

Abstract
Classic approaches to mapping the developmental history of cells in vivo have relied on techniques that require complex interventions and often capture only a single trajectory or moment in time. We have previously described a developmental barcoding system to address these issues using synthetically induced mutations to record information about each cell’s lineage in its genome. This system uses MARC1 mouse lines which have multiple homing guide RNAs that each generate hundreds of mutant alleles and combine to produce an exponential diversity of barcodes. Here, we detail two MARC1 lines which are available from a public repository. We describe strategies for using MARC1 mice and experimental design considerations. We provide a 2-day protocol for barcode retrieval and sequencing as well as the analysis of the sequencing data. This protocol generates barcodes based on synthetically-induced mutations in mice to enable lineage analysis.
Keywords: lineage barcoding, combinatorial barcoding, homing CRISPR mouse, MARC1 mouse, barcoding mouse, homing guide RNA, hgRNA
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary associated with this article.
Introduction
The ability to track cells and their descendants through complicated biological processes is essential for understanding development, regeneration, and cancer among others. Marking cells with uniquely distinguishable dyes or genetic elements has long been used for marking their lineages. New strategies have emerged in recent years that incorporate advances in genome engineering technologies to enable high-throughput tracing of cellular histories through gradual barcoding of their genomes with heritable modifications1,2. These approaches not only enable the descendants of a large number of cells to be traced, they further promise to delineate the exact relationship between all those descendants, a concept known as the lineage tree3,4. In theory, such a system implemented in the zygote can map the lineage tree of an entire organism.
These barcoding strategies have been implemented in a variety of forms and in multiple model systems in recent years5–11. Implementation in the mouse has been particularly challenging due to the complexities associated with transgenesis in this organism. Nonetheless, recent advances provide tools that make access to barcoding generally available in this model system7,10,12,13. One of these resources is the Mouse for Actively Recording Cells! (MARC1), a stable line with complex genetics that can be crossed with a Cas9 line to result in combinatorial and cumulative developmental barcoding in its offspring7. The MARC1 system is unique in that it creates an exponential diversity of barcodes, has robust barcoding activity across cell types, is less susceptible to large deletions that erase barcode information, and imposes a low burden on cells and thus minimally disturbs development. Here we describe this system and the principles underlying its performance. We introduce a new MARC1 strain and detail its performance. Finally, we provide protocols for obtaining, maintaining, and using MARC1 lines.
Barcoding Principle
The principle of developmental barcoding rests upon a number of loci in the genome that gradually undergo random, irreversible changes in their sequence (Figure 1a). We refer to each locus as a “barcoding element” and to the combination of all such elements in a cell as a “barcoding set.” At the outset of the experiment, all elements are in their non-mutated and active state. As cells divide, each element undergoes random mutations that eventually inactivate it, resulting in a fixed mutant allele. This allele will be inherited by all the offspring of the cell where it first appeared, leading to a mark that distinguishes these offspring from all other cells. Over time, additional barcoding elements accrue fixed and inactive mutant alleles, further labeling the offspring. In the end, as closely related cells will share a higher fraction of fixed alleles in their barcoding set than those more distantly related, the combination of mutant alleles across elements of the set in each cell—the cell’s barcode—represents the history of that cell with respect to all cells. While this system functions at the level of single cells, collective barcodes of cells within a population can be compared to those of other populations as a measure of relatedness in the lineage tree and, if said populations are of distinct lineage origins, used to decipher lineage relationships between them. Under appropriate circumstances, these barcodes can thus be used to resolve the entire lineage tree that connects all cells.
Figure 1.
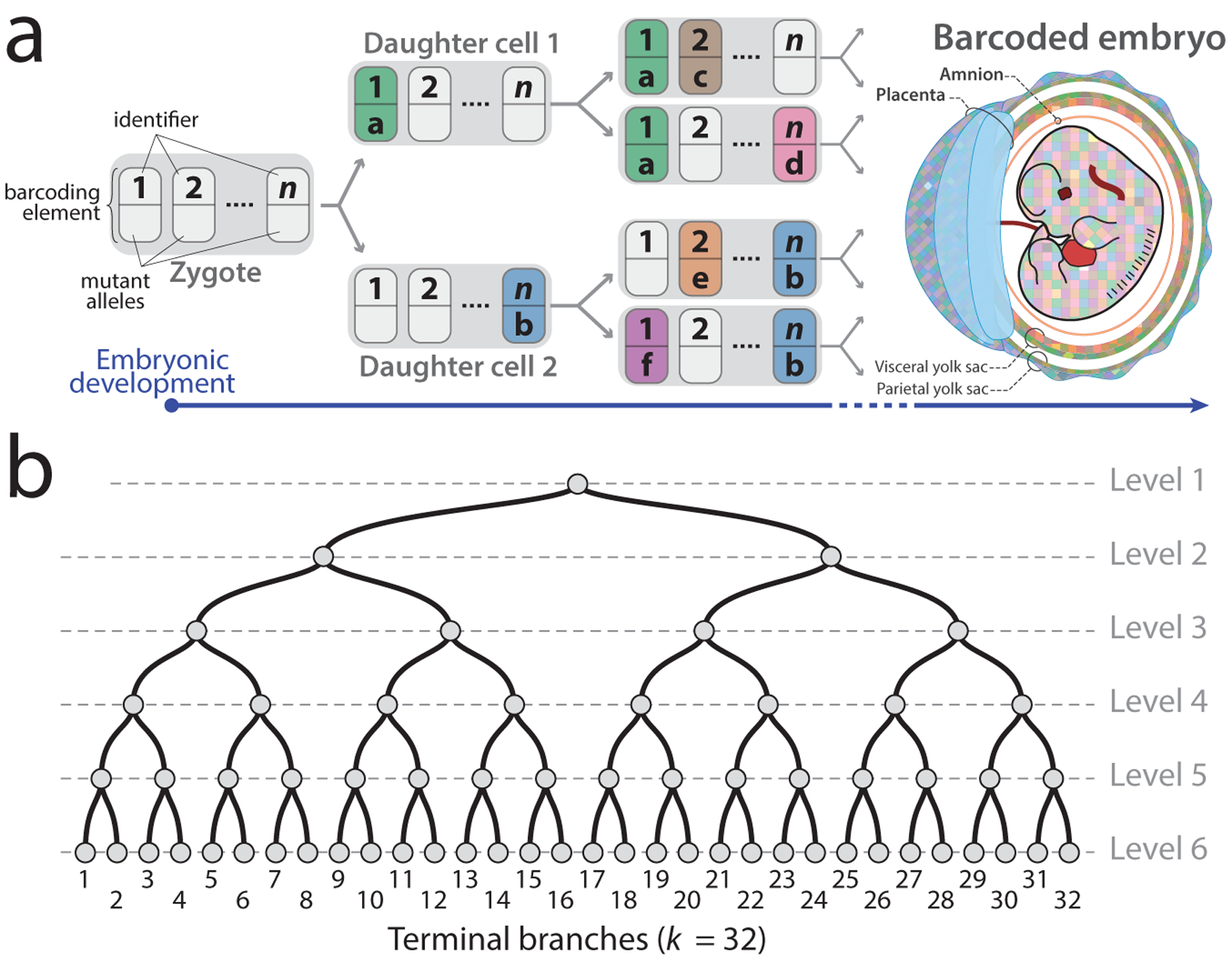
Recording lineages using synthetically induced somatic mutations in the genome. (a) A number of loci (n) accumulate heritable mutations as cells divide, thereby recording the lineage relationship of the cells in an array of mutational barcodes across an embryo. Gray domino, active barcoding elements; colored dominos, fixed mutant barcoding elements. (b) A cell division tree with the number of terminal branches (k) and levels shown.
The properties of the barcoding set determine its capacity for labeling complex lineages. In a system where the barcoding set comprises n independent elements and each element is capable of producing m alleles, a total of mn barcodes are possible, delineating the maximum number of distinct terminal branches that may be distinguished (Figure 1b). Furthermore, such a system can barcode a maximum of n successive cell divisions4,14, capping the total number of levels in the tree to n +1 (Figure 1b). From this we derive that resolving a complete bifurcating lineage tree of k cells that originated from a single cell requires a system where k ≤ mn and . It is important to emphasize that these equations establish the upper theoretical bounds of barcoding capacity; in practice, effective barcoding capacity would be lower because multiple elements may mutate at the same time or a cell may divide without any mutation. Nevertheless, these equations show that meaningful lineage analysis will require more than one barcoding element since n = 1 results in a tree with only two levels. In other words, only the combination of multiple barcoding elements can be used to decipher lineage information. Similarly, if barcoding elements create a very limited number of mutant alleles, such as m = 2, unique barcoding of each single cell will not be achievable.
Homing guide RNAs
In the MARC1 system, the barcoding elements are homing guide RNAs (hgRNAs). hgRNAs are modified versions of canonical single guide RNAs (sgRNAs) that target their own loci to induce double-stranded breaks15,16 (Figure 2a,b). The error-prone non-homologous end-joining (NHEJ) process17 can then introduce mutations in the hgRNA sequence (Figure 2b). Compared to sgRNAs, hgRNAs have a few important features as barcoding elements. First, as they target their own loci, they obviate the need for a separate target locus for each barcoding element (Figure 2a,b). Accordingly, more independent barcoding elements can be created by using multiple hgRNAs with distinct spacer sequences. In other words, it is straightforward to increase n. Second, hgRNAs can produce a substantially larger amount of allelic diversity compared to sgRNAs. In other words, hgRNAs generate a higher m. Previous measurements suggest this increase is approximately tenfold15. In the combinatorial context of n barcoding elements, a tenfold expansion results in a drastic increase in the total number of barcodes. For example, with only five barcoding elements, a ten-fold expansion of each element’s allelic diversity leads to a 100,000-fold expansion of the barcode diversity space.
Figure 2.
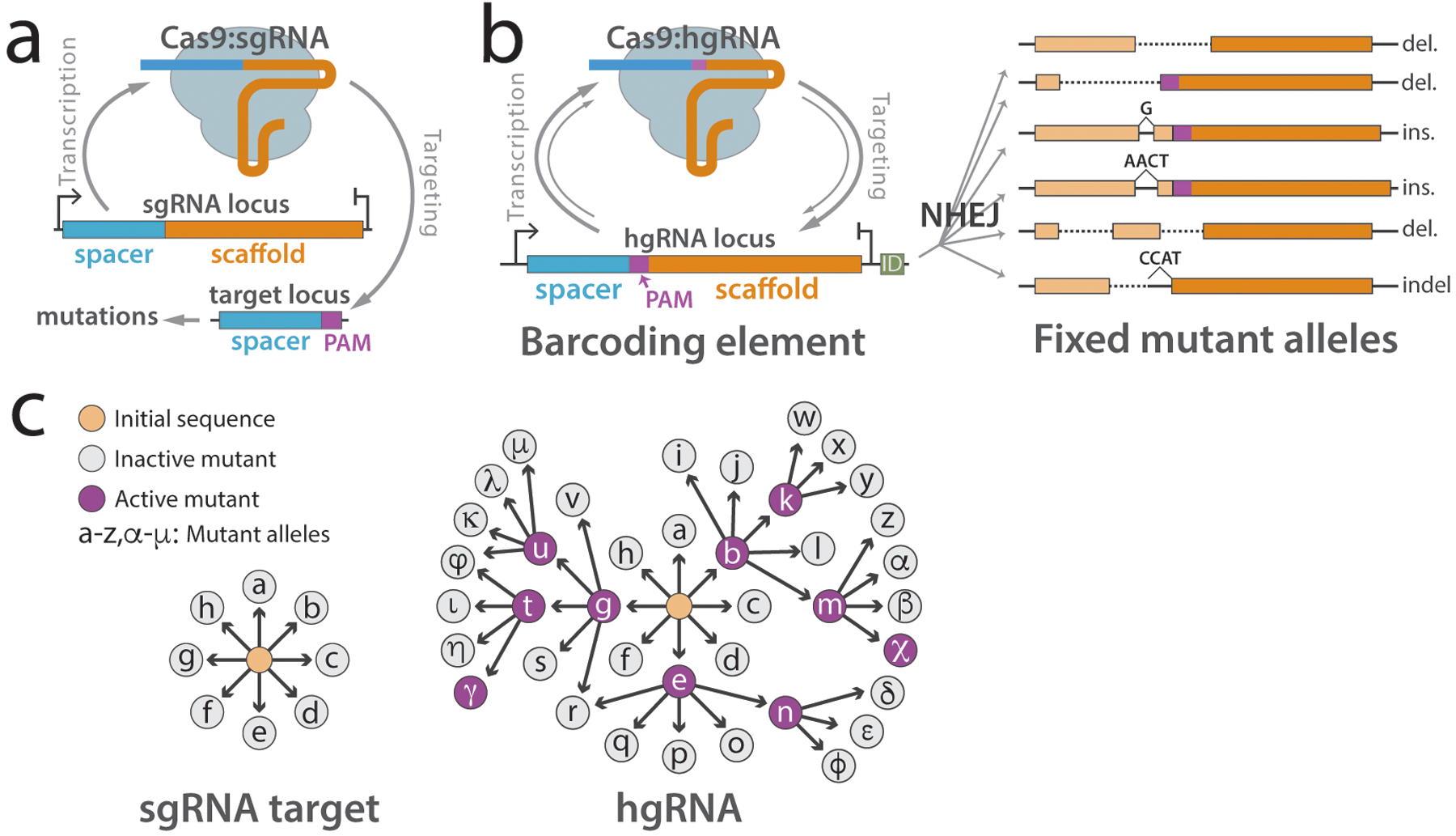
Principles of the homing CRISPR system. (a) Canonical CRISPR sgRNAs and Cas9 protein target the locus containing a spacer and PAM leading to mutations in the target locus. (b) Homing CRISPR system barcoding element. The hgRNA transcript expressed by the locus forms a complex with Cas9 protein and targets the locus for double-strand break. As the NHEJ repair system repairs the cut, it introduces mutations in the hgRNA locus. These random mutations can effectively act as barcodes. (c) sgRNAs (left) are limited to a single round of targeting and mutagenesis. hgRNAs (right) can undergo multiple rounds of targeting and mutagenesis, expanding their allelic diversity. Panel b is adapted with permission from ref. 7.
The increased allelic diversity of hgRNAs compared to sgRNAs is likely a consequence of the non-randomness of NHEJ outcomes. Several studies have demonstrated that NHEJ outcomes of Cas9-induced double-strand breaks in standard gene targeting applications lead to a limited and predictable set of mutations18–22. For instance, we and others have shown that single-base duplication of a specific position in the target site that results from staggered cutting by Cas9 is an overwhelmingly common outcome7,23–25. This non-randomness greatly limits the effective diversity of mutant alleles per barcoding element (m) that can be produced by an sgRNA at its target site (Figure 2c). However, unlike sgRNAs, which cease to match their target after the first round of mutations, hgRNAs can proceed for multiple rounds of mutagenesis before being inactivated by losing the protospacer adjacent motif (PAM) or another essential part. This allows hgRNAs to develop beyond the initial limited set of mutant alleles and create a larger diversity of mutant alleles than canonical sgRNAs (Figure 2c). We currently only use the final observed mutant hgRNA sequence as the mutant allele of the barcoding element without making any assumptions about its prior history. However, future work may make it possible to decipher the evolutionary path of an hgRNA sequence through multiple rounds of self-targeting and mutagenesis, thus expanding the lineage information that can be extracted from each barcoding element.
MARC1 founders and their lines
We have created two similar mouse lines with hgRNA barcoding elements which we refer to as MARC1 lines. Each line carries multiple distinct hgRNA barcoding elements that are randomly scattered throughout the genome. These hgRNAs are dormant, but when a MARC1 mouse is crossed with a mouse that expresses the Cas9 nuclease, they are activated in the progeny and lead to combinatorial barcoding.
Each MARC1 line was started by a single transgenic founder that carried multiple hgRNAs in the heterozygous state. To create the MARC1 founders, a library of hgRNAs with variable spacer lengths and sequences was assembled (Figure 3a). The library contained a 10-base identifier downstream of the hgRNA scaffold to allow for hgRNA identification in the case of a large deletion. It also contained common primer-binding sites to allow for easy amplification of all hgRNAs simultaneously. This library was transfected to mouse embryonic stem (mES) cells to result in multiple integrations per cell7. Transfected mES cells were injected into blastocysts to generate several chimeric male mice (Figure 3b). Two of these chimeric males, 1763PB3 and 1763PB7, had more than fifty hgRNAs integrated into their germlines’ genomes and were chosen as the MARC1 founders; their progeny respectively became the PB3 and PB7 MARC1 lines. While the PB7 founder has been previously described7, the PB3 founder is introduced here. Features of both founders are detailed here to provide a comprehensive picture.
Figure 3.
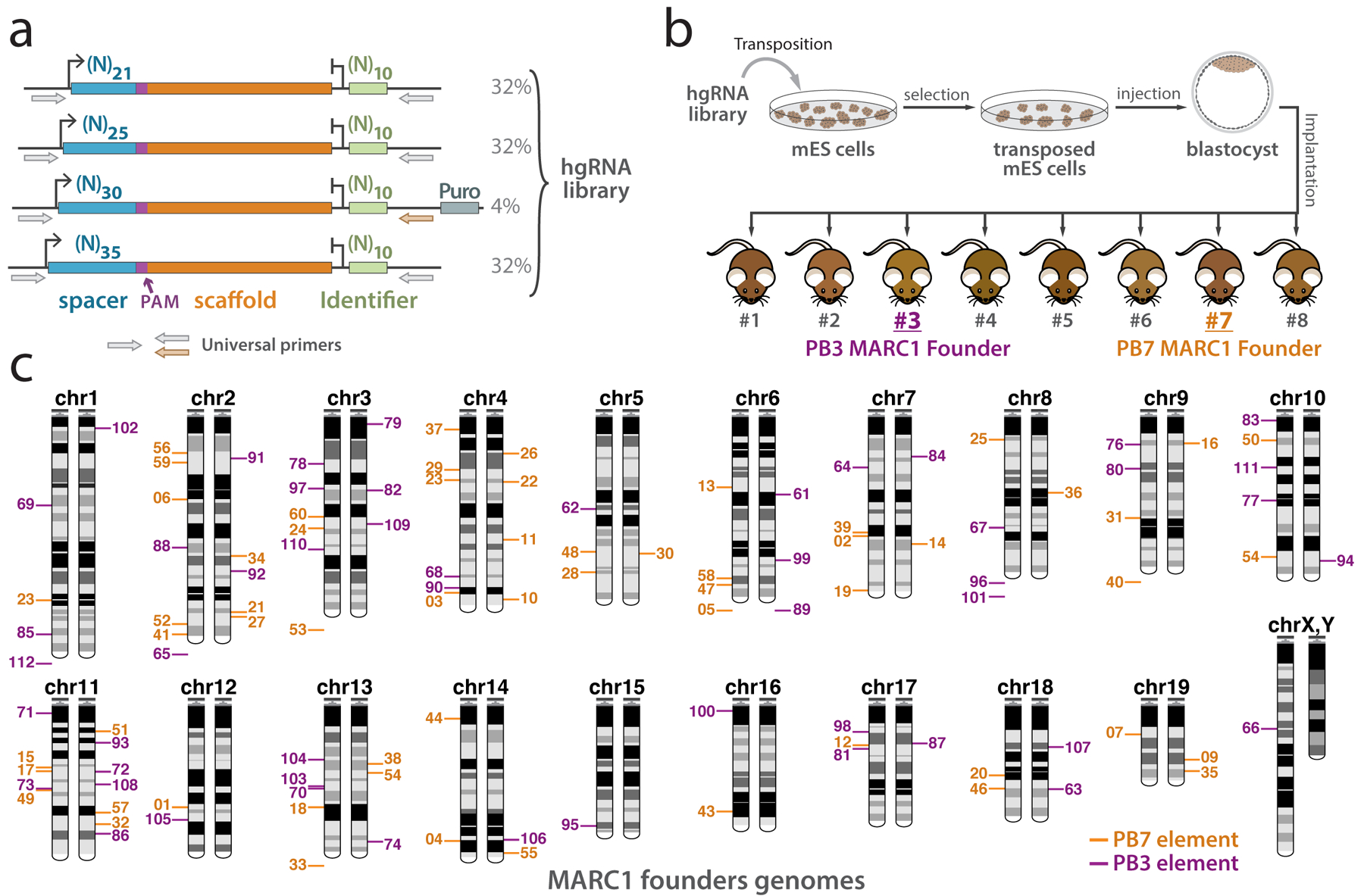
Strategy to generate mice with multiple hgRNA integrations. (a) Design of the PiggyBac hgRNA library for creating a transgenic mouse. Four hgRNA sublibraries with 21, 25, 30, and 35 bases of distance between transcription start site (TSS) and scaffold PAM were constructed and combined. The spacer sequence (blue box) and the identifier sequence (green box) were composed of degenerate bases. (b) Blastocyst injection strategy for producing hgRNA mice. The hgRNA library was transposed into mES cells. Cells with a high number of transpositions were enriched using puromycin selection and injected into E3.5 mouse blastocysts to obtain chimeras. Chimeras 3 and 7 were chosen as PB3 and PB7 MARC1 founders respectively. (c) Chromosomal positions of all 54 PB7 (orange) and 47 PB3 (purple) hgRNAs whose genomic position was deciphered in the MARC1 founders. Bars on the left or right copy of the chromosome indicate the hgRNAs that were linked on the same homologous copy. hgRNAs whose exact genomic position is not known but whose chromosome can be determined on the basis of linkage are shown below the chromosome. Puro, puromycin resistance. Panels b and c are adapted with permission from ref. 7.
Each MARC1 founder was characterized by first sequencing its somatic hgRNA loci. 60 different hgRNAs were identified in the PB7 founder and another 52 in the PB3 founder (Supplementary Table 1). PB7 hgRNAs were numbered from 1 to 60 and PB3 hgRNAs were numbered from 61 to 112. Each hgRNA has a unique 10-base identifier and a different spacer sequence. To identify germline-integrated hgRNAs, we crossed founders with multiple females and analyzed more than one hundred F1 offspring for each founder. For both founders, all hgRNAs were transmitted through the germline. For PB7 hgRNAs, 58 of the 60 showed a Mendelian inheritance pattern, appearing in about 50% of the offspring (Supplementary Table 1). The remaining two were inherited to about 75% of the offspring, which can be explained by the integration of the same hgRNAs in two unlinked loci in the genome. To determine how many injected stem cells gave rise to each founder’s germline, we also compared the co-inheritance frequencies of the hgRNAs (Supplementary Figure 1). We found no mutually-exclusive co-segregating groups of hgRNAs in either founder (Supplementary Figure 1a), indicating that the entire germline in each founder was derived from a single injected stem cell and is thus genetically homogeneous.
The co-inheritance analysis also revealed groups of hgRNAs that deviate from an independent segregation pattern, suggesting linkage on a chromosome (Supplementary Figure 1b). To determine the exact genomic location of hgRNAs, we sequenced the regions immediately flanking the hgRNAs in both founders as described in the previous publication7, allowing us to determine the genomic positions of 54 of the hgRNAs in PB7 and 47 of the hgRNAs in PB3 with varying levels of confidence (Supplementary Table 1). By analyzing the linkage disequilibrium (Supplementary Figure 1b), we were able to phase hgRNA haplotypes (Supplementary Figure 1c). Combined, this data allowed us to decipher the cytogenetic location of most hgRNAs in each MARC1 founder with a high degree of confidence (Figure 3c).
MARC1 founders and their progeny were fertile, had normal litter sizes, and presented no morphological abnormalities. Their hgRNAs are inherited at the expected Mendelian fractions and are not positioned in coding exons. These observations suggest that hgRNA insertions are not deleterious in the heterozygous state. Therefore, each founder was used to establish a separate line. To start these lines, we first crossed each founder with multiple wild-type females to obtain a first generation (F1). To obtain the following generation (F2), we genotyped all F1 progeny, and arranged crosses between multiple F1 pairs to maximize the number of expected hgRNAs in F2 progeny. The same strategy was applied to F2 to obtain F3 and continues to maintain each founder’s line separately. As the wild-type females used to create F1 were from both CD1 IGS and C57BL/6J strains, MARC1 mice have a mixed background. Furthermore, the animals will only have a subset of the hgRNAs from their founder.
MARC1 hgRNA activity profiles
To analyze the activity of MARC1 hgRNAs in the presence of Cas9, we crossed MARC1 males with Rosa26-Cas9 knock-in females26, which constitutively express Cas9, and collected samples from the resulting progeny at various embryonic stages or the adult stage (Supplementary Table 2). The hgRNA loci in each sample were then amplified and sequenced as a pool. The identifier sequence was used to group the sequencing reads corresponding to the same hgRNA and analyze the fraction of mutated spacers (Figure 4a). The results show a wide range of activity among the hgRNAs in PB3 and PB7 founders, with some mutating quickly after Cas9 introduction in almost all cells while some others show no activity (Figure 4a, Supplementary Table 3). Based on these activity profiles, we classified hgRNAs into four general groups (Figure 4b): “Fast” hgRNAs are mutated in at least 50% of the cells in each sample by E3.5 and are mutated in almost all (>95%) cells by E8.5; “slow” hgRNAs are mutated in a minority of cells even in the adult stage; “mid” hgRNAs are intermediates between “fast” and “slow” as they continue to mutate throughout embryonic development and are mutated in almost all cells only in later embryonic stages or in the adult animals; the remaining hgRNAs appear to be inactive with this level of Cas9 expression, with a mutation rate below 2% in all samples (Supplementary Table 3). Because the mutagenesis process is stochastic, this classification only describes the average behavior of each hgRNA. Accordingly, in a given barcoded mouse, a “mid” hgRNA may mutate early in some cells and late in others. However, we observe consistent average behavior for each hgRNA between different embryos (Supplementary Figure 2). Moreover, different tissues within the same embryo show a consistent average of hgRNA mutation levels (Supplementary Figure 3).
Figure 4.
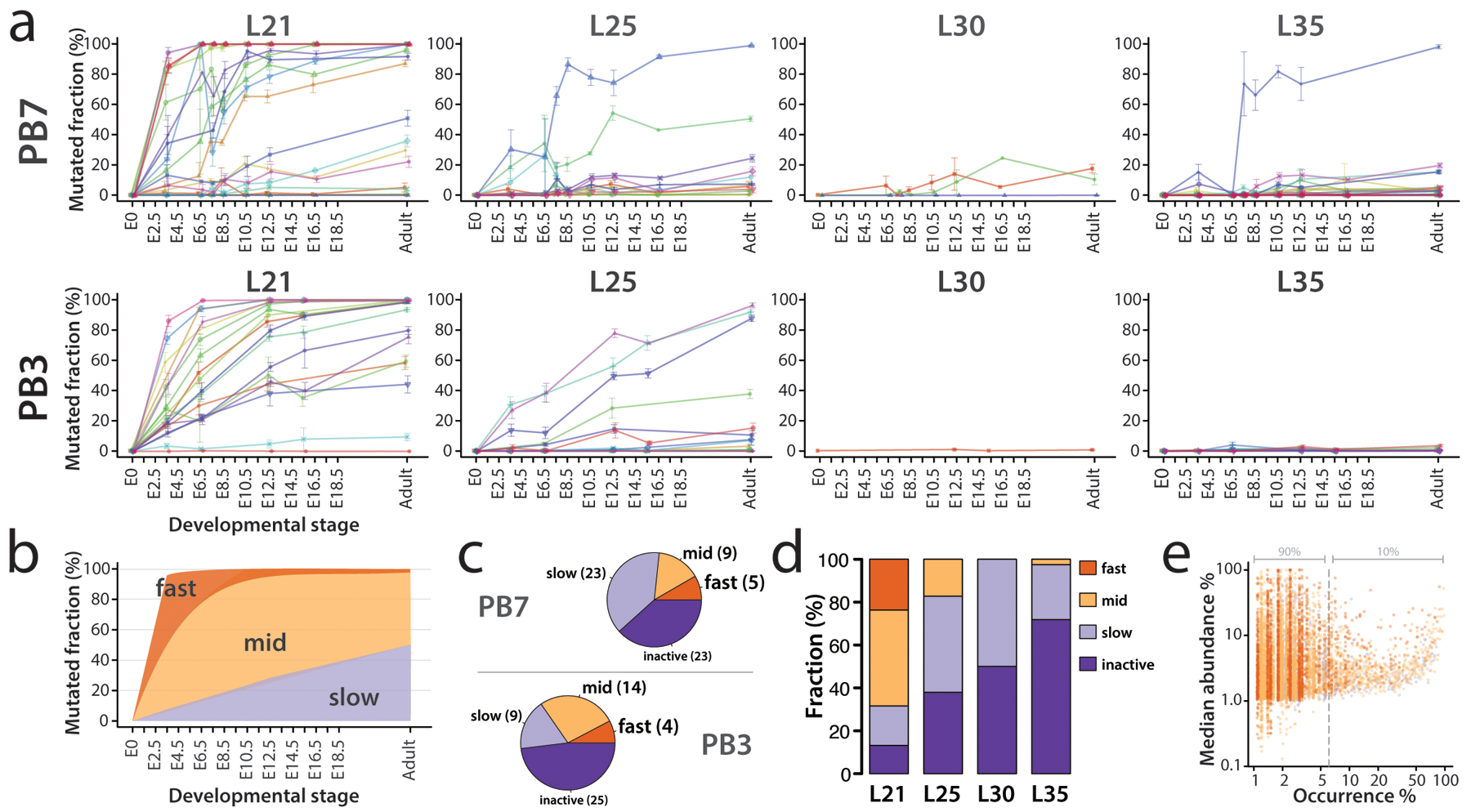
Activity of MARC1 hgRNAs. (a) Activity profiles of all 60 PB7 and 54 PB3 hgRNAs in embryonic and adult progenies of the MARC1 founder crossed with Cas9 knock-in females, broken down by hgRNA length. The fraction of mutated spacers in each hgRNA was measured. Lines connect the observed average mutation rates of the same hgRNA at different timepoints. Means ± SEMs are shown (N is different for each value; see Supplementary Table 2). See Supplementary Table 3 for numerical values of the plot. Adult mice were sampled via ear punch at 21 days old. (b) Range of activity profiles of each hgRNA class across progeny of both MARC1 founders crossed with constitutive Cas9 knock-in females. (c) The breakdown of hgRNA classes in the PB7 and PB3 lines; parentheticals represent absolute counts of each class. (d) Classification of combined PB3 and PB7 hgRNAs based on their activity profile in (a), broken down by length. (e) Observed distribution of all mutant alleles for all MARC1 hgRNAs shows that a majority of mutant alleles are unique. Each dot represents an observed mutant allele of a given hgRNA. Results for the combined set of all 112 hgRNAs are overlaid. The x-axis corresponds to the percentage of mice in which a specific allele was observed. The y-axis corresponds to the abundance of each allele when present. For example, a value of (5%, 10%) would indicate that a given mutant allele was observed in 5% of all barcoded mice inheriting the corresponding hgRNA identifier and with an abundance of 10%, on median. Dots are colored based on hgRNA activity profiles in accordance with the color key in d. Supplementary Table 4 contains the numeric breakdown for all observed alleles. L21, L25, L30, L35: hgRNAs with spacers of length 21, 25, 30, and 35 bases respectively. Top row of plots in panel a is adapted from ref. 7 with permission.
The breakdown of hgRNA classification shows a different distribution for the two founders (Figure 4c) with PB7 having more “fast” and “slow” and PB3 having more “mid” hgRNAs. In general, hgRNA activity shows an association with its length (Figure 4d), with longer lengths resulting in reduced activity levels. Additionally, a high diversity of mutant alleles is produced by each hgRNA (Supplementary Table 4). About 250 distinct mutant alleles were observed per “fast” and “mid” hgRNA with about 77% of these alleles observed in only one barcoded mouse out of an average of fifty-six (Supplementary Table 4). In all, the high diversity of hgRNA mutation outcomes resulted in the majority of possible alleles being detected in only a small fraction of mice analyzed (Figure 4e). These results indicate that a MARC1 mouse can be crossed with a line carrying Cas9 to obtain developmentally barcoded mice in embryonic and adult stages.
Obtaining and maintaining MARC1 mice
PB3 and PB7 MARC1 lines can be obtained from the Mutant Mouse Resource and Research Center (MMRRC) as MARC1-PB3 RRID:MMRRC_065812-UCD and MARC1-PB7 RRID:MMRRC_065424-UCD, respectively. The MMRRC follows the procedures described here to maximize the number of active hgRNAs in their MARC1 colonies, in each generation prioritizing, in order, fast, mid, and slow hgRNAs, followed by homozygosity in those hgRNAs in the same order. Animals provided by the MMRRC will be accompanied with genotype information that outlines their hgRNA combination. Each hgRNA may be in a homozygous or heterozygous state. These mice can be readily crossed with Cas9 mice for barcoding experiments and re-ordered as needed. Groups that require their own MARC1 colony in-house can start by obtaining a large number of MARC1 males and females from the MMRRC. We recommend crossing these mice to generate at least 30 progeny in each generation. All animals should then be genotyped according to the protocols described below. Based on the genotypes, an optimal set of crosses can be calculated that maximize the inheritance of the desired hgRNAs to the next generation. This procedure can be repeated for every generation to perpetuate the line.
Comparison with other methods; advantages and limitations
An important feature of the MARC1 system is the genetic integration of all necessary barcoding constructs in stable mouse lines. A number of the existing methods for CRISPR-based lineage tracing require injecting individual embryos with the necessary constructs (e.g. sgRNAs, Cas9, transposons) for barcoding5,6,8,10. In these strategies, the dilution of Cas9 and other elements caused by degradation and cell division may limit barcoding timespan and result in different levels of barcoding in different cells. Additionally, injecting mouse embryos for each experiment is technically challenging and requires expertise not available in many labs. MARC1 barcoding, on the other hand, only requires crossing animals—a process accessible to most labs—while ensuring identical barcoding elements and similar barcoding activity in all cells (Supplementary Figures 2,3).
Another feature of the MARC1 system is the distributed nature of barcoding elements in the genome, which minimizes unwanted loss of mutant alleles as a result of large deletions. This results in the independent accumulation of mutations in each element and combinatorial expansion of barcode diversity in this system. Some methods rely on an array of CRISPR targets in tandem5,6,10,12,27. While conferring the advantage of fewer loci for genotyping, tandem arrays are susceptible to large deletions that frequently span multiple target sites and delete previously recorded mutations. However, a drawback of MARC1 distributed array is the challenges associated with maintaining these independently-segregating loci in the line. The availability of the lines from a public repository reduces this burden for the end-user. Moreover, the behavior of individual MARC1 barcoding elements (e.g., mutation rate and mutational outcomes) are, to our knowledge, characterized more comprehensively than in any other system.
Another quality of the MARC1 system is the low transcriptional burden it imposes on cells because it mainly uses the RNA Polymerase III promoter to express short transcripts. As a result, barcoding elements are stably inherited with minimal effect on normal development. However, this design results in hgRNA transcripts lacking polyadenylation, which precludes their integration in the common single-cell sequencing platforms that only capture polyadenylated RNA. Many of the other CRISPR-based lineage barcoding methods have placed barcode arrays under RNA Polymerase II promoters, thus enabling simultaneous single-cell mRNA and barcode sequencing using broadly available high-throughput workflows8,10,12,27. Joint single-cell lineage and transcriptome analysis in the MARC1 system can be performed by sorting single cells into single wells and amplifying barcodes from genomic DNA followed by conversion of mRNA to cDNA as previously described6. However, this strategy suffers from a lower throughput and a higher degree of technical difficulty.
Finally, the novelty and complexity of developmental barcoding technologies combined with the intricacies of vertebrate development inspire questions about the best ways to apply them and the exact limits of their power. These questions can be fully addressed only after broader application of these approaches by the community. We hope that this manuscript aids the adoption of the MARC1 system as well as other barcoding strategies.
Experimental Design
Choosing a cross.
To carry out barcoding experiments, a MARC1 mouse should be crossed with a mouse carrying a Cas9 transgene to activate barcoding in the progeny. Design considerations should be given to both the Cas9-expressing and the MARC1 mouse to maximize the chance of obtaining desirable outcomes. For the Cas9-expressing mouse, lines that express this transgene at high levels in all tissues of interest should be considered. We routinely use the The Jackson Laboratory 024858 strain26 (JAX 024858) and its derivatives, which constitutively express Cas9. Using this strain activates barcoding very early in embryogenesis (likely the 2-cell stage7,28). We have obtained lower levels of barcoding when using the JAX 027650 strain (data not shown) but have not characterized other strains. For all studies, we recommend the MARC1–JAX024858 cross as the starting point because it typically leads to sustained barcoding throughout embryogenesis. Furthermore, the behavior of each hgRNA in this cross has been fully characterized (Figure 4).
Follow-up studies may be carried out with lineage-specific activation of Cas9, for example using Cre drivers in conjunction with a Cre-dependent Cas9 such as JAX 02655626, a floxed-STOP cassette Cas9 knock-in line (fCas9). In such cases, we recommend breeding the fCas9 line with the Cre driver line of choice to obtain a hybrid mouse with both Cre and fCas9. Crossing this Cre/fCas9 hybrid with a MARC1 mouse will result in barcoding of the progeny in lineages that express the Cre driver. However, this strategy may not always be possible. For example, if the Cre driver and fCas9 transgenes are in the same chromosomal locus, the Cre/fCas9 hybrid will not pass both transgenes to its offspring. If the Cre driver transgene has specific or non-specific expression in the germline, it will lead to Cas9 activation in the germline of the Cre/fCas9 hybrid and subsequent zygotic activation of barcoding when the hybrid is crossed with MARC1. In these cases, alternative arrangements should be considered, such as obtaining strains with Cas9 under a lineage-specific promoter.
After selecting the appropriate breeder on the Cas9 side of the cross, the appropriate MARC1 breeder should be obtained. To select the appropriate MARC1 breeder(s), the hgRNA composition of the mice should be considered. Because the line is genetically heterogeneous, each breeder will have a different number of fast, mid, and slow hgRNAs. This composition is either provided by the vendor (MMRRC) or can be determined by genotyping (see Procedures below). In all cases, inactive hgRNAs do not carry any information while a higher number of active hgRNAs leads to better outcomes7. For experiments where barcoding over an extended period of time is desirable, a higher fraction of mid and slow hgRNAs may be more appropriate.
To study specific genetic perturbations or mice with different genetic backgrounds for barcoding analysis, choosing the appropriate cross will depend on the number of mutant loci and their mode of inheritance. The more straightforward cases are those of autosomal dominant modifications. In such cases, the Cas9 line can be modified—by breeding or transgenesis—with the necessary alterations and crossed with MARC1 to obtain barcoded progeny with the desirable genetic modification. Recessive modifications, by contrast, would further necessitate modifying the MARC1 line. In such cases, breeding MARC1 with another line to introduce the desirable modification(s) can result in loss of hgRNAs unless a backcrossing scheme is implemented.
Setting up the cross.
When both sides of the cross have been selected, the cross has to be set up. We typically cross MARC1 males with multiple Cas9 females. This arrangement has two advantages. One advantage is that if embryos are being obtained, contamination with maternal tissue would not affect hgRNA barcode readouts because Cas9 females lack hgRNAs. Another advantage is that as MARC1 animals are often the more limited resource, multiple females can be crossed with the same MARC1 male simultaneously.
Irrespective of the exact arrangement of the cross, we recommend using multiple crosses and analyzing multiple progeny. In addition to the fundamental requirement to carry out experiments in replicates to establish rigor and reproducibility, several stochastic factors make it imprudent to rely on a single barcoded progeny for drawing conclusions. First, each heterozygous hgRNA in the MARC1 breeder has a 50% chance of segregating to the barcoded progeny; a particular progeny may therefore have far too few hgRNAs or an insufficient fraction of a specific hgRNA type for the lineages under analysis. Analyzing multiple progeny maximizes the chance of obtaining progeny with the appropriate number and composition of hgRNAs. Second, mutagenesis in hgRNAs is stochastic: a fast hgRNA may mutate later than expected or a slow hgRNA may mutate very early. This stochasticity means informative mutagenesis events may or may not take place at the appropriate time points for the lineages of interest in a single progeny. Analyzing multiple barcoded progeny increases the chance that several of them contain mutations that took place in an appropriate window of time with respect to the lineages under study. As an added advantage when analyzing multiple progeny, if the same set of samples are obtained from each barcoded mouse, and under the assumption that the lineages under investigation do not vary between mice, barcoding results from multiple progeny can be combined for calculating lineage trees.
Choosing samples to collect.
Choosing an appropriate set of samples to collect from each barcoded progeny is an aspect of experimental design that deserves extensive consideration. This choice is highly dependent on the biological process under investigation as well as the available methods of cell isolation and sequencing. It is important to consider samples that are not only practical to obtain but also informative in the context of the final output of the analysis. The final output of the experiment is information about the lineage relationship of samples. Most often, this information represents a phylogenetic-style tree in which each sample is a terminal branch. Whether the samples are single cells or populations of cells, barcoding experiments only address the relationship between branches — or the topology of the tree. Therefore, samples should be chosen such that the topology of this tree can address the hypothesis that drives the investigation.
Internal control samples.
In addition to the choice of samples being investigated, it is important to obtain samples of known lineage relationships as internal controls for each analyzed mouse. We recommend sampling control groups of cells whose lineage relationship to each other and to the cells under investigation is known. In most experiments, these “outgroup”29 controls can be readily obtained. We further recommend collecting duplicate samples of each population. This can be simply accomplished by splitting the obtained sample in two tubes after collection and processing them in parallel. In the end, these replicate samples should cluster together across the board. Together, these samples will serve as internal controls to validate the experimental and analytical aspects of the experiment. They further help determine if a particular embryo contained an adequate number of hgRNAs or mutated at an appropriate juncture. Finally, they enable a more straightforward interpretation of the lineage trees by grounding them in known lineage events.
Technical replicates.
Once samples of interest are obtained, genomic DNA should be extracted. The hgRNA loci can then be amplified using the Procedures outlined below. We recommend performing amplification and sequencing in two or three replicates. Ideally, these technical replicates would be entirely independent (i.e., separate amplification and sequencing). Such replicates are important because library amplification and sequencing are subject to a variety of PCR and sequencing errors and biases. Barcoding analysis is sensitive to these errors and biases, including cross contamination between samples, template switching during PCR, and index-swapping during sequencing. In our experience, sequencing all samples in replicates can effectively rule out or mitigate such issues. Once validated by comparison with each other, technical replicates can be combined to obtain deeper coverage of the sample they represent.
Procedure controls.
For the experimental procedure outlined below, we recommend that at least one no-template control sample, in which the first PCR reaction receives an empty buffer instead of a DNA template, be carried through the entire procedure in parallel to the samples and included in the final sequencing library. The sequencing results from the no-template controls, together with technical replicates described above, can help rule out or identify contamination during amplification steps or extensive index-swapping during sequencing.
Experimental design case study.
To help clarify the points above, we here describe a previously successful experiment as an example. In a previous publication7, we analyzed the order of commitment of brain progenitors to embryonic anterior–posterior and left–right axes (Figure 5). The neuronal cells under analysis in this case are derived from ectoderm. Therefore, we obtained samples of mesoderm-derived tissues (blood, muscle) as known outgroups. The fact that these outgroups formed a separate cluster as expected (Figure 5a) indicated that meaningful barcoding had taken place before our lineages of interest began to commit in the mouse under analysis. In addition to these outgroups, we collected duplicate samples from each population of interest (neurons from the left and right hindbrain, midbrain, and forebrain) by separating it in two tubes (s1 and s2). The fact that the s1 and s2 samples corresponding to each region of the brain clustered together across the board (Figure 5a,b) further indicated that meaningful barcoding had taken place during lineage commitments, and that sequencing of the barcodes was reproducible and not confounded by noise. Additionally, we obtained the same lineage tree in the other barcoded mouse we analyzed7. Together, these provisions in experimental design—duplicated sampling, outgroup controls, and analysis of multiple mice—enabled us to conclude that mice were properly barcoded and the obtained lineage tree was accurate.
Figure 5.
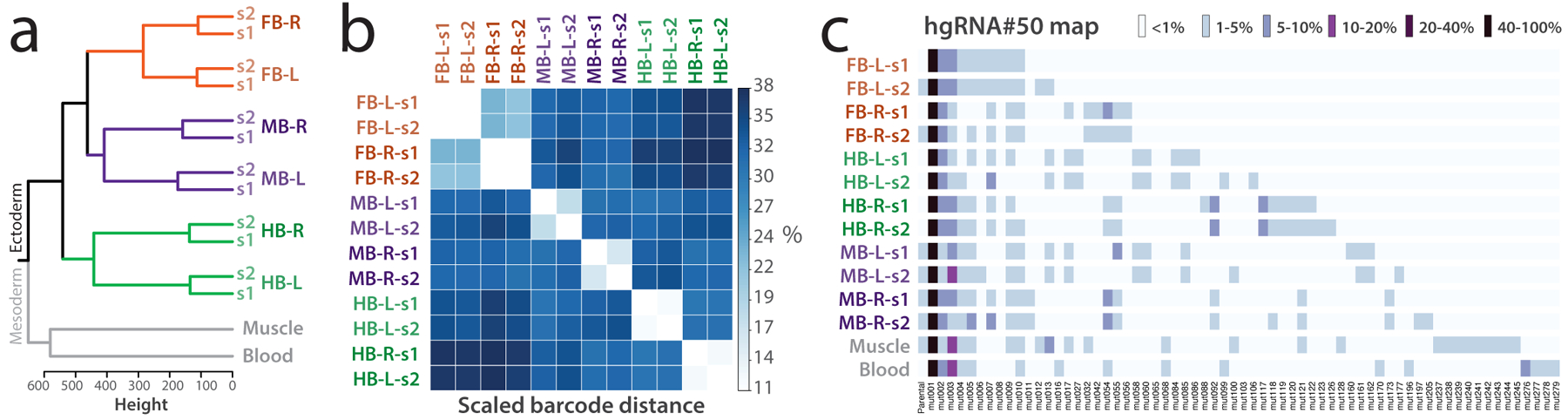
Example experimental design and results. (a) Outgroup selection and replicates provide internal controls. Clustering barcodes from 30 active hgRNAs in a twelve-month-old female mouse show the order of commitment to brain axes by neuronal progenitors. Mesoderm outgroups, in gray, clustered separately from ectodermal neuronal samples; duplicate samples (s1, s2) consistently clustered together, confirming consistency of barcoding across the board. Hierarchical clustering was carried out based on Manhattan distance of each sample’s full barcode (Supplementary Table 8) using Ward’s minimum variance method with squared dissimilarities (ward.D2)7. FB: Forebrain; MB: Midbrain; HB: Hindbrain; R: Right; L: Left. (b) Heatmap of the scaled pairwise barcode distances between all samples shown in a. The barcode distance is scaled such that a value of 0% corresponds to the presence of the same alleles at the exact same frequencies and a value of 100% corresponds to entirely non-overlapping sets of alleles. (c) Visual representation of a part of the barcode table showing the allele composition of one of the 30 active hgRNAs (hgRNA#50, ID: GTACACAATT) that contribute to the clustering in a. Only the 65 mutant alleles with 1% or more abundance in at least one sample are shown (among 302 in total). Full barcodes for all samples can be found in Supplementary Table 8. Panel a is reproduced with permission from ref. 7.
At a technical level, each sample (e.g., Forebrain-R-s1) was amplified and sequenced in three replicates which had a high level of agreement. Data from these technical replicates were then pooled for final analysis. Additionally, one no-template control was included for every ten amplification reactions, which verified an absence of cross contamination between samples. These technical controls ruled out various concerns with sample collection, amplification, sequencing, and analysis pipelines.
DNA extraction (Procedure Steps 2–4).
While we have recommended two DNA extraction strategies based on the size of the initial sample, any other strategy to extract and adequately clean up genomic DNA to allow for PCR amplification would be effective. When handling a small number of cells (less than 100) it is important to design the extraction procedure to maximize the amount of DNA that can be used as a template for PCR.
PCR amplification and sequencing (Procedure Steps 5–18).
The experimental procedures use PCR to amplify hgRNAs from genomic DNA, append sequencing adaptors to each hgRNA, and sequence them. The same experimental workflow is used both for genotyping purposes and barcode retrieval experiments. Based on the starting amount of DNA, one or two rounds of PCR may be required to amplify hgRNAs (Supplementary Figure 4a). We use a single PCR to amplify hgRNAs and add partial sequencing adaptors from template amounts corresponding to 100 cells or more (see Procedure Steps 9–11). For template amounts corresponding to 1–100 cells, we use an additional PCR for preamplification (see Procedures Steps 5–8). After hgRNA amplification, we carry out an indexing PCR for completing sequencing adaptors and giving each sample a unique index for pooled sequencing (see Procedure Steps 12 and 13). When the outlined procedures are carried out successfully, all of these reactions produce only specific products, obviating the need for any type of size-selection or purification between PCR amplifications. To achieve this outcome, and to minimize other PCR artifacts, it is critical to use real-time monitoring and halt reactions during the mid-exponential phase before the curve plateaus (Supplementary Figure 4b,c).
Data processing (Procedure Steps 19–26).
The data analysis steps process the sequencing output to determine the hgRNAs in each sample, as well as the mutant alleles for each hgRNA in each sample, and the relative abundance of each allele across all the samples analyzed in a given run. We have provided a pipeline for data analysis that takes raw sequencing results (FASTQ files) and converts them to a list of identifier–spacer pairs and indicates the frequency with which they were observed. This process is carried out in several steps that adjust for sequencing errors and amplification artifacts to distill the spacer mutations caused by NHEJ. The pipeline comprises 5 folders, numbered from 0 to 4 according to their order in the pipeline. It is designed to automatically act on all sample files in succession. The FASTQ sequencing files should be copied into the first folder (0) which already contains sequencing results from each of PB3 and PB7 founders as reference. There are filename requirements for the FASTQ files that are outlined in the Procedures below. Each of the folders after 0 has a code that acts on the files in the previous folders and creates outputs in its own folder (e.g., the code in folder 1 acts on files in folder 0 to create its output in folder 1). For simple genotyping purposes, a table containing the identifiers and their most commonly observed spacers are created in folder 3 (Supplementary Tables 5 and 6). If necessary, presence of a specific hgRNA in a sample can be confirmed using PCR with specific primers (Supplementary Table 7). For barcoding purposes, the table describing the filtered set of identifier–spacer allele pairs and their relative counts is produced in folder 4, as well as an output that describes what fraction of the spacers for each identifier in each sample were mutated compared to the founder mice. The pipeline compiles a barcode table with the abundance of each mutant allele in each sample (Figure 5c, Supplementary Table 8). In this table, each row corresponds to a sample and each column to a mutant allele of an hgRNA. The value of each cell is the abundance of the mutant allele in the sample. Therefore, each sample is represented by a vector. The pairwise distances between these vectors (e.g., Manhattan distance) represent the lineage distance of their corresponding samples. This table can thus form the basis for lineage tree derivation using neighbor joining7, maximum parsimony5, maximum likelihood30, or a combination of these approaches31. The choice of appropriate tree calculation strategy may differ for each application depending on the nature and number of samples and whether single cells or cell populations are being analyzed.
The Procedure provides detailed instructions for novice programmers to apply the pipeline to their results. Experienced users may want to take advantage of multithreading on a computing cluster to speed up data processing. The current version of the pipeline is provided as Supplementary Software. More detailed instructions, in addition to the most updated version of the pipeline, are maintained at our repository: https://github.com/Kalhor-Lab/MARC1-Pipeline. The repository also includes an executed example of the pipeline on a sample dataset as well as instructions for clustering the barcode table using a neighbor joining approach.
Materials
Biological Materials
MARC1 mice (Mutant Mouse Resource and Research Center; stock #065424 or #065812)
-
Cas9 mouse line of choice (e.g. Jackson Laboratories, stock #024858)
▲CAUTION All animal studies should be carried out in compliance with relevant government and institutional guidelines.
CRITICAL: All animal procedures were approved by the Johns Hopkins University Animal Care and Use Committee (ACUC) or Harvard University Institutional Animal Care and Use Committee (IACUC). Mice were housed in barrier cages free of excluded pathogens on a 12 hour light-dark cycle, accessed water at will through a drip and were fed onPicoLab® Rodent Diet 20 5053. Animals were monitored by facility staff and researchers and attended by on-call veterinarians. They were euthanized in their home cages according to institutional guidelines, and death ensured with a secondary means before tissue collection as appropriate.
Reagents
Ethanol (Sigma Aldrich, cat. no. E7023–500ML)
Primers (Integrated DNA Technologies, see Table 1 for details)
- DNA extraction kit
- Qiagen DNeasy Blood & Tissue Kit (Qiagen, cat. no. 69504)
- Lucigen QuickExtract™ DNA Extraction Solution (Lucigen, cat. no. QE09050)
Qubit dsDNA HS Assay Kit (ThermoFisher, cat. no. Q32851)
Qubit dsDNA BR Assay Kit (Thermo Fisher Scientific, cat. no. Q32850)
Qubit Assay Tubes (Thermo Fisher, cat. no. Q32856)
Water (UltraPure, nuclease-free; Thermo Fisher Scientific, cat. no. 10977023)
KAPA SYBR FAST qPCR Master Mix (2x; Roche, cat. no. 07959389001)
-
NEBNext Ultra II Q5 Master Mix (2x; New England Biolabs cat. no. M0544S).
▲CRITICAL STEP Alternate high-fidelity enzyme mixes can be adversely affected by the inclusion of SYBR Green at a 1x concentration.
SYBR Green I Nucleic Acid Gel Stain (10,000x; Invitrogen cat no. S7563)
-
Dimethyl sulfoxide (DMSO; Sigma-Aldrich; cat. no. 276855)
▲CAUTION DMSO readily penetrates skin and may be harmful by inhalation, ingestion, or skin absorption. Wear appropriate personal protective clothing and avoid contact.
NEBNext Multiplex Oligos for Illumina, 96 Unique Dual Index Primer Pairs (New England BioLabs, cat. no. E6440S)
E-Gel Agarose Gels with SYBR Safe DNA Gel Stain, 4% (Thermo Fisher Scientific, cat. no. A42136)
DNA Clean & Concentrator-5 (uncapped; Zymo Research, cat. no. D4003)
100 bp DNA ladder (New England BioLabs, cat. no. N0551S) or 50 bp DNA ladder (New England BioLabs, cat. no. N3236S)
PhiX Control Kit v3 (Illumina; cat. no. FC-110–3001)
-
MiSeq Reagent Kit v2, 300-cycles (Illumina, cat. no. MS-102–2002)
▲CAUTION For instruments and reagents required for MiSeq sequencing, refer to the manufacturer’s instructions or consult your sequencing core facility.
Table 1.
Primer sequences for PCR#0 and PCR#1
| Name | Sequence (5ʹ–3ʹ) |
|---|---|
| PBLib-preamp-F | aagtaataatttcttgggtagtttgcag |
| PBLib-preamp-R | gaaaaagccataccaatgggc |
| SBS3-PBLib-F | acactctttccctacacgac gctcttccgatctatggactatcatatgcttaccgt |
| trSBS3 | ctacactctttccctacacgac |
| SBS9-PBLib-R | tgactggagttcagacgtgtgctcttccgatct gccataccaatgggcccgaa |
| trSBS9 | gtgactggagttcagacgtg |
| SBS9-PBLib30-R | tgactggagttcagacgtgtgctccttccgatct ggagcgatcgcagatccttc |
Equipment
Pipettes
Sterile filter pipette tips
Real-time PCR system (StepOnePlus™ Real-Time PCR System; Thermo Fisher, cat. no. 4376600)
Real-time compatible PCR plates (MicroAmp™ Fast Optical 96-Well Reaction Plate, 0.1 mL; Thermo Fisher Scientific, cat. no. 4346907)
Optically clear real-time PCR compatible sealing films (Microseal ‘B’ Adhesive Sealing Film; Bio-Rad, cat. no. MSB1001)
Qubit fluorometer (Thermo Fisher Scientific, cat. no. Q33238)
E-Gel Power Snap Electrophoresis Device (Thermo Fisher Scientific, cat. no. G8100)
Benchtop microcentrifuge
Plate centrifuge
Illumina Miseq (Illumina, cat. no. SY-410-1003)
Software
A Unix based system such as OSX or Linux.
A standard installation of the R statistical software (version 3.6.1)
- R software packages
- VGAM (version 1.1.1)
- stringdist (version 0.9.5.1)
Analysis pipeline repository (Supplementary Software or https://github.com/Kalhor-Lab/MARC1-Pipeline for the most up-to-date version).
Reagent setup
Qubit dsDNA HS assay
Follow the instructions according to the manufacturer.
▲CRITICAL Use only freshly prepared calibrating solutions to measure accurate concentrations.
Oligos and primers
Resuspend IDT primers (Tables 1 and 2) to 100 μM in nuclease-free water. Dilute all primer solutions to working concentration of 10 μM in nuclease-free water or a primer dilution buffer of choice. Combine primers for PCR#1 as specified in Table 2. Primers can be stored at −20℃ for at least two years.
Table 2.
Primer mix compositions for PCR#1.
| Primer Mix | Primer | μM in primer mix |
|---|---|---|
| PBLib-F (10 μM) | SBS3-PBLib-F | 2.5 |
| trSBS3 | 7.5 | |
| PBLib-R (10 μM) | SBS9-PBLib-R | 1.67 |
| trSBS9 | 7.5 | |
| SBS9-PBLib30-R | 0.83 |
Q5 High-Fidelity Mix with SYBR Green
Mix 10 μL 10,000x SYBR Green with 90 μL DMSO to make 1,000x SYBR Green. Add 1,000x SYBR Green to a final concentration of 2x in NEBNext Ultra II Q5 Master Mix. Both solutions can be stored at −20℃ for up to 6 months, protected from light.
DNA Clean & Concentrator −5
Add 24 ml 100% ethanol to the 6 ml DNA Wash Buffer concentrate before use. Follow manufacturer’s instructions.
Procedure
▲CRITICAL This protocol is highly sensitive to cross-contamination between samples and between successive library preparations. We recommend strict separation of pre- and post-PCR areas and the use of 10% bleach or similar DNA decontamination solutions on work spaces before starting and after ending work.
▲CRITICAL All PCR steps should be monitored in real-time and halted at or before the midpoint of the exponential phase (Supplementary Figure 4b,c). Over-amplifying—that is allowing any reaction to proceed past the mid-exponential point—will increase PCR artifacts such as template-switching leading to spurious barcodes.
Sample collection
-
1
Collect an appropriate set of samples for analysis. For genotyping purposes, a toe or tail clipping of young mice (<14 days) or a 2mm of the ear pinna of older (>14 days) mice provides more than adequate genomic material. For barcode analysis, sample collection must be tailored to the application, but any strategy that results in enough material for later extraction of DNA, including Fluorescence-activated Cell Sorting (FACS) and tissue dissection, among others, would be appropriate. Examples of sample collection using FACS or dissection can be found in ref. 7.
DNA extraction and quantification
⚈ TIMING 1h hands-on
-
2To extract genomic DNA from each sample, follow option A if the sample consists of more than 1,000 cells or 1 μg tissue; follow option B if the sample consists of less than 1,000 cells or 1 μg tissue.
- A sample of more than 1,000 cells or 1 μg tissue:
- Extract DNA using the Qiagen DNeasy Blood & Tissue Kit (or equivalent) according to the manufacturer protocol.
- A sample of less than 1,000 cells or 1 μg tissue:
- Extract DNA using the Lucigen QuickExtract™ DNA Extraction Solution (or equivalent) according to the manufacturer protocol.
-
3
Quantify genomic DNA using the Qubit dsDNA BR Assay Kit or the Qubit dsDNA HS Assay Kit.
-
4
If sample concentration is greater than 5 ng/μL, dilute to 0.1–5 ng/μL with nuclease-free water.
▩ PAUSE POINT Samples can be stored in −20°C indefinitely.
Pre-amplification (PCR#0)
⚈ TIMING 2 hours
-
5
If template DNA concentration from previous steps is greater than 0.1 ng/μL, skip to step 9 (PCR#1).
-
6
Prepare PCR#0 on ice as indicated below in a 96-well plate compatible with your real-time thermocycler machine. Use a mastermix containing all components but genomic DNA for all samples and controls.
▲CRITICAL STEP We have found other commercially available PCR or real-time PCR mixes may perform differently in this experiment. Such mixes would require optimization.
▲CRITICAL STEP Include a genomic DNA negative control, using the solvent used in DNA extraction method above without any template DNA in it. This control will enable you to assess contamination.Component Amount (μL) Final Nuclease-free water to 10 μL KAPA SYBR FAST qPCR Master Mix (2X) 10 1X PBLib-preamp-F1, 10 μM 0.8 0.4 μM PBLib-preamp-R1, 10 μM 0.8 0.4 μM Genomic DNA 6 pg – 0.2 ng 6 pg – 0.2 ng Total volume 20 -
7
Run the PCR program as indicated in the table below in a real-time thermocycler with the lid heated at 105 °C.
▲CRITICAL STEP Monitor the amplification curves on the PCR machine and stop amplification during extension in the cycle when any sample reaches the mid-exponential phase of the amplification. In our hands, even for amplification from a single cell, no more than 30 cycles are necessary.Cycle Number Denature Anneal Extend 1 95°C, 3 min – – Repeat (2–31 or less) 95°C, 20 sec 60°C, 30 sec (read SYBR Green) 72°C, 10 sec -
8
Dilute each sample 10 to 100 fold in water. It will be helpful to equalize sample concentrations during this dilution. For example, samples that reach the mid-exponential phase should be diluted 100 fold. Others can be diluted inversely proportional to their amplification. The no-template negative control and samples whose curves do not show amplification should be diluted 10-fold. Diluted PCR products serve as templates in the next amplification step.
▩ PAUSE POINT PCR products can be stored in −20°C indefinitely.
hgRNA locus amplification (PCR#1)
⚈ TIMING 2 hours
-
9
Prepare PCR#1 on ice as indicated below in a 96-well plate compatible with your real-time thermocycler machine. Use a mastermix involving all components but genomic DNA for all samples and controls.
▲CRITICAL STEP We have found other commercially available PCR or real-time PCR mixes may perform differently in this experiment and would require additional optimization.
▲CRITICAL STEP Include a genomic DNA negative control, involving the solvent of DNA based on extraction method above. This control will enable you to assess cross-contamination.
▲CRITICAL STEP PCR#1 primers append the hgRNA amplicons with the necessary adaptors for subsequent indexing for Illumina sequencing in PCR#2.Component Amount (μL) Final Nuclease-free water to 10 μL KAPA SYBR FAST qPCR Master Mix (2X) 5 1X PBLib-F primer mix, 10 μM 0.25 0.25 μM PBLib-R primer mix, 10 μM 0.25 0.25 μM Genomic DNA
or
Diluted PCR#0 product2 0.2 ng – 10 ng
or
N/ATotal volume 10 -
10
Run the PCR program as indicated in the table below in a real-time thermocycler with the lid heated at 105 °C.
▲CRITICAL STEP Prevent overamplification by monitoring the amplification curves on the PCR machine, and stop amplification during extension in the cycle when any of the samples reaches the mid-exponential phase of the amplification. In our hands, no more than 28 cycles are necessary, with genomic DNA samples amplifying in as early as 20 cycles and pre-amplified PCR#0 products amplifying in as few as 10 cycles.Cycle Number Denature Anneal Extend 1 95°C, 3 min – – Repeat (2–29 or less) 95°C, 20 sec 64°C, 20 sec (read SYBR Green) 72°C, 10 sec ? TROUBLESHOOTING
-
11
Dilute each sample at least 10 and up to 100 fold in water. It will be helpful to equalize sample concentrations during this dilution. Samples can be diluted inversely proportional to their amplification; for examples, samples that reach mid-exponential should be diluted 100 fold. The no-template negative control and samples whose curves do not show amplification should be diluted 10 fold. Diluted PCR products will serve as templates in the next amplification step.
▩ PAUSE POINT PCR products can be stored in −20°C indefinitely.
Indexing PCR (PCR#2)
⚈ TIMING 2 hours
-
12
Prepare PCR#2 on ice as indicated below in a 96-well plate compatible with your real-time thermocycler machine. Use a mastermix involving all components but indexing primers and genomic DNA for all samples and controls.
▲CRITICAL STEP Include a no-template control carried through from the previous PCR.This control will enable you to assess contamination.
▲CRITICAL STEP Index each well uniquely by adding to it a different combination of forward and reverse indexing primers. Take note of the index combination each sample receives.
▲CRITICAL STEP If you have fewer than 8 samples in total, it is important to use the appropriate combination of indexes, as recommended by your indexing kit, to ensure proper color diversity and base-calling of indexes during sequencing.Component Amount (μL) Final Nuclease-free water 2.5 NEBNext Ultra II Q5 2x Master Mix with 2x SYBR Green 5 1X Dual Index Primer Pair (from NEBNext® 96 Unique Dual Index Primer Pairs for Illumina®), 10 μM 0.5 0.5 μM
(0.25 μM each primer)Diluted PCR#1 product 2 Total volume 10 -
13
Run the PCR program as indicated in the table below in a real-time thermocycler with the lid heated at 105 °C.
▲CRITICAL STEP Prevent overamplification by monitoring the amplification curves on the PCR machine and stop amplification during extension in the cycle when any of the samples reaches the mid-exponential phase of the amplification. In our hands, between a minimum of 10 and a maximum of 15 cycles is adequate for this step.Cycle Number Denature Anneal Extend 1 98°C, 30 sec – – Repeat (10–15) 98°C, 10 sec 64°C, 20 sec (read SYBR Green) 72°C, 10 sec ▩ PAUSE POINT PCR products can be stored in −20°C indefinitely.
? TROUBLESHOOTING
Library pooling and purification
⚈ TIMING .5 – 1h
-
14
Pool all PCR#2 products in one tube. You may equalize the representation of different samples by taking from each sample an amount inversely proportional to its amplification in PCR#2.
-
15
Purify the pooled library using the Zymo DNA Clean & Concentrator-5 kit, according to manufacturer instructions for PCR products.
▲CRITICAL STEP For elution of libraries with more than 20 samples, elute the final library in 25 μL. For pools of less than 20 samples, lower elution volumes of 10–15 μL may be appropriate to achieve reasonable final concentrations.
▩ PAUSE POINT The purified library can be stored in −20°C indefinitely.
Library quantification and sequencing
⚈ TIMING 20 hours (1–2h hands-on)
-
16
Measure the concentration of the library with the Qubit dsDNA HS Assay kit by following the kit manual.
-
17
Dilute 20–40 ng of the library in 20 μL of water. Also dilute 20–40 ng of a 50 or 100 bp ladder in 20 μL water. Load diluted library and ladder side-by-side on a 4% Agarose E-Gel with SYBR Safe and run the gel using E-Gel electrophoresis device for 30 minutes. Evaluate library size using the E-Gel electrophoresis device transilluminator. Libraries are 330–380 bp in size. Incorrectly sized bands larger or smaller than expected may indicate heteroduplex formation or primer artifacts, respectively. Examples of successful, less successful, and failed library prep reactions and purifications are shown in Supplementary Figure 4d.
? TROUBLESHOOTING
-
18
Submit samples to the sequencing facility or sequence using an in-house setup. We typically sequence the samples on Illumina MiSeq using a V2 kit with 190 bp for Read 1 and 60 bp for Read 2. We load the libraries at a final concentration of 12 pM, and use a 20–25% PhiX spike-in to ensure adequate color diversity and base calling. We aim to obtain at least 10,000 reads per sample for genotyping and at least 100,000 reads per sample for barcoding; this is equivalent to approximately 2000 reads per barcoding element per sample.
▲CRITICAL STEP hgRNA amplicon libraries have a very low diversity of sequences compared to standard libraries, which challenges proper base-calling and cluster filtering by the Illumina analysis pipeline. It is critical to use no less than 10% Phi-X spike-in.
▲CRITICAL STEP Read 1 can be no shorter than 125 bp in order to fully cover the spacer region of the hgRNA and be compatible with our analysis pipeline. Read 2 should be no shorter than 40bp in order to fully cover the identifier region and be compatible with our analysis pipeline.
▲CRITICAL STEP Provide each sample’s index combination to the core facility or your sequencing pipeline to obtain demultiplexed FASTQ files after your run.
? TROUBLESHOOTING
Data download and processing
⚈ TIMING 30 minutes per sample
▲CRITICAL: These instructions are also reproduced in the README of the associated repository at https://github.com/Kalhor-Lab/MARC1-Pipeline. The repository also includes an executed version of the pipeline on an example dataset inside the ‘exampleRun’ folder.
▲CRITICAL The following analysis pipeline has been designed to run on a Unix-based system such as Linux or OS X.
-
19Using a terminal in your workstation, create a directory for running the analysis pipeline using the following command:
~$ mkdir analysis ~$ cd analysis
-
20Download the latest version of the data analysis pipeline manually from the public GitHub repository or clone with the following command in terminal:
~$ git clone https://github.com/Kalhor-Lab/MARC1-Pipeline.git ~$ cd MARC1-Pipeline/
-
21
Download the demultiplexed FASTQ files into the 0-raw_data subfolder of your analysis folder. For each sample, you should obtain one FASTQ file for Read 1 and another for Read 2.
▲CRITICAL STEP The Read 1 and Read 2 filenames are expected to have the same name followed by “_R1_001.fastq.gz” for Read 1 and “_R2_001.fastq.gz” for Read 2. For example: “[sampleName1]_R1_001.fastq.gz” and “[sampleName1]_R2_001.fastq.gz” would correspond to the same sample. If your analysis pipeline has produced a different arrangement, adjust the names accordingly.
▲CRITICAL STEP The 0-raw_data subfolder in the analysis pipeline comes with four reference fastq files: the Read 1 and Read 2 files from each of PB3 and PB7 founders. Process alongside your samples, as later in the pipeline these files will be used to measure mutation levels in each sample.
-
22If FASTQ files are compressed, run the following commands to decompress them.
$ cd 0-raw_data_PB $ gunzip *.gz
-
23Compile Read 1 and Read 2 sequences from each sample to a list of paired identifiers and their associated spacer sequences with the following commands.
$ cd ../1-pair_counting $ chmod +x submit.sh $ ./submit.sh
? TROUBLESHOOTING
-
24To correct the sequencing errors associated with the identifiers and find true identifiers in each sample32, run the following commands:
$ cd ../2-ID_err_correction $ Rscript Filter-identifiers.R
? TROUBLESHOOTING
-
25To correct for the sequencing errors in the spacer region and compile complete lists of identifier-spacer counts, run the following commands:
$ cd ../3-SP_err_correction $ Rscript Filter-spacers.R
▲CRITICAL STEP This analysis creates two important files for each sample. [sampleName]_genotypes.txt lists each identifier and the spacer most frequently associated with it. This file may be used for genotyping applications. [sampleName]_allpairs.txt lists all spacers associated with each identifier together with the number of times they were observed together. This file includes all observed pairs in sequencing results and does not apply any subjective filtering criteria. It may be used downstream for barcoding analysis applications. However, applying simple filtering criteria, as seen in the next step, can lead to a more appropriate representation of the dataset.
-
26To obtain a list of filtered identifier-spacer pairs and their counts, a report of mutation level in each hgRNA in each sample, and a table containing each sample’s full barcode (represented by the abundance of each hgRNA allele in it) run the following commands:
$ cd ../4-pair_filtering/ $ Rscript Final-Filtering.R
▲CRITICAL STEP The filtering criteria in this step were designed based on our experience and represent our best current understanding of how data should be filtered to account for sequencing and amplification errors. These filtering criteria and their parameters have been described in the headers of the source code and can be modified.
▲CRITICAL STEP The Final-Filtering.R file must be modified based on whether you are using the PB3 or the PB7 line. Instructions are noted in both the code and the GitHub readme.
▲CRITICAL STEP In some cases, some identifiers might get truncated by large deletions and the pipeline cannot automatically match them to an ID in PB3 or PB7 founders. In these cases, the pipeline will generate an ERROR.txt file and lists all such orphan IDs. Find the correct ID by comparing partial spacer and ID sequence to those of founders, then modify the Final-Filtering.R script following the instructions provided in the ERROR.txt and re-run step 26. If the truncated IDs remain unresolved, the pipeline excludes them from mutation level and barcode table files.
? TROUBLESHOOTING
Timing
Steps 2–4, DNA extraction: 1h
Steps 5–8, PCR#0: 2h
Steps 9–11, PCR#1: 2h
Steps 12–13, PCR#2: 2h
Steps 14–15, library pooling and purification: 0.5–1h
Steps 16–18, library quantification and sequencing: 20h (1–2 h hands on)
Steps 19–26, data download and processing: 30mins/sample
Anticipated results
PCR#0 and PCR#1
Real time PCR amplification should show a characteristic exponential curve by cycle 30 and typically earlier, and the no-template controls should remain flat.
PCR#2
Indexing samples should have reached the mid exponential phase by cycle 15 at the latest, and typically by cycles 10 to 12.
DNA quantification and sequencing.
Final libraries should be between 330–380 bp in length (Supplementary Figure 4d), and purified library pools should yield at least 1 ng/μL and typically 5–10 ng/μL, depending on sample pooling and elution volume after column purification.
Analysis of sequencing data
The analysis pipeline generates three important files, along with intermediates, from each pair of paired-end sequencing files that correspond to one sample. These important files are (1) ‘allpairs’, listing all identifier-spacer observations and their frequencies without any filtering, (2) ‘genotype’, listing each identifier and the most commonly observed associated spacer, (3) ‘filteredpairs’, listing high-confidence pairs and their abundances. The ‘allpairs’ files represent raw barcoding data tabulated from sequencing files. The ‘genotype’ files are useful for determining the hgRNAs that are present in each sample when genotyping MARC1 breeders or barcoded mice. An example of genotype is provided in Supplementary Table 5. The ‘filteredpairs’ files, which are filtered versions of ‘allpairs’ files, are an appropriate starting point for analysis of barcodes. An example of filtered pairs is provided in Supplementary Table 6. Complete example outputs of all files are included in the software repository.
The pipeline also generates files that combine information from multiple samples. The most important such files are the (1) ‘hgRNA-mutation-levels.txt’ file and the (2) ‘BarcodeTable.txt’ file. The mutation file reports the fraction of mutated spacers for each hgRNA in each sample, based on comparison with reference sequences from the PB3 or PB7 founder. This file can be helpful to determine the level of barcoding in each mouse. The barcode table file contains the abundance of all alleles across all samples. Figure 5c visualizes, as a heatmap, the part of the full table in our case study that corresponds to one hgRNA. In this table, rows correspond to samples and columns correspond to the union of all observed alleles for all hgRNAs in all samples (Supplementary Table 8). The value of each cell represents the abundance of the corresponding allele in the corresponding sample. If an allele is not observed in a sample, its corresponding abundance will be 0. The abundances of all alleles of the same hgRNA in each sample add to unity. When samples are single cells, only one allele should be present for each hgRNA and all the other alleles would have an abundance of 0. Therefore, each row in this table is a vector representing allele abundances in each sample. The Manhattan distances between two samples’ vectors can represent their lineage distance7 (Figure 5b). At the most basic level, this data can be used to identify each sample’s closest lineage relative based on its vector’s Manhattan distance from all the other samples.
In interpreting these barcoding data, it is important to consider that both the time and outcome of mutagenesis for each hgRNA are stochastic. An hgRNA may mutate in some cells very early during development, leading to mutant alleles that are abundant in many lineages (Figure 5c). In other cells, the same hgRNA may mutate in later stages, leading to lower frequency alleles that are specific to some lineages. Furthermore, certain mutant alleles are more likely to occur (Supplementary Table 4), leading to the possibility that independent mutagenesis events lead to the same outcome. The combination of these stochastic outcomes can inform details of lineage relationships between samples when considered within an appropriate statistical framework. For instance, neighbor joining (see ref. 7) (Figure 5a), maximum parsimony (see ref. 5), maximum likelihood (see ref. 30), or a combination of these approaches (see ref. 31) can derive lineage trees based on the barcode table. The choice of appropriate tree derivation strategy may differ for each application depending on the nature and number of samples and whether single cells or cell populations are analyzed. The repository includes instructions for clustering the barcode table using a neighbor joining approach based on Manhattan distances.
TROUBLESHOOTING
Troubleshooting advice can be found in Table 3.
Table 3.
Troubleshooting.
| Step | Problem | Possible reason(s) | Solution(s) |
|---|---|---|---|
| 10, 13 | Signal from negative controls during real time PCR monitoring | Cross contamination from current samples or previous libraries; primer artifact formation | Rigorous cleaning with 10% bleach of all work areas; reprepare library with additional care and maintain separate pre- and post-PCR areas; ensure template concentration is high enough that PCR#1 is run for fewer than 30 cycles and PCR#2 is run for fewer than 20 cycles. |
| 17 | Library size smaller than expected | Insufficient template in PCR#1 leading to amplification of a primer artifact instead of target amplicon; contamination | Increase template DNA for PCR#1 or incorporate PCR#0 into the workflow if working with small template amounts. Increase amplification cycles in PCR#1. Assess product size from PCR#1, and if applicable, PCR#0 to confirm amplification of the correct target. Ensure that PCR#1 and PCR#2 reach the early or mid-exponential phase of the amplification. Use negative controls to rule out contamination. |
| 17 | Library size larger than expected | Overamplification in PCR#2; contamination | Reduce amplification cycles in PCR#2. Use negative controls to rule out contamination. |
| 18 | Large variation in sample coverage | Inaccurate pooling strategy | Prepare sample pooling based on real-time PCR data; quantify separately before pooling for sequencing. |
| 18 | Substantially fewer sequencing reads than calculated | Insufficient cycles to add index adapters; inaccurate library quantification | Ensure PCR2 runs for at least 10 cycles; ensure Qubit quantification is not substantially over-estimating library concentration due to non-specific product formation. |
| 18 | Large fraction of unidentified reads in Illumina data | contamination by previous library preparations; poor read quality | Rigorous cleaning with 10% bleach of all work areas; reprepare library with additional care and attention to cross contamination. |
| 23 | Compilation of Read 1 and Read 2 sequences takes an unreasonably long time | Analysis entails a number of read-write operations | This step entails a large number of read-write operations. Choose a drive with faster I/O capabilities for analysis to shorten run time. |
| 23 | Compilation of Read 1 and Read 2 sequences fails | Analysis-provided Blat version may be incorrect | The pipeline includes the version of Blat that is current at the time of this publication. Future versions of Linux and OSX operating systems may require Blat to be updated to the latest version. Check the blat version provided against your operating system, and download the newest version of blat from http://hgdownload.soe.ucsc.edu/admin/exe/. Depending on your operating system, replace blat_linux or blat_osx file in the 1-pair_counting folder of the pipeline with the updated and renamed version. |
| 24 | Rscript for filtering identifier fails | Lack of compatible R versions | Check that R and R package version numbers are correct. |
| 26 | Rscript for final filtering reports orphan barcodes | Large deletions truncate part of an identifier | Check that the correct founder is selected for this sample. Additional orphan barcodes can be manually corrected in Final-Filtering.R. If the problem is pervasive, this may reflect large errors in the library preparation process or in Illumina sequencing, or cross contamination between samples or previous libraries. |
Supplementary Material
Supplementary Figure 1. Linkage disequilibrium of PB3 founder hgRNAs. (a) Coinheritance enrichment of all PB3 hgRNAs pairs, indicating elevated (purple color gradient) or diminished (blue color gradient) probabilities that two hgRNA are passed on to the same progeny. (b) Linkage index of all pairs for hgRNAs with uniquely identified genomic positions, indicating their possibility of residing less than 50 centimorgans on the same chromosome. (c) Coinheritance enrichment among linked hgRNAs on each chromosome. For the same data on PB7 founder hgRNAs, as well as derivation methods, see the original reference (Kalhor et al., Science, 2018).
Supplementary Figure 2. Variation in hgRNA mutation levels in different barcoded mice. Each data point in each hgRNA’s plot corresponds to the average mutated fraction of that hgRNA (Y-axis) in the samples taken from a single barcoded mouse at a given stage in embryogenesis (X-axis).
Supplementary Figure 3. Variation in hgRNA mutation levels within barcoded mice. Each column in each hgRNA’s plot corresponds to a single barcoded mouse at a given stage of embryogenesis (X-axis); each dot within a column corresponds to the observed mutated fraction of that hgRNA (Y-axis) in a different sampled tissue. The line connecting the dots indicates range. Vertical dashed lines separate embryos in different stages.
Supplementary Figure 4. Amplification layout and expected outputs for real time PCR and library preparation. (a) Layout of hgRNA template in genomic DNA together with the binding site of primers for different PCR steps. (b) Representative standard curve for genomic DNA concentrations without pre-amplification. We observe consistent non-specific product formation in no-template controls (NTCs) above cycle 30 (dashed line), likely due to primer length, and suggest loading at least 0.2 ng of template such that all samples amplify well before these products form. (c) Representative SYBR Green data from a PCR#2 of 7 different samples and 1 NTC. Blue vertical line denotes an appropriate cutoff point; dashed lines represent overamplification phase. (d) 4% agarose gel of pooled and purified libraries. Lane 1: over-amplified reactions with both correctly sized band and heteroduplex formation, Lane 2: correctly amplified reactions, Lane 3: non-specific PCR#1 products carried through to PCR#2. Lane 4: 100 bp ladder, bands at 100bp intervals.
Supplementary Table 2. Sample numbers for various developmental stages. Each sample per embryo represents the total number of unique tissue samples within that embryo. Values indicate the number of embryos sampled at each stage. When multiple samples were obtained from an embryo, different regions of both embryonic and extraembryonic tissues were targeted. For adult mice, samples correspond to P18–P25 stage and comprise ear notches. For E3.5 and E6.5, all samples are whole conceptus. For E7.5 samples are either whole conceptus or conceptus divided in 2 or 3 segments along its anterior–posterior axis. For E8.5 through E16.5, the placenta, parietal yolk sac, visceral yolk sac, head, heart, limb, or tail, or a combination thereof were collected. For these E8.5–E16.5 samples, cases with more than a single sample represent at least one sample from embryonic and another from extraembryonic tissues.
Supplementary Table 1. List of all hgRNAs in the MARC1 founder males. For each hgRNA, its assigned number, ID sequence, spacer sequence, TSS to PAM length, spacer length, observed inheritance probability, chromosomal location, and confidence level of the chromosomal location are given. A “+” sign in the location column indicates that the hgRNA is transcribed in the same direction as the positive strand of the mm10 reference sequence.
Supplementary Table 3. Average observed mutation level of each hgRNA during different stages of development. Values indicate percent mutated while “NA” indicates values that were not measured. Functional category based on this mutation level is also shown. Barcodes with less than 2% maximum mutation rate in all samples were assigned as “inactive”, those with 2–50% maximum mutation rate in all samples were assigned as “slow”, those with more than 90% average mutation frequency at any point before E9 were assigned as “fast”, and the rest were assigned as “mid.”
Supplementary Table 4. For each mutant spacer allele of each hgRNA, its sequence, occurrence probability (fraction of mice with that hgRNA which contained the allele), occurrence (number of mice out of total with that hgRNA which contained the allele), and median abundance (median of its abundance fraction in all mice in which it was observed) are shown.
Supplementary Table 5. Example genotyping output of the analysis pipeline, corresponding to a mutated sample. A [sampleName]_genotypes.txt file is produced for each sample It provides each identifier (# and ID) and the most commonly-observed associated spacer sequence (SP), forming an identifier–spacer pair; the absolute count of observations of this pair (count); the percent of this pair relative to all observed pairs in a sample; and the percent of this pair relative to all pairs with that identifier.
Supplementary Table 6. Example filtered pairs output of the analysis pipeline on a mutated sample. A [sample]_filteredpairs.txt file, produced for each sample, contains information on mutation levels and all observed pairs. This table contains the observed identifier with a corresponding observed spacer, the total observation count for that pair, the percent of that pair within all observations of that identifier, and the percent of that pair within all sample observations.
Supplementary Table 7. Suggested primer sets for specific amplification of each hgRNA in non-barcoded mice. Each pair is expected to produce an amplicon of 141–155 bp in length. Using each primer pair in a standard PCR on genomic DNA of MARC1 mice can establish the presence of the targeted hgRNA if an amplicon of the correct size appears. Absence of the correct amplicon in the PCR product would indicate absence of the hgRNA. These primer sets are not experimentally validated and may fail to amplify mutated hgRNA sequences, as they depend on the spacer region to bind.
Supplementary Table 8. Table of observed mutant allele frequencies in each sample in Figure 5. Rows correspond to samples and columns to each of the mutant spacer alleles that were observed. Alleles with less than 0.25% abundance in each sample are not included. parSP indicates spacer sequence observed in founder, mutSP indicates mutant spacer sequences observed in samples under analysis.
Acknowledgements
The authors would like to acknowledge Joel Dapello for comments on the manuscript and Leo Mejia for assistance in preparing sequencing libraries. The authors would further like to acknowledge the team at MMRRC and UC-Davis Mouse Biology Program for making it possible to distribute MARC1 lines. This work was supported by Johns Hopkins Institutional Funds, grants from the Simons Foundation (SFARI 606178, R.K.), the NIH (MH103910, G.M.C. and R01GM123313, P.M.) and the Intelligence Advanced Research Projects Activity (IARPA) via the Department of Interior/Interior Business Center (DoI/IBC) contract number D16PC00008.
Footnotes
Competing Financial Interests
The authors declare no competing financial interest.
Data Availability Statement
All sequencing data are available in the Sequence Read Archive under accession SRP155997; the parts of this dataset that correspond to the PB7 line were previously published7. Figures 3, 4, and 5 contain associated raw data for both PB3 and PB7 lines which are available in Supplementary Tables 1–4 and SRP155997.
Supplementary Software
Analysis pipeline is provided as a compressed file. See the manuscript for instruction on how to use this pipeline. The latest version of this pipeline may be obtained from https://github.com/Kalhor-Lab/MARC1-Pipeline.
References
- 1.Baron CS & van Oudenaarden A Unravelling cellular relationships during development and regeneration using genetic lineage tracing. Nat. Rev. Mol. Cell Biol 20, 753–765 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Wagner DE & Klein AM Lineage tracing meets single-cell omics: opportunities and challenges. Nat. Rev. Genet (2020) doi: 10.1038/s41576-020-0223-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sulston JE, Schierenberg E, White JG & Thomson JN The embryonic cell lineage of the nematode Caenorhabditis elegans. Developmental Biology vol. 100 64–119 (1983). [DOI] [PubMed] [Google Scholar]
- 4.Frumkin D, Wasserstrom A, Kaplan S, Feige U & Shapiro E Genomic variability within an organism exposes its cell lineage tree. PLoS Comput. Biol 1, e50 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKenna A et al. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 353, aaf7907 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alemany A, Florescu M, Baron CS, Peterson-Maduro J & van Oudenaarden A Whole-organism clone tracing using single-cell sequencing. Nature 556, 108–112 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Kalhor R et al. Developmental barcoding of whole mouse via homing CRISPR. Science 361, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spanjaard B et al. Simultaneous lineage tracing and cell-type identification using CRISPR-Cas9-induced genetic scars. Nat. Biotechnol 36, 469–473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frieda KL et al. Synthetic recording and in situ readout of lineage information in single cells. Nature 541, 107–111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan MM et al. Molecular recording of mammalian embryogenesis. Nature 570, 77–82 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Askary A et al. Publisher Correction: In situ readout of DNA barcodes and single base edits facilitated by in vitro transcription. Nat. Biotechnol 38, 245 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Bowling S et al. An Engineered CRISPR-Cas9 Mouse Line for Simultaneous Readout of Lineage Histories and Gene Expression Profiles in Single Cells. Cell (2020) doi: 10.1016/j.cell.2020.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalhor K & Church GM Single-Cell CRISPR-Based Lineage Tracing in Mice. Biochemistry 58, 4775–4776 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Behjati S et al. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature 513, 422–425 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kalhor R, Mali P & Church GM Rapidly evolving homing CRISPR barcodes. Nat. Methods 14, 195–200 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perli SD, Cui CH & Lu TK Continuous genetic recording with self-targeting CRISPR-Cas in human cells. Science 353, (2016). [DOI] [PubMed] [Google Scholar]
- 17.Lieber MR & Wilson TE SnapShot: Nonhomologous DNA end joining (NHEJ). Cell 142, 496–496.e1 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Allen F et al. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol (2018) doi: 10.1038/nbt.4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen W et al. Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair. Nucleic Acids Res. 47, 7989–8003 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shou J, Li J, Liu Y & Wu Q Precise and Predictable CRISPR Chromosomal Rearrangements Reveal Principles of Cas9-Mediated Nucleotide Insertion. Mol. Cell 71, 498–509.e4 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Guo T et al. Harnessing accurate non-homologous end joining for efficient precise deletion in CRISPR/Cas9-mediated genome editing. Genome Biol. 19, 170 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen MW et al. Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 563, 646–651 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zuo Z & Liu J Cas9-catalyzed DNA Cleavage Generates Staggered Ends: Evidence from Molecular Dynamics Simulations. Sci. Rep 5, 37584 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lemos BR et al. CRISPR/Cas9 cleavages in budding yeast reveal templated insertions and strand-specific insertion/deletion profiles. Proc. Natl. Acad. Sci. U. S. A 115, E2040–E2047 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taheri-Ghahfarokhi A et al. Decoding non-random mutational signatures at Cas9 targeted sites. Nucleic Acids Res. 46, 8417–8434 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Platt RJ et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raj B et al. Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat. Biotechnol 36, 442–450 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee MT, Bonneau AR & Giraldez AJ Zygotic Genome Activation During the Maternal-to-Zygotic Transition. Annual Review of Cell and Developmental Biology vol. 30 581–613 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilberg EW What’s in an Outgroup? The Impact of Outgroup Choice on the Phylogenetic Position of Thalattosuchia (Crocodylomorpha) and the Origin of Crocodyliformes. Systematic Biology vol. 64 621–637 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Feng J et al. Estimation of cell lineage trees by maximum-likelihood phylogenetics. BioRxiv doi: 10.1101/595215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones MG et al. Inference of single-cell phylogenies from lineage tracing data using Cassiopeia. Genome Biol. 21, 92 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beltman JB et al. Reproducibility of Illumina platform deep sequencing errors allows accurate determination of DNA barcodes in cells. BMC Bioinformatics 17, 1–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Linkage disequilibrium of PB3 founder hgRNAs. (a) Coinheritance enrichment of all PB3 hgRNAs pairs, indicating elevated (purple color gradient) or diminished (blue color gradient) probabilities that two hgRNA are passed on to the same progeny. (b) Linkage index of all pairs for hgRNAs with uniquely identified genomic positions, indicating their possibility of residing less than 50 centimorgans on the same chromosome. (c) Coinheritance enrichment among linked hgRNAs on each chromosome. For the same data on PB7 founder hgRNAs, as well as derivation methods, see the original reference (Kalhor et al., Science, 2018).
Supplementary Figure 2. Variation in hgRNA mutation levels in different barcoded mice. Each data point in each hgRNA’s plot corresponds to the average mutated fraction of that hgRNA (Y-axis) in the samples taken from a single barcoded mouse at a given stage in embryogenesis (X-axis).
Supplementary Figure 3. Variation in hgRNA mutation levels within barcoded mice. Each column in each hgRNA’s plot corresponds to a single barcoded mouse at a given stage of embryogenesis (X-axis); each dot within a column corresponds to the observed mutated fraction of that hgRNA (Y-axis) in a different sampled tissue. The line connecting the dots indicates range. Vertical dashed lines separate embryos in different stages.
Supplementary Figure 4. Amplification layout and expected outputs for real time PCR and library preparation. (a) Layout of hgRNA template in genomic DNA together with the binding site of primers for different PCR steps. (b) Representative standard curve for genomic DNA concentrations without pre-amplification. We observe consistent non-specific product formation in no-template controls (NTCs) above cycle 30 (dashed line), likely due to primer length, and suggest loading at least 0.2 ng of template such that all samples amplify well before these products form. (c) Representative SYBR Green data from a PCR#2 of 7 different samples and 1 NTC. Blue vertical line denotes an appropriate cutoff point; dashed lines represent overamplification phase. (d) 4% agarose gel of pooled and purified libraries. Lane 1: over-amplified reactions with both correctly sized band and heteroduplex formation, Lane 2: correctly amplified reactions, Lane 3: non-specific PCR#1 products carried through to PCR#2. Lane 4: 100 bp ladder, bands at 100bp intervals.
Supplementary Table 2. Sample numbers for various developmental stages. Each sample per embryo represents the total number of unique tissue samples within that embryo. Values indicate the number of embryos sampled at each stage. When multiple samples were obtained from an embryo, different regions of both embryonic and extraembryonic tissues were targeted. For adult mice, samples correspond to P18–P25 stage and comprise ear notches. For E3.5 and E6.5, all samples are whole conceptus. For E7.5 samples are either whole conceptus or conceptus divided in 2 or 3 segments along its anterior–posterior axis. For E8.5 through E16.5, the placenta, parietal yolk sac, visceral yolk sac, head, heart, limb, or tail, or a combination thereof were collected. For these E8.5–E16.5 samples, cases with more than a single sample represent at least one sample from embryonic and another from extraembryonic tissues.
Supplementary Table 1. List of all hgRNAs in the MARC1 founder males. For each hgRNA, its assigned number, ID sequence, spacer sequence, TSS to PAM length, spacer length, observed inheritance probability, chromosomal location, and confidence level of the chromosomal location are given. A “+” sign in the location column indicates that the hgRNA is transcribed in the same direction as the positive strand of the mm10 reference sequence.
Supplementary Table 3. Average observed mutation level of each hgRNA during different stages of development. Values indicate percent mutated while “NA” indicates values that were not measured. Functional category based on this mutation level is also shown. Barcodes with less than 2% maximum mutation rate in all samples were assigned as “inactive”, those with 2–50% maximum mutation rate in all samples were assigned as “slow”, those with more than 90% average mutation frequency at any point before E9 were assigned as “fast”, and the rest were assigned as “mid.”
Supplementary Table 4. For each mutant spacer allele of each hgRNA, its sequence, occurrence probability (fraction of mice with that hgRNA which contained the allele), occurrence (number of mice out of total with that hgRNA which contained the allele), and median abundance (median of its abundance fraction in all mice in which it was observed) are shown.
Supplementary Table 5. Example genotyping output of the analysis pipeline, corresponding to a mutated sample. A [sampleName]_genotypes.txt file is produced for each sample It provides each identifier (# and ID) and the most commonly-observed associated spacer sequence (SP), forming an identifier–spacer pair; the absolute count of observations of this pair (count); the percent of this pair relative to all observed pairs in a sample; and the percent of this pair relative to all pairs with that identifier.
Supplementary Table 6. Example filtered pairs output of the analysis pipeline on a mutated sample. A [sample]_filteredpairs.txt file, produced for each sample, contains information on mutation levels and all observed pairs. This table contains the observed identifier with a corresponding observed spacer, the total observation count for that pair, the percent of that pair within all observations of that identifier, and the percent of that pair within all sample observations.
Supplementary Table 7. Suggested primer sets for specific amplification of each hgRNA in non-barcoded mice. Each pair is expected to produce an amplicon of 141–155 bp in length. Using each primer pair in a standard PCR on genomic DNA of MARC1 mice can establish the presence of the targeted hgRNA if an amplicon of the correct size appears. Absence of the correct amplicon in the PCR product would indicate absence of the hgRNA. These primer sets are not experimentally validated and may fail to amplify mutated hgRNA sequences, as they depend on the spacer region to bind.
Supplementary Table 8. Table of observed mutant allele frequencies in each sample in Figure 5. Rows correspond to samples and columns to each of the mutant spacer alleles that were observed. Alleles with less than 0.25% abundance in each sample are not included. parSP indicates spacer sequence observed in founder, mutSP indicates mutant spacer sequences observed in samples under analysis.