

Abstract
Cardiotoxicity is one of the most serious side effects of cancer chemotherapy. Current approaches to monitoring of chemotherapy-induced cardiotoxicity (CIC) as well as model systems that develop in vivo or in vitro CIC platforms fail to notice early signs of CIC. Moreover, breast cancer (BC) patients with preexisting cardiac dysfunctions may lead to different incident levels of CIC. Here, we present a model for investigating CIC where not only induced pluripotent stem cell (iPSC)-derived cardiac tissues are interacted with BC tissues on a dual-organ platform, but electrochemical immuno-Aptasensors can also monitor cell-secreted multiple biomarkers. Fibrotic stages of iPSC-derived cardiac tissues have been promoted with a supplement of transforming growth factor-β1 to assess the differential functionality in healthy and fibrotic cardiac tissues after treatment with doxorubicin (DOX). The production trend of biomarkers evaluated by using the immuno-Aptasensors well-matches the outcomes from conventional enzyme-linked immunosorbent assay, proving the accuracy of our sensing platform with much higher sensitivity and lower detection limits for early monitoring of CIC and BC progression. Furthermore, the versatility of this platform is demonstrated by applying a nanoparticle-based DOX-delivery system. Our proposed platform would potential help allow early detection and prediction of CIC in individual patients in the future.
Keywords: organs-on-a-chip, electrochemical biosensor, iPSC-cardiac tissue, breast cancer, cardiotoxicity
Graphical Abstract
In this paper, a cardiotoxicity-on-a-chip platform containing iPSC-derived cardiac tissue communicated with breast cancer tissue and electrochemical immuno-Aptasensors for non-invasive monitoring of cell secreted multiple biomarkers. The suggested platform is capable of differentiating functionality and toxicity in healthy and fibrotic cardiac tissue after treatment with chemotherapy to step toward early detection and prediction of cardiotoxicity in individual patients.
1. Introduction
Breast cancer (BC) remains the most common malignancy in women and is currently a major health problem worldwide.[1] In this respect, several diverse efforts have been made over the years towards finding better approaches, such as developing new therapies and improving efficacy of chemotherapy or radiation therapy, for detection and treatment of BC.[2,3] As a result, death rates from BC have notably declined, and thus it seems now to be considered as a treatable disease.[4] However, BC treatments—especially chemotherapy (e.g. anthracyclines and targeted agents)—have toxic effects in various body systems, particularly leading to the risk of cardiotoxicity.[5,6] Cardiotoxicity is known as one of the most troubling side effect of BC chemotherapy and responsible for subclinical ventricular dysfunction or clinical heart failure, leading to substantial increase in morbidity and mortality for those undergoing the chemotherapy. For example, decrease in the pumping ability of the heart was observed in BC patients with common chemotherapy drugs such as doxorubicin (DOX, Adriamycin: cardiotoxicity in ~3–26 % of treated patients) and trastuzumab (Herceptin: cardiotoxicity in ~2–28 % of treated patients).[7] These risks of chemotherapy-induced cardiotoxicity (CIC) may result in limited therapeutic options including reduced doses-lowered efficacy in BC treatment. However, early detection of CIC including cardiomyopathy and arrhythmias seems difficult to achieve with current approaches, and their mechanisms still remain elusive.[8–10]
Several approaches such as myocardial biopsy, echocardiography, and magnetic resonance imaging have been employed to monitor variations in cardiac functions and structures during cancer chemotherapy.[9] However, using a biopsy is highly invasive, and all other approaches described above only allow the detection of late-stage cardiac failure, which would be irreversible. In this respect, detecting/monitoring biomarkers could be an interesting tool as a non-invasive, simple, and time-efficient way for early monitoring of CIC and BC progression before irreversible cardiac failure occurs.[11] Cardiac Troponin T and creatine kinase-MB isoenzyme (CK-MB) are widely employed biomarkers for monitoring of cardiac functionality.[12] Since it has been shown that changes in production rates of Troponin T and CK-MB are associated with myocardial injury, these biomarkers could be used for the early identification and prediction of CIC.[13–15] For example, a previous study—employing these biomarkers to prove the possibility of monitoring CIC in patients after the 2-week treatment with DOX—showed the production rate of both Troponin T and CK-MB increased.[15] In addition, overexpression of human epidermal growth factor receptor 2 (HER-2) is present in around 20 to 30% of BC cells, and these HER-2-positive BC cells have been shown to grow and spread faster compared to those negatively expressing HER-2.[16] Since the detection and inhibition of HER-2 could be a promising way to efficiently manage aggressive BC cells, it could be used as a potential biomarker for monitoring of the development and progression of BC.[17] Together, evaluating dynamic behaviors of essential biomarkers in response to treatment with drugs/agents could be of great importance in the early-stage detection and prediction of CIC and BC progression. Nevertheless, to achieve the goal, the development of robust, sensitive, and accurate bioassay platforms to assess multiple critical biomarkers is strongly required.
Organs-on-a-chip models have emerged for replacement of current treatment and investigation options based on pre-clinical in vivo and simple in vitro models that may not be reproducible, time-efficient, or relevant in humans and thus result in the failure to monitor serial assessments of diseases and their side effects.[18] These models—using three-dimensional (3D) microfluidic-based platforms for culturing living human cells—enable stimulating the activities, mechanics, and physiological response of different organ systems to step toward personalized medicine. Recently, cardiomyocytes derived from embryonic stem cells (ESCs) or induced pluripotent stem cells (iPSCs) have been employed in several CIC models,[19] suggesting that it may be possible to achieve improved clinical outcomes with minimal side effects in individual BC patients. In most cardiac models, however, little effort has been made to develop more advanced disease models such as considering individual patients with unknown cardiovascular history whose CIC may have different paces compared to those without cardiovascular dysfunctions.[20] Therefore, we reasoned that examining differential functionality and toxicity in healthy and diseased cardiac tissues after treatment with chemotherapy would provide opportunities to move toward a more personalized approach for the reliable prediction of early CIC. To this end, we developed a novel platform where an iPSC-derived healthy or diseased cardiac tissue is intercommunicated with a BC tissue and electrochemical (EC) immuno-Aptasensors can non-invasively monitor critical biomarkers from those tissues in response to chemotherapeutic drugs to verify the hypothesis that our proposed platform could be potentially suitable for early detection and prediction of CIC and BC progression. Furthermore, the platform was challenged to evaluate the performance of a class of new drug nanocarriers, the graphene-based yolk-shell magnetic nanoparticles (GYSM-NPs), as a smart and on-demand drug delivery system with pH- and temperature-dependent controlled release.
2. Results and Discussion
2.1. Design of the Heart-BC-on-a-chip Platform with the EC Immuno-Aptasensing System
3D microtissue culture systems based on polymeric scaffolds provide an opportunity to build in vitro models that can mimic important in vivo features including direct cell-cell contacts and biophysical cues present in the extracellular microenvironment.[21] In addition, culturing them in microfluidics-based perfusion systems allows investigating multi-organ interactions under more in vivo-mimicking conditions over simple in vitro and preclinical in vivo models.[22] To build the integrated cardiac-BC-on-a-chip platform, human iPSCs-derived cardiac spheroids composed of key cell types in the heart (cardiomyocytes, fibroblasts, and myofibroblasts) were prepared to encapsulate them into gelatin methacryloyl (GelMA) hydrogels (Figure 1a). To explore how the degree of cardiac fibrosis could influence different incident levels of CIC, we employed healthy cardiac tissues as well as fibrotic cardiac tissues generated with a supplement of transforming growth factor-beta 1 (TGFβ1), which is known to play a crucial role in the fibrosis induction.[23] In addition, BC spheroids were generated and cultured in soft hydrogel matrices mimicking mechanical properties of the in vivo breast tissues.[24] These BC tissues were interconnected with the healthy or fibrotic cardiac tissues using a microfluidics-based perfusion system. Here, we hypothesized that individual BC patients with preexisting cardiac fibrosis may have different incident levels of CIC compared to healthy BC patients. In order to examine our hypothesis, we first developed a dual-organ system composed of iPSC-derived cardiac and BC tissues for disease modeling of cardiac fibrosis (Figure 1a and b). To promote the fibrotic stage of cardiac tissues, TGFβ1 was supplemented to determine the differentiated functionality and toxicity in healthy and fibrotic cardiac tissues after treatment with chemotherapy. Moreover, early-stage detection and prediction of CIC are important to reduce the risks of CIC while maximizing treatment efficacy for BC. To this end, we also developed EC immuno-Aptasensors for highly sensitive, reliable, and low-volume monitoring of cell-secreted multiple biomarkers (Figure 1c). Since our sensors were based on a multiplexed sensing array, secretion rates of multiple biomarkers could be measured at the same time. Moreover, EC immuno-Aptasensing chips could be easily replaced on demand, so that continuous and non-invasive detection could be realized using multiple chips.
Figure 1. Design of the cardiac-breast cancer-on-a-chip platform with the EC immuno-Aptasensing system using multiplexed microelectrode array.
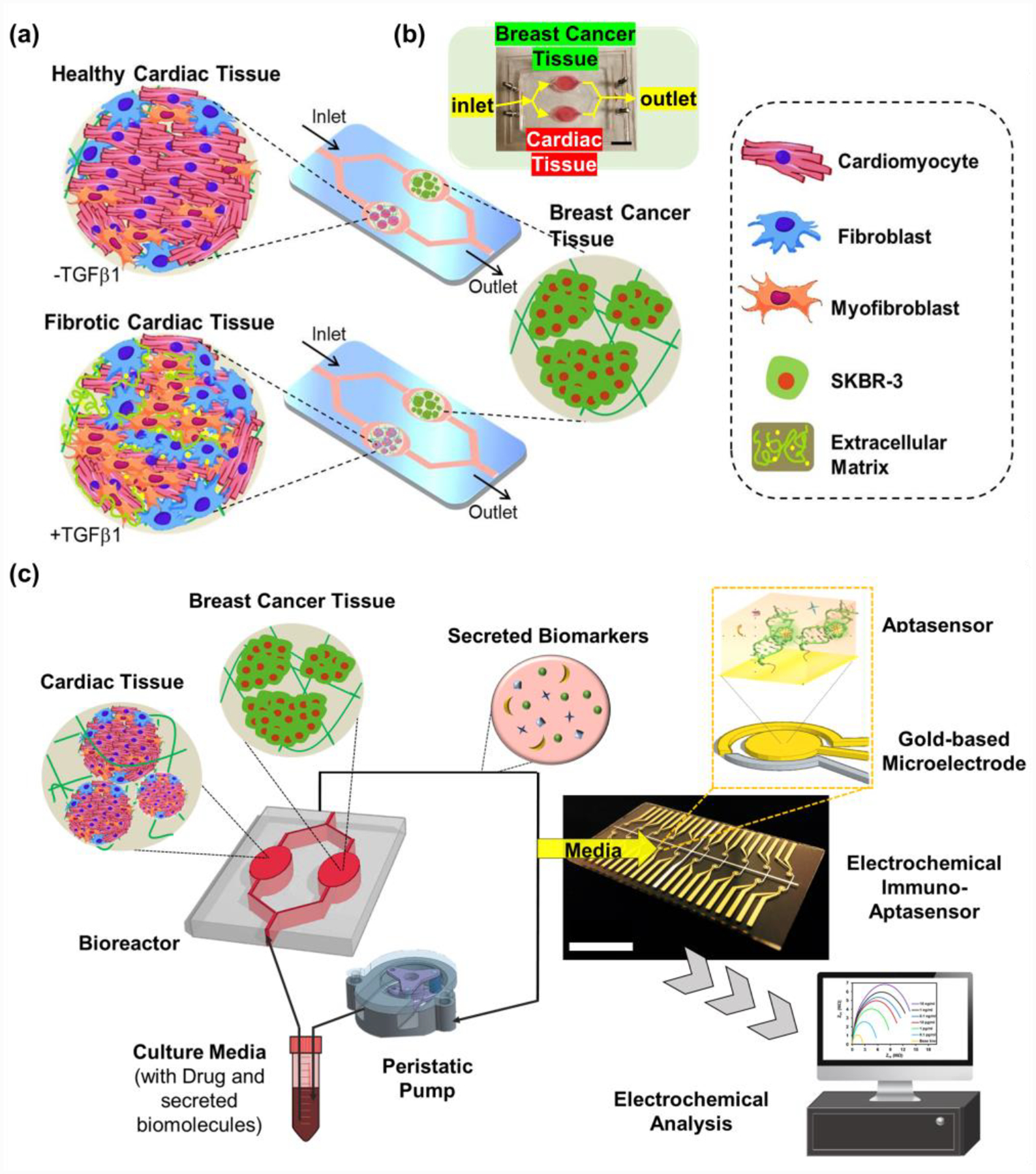
a) Schematic illustrating the development of healthy and disease cardiac model on chip for addressing the induced cardiotoxicity as a result of BC chemotherapy. b) A photograph of the fabricated microfluidic device having iPSC-derived cardiac tissues and BC tissues. c) Schematic illustration of the integrated cardiac-breast cancer-on-a-chip platform with the EC immuno-Aptasensing system using aptamers-functionalized biosensors on microelectrodes. Scale bars: 2 cm.
2.2. Engineering of Physiologically Relevant BC Tissue Models
Development of more physiologically relevant tumor tissue models plays a critical role in improving productivity of drug development with better predictivity of the efficacy and toxicity.[24] To engineer a physiologically relevant BC tissue model, BC spheroids were generated after culturing cells in microwells to achieve 3D culture with direct cell-cell interactions (Figure 2a). These produced spheroids were then encapsulated in hydrogels to generate our BC tissue model, which is physiologically relevant to the native breast tumor tissue. Here, we employed SK-BR-3—a HER-2-overexpressing human BC cell line—as a model cell line to build the BC-on-a-chip platform.[25] Since HER-2 overexpression has been generally associated with metastasis and poor prognosis of BC,[26] monitoring of HER-2 as a BC biomarker would be crucial for predicting responses and progression of BC cells with chemotherapies.
Figure 2. Engineering of BC tissue models where breast spheroids are encapsulated in hydrogels mimicking mechanical properties of native breast tissues.
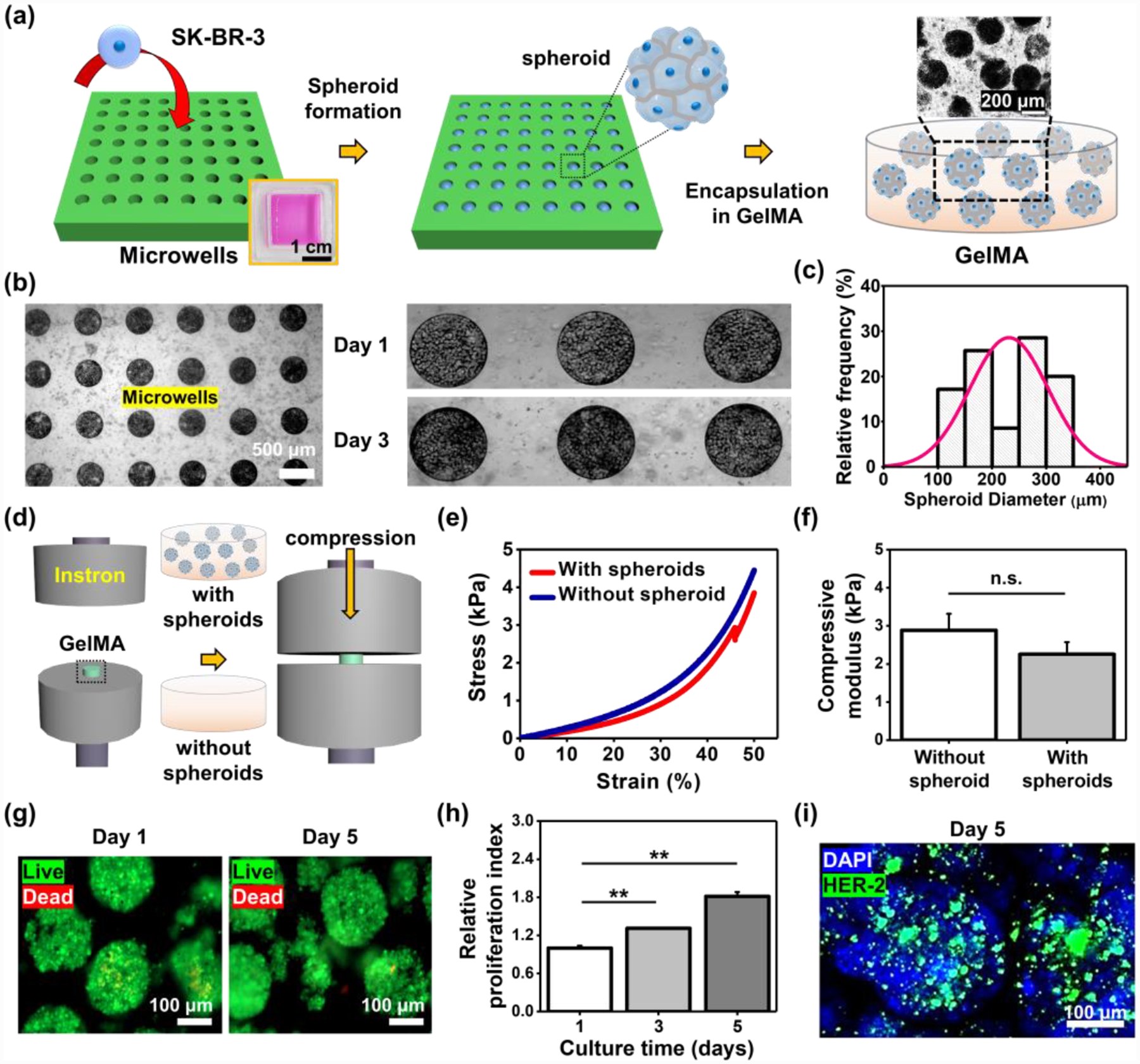
a) Schematic of the procedure to make physiologically relevant BC tissue models. b) BC spheroids cultured for 1 and 3 days in the microwells. c) Histogram of the spheroid dimeters after collecting them from the microwells on day 5 (N=80). d) Schematic illustration of measurement of the compressive elastic modulus of GelMA hydrogels with or without a presence of BC spheroids. e) Stress-strain curves of GelMA with or without BC spheroids. f) Measured compressive elastic modulus of GelMA with or without BC spheroids (N=3). g) Representative immunofluorescence images of live/dead staining for BC cells in spheroids after 1 and 5 days in culture. h) Proliferation index for BC cells in spheroids cultured for 5 days in GelMA hydrogels (N=3). i) Representative immunofluorescence images of DAPI/HER-2 staining of spheroids on day 5 after culturing in GelMA hydrogels. (One-way ANOVA with Tukey significant difference post-hoc test; **p<0.005). Error bars represent standard deviation.
First, we fabricated an array of polydimethylsiloxane (PDMS) microwells (300 μm in diameter and 250 μm in depth) using conventional photolithography techniques. To generate the BC spheroids, a concentration of 106 cells in 1 mL of medium was seeded onto the microwells, and the seeded cells were allowed to settle in the wells for 30 min. Non-adherent cells and adhered cells on the surface areas between the microwells were removed after washing with medium several times, so that only sedimented cells inside the microwells were cultured for 5 days to form spheroids (Figure 2b). Cells reached confluence in the microwells on day 1 and then aggregated with each other for the rest of days in culture. Spheroids were then collected from the microwells by pipetting multiple times on day 5, and the size distribution histogram of the collected spheroids was assessed. The result displayed that the average diameter of the spheroids was ~231 μm and their sizes were not monodispersed with a wide distribution, indicating the collection process with multiple pipetting might have an effect on the morphology of spheroids (Figure 2c). Previous reports revealed that the fabrication of spheroids within a range of 200–300 μm in diameter was desired to avoid the necrosis core, which is in line with the diameter range of our BC spheroids.[27] Next, the harvested spheroids were encapsulated in the photocrosslinkable GelMA hydrogels, and the compressive modulus of the GelMA hydrogels with or without encapsulated spheroids were then obtained using Instron (Figure 2d). The result exhibited no significant difference between hydrogels with and without the presence of spheroids (without spheroid: ~2.9 kPa and with spheroids: ~2.3 kPa) (Figure 2e and f). These measured values are within the range of mechanical properties of in vivo breast tissues,[24] indicating our suggested model is physiologically relevant for building BC tissues. GelMA is known to provide an excellent biological microenvironment to cells.[28] We then examined cell viability and proliferation in GleMA hydrogels. The encapsulated BC cells showed a high level of cell viability in GelMA, indicating the photoencapsulation process, the GelMA hydrogels, and their degradation byproducts had minimal effects on BC cell survival (Figure 2g). Moreover, BC spheroids encapsulated in GelMA showed increased proliferation over the 5 days (~1.3-fold and ~1.8-fold higher on day 3 and 5, respectively, relative to that on day 1) (Figure 2h). Furthermore, HER-2 staining for the spheroids in GelMA demonstrated that SK-BR-3 employed in this study displayed a high expression level of HER-2 (Figure 2i), which corresponds to the previous reports.[25]
2.3. Modeling of Healthy and Fibrotic Cardiac Tissues
As we hypothesized above, healthy and fibrotic cardiac tissues may have different levels of functionality and toxicity with chemotherapy. To test this hypothesis, iPSCs were differentiated into cardiac cells including cardiomyocytes and cardiac fibroblasts according to a previously established protocol.[29] The ratio between the differentiated cardiomyocytes and cardiac fibroblasts from iPSCs was examined using flow cytometry analysis (Figure S1, Supporting Information). Differentiated cells were stained with one of the representative cardiac fibroblast makers (vimentin) and the population of cardiac fibroblast phenotype were quantified based on the threshold level of unstained cells. We observed that ~46.5% of the differentiated cells were cardiac fibroblast phenotype, which is close to the cardiac fibroblast population in the native cardiac tissues (~60%).[30] It is known that the crosstalk between cardiomyocytes and fibroblasts is crucial for cardiac development, functionality, and remodeling.[31] Thus, cardiac spheroids were then generated according to a previous protocol to design a better cardiac tissue model.[32] These cardiac spheroids were encapsulated in 7% GelMA hydrogels with a Young’s modulus of ~12 kPa[33], which mimics those of the native human heart tissues (~10 kPa)[34] (Figure 3a). The histogram of cardiac spheroid diameters displayed an average dimeter of ~180 μm (Figure 3b).
Figure 3. Development of healthy and fibrotic iPSC-derived cardiac spheroids encapsulated in GelMA hydrogels resembling mechanical properties of native cardiac tissues.
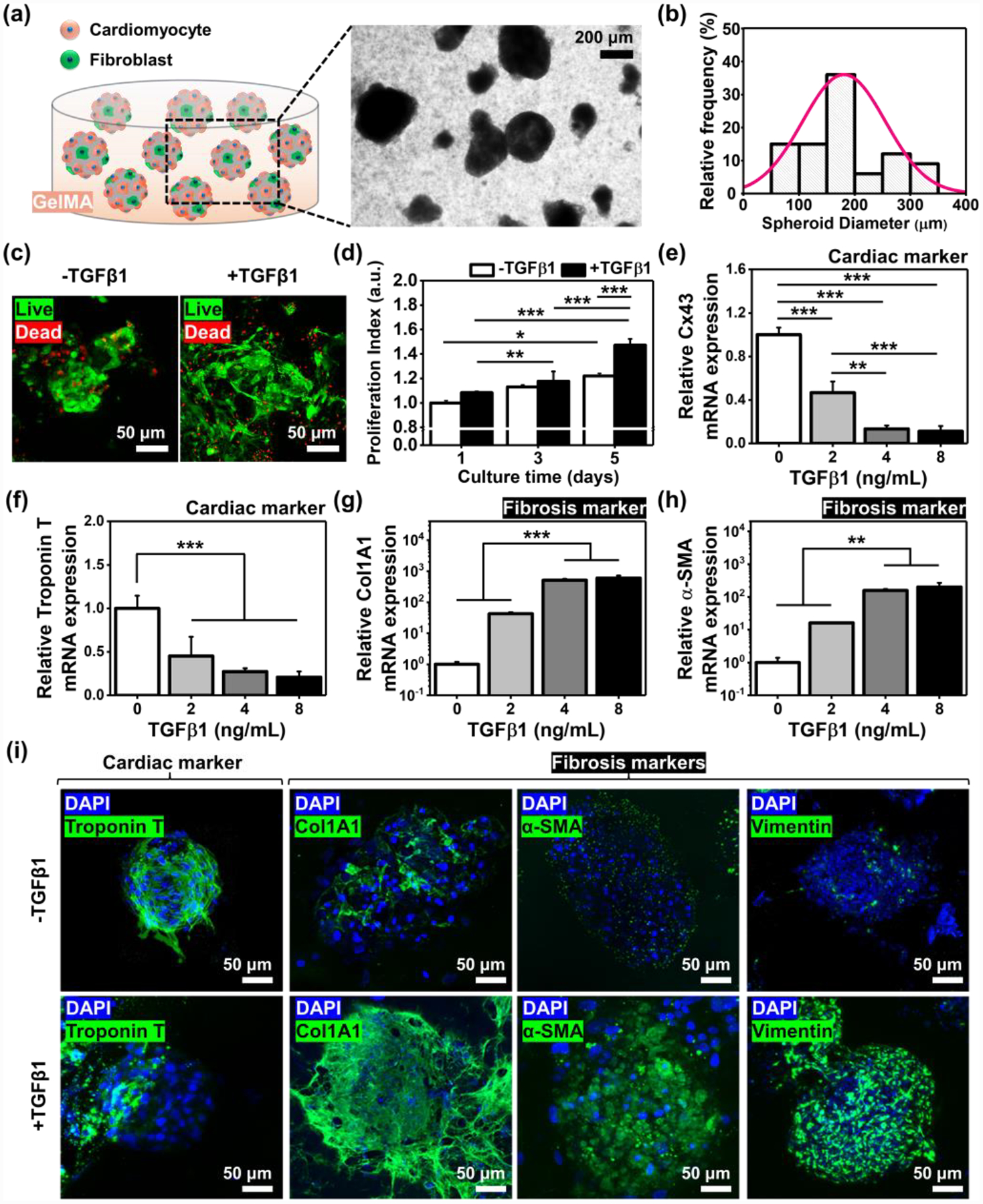
a) A schematic and photograph of engineered cardiac tissue models. b) Histogram of the spheroid dimeters after collecting them from the microwells on day 5 (N=80). c) Representative immunofluorescence images of live/dead staining for iPSC-derived cardiac tissues after 5 days in culture with or without a supplement of TGFβ1 (8 ng/mL). d) Proliferation index for cardiac tissues cultured for 5 days in GelMA hydrogels (N=3). Outcomes of qRT-PCR to measure the gene expression of cardiac specific transcripts, e) Cx43 and f) Troponin T, and cardiac fibrosis markers, g) Col1A1 and h) α-SMA, with the dose-dependent induction of TGFβ1 in the iPSC-derived cardiac tissues cultured in GelMA hydrogels (N= 3). i) Representative immunofluorescence images of DAPI/Troponin T, /Col1A1, /α-SMA, and /Vimentin staining of cardiac tissues on day 5 after culturing in GelMA hydrogels with or without a supplement of TGFβ1 (8 ng/mL). (One-way ANOVA with Tukey significant difference post-hoc test; *p<0.05, **p<0.005, and ***p<0.0005). Error bars represent standard deviation.
To generate a cardiac fibrosis model, TGFβ1 was supplemented to the cardiac spheroids in GelMA hydrogels. Addition of TGFβ1 (8 ng/mL) to the cardiac fibroblast culture plays a crucial role in the fibrosis-induction, and we did not observe notably different level of viability between cardiac spheroids cultured with and without TGFβ1 supplement, indicating the cardiac fibrosis induction using the range of TGFβ1 may not be harmful (Figure 3c). Cardiac spheroids supplemented with TGFβ1 (8 ng/mL) displayed a higher rate of proliferation for 5 days compared to those without TGFβ1 induction (~1.08-fold, ~1.04-fold, and ~1.21-fold higher on days 1, 3, and 5, respectively) (Figure 3d). TGFβ1 can lead to activation of fibroblasts but also accumulation of cells found in a number of connective tissues; degranulation of these cells releases histamine, which in turn stimulates fibroblast proliferation and collagen synthesis.[35] Thus, addition of TGFβ1 to the cardiac tissue could lead to increased levels of cardiac fibroblast proliferation unlike the non-proliferative cardiomyocytes.[36] This is in agreement with our results where we showed a higher proliferative index for cardiac spheroids incubated with TGFβ1. Cardiac fibrosis is known as a process where cardiac fibroblasts could transdifferentiate into the myofibroblast phenotype; this fibroblast-to-myofibroblast transition involves (1) remodeling of extracellular matrix (ECM) proteins such as fibronectin and collagen type I, (2) incorporation of α–smooth muscle actin (α-SMA) in stress fibers, and (3) increased rate of cell proliferation.[37] The cardiac functionality and phenotype shifting toward myofibroblasts were evaluated with a supplement of different concentrations of TGFβ1 (0, 2, 4, and 8 ng/mL) by gene expression analysis using real-time quantitative reverse transcription-polymerase chain reaction (qRT-PCR) for a panel of markers associated with cardiac functionality (Cx43 and Troponin T) and fibrosis (Col1A1 and α-SMA) (Figure 3e–h). It was expected that the induction of fibrosis leads to decreased levels of functionality and increased levels of fibrosis markers. In line with the expectation, the qRT-PCR results showed that cardiac spheroids with higher levels of fibrosis had significantly decreased expression levels of Cx43 (~0.47-fold, ~0.13-fold, and ~0.11-fold when supplemented with 2, 4, and 8 ng/mL of TGFβ1, respectively) and Troponin T (~0.45-fold, ~0.27-fold, and ~0.21-fold when supplemented with 2, 4, and 8 ng/mL of TGFβ1, respectively) compared to those without TGFβ1 supplement. In addition, it was shown notably higher expression levels of Col1A1 (~42.5-fold, ~507.4-fold, and ~595.7-fold when supplemented with 2, 4, and 8 ng/mL of TGFβ1, respectively) and α-SMA (~16.1-fold, ~156.5-fold, and ~200.7-fold when supplemented with 2, 4, and 8 ng/mL of TGFβ1, respectively) for fibrosis-induced cardiac tissues compared to the healthy ones. We found that the trends for the expression levels of functionality and fibrotic markers were saturated when the concentration reached to 8 ng/mL of TGFβ1, and thus, we decided to supplement the 8 ng/mL of TGFβ1 for modeling of our in vitro fibrotic cardiac tissues. To verify the outcomes obtained from qRT-PCR, healthy and fibrotic cardiac tissues were stained with a representative cardiac functional marker (Troponin T) and multiple fibrosis markers (Col1A1, α-SMA, vimentin) on day 5, revealing that our fibrotic cardiac model may be suitable to investigate the differential functionality from the healthy model after treatment with chemotherapeutic drugs.
2.4. Design of EC Immuno-Aptasensors for Monitoring of Multiple Biomarkers
Microfluidic platforms provide exquisite capabilities in regulating fluid flow and mass transport, and numerical simulation of their regulations is a valuable tool for visualizing and investigating the distribution of concentrated species in those platforms. We employed a computational model to simulate the distribution of flow rate, oxygen level, and drug concentration in our microfluidic device where both BC and health/fibrotic cardiac tissues were located and communicated (modeling domain I: cardiac tissues, II: BC tissues, and III: fluids outside of tissues (laminar flow)) (Figure 4a). Here, we assumed that the density and average size of BC and cardiac spheroids were constant, and they were homogeneously distributed in GelMA hydrogels. In addition, for simplicity, the hydrogels were assumed as solids in the model; the flow rate within hydrogels was set as zero, and only diffusion inside the hydrogels was considered. To calculate the velocity field from convection, we employed the incompressible Navier-Stokes model for Newtonian flow (fixed viscosity) with the flow rate of 79.4 μL/min in a total volume of 5 mL of medium. The result showed decreased flow components toward the hydrogels (Figure 4b). Moreover, in the domains I and II where the cells reside, we considered that the oxygen consumption of the cells occurred based on Michaelis-Menten kinetics and assumed that oxygen transportation happened only via diffusion, exhibiting the concentration gradient of oxygen allowing for gas exchange during cultures (Figure 4c). For the DOX, we assumed that the cardiac tissues had much higher levels of DOX concentration compared to the BC tissues due to their smaller consumption rate of DOX (Figure 4d).[38,39] However, the transport of DOX within cells such as cytosolic region and intracellular organelles or reversible binding sites in the intracellular space was omitted.[40]
Figure 4. Development of the EC aptamers-functionalized biosensors for detection of multiple biomarkers produced from cardiac or BC tissues.
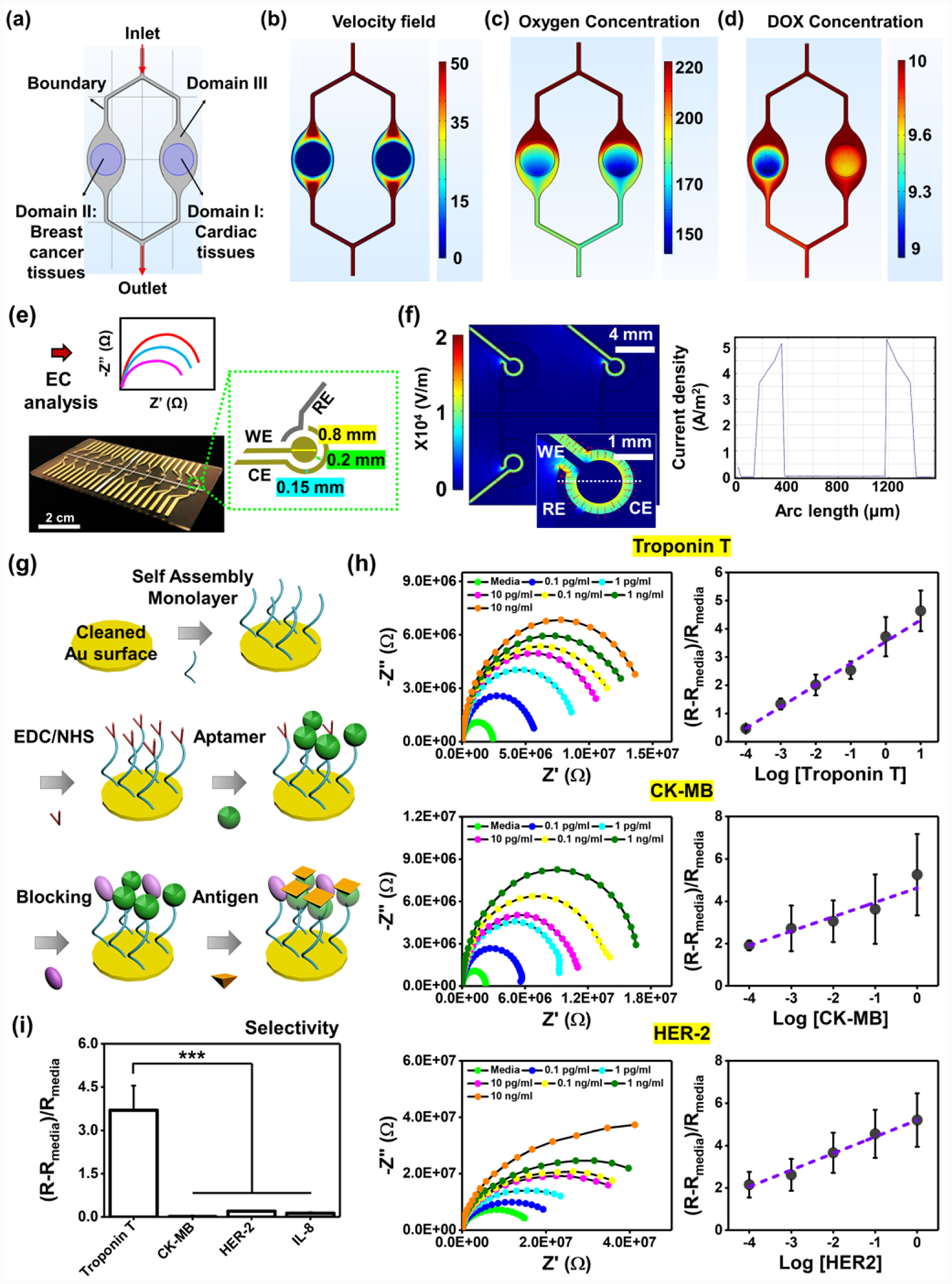
Mathematical model of a) geometry consisting of three different domains (Domain I: cardiac tissues, Domain II: BC tissues, and Domain III: fluidic channels) for simulating b) velocity field, c) oxygen concentration, and d) DOX concentration. e) Multielectrode array design, fabrication and plug-in cable connector: the microelectrode containing RE (Ag), WE (Au), and CE (Au). f) A map plot of the electric fields using a COMSOL simulation (top) and current density plot along the cut line as shown in electric field map. The first peak refers to the current between RE and WE, the second peak between WE and CE. g) A schematic illustrating immobilization of aptamers-functionalized biosensors on the surface of the microelectrodes. h) Nyquist and standard curves of multielectrode array chip for representative cardiac biomarkers, Troponin T and CK-MB, and BC biomarker, HER-2 (N= 3). i) Selectivity of Troponin T sensors in the presence of multiple unspecific biomarkers at a concentration of 1 ng/mL. (One-way ANOVA with Tukey significant difference post-hoc test; ***p<0.0005). Error bars represent standard deviation.
To non-invasively monitor secreted biomarkers from the cardiac and BC spheroids, the sensor chips based on EC impedance spectroscopy (EIS) measurements for samples from bioreactors were employed as previously reported.[41] Basically, a counter electrode (CE), a reference electrode (RE), and a working electrode (WE) composed a microelectrode set, and we custom-designed this set for easy and simple integration with our EC sensing platform (Figure 4e). The microfabricated electrode array contained a RE and 20 WEs and CEs (each RE had their own CE). We employed gold (Au) as the electrode materials for CE and WE due to its benefits such as good stability, high electrical conductivity, and convenient covalent bonding on the surface. Briefly, an electron beam deposition through a shadow mask was used to deposit the metal layers; 15-nm-thick titanium (Ti) was initially deposited on a piece of cleaned glass, 15-nm-thick palladium (Pd) and 300 nm-thick silver (Ag) were then selectively deposited to prevent the diffusion between the Ti and Au layers during the annealing process, and 300-nm-thick Au was finally deposited on the Ti-Pd layers. Using COMSOL two-dimensional (2D) simulation of electric field distribution along the sensor chip, we showed the instantaneous amplitude of the electric field with a maximum field of ~1.2 × 104 V/m and a minimum field of ~0.8 × 104 V/m (Figure 4f). The current density distribution along the cut line in the electric field map displays that the current density between the RE and WE was ~5.2 A/m2 and the second peak between WE and CE was ~5.4 A/m2. Next, we immobilized aptamers onto the surface of Au electrodes (Figure 4g). Briefly, a self-assembled monolayer (SAM) with thiol and carboxyl groups on each terminal was first immobilized on the Au surface (Au-S bonding). Aptamers with an amine end group were covalently bonded to the carboxyl groups of SAM using the N-ethylcarbodiimide (EDC)/N-hydroxysuccinimide (NHS)-based carbodiimide chemistry, and unreacted carboxyl groups of the SAM were blocked. These simple and efficient immobilization steps of aptamer functionalization onto the microelectrodes enabled performing stable, continuous, and reproducible measurements of biomarkers. We could further multiplex our EC Aptasensor platform to detect multiple biomarkers at the same time. Their calibration curves obtained using biochemical sensing modules to indicate cellular performances could be detected as low as 0.1 pg/mL for all markers and sensitivities of ~0.8, ~1, and ~0.9 (log(ng/mL))−1 for Troponin T, CK-MB, and HER-2, respectively. Compared to the conventional enzyme-linked immunosorbent (ELISA) assay for these three markers having a detection limit of several hundreds of pg/mL, our microfluidic EC sensor exhibited much better performances in terms of detection limit and sensitivity while it requires orders of less sample volumes. In addition, the selectivity of biomolecules is considered as one of the most important factors for revealing the accuracy of the sensing platform, and it is known that aptamer-based biosensors have high selectivity.[42] We functionalized Troponin T-specific aptamers on microelectrodes, as a representative example, to examine the selectivity of our sensing platform. Other biomolecules such as CK-MB, HER-2, and interleukin 8 (IL-8) in cell culture media, which included high concentrations of non-specific proteins and other biomolecules, were also exposed together with Troponin T molecules, showing that Troponin T-specific aptamers could dominantly detect the Troponin T molecules and the other biomolecules negligibly influenced the Troponin T detection. This result revealed the high selectivity of our sensing platform (Figure 4i).
2.5. Monitoring of Biomarkers: Role of Communications between Cardiac and BC Tissues
Before investigating the different incident levels of CIC from healthy and fibrotic cardiac tissues, we questioned whether the cardiac-BC interactions that mimic cellular crosstalk in the native microenvironment may have the potential to regulate their secretions of biomolecules associated with cardiac functionality (Troponin T and CK-MB) and BC progression (HER-2).
To answer the question, we employed (1) single- and (2) dual-organ systems for cardiac and BC tissues to assess the effect of their interactions on production rates of the biomarkers (Figure 5a). We used the same dual-organ chip for both single (only one of the two chambers was filled with either cardiac or BC tissue) and dual (the two chambers were separately filled with the cardiac and BC tissues) systems to keep the identical flow rate and other parameters. We first subjected either the single system or the dual system to physiologically relevant interstitial perfusion. Next, these tissues were cultured for 5 days to monitor the relevant biomarkers using ELISA (Figure 5b–g and S3, Supporting Information). We obtained several important outcomes from the experiment. (1) Production rates of Troponin T from healthy cardiac tissues increased in both single- and dual-chips, but the secretion rates from healthy cardiac tissues in the dual-chip were lower compared to those without interactions with BC tissues until day 3 in culture (~0.61-fold, ~0.74-fold, and ~0.87-fold on days 1, 3, and 5, respectively) (Figure 5b). Similar to the results from healthy cardiac tissues, the fibrotic cardiac tissues communicating with BC tissues displayed lower production rates of Troponin T compared to those without the interactions (~0.81-fold, ~0.83-fold, and ~0.62-fold on days 1, 3, and 5, respectively), but their rates of Troponin T production in the presence of the BC interaction stopped increasing on day 5 (~0.82-fold difference relative to the rates on day 3) (Figure 5e). (2) CK-MB secretion rates for the healthy cardiac tissues in the dual-chips were lower on days 1 (~0.056-fold) and 5 (~0.63-fold) compared to those cultured in the single-chips, in which the trend seemed similar to their Troponin T production (Figure 5c). However, the fibrotic cardiac tissues cultured for 5 days in both single- and dual-chips exhibited decreased production rates of CK-MB, in which the trend was opposite from that of the healthy cardiac tissues (Figure 5f). Nevertheless, these CK-MB production rates from the fibrotic cardiac tissues with or without communications with BC cells were not significantly different. (3) BC cells connected with the healthy cardiac tissues exhibited lower secretion rates of HER-2 on day 1 (~0.39-fold), but their production rates became higher when compared to those without the connection with healthy cardiac tissues on days 3 (~1.21-fold) and 5 (~1.63-fold) (Figure 5d). Similar trends were observed for HER-2 production rates from BC cells connected with the fibrotic cardiac tissues (~0.66-fold, ~2.19-fold, and ~1.37-fold on days 1, 3, and 5, respectively) (Figure 5g). Together, it has been demonstrated that the communications between healthy/fibrotic cardiac and BC cells play an important role in the secretion rate of biomarkers. To closely mimic the cellular crosstalk in the native microenvironment and obtain more precise results, we decided to culture both tissues in the dual-chip configuration for further experiments.
Figure 5. Crucial role of interaction between BC and cardiac tissues in regulating production rates of biomarkers.
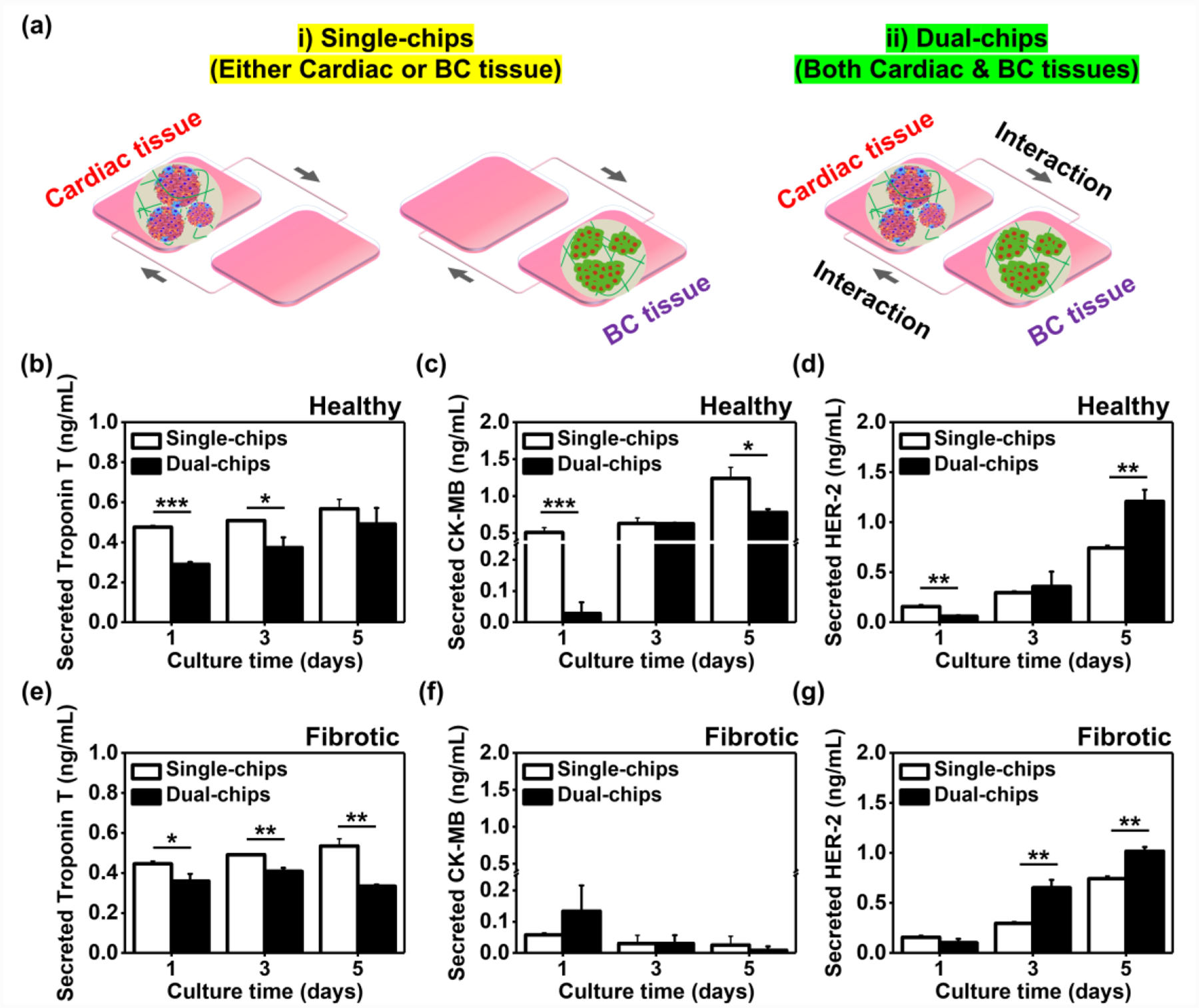
a) Schematic illustration displaying i) disconnected and ii) connected breast cancer-cardiac tissues on chip. b) Troponin T and c) CK-MB production rates in iPSC-derived healthy cardiac tissues for 5 days on single or dual chips using ELISA (N= 3). d) ELISA measurement of HER-2 rate in BC tissues on single or dual (with healthy cardiac tissues) chips (N= 3). e) Troponin T and f) CK-MB production rates in iPSC-derived fibrotic cardiac tissues for 5 days on single or dual chips using ELISA (N= 3). g) ELISA measurement of HER-2 rate in BC tissues on single or dual (with fibrotic cardiac tissues) chips (N= 3). (One-way ANOVA with Tukey significant difference post-hoc test; *p<0.05, **p<0.005, and ***p<0.0005). Error bars represent standard deviation.
2.6. Monitoring of Multiple Biomarkers using EC Immuno-Aptasensors
We showed that the interactions between BC and cardiac tissues were required to accurately reflect the levels of biomarkers associated with cardiac functionality and BC progression. To examine our hypothesis on requiring different prediction and management of CIC for the fibrotic cardiac tissues vs. the healthy cardiac tissues, we employed DOX as a model chemotherapeutic drug since it is the drug that has been widely used and standardized for BC patients at different stages.[43] First, we assessed the viability of healthy/fibrotic cardiac and BC cells with or without addition of DOX (Figure 6a). While the both cell types showed high levels of viability without DOX (Figure 2g), numerous dead cells were observed across the conditions when cultured for 5 days with the DOX treatment (10 μM). In addition, relatively spontaneous beating rates of healthy and fibrotic cardiac tissues were assessed to study their electrophysiological functions in response to DOX (Figure S4, Supporting Information). We found notably decreased beating frequencies for both healthy and fibrotic cardiac tissues cultured for 5 days with the treatment of DOX. These results from both cell viability and beating frequency indicated that our physiologically relevant tissue models on the dual-organ system could be reliable and reproducible for the study of CIC.
Figure 6. Continual monitoring of biomarkers associated with cardiotoxicity and tumor progression using EC biosensors.
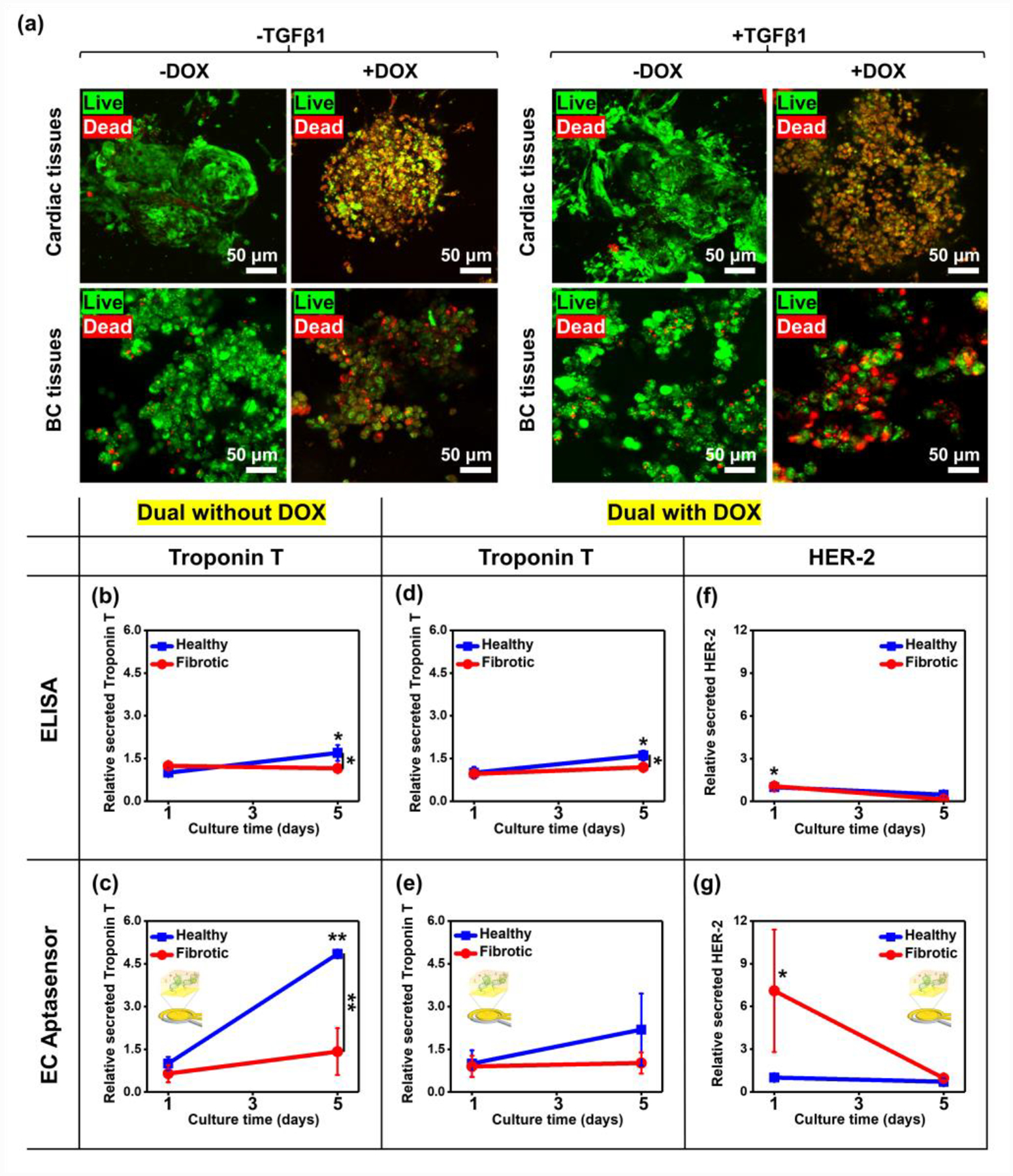
a) Representative immunofluorescence images of live/dead staining for iPSC-derived healthy or fibrotic cardiac and BC tissues after 5 days in culture with or without a supplement of DOX. b) ELISA and c) EC measurement of Troponin T rates in healthy or fibrotic cardiac tissues on dual chips without DOX (N= 3). d) ELISA and e) EC measurement of Troponin T rates in healthy or fibrotic cardiac tissues on dual chips with the DOX treatment (N= 3). f) ELISA and g) EC measurement of HER-2 rates in healthy or fibrotic cardiac tissues on dual chips with the DOX treatment (N= 3). To compare fold-changes between outcomes from ELISA and EC Aptasensors, each group was plotted with the same scale. (One-way ANOVA with Tukey significant difference post-hoc test; *p<0.05 and **p<0.005). Error bars represent standard deviation.
We next monitored the production rate of biomolecules from the cells using our EC immuno-Aptasensors to compare the trends with outcomes from ELISA (Figures 6b–g and S5, Supporting Information). Since Troponin T was demonstrated as a more specific biomarker for myocardial injuries compared to CK-MB,[44] we decided to monitor the production rate of Troponin T when the cells were treated with or without DOX. To monitor Troponin T secretion levels, sample solutions were collected on days 1 and 5 from the circulating media reservoir and manually applied to the EC immuno-Aptasensors. From ELISA, we observed the significantly increased rates of Troponin T production from day 1 through day 5 for the healthy cardiac tissues (~1.69-fold), whereas no detectable changes were observed for fibrotic cardiac tissues without DOX treatment (~0.95-fold) (Figure 6b). Moreover, notably different rates of secreted Troponin T were observed between healthy and fibrotic cardiac tissues on day 5 (~1.47-fold). In line with the results from ELISA, Troponin T production rates measured by the EC immuno-Aptasensors without DOX treatment were significantly different not only for the healthy cardiac cells cultured from day 1 through day 5 (~4.85-fold) but also between healthy and fibrotic cardiac cells on day 5 (~3.42-fold) (Figure 6c). Interestingly, the amounts of detected Troponin T from our EC Aptasensors (~0.01 pg/mL on day 1 in Figure S5a) was ~104 times less than those measured from ELISA (~0.3 ng/mL on day 1 in Figure 5b), demonstrating the much lower detection limits of the measurements using our EC sensors. Similarly, the sensitivity of our sensors (~4.85-fold changes for the healthy cardiac tissues on day 5) was much better than that from ELISA (~1.69-fold changes for the healthy cardiac tissues on day 5), revealing the higher sensitivity of our sensors compared to ELISA. Next, we treated the cells with DOX to evaluate the variation rates of produced biomolecules. We found that the DOX treatment led to the same trend but decreased variations of Troponin T production rates for both healthy and fibrotic cardiac tissues relative to the outcomes without DOX (Figure 6d and e). Although, the differences measured in our Aptasensors were not significant, the obtained changes from our sensors were higher than those from ELISA (~2.19-fold from Aptasensors vs. ~1.61-fold from ELISA for the healthy cardiac tissues on day 5) with low detection limits ranging from ~0.0003 to ~0.003 pg/mL (Figure S5b, Supporting Information). Furthermore, we measured production rates of CK-MB using ELISA, which is one of early cardiac biomarkers for monitoring cardiac functionality and known for the early identification and prediction of CIC.[13–15] We found that the CK-MB production trends from healthy and fibrotic cardiac tissues with or without DOX treatment corresponded to those of Troponin T production in the respective tissues (Figure S6, Supporting Information).
It was demonstrated that elevated HER-2 levels were observed in BC patients with invasive BC cells, worse survival, and even recurrence after treatment,[45] and thus we also monitored HER-2 production levels from BC cells cultured with healthy or fibrotic cardiac tissues for 5 days with DOX treatment. The subtle decreased secretion rates of HER-2 from BC cells communicating with the healthy cardiac tissues were observed for 5 days in culture by using both ELISA and our Aptasensors. Moreover, BC cells interacting with the fibrotic cardiac tissues produced lower levels of HER-2 on day 5 compared to the levels on day 1 (Figure 6f and g). We showed ~1.07-fold higher HER-2 was produced from BC cells with fibrotic tissues using ELISA compared to those with healthy tissues on day 1. However, using our sensor platform, we observed ~7.10-fold higher HER-2 productions, followed by decreased HER-2 secretion to around the levels of BC cells communicating with the healthy cardiac cells on day 5 (Figure 6g and S5c, Supporting Information). Nevertheless, we showed that the production trend of biomarkers (Troponin T and HER-2) evaluated by using the immuno-Aptasensors well-matched the results from ELISA, proving the accuracy of our sensing platform with much higher sensitivity and lower detection limits for early monitoring of CIC and BC progression.
2.7. Predicted Production Rate Changes of Biomarkers for Early Detection and Prediction of CIC and BC Progression
The early-stage prediction of imminent cardiac dysfunctions and/or suppression of BC progression after the treatment of chemotherapy could play a critical role in preventing CIC and increasing the treatment efficacy for BC. We showed that (1) the communications between the cardiac and BC cells, (2) the fibrotic stage of the cardiac tissues, and (3) the DOX treatment, led to different production rates of biomolecules associated with cardiac functionality and BC progression. To discover whether these findings using iPSC-derived cardiac tissues could help to better detect and predict early CIC and BC progression, the trends of production rates of biomarkers associated with cardiac functionality and BC progression were summarized (Figure 7a and b). For the cardiac functionality, we observed that healthy cardiac cells produced higher levels of Troponin T compared to the levels of fibrotic cardiac cells. Moreover, addition of DOX led to the decreased Troponin T production rate and no significant changes for both healthy and fibrotic cardiac cells, respectively. The increased production of Troponin T for 5 days in culture without addition of DOX may be caused by the proliferating cardiac cells in culture (Figure 3d), and the reduced rates of Troponin T production for the fibrotic cardiac tissues relative to the healthy ones corresponded to the qRT-PCR and immunofluorescence analysis (Figure 3f and i). In addition, the number of dead cells after treatment with DOX may result in the decreased Troponin T secretion for both healthy and fibrotic cardiac tissues (Figure 6a). However, the fact that no major changes in Troponin T production rates from fibrotic cardiac cells indicated that early detection and prediction of CIC may be difficult in individual BC patients with preexisting cardiac fibrosis and they might need different biomarkers or approaches in detecting early CIC.
Figure 7. Predicted production rate changes of biomarkers associated with cardiac functionality and BC progression, which are regulated by the preexistence of cardiac fibrosis and BC chemotherapy using DOX.
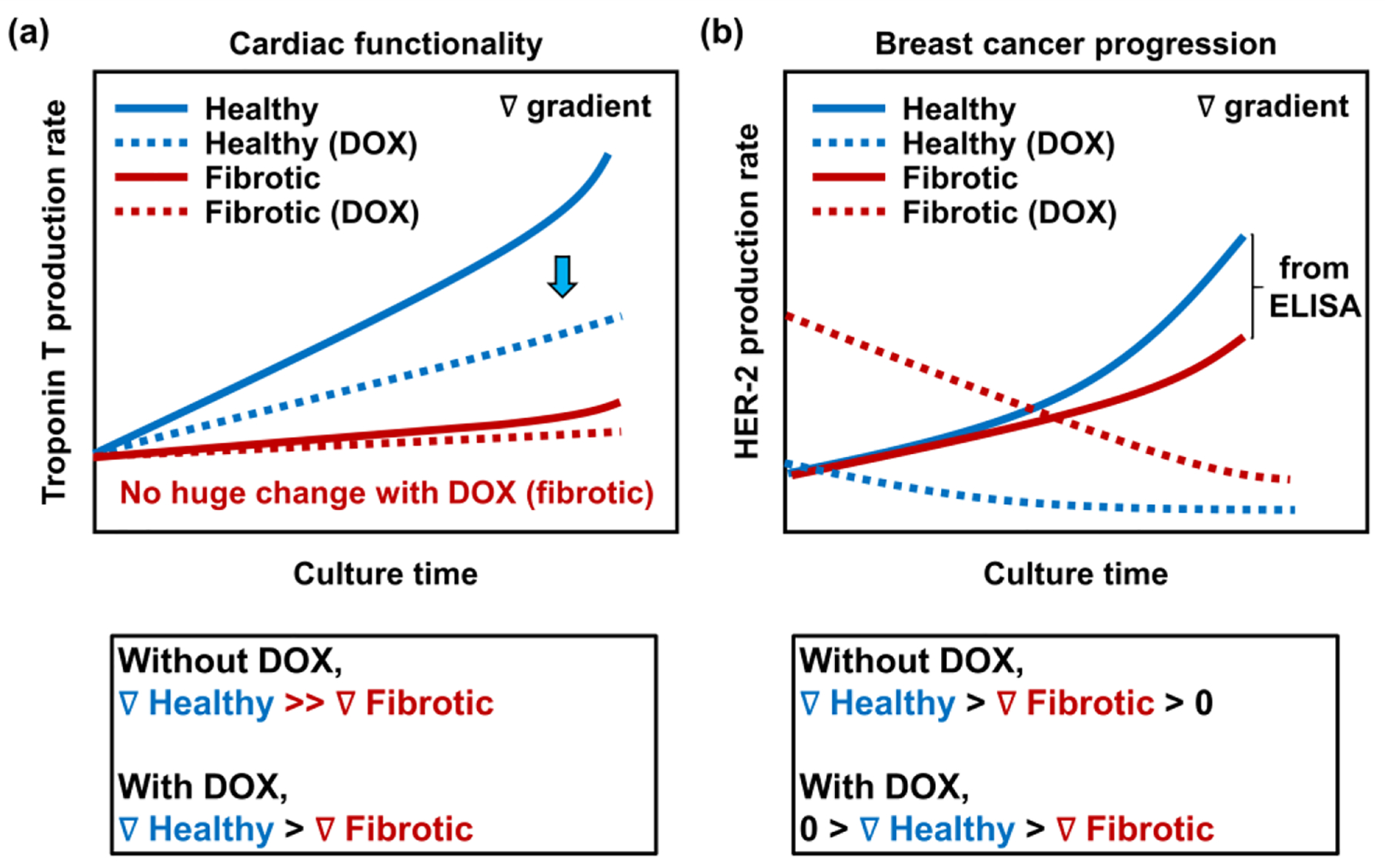
a) Predicted Troponin T rate in healthy and fibrotic cardiac tissues on dual chips with and without an addition of DOX. Cardiac tissues with preexisting cardiac fibrosis show lower production levels of a representative cardiac functionality marker, Troponin T compared to healthy cardiac tissues, regardless of the DOX treatment. In addition, healthy cardiac tissues exhibit a sudden decrease in Troponin T secretions when treated with DOX, while fibrotic cardiac tissues display no huge change between Troponin T productions with and without DOX. b) Predicted HER-2 rate in healthy and fibrotic cardiac tissues on dual chips with and without the treatment of DOX. Positive gradient of HER-2 production rate is shown for both healthy and fibrotic cardiac tissues without DOX, while negative gradient of HER-2 secretion rate is observed for the both conditions treated with DOX. Healthy cardiac tissues display higher gradient of HER-2 production rates with and without the treatment of DOX compared to fibrotic cardiac tissues.
Monitoring HER-2 production rates from BC cells interacting with healthy or fibrotic cardiac cells could also provide useful information regarding BC development and progression after the treatment with DOX. The increased HER-2 production may be due to the proliferating BC cells during culture (Figure 2h). However, BC cells cultured with the healthy cardiac cells without DOX showed higher production levels of HER-2 compared to the levels from those with the fibrotic cardiac cells (Figure S7, Supporting Information). In addition, we found the increased production level of HER-2 from BC cells communicating with the fibrotic cardiac cells with the addition of DOX. The level notably decreased for 5 days in culture (Figure 6g), which also showed the different trend from BC cells interacting with the healthy cardiac cells. These results suggested that the BC cells communicating with cardiac cells under different stages of cardiac fibrosis could influence their HER-2 biomarker production with and without DOX treatment. Although we verified that our proposed sensing platform could be potentially suitable for early detection and prediction of CIC, we acknowledge further validation studies are required such as (1) employing various iPSC-derived cardiac tissues from different patients, (2) enabling long-term culture to cross-compare between multiple biomarkers for the accumulation of meaningful data, and (3) comparing the results with different approaches in patients. Nevertheless, we demonstrated that BC patients with preexisting cardiac fibrosis may need different approaches to early detection and prediction of CIC and BC progression from healthy patients. Furthermore, we believe that our suggested platform would guide to design better CIC models to provide crucial information for individual BC patients via evaluating essential biomarkers from patient-derived and disease-modeled cardiac tissues when interacted with multiple surrounding tissues.
2.8. Heart-BC-on-a-chip Platform with Nanoparticles (NPs)-based Drug Delivery System
Various types of NPs have been employed as carrier systems for chemotherapeutic drugs to improve anticancer efficiency through reversing drug resistance.[46] To validate the applicability of our platform to continuously and non-invasively monitor multiple biomarkers using our EC Aptasensors, production rates of biomarkers from the cardiac and BC cells were assessed when treated with DOX using the GYSM-NPs. First, GYSM-NPs were fabricated as previously reported (Figure 8a),[47] and DOX-loaded GYSM-NPs were introduced into the dual-chip platform where both cardiac and BC tissues were placed and cultured to communicate each other. GYSM-NPs were previously reported as a stimuli-responsive drug delivery platform with an excellent drug loading capacity (~91%) as well as controlled release by both pH and temperature under an alternating current (AC) magnetic field.[47] To examine the improved anticancer efficiency, we first defined the concentration of DOX loaded in the GYSM-NPs when compared with the concentration of DOX without using NPs (free DOX) (Figure S8, Supporting Information). Among the wide range of DOX concentrations loaded in the GYSM-NPs (15–60 μg/mL), we found that the released amount of DOX (30 μg/mL) was comparable to the concentration of free DOX (10 μM). Then, the viability and proliferation of cardiac and BC cells and their production rates of biomolecules were assessed. Previously, the amount of DOX released from the GYSM-NPs displayed around 6.8%, 30%, and 45.6% of DOX release at pH 7.4, 6.0, and 4.5, respectively, after 30 min under the AC magnetic field.[47] Since extracellular pH of cancer cells is more acidic compared to normal tissues, it is expected that better release of DOX could be achieved at the BC tissues. Indeed, we observed more live cells in the cardiac tissues and more dead cells in the BC tissues with DOX delivery using the GYSM-NPs (Figure 8b), whereas small numbers of live cells were observed for both cardiac and BC tissues when treated with free Dox. Next, proliferation profiles of cardiac and BC cells were accessed (Figure 8c and d). Both cardiac and BC tissues treated with free DOX and GYSM-NPs-DOX showed notably decreased proliferation for 5 days (cardiac: ~0.82-fold with free DOX and ~0.87-fold with NP-DOX on day 5; BC: ~0.40-fold with free DOX and ~0.63-fold with NP-DOX on day 5) unlike tissues cultured with untreated and GYSM-NPs without DOX-loading (cardiac: ~1.13-fold with untreated and ~1.07-fold with GYSM-NPs only on day 5 & BC: ~1.82-fold with free DOX and ~1.64-fold with NP-DOX on day 5). In addition, production rates of markers associated with cardiac functionality (Troponin T and CK-MB) and BC progression (HER-2) were obtained from tissues treated with free DOX or NP-DOX by using our sensor platform (Figure 8e–g). It was shown that healthy cardiac cells exhibited decreased production rates of functionality markers when treated with free DOX relative to those without the treatment (Figure 7a). We found that healthy cardiac cells displayed notably less secretion rates of Troponin T and CK-MB when treated with NP-DOX compared to those treated with free DOX, which may indicate the increased risk of CIC. For proving the effect of NP-based drug delivery systems on the risk of CIC, further works will be conducted using our cardiotoxicity-on-a-chip platform by applying various types of NP-based systems. In addition, we showed that BC cells interacting with the healthy cardiac cells produced decreased levels of HER-2 with addition of free DOX (Figure 7b), whereas, we observed that BC cells produced significantly decreased rate of HER-2 when treated with NP-DOX, indicating the better effectiveness of targeted cancer therapy using GYSM-NPs as the drug carriers.
Figure 8. Application of our cardiac-breast cancer-on-a-chip platform for further studying cardiotoxicity with NP-based drug delivery system.
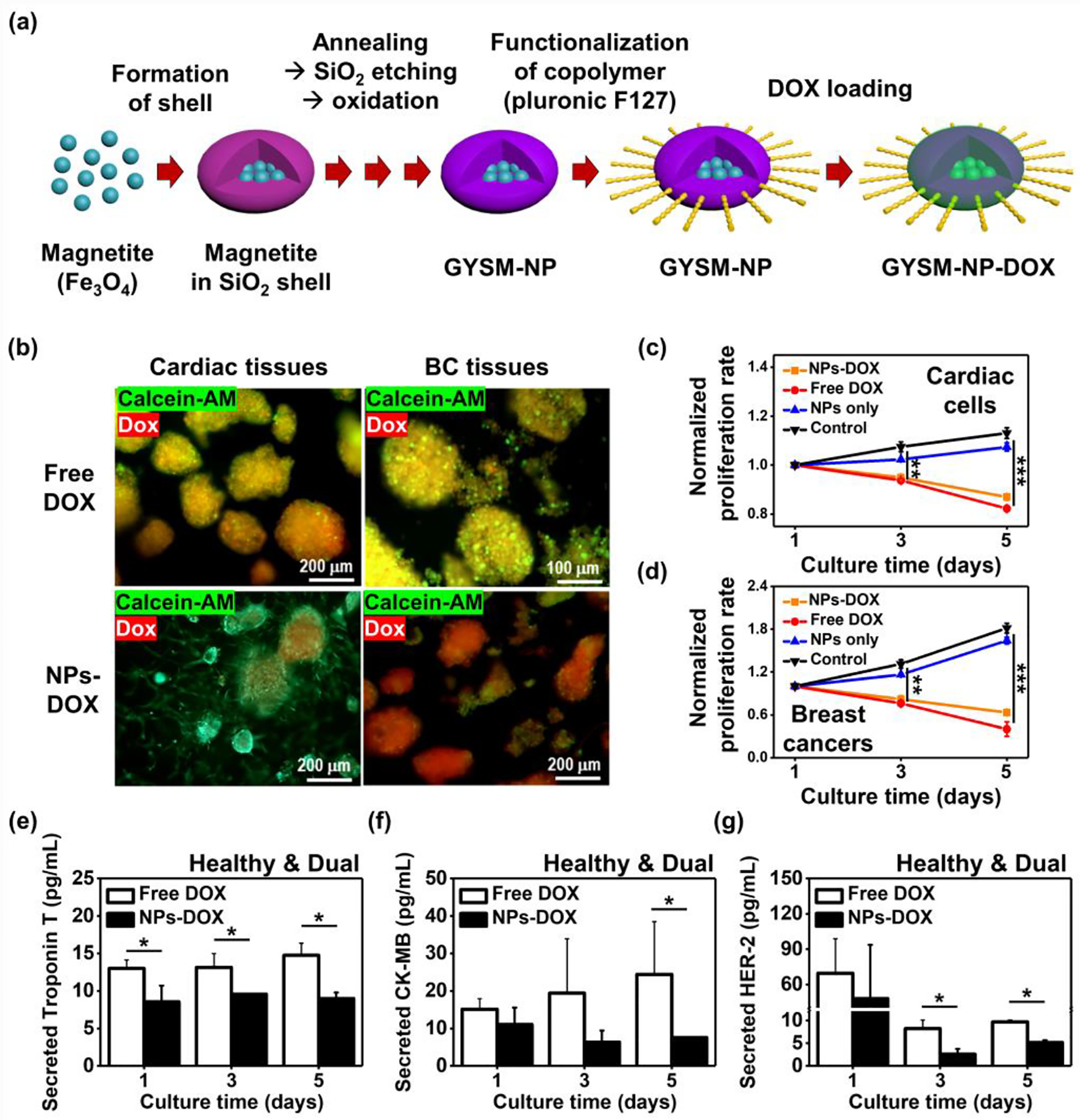
a) Schematic of the procedure to generate GYSM-NP-DOX. b) Representative immunofluorescence images of live/dead staining for iPSC-derived healthy or fibrotic cardiac and BC tissues after 6 days in culture with a supplement of free DOX or NP-conjugated DOX. Cytotoxicity quantification of free DOX, NPs and NP+DOX on c) cardiac and d) BC cells for 5 days in culture using Prestoblue (N=4). EC measurement of e) Troponin T, f) CK-MB, and g) HER-2 rate in the cardiac-breast cancer-on-a-chip with a supplement of free DOX or NP-conjugated DOX (N=3). (One-way ANOVA with Tukey significant difference post-hoc test; *p<0.05, **p<0.005, and ***p<0.0005). Error bars represent standard deviation.
3. Conclusion
To address the lack of model systems recapitulating the complicated and patient-specific cardiac functions for detecting early symptoms of CIC, we developed a cardiotoxicity-on-a-chip platform containing iPSC-derived cardiac tissues communicating with BC tissues as well as multiplexed EC immuno-Aptasensors for non-invasive monitoring of cell secreted multiple biomarkers. Using the platform, the effect of DOX on functionality and toxicity of iPSCs-derived cardiac tissues with preexisting cardiac fibrosis could be compared with healthy cardiac tissues to step toward early detection and prediction of CIC in individual patients. Production trends of multiple biomarkers evaluated using the EC immuno-Aptasensors matched well with the outcomes from ELISA, demonstrating the excellent accuracy and high sensitivity of the sensing platform with much lower detection limits for monitoring of CIC and BC progression. Furthermore, the applicability of our platform was demonstrated by comparing production rates of biomarkers from cardiac and BC tissues when treated with DOX using GYSM-NPs. It is expected that the suggested platform will be broadly applicable across other individualized disease modeling where (1) tissue-tissue communications play a crucial role in the functionality of multicellular tissues as well as (2) early developmental monitoring of targeted diseases is highly required with high sensitivity, low detection limit, and excellent reproducibility.
4. Experimental Section
Materials:
Potassium ferricyanide (K3Fe(CN)6, 99 wt.%), sulfuric acid (H2SO4, 95 wt.%), Dulbecco’s phosphate buffered saline (DPBS), 11-mercaptoundecanoic acid (11-MUA, 95 wt.%), ethanol absolute (C2H6O, 99.8 wt.%), N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC, 97 wt.%), N-hydroxysuccinimide (NHS, 97 wt.%) and gelatin Type A from porcine skin were obtained from Sigma-Aldrich (MO, USA). Doxorubicin hydrochloride (DOX, 98 wt.%) and PDMS/curing agent were acquired from Discovery Fine Chemicals (Wimborne, UK) and Dow Corning (Sylgard 184, MI, USA), respectively. Prestoblue cell viability assay was obtained from ThermoFisher scientific (MA, USA). Live/Dead viability assays were obtained from Invitrogen (CA, USA). CK-MB and cardiac Troponin T ELISA kits were acquired from Abcam (Cambridge, UK). HER-2 ELISA kit was obtained from LS Biosciences. High capacity reverse transcription (HCRT) kit and SYBR green was obtained from Lifescience Technologies (USA). All antibodies were purchased from Abcam. Ultrapure water (18.2 MΩ.cm at 25 °C)—produced in a Milli-Q system (Millipore)—was used to prepare all aqueous solutions.
Preparation of GelMA:
GelMA was fabricated based on a previously stated protocol.[48] Briefly, a gelatin solution (10% w/v) in DPBS was heated to 50 °C for 1 h, followed by addition of methacrylic anhydride (400 μL/g of gelatin) to allow a reaction under continuous stirring for 2 h. Then, twice the volume of DPBS was added to the above described gelatin-methacrylate mixture to conclude the reaction. A 12–14 kDa MWCO Spectra/por dialysis membrane was employed to dialyze the last solution against ultrapure water for 5 days at 40 °C. The final produces were freeze-dried and then dissolved in DPBS for further use.
Formation of breast cancer spheroids:
A PDMS micro-well platform was employed to generate BC spheroids using SK-BR-3 cell line obtained from ATCC. Briefly, to fabricate a PDMS replica, conventional lithography-techniques was employed to make a negative silicon mold having pillars (300 μm-diameter and 250 μm-depth), followed by curing PDMS (10:1 weight ratio of base & curing agent) for 1 h at 80 °C. Micro-wells were washed with 70% ethanol under the vacuum to remove all the micro-bubbles. Once, sterilized the micro-wells were conditioned overnight in Maccoy’s 5A medium supplemented with 10% FBS and 1% penicillin/streptomycin. SK-BR-3 cells (106/mL) were seeded on PDMS mold containing 24 × 24 micro-wells and allowed to settle down and aggregate in the microwells for 30 min. Medium was changed daily for 5 days, and then spheroids formed in microwells were collected. A small portion of collected spheroids were trypsinized and dissociated into single cells to calculate the cellular density of the spheroids.
Formation of human iPSC derived cardiac spheroids:
Directed cardiac differentiation of iPSCs to cardiac spheroids was performed using a previously described established protocol with modification to optimize yield in the iPSC line.[32] Briefly, dissociation of iPSCs reaching 70–80% confluency into single cells were performed with StemPro™ Accutase™ on day 0 and cultured in suspension with E8 supplemented with Matrigel™, BMP4 (1 ng/mL) and Y-27632 (10 μM) under 37 °C and 5% O2 hypoxic conditions. On day 1, the medium was changed to StemPro™−34 SFM (SP34) complemented with GlutaMAX, Ascorbic acid (50 μg/mL), Activin A (10 ng/mL), and BMP4 (10 ng/mL) for mesodermal induction in the embryoid bodies. On day 4, BMP4 and Activin A were replaced with IWR-1(5 μM) in StemPro™−34 SFM (SP34) and ascorbic acid to direct mesodermal cells to a predominantly ventricular subtype. On day 8, IWR-1 was removed and the resultant cardiospheres were placed in StemPro™−34 SFM (SP34) and ascorbic acid under 37 °C and 5% CO2 normoxic condition, with its medium replaced twice a week.
Mechanical stability measurements:
GelMA hydrogels with and without encapsulating spheroids in a DPBS bath were located onto an Instron (5944, 1 N load cell) at room temperature (RT) to obtain the compressive modulus of the GelMA using measured compressive stress-strain values. The strain rate (0.5 mm/min) was employed, and compression was carried out around 50% deformation. The gradient of the initial 10% strain in stress-strain curves was employed to calculate compressive modulus.
Induction of fibrotic condition in cardiac spheroids:
Healthy cardiac organoids were fabricated by encapsulating the iPSC derived cardiac spheroids within 7% GelMA hydrogels. At day 0, the cardiac organoids were cultured in Stem Pro medium. At day 1, fibrosis was induced in healthy organoids by treating the them with 8 ng/mL of TGFβ1. Media changes were made every alternate day up to day 5 to keep the amount of drug constant.
Analysis of gene expression:
Trizol method was used for extracting RNA from each hydrogel sample (N=3) on day 5. The extracted RNA was converted into corresponding cDNA using HCRT kit in a PCR thermal cycler (Applied Biosystems), and then qPCR was performed using forward/reverse primers of cardiac specific genes such as Cx43, Troponin T, Col1A1, and α-SMA with SYBR Green dye following vendor’s protocol. The expression levels were quantified using a comparative 2-ΔCt method and normalized by one of the well-known housekeeping gene, glyceraldehyde-3-phosphate-dehydrogenase (GAPDH). Primer sequences for Cx43, Troponin T, Col1A1, α-SMA, and GAPDH are listed in Table S2 (Supporting Information).
Characterization of cellular viability and proliferation:
Viability and proliferation of cells were examined using Live/dead and Prestoblue assays, respectively. For the viability assay, hydrogels encapsulating cardiac or BC cells were rinsed two times with DPBS, followed by addition of calcein-AM (40 nM)/ethidium homodimer (20 nM) to the hydrogels for staining cells at a humid chamber (37 °C and 5% CO2) for 30 min. After the incubation, the samples were rinsed two times with DPBS and were imaged using a ZEISS LSM 880 with Airyscan Microscope (Carl Zeiss, Jena, Germany) or a Nikon Eclipse Ti-S Microscope (Nikon, Tokyo, Japan).
Immunostaining:
GelMA hydrogels encapsulating spheroids were gently washed three times with DPBS, and cells in hydrogels were fixed with 4% paraformaldehyde for 20 min. Cell permeabilization was done using 0.1 v/v% Triton X-100 in DPBS for 20 min, and blocking was performed with 1% BSA and 0.2% Tween in DPBS for 1 h. The conjugation of primary antibodies—such as rabbit polyclonal anti-Troponin T, rabbit polyclonal anti-vimentin, rabbit polyclonal anti-collagen 1A1 and mouse monoclonal α-SMA (1:100, Abcam) for cardiac tissues and anti-Her-2/Erb2 for BC spheroids—was performed overnight at 4 °C, followed by rinsing three times for the constructs with DPBS. Then, the constructs were incubated with secondary antibodies (1:200 dilution) for 3 h and 4’,6-diamidino-2-phenylindole (DAPI; 1:500 dilution) for 1.5 h, followed by imaging samples with a confocal microscope.
Fabrication of Single- and dual-organ bioreactors:
The drawing of the dual bioreactor was performed with the software Solidworks and the PMMA mask produced with a laser cutting machine. To fabricate the PDMS mold, the PMMA mask was covered with PDMS (1:10, w/w), followed by curing the mixture in the oven (80 °C) for around 1 h. For each dual bioreactor chip, the cured PDMS was detached from the mask, and two pieces of the cured PDMS containing the dual-organ geometry were used. Finally, the PDMS molds were placed between two PMMA plates and fixed with 8 screws to avoid leakages and contaminations, as shown in Figure 4A. Each bioreactor was connected by tubing to a microfuidic pump (Golander Pump, Norcross, GA, USA) and media reservoir containing the appropriate working fluid in test. Flow was initiated at 79.4 μL/min to fill the systems and maintained during the all experiments.
COMSOL Multiphysics simulation of dual-organ bioreactors:
The flow velocity of culture medium, oxygen concentration and DOX distribution were simulated using COMSOL Multiphysics software, considering free tetrahedral meshes with a maximum element size of 0.2 mm. To compute the velocity from convection, the incompressible Navier-Stokes model for Newtonian flow (fixed viscosity) was employed (Equation 1).
| Equation 1 |
Where the density of culture medium (ρmedium), and viscosity of culture medium (μ) were extracted from the literature and flow velocity (v) was set according to the experimental setup (79.4 μL/min).
In the microfluidic domain III in Figure 4c, oxygen transport is presumed to happen via convection and diffusion, thus the governing equation for the oxygen concentration (C) can be written as Equation 2.
| Equation 2 |
In which, oxygen diffusion coefficient in medium (Dmedium) was extracted from the literature.
In the sample domain (where cells reside), oxygen transport is expected to happen only through diffusion. The central calculation for the oxygen profile in the sample domain (Equations 3 and 4 using Michaelis-Menten Kinetics Equation) was based on the oxygen consumption of cells (Michaelis-Menten kinetics). Cell density (including cancer cells) is assumed to be constant, without considering the proliferation. Cell spheroids were simplified to be that cells were homogeneously distributed within GelMA, due to its complexity with considering the sphere sizes distribution. The gas permeability of PDMS was omitted by replacing the wall condition with no influx due to the lack of the information about the environment.
| Equation 3 |
| Equation 4 |
Where the oxygen diffusion coefficient in GelMA (DGelMA), cell density (ρc(z)), and maximum uptake rate () and Michaelis-Menten constant (Km) of oxygen in either of cardiac or cancer cells were extracted from the literature.[49–51] The transportation of doxorubicin within cells such as cytosolic region and intracellular organelles or reversible binding sites in the intracellular space was omitted. The pharmacokinetic modeling of doxorubicin was described by Michaelis-Menten equation.[40] Therefore, for simulating DOX concentration over the bioreactors, the aforementioned Equations 3 and 4 were used and the corresponding parameters were accordingly extracted from the literature.[38–40,52,53]
Design and microfabrication of electrode arrays:
A new multielectrode array chip was designed using AutoCAD (version N.49.M.324) based on the previously reported single biosensors.[41] Briefly, 20 WE and CE were aligned along a common and single RE, so that the three-electrode configuration was preserved while optimizing space for connections. This configuration allowed functionalization of individual electrodes for target specific sensing. The electrodes were spaced to avoid electrical interference from adjacent sensors. In order to further investigate the electrical interference, the setup was simulated with COMSOL (version 5.2) to check how the electric fields were distributed along the chip. A simplified model with a dielectric field was tested. The simulation was carried out with an AC/DC module, where the static measurements were performed at the highest frequency used for EIS (100 kHz) and water was used for dielectric material. Chambers were separated by PDMS.
Single and multi-array microelectrodes were fabricated using the same procedure reported elsewhere.[41] Briefly, the microelectrode set was fabricated with a silver RE, gold CE and gold WE to accomplish a robust biosensing system. The gap among the WE (800 μm-diameter) and the other two electrodes (CE and RE with 150 μm-width) is 200 μm. The detection area shows about 1500 μm in diameter. WE and CE electrodes (Ti: 15 nm-thick, Pd: 15 nm-thick, and Au: 300 nm-thick) were fabricated on cleaned glass through a shadow mask using an e-beam evaporator (EE-Denton machine), and RE electrode (Ti: 15 nm-thick, Pd: 15 nm-thick, and Ag: 300 nm-thick) was deposited onto the Au-deposited glass subtract, followed by annealing them in a furnace (300 °C) for 6 h.
Functionalization and calibration of EC biosensors:
The aptamer immobilization procedure was performed on the electrodes (Table S1, Supplementary Information).[41] Immuno-Aptasensors for monitoring of biomarkers associated with cardiac tissues (Troponin T and CK-MB) and BC tissues (HER-2) were prepared. Briefly, prior to self-assembly monolayer (SAM) coating, the fabricated microelectrodes were first cleaned using H2SO4 (10 mM) and DPBS solutions three times. Then, the microelectrodes were coated with 30 μL of 11-MUA solution (10 mM) for 1 h to generate a thiol terminated SAM. The microelectrodes were rinsed with ethanol and DPBS to remove the unbounded 11-MUA molecules. In the following step, 60 μL of the solution (50 mM of EDC mixed with 50 mM of NHS) was applied to the electrode surface for 15 min. In this process, the activated NHS ester reacts with the primary amine groups of aptamers by the formation of an amide bond. Then, the selected aptamer (10 μg/mL) was incubated for 30 min and the unbounded molecules rinsed with DPBS. The unspecific sites were blocked with cell culture media for 30 min, which also served as baseline. Finally, the antigens of the selected biomarkers were measured after flowed into the biosensors and being incubated for 30 min.
The same procedure was used to perform the calibration curves of the selected biomarkers. In this case, different concentrations of the antigens were successively incubated for 30 min and the Nyquist curves obtained. Then, to eliminate any electrochemical difference that could exist among the different electrodes, the electron-transfer resistance (Rct) was normalized as Rct antigen/Rct medium. Each experiment was performed at least in triplicate using independent biosensors.
Biomarkers evaluation by EC biosensors:
Mediums containing biomarkers were collected on day 1, 3, and 5 from bioreactors and stored at −80 °C for later measurements with biosensors. Measurements of electrochemical impedance spectroscopy (EIS) were achieved with an electrochemical workstation (CHI660E, CH instrument, Inc.). The initial potential (0.1 V) and the frequencies (between 0.1 Hz and 100 kHz at 5 mV of amplitude) were employed for the EIS technique. Potassium ferricyanide (K3Fe(CN)6) electrolyte solution (50 mM) was used for all measurements.
Doxorubicin-induced cardiotoxicity analysis using nano-carriers:
(a) Static mode-Prior to the microfluidic bioreactor study using the organ-on-a-chip platform, the drug nanocarrier was tested in vitro by the 2D cellular studies. These studies allowed to: (i) obtain the optimal concentration of the GYSM-NPs-DOX in order to deliver equal amount of the free chemotherapeutic drug (DOX, 10 μM); (ii) determine the biocompatibility of unloaded GYSM-NPs in healthy induced iPSC-CM and SK-BR-3; and (iii) determine the toxicological effect of the GYSM-NPs-DOX on this same cell lines. The nanosystem GYSM-NPs was synthesized and characterized by some of the authors of this study as pH-dependent drug nanocarrier, as previously published.[47] The optimal concentration of GYSM-NPs-DOX to release equal amount of free DOX (10 μM) was verified by ranging the drug nanocarrier concentrations between 15 and 60 μg/mL, which correspond to a total loaded DOX of 10 and 40 μM. SK-BR-3 cell line was used to assess the cellular uptake of drug delivery by the nanocarriers. Briefly, 100 μL of McCoy’s 5a mediums with different concentrations of GYSM-NPs-DOX were supplemented in 96 well tissue culture plates containing BC cells (1.0 × 104), whereas untreated cells were used as blank. On another well seeded with cells, free DOX was added at 10 μM to be used as comparison drug control (N=4 across the conditions). These threated (with or without the drug carrier) or untreated cells were cultured in 5% CO2, at 37 °C for 24 h. The biocompatibility and toxicological studies of DOX-loaded or -unloaded were conducted in cardiac and BC tissues using Prestoblue colorimetric assay. Briefly, 100 μL of Maccoy’s 5A medium modified containing unloaded GYSM-NPs (16.6 μg/mL), GYSM-NPs-DOX (30 μg/mL) and free DOX (10 μM), were seeded in 96 well tissue culture plates containing the cardiac or BC tissues. After 24 h (day 1), 30 μL of Prestoblue was supplemented for 30 min in a humid chamber (37 °C and 5% CO2), followed by reading the absorbance at 570 and 600 nm. The procedure was repeated on day 3 and 5 as well.
(b) Dynamic mode cardiac and breast organoids containing total number of 1.0 × 106 cells, were cultured in the bioreactors for up to 5 days at a constant flow rate of 79.4 μL/min in a total volume of 5 mL of McCoy’s 5a medium modified. To monitor the level of biomarkers secreted during the experiments (free DOX and GYSM-NPs-DOX), 500 μL of medium was collected at day 1, 3 and 5. All samples were maintained at −80 °C for sample preservation until testing. The viability of the cardiomyocytes and BC were further analyzed using a live/DOX assay.
Statistical analysis:
The statistical analysis was conducted using analysis of variance (ANOVA) with Tukey HSD Post-hoc testing. p values (*p<0.05, **p<0.005, and ***p<0.0005) were considered to be statistically significant. The data were presented with standard deviation of at least triplicate experiments.
Supplementary Material
Acknowledgements
This paper was sponsored by the Office of the Secretary of Defense and was accomplished under Agreement Number W911NF-17-3-003. The views and conclusions contained in this document are those of the authors and should not be interpreted as representing the official policies, either expressed or implied, of the Office of the Secretary of Defense or the U.S. Government. The U.S. Government is authorized to reproduce and distribute reprints for Government purposes notwithstanding any copyright notation herein. The authors gratefully acknowledge funding by the Center for Nanoscale systems (CNS) at Harvard university. S.M. acknowledges funding and support from Fulbright Nehru Doctoral Research Fellowship (FNDR), MHRD (India) and IIE (U.S.A.) to carry out the research work. E.Z. acknowledges Vahabzadeh scholarship in Switzerland. B.B.M. acknowledges generous funding from Department of Biotechnology (DBT) and Department of Science and Technology (DST), Govt. of India. We also acknowledge BWH Neuroscience Department for Confocal Microscope Facility and thank Dr. Kiho Im and Kamyar Mehrabi for providing advices on the MATLAB code for analyzing the beating behaviors.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Junmin Lee, Division of Engineering in Medicine, Brigham and Women’s Hospital, Department of Medicine, Harvard Medical School, Cambridge, MA 02139, USA; Harvard-MIT Division of Health Sciences and Technology Massachusetts Institute of Technology Cambridge, MA 02139, USA; Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California-Los Angeles, Los Angeles, CA 90095, USA; Terasaki Institute for Biomedical Innovation, Los Angeles, CA 90064, USA.
Shreya Mehrotra, Division of Engineering in Medicine, Brigham and Women’s Hospital, Department of Medicine, Harvard Medical School, Cambridge, MA 02139, USA; Harvard-MIT Division of Health Sciences and Technology Massachusetts Institute of Technology Cambridge, MA 02139, USA; Department of Biosciences and Bioengineering, Indian Institute of Technology Guwahati, Guwahati-781039, Assam, India.
Elaheh Zare-Eelanjegh, Division of Engineering in Medicine, Brigham and Women’s Hospital, Department of Medicine, Harvard Medical School, Cambridge, MA 02139, USA; Harvard-MIT Division of Health Sciences and Technology Massachusetts Institute of Technology Cambridge, MA 02139, USA.
Raquel Rodrigues, Division of Engineering in Medicine, Brigham and Women’s Hospital, Department of Medicine, Harvard Medical School, Cambridge, MA 02139, USA; Harvard-MIT Division of Health Sciences and Technology Massachusetts Institute of Technology Cambridge, MA 02139, USA; Center for MicroElectromechanical Systems (CMEMS-UMinho), University of Minho, Campus de Azurém, 4800-058 Guimarães, Portugal.
Alireza Akbarinejad, Division of Engineering in Medicine, Brigham and Women’s Hospital, Department of Medicine, Harvard Medical School, Cambridge, MA 02139, USA; Harvard-MIT Division of Health Sciences and Technology Massachusetts Institute of Technology Cambridge, MA 02139, USA; Department of Chemistry, Faculty of Basic Sciences, Tarbiat Modares University, P.O. Box 14115-175, Tehran, Iran.
David Ge, Division of Engineering in Medicine, Brigham and Women’s Hospital, Department of Medicine, Harvard Medical School, Cambridge, MA 02139, USA; Harvard-MIT Division of Health Sciences and Technology Massachusetts Institute of Technology Cambridge, MA 02139, USA.
Luca Amato, Division of Engineering in Medicine, Brigham and Women’s Hospital, Department of Medicine, Harvard Medical School, Cambridge, MA 02139, USA; Harvard-MIT Division of Health Sciences and Technology Massachusetts Institute of Technology Cambridge, MA 02139, USA.
Kiavash Kiaee, Division of Engineering in Medicine, Brigham and Women’s Hospital, Department of Medicine, Harvard Medical School, Cambridge, MA 02139, USA; Harvard-MIT Division of Health Sciences and Technology Massachusetts Institute of Technology Cambridge, MA 02139, USA; Department of Mechanical Engineering, Stevens Institute of Technology, Hoboken, NJ, 07030 USA.
YongCong Fang, Division of Engineering in Medicine, Brigham and Women’s Hospital, Department of Medicine, Harvard Medical School, Cambridge, MA 02139, USA; Harvard-MIT Division of Health Sciences and Technology Massachusetts Institute of Technology Cambridge, MA 02139, USA; Department of Mechanical Engineering, Tsinghua University, Beijing, 100084, China.
Aliza Rosenkranz, Division of Engineering in Medicine, Brigham and Women’s Hospital, Department of Medicine, Harvard Medical School, Cambridge, MA 02139, USA; Harvard-MIT Division of Health Sciences and Technology Massachusetts Institute of Technology Cambridge, MA 02139, USA.
Wendy Keung, Dr. Li Dak Sum Research Centre, The University of Hong Kong, Pokfulam, Hong Kong.
Biman B. Mandal, Department of Biosciences and Bioengineering, Indian Institute of Technology Guwahati, Guwahati-781039, Assam, India Centre for Nanotechnology, Indian Institute of Technology Guwahati, Guwahati-781039, Assam, India.
Ronald A Li, Dr. Li Dak Sum Research Centre, The University of Hong Kong, Pokfulam, Hong Kong; Department of Paediatrics and Adolescent Medicine, LKS Faculty of Medicine, The University of Hong Kong, Pokfulam, Hong Kong; Ming Wai Lau Centre for Reparative Medicine, Karolinska Institutet, Shatin, Hong Kong.
Ting Zhang, Division of Engineering in Medicine, Brigham and Women’s Hospital, Department of Medicine, Harvard Medical School, Cambridge, MA 02139, USA; Harvard-MIT Division of Health Sciences and Technology Massachusetts Institute of Technology Cambridge, MA 02139, USA; Department of Mechanical Engineering, Tsinghua University, Beijing, 100084, China.
HeaYeon Lee, Division of Engineering in Medicine, Brigham and Women’s Hospital, Department of Medicine, Harvard Medical School, Cambridge, MA 02139, USA; Harvard-MIT Division of Health Sciences and Technology Massachusetts Institute of Technology Cambridge, MA 02139, USA; MARA Nanotech New York, inc. New York, NY 10031-9101, US.
Mehmet Remzi Dokmeci, Division of Engineering in Medicine, Brigham and Women’s Hospital, Department of Medicine, Harvard Medical School, Cambridge, MA 02139, USA; Harvard-MIT Division of Health Sciences and Technology Massachusetts Institute of Technology Cambridge, MA 02139, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California-Los Angeles, Los Angeles, CA 90095, USA; Terasaki Institute for Biomedical Innovation, Los Angeles, CA 90064, USA; Department of Radiology, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Yu Shrike Zhang, Division of Engineering in Medicine, Brigham and Women’s Hospital, Department of Medicine, Harvard Medical School, Cambridge, MA 02139, USA; Harvard-MIT Division of Health Sciences and Technology Massachusetts Institute of Technology Cambridge, MA 02139, USA.
Ali Khademhosseini, Division of Engineering in Medicine, Brigham and Women’s Hospital, Department of Medicine, Harvard Medical School, Cambridge, MA 02139, USA; Harvard-MIT Division of Health Sciences and Technology Massachusetts Institute of Technology Cambridge, MA 02139, USA; Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California-Los Angeles, Los Angeles, CA 90095, USA; Terasaki Institute for Biomedical Innovation, Los Angeles, CA 90064, USA; Department of Radiology, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA; Department of Chemical and Biomolecular Engineering, Henry Samueli School of Engineering and Applied Sciences, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Su Ryon Shin, Division of Engineering in Medicine, Brigham and Women’s Hospital, Department of Medicine, Harvard Medical School, Cambridge, MA 02139, USA; Harvard-MIT Division of Health Sciences and Technology Massachusetts Institute of Technology Cambridge, MA 02139, USA.
References
- [1].Ginsburg O, Bray F, Coleman MP, Vanderpuye V, Eniu A, Kotha SR, Sarker M, Huong TT, Alliance C, Hutchinson F, Lancet 2017, 389, 847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Parhi P, Mohanty C, Sahoo SK, 2012, 17. [DOI] [PubMed] [Google Scholar]
- [3].Hassan MSU, Ansari J, Spooner D, Hussain SA, 2010, 1121. [DOI] [PubMed]
- [4].Kramer BS, 2016, DOI 10.1056/NEJMoa1600249. [DOI]
- [5].Sawaya H, Sebag IA, Plana JC, Januzzi JL, Ky B, Cohen V, Gosavi S, Carver JR, Wiegers SE, Martin RP, Picard MH, Gerszten RE, Halpern EF, Passeri J, Kuter I, Scherrer-crosbie M, 2011, DOI 10.1016/j.amjcard.2011.01.006. [DOI] [PMC free article] [PubMed]
- [6].Martel S, Maurer C, Lambertini M, Pondé N, Expert Opin. Drug Saf 2017, 16, 1021. [DOI] [PubMed] [Google Scholar]
- [7].Yeh ETH, Bickford CL, 2009, 53, DOI 10.1016/j.jacc.2009.02.050. [DOI] [Google Scholar]
- [8].Koutsoukis A, Ntalianis A, Repasos E, Kastritis E, 2018, 64. [DOI] [PMC free article] [PubMed]
- [9].Medicine C, 2013, 8, 59. [Google Scholar]
- [10].Ii VCS, Duffy SA, Dadlani GH, Miller TL, Lipshultz SE, 2005, 7, 187. [DOI] [PubMed] [Google Scholar]
- [11].Januzzi JL, Sebag IA, Plana C, Cohen V, Banchs J, Carver JR, Wiegers SE, Martin RP, Picard MH, Gerszten RE, Halpern EF, Passeri J, Kuter I, Scherrer-crosbie M, D PH, 2014, 63, DOI 10.1016/j.jacc.2013.10.061. [DOI] [Google Scholar]
- [12].Pongprot Y, Sittiwangkul R, 2012, 34, 589. [DOI] [PubMed] [Google Scholar]
- [13].Simões R, Maria L, Luiz A, Mussi V, Gomes V, De Paula A, Braga K, Biomed. Pharmacother 2018, 107, 989.30257411 [Google Scholar]
- [14].Kilickap S, Barista I, Akgul E, Aytemir K, Aksoyek S, Aksoy S, Celik I, Kes S, Tekuzman G, 2005, 798. [DOI] [PubMed]
- [15].Zilinyi R, Czompa A, Czegledi A, Gajtko A, Pituk D, Lekli I, Tosaki A, 2018, 1. [DOI] [PMC free article] [PubMed]
- [16].Mitri Z, Constantine T, O’Regan R, Chemother. Res. Pract 2012, 2012, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].de Paula Costa Monteiro I, Madureira P, de Vasconscelos A, Humberto Pozza D, Andrade de Mello R, Pharmacogenomics 2015, 16, 257. [DOI] [PubMed] [Google Scholar]
- [18].Polini A, Prodanov L, Bhise NS, Manoharan V, Dokmeci MR, Khademhosseini A, 2014, 0441, DOI 10.1517/17460441.2014.886562. [DOI] [PubMed] [Google Scholar]
- [19].Burridge PW, Li YF, Matsa E, Wu H, Ong S, Sharma A, Holmström A, Chang AC, Coronado MJ, Ebert AD, Knowles JW, Telli ML, Witteles RM, Blau HM, Bernstein D, Altman RB, Wu JC, 2016, 22, DOI 10.1038/nm.4087. [DOI] [Google Scholar]
- [20].Leopold JA, Loscalzo J, Circ Res. 2018, 122, 1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fang Y, Eglen RM, 2017, DOI 10.1177/1087057117696795. [DOI]
- [22].Wagner I, Materne EM, Brincker S, Süßbier U, Frädrich C, Busek M, Sonntag F, Sakharov DA, Trushkin EV, Tonevitsky AG, Lauster R, Marx U, Lab Chip 2013, 13, 3538. [DOI] [PubMed] [Google Scholar]
- [23].Schultz JEJ, Kimball TR, Invest JC, Schultz JEJ, Witt SA, Glascock BJ, Nieman ML, Reiser PJ, Nix SL, Kimball TR, Doetschman T, 2002, 109, 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cox TR, Erler JT, 2011, 178, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Henjes F, Bender C, Von Der Heyde S, Braun L, Mannsperger HA, Schmidt C, Wiemann S, Hasmann M, Aulmann S, Beissbarth T, Oncogenesis 2012, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Catarina A, Ades F, De Azambuja E, Piccart-gebhart M, 2013, 22, 152. [Google Scholar]
- [27].Bhise NS, Manoharan V, Massa S, Tamayol A, Ghaderi M, Miscuglio M, Lang Q, Zhang YS, Shin SR, Calzone G, Annabi N, Shupe TD, Bishop CE, Atala A, Dokmeci MR, Khademhosseini A, Biofabrication 2016, 8, DOI 10.1088/1758-5090/8/1/014101. [DOI] [PubMed] [Google Scholar]
- [28].Nichol JW, Koshy ST, Bae H, Hwang CM, Yamanlar S, Khademhosseini A, Biomaterials 2010, 31, 5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Weng Z, He J, Zi M, Chow Y, Mok CF, Keung W, Chow H, Leung AYH, Hajjar RJ, Li RA, Chan CW, 2014, 23, 1704. [Google Scholar]
- [30].Polonchuk L, Chabria M, Badi L, Hoflack JC, Figtree G, Davies MJ, Gentile C, Sci. Rep 2017, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Daniela Tirziu MS, Giordano Frank J., Circulation 2010, 122, 928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Weng Z, Kong CW, Ren L, Karakikes I, Geng L, He J, Chow MZY, Mok CF, Harvey C, Sarah W, Keung W, Chow H, Andrew M, Leung AYH, Hajjar RJ, Li RA, Chan CW, Stem Cells Dev. 2014, 23, 1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sadeghi AH, Shin SR, Deddens JC, Fratta G, Mandla S, Yazdi IK, Prakash G, Antona S, Demarchi D, Buijsrogge MP, Sluijter JPG, Hjortnaes J, Khademhosseini A, 2017, 1601434, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Engler AJ, Carag-krieger C, Johnson CP, Raab M, Tang H, Speicher DW, Sanger JW, Sanger JM, Discher DE, 2008, 3794. [DOI] [PMC free article] [PubMed]
- [35].Hinderer S, Schenke-Layland K, Adv. Drug Deliv. Rev 2019, 146, 77. [DOI] [PubMed] [Google Scholar]
- [36].Leone M, Engel FB, Clin. Sci 2019, 133, 1229. [DOI] [PubMed] [Google Scholar]
- [37].Santiago J, Dangerfield AL, Rattan SG, Bathe KL, Cunnington RH, Raizman JE, Bedosky KM, Freed DH, Kardami E, Dixon IMC, 2010, 1573. [DOI] [PubMed]
- [38].Eikenberry S, Theor. Biol. Med. Model 2009, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kang W, Weiss M, Drug Metab. Dispos 2003, 31, 462. [DOI] [PubMed] [Google Scholar]
- [40].Weiss M, Kang W, J. Pharmacol. Exp. Ther 2002, 300, 688. [DOI] [PubMed] [Google Scholar]
- [41].Shin SR, Zhang YS, Kim D, Manbohi A, Avci H, Silvestri A, Aleman J, Hu N, Kilic T, Keung W, Righi M, Assawes P, Alhadrami HA, Li RA, Dokmeci MR, Khademhosseini A, 2016, DOI 10.1021/acs.analchem.6b02028. [DOI] [PMC free article] [PubMed]
- [42].Song S, Wang L, Li J, Fan C, Zhao J, TrAC - Trends Anal. Chem 2008, 27, 108. [Google Scholar]
- [43].Khasraw M, Bell R, Dang C, Breast 2012, 21, 142. [DOI] [PubMed] [Google Scholar]
- [44].Collinson PO, Eur. Heart J 1998, 19 Suppl N, N16–24. [PubMed] [Google Scholar]
- [45].Lian Lam LS, Czerniecki Brian J., Fitzpatrick Elizabeth, Xu Shuwen, Xu H. Z. Xiaowei, J Mol Biomark Diagn 2012, 4, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].De Jong WH, Borm PJA, Int. J. Nanomedicine 2008, 3, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Rodrigues RO, Baldi G, Doumett S, Garcia-Hevia L, Gallo J, Bañobre-López M, Dražić G, Calhelha RC, Ferreira ICFR, Lima R, Gomes HT, Silva AMT, Mater. Sci. Eng. C 2018, 93, 206. [DOI] [PubMed] [Google Scholar]
- [48].Lee J, Manoharan V, Cheung L, Lee S, Cha BH, Newman P, Farzad R, Mehrotra S, Zhang K, Khan F, Ghaderi M, Lin YD, Aftab S, Mostafalu P, Miscuglio M, Li J, Mandal BB, Hussain MA, Wan KT, Tang XS, Khademhosseini A, Shin SR, ACS Nano 2019, 13, 12525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Brown J. C. Y. D. David A., MacLellan W. Robb, Laks Hillel, Wu R. E. B. Benjamin M., Biotechnol. Bioeng 1996, 189 ( Pt 3, 503. [Google Scholar]
- [50].van Stroe-Biezen SAM, Everaerts FM, Janssen LJJ, Tacken RA, Anal. Chim. Acta 1993, 273, 553. [Google Scholar]
- [51].Astolfi M, Péant B, Lateef MA, Rousset N, Kendall-Dupont J, Carmona E, Monet F, Saad F, Provencher D, Mes-Masson AM, Gervais T, Lab Chip 2016, 16, 312. [DOI] [PubMed] [Google Scholar]
- [52].Weinberg BD, Patel RB, Exner AA, Saidel GM, Gao J, Control J. Release 2007, 124, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zhan W, Xu XY, J. Drug Deliv 2013, 2013, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.