | 6.82 dd 1,2 = 3.8 1,F = 16.2 |
5.74 ddd 2,1 = 3.8 2,F = 52.1 2,3 = 4.7 |
5.67 ddd 3,2 = 4.7 3,F = 16.1 3,4 = 3.9 |
6.08 dd 4,3 = 3.9 4,F = 1.2 |
H-8: 8.55 d 8,F = 2.5 |
 |
D2O g | 6.02 d 1,2 = 6.5 |
4.97 ddd 2,1 = 6.5 2,3 = 4.4 2,4 = 0.6 |
4.37 dt 3,2 = 4.4 3,4 = 0.6 3,P = 0.6 |
5.21 t 4,3 = 0.6 4,2 = 0.6 |
H-8: 8.09 s |
 |
D2O h | 6.42 d 1,2 = 5.8 |
5.55 dd 2,1 = 5.8 2,3 = 4.6 |
4.52 dd 3,2 = 4.6 3,4 = 0.7 |
5.31 d 4,3 = 0.7 |
H-2: 8.34 s |
 |
D2O i | 6.20 d 1,2 = 6.1 |
5.39 dd 2,1 = 6.1 2,3 = 4.6 |
4.46 dd 3,2 = 4.6 3,4 = 0.8 |
5.27 d 4,3 = 0.8 |
– |
 |
DMSO j | 6.87 d 1,2 = 5.7 |
6.51 dd 2,1 = 5.7 2,3 = 5.0 |
5.96 dd 3,2 = 5.0 3,4 = 1.1 |
5.70 d 4,3 = 1.1 |
H-2: 8.78 s H-8: 8.71 s |
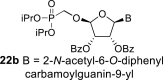 |
DMSO k | 6.71 d 1,2 = 5.7 |
6.46 dd 2,1 = 5.7 2,3 = 5.0 |
5.94 dd 3,2 = 5.0 3,4 = 1.2 |
5.68 d 4,3 = 1.2 |
H-8: 8.645 s |
 |
DMSO l | 6.245 d 1,2 = 6.4 |
5.22 td 2,1 = 6.4 2,3 = 4.4 2, OH = 6.7 |
4.15 td 3,2 = 4.4 3,4 = 1.1 3, OH = 4.5 |
5.68 d 4,3 = 1.1 |
H-2: 8.33 s NH2: 8.53 br and 8.18 br |
 |
DMSO m | 6.59 d 1,2 = 4.7 |
6.52 t 2,1 = 4.7 2,3 = 4.8 |
5.93 dd 3,2 = 4.7 3,4 = 2.1 |
5.72 d 4,3 = 2.1 |
CO–NH: 11.15 bs |
 |
DMSO n | 6,94 d 1,2 = 4.0 |
6.56 dd 2,1 = 4.0 2,3 = 5.1 |
5.95 dd 3,2 = 5.1 3,4 = 2.3 |
5.765 d 4,3 = 2.3 |
CO–NH: 11.47 bs NH2: 6.60 br |
 |
DMSO o | 6.95 d 1,2 = 4.2 |
6.70 q 2,1 = 4.2 2,3 = 5.1 |
6.03 dd 3,2 = 5.1 3,4 = 2.2 |
5.76 d 4,3 = 2.2 |
H-2: 8.36 s NH2: 8.67 br and 8.26 br |
 |
DMSO p | 7.14 d 1,2 = 6.4 |
6.18 t 2,1 = 6.4 2,3 = 6.0 |
5.73 dd 3,2 = 6.0 3,4 = 1.3 |
6.10 d 4,3 = 1.3 |
H-2: 8.295 s NH2: 8.37 br and 8.15 br |
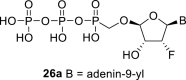 |
D2O q | 6.52 dd 1,2 = 4.3 1,F = 14.5 |
5.78 dt 2,F = 50.8 2,1 = 4.3 2,3 = 4.5 |
4.65 ddd 3,2 = 4.5 3,4 = 1.8 3,F = 7.0 |
5.40 t 4,3 = 1.8 4,F = 1.8 |
H-2: 8.24 s H-8: 8.42 s |
 |
D2O r | 6.32 dd 1,2 = 4.5 1,F = 15.1 |
5.77 dt 2,F = 51.0 2,1 = 4.5 2,3 = 4.5 |
4.66 ddd 3,2 = 4.5 3,4 = 1.8 3,F = 6.7 |
5.36 t 4,3 = 1.8 4,F = 1.9 |
H-8: 8.05 s |
 |
D2O s | 6.04 d 1,2 = 6.7 |
5.01 ddd 2,1 = 6.7 2,3 = 4.5 2,4 = 0.5 |
4.42 dt 3,2 = 4.5 3,4 = 0.6 3,P = 0.6 |
5.27 t 4,3 = 0.6 4,2 = 0.5 |
H-8: 8.09 s |
 |
D2O t | 6.43 d 1,2 = 6.2 |
5.56 dd 2,1 = 6.2 2,3 = 4.6 |
4.555 dd 3,2 = 6.2 3,4 = 0.9 |
5.395 d 4,3 = 0.9 |
H-2: 8.37 s |
 |
D2O u | 6.235 d 1,2 = 6.6 |
5.43 dd 2,1 = 6.6 2,3 = 4.6 |
4.49 bd 3,2 = 4.6 3,4 < 1.0 |
5.34 bs 4,3 < 1.0 |
– |
 Mixture of diastereomers 53:47 |
DMSO v Major |
6.39 dd 1,2 = 5.0 1,F = 14.7 |
5.81 dt 2,F = 51.3 2,1 = 5.0 2,3 = 4.5 |
4.37 qd 3,2 = 4.5 3,4 = 1.7 3,F = 5.0 3, OH = 5.0 |
5.16 t 4,3 = 1.7 4,F = 1.7 |
H-2: 8.17 s H-8: 8.25 s |
| Minor | 6.38 dd 1,2 = 5.0 1,F = 14.7 |
5.78 dt 2,F = 51.3 2,1 = 5.0 2,3 = 4.5 |
4.355 qd 3,2 = 4.5 3,4 = 1.7 3,F = 5.0 3, OH = 5.0 |
5.02 t 4,3 = 1.7 4,F = 1.7 |
H-2: 8.17 s H-8: 8.21 s |
|
 Mixture of diastereomers 57:43 |
DMSO w Major |
6.175 dd 1,2 = 5.4 1,F = 14.3 |
5.605 dt 2,F = 51.6 2,1 = 5.4 2,3 = 4.3 |
4.27 qd 3,2 = 4.3 3,4 = 1.7 3,F = 5.0 3, OH = 5.1 |
5.11 t 4,3 = 1.7 4,F = 1.6 |
H-8: 7.79 s |
| minor | 6.16 dd 1,2 = 5.6 1,F = 14.2 |
5.58 dt 2,F = 51.7 2,1 = 5.6 2,3 = 4.3 |
4.25 qd 3,2 = 4.3 3,4 = 1.6 3,F = 5.0 3, OH = 5.1 |
4.96 t 4,3 = 1.6 4,F = 1.5 |
H-8: 7.77 s |
Substituents.
O–CH2–P = O(OH)2: 3.81 dd, J = 12.9, 8.9 Hz and 3.62 dd, J = 12.9, 9.7 Hz.
O–CH2–P = O(OH)2: 3.76 m and 3.53 m.
3-OBz: 8.10 m (2x o-ArH), 7.62 m (2x m-ArH), 7.465 m (p-ArH); O–CH2–P = O(OiPr)2: 3.97 dd, J = 13.9; 9.1 Hz and 3.92 dd, J = 13.9; 9.1 Hz (P–CH2–O), 4.54 m (2x O–CH<), 1.204 d, J = 6.2 Hz, 1.207 d, J = 6.2 Hz, 1.233 d, J = 6.2 Hz, 1.238 d, J = 6.2 Hz (2x OiPr); NHBz: 11.30 br (NH), 8.05 m (2x o-ArH), 7.56 m (2x m-ArH), 7.56 m (p-ArH).
NHAc: 10.82 s (NH), 2.18 s (CH3); 3-OBz: 8.07 m (2x o-ArH), 7.61 m (2x m-ArH), 7.75 m (p-ArH); O–CH2–P = O(OiPr)2: 3.95 d, J = 9.5 Hz (P–CH2–O), 4.57 m (2x O–CH<), 1.121 d, J = 6.2 Hz, 1.156 d, J = 6.2 Hz, 1.160 d, J = 6.2 Hz, 1.186 d, J = 6.2 Hz (2x OiPr); O–CO–N(C6H5)2: 7.51 m (4x o-ArH), 7.45 m (4x m-ArH), 7.33 (2x p-ArH).
3-OBz: 8.05 m (2x o-ArH), 7.60 m (2x m-ArH), 7.735 m (p-ArH); O–CH2–P = O(OiPr)2: 4.08 dd, J = 14.0; 9.2 Hz and 4.02 dd, J = 14.0; 9.4 Hz (P–CH2–O), 4.64 m (2x O–CH<), 1.234 d, J = 6.2 Hz, 1.240 d, 6H, J = 6.2 Hz and 1.254 d, J = 6.2 Hz (2x OiPr); NHBz: 11.29 bs (NH), 8.055 m (2x o-ArH), 7.56 m (2x m-ArH), 7.65 (p-ArH).
3-OBz: 8.00 m (2x o-ArH), 7.57 m (2x m-ArH), 7.72 m (p-ArH); O–CH2–P = O(OiPr)2: 4.05 dd, J = 13.9; 9.0 Hz and 4.035 dd, J = 13.9; 9.1 Hz (P–CH2–O), 4.63 m (2x O–CH<), 1.214 d, J = 6.2 Hz, 1.225 d, J = 6.2 Hz, 1.226 d, J = 6.2 Hz and 1.239 d, J = 6.2 Hz (2x OiPr); NHAc: 10.76 s (NH), 2.23 s (CH3); O–CO–N(C6H5)2: 7.49 br m (4x o-ArH), 7.44 m (4x m-ArH), 7.32 (2x p-ArH).
O–CH2–P = O(OH)2: 3.75 dd, J = 12.8, 8.6 Hz and 3.50 dd, J = 12.8, 9.8 Hz.
O–CH2–P = O(OH)2: 3.62 dd, J = 13.4, 8.4 Hz and 3.50 dd, J = 13.4, 9.5 Hz.
O–CH2–P = O(OH)2: 3.67 dd, J = 13.1, 8.1 Hz and 3.46 dd, J = 13.1, 9.7 Hz.
3-OBz: 7.82 m (2x o-ArH), 7.43 m (2x m-ArH), 7.625 m (p-ArH); 3-OBz: 8.045 m (2x o-ArH), 7.57 m (2x m-ArH), 7.72 m (p-ArH); O–CH2–P = O(OiPr)2: 3.98 m (P–CH2–O), 4.65 m (2x O–CH<), 1.247 d, J = 6.2 Hz, 1.255 d, J = 6.2 Hz, 1.266 d, 6H, J = 6.4 Hz (2x OiPr); NHBz: 11.27 br (NH), 8.055 m (2x o-ArH), 7.555 m (2x m-ArH), 7.65 m (p-ArH).
2-OBz: 7.80 m (2x o-ArH), 7.41 m (2x m-ArH), 7.61 m (p-ArH); 3-OBz: 8.005 m (2x o-ArH), 7.56 m (2x m-ArH), 7.71 m (p-ArH); O–CH2–P = O(OiPr)2: 3.97 d, J = 9.4 Hz (P–CH2–O), 4.65 m (2x O–CH<), 1.236 d, J = 6.2 Hz, 1.256 d, J = 6.2 Hz, 1.263 d, J = 6.0 Hz and 1.273 d, J = 6.0 Hz (2x OiPr); NHAc: 10.76 s (NH), 2.215 s (CH3); O–CO–N(C6H5)2: 7.505 m (4x o-ArH), 7.44 m (4x m-ArH), 7.32 (2x p-ArH).
O–CH2–P = O(OH)2: 3.69 d, 2H, J = 8.8 Hz; 2-OH: 5.66 d, J = 6.7 Hz; 3-OH: 5.73 d, J = 4.5 Hz.
2-OBz: 7.97 m (2x o-ArH), 7.53 m (2x m-ArH), 7.70 m (p-ArH); 3-OBz: 7.85 m (2x o-ArH), 7.45 m (2x m-ArH), 7.64 m (p-ArH); O–CH2–P = O(OiPr)2: 3.88 d, 2H, J = 9.0 Hz (P–CH2–O), 4.58 m (2x O–CH<), 1.196 d, J = 6.2 Hz, 1.209 d, J = 6.2 Hz, 1.222 d, J = 6.2 Hz and 1.235 d, J = 6.2 Hz (2x OiPr).
2-OBz: 7.94 m (2x o-ArH), 7.51 m (2x m-ArH), 7.69 m (p-ArH); 3-OBz: 7.87 m (2x o-ArH), 7.46 m (2x m-ArH), 7.65 m (p-ArH); O–CH2–P = O(OiPr)2: 3.88 dd, J = 13.9; 9.4 Hz and 3.80 dd, J = 13.9; 8.5 Hz, (P–CH2–O), 4.56 m (2x O–CH<), 1.165 d, J = 6.2 Hz 1.196 d, J = 6.2 Hz, 1.202 d, J = 6.2 Hz and 1.208 d, J = 6.2 Hz (2x OiPr).
2-OBz: 7.86 m (2x o-ArH), 7.44 m (2x m-ArH), 7.645 m (p-ArH); 3-OBz: 8.00 m (2x o-ArH), 7.52 m (2x m-ArH), 7.70 m (p-ArH); O–CH2–P = O(OiPr)2: 3.88 dd, J = 13.9; 9.4 Hz and 3.78 dd, J = 13.9; 8.6 Hz, (P–CH2–O), 4.55 m (2x O–CH<), 1.141 d, J = 6.2 Hz, 1.185 d, J = 6.2 Hz, 1.187 d, J = 6.2 Hz and 1.202 d, J = 6.2 Hz (2x OiPr).
2-OBz: 7.24 m (2x o-ArH), 7.15 m (2x m-ArH), 7.46 m (p-ArH); 3-OBz: 8.00 m (2x o-ArH), 7.45 m (2x m-ArH), 7.64 m (p-ArH); O–CH2–P = O(OiPr)2: 4.14 dd, J = 14.0; 9.0 Hz and 4.12 dd, J = 14.0; 8.8 Hz, (P–CH2–O), 4.68 m (2x O–CH<), 1.282 d, J = 6.2 Hz, 1.285 d, J = 6.2 Hz and 1.290 d, 6H, J = 6.2 Hz (2x OiPr).
O–CH2–P( = O)(OH)–O–P( = O)(OH)–O–P( = O)(OH)2: 3.99 dd, J = 13.3; 8.5 Hz and 3.89 dd, J = 13.2; 10.1 Hz (P–CH2–O).
O–CH2–P( = O)(OH)–O–P( = O)(OH)–O–P( = O)(OH)2: 3.98 dd, J = 13.3; 8.4 Hz and 3.86 dd, J = 13.3; 10.3 Hz (P–CH2–O).
O–CH2–P( = O)(OH)–O–P( = O)(OH)–O–P( = O)(OH)2: 3.95 dd, J = 13.3; 8.2 Hz and 3.82 dd, J = 13.3; 10.3 Hz (P–CH2–O).
O–CH2–P( = O)(OH)–O–P( = O)(OH)–O–P( = O)(OH)2: 3.775 dd, J = 13.8; 7.7 Hz and 3.75 dd, J = 13.8; 9.1 Hz (P–CH2–O).
O–CH2–P( = O)(OH)–O–P( = O)(OH)–O–P( = O)(OH)2: 3.875 dd, J = 13.6; 7.4 Hz and 3.765 dd, J = 13.6; 9.9 Hz (P–CH2–O).
Major diastereomer: 3-OH: 6.165 d, J = 5.0 Hz; Bu-O-CH(CH2C6H5)–CO–NH–P( = O)(O–C6H5)–CH2–O: 0.81 t, 3H, J = 7.4 Hz, 1.18 m, 2H, 1.39 m, 2H, 3.92 m, 2H (Bu-O-); 4.05 m, 1H, 2.94 m, 1H and 2.735 m, 1H (O–CH–CH2); 7.38 br, 1H (NH); 7.00–7.30 m, 10H (10x ArH); 3.45 dd, 1H, J = 13.5; 8.8 Hz and 3.405 dd 1H, J = 13.5; 8.2 Hz (P–CH2–O); minor diastereomer: 3-OH: 6.155 d, J = 5.0 Hz; Bu-O-CH(CH2C6H5)–CO–NH–P( = O)(O–C6H5)–CH2–O: 0.785 t, 3H, J = 7.4 Hz, 1.18 m, 2H, 1.39 m, 2H, 3.90 m, 2H (Bu-O-); 4.07 m, 1H, 2.91 m, 1H and 2.72 m, 1H (O–CH–CH2); 7.395 br, 1H (NH); 7.00–7.30 m, 10H (10x ArH); 3.84 dd, 1H, J = 13.7; 8.8 Hz and 3.69 dd 1H, J = 13.7; 7.0 Hz (P–CH2–O).
Major diastereomer: 3-OH: 6.13 d, J = 5.1 Hz; Bu-O-CH(CH2C6H5)–CO–NH–P( = O)(O–C6H5)–CH2–O: 0.805 t, 3H, J = 7.4 Hz, 1.195 m, 2H, 1.41 m, 2H, 3.93 m, 2H (Bu-O-); 4.07 m, 1H, 2.915 ddd, 1H, J = 13.6; 6.0; 2.0 Hz and 2.72 dd, 1H, J = 13.6; 8.5 Hz (O–CH–CH2); 7.38 br, 1H (NH); 7.025–7.275 m, 10H (10x ArH); 3.88 dd, 1H, J = 13.5; 9.1 Hz and 3.695 dd 1H, J = 13.5; 7.2 Hz (P–CH2–O); minor diastereomer: 3-OH: 6.12 d, J = 5.1 Hz; Bu-O-CH(CH2C6H5)–CO–NH–P( = O)(O–C6H5)–CH2–O: 0.805 t, 3H, J = 7.4 Hz, 1.195 m, 2H, 1.41 m, 2H, 3.905 m, 2H (Bu-O-); 4.07 m, 1H, 2.96 ddd, 1H, J = 13.5; 6.2; 1.5 Hz and 2.75 dd, 1H, J = 13.5; 9.1 Hz (O–CH–CH2); 7.395 br, 1H (NH); 7.025–7.275 m, 10H (10x ArH); 3.46 dd, 1H, J = 13.5; 8.3 Hz and 3.43 dd, 1H, J = 13.5; 8.9 Hz (P–CH2–O).
Coupling constants are written in italics in a shortened form (e.g. instead J(1‘,2‘) = 4.3 Hz we type simply 1,2 = 4.3).
Table IIb.
13C and31P NMR data of compounds 5, 6, 16 and 22–27x
| Compound | Solvent | C-1‘ | C-2‘ | C-3‘ | C-4‘ | Base | 31P | 19F |
|---|---|---|---|---|---|---|---|---|
 |
D2O a | 88.14 1,F = 34.0 |
96.60 2,F = 192.0 |
75.51 3,F = 15.1 |
112.10 4,P = 12.1 4,F = 3.2 |
C-2: 155.51 C-4: 151.41 C-5: 121.14 C-6: 158.18 C-8: 142.68 |
14.93 | −210.06 |
 |
D2O b | 87.78 | 96.18 | 75.27 | 111.87 | C-2: n.d. C-4: 154.37 C-5: 118.81 C-6: n.d. C-8: 140.52 |
14.15 | −209.59 |
 |
DMSO c | 86.31 1,F = 34.4 |
91.24 2,F = 193.1 |
74.45 3,F = 14.0 |
106.69 4,P = 12.4 4,F = 1.1 |
C-2: 152.62 C-4: 152.14 C-5: 125.77 C-6: 150.94 C-8: 143.32 |
18.29 | −204.85 |
 |
DMSO d | 86.57 1,F = 15.0 |
91.14 2,F = 192.6 |
74.33 3,F = 13.9 |
106.95 4,P = 13.2 |
C-2: 150.19 C-4: 154.35 C-5: 120.32 C-6: 152.57 C-8: 144.37 |
18.93 | −203.23 |
 |
DMSO e | 82.10 1,F = 15.6 |
88.90 2,F = 197.1 |
74.78 3,F = 14.0 |
105.04 4,P = 11.7 |
C-2: 152.33 C-4: 152.13 C-5: 124.96 C-6: 150.75 C-8: 142.98 8,F = 5.5 |
19.18 | −207.71 |
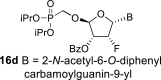 |
DMSO f | 82.59 1,F = 15.7 |
88.63 2,F = 197.5 |
74.61 3,F = 16.1 |
105.42 4,P = 12.2 |
C-2: 150.19 C-4: 154.55 C-5: 119.67 C-6: 152.75 C-8: 144.05 8,F = 5.1 |
19.09 | −206.65 |
 |
D2O g | 89.27 | 77.15 | 76.56 | 111.95 4,P = 11.8 |
C-2: 156.79 C-4: 154.74 C-5: 118.76 C-6: 161.68 C-8: 140.52 |
14.35 | – |
 |
D2O h | 91.41 | 75.65 | 76.69 | 112.28 4,P = 11.0 |
C-2: 159.86 C-4: 152.00 C-5: 127.19 C-6: 158.94 |
15.93 | – |
 |
D2O i | 91.09 | 76.23 | 76.57 | 112.17 4,P = 10.9 |
C-2: 158.82 C-4: 155.02 C-5: 127.40 C-6: 160.97 |
15.38 | – |
 |
DMSO j | 85.82 | 74.08 | 74.44 | 106.20 4,P = 12.4 |
C-2: 152.16 C-4: 152.38 C-5: 125.72 C-6: 150.80 C-8: 149.09 |
19.16 | – |
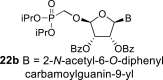 |
DMSO k | 86.12 | 73.82 | 74.43 | 106.34 4,P = 13.4 |
C-2: 150.23 C-4: 154.72 C-5: 120.36 C-6: 152.70 C-8: 144.24 |
19.42 | – |
 |
DMSO l | 88.35 | 72.46 | 73.92 | 108.70 4,P = 11.8 |
C-2: 157.39 C-4: 149.95 C-5: 124.14 C-6: 156.38 |
19.77 | – |
 |
DMSOm | 86.13 | 73.22 | 74.63 | 106.54 4,P = 12.9 |
C-2: 155.69 C-4: 152.30 C-5: 124.66 C-6: 156.25 |
19.06 | – |
 |
DMSO n | 89.10 | 73.65 | 74.75 | 107.15 4,P = 12.4 |
C-2: 154.29 C-4: 153.72 C-5: 113.66 C-6: 161.55 |
18.70 | – |
 |
DMSO o | 86.94 | 73.36 | 74.41 | 106.97 4,P = 12.5 |
C-2: 157.61 C-4: 149.56 C-5: 124.12 C-6: 156.46 |
18.20 | – |
 |
DMSO p | 91.99 | 70.82 | 72.63 | 107.40 4,P = 11.7 |
C-2: 157.80 C-4: 157.12 C-5: 126.12 C-6: 158.00 |
n.d. | – |
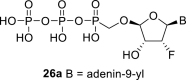 |
D2O q | 88.24 1,F = 34.1 |
96.51 2,F = 192.3 |
75.42 3,F = 14.9 |
112.14 4,P = 12.2 4,F = 3.2 |
C-2: 155.49 C-4: 151.68 C-5: 121.36 C-6: 158.30 C-8: 142.89 |
Pα: 8.39 α,β = 25.6 Pβ: 22.10 β,α = 25.6 β,γ = 19.7 Pγ: 9.48 γ,β = 19.7 |
−210.72 |
 |
D2O r | 87.85 1,F = 33.9 |
95.38 2,F = 191.8 |
75.11 3,F = 14.9 |
111.72 4,P = 12.0 4,F = 3.2 |
C-2: 156.71 C-4: 154.34 C-5: 118.85 C-6: 161.67 C-8: 140.50 |
Pα: 8.62 α,β = 25.7 Pβ: 22.02 β,α = 25.7 β,γ = 16.7 Pγ: 9.60 γ,β = 16.7 |
−210.89 |
 |
D2O s | 89.36 | 76.78 | 76.41 | 111.82 4,P = 12.0 |
C-2: 156.79 C-4: 154.81 C-5: 118.84 C-6: 161.75 C-8: 140.59 |
Pα: 8.36 α,β = 25.8 Pβ: 22.51 β,α = 25.8 β,γ = 19.6 Pγ: 9.82 γ,β = 19.6 |
– |
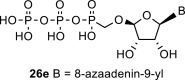 |
D2O t | 91.38 | 75.54 | 76.52 | 112.34 4,P = 9.8 |
C-2: 159.95 C-4: 152.13 C-5: 127.33 C-6: 159.08 |
Pα: 9.06 α,β = 26.2 Pβ: 22.03 β,α = 26.2 β,γ = 19.4 Pγ: 9.27 γ,β = 19.4 |
– |
 |
D2O u | 91.34 | 74.71 | 76.23 | 111.88 4,P = 10.5 |
C-2: 160.82 C-4: 154.82 C-5: 127.39 C-6: 158.54 |
Pα: 9.21 α,β = 26.2 Pβ: 22.30 β,α = 26.2 β,γ = 19.4 Pγ: 9.84 γ,β = 19.4 |
– |
 Mixture of diastereomers 53:47 |
DMSO v Major |
84.73 1,F = 33.0 |
93.16 2,F = 191.1 |
72.35 3,F = 15.0 |
108.30 4,P = 10.8 4,F = 4.1 |
C-2: 153.24 C-4: 149.67 C-5: 118.93 C-6: 156.33 C-8: 139.06 |
23.18 | −211.17 |
| Minor | 84.63 1,F = 33.2 |
93.30 2,F = 191.2 |
72.30 3,F = 15.0 |
108.38 4,P = 11.8 4,F = 3.2 |
C-2: 153.24 C-4: 149.68 C-5: 118.88 C-6: 156.33 C-8: 139.03 |
22.74 | −211.28 | |
 Mixture of diastereomers 57:43 |
DMSOw Major |
84.14 1,F = 32.5 |
93.37 2,F = 191.7 |
72.22 3,F = 15.0 |
108.00 4,P = 4.2 |
C-2: 154.22 C-4: 151.52 C-5: 116.69 C-6: 156.85 C-8: 135.00 |
23.29 | −212.34 |
| minor | 84.11 1,F = 32.5 |
93.20 2,F = 191.6 |
72.16 3,F = 15.0 |
108.08 4,P = 4.2 |
C-2: 154.19 C-4: 151.48 C-5: 116.78 C-6: 156.88 C-8: 135.23 |
22.95 | −212.81 |
Substituents.
O–CH2–P = O(OH)2: 68.13 d, J = 155.9 Hz.
O–CH2–P = O(OH)2: 68.59.
O–CH2–P = O(OiPr)2: 62.62 d, J = 166.0 Hz (P–CH2–O), 71.02 d, J = 6.3 Hz (2x O–CH<), 23.97 d, J = 3.5 Hz, 23.99 d, J = 3.5 Hz, 24.04 d, J = 4.4 Hz, 24.12 d, J = 3.8 Hz (2x OiPr); 3-OBz: 164.78 (C O), 128.50 (i-ArC), 129.86 (2x o-ArC), 129.32 (2x m-ArC), 134.52 (p-ArC); NHBz: 165.98 (C O), 133.55 (i-ArC), 128.78 (2x o-ArC and 2x m-ArC), 132.82 (p-ArC).
NHAc: 168.71 (C O), 24.71 (CH3); 3-OBz: 164.45 (C O), 128.53 (i-ArC), 129.70 (2x o-ArC), 129.22 (2x m-ArC), 134.35 (p-ArC); O–CH2–P = O(OiPr)2: 62.24 d, J = 166.3 Hz (P–CH2–O), 70.85 d, J = 6.3 Hz (2x O–CH<), 23.69 d, J = 6.3 Hz, 23.72 d, J = 4.4 Hz, 23.86 d, J = 4.0 Hz, 23.89 d, J = 4.0 Hz (2x OiPr); O–CO–N(C6H5)2: 155.54 (C O), 141.75 (2x i-ArC), 126.67 (4x o-ArC), 129.61 (4x m-ArC), 127.10 (2x p-ArC).
O–CH2–P = O(OiPr)2: 62.31 d, J = 165.8 Hz (P–CH2–O), 70.80 d, J = 6.2 Hz and 70.81 d, J = 6.2 Hz (2x O–CH<), 23.84 d, J = 4.6 Hz, 23.85 d, J = 4.4 Hz and 23.98 d, 2C, J = 3.8 Hz (2x OiPr); 3-OBz: 164.64 (C O), 128.42 (i-ArC), 129.72 (2x o-ArC), 129.23 (2x m-ArC), 134.40 (p-ArC); 6-NHBz: 165.85 (C O), 133.45 (i-ArC), 128.71 (2x o-ArC), 128.68 (2x m-ArC), 132.72 (p-ArC).
O–CH2–P = O(OiPr)2: 62.47 d, J = 166.0 Hz (P–CH2–O), 70.77 d, J = 5.9 Hz (2x O–CH<), 23.79 d, J = 4.5 Hz, 23.81 d, J = 4.5 Hz and 23.95 d, 2C, J = 3.7 Hz (2x OiPr); 3-OBz: 164.62 (C O), 128.40 (i-ArC), 129.67 (2x o-ArC), 129.17 (2x m-ArC), 134.34 (p-ArC); NHAc: 169.19 (C O), 24.86 (CH3); O–CO–N(C6H5)2: 155.45 (C O), 141.71 (2x i-ArC), 127.17 (4x o-ArC), 129.60 (4x m-ArC), 127.53 (2x p-ArC).
O–CH2–P = O(OH)2: 67.95 d, J = 154.5 Hz.
O–CH2–P = O(OH)2: 66.72 d, J = 156.7 Hz.
O–CH2–P = O(OH)2: 67.37 d, J = 154.8 Hz.
O–CH2–P = O(OiPr)2: 62.09 d, J = 166.3 Hz (P–CH2–O), 70.91 d, J = 6.3 Hz and 70.92 d, J = 6.3 Hz (2x O–CH<), 23.82 d, 2C, J = 4.6 Hz, 23.94 d, J = 4.1 Hz and 23.97 d, J = 4.1 Hz (2x OiPr); 2-OBz: 164.63 (C O), 128.31 (i-ArC), 129.49 (2x o-ArC), 128.95 (2x m-ArC), 134.20 (p-ArC); 3-OBz: 164.66 (C O), 128.53 (i-ArC), 129.67 (2x o-ArC), 129.16 (2x m-ArC), 134.34 (p-ArC); NHBz: 165.82 (C O), 133.50 (i-ArC), 128.69 (2x o-ArC), 128.66 (2x m-ArC), 132.67 (p-ArC).
O–CH2–P = O(OiPr)2: 61.97 d, J = 167.1 Hz (P–CH2–O), 70.99 d, J = 6.3 Hz and 71.09 d, J = 6.3 Hz (2x O–CH<), 23.79 d, J = 4.4 Hz, 23.81 d, J = 4.4 Hz and 23.94 d, 2C, J = 3.6 Hz (2x OiPr); 2-OBz: 164.63 (C O), 128.32 (i-ArC), 129.53 (2x o-ArC), 128.93 (2x m-ArC), 134.20 (p-ArC); 3-OBz: 164.61 (C O), 128.56 (i-ArC), 129.60 (2x o-ArC), 129.18 (2x m-ArC), 134.36 (p-ArC); NHAc: 169.06 (C O), 24.76 (CH3); O–CO–N(C6H5)2: 155.53 (C O), 141.76 (2x i-ArC), 127.12 (4x o-ArC), 129.60 (4x m-ArC), 127.54 (2x p-ArC).
O–CH2–P = O(OiPr)2: 61.20 d, J = 165.2 Hz (P–CH2–O), 70.50 d, J = 6.2 Hz and 70.65 d, J = 6.2 Hz (2x O–CH<), 23.81 d, J = 4.7 Hz, 23.82 d, J = 4.5 Hz, 23.96 d, J = 3.7 Hz and 24.00 d, J = 3.7 Hz (2x OiPr).
O–CH2–P = O(OiPr)2: 61.94 d, J = 164.2 Hz (P–CH2–O), 70.87 d, J = 6.2 Hz and 70.92 d, J = 6.2 Hz (2x O–CH<), 23.75 d, J = 4.7 Hz, 23.78 d, J = 4.6 Hz, 23.93 d, J = 3.8 Hz and 23.94 d, J = 3.9 Hz (2x OiPr); 2-OBz: 164.69 (C O), 128.49 (i-ArC), 129.61 (2x o-ArC), 129.14 (2x m-ArC), 134.34 (p-ArC); 3-OBz: 164.66 (C O), 128.33 (i-ArC), 129.56 (2x o-ArC), 129.03 (2x m-ArC), 134.27 (p-ArC).
O–CH2–P = O(OiPr)2: 62.22 d, J = 165.0 Hz (P–CH2–O), 70.77 d, J = 6.2 Hz and 70.88 d, J = 6.2 Hz (2x O–CH<), 23.73 d, J = 4.5 Hz, 23.76 d, J = 4.5 Hz, 23.91 d, J = 3.5 Hz and 23.93 d, J = 3.5 Hz (2x OiPr); 2-OBz: 164.67 (C O), 128.46 (i-ArC), 129.56 (2x o-ArC), 129.13 (2x m-ArC), 134.33 (p-ArC); 3-OBz: 164.62 (C O), 128.34 (i-ArC), 129.59 (2x o-ArC), 129.02 (2x m-ArC), 134.27 (p-ArC).
O–CH2–P = O(OiPr)2: 62.13 d, J = 165.7 Hz (P–CH2–O), 70.70 d, J = 6.2 Hz and 70.83 d, J = 6.2 Hz (2x O–CH<), 23.69 d, J = 4.7 Hz, 23.74 d, J = 4.6 Hz, 23.89 d, J = 3.8 Hz and 23.92 d, J = 3.8 Hz (2x OiPr); 2-OBz: 164.67 (C O), 128.37 (i-ArC), 129.59 (2x o-ArC), 128.97 (2x m-ArC), 134.22 (p-ArC); 3-OBz: 164.72 (C O), 128.48 (i-ArC), 129.68 (2x o-ArC), 129.10 (2x m-ArC), 134.31 (p-ArC).
O–CH2–P = O(OiPr)2: 62.14 d, J = 166.0 Hz (P–CH2–O), 71.04 d, J = 6.3 Hz and 71.06 d, J = 6.3 Hz (2x O–CH<), 23.96 d, 2C, J = 4.4 Hz, 24.07 d, J = 3.6 Hz and 24.08 d, J = 3.8 Hz (2x OiPr); 2-OBz: 163.83 (C O), 127.81 (i-ArC), 129.03 (2x o-ArC), 128.74 (2x m-ArC), 134.22 (p-ArC); 3-OBz: 164.81 (C O), 128.47 (i-ArC), 129.94 (2x o-ArC), 128.99 (2x m-ArC), 134.22 (p-ArC).
O–CH2–P( = O)(OH)–O–P( = O)(OH)–O–P( = O)(OH)2: 67.24 d, J = 164.5 Hz (P–CH2–O).
O–CH2–P( = O)(OH)–O–P( = O)(OH)–O–P( = O)(OH)2: 66.96 d, J = 164.9 Hz (P–CH2–O).
O–CH2–P( = O)(OH)–O–P( = O)(OH)–O–P( = O)(OH)2: 66.54 d, J = 164.8 Hz (P–CH2–O); ’
O–CH2–P( = O)(OH)–O–P( = O)(OH)–O–P( = O)(OH)2: 66.39 d, J = 163.7 Hz (P–CH2–O).
O–CH2–P( = O)(OH)–O–P( = O)(OH)–O–P( = O)(OH)2: 66.18 d, J = 164.2 Hz (P–CH2–O).
Major diastereomer: Bu-O-CH(CH2C6H5)–CO–NH–P( = O)(O–C6H5)–CH2–O: 13.73, 18.62, 30.16, 64.49 (Bu-O-); 55.41, 39.74 (O–CH–CH2); 150.30 and 137.10 (2x i-ArC), 120.7–129.7 (10x ArC); 172.49 (C O), 63.34 d, J = 156.5 Hz (P–CH2–O); minor diastereomer: Bu-O-CH(CH2C6H5)–CO–NH–P( = O)(O–C6H5)–CH2–O: 13.68, 18.62, 30.19, 64.37 (Bu-O-); 55.35, 39.46 (O–CH–CH2); 150.25 and 137.19 (2x i-ArC), 120.7–129.7 (10x ArC); 172.71 (C O), 63.07 d, J = 154.7 Hz (P–CH2–O).
Major diastereomer: Bu-O-CH(CH2C6H5)–CO–NH–P( = O)(O–C6H5)–CH2–O: 13.66, 18.61, 30.15, 64.48 (Bu-O-); 55.37, 39.46 (O–CH–CH2); 137.06 (i-ArC), 129.41 (2x o-ArC), 128.37 (2x m-ArC), 124.56 (p-ArC) (C6H5), 172.68 d, J = 2.6 Hz (C O), 150.20 d, J = 9.2 Hz (i-ArC), 120.68 d, J = 4.5 Hz (2x o-ArC), 129.65 (2x m-ArC), 126.67 (p-ArCH) (C6H5); 62.96 d, J = 154.9 Hz (P–CH2–O); minor diastereomer: Bu-O-CH(CH2C6H5)–CO–NH–P( = O)(O–C6H5)–CH2–O: 13.70, 18.60, 30.17, 64.36 (Bu-O-); 55.33, 39.70 (O–CH–CH2); 137.18 (i-ArC), 129.52 (2x o-ArC), 128.29 (2x m-ArC), 124.62 (p-ArC) (C6H5), 172.48 d, J = 2.5 Hz (C O), 150.27 d, J = 9.2 Hz (i-ArC), 120.74 d, J = 4.5 Hz (2x o-ArC), 129.63 (2x m-ArC), 126.71 (p-ArC) (C6H5); 63.25 d, J = 157.6 Hz (P–CH2–O).
Coupling constants are written in italics in a shortened form (e.g. instead J(C1‘,F) = 34.0 Hz we type simply 1,F = 34.0).
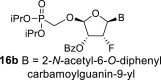
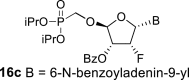
4.10. (2R,3R,4R,5R)-5-(2-acetamido-6-((diphenylcarbamoyl)oxy)-9H-purin-9-yl)-2-((diisopropoxyphosphoryl)methoxy)-4-fluorotetrahydrofuran-3-yl benzoate (16b)
Nucleoside phosphonate 16b was prepared according to the synthesis of compound 16a, starting from the acetate 15 (2 g; 4.33 mmol) and N2-acetyl-O6-(diphenylcarbamoyl)guanine (550 mg; 1.4 mmol). The reaction afforded a mixture of compounds 16b and 16d in a 1:1 ratio. Compound 16b was isolated in a yield of 662 mg (19%) as a faster eluting isomer. HRMS (M + Na)+ for C38H40O10N6FNaP calculated: 813.24198; measured: 813.24189; IR (coating, cm−1): 3219, 3094, 3065, 3040, 2980, 2873, 1738, 1696, 1622, 1591, 1525, 1492, 1452, 1412, 1387, 1374, 1335, 1271, 1249, 1170, 1107, 989, 713, 700. For NMR data, see Table IIa, Table IIbb.
4.11. (2R,3R,4R,5S)-5-(6-benzamido-9H-purin-9-yl)-2-((diisopropoxyphosphoryl)methoxy)-4-fluorotetrahydrofuran-3-yl benzoate (16c)
Nucleoside phosphonate 16c was prepared as a side product of the preparation of nucleoside phosphonate 16a.
Alpha anomer was isolated as a slower eluting isomer in a yield of 980 mg (23%). HRMS (M + Na)+ for C30H33O8N5FNaP calculated: 664.19430; measured: 664.19348; IR (coating, cm−1): 3416, 3238, 3164, 3064, 2980, 2934, 2875, 1733, 1700, 1603, 1583, 1511, 1490, 1452, 1386, 1375, 1340, 1316, 1296, 1275, 1252, 1178, 1118, 1105, 1080, 1027, 996, 904, 889, 798, 753, 710, 686, 644, 506. For NMR data, see Table IIa, Table IIbb.
4.12. (2R,3R,4R,5S)-5-(2-acetamido-6-((diphenylcarbamoyl)oxy)-9H-purin-9-yl)-2-((diisopropoxyphosphoryl)methoxy)-4-fluorotetrahydrofuran-3-yl benzoate (16d)
Nucleoside phosphonate 16d was prepared as a side product of the preparation of phosphonate nucleoside 16b. Alpha anomer was isolated in a yield of 430 mg (13%) as a slower eluting isomer. HRMS (M + Na)+ for C38H40O10N6FNaP calculated: 813.24198; measured: 813.24215; IR (coating, cm-1): 3217, 3064, 2980, 2870, 2854, 1738, 1700, 1623, 1600, 1591, 1511, 1492, 1452, 1386, 1375, 1335, 1316, 1272, 1246, 1179, 1170, 1135, 1121, 1106, 1078, 1061, 1026, 1003, 991, 907, 889, 758, 713, 700, 695, 666, 641, 532, 510. For NMR data, see Table IIa, Table IIbb.
4.13. ((((2R,3R,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-fluoro-3-hydroxytetrahydrofuran-2-yl)oxy)methyl)phosphonic acid (5a)
Bromotrimethylsilane (410 μL; 3.1 mmol) was added to the phosphonate 16a (200 mg; 0.31 mmol) in pyridine (5 mL), and the mixture was stirred for 8 h at room temperature and concentrated. The residue was diluted with saturated NH3 in 50% MeOH/H2O (20 mL), stirred for 16 h at room temperature, and then concentrate. The nucleotide 5a was isolated by reverse phase chromatography (first 10 min of isocratic elution with 0.1 M TEAB, then 40 min gradient 0–15% MeOH in 0.1 M TEAB) in a yield of 100 mg (92%): HRMS (M − H)- for C10H12O6N5FP calculated: 348.05147; measured: 348.05117; IR (KBr, cm-1): 3388, 3273, 2755, 2679, 2530, 2492, 1687, 1642, 1575, 1474, 1421, 1296, 1245, 1212, 1087, 1050, 796, 720, 642. For NMR data, see Table IIa, Table IIbb.
4.14. ((((2R,3R,4R,5R)-5-(2-amino-6-hydroxy-9H-purin-9-yl)-4-fluoro-3-hydroxytetrahydrofuran-2-yl)oxy)methyl)phosphonic acid (5b)
The compound 5b was prepared according to the synthesis of compound 5a, starting from 16b (586 mg; 0.74 mmol) in a yield of 229 mg (84%): HRMS (M − H)- for C10H12O7N5FP calculated: 364.04639; measured: 364.04587; IR (coating, cm−1): 3466, 3129, 2958, 2934, 2874, 2743, 1693, 1654, 1610, 1532, 1488, 1467, 1377, 1142, 1106, 1068, 889, 796, 779, 688. For NMR data, see Table IIa, Table IIbb.
4.15. 1,2,3,5-Tetraacetyl-l-ribofuranose (18)
Acetyl chloride (3.5 mL; 50 mmol) in 100 mL of dry MeOH was added to a solution of l-ribose 17 (100 g; 0.67 mol) in dry MeOH (1200 mL), and the reaction mixture was stirred for 16 h at 4 °C. Afterwards, it was filtered through the column of Dowex® 1 × 4 (150 mL; OH− cycle) and evaporated. The crude mixture was diluted with dioxane and co-evaporated (3 × 100 mL). H2SO4 (3 mL; 56 mmol) in Ac2O (20 mL) was added dropwise to the solution of crude methyl l-ribofuranoside in AcOH (250 mL) and Ac2O (500 mL) at 0 °C, and the reaction mixture was stirred for 2 h at room temperature. Next, the reaction was quenched by the addition of 100 mL of dry MeOH, and after 5 min of stirring, AcONa (6 g, 73 mmol) was added. The reaction product was evaporated, diluted with toluene (1 L) and washed with brine (500 mL) and sat. NaHCO3 (aq., 500 mL). The organic layer was dried over MgSO4, filtrated and evaporated. Crude acetate 18 was used for the next step without further purification.
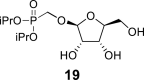
4.16. Diisopropylphosphonomethyl β-l-ribofuranoside (19)
Hexamethyldisilazane (400 mL) and a catalytic amount of saccharin were added to diisopropyl (hydroxymethyl)phosphonate (137 g; 700 mmol), and the reaction mixture was stirred for 8 h at 100 °C. Hexamethyldisilazane was evaporated, and the silylated phosphonate was coevaporated with toluene (3 × 100 mL). The crude acetate 18 was added to the silylated phosphonate in dry ACN (1.5 L). Finally, SnCl4 (100 mL; 850 mmol) was carefully added. The reaction mixture was stirred for 1 h at 55 °C, then quenched with 280 mL of pyridine, filtered and concentrated. Next, the crude mixture was dissolved in sat. NH3 in MeOH (1 L), and stirred for 16 h at room temperature. Product 19 was isolated by chromatography on a silica gel (0–10% MeOH in CHCl3) in a yield of 195 g (90% over 4 steps): HRMS (M + Na)+ for C12H25O8NaP calculated: 351.11793; measured: 351.11835; IR (CHCl3, cm−1): 3355, 2981, 2934, 2878, 1467, 1454, 1387, 1376, 1234, 1180, 1104, 1054, 996, 891, 721. For NMR data, see Table Ia, Table Ibb.
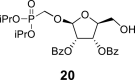
4.17. Diisopropylphosphonomethyl 2,3-O-dibenzoyl-β-l-ribofuranoside (20)
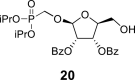
TBDPSCl (20 mL; 77 mmol) was added dropwise to the solution of phosphonate 19 (22.7 g; 69 mmol) in dry pyridine (500 mL), and the reaction mixture was stirred for 16 h at room temperature. Next, Et3N (20 mL; 140 mmol) and DMAP (1 g; 8 mmol) were added, followed by the dropwise addition of BzCl (17 mL; 140 mmol). The mixture was then stirred for another 8 h at room temperature. The reaction was quenched by the addition of water (50 mL), and concentrated. The residue was diluted with Et2O (1 L), and extracted with a saturated solution of brine (500 mL) and sodium bicarbonate (2 × 500 mL). The organic phase was dried over Na2SO4, filtered, concentrated and dried by co-evaporation with toluene (3 × 100 mL). Next, the residue was diluted with THF (400 mL), TBAF × 3H2O was added (25.3 g; 80 mmol), and the mixture was stirred for 1 h at room temperature. The reaction was quenched by the addition of 20 mL of water, concentrated, diluted with Et2O (500 mL) and extracted between Et2O and saturated aqueous NH4Cl (3 × 300 mL). The organic phase was dried over Na2SO4, filtered and concentrated. Product 20 was isolated by chromatography on a silica gel (0–50% EtOAc in toluene) in a yield of 28.85 g (78%): HRMS (M + Na)+ for C42H51O10NaPSi calculated: 797.28813; measured: 797.28842; IR (CHCl3, cm−1): 2978, 2892, 1732, 1602, 1585, 1491, 1472, 1464, 1452, 1428, 1386, 1375, 1362, 1316, 1276, 1178, 1125, 1113, 1070, 1028, 991, 938, 889, 708, 615, 505. For NMR data, see Table Ia, Table Ibb.
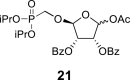
4.18. (3R,4S,5R)-2-acetoxy-5-((diisopropoxyphosphoryl)methoxy)tetrahydrofuran-3,4-diyl dibenzoate (21)
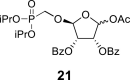
2,2,6,6-Tetramethylpiperidine 1-oxyl (156 mg; 1 mmol) and (diacetoxyiodo)benzene (6.5 g; 20 mmol) were added to the phosphonate 20 (5.3 g; 10 mmol) in 30% water in ACN (100 mL), and the mixture was stirred for 16 h at room temperature. The reaction was quenched by the addition of 20 mL of EtOH, evaporated and co-evaporated with water (5 × 50 mL) and then with toluene (3 × 30 mL). The reaction intermediate was used in the next step without further purification.
Pb(OAc)4 (5.5 g; 12.3 mmol) was added to crude carboxylic acid dissolved in 100 mL dry THF. The reaction mixture was stirred for 2 h at room temperature, filtered and concentrated. Product 21 was isolated by chromatography on a silica gel (0–30% EtOAc in toluene) in a yield of 2.95 g (53% over two steps): HRMS (M + Na)+ for C27H33O11NaP calculated: 587.16527; measured: 587.16534; IR (CHCl3, cm−1): 2981, 2878, 1734, 1602, 1492, 1467, 1452, 1386, 1375, 1364, 1281, 1263, 1224, 1179, 1163, 1123, 1071, 1024, 991, 981, 888, 711. For NMR data, see Table Ia, Table Ibb.
4.19. (2R,3R,4S,5R)-2-(6-benzamido-9H-purin-9-yl)-4-benzoyl-5-((diisopropoxyphosphoryl)methoxy)tetrahydrofuran-3-yl benzoate (22a)
Bis(trimethylsilyl)acetamide (1.1 mL; 4.5 mmol) was added to 6N-benzoyladenine (335 mg; 1.4 mmol) in 1,2-dichloroethane (14 mL), and the reaction mixture was stirred for 1 h at 60 °C. The mixture was concentrated and co-evaporated with dry toluene (2 × 20 mL). Next, acetate 21 (565 mg; 1 mmol) in ACN (10 mL) was added, followed by SnCl4 (600 μL; 5.1 mmol). The mixture was stirred for 2 h at room temperature. The reaction was then quenched by the addition of 1 mL of pyridine, filtered and concentrated. Product 22a was isolated by chromatography on a silica gel (0–5% methanol in chloroform) in a yield of 160 mg (50%). HRMS (M + Na)+ for C37H38O10N5NaP calculated: 766.22485; measured: 766.22449; IR (coating, cm-1): 3227, 3090, 3065, 3032, 2978, 2925, 2870, 2854, 1733, 1700, 1610, 1602, 1583, 1512, 1490, 1452, 1386, 1376, 1334, 1316, 1269, 1252, 1178, 1158, 1126, 1105, 1063, 1024, 990, 937, 889, 711, 672, 642, 528. For NMR data, see Table IIa, Table IIbb.
4.20. (2R,3R,4S,5R)-2-(2-acetamido-6-((diphenylcarbamoyl)oxy)-9H-purin-9-yl)-5-((diisopropoxyphosphoryl)methoxy)tetrahydrofuran-3,4-diyl dibenzoate (22b)
Bis(trimethylsilyl)acetamide (1.1 mL; 4.5 mmol) was added to N2-acetyl-O6-(diphenylcarbamoyl)guanine (550 mg; 1.4 mmol) in 1,2-dichloroethane (14 mL), and the reaction mixture was stirred for 1 h at 60 °C. The mixture was concentrated and co-evaporated with dry toluene (3 × 20 mL). Then, the acetate 21 (565 mg; 1 mmol) in ACN (10 mL) was added, followed by SnCl4 (600 μL; 5.1 mmol). The mixture was stirred for 2 h at room temperature. The reaction was then quenched by the addition of 1 mL of pyridine, filtered and concentrated. Product 22b was isolated by chromatography on a silica gel (0–100% EtOAc in toluene) in a yield of 340 mg (42%). HRMS (M + Na)+ for C45H45O12N6NaP calculated: 915.27253; measured: 915.27259; IR (CHCl3, cm−1): 3318, 3185, 1737, 1699, 1618, 1598, 1591, 1519, 1511, 1492, 1452, 1386, 1374, 1315, 1298, 1273, 1219, 1180, 1168, 1123, 1106, 1023, 989, 907, 887, 805, 728, 719, 713, 641, 531. For NMR data, see Table IIa, Table IIbb.
4.21. Diisopropyl ((((2R,3S,4R,5R)-5-(7-amino-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)-3,4-dihydroxytetrahydrofuran-2-yl)oxy)methyl)phosphonate (22c)
SnCl4 (2.6 mL; 22 mmol) was added dropwise to the heterogenic mixture of the acetate 21 (4.15 g; 7.35 mmol) and 8-azaadenine (1 g; 7.35 mmol) in dry acetonitrile (30 mL). The mixture was warmed to 60 °C and stirred for 3 h. The reaction was then quenched by the addition of pyridine (3 mL), filtered and adsorbed on a silica gel. The product of nucleosidation was isolated by chromatography on a silica gel (0–6% methanol in chloroform) as a mixture of 23 and 25 regioisomers (in ratio 1:3 according NMR) in a yield of 3.06 g (65%) as faster eluting derivatives followed by compound 24 in a yield of 850 mg (18%) as a slower eluting regioisomer. The mixture of regioisomers 23 and 25 was then stirred in sat. NH3 in 50% aq. MeOH (30 mL) for 16 h at room temperature, concentrated, dissolved in 80% aq. AcOH (20 mL) and stirred for 1 day at room temperature. The 8-azaadenine-7-yl derivative de-purinated, and product 22c was isolated by chromatography on a silica gel (0–12% methanol in chloroform) in a yield of 500 mg (16%). HRMS (M + Na)+ for C15H25O7N6NaP calculated: 455.14145; measured: 455.14099; IR: 3449, 2980, 1700, 1652, 1574, 1466, 1409, 1376, 1335, 1264, 1226, 1178, 1139, 1106, 1081, 1040, 992, 926, 889. For NMR data, see Table IIa, Table IIbb.
4.22. (2R,3R,4S,5R)-2-(5-amino-7-hydroxy-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)-5-((diisopropoxyphosphoryl)methoxy)tetrahydrofuran-3,4-diyl dibenzoate (22d)
Bis(trimethylsilyl)acetamide (3 mL; 12 mmol) was added to 8-azaguanine (400 mg; 2.6 mmol) in 1,2-dichloroethane (18 mL), and the reaction mixture was stirred for 1 h at 60 °C. The mixture was concentrated, co-distilled with dry toluene (2 × 20 mL), and then added to the acetate 21 (1.52 g; 2.7 mmol) in ACN (20 mL). Finally, SnCl4 (2 mL; 17 mmol) was added in one portion, and the mixture was stirred for 2 h at room temperature. The reaction was then quenched by the addition of 2 mL of pyridine, filtered and concentrated. The reaction afforded a mixture of compounds 22d and 22e in a 1:5 ratio. Product 22d was isolated by chromatography on a silica gel (0–5% methanol in DCM) in a yield of 160 mg (10%) as a faster eluting regioisomer. HRMS (M + Na)+ for C29H33O10N6NaP calculated: 679.18880; measured: 679.18901; IR (CHCl3, cm-1): 3319, 3165, 2980, 2875, 1733, 1706, 1643, 1601, 1493, 1466, 1452, 1386, 1376, 1316, 1274, 1243, 1179, 1121, 1106, 1026, 996, 891, 774, 712, 685. For NMR data, see Table IIa, Table IIbb.
4.23. (2R,3R,4S,5R)-2-(5-amino-7-hydroxy-1H-[1,2,3]triazolo[4,5-d]pyrimidin-1-yl)-5-((diisopropoxyphosphoryl)methoxy)tetrahydrofuran-3,4-diyl dibenzoate (22e)
Nucleoside phosphonate 22e was prepared as an undesired product of the preparation of nucleoside phosphonate 22d. Product 22e was isolated by chromatography on a silica gel (0–5% methanol in DCM) in a yield of 830 mg (50%) as a slower regioisomer. For NMR data, see Table IIa, Table IIbb.
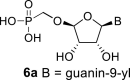
4.24. ((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)oxy)methyl)phosphonic acid (1)
Bromotrimethylsilane (490 μL; 3.7 mmol) was added to 22a (275 mg; 0.37 mmol) in pyridine (5 mL), and the mixture was stirred for 6 h and concentrated. The residue was diluted with saturated NH3 in 50% MeOH/H2O (20 mL), stirred for 16 h at room temperature and then concentrated. Nucleotide 1 was isolated by reverse phase chromatography (first 15 min of isocratic elution with 0.1 M TEAB, then 35 min gradient 0–15% MeOH in 0.1 M TEAB) in a yield of 134 mg (80%).
Spectral data were in accordance with literature values [5].
4.25. ((((2R,3S,4R,5R)-5-(2-amino-6-hydroxy-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)oxy)methyl)phosphonic acid (6a)
Bromotrimethylsilane (490 μL; 3.7 mmol) was added to 22b (330 mg; 0.37 mmol) in pyridine (5 mL), the mixture was stirred for 6 h, and then concentrated. The residue was diluted with saturated NH3 in 50% MeOH/H2O (20 mL), stirred for 16 h at room temperature and concentrated. Nucleotide 6a was isolated by reverse phase chromatography (first 15 min of isocratic elution with 0.1 M TEAB, then 35 min gradient 0–15% MeOH in 0.1 M TEAB) in a yield of 138 mg (80%). HRMS (M − H)- for C10H13O8N5P calculated: 362.05072; measured: 362.05020; IR (CHCl3, cm−1): 3402, 3153, 2823, 2739, 2680, 2492, 1693, 1645, 1605, 1571, 1480, 1451, 1398, 1229, 1162, 1093, 1038, 999, 965, 783, 682, 574. For NMR data, see Table IIa, Table IIbb.
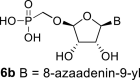
4.26. ((((2R,3S,4R,5R)-5-(7-amino-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)-3,4-dihydroxytetrahydrofuran-2-yl)oxy)methyl)phosphonic acid (6b)
Bromotrimethylsilane (1.6 mL; 11.6 mmol) was added to 22c (500 mg; 1.16 mmol) in pyridine (20 mL). The mixture was stirred for 6 h and concentrated. Nucleotide 6b was isolated by reverse phase chromatography (first 15 min of isocratic elution with 0.1 M TEAB, then 35 min gradient 0–15% MeOH in 0.1 M TEAB) in a yield of 980 mg (84%). HRMS (M − H)- for C9H12O7N6P calculated: 347.05106; measured: 347.05098; IR (coating MeOH, cm−1): 3395, 3325, 3165, 2738, 2677, 2571, 2491, 2349, 1660, 1607, 1577, 1468, 1266, 1140, 1061, 1061, 1061, 913, 849, 799, 684, 645. For NMR data, see Table IIa, Table IIbb.
4.27. ((((2R,3S,4R,5R)-5-(5-amino-7-hydroxy-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)-3,4-dihydroxytetrahydrofuran-2-yl)oxy)methyl)phosphonic acid (6c)
Bromotrimethylsilane (330 μL; 2.5 mmol) was added to 22d (160 mg; 0.24 mmol) in pyridine (5 mL), the mixture was stirred for 6 h, and then concentrated. The residue was diluted with saturated NH3 in 50% MeOH/H2O (10 mL), stirred for 16 h at room temperature, and then concentrated. Nucleotide 6c was isolated by reverse phase chromatography (first 15 min of isocratic elution with 0.1 M TEAB, then 35 min gradient 0–15% ACN in 0.1 M TEAB) in a yield of 100 mg (90%). HRMS (M − H)- for C9H12O8N6P calculated: 363.04597; measured: 363.04563; IR (CHCl3, cm−1): 3419, 3167, 2686, 2491, 1711, 1639, 1532, 1457, 1240, 1112, 1056, 1039, 788, 682; NMR: For NMR data, see Table IIa, Table IIbb.
4.28. ((((2R,3R,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-fluoro-3-hydroxytetrahydrofuran-2-yl)oxy)methyl)phosphonic diphosphoric anhydride (26a)
Triethylammonium salt of phosphonate 5a (90 mg; 0.2 mmol) was converted to tetrabutylammonium salt (Dowex® 50WX 8 in tetrabutylammonium cycle) and dried by co-evaporation with anhydrous pyridine. The mixture of phosphonate salt, imidazole (211 mg; 3.1 mmol), and tri-N-octylamine (0.57 mL; 1.3 mmol) was dried by co-evaporation with anhydrous DMF (2 × 10 mL). The semi-solid residue was dissolved in anhydrous DMF (12 mL), triphenylphosphine (341 mg; 1.3 mmol), and 2,2′-dipyridyldisulfide (Aldrithiol™, 286 mg; 1.3 mmol) were added, and the mixture was stirred for 16 h at room temperature.
The reaction mixture was added dropwise to the precipitation solution: sodium perchlorate monohydrate (702 mg; 5 mmol) and triethylamine (4 mL) in peroxide free mixture of acetone (60 mL) and diethylether (36 mL) at 0 °C. The solution was allowed to precipitate at 0 °C for about 30 min. The precipitate was then separated by centrifugation (10000 RPM, 3 °C, 20 min), washed with the precipitation solution, and then with the dry diethylether. Solid imidazolide was dried in vacuo. Tributylammonium pyrophosphate (0.5 M solution in DMSO, 1.2 mL; 0.6 mmol) was added to imidazolide, and the solution was kept at room temperature for 48 h. The phosphonodiphosphate was purified by column chromatography on reverse phase (Phenomenex Luna C18 5 μm), using a linear gradient of acetonitrile (0–5%) in triethylamine bicarbonate buffer (0.1 M).
Triethylammonium salt of the product was converted to sodium salt using Dowex® 50WX 8, Na+ cycle yielding 93 mg (65%) of desired triphosphate analogue 26a. HRMS (M − H)- for C10H14O12N5FP3 calculated: 507.98413; measured: 507.98370; IR (KBr, cm−1): 3428, 1695, 1647, 1579, 1479, 1424, 1332, 1247, 1077, 1042, 899, 842, 719. For NMR data, see Table IIa, Table IIbb.
4.29. ((((2R,3R,4R,5R)-5-(2-amino-6-hydroxy-9H-purin-9-yl)-4-fluoro-3-hydroxytetrahydrofuran-2-yl)oxy)methyl)phosphonic diphosphoric anhydride (26b)
Phosphonodiphosphate 26b was prepared according to the procedure described for compound 23a, starting from 5b (45 mg; 0.12 mmol) in a yield of 14 mg (20%). HRMS (M − H)- for C10H14O13N5FP3 calculated: 523.97905; measured: 523.97839; IR (KBr, cm−1): 3455, 3303, 3126, 2963, 2876, 2760, 2361, 1696, 1654, 1606, 1534, 1378, 1252, 1215, 1129, 1094, 1077, 1003, 932, 799, 688, 637, 529, 479. For NMR data, see Table IIa, Table IIbb.
4.30. ((((2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)oxy)methyl)phosphonic diphosphoric anhydride (26c)
Phosphonodiphosphate 26c was prepared according to the procedure described for compound 26a, starting from 1 (90 mg; 0.2 mmol) in a yield of 50 mg (45%).
Spectral data were in accordance with values from the literature [14].
4.31. ((((2R,3S,4R,5R)-5-(2-amino-6-hydroxy-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)oxy)methyl)phosphonic diphosphoric anhydride (26d)
Phosphonodiphosphate 26d was prepared according to the procedure described for compound 26a, starting from 6a (180 mg; 0.39 mmol) in a yield of 150 mg (68%). HRMS (M − H)- for C10H15O14N5P3 calculated: 521.98338; measured: 521.98242; IR (coating MeOH, cm−1): 3432, 3313, 3098, 2346, 1695, 1650, 1533, 1449, 1232, 1128, 1073, 1032, 999, 895, 792, 688. For NMR data, see Table IIa, Table IIbb.
4.32. ((((2R,3S,4R,5R)-5-(7-amino-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)-3,4-dihydroxytetrahydrofuran-2-yl)oxy)methyl)phosphonic diphosphoric anhydride (26e)
Phosphonodiphosphate 26e was prepared according to the procedure described for the compound 26a, starting from 6b (100 mg; 0.28 mmol) in a yield of 43 mg (28%). HRMS (M − H)- for C9H13N6Na2O13P3 calculated: 506.98372; measured: 506.98343; IR (KBr, cm-1): 3344, 3285, 3285, 3190, 1662, 1579, 1453, 1420, 1334, 1248, 1126, 1075, 1037, 1016, 904, 687, 648. For NMR data, see Table IIb, Table IIab.
4.33. ((((2R,3S,4R,5R)-5-(5-amino-7-hydroxy-3H-[1,2,3]triazolo[4,5-d]pyrimidin-3-yl)-3,4-dihydroxytetrahydrofuran-2-yl)oxy)methyl)phosphonic diphosphoric anhydride (26f)
Phosphonodiphosphate 26f was prepared according to the protocol developed for the compound 26a, starting from 6c (100 mg; 0.27 mmol) in a yield of 110 mg (80%). HRMS (M − H)- for C9H14O14N6P3 calculated: 522.97863; measured: 522.97760; IR (KBr, cm−1): 3402, 3387, 3163, 2493, 1709, 1644, 1533, 1456, 1227, 1109, 1070, 1036, 1001, 930, 788, 682; NMR: For NMR data, see Table IIa, Table IIbb.
4.34. Butyl (((((2R,3R,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-fluoro-3-hydroxytetrahydrofuran-2-yl)oxy)methyl)(phenoxy)phosphoryl)-l-phenylalaninate (27a)
Triethylammonium salt of phosphonate 5a (135 mg; 0.3 mmol) was converted to tetrabutylammonium salt (Dowex® 50WX 8 in tetrabutylammonium cycle) and dried by the co-evaporation with anhydrous pyridine. The solution of 5a, l-phenylalanine butyl ester hydrochloride (137 mg; 0.53 mmol), phenol (125 mg; 1.3 mmol) and Et3N (445 μL; 3.18 mmol) in dry pyridine (12 mL) was stirred for 15 min at 60 °C. Next, the solution of triphenylphosphine (412 mg; 1.57 mmol) and Aldrithiol™ (485 mg; 7.34 mmol) in dry pyridine (6 mL) was added to the mixture. The mixture was stirred for 16 h at 60 °C, and then concentrated. The solid residue was adsorbed on a silica gel in acetone, and phosphonoamidate 27a was isolated by chromatography on a silica gel (0–8% EtOH in CHCl3) in a yield of 85 mg (45%) as a mixture of epimers (estimated ratio by 31P NMR 1:1). HRMS (M + Na)+ for C29H34FN6O7P calculated: 651.21028; measured: 651.20978; IR (CHCl3, cm−1): 3601, 3413, 3062, 2963, 2931, 2875, 2856, 1631, 1602, 1589, 1491, 1471, 1456, 1420, 1330, 1294, 1244, 1069, 1032, 901, 702, 690. For NMR data, see Table IIa, Table IIbb.
4.35. Butyl (((((2R,3R,4R,5R)-5-(2-amino-6-hydroxy-9H-purin-9-yl)-4-fluoro-3-hydroxytetrahydrofuran-2-yl)oxy)methyl)(phenoxy)phosphoryl)-l-phenylalaninate (27b)
Phosphonoamidate 27b was prepared according to the procedure described for the compound 27a, starting from 5b (50 mg; 0.1 mmol) in a yield of 28 mg (43%) as a mixture of epimers. HRMS (M + Na)- for C29H34O8N6FNaP calculated: 667.20520; measured: 667.20472; IR (CHCl3, cm−1): 3469, 3307, 3066, 2963, 2935, 2877, 1693, 1654, 1604, 1591, 1533, 1491, 1466, 1456, 1379, 1379, 1238, 1175, 1071, 1036, 701, 690, 642. For NMR data, see Table IIa, Table IIbb.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The work was supported by the European Regional Development Fund; OP RDE; Project: “Chemical biology for drugging undruggable targets (ChemBioDrug)" (No. CZ.02.1.01/0.0/0.0/16_019/0000729).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.tet.2021.132159.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.De Clercq E. A 40-year journey in search of selective antiviral chemotherapy. Annu. Rev. Pharmacol. Toxicol. 2011;51:1–24. doi: 10.1146/annurev-pharmtox-010510-100228. [DOI] [PubMed] [Google Scholar]
- 2.Polakova I., Budesinsky M., Tocik Z., Rosenberg I. Tetrofuranose nucleoside phosphonic acids: synthesis and properties. Collect. Czech Chem. Commun. 2011;76(5):503–536. [Google Scholar]
- 3.Králíková Š., Buděšínký M., Masojídková M., Rosenberg I. Nucleoside 5′-C-phosphonates: reactivity of the α-hydroxyphosphonate moiety. Tetrahedron. 2006;62(20):4917–4932. [Google Scholar]
- 4.Simak O., Pachl P., Fabry M., Budesinsky M., Jandusik T., Hnizda A., Sklenickova R., Petrova M., Veverka V., Rezacova P., Brynda J., Rosenberg I. Conformationally constrained nucleoside phosphonic acids - potent inhibitors of human mitochondrial and cytosolic 5[prime or minute](3[prime or minute])-nucleotidases. Org. Biomol. Chem. 2014;12(40):7971–7982. doi: 10.1039/c4ob01332h. [DOI] [PubMed] [Google Scholar]
- 5.Kim C.U., Luh B.Y., Martin J.C. Regiospecific and highly stereoselective electrophilic addition to furanoid glycals: synthesis of phosphonate nucleotide analogs with potent activity against HIV. J. Org. Chem. 1991;56(8):2642–2647. [Google Scholar]
- 6.De S., De Jonghe S., Herdewijn P. Synthesis of a 3’-Fluoro-3’-deoxytetrose adenine phosphonate. J. Org. Chem. 2017;82(18):9464–9478. doi: 10.1021/acs.joc.7b01482. (Ahead of Print) [DOI] [PubMed] [Google Scholar]
- 7.Li Q., Groaz E., Herdewijn P. Synthesis of tetradialdose phosphonate nucleosides as mimics of l-nucleotides. Tetrahedron. 2019;75(37):130497. [Google Scholar]
- 8.Boojamra C.G., Mackman R.L., Markevitch D.Y., Prasad V., Ray A.S., Douglas J., Grant D., Kim C.U., Cihlar T. Synthesis and anti-HIV activity of GS-9148 (2′-Fd4AP), a novel nucleoside phosphonate HIV reverse transcriptase inhibitor. Bioorg. Med. Chem. Lett. 2008;18(3):1120–1123. doi: 10.1016/j.bmcl.2007.11.125. [DOI] [PubMed] [Google Scholar]
- 9.Gentile I., Borgia F., Buonomo A.R., Castaldo G., Borgia G. A novel promising therapeutic option against hepatitis C virus: an oral nucleotide NS5B polymerase inhibitor sofosbuvir. Curr. Med. Chem. 2013;20(30):3733–3742. doi: 10.2174/09298673113209990178. [DOI] [PubMed] [Google Scholar]
- 10.Ren H., An H., Hatala P.J., Stevens W.C., Jr., Tao J., He B. Versatile synthesis and biological evaluation of novel 3’-fluorinated purine nucleosides. Beilstein J. Org. Chem. 2015;11:2509–2520. doi: 10.3762/bjoc.11.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sivets G.G., Amblard F., Schinazi R.F. Synthesis of 2-fluoro-substituted and 2,6-modified purine 2′,3′-dideoxy-2′,3′-difluoro-d-arabinofuranosyl nucleosides from d-xylose. Tetrahedron. 2019;75(13):2037–2046. doi: 10.1016/j.tet.2019.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Käppi R., Kazimierczuk Z., Seela F., Lönnberg H. Kinetics and mechanism for acid-catalyzed hydrolysis of regioisomeric 2′-deoxyribonucleosides of 8-azaadenine and substituted benzotriazoles. Nucleos Nucleot. 1991;10(1–3):571–572. [Google Scholar]
- 13.Dyatkina N.B., Theil F., von Janta-Lipinski M. Stereocontrolled synthesis of the four stereoisomeric diphosphorylphosphonates of carbocyclic 2′,3′-dideoxy-2′,3′-didehydro-5′-noradenosine. Tetrahedron. 1995;51(3):761–772. [Google Scholar]
- 14.Koh Y.-h., Shim J.H., Wu J.Z., Zhong W., Hong Z., Girardet J.-L. Design, synthesis, and antiviral activity of adenosine 5‘-phosphonate analogues as chain terminators against hepatitis C virus. J. Med. Chem. 2005;48(8):2867–2875. doi: 10.1021/jm049029u. [DOI] [PubMed] [Google Scholar]
- 15.Klejch T., Keough D.T., Chavchich M., Travis J., Skácel J., Pohl R., Janeba Z., Edstein M.D., Avery V.M., Guddat L.W., Hocková D. Sulfide, sulfoxide and sulfone bridged acyclic nucleoside phosphonates as inhibitors of the Plasmodium falciparum and human 6-oxopurine phosphoribosyltransferases: synthesis and evaluation. Eur. J. Med. Chem. 2019;183:111667. doi: 10.1016/j.ejmech.2019.111667. [DOI] [PubMed] [Google Scholar]
- 16.Mackman R.L., Ray A.S., Hui H.C., Zhang L., Birkus G., Boojamra C.G., Desai M.C., Douglas J.L., Gao Y., Grant D., Laflamme G., Lin K.-Y., Markevitch D.Y., Mishra R., McDermott M., Pakdaman R., Petrakovsky O.V., Vela J.E., Cihlar T. Discovery of GS-9131: design, synthesis and optimization of amidate prodrugs of the novel nucleoside phosphonate HIV reverse transcriptase (RT) inhibitor GS-9148. Bioorg. Med. Chem. 2010;18(10):3606–3617. doi: 10.1016/j.bmc.2010.03.041. [DOI] [PubMed] [Google Scholar]
- 17.Birkus G., Kutty N., Frey C.R., Shribata R., Chou T., Wagner C., McDermott M., Cihlar T. Role of cathepsin A and lysosomes in the intracellular activation of novel antipapillomavirus agent GS-9191. Antimicrob. Agents Chemother. 2011;55(5):2166. doi: 10.1128/AAC.01603-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hercík K., Kozak J., Šála M., Dejmek M., Hřebabecký H., Zborníková E., Smola M., Ruzek D., Nencka R., Boura E. Adenosine triphosphate analogs can efficiently inhibit the Zika virus RNA-dependent RNA polymerase. Antivir. Res. 2017;137:131–133. doi: 10.1016/j.antiviral.2016.11.020. [DOI] [PubMed] [Google Scholar]
- 19.Cho A., Zhang L., Xu J., Lee R., Butler T., Metobo S., Aktoudianakis V., Lew W., Ye H., Clarke M., Doerffler E., Byun D., Wang T., Babusis D., Carey A.C., German P., Sauer D., Zhong W., Rossi S., Fenaux M., McHutchison J.G., Perry J., Feng J., Ray A.S., Kim C.U. Discovery of the first C-nucleoside HCV polymerase inhibitor (GS-6620) with demonstrated antiviral response in HCV infected patients. J. Med. Chem. 2014;57(5):1812–1825. doi: 10.1021/jm400201a. [DOI] [PubMed] [Google Scholar]
- 20.Mejdrová I., Chalupská D., Kögler M., Šála M., Plačková P., Baumlová A., Hřebabecký H., Procházková E., Dejmek M., Guillon R., Strunin D., Weber J., Lee G., Birkus G., Mertlíková-Kaiserová H., Boura E., Nencka R. Highly selective phosphatidylinositol 4-kinase IIIβ inhibitors and structural insight into their mode of action. J. Med. Chem. 2015;58(9):3767–3793. doi: 10.1021/acs.jmedchem.5b00499. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.