

Abstract
Electrochemical techniques have long been heralded for their innate sustainability as efficient methods for achieving redox reactions. Carbonyl desaturation, as a fundamental organic oxidation, is an oft-employed transformation to unlock adjacent reactivity through the formal removal of two hydrogen atoms. To date, the most reliable methods for achieving this seemingly trivial reaction have relied on transition metals (Pd/Cu) or stoichiometric reagents based on I, Br, Se, or S. Herein we report an operationally simple pathway to access such structures from enol silanes and phosphates using electrons as the primary reagent. This electrochemically driven desaturation exhibits a broad scope across an array of carbonyl derivatives, is easily scalable (1–100g), and can be predictably implemented into synthetic pathways using experimentally or computationally derived NMR shifts. Systematic comparisons to state-of-the-art techniques reveal that this method can uniquely desaturate a wide array of carbonyl groups. Mechanistic interrogation suggests a radical-based reaction pathway.
Graphical Abstract

The removal of one molecule of hydrogen adjacent to a carbonyl compound is one of the simplest organic oxidation reactions known and is a widely employed tactic in synthesis.1–3 Classic methods for accomplishing this transformation involve indirect α-functionalization approaches traversing through halide, sulfur, and selenium derivatives.4–8 Chemoselective methods that directly afford enones from ketones are indeed more desirable and have been extensively explored (Figure 1). Amongst them, the Saegusa-Ito reaction, discovered in 1978, remains the most oft-applied method for such applications.9 In its canonical implementation, formation of a silyl enol ether, followed by exposure to stoichiometric (from 0.5–1.0 equiv.) quantities of palladium delivers the desired α,β-desaturated product.9 Variants that employ a co-oxidant (e.g. O2, quinone or [Cu]) to lower the [Pd]-levels have also been reported.10,11 Another popular approach involves the use of stoichiometric IBX through an SET-based process.12,13 Recently, two new methods have also appeared from the Newhouse and Dong groups that allow the use of catalytic amounts of palladium and copper, respectively.14–20 These methods expand the scope of available desaturation methods to nitriles, esters, lactones, and lactams and do not require the preparation of enol ethers. Since the essence of this reaction involves a formal 2-electron oxidation, it stands to reason that even simpler redox approaches might be developed. Indeed, in 1973, the Shono group demonstrated that enol acetates can undergo anodic oxidation in AcOH as solvent to afford the corresponding enone. In order to deliver synthetically useful yields of product, α-substitution was required with simple cyclohexanone-substrates providing <10% enone.21,22 Similar reactivity was also observed by Moeller and co-workers in their studies of silyl enol ether alkylation where trace amounts of enone were isolated as a by-product (<5% yield).23–25 Building on these encouraging studies, we report herein an electrochemically driven approach to elicit desaturation that requires no metals or chemical oxidants and features a broad substrate scope with inherent scalability. The utility of this method is placed in the context of the most popular and recently disclosed methods, and a simple method for predicting reactivity is also described.
Fig. 1. α,β-desaturation of carbonyl and enol compounds, state-of-the-art and design of this work.

a) Chemical approaches: The most used chemical approaches for the desaturation of carbonyl and enol ether compounds. Saegusa and Ito reported a method using stoichiometric palladium, which is limited to ketones and aldehydes9. Newhouse also reported studies using palladium, which also require expensive Zn(TMP)2, and elevated temperature14–16. Studies by Dong require copper, peroxide, and elevated temperature and have limited scope18. Previous studies from Nicolaou lab use stoichiometric IBX, strong oxidant, and are limited to ketones and aldehydes12. b) Electrochemical precedents: The Shono group demonstrated that enol acetates can undergo anodic oxidation to afford the corresponding enone. Moeller and co-workers observed that silyl enol ether can similarly undergo direct anodic oxidation to form trace amounts of enone under the described conditions. In both cases, the methods are limited to ketones, have shown limited functional group tolerance, and low yields were obtained with non-substituted ketones, c) Electrochemically Driven Desaturation (EDD). Metal, chemical oxidant free and scalable electrochemical desaturation method is described. EDD is applicable with various types of carbonyls; ketones, esters, lactams, and aldehydes.
RESULTS AND DISCUSSION
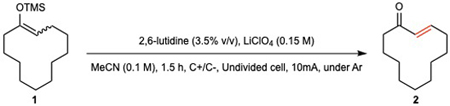
The TMS-enol ether of cyclododecanone 1 was chosen for initial optimization of electrochemically driven desaturation (EDD); an abbreviated summary is depicted in Table 1 (See Supplementary Table 1–5). Trial runs using the literature conditions noted above provided only trace quantities of product. Our prior experiences for electrochemical reaction development served as a template for this study.26–33 A myriad of electrolytes, electrodes, and solvents were evaluated. First, an electrolyte screen revealed that inorganic non-nucleophilic salts proved optimal (entries 4–7) with NaSbF6 ($0.56/gram) delivering the highest conversion. The use of a graphite anode was found to be essential whereas several materials were suitable for the cathode (entries 8–11). Ultimately the low cost (ca. $0.1/cm2) and efficiency of graphite motivated its selection for both electrode materials. Of all solvents screened, MeCN, acetone, DMA, and DMF could be employed but MeCN gave the highest yield across a broad range of substrates. A variety of bases were also tested with heteroaromatic amines proving most promising (entries 15–16). 2,4,6-Collidine (30% v/v – entry 18) emerged as optimum providing the desired product 2 in 62% isolated yield. The final set of EDD conditions tolerates exogenous air and moisture, lead to completion in 90 minutes or less, and can be setup in minutes, using a simple undivided cell and a commercial potentiostat.
Table 1.
Optimization of the EDD reaction.
 | ||
|---|---|---|
| Entry | Deviation from above | Yield (%)a |
| 1 | None | 22 (17) |
| 2 | TEAOTs, AcOK in acetic acid (Shono. enol acetate, ref 21) | n.d. |
| 3 | LiClO4, 3.5% lutidine in MeCN/PrOH (1:1) (Moeller, ref 23/25) | 8 |
| 4 | TBABF4 instead of LiClO4 | n.d. |
| 5 | LiBF4 instead of LiClO4 | 12 |
| 6 | NaOTs instead of LiClO4 | 14 |
| 7 | NaSbF6 instead of LiClO4 | 30 |
| 8 | RVC anode. NaSbF6 instead of LiClO4 | <5% |
| 9 | Platinium anode, NaSbF6 instead of LiClO4 | n.d. |
| 10 | Nickel foam anode, NaSbF6 instead of LiClO4 | 19 |
| 11 | RVC cathode, NaSbF6 instead of LiClO4 | 12 |
| 12 | Acetone instead of MeCN, NaSbF6 instead of LiClO4 | 14 |
| 13 | DMF instead of MeCN, NaSbF6 instead of LiClO4 | 11 |
| 14 | DMA instead of MeCN, NaSbF6 instead of LiClO4 | 18 |
| 15 | TEA instead of lutidine, NaSbF6 instead of LiClO4 | n.d. |
| 16 | Collidine instead of lutidine, NaSbF6 instead of LiClO4 | 45 (39) |
| 17 | 20% collidine., NaSbF6 instead of LiClO4 | 55 |
| 18 | 30% collidine, NaSbF6 instead of LiClO4 | 70 (62) |
| 19 | 50% collidine, NaSbF6 instead of LiCI04 | 68 |
| 20 | Under air | 65 |
| 21 | Non-anhydrous MeCN | 64 |
| 22 | No 2,4,6-collidine | n.d. |
| 23 | No NaSbF6 | n.d. |
| 24 | No electrolysis | n.d. |
Isolated yield. n.d.: not detected.
EDD could be applied to a diverse set of carbonyl derivatives as illustrated in Table 2. As there are numerous desaturation methods available to the practitioner all of the results are placed into context with direct comparison to the powerful Pd, Cu, and hypervalent iodine-based systems. With regards to ketone substrates, both cyclic (from 5–15 membered rings) and acyclic derivatives could be employed (2–17). This stands in contrast to recently developed catalytic methods that operate smoothly on cyclic systems but fail on acyclic ones (14 and 15).15,18 As EDD of ketones is reliant on formation of a silyl enol ether, regioselective desaturation is possible simply by tuning conditions (i.e. 7 vs. 8). Substituents at the α-, β-, and γ-position are all tolerated as well as Lewis-basic heteroatoms (16), alkynes (10), proximal cyclopropanes (15), esters (16 and 17), TBS protected alcohols (11), and acid-labile ketals (6). A two-step in situ EDD protocol was also developed to afford enones 6, 12 and 17 in decent yields directly from the respective ketone starting material. Esters and lactones, substrate classes that have only recently succumbed to direct dehydrogenation,14,17,18 can also be subjected to EDD using the corresponding diphenylphosphate ester derivatives (18–31). Such enol derivatives are easily prepared and hydrolytically stable unlike the corresponding silyl ketene acetals. Simple lactones and benzolactones, which are outside the substrate scope of IBX and Saegusa methodologies can be smoothly dehydrogenated. As with EDD of ketones, the functional group tolerance here is also broad including aryl halides (22 and 24), CF3 (30), oxidizable anisoles (23), tosyl protected amines (29), and alkenes (32). In addition, α-aryl lactones also afforded the desired products in satisfactory yields (21–23 and 25). The difficulty of desaturating such substrates has been documented by Dong. They are often alternatively accessed through cross-coupling on the corresponding vinyl halide derivatives or through α-bromination/elimination sequences.34,35 It worth noting that while comparing EDD with other precedented methods, the set of conditions reported by the Newhouse group was found to provide the desaturated α-aryl lactones in moderate yields. Next, the particularly difficult class of aldehydes were investigated (32–34, silyl enol ethers employed). Due to the instability of such desaturated products the yields observed were moderate (and accompanied by 5–11% of recovered parent aldehyde). Other direct catalytic methods for this dehydrogenation are not applicable, with the Saegusa protocol being the only other option. Lactams, a similarly challenging class of carbonyls, were surveyed as diphenylphosphate-ketenimine acetals, and in select cases (35–37) were viable.
Table 2.
Scope of the EDD reaction.
 |
Yield based on GCMS conversion.
Using in-situ protocol.
Yield based on NMR conversion.
Prepared from phenyl acetate-keteniminyl acetals. n/a: not applicable. n.d.: not detected.
The scalability of the method was evaluated using cyclopentadecanone-derived silyl enol ether 38 on a 4-gram scale (Figure 2A) to afford enone 12, a key intermediate in the synthesis of (R)-muscone 39, a valuable ingredient in the fragrance industry.36 A simple increase of current (from 10 mA to 300 mA) and the use of alternating polarity (to avoid any accumulation of material at the anode) enabled the standard EDD reaction to smoothly deliver compound 12 in 66% yield. To increase scale further, the design and assembly of a flow apparatus containing six reaction cells was undertaken (Figure 2A). After optimization, 100 grams of 38 were successfully converted to compound 12 by increasing the current value to 3.6 A (compared to 300 mA in batch) to obtain 61% isolated yield and 27% recovered starting material 38.
Fig. 2. Scale-up and mechanistic study of EDD reaction.

a) Batch (4 gram) and flow (100 gram) scale-up of a key intermediate in the synthesis of (R)-muscone. b) Proposed mechanism of the EDD reaction based on observations. It is postulated that EDD proceeds through three elementary steps: (1) formation of the radical cation intermediate through direct anodic oxidation; 2) deprotonation of the radical anion intermediate to afford the allylic radical; 3) a second oxidation to form an oxonium intermediate. c) Experimental evidence for step 1. d) Experimental evidence for step 2. e) Experimental evidence for step 3. f) Prediction of reactivity based on 1H NMR chemical shift. aYield based on LCMS conversion.
From a mechanistic standpoint, the EDD reaction accomplishes the formal removal of two electrons and one proton from the corresponding silyl enol ether. The electrochemical oxidation of silyl enol ethers has been previously disclosed by Moeller and studied mechanistically by Wright.25 These studies demonstrated that initial anodic oxidation leads to the formation of an enol ether radical cation intermediate by using a cyclopropyl ring-opening clock. A similar conclusion was made by Moeller and co-workers when they oxidized various alkyl-enol ethers and thio-enol ethers.37 It is therefore postulated that EDD proceeds through three elementary steps: (1) formation of the radical cation intermediate 40; (2) deprotonation to afford 41; (3) a second oxidation to form oxonium 42 which affords the desired enone product 5 (Figure 2B). To provide empirical support for the proposed mechanism, three control experiments were designed and tested (Figure 2C–E). First, the standard reaction conditions under air revealed the formation of the 1,2-diketone side product 44 in 9% yield. The amount of this by-product decreases to 4% when the reaction performed under inert atmosphere. In addition, the parent ketone is the only product observed when water is added to the reaction. These results suggest that formation of compound 44 derived from molecular oxygen via a radical type mechanism. Next, to support the crucial role of the base in the deprotonation event, the EDD reaction conditions were applied to compound 1 with various amounts of 2,4,6-collidine (Figure 2D). No desired product was obtained when base was excluded, reinforcing the importance of the base for the EDD reaction and its implication in the deprotonation step 2 (Figure 2B). Furthermore, a noticeable improvement was observed between the reaction efficiency and the base concentration. Finally, a third experiment was conducted to explore the formation of an oxonium intermediate (Figure 2E). Compound 45 was subjected to the reaction conditions affording naphthalene 47 in 49% yield. The formation of this product is in accordance with the formation of intermediate (46), which after elimination would deliver 47. Similarly, when the diethylphosphate 48 derived from dehydroepiandrosterone was subjected to EDD, diene 50 was also observed as a side-product.
During these studies it was empirically noted that ketone, lactam and lactone substrates whose vinylic proton shifts registered between 4.6 and 5.1 ppm showed good conversion to the desaturated product. However, compounds with NMR shifts lower than 4.6 ppm led to the formation of dimer, hydrolysis, and other by-products. Compounds with NMR shifts higher than 5.1 ppm showed low reactivity toward oxidation. Based on these findings and Moeller’s reports,37 it is possible to identify a trend between the oxidation potential of the enol ether (to form a radical cation intermediate similar to 40) and the electron density of the π-system (Figure 2F, see Supplementary Figure 10 for details). Compounds such as dehydroepiandrosterone-phosphate 51 and the 8-membered lactam 57 cannot be oxidized under the EDD reaction conditions due to the deshielded vinylic proton correlating to a higher oxidation potential (CV = 2.06 to 2.47 V). When the vinylic proton is shielded, the oxidation potential becomes lower and reactivity toward EDD is observed (CV = 1.54 to 1.72 V). On the other hand, compounds such as dehydroepiandrosterone-TMS 54, lactone-TMS 56, and lactam-TMS 59 whose vinylic protons that are too shielded, afforded low amounts of the desaturated product (CV = 1.32 to 1.4 V). In this case, the enol derivatives react as nucleophilic radicals rather than electrophilic radical cations due to greater cationic stabilization, which renders the EDD pathway less favorable.38,39 Accordingly, this explains why electron withdrawing enol-phosphates are required for esters and lactams in EDD rather than the corresponding silyl enol ethers.
Based on these observations, one could imagine predicting the outcome of the EDD reaction by calculating the NMR shift of the TMS enol ethers of interest (Figure 3, Step 1). With this idea in mind, a simple protocol using GAUSSIAN16, a quantum chemical calculation software, was developed. This protocol gives access to a calculated NMR shift based on the shielding constants using the gauge-including atomic orbital (GIAO) method in the WP04 database (functional) with an aug-cc-pVDZ basis set using tetramethylsilane as a reference (Figure 3, Step 2).40,41 Next, to obtain a more accurate value, the NMR shift value was corrected using an experimentally generated linear regression (Figure 3, Step 3 and See Supplementary section “Computational calculations using GAUSSIAN16” for detailed graphical step-by-step guide). Finally, the corrected NMR shift can be used to predict the efficiency of the EDD reaction (Figure 3, Step 4). To our knowledge, this is a rare example of using calculated NMR shift to predict the scope of an organic methodology.42
Fig. 3. Gaussian computational experiment to assess the feasibility of the EDD reaction; Case study of TMS-enolates.
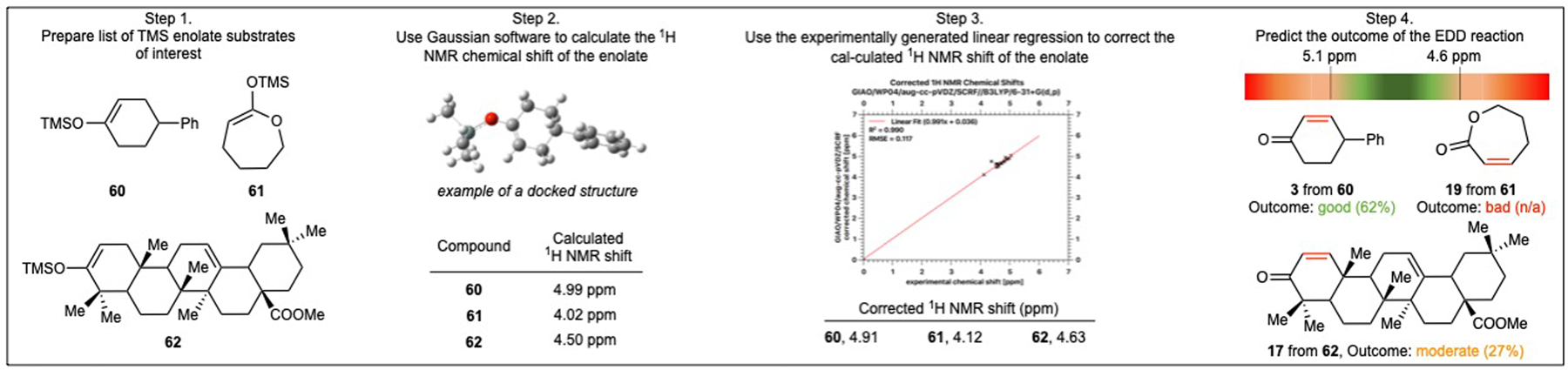
A simple four step protocol using GAUSSIAN16 is described to predict the efficiency of the EDD reaction; Step 1) Prepare a list of TMS enol ether substrates of interest. Step 2) Use Gaussian software, “GIAO/WP04/aug-cc-pVDZ//B3LYP/6–31+G(d,p)” functional, to calculate the 1H NMR chemical shift of the desired enol ether. Step 3) Use the experimentally generated linear regression to correct the calculated 1H NMR shift of the enol ether. Step 4) Predict the outcome of the EDD reaction.
The desaturation of carbonyl derivatives is a basic reaction of utmost utility in organic chemistry as it unlocks a variety of useful downstream transformations. Studies in this area continue to the present day; the contribution reported herein affords a potentially simple solution to this problem. Drawing from early studies in electrosynthesis and more recent mechanistic studies of anodic enol-oxidation, a useful protocol for EDD has been uncovered. This oxidation protocol can be performed in an undivided cell, on multiple scales, without strict removal of air or water, and in the absence of expensive metals, ligands, or stoichiometric organic oxidants. As with the oxidation of alcohols, for which numerous methods are available, this desaturation study has been placed into context with the most powerful methods currently available to aid the practitioner. Finally, a simple 1H NMR-based rubric was created to allow users to experimentally or computationally predict which substrates are suitable for EDD, which should facilitate its rapid adoption.
Supplementary Material
ACKNOWLEDGMENT
Financial support for this work was provided by NIH Grant (GM-118176), the NSF (#1740656), and AGC Inc. (to Y.T.). S.G. thanks the Council for Higher Education, Fulbright Israel and Yad Hanadiv for the generous fellowships. Authors are grateful to Dr. Dee-Hua Huang and Dr. Laura Pasternack (Scripps Research) for assistance with nuclear magnetic resonance (NMR) spectroscopy, to Dr. Jason Chen, Brittany Sanchez and Emily Sturgell (Scripps Automated Synthesis Facility) for assistance with HPLC, HRMS and LCMS. For any questions contact P.S.B.
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article and its Supplementary Information.
REFERENCES
- (1).Muzart J One-Pot Syntheses of α, β-Unsaturated Carbonyl Compounds through Palladium-Mediated Dehydrogenation of Ketones, Aldehydes, Esters, Lactones and Amides. Eur. J. Org. Chem 20, 3779–3790 (2010). [Google Scholar]
- (2).Otera J Modern Carbonyl Chemistry. Wiley-VCH: Weinheim, Germany: (2000). [Google Scholar]
- (3).Turlik A, Chen Y & Newhouse TR Dehydrogenation Adjacent to Carbonyls Using Palladium–Allyl Intermediates. Synlett 27, 331–336 (2016). [Google Scholar]
- (4).Reich HJ, Reich IL & Renga JM Organoselenium chemistry: alpha-Phenylseleno carbonyl compounds as precursors for alpha, beta-unsaturated ketones and esters. J. Am. Chem. Soc 95, 5813–5815 (1973). [Google Scholar]
- (5).Sharpless KB & Gordon KM Selenium dioxide oxidation of ketones and aldehydes. Evidence for the intermediacy of beta-ketoseleninic. J. Am. Chem. Soc 98, 300–301 (1976,). [Google Scholar]
- (6).Trost BM, Salzmann TN & Hiroi K New synthetic reactions. Sulfenylations and dehydrosulfenylations of esters and ketones. J. Am. Chem. Soc 98, 4887–4902 (1976). [Google Scholar]
- (7).Trost BM & Salzmann TN New synthetic reactions. Sulfenylation-dehydrosulfenylation as a method for introduction of unsaturation. J. Am. Chem. Soc 95, 6840–6842 (1973). [Google Scholar]
- (8).Larock RC Comprehensive Organic Transformations. VCH Publishers: New York, 129–131 (1989). [Google Scholar]
- (9).Ito Y, Hirao T & Saegusa T Synthesis of α,β-unsaturated carbonyl compounds by palladium (II)-catalyzed dehydrosilylation of silyl enol ethers. J. Org. Chem 43, 1011–1013 (1978). [Google Scholar]
- (10).Shimizu I & Tsuji J Palladium-Catalyzed Decarboxylation-Dehydrogenation of Allyl β-Keto Carboxylates and Allyl Enol Carbonates as a Novel Synthetic Method for α-Substituted α,β-Unsaturated Ketones. J. Am. Chem. Soc 104, 5844–5846 (1982). [Google Scholar]
- (11).Diao T & Stahl SS Synthesis of cyclic enones via direct palladium-catalyzed aerobic dehydrogenation of ketones. J. Am. Chem. Soc 133, 14566–14569 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Nicolaou KC, Zhong YL & Baran PS A new method for the one-step synthesis of α,β-unsaturated carbonyl systems from saturated alcohols and carbonyl compounds. J. Am. Chem. Soc 122, 7596–7597 (2000). [Google Scholar]
- (13).Nicolaou KC, Montagnon T, Baran PS & Zhong YL Iodine (V) Reagents in Organic Synthesis. Part 4. o-Iodoxybenzoic Acid as a Chemospecific Tool for Single Electron Transfer-Based Oxidation Processes J. Am. Chem. Soc 124, 2245–2258 (2002). [DOI] [PubMed] [Google Scholar]
- (14).Chen Y, Romaire JP & Newhouse TR Palladium-catalyzed α,β-dehydrogenation of esters and nitriles. J. Am. Chem. Soc 137, 5875–5878 (2015). [DOI] [PubMed] [Google Scholar]
- (15).Huang D, Zhao Y & Newhouse TR Synthesis of Cyclic Enones by Allyl-Palladium-Catalyzed α,β-Dehydrogenation. Org. Lett 20, 684–687 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Chen Y, Turlik A & Newhouse TR Amide α,β-dehydrogenation using allyl-palladium catalysis and a hindered monodentate anilide. J. Am. Chem. Soc 138, 1166–1169 (2016). [DOI] [PubMed] [Google Scholar]
- (17).Huang D, Szewczyk SM, Zhang P & Newhouse TR Allyl-Nickel Catalysis Enables Carbonyl Dehydrogenation and Oxidative Cycloalkenylation of Ketones. J. Am. Chem. Soc 141, 5669–5674, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chen M & Dong G Copper-catalyzed desaturation of lactones, lactams, and ketones under pH-neutral conditions. J. Am. Chem. Soc 141, 14889–14897 (2019). [DOI] [PubMed] [Google Scholar]
- (19).Chen M & Dong G Direct catalytic desaturation of lactams enabled by soft enolization. J. Am. Chem. Soc 139, 7757–7760 (2017). [DOI] [PubMed] [Google Scholar]
- (20).Chen M, Rago AJ & Dong G Platinum-Catalyzed Desaturation of Lactams, Ketones, and Lactones. Angew. Chem. Int. Ed 130, 16437–16441 (2018). [DOI] [PubMed] [Google Scholar]
- (21).Shono T, Matsumura Y & Nakagawa Y Electroorganic chemistry. XII. Anodic oxidation of enol esters. J. Am. Chem. Soc 96, 3532–3536 (1974). [Google Scholar]
- (22).Shono T, Okawa M & Nishiguchi I Electroorganic chemistry. XXI. Selective formation of. alpha-acetoxy ketones and general synthesis of 2, 3-disubstituted 2-cyclopentenones through the anodic oxidation of enol acetates. J. Am. Chem. Soc 97, 6144–6147 (1975). [Google Scholar]
- (23).Reddy SHK, Chiba K, Sun Y & Moeller KD Anodic oxidations of electron-rich olefins: radical cation based approaches to the synthesis of bridged bicyclic ring skeletons. Tetrahedron 57, 5183–5197 (2001). [Google Scholar]
- (24).Perkins RJ, Feng R, Lu Q & Moeller KD Anodic Cyclizations, Seven-Membered Rings, and the Choice of Radical Cation vs. Radical Pathways. Chin. J. Chem 37, 672–678 (2019). [Google Scholar]
- (25).Sperry JB, Whitehead CR, Ghiviriga I, Walczak RM & Wright DL Electrooxidative coupling of furans and silyl enol ethers: application to the synthesis of annulated furans. J. Org. Chem 69, 3726–3734 (2004). [DOI] [PubMed] [Google Scholar]
- (26).Kingston C et al. A Survival Guide for the “Electro-curious” Acc. Chem. Res 53, 72–83 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Xiang J et al. Hindered Dialkyl Ether Synthesis via Electrogenerated Carbocations. Nature 573, 398–401 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Takahira Y et al. Electrochemical C(sp3)-H Fluorination. Synlett 30, 1178–1182 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kawamata Y et al. Electrochemically Driven, Ni-Catalyzed Aryl Amination: Scope, Mechanism, and Applications. J. Am. Chem. Soc 141, 6392–6402 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Peters BK et al. Scalable and Safe Synthetic Organic Electroreduction Inspired by Li-Ion Battery Chemistry. Science 363, 838–845 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Yan M, Kawamata Y & Baran PS Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. Chem. Rev 117, 13230–13319 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Li C et al. Electrochemically Enabled, Nickel-Catalyzed Amination. Angew. Chem. Int. Ed 56, 13088–13093 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kawamata Y et al. Scalable, electrochemical oxidation of unactivated C–H bonds. J. Am. Chem. Soc 139, 7448–7451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Rosen JD, Nelson TD, Huffman MA & McNamara JM A convenient synthesis of 3-aryl-δ-lactones. Tet. Lett 44, 365–368 (2003). [Google Scholar]
- (35).Cai XC & Snider BB Synthesis of the Spiroiminal Moiety and Approaches to the Synthesis of Marineosins A and B. J. Org. Chem 78, 12161–12175 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Tanaka K, Ushio H, Kawabata Y & Suzuki H Asymmetric synthesis of (R)-(−)-and (S)-(+)-muscone by enantioselective conjugate addition of chiral dimethylcuprate to (E)-cyclopentadec-2-en-1-one. J. Chem. Soc., Perkin Trans 1, 1445–1452 (1991). [Google Scholar]
- (37).Feng R, Smith JA & Moeller KD Anodic cyclization reactions and the mechanistic strategies that enable optimization. Acc. Chem. Res 50, 2346–2352 (2017). [DOI] [PubMed] [Google Scholar]
- (38).Murphy JA, Khan TA, Zhou S, Thomson DW & Mahesh M Highly Efficient Reduction of Unactivated Aryl and Alkyl Iodides by a Ground-State Neutral Organic Electron Donor. Angew. Chem. Int. Ed 44, 1356–1360 (2005). [DOI] [PubMed] [Google Scholar]
- (39).Tang F & Moeller KD Anodic oxidations and polarity: exploring the chemistry of olefinic radical cations. Tetrahedron 65, 10863–10875 (2009). [Google Scholar]
- (40).Wiitala KW, Hoye TR & Cramer CJ Hybrid density functional methods empirically optimized for the computation of 13C and 1H chemical shifts in chloroform solution. J. Chem. Theory Comput 2, 1085–1092 (2006). [DOI] [PubMed] [Google Scholar]
- (41).Jain R, Bally T & Rablen PR Calculating accurate proton chemical shifts of organic molecules with density functional methods and modest basis sets. J. Org. Chem 74, 4017–4023 (2009). [DOI] [PubMed] [Google Scholar]
- (42).Handy ST & Zhang Y A simple guide for predicting regioselectivity in the coupling of polyhaloheteroaromatics. Chem. Comm 3, 299–301 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available within the article and its Supplementary Information.