

Abstract
In cells, myriad membrane-interacting proteins generate and maintain curved membrane domains with radii of curvature around or below 50 nm. To understand how such highly curved membranes modulate specific protein functions, and vice versa, it is imperative to use small liposomes with precisely defined attributes as model membranes. Here, we report a versatile and scalable sorting technique that uses cholesterol-modified DNA “nanobricks” to differentiate hetero-sized liposomes by their buoyant densities. This method separates milligrams of liposomes, regardless of their origins and chemical compositions, into 6–8 homogeneous populations with mean diameters of 30–130 nm. We show that these uniform, leak-resistant liposomes serve as ideal substrates to study, with an unprecedented resolution, how membrane curvature influences peripheral (ATG3) and integral (SNARE) membrane protein activities. Compared with conventional methods, our sorting technique represents a streamlined process to achieve superior liposome size uniformity, which benefits research in membrane biology and development of liposomal drug-delivery systems.
Vesicles — tiny bubbles of fluid wrapped by membranes — are abundant in cells and extracellular space, performing critical tasks including nutrient uptake, cargo transport, and waste confinement. Vesicles smaller than ~150 nm in diameter, such as many transport and secretory vesicles, are usually recognized and operated upon by proteins based on their membrane curvatures and chemical compositions1,2. Therefore, uniformly-sized liposomes (vesicles made of synthetic components) within this size range are uniquely suitable to accurately recapitulate the cellular vesicles’ structure and behavior. Classical methods for controlling the size of sub-150-nm liposomes, including the small unilamellar vesicles (SUVs), rely on liposome formation conditions3–5 (e.g., lipid composition and solvent-to-water mixing ratio) as well as post-formation homogenization6–9 (e.g., extrusion and sonication) and purification10,11 (e.g., centrifugation and size-exclusion chromatography). The production outcome is tied to a set of empirically determined parameters that may not be independently tunable, thus limiting users’ ability to selectively vary the liposome size and composition. Microfluidic-based systems provide a way to tune liposome size and dispersity, but often require nonstandard devices built in-house12–14. Additionally, the capability of microfluidic-based methods to incorporate functional membrane proteins into SUVs has yet to be examined. Another promising approach is to guide lipid-bilayer self-assembly by DNA nanotemplates15–17. While effective in forming size-controlled liposomes with programmable membrane-protein stoichiometry, this approach is cost-ineffective for mass production due to the requirement of a unique DNA template for each liposome configuration and the relatively low lipid recovery. Moreover, the use of detergent limits the selection of compatible cargo molecules.
To overcome these problems, here we devised a liposome sorting strategy (Fig. 1a, b) that can be used in conjunction with an assortment of liposome manufacturing methods. Although typical lipid bilayers are lighter than aqueous solutions, liposomes that are different in size but identical in membrane and internal contents differ only slightly in buoyant density, because a liposome’s aqueous lumen constitutes the bulk of its mass. However, the surface-area-to-volume ratio (S/V) of a spherical liposome decreases rapidly with increasing size (S/V is inversely proportional to radius), affording the opportunity to amplify the buoyant density difference among liposomes by ubiquitously coating them with a dense material (similar to attaching bricks to helium balloons). In theory, smaller liposomes will gain more density than larger ones when coated by such molecular bricks (Fig. 1c), allowing liposome separation by isopycnic centrifugation.
Figure 1.
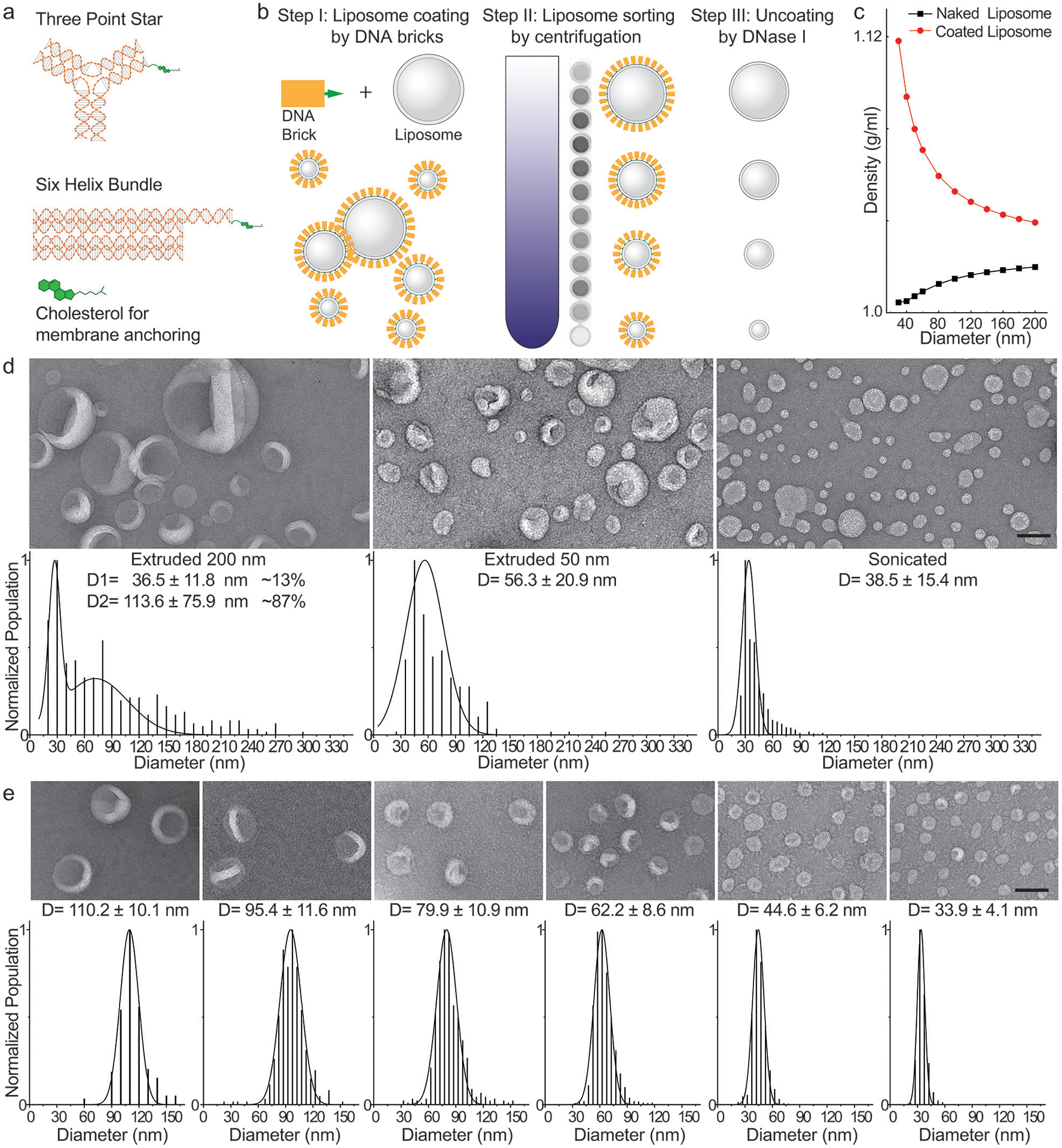
DNA-brick-assisted liposome sorting scheme and results. (a) Schematic diagrams of cholesterol-labeled DNA bricks. Note the absence of sticky ends on DNA bricks. (b) Steps for brick-assisted liposome sorting — liposome coating by DNA bricks, separation of DNA-coated liposomes by isopycnic centrifugation, and removal of DNA bricks from the sorted liposomes. A monochromic fluorescence image of 12 fractions recovered after centrifugation (Step II) shows the spread of liposomes in the density gradient. (c) A plot showing buoyant densities of naked and DNA-brick coated liposomes of various sizes. The theoretical values were calculated assuming the buoyant density, footprint, and molecular weight of a six-helix bundle DNA brick to be 1.7 g/cm3, 189 nm2 and 189 kD, respectively, and only meant to illustrate the general trends of liposome density versus size in the presence and absence of DNA coating. (d) Negative-stain TEM images (top) and size distributions (bottom, shown as D=mean±SD, n=390, 251, 1350 from left to right) of liposomes produced by extrusion through polycarbonate filters with 200 nm and 50 nm pores as well as by sonication. (e) A 1:1:1 mixture of extruded (through 200 nm and 50 nm pores) and sonicated liposomes are sorted into distinct sizes with the help of the six-helix-bundle DNA bricks. Representative negative-stain TEM images are shown above the corresponding histograms (shown as D=mean±SD, n=164, 353, 515, 1690, 1240, 1375 from left to right) fitted by Gaussian functions. Liposomes are made of ~59.2% 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 30% 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 10% 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS), and 0.8% 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (rhodamine-DOPE). Scale bars: 100 nm.
Results
DNA-brick assisted liposome sorting by centrifugation.
We chose DNA as the coating material for its high buoyant density (~1.7 g/mL in CsCl medium)18, excellent solubility, programmable self-assembly behaviors19, and easiness to conjugate with hydrophobic molecules20. Previously, designer DNA nanostructures bearing hydrophobic moieties have shown promise in functionalizing and deforming liposomes21–23. In this work, we built two DNA structures (Fig. 1a, Supplementary Fig. 1 and Supplementary Table 1), a three-point star24 (3PS, ~86 kD) and a six-helix-bundle rod25 (6HB, ~189 kD), with a single cholesterol at the end of each DNA structure as the membrane anchor. Placing only one hydrophobic molecule per structure minimizes the brick’s footprint on the liposome surface and limits aggregation and membrane deformation. To facilitate analysis, we labeled ~10% of DNA bricks with Cy5 fluorophore. After assembling the cholesterol-modified DNA bricks by thermal annealing and purifying them by rate-zonal centrifugation (Supplementary Fig. 2), we incubated them with liposomes (59.2% DOPC, 30% DOPE, 10% DOPS, and 0.8% rhodamine-DOPE, see Supplementary Table 2) at the brick:lipid molar ratio of 1:375. To test how the method performed starting from a very polydisperse sample, we mixed sonicated liposomes with two sets of extruded ones (through filters with 200-nm and 50-nm pores) at equal lipid molar ratio. As previously reported6–8, the liposomes produced by extrusion, despite having expected mean sizes, showed broad size distributions (coefficient of variation >0.34, Fig. 1d and Supplementary Fig. 3). Centrifuging these DNA-coated liposomes in a gradient of isosmotic density medium (0%−22.5% iodixanol, ~5 mL per tube) at a maximum of ~300k-rcf for 4.5 hours spread the liposomes into a smeared band spanning the central two-thirds of the gradient. Analyzing the gradient fractions (~200 μL each, named F1–F24 from top to bottom) by SDS-Agarose gel electrophoresis confirmed the coexistence of DNA bricks and liposomes in the middle portion of the gradient, and revealed free DNA bricks at the very bottom, suggesting the bricks may have saturated the surface of liposomes (Supplementary Fig. 4). Negative-stain transmission electron microscopy (TEM) study showed that F6–F18 each contained uniformly-sized liposomes with coefficient of variation less than 0.15 (Fig. 1e and Supplementary Fig. 5), on par with the size homogeneity achieved through DNA-template guided lipid self-assembly15–17. This finding was corroborated by cryo-electron microscopy (cryo-EM), which further showed 77% of liposomes as unilamellar (Supplementary Fig. 6). The multi-lamellar liposomes were most likely generated when extruding liposomes through filters with 200-nm pores6 before sorting. Importantly, the recovered fractions contained liposomes with quasi-continuous mean diameters in the range of 30–130 nm (larger liposomes found in lighter fractions), allowing us to select or bin any fractions for particular liposome sizes needed in downstream applications. By and large, coating liposomes with the two types of DNA bricks yielded comparable separation resolutions, while uncoated liposomes remained inseparable after centrifugation (Fig. 1e and Supplementary Fig. 7–8). The heavier rod-shaped brick performed better when used to sort the >100-nm liposomes and the three-point-star brick led to a finer separation of liposomes smaller than 40 nm.
To demonstrate that efficient liposome separation necessitates sufficient DNA-brick coating, we performed two additional control experiments. First, replacing DNA bricks with cholesterol-labeled 42-mer DNA oligonucleotides resulted in the co-migration of liposomes of different sizes in the density gradient (Supplementary Fig. 9), even at a very high DNA:lipid molar ratio (1:62.5). Second, coating liposomes with brick:lipid molar ratios less than 1:500 led to less efficient liposome separation (Supplementary Fig. 10). Together, these results highlight the importance of high membrane coverage by DNA bricks. In a typical sorting experiment (brick:lipid = 1:375), we estimated that on average, an SUV is coated by ~25–300 DNA bricks, with smaller liposomes coated more densely (Supplementary Fig. 7). Similar curvature-dependent surface labeling has been reported for DNA-functionalized gold nanoparticles26, and here we attribute this phenomenon to more lipid packing defects and less spatial hindrance between DNA bricks on highly curved membranes. Interestingly, we observed that small amounts of DNA bricks can detach from sorted liposomes (Supplementary Fig. 11), suggesting a dynamic equilibrium between membrane-bound and free bricks. Nevertheless, under our optimized sorting conditions, the liposome separation resolution and recovery yield (typically >90%) were consistent from batch to batch, at different separation scales (11 μg–1.3 mg), and across a spectrum of lipid compositions, as long as the liposome surface was not overcrowded with polyethylene glycol (Supplementary Fig. 8, 12 and 13, Supplementary Table 2–3). Additionally, the dense layer of DNA bricks (clearly visible by electron microscopy in the case of six-helix bundle rods) prolonged the shelf life of sorted liposomes (up to 20 weeks at room temperature, Supplementary Fig. 14), despite reduced concentrations of smaller liposomes, likely due to surface adsorption, after long-term storage. Finally, the DNA bricks are readily removable by DNase I digestion (Supplementary Fig. 15), leaving only short oligonucleotides on liposome surfaces. Once uncoated, liposomes started to aggregate and fuse in ~2 days (Supplementary Fig. 16).
Sorting liposomes containing nucleic acid cargos.
The well-maintained monodispersity after long-term storage and the clear, intact boundaries observed by cryo-EM were promising signs of membrane integrity of sorted liposomes. To confirm this, we used 6-helix-bundle bricks to assist the sorting of extruded liposomes (a 1:1 mixture of liposomes passed through filters with 200-nm and 50-nm pores) loaded with fluorescein-labeled class I deoxyribozymes (I-R1a), which self-cleave in minutes upon exposure to ~1 mM Zn2+ at near-neutral pH (Fig. 2a)27. Similar to the plain liposomes, most deoxyribozyme-loaded liposomes with DNA-brick coatings were sorted into six homogeneous populations with mean diameters from 64 to 129 nm (Fig. 2b and Supplementary Fig. 17, few smaller liposomes recovered due to their low abundance in the extruded liposomes). The narrow size distribution of each sorted fraction contrasts with the heterogeneous populations generated by filter-driven homogenization (Fig. 1d, Supplementary Fig. 3 and 8), again highlighting the effectiveness and necessity of DNA-assisted sorting. The molar ratio between deoxyribozyme and lipid (determined by the fluorescence of fluorescein and rhodamine, respectively) was proportional to liposome diameter, as expected from V/S of a sphere (Fig. 2c). Likewise, the number of encapsulated I-R1a per liposome increase linearly with the liposome volume, from ~1 per 64-nm liposome to ~8 per 129-nm liposome, indicating the unbiased cargo load in all sizes of liposomes (Supplementary Fig. 18). Moreover, the liposomes, sorted or not, were impermeable to Zn2+ (2 mM) and deoxyribozyme (1 μM), showing no detectable I-R1a self-cleavage when incubated with Zn2+-containing solutions for over 12 hours, until we lysed liposomes with detergent (1% OG, octyl β-D-glucopyranoside, Fig. 2d).
Figure 2.
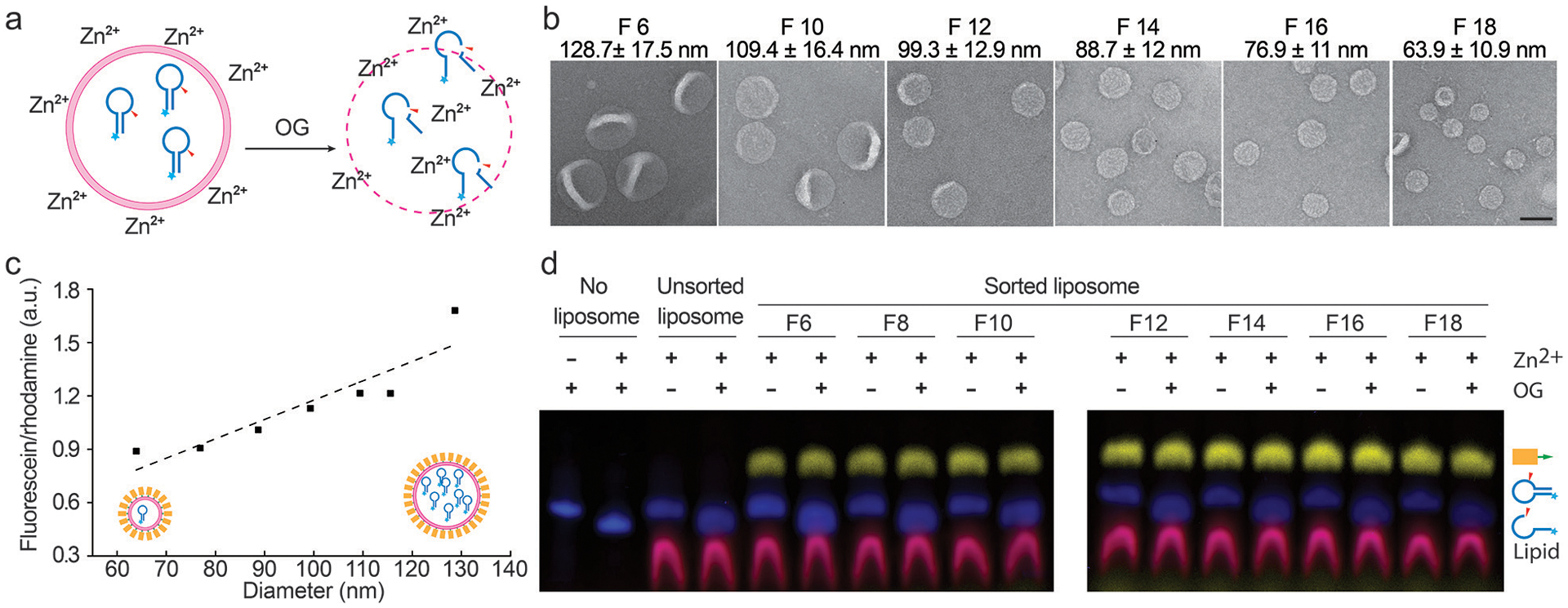
Sorting liposomes containing self-cleaving deoxyribozymes. (a) A schematic drawing of the leakage assay used to assess membrane permeability. Fluorescein-labeled deoxyribozymes undergo site-specific hydrolysis when exposed to Zn2+ outside of the liposomes. (b) Representative TEM images of sorted liposomes containing deoxyribozymes. Fraction numbers (e.g., F6) and liposome diameters (mean±SD, n=212, 436, 391, 376, 555, 621 from left to right) are noted above the corresponding images. Scale bar: 100 nm. (c) A plot showing the deoxyribozyme-to-lipid ratios in sorted liposomes fitted via linear regression (dashed line). (d) Permeability of liposomes characterized by SDS-polyacrylamide gel electrophoresis following the deoxyribozyme-based leakage assay. Prior to the leakage assay, free deoxyribozymes were removed from unsorted liposomes by isopycnic centrifugation without DNA-brick coating. Pseudo-colors: Cy5 (on DNA bricks) = yellow; fluorescein (on deoxyribozymes) = blue; rhodamine (on liposomes) = red. Liposomes are made of 59.2% DOPC, 30% DOPE, 10% DOPS, and 0.8% rhodamine-DOPE. Liposomes prepared in the same experiment were loaded in two gels that were run and processed in parallel. This experiment was repeated 3 times with similar results.
Studying curvature-sensitive GL1-lipid conjugation.
In cells, membranes are shaped into various curvatures that localize biochemical reactions and modulate membrane remodeling. Liposomes with a fine gradient of sizes provide an ideal platform to study such curvature-dependent activities in vitro in a systematic and precise manner. Here we applied the liposome size sorting technique to revamp two classical assays, highlighting the benefit of using uniform-size liposomes for the experimental modeling of lipid biochemistry and membrane dynamics.
We first studied the curvature-sensing capability of a conjugating enzyme that works on the membrane surface of the autophagosome. As the autophagosome grows, GABARAP-L1 (GL1) and its homologs become covalently attached to phosphatidylethanolamine (PE) lipids on the membrane surface through the serial actions of the ATG7 and ATG3 enzymes28. ATG3 catalyzes the final step in this cascade and its activity depends upon an amphipathic helix that senses lipid packing defects in highly curved membranes, suggesting that this protein may specifically target the rim of the cup-shaped autophagosome as a unique intracellular morphology. Previous in vitro studies revealed a curvature dependence of ATG3 activity (higher activities with 30 nm diameter liposomes than 800 nm ones)29, but with extruded liposome preparations and/or sonication, it was not possible to collect curvature sensing information across the biologically relevant range of 25–60 nm, where vesicles, tubules and the autophagic rim are found. Using sorted liposomes (59.2% DOPC, 30% DOPE, 10% DOPS, and 0.8% rhodamine-DOPE) of eight selected sizes (mean diameter: 30, 40, 55, 77, 90, 98, 105, and 122 nm) for ATG3-catalyzed reactions, we confirmed that the lipidation of GL1 in general favored smaller liposomes possessing higher curvature. Specifically, our data revealed a circa 5× enrichment of GL1-PE conjugates in liposomes that are 30–55 nm in diameter in comparison to larger liposomes, with the lipidation peaking on liposomes with ~40-nm diameter (Fig. 3, Supplementary Fig. 19 and 20). This curvature range is reminiscent of the typical autophagosome rim (20–50 nm lamellar spacing)28, the inferred hotspot of ATG3-dependent lipidation in vivo. As ATG3 is a peripheral protein, it must gain access to the membrane surface, and thus a potential concern of using sorted liposomes is that the DNA bricks might directly impede lipidation. To maximize membrane accessibility, we treated the sorted liposomes with DNase I before the lipidation assay. We note that GL1-lipidation activities measured here are across-the-board lower than those previously reported29, which may be caused by a combination of factors including the halved lipid concentration and the ~9-fold higher Mg2+ concentration, as well as the residual short DNA oligonucleotides after nuclease treatment. Overall, homogeneous liposomes improved the precision of the in vitro lipidation assay, enabling a granular analysis of the curvature-dependent ATG3/ATG7 ligation cascade.
Figure 3.
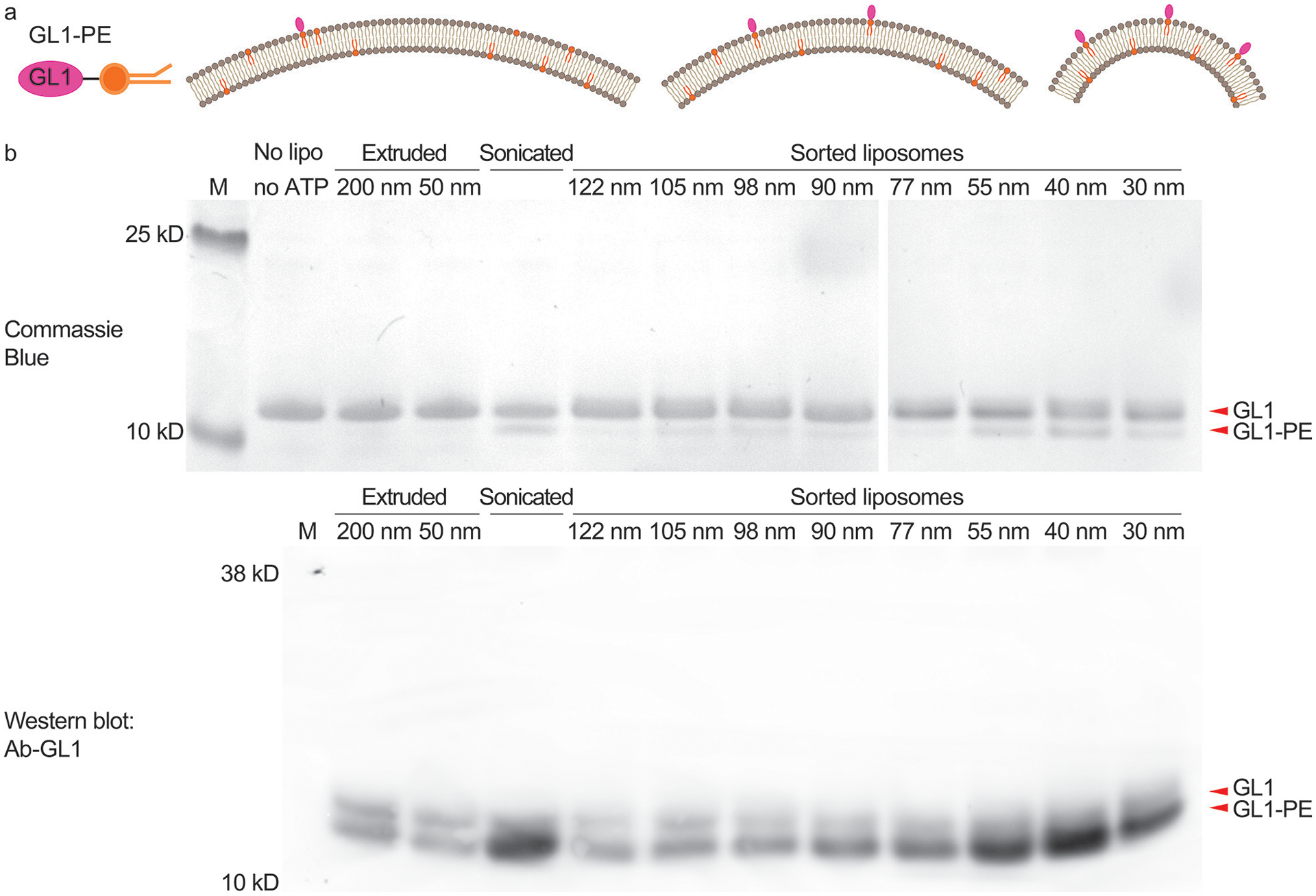
Atg3-catalyzed GL1 lipidation reaction studied using uniform-size liposomes. (a) Schematic illustrations of GL1-DOPE conjugate (left) and the expected lipidation outcomes on liposomes with differential membrane curvatures (right, radii of curvature = 70 nm, 43 nm, and 18 nm). (b) GL1-lipidation efficiencies on extruded, sonicated and sorted liposomes (59.2% DOPC, 30% DOPE, 10% DOPS, and 0.8% rhodamine-DOPE) characterized by gel electrophoresis (top row, stained by Coomassie Blue) and immunoblot against GL1 with an antibody that preferentially recognizes the GL1-PE conformation (bottom row). The numbers (in nm) above lanes represent the nominal pore size of the filters (extruded liposomes) or measured mean diameters (sorted liposomes). Reaction mixtures from the same experiment were loaded in two gels that were run and processed in parallel. This experiment was repeated 5 times with similar results (see Supplementary Fig. 28).
Studying the effect of curvature on membrane fusogenicity.
We next turned our attention to how DNA-brick mediated sorting might work with transmembrane proteins. Soluble NSF attachment protein receptors (SNAREs) are a family of proteins that fuel membrane fusion in many intracellular trafficking routes, including the vesicular release of neurotransmitters and hormones30–32. Two types of SNAREs, v-SNAREs on the vesicle and t-SNAREs on the target membrane, assemble into four-helix bundles to force the membranes into proximity and eventually drive fusion. Previous experimental33,34 and theoretical35 work suggests that membrane curvature may be a critical factor in determining the kinetics of fusion and the number of SNARE complexes required. However, past experiments measured the fusion rates of proteoliposomes with only one or two sizes, due to constraints in preparation of protein-reconstituted liposomes33,34,36. In addition, the preparation methods often produce liposomes with broad size distributions37. These limitations prevented systematic studies of the curvature dependence of fusion rates. Thus, it is highly desirable to develop methods that can produce proteoliposomes with sharp size distributions.
In previous work, we addressed this issue by building DNA-ring templated liposomes displaying a predetermined number of SNARE proteins38. Despite the uniform and controllable proteoliposome size, an exhaustive examination of the impact of membrane curvature on fusion rate was impractical, because the obligated redesign of DNA templates for each liposome size and the small preparation scale (typically less than a few micrograms) limited the throughput of our fusion assay. To address this challenge, here we applied DNA-brick assisted size-sorting to produce proteoliposomes with well-defined sizes, but only at 1/10 of the cost of DNA-templated liposomes (see Supplementary Note 1). We reconstituted the neuronal/exocytotic v-SNARE VAMP2 into liposomes (lipid:VAMP2 ≈ 200:1) containing FRET-dye-labeled lipids (NBD- and rhodamine-DOPE) and performed DNA-brick assisted sorting on 440 μg of such proteoliposomes. The pre-existence of proteins in vesicle membranes did not compromise the separation effectiveness, as confirmed by negative-stain TEM (Supplementary Fig. 21). After enzymatic digestion of DNA bricks (unnecessary in hindsight as the DNA bricks did not affect fusion, see Supplementary Fig. 22), we mixed VAMP2-embedded liposomes of eight different diameters (37–104 nm) with unlabeled (and unsorted) liposomes carrying cognate t-SNAREs in separate test tubes; the mixtures (lipid concentration = 3 mM) were kept at 4°C for 2 hours, a temperature that allows vesicle docking but little to no fusion (Supplementary Fig. 23). Finally, we warmed the pre-docked liposomes to 37°C and monitored NBD fluorescence for 2 hours using a fluorescence microplate reader. Merging of liposome membranes increases the distance between NBD dyes and their rhodamine quenchers, providing a read-out of lipid mixing kinetics (Fig. 4a). Consistent with previous findings30,33,36, we showed that the membrane fusion is SNARE-dependent and can be inhibited by the cytosolic domain of VAMP2 (Supplementary Fig. 24). However, unlike the conventional assays, our setup discerned the lipid mixing kinetics as a function of vesicle size (Fig. 4b). Because SNARE density is a known determinant of SNARE-mediated lipid mixing36, we sorted two additional sets of liposomes reconstituted with 300:1 and 400:1 lipid:VAMP2 ratios, and determined the v-SNARE densities on the sorted and unsorted liposomes (Fig. 4c). Interestingly, VAMP2 density differed significantly (up to 4–6 fold) among sorted liposomes of the same origin and peaked for liposomes of ~40 nm diameter, an overlooked phenomenon in conventional assays due to large dispersion in liposome sizes. Sorted liposomes that were initially reconstituted with different VAMP2:lipid ratios showed similar size-dependent lipid-mixing kinetics: liposomes with mean diameters of 40–50 nm appeared to fuse most rapidly, with the most and least fusogenic vesicles showing ~3–6-fold differences in the final NBD fluorescence (Fig. 4d), which correlated well with the v-SNARE density on liposomes (Fig. 4e). This result, though consistent with previous findings36, does not tease apart the effect of SNARE density from that of membrane curvature, because VAMP2 density is liposome size dependent after sorting. To tackle this problem, we calculated the rounds of fusion that an average liposome of a particular size underwent in 2 hours, using a calibration curve that correlates NBD fluorescence with lipid dilution factors after fusion39 (Fig. 4f, also see Supplementary Methods and Supplementary Fig. 25). As expected, the rounds of fusion positively correlated with the number of v-SNAREs per liposome (Fig. 4g–4h), as more v-SNAREs allowed for engagement with more t-SNARE-bearing liposomes for additional rounds of fusion. The most striking result emerged when we normalized rounds of fusion by membrane area and v-SNARE number (μm2 × v-SNARE)−1, revealing the true effect of membrane curvature on lipid-mixing kinetics and vesicle fusogenicity (Fig. 4i–4j, see Supplementary Note 2 for further discussions). That is, higher membrane curvature promotes fusion, a trend that is predictable from membrane bending energy33,40 and the coupling between membrane curvature and entropic forces acting on SNARE complexes bridging membranes35. However, testing these predictions rigorously was only possible with precise control of liposome sizes.
Figure 4.
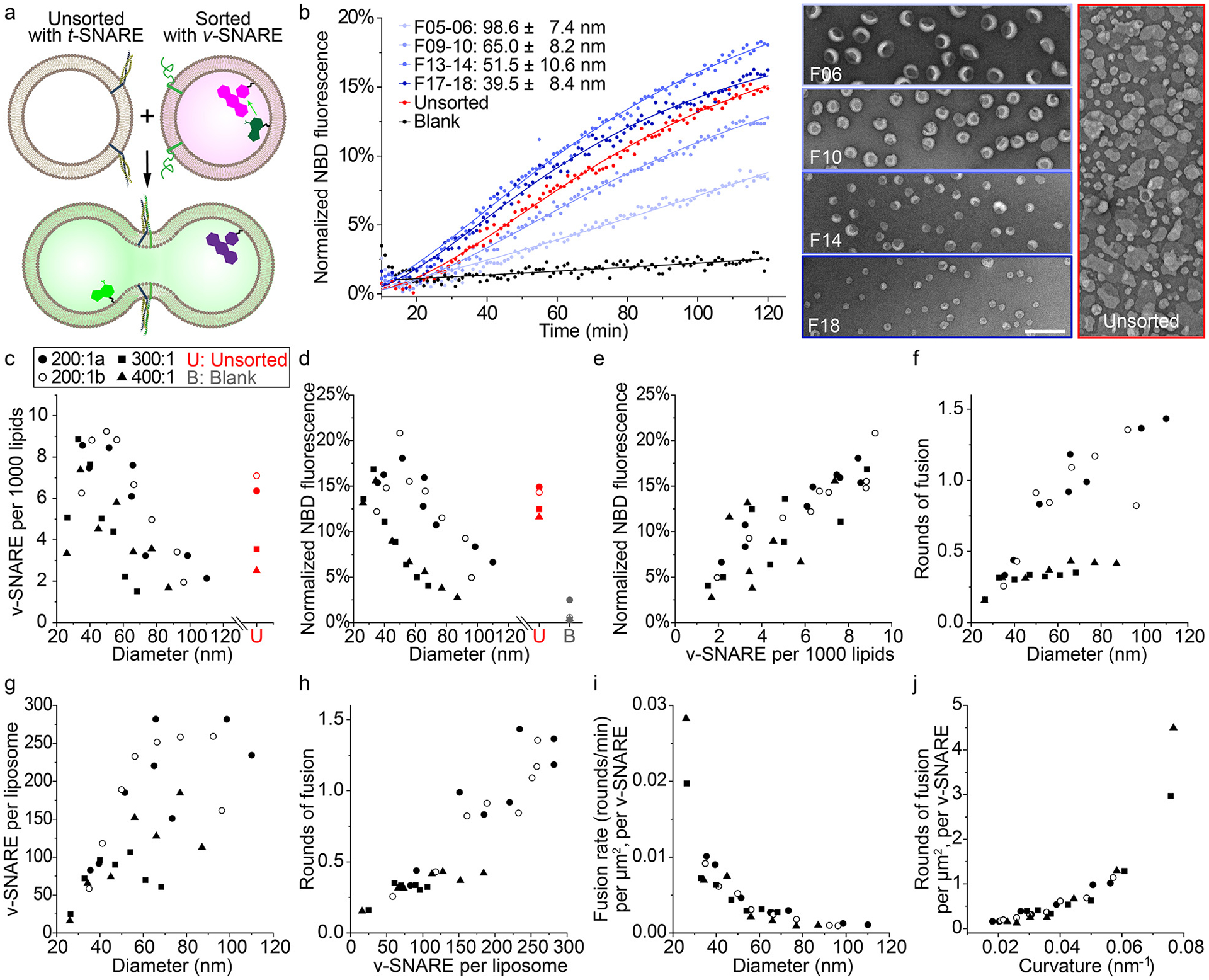
SNARE-mediated membrane fusion studied using uniform-size liposomes. (a) A schematic illustration of the lipid-mixing assay used to monitor membrane fusion. Initially quenched NBD dyes (green) fluoresce following membrane fusion due to a decrease in FRET with rhodamine dyes (magenta). SNARE proteins are shown as blue, yellow (t-SNAREs) and green (VAMP2, v-SNARE) ribbons on the membranes. Models of liposomes, proteins and dyes are not drawn to scale. (b) Left: representative fluorescence traces showing the kinetics of fusion between unsorted liposomes bearing t-SNAREs and unsorted (red) or sorted (different shades of blue, diameters marked as mean±SD, n=603, 488, 345, 477 from top to bottom) liposomes bearing v-SNAREs. Protein-free liposomes are mixed with v-SNARE bearing liposomes as a negative control (black). The solid curves are a guide to the eye. Right: representative TEM images of sorted and unsorted v-SNARE-bearing liposomes. Scale bar: 200 nm. Liposomes with v-SNAREs are reconstituted with 82% 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC), 12% DOPS, 1.5% Rhodamine-DOPE, 1.5% 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2–1,3-benzoxadiazol-4-yl) (NBD-DOPE), and a lipid:protein molar ratio of 200:1. Liposomes with t-SNAREs are reconstituted with 58% POPC, 25% DOPS, 15% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), 2% phosphatidylinositol 4,5-bisphosphate (PIP2) and a lipid:protein molar ratio of 400:1. (c) v-SNARE density on sorted and unsorted liposomes reconstituted with lipid:VAMP2 molar ratios of 200:1 (two trials, labeled with a and b), 300:1, and 400:1. Mean liposome diameters are measured from TEM images for unsorted liposomes and all fractions of sorted liposomes with initial lipid:VAMP2 ratio of 200:1, but only for 3 (out of 7) fractions of those with initial lipid:VAMP2 ratio of 300:1 or 400:1. Sizes of other fractions are interpolated. (d) Lipid mixing after 2 hours of fusion reactions (measured by NBD fluorescence, as shown in (b)) plotted against the average diameters of sorted v-SNARE-bearing liposomes. (e) Correlation between NBD fluorescence and v-SNARE density. (f) Rounds of fusion that sorted v-SNARE-bearing liposomes undergo in 2 hours (see Methods for details). (g) v-SNARE copy numbers per liposome measured from sorted liposomes. (h) Correlation between rounds of fusion and v-SNARE copy number per liposome. (i) The rates for rounds of fusion (between 30 and 60 min) normalized by surface area of v-SNARE-bearing liposomes and the corresponding v-SNARE copy numbers, as a function of mean diameter of sorted liposomes. (j) Rounds of fusion normalized by surface area of v-SNARE-bearing liposomes and the corresponding v-SNARE copy numbers, as a function of the mean curvature (1/radius) of sorted liposomes.
In neurons, synaptic vesicle sizes are highly homogeneous and regulated41,42. Here we only studied the minimal fusion machinery (SNAREs) to prove the concept. However, the platform can in principle be adapted to model more physiological conditions, where additional proteins (e.g., Synaptotagmin-1 or Munc18) affect the fate of vesicles.
Discussion
Self-assembled DNA nanostructures have been interfaced with lipid bilayers in a number of unconventional ways towards the goal of programmable membrane engineering21–23. In the past, this took one of two forms. The first approach is to scaffold liposome formation with DNA templates, which excels at precision but any pre-existing membrane needs to be micellized before reassembly15–17. The second strategy is to reshape the membrane landscape of liposomes with DNA devices that oligomerize or reconfigure on command, which may preserve certain pre-existing membrane features (e.g., lipid composition, internal content) but the end products tend to be less homogeneous43–45. By bridging this gap, the DNA-brick assisted liposome sorting method further advances the membrane engineering capability of DNA nanotechnology. Specifically, the method separates liposomes from virtually any source into a range of narrowly distributed sizes with minimal impact on the original membrane properties. Further, two DNA structures composed of a handful of oligonucleotides fulfilled various sorting tasks. The scalability and robustness of the technique make it readily adaptable by any biochemical laboratory with access to research-grade ultracentrifuges (Supplementary Fig. 26). Currently, the most time-consuming steps in the entire sorting workflow (~3 days) are the manual fraction recovery and the TEM characterization of liposomes. The former can be automated using a gradient station. The latter, though essential for quality control, does not have to be repeated for every preparation. For labs to develop a new sorting protocol, theoretical analyses (Fig. 1c) and the sorting results reported here (Supplementary Table 4) should serve as a good starting point. Once the sorting conditions (e.g., DNA brick mass, brick:lipid ratio, and concentrations of gradient media) are optimized, the resulting liposome size distributions should be highly reproducible, so long as the liposome source remains unchanged. Future method development will benefit from the programmability of DNA nanostructures. For example, coating liposomes with more massive DNA bricks could facilitate the separation of larger liposomes; using chemically or photo-cleavable linkers between cholesterol and DNA brick would facilitate nuclease-free removal of DNA coat from sorted liposomes; changing cholesterol anchors to protein-specific ligands could enable the sorting of natural vesicles by their surface markers. In addition to the utilities in basic research, we envision the method (in its current or adapted forms) finding applications in biotechnology, such as in aiding the development of drug-delivering liposomes as well as isolating disease-specific extracellular vesicles.
Methods
DNA oligonucleotides (oligos) were synthesized by Integrated DNA Technologies. Chemically modified oligos were purified via High-performance liquid chromatography (HPLC) by manufacturer, while unmodified oligos were purified via polyacrylamide gel electrophoresis (PAGE) in house (see Supplementary Table 1 for oligo sequences). DNA brick designs (rendered in Tiamat46) are shown in Supplementary Figure 1, along with the PAGE analyses of the assembly products.
All lipids were purchased from Avanti Polar Lipids. For general sorting experiments, leakage assay, and lipidation assay, liposomes were prepared in Buffer X. For the vesicle fusion study, reconstituted proteoliposomes were enriched in Buffer Y before sorting (Method 7). To avoid osmolality shock, DNA bricks were prepared in the same buffer (X or Y) as the liposomes (see Methods 1 and 2). Lipid and buffer compositions are listed in Supplementary Table 2.
1. DNA brick preparation
1a. Assembly
PAGE or HPLC purified oligos were dissolved in deionized, Milli-Q water (Millipore) with concentrations normalized to 120 μM each. To assemble the 3PS and 6HB DNA bricks, various amounts of cholesterol-modified oligos (1–2.5 μM) and a stoichiometric amount of unmodified oligos (1 μM each) were mixed in Buffer X and underwent thermal annealing from 95 to 4°C (held at 95, 65, 50, 42, 37, 22, and 4°C for 5 min each). The assembly products were electrophoresed in a non-denaturing 6% polyacrylamide gel under 15V/cm for 70 min in 1×TAE, 10 mM MgCl2. The optimal molar ratio between modified and unlabeled oligos, which gave rise to a sharp, distinct band after Sybr Gold staining, was chosen for DNA brick assembly for the rest of this study. Optionally, 10% of an unmodified oligo (C in 3PS and 6hb-S3 in 6HB, Supplementary Table 1) was replaced with a Cy5-labeled oligo for staining-free visualization of DNA bricks on gels.
1b. Purification and characterization
Large scale (400 μL of 5 μM) DNA-brick assemblies were placed on top of a 5%–20% glycerol gradient in a 5-mL ultracentrifugation tube (Beckman Coulter, Cat# 344057). The sample-loaded density medium was spun at 55,000 rpm and room temperature (RT) for 4.5 hr in an SW55-Ti rotor (Beckman Coulter) before fractionated into 200-μL fractions. Five microliters of each fraction were electrophoresed in a 3.5% agarose gel containing 0.05% ethidium bromide under 10 V/cm for 1.5 hr in 0.5×TBE, 10 mM MgCl2 (see an example in Supplementary Figure 2). Fractions containing well-formed bricks (e.g., fractions 8–10 in Supplementary Figure 2) were combined and concentrated to 50–100 μL by centrifugation (10 min at 10,000 rcf) on Amicon filtration units (Millipore) with 10 kD nominal molecular weight limit (NMWL). The concentrated sample was diluted in Buffer X or Y to 500 μL and concentrated again for a total of four times. The DNA brick concentration was determined by OD260 measurement of a NanoDrop spectrometer (Thermo Fisher Scientific). The purified bricks were diluted to 5 μM in Buffer X or Y and stored at −20°C.
2. Liposome preparation
2a. Solvent evaporation and lipid rehydration
To prepare 1 mL of liposomes containing 3 μmol total lipids (final Clipid = 3 mM) of a specific composition (Supplementary Table 2), appropriate volumes of lipid stocks (dissolved in chloroform) were mixed in a round-bottom glass tube. The mixture was blown-dried under N2 for at least 1/2 hour. The resulting lipid film at the tube bottom was further dried overnight in a desiccator under vacuum. Unless noted otherwise, 1 mL of Buffer X (Supplementary Table 2) was added into the tube and agitated for 1/2 hour. To prepare for the leakage assay, 1 mL of 10 μM FAM-modified I-R1a deoxyribozyme (dissolved in Buffer X) was used instead for rehydration. The glass tubes were wrapped with aluminum foil to reduce photobleaching of fluorescent labels.
2b. Sequential extrusion (to produce liposome with nominal diameters of 50–200 nm)
The rehydrated lipid suspension was transferred into a 1.5 mL centrifuge tube and thermo-cycled between a liquid-nitrogen bath and a 37°C-water bath for 7–10 times. The frozen-thawed suspension was then sequentially extruded through polycarbonate filters of nominal pore sizes of 400 nm, 200 nm, and 50 nm (>60 passes each), using a Mini Extruder (Avanti Polar Lipids) at RT (well above the transition temperatures of constituent lipids) following a protocol recommended by the manufacture. The extruded liposomes after passing through 200-nm and 50-nm filters (typically 300 μL each) were stored at 4°C; the remaining 400 μL of liposomes was sonicated as described in Method 2c. Typical results are shown in Figure 1d. Additionally, liposomes rehydrated in Buffer Y were extruded sequentially through 400, 200, 100, 50, and 30 nm filters for comparison (Supplementary Figure 3). Cryogenic electron microscopy (cryo-EM) images of sequentially extruded liposomes can be found in our previous work15.
2c. Sonication (to produce liposome with nominal diameters <50 nm)
Extruded liposomes (~400 μL) were sonicated using a Qsonica Q125 dip-probe sonicator for 1 min (10 cycles of 1-s on, 1-s off) while sitting on an ice-water bath.
3. DNA-brick assisted liposome sorting
3a. Liposome coating
For small scale sorting, 40 μL of purified 3PS or 6HB brick (cholesterol-labeled, 1 μM) and 5 μL of liposome (3 mM lipid) were mixed in a 200 μL tube and incubated at RT for 1–2 hr under continuous agitation. The brick:lipid ratio of 1:375 is empirically determined to be sufficient for subsequent liposome sorting (see 4a and Supplementary Figure 10). In the case of suboptimal sorting, a higher concentration of DNA brick may be used for liposome coating. When sorting larger quantities of liposomes, the amount of DNA brick and liposome was increased proportionally; the DNA brick concentration may be adjusted as appropriate. Supplementary Table 3 provides some guidelines. For example, our largest scale preparation started with >1 mg liposome (1.8 μmol total lipid), which was split into six 5-mL ultracentrifuge tubes after DNA-coating for isopycnic centrifugation.
3b. Liposome sorting by isopycnic centrifugation
Iodixanol density gradient was prepared from stock solutions of 45%, 18%, 15%, 12%, 9%, 6%, 3% and 0% (v/v) iodixanol (Stemcell Technologies) in Buffer X.
DNA-coated liposomes were mixed with an equal volume of 45% iodixanol, forming a 22.5% iodixanol solution at the bottom of an ultracentrifugation tube. For the small scale separation (1× in Supplementary Table 3), 80 μL of such a solution was pipetted to an 800-μL tube (Beckman Coulter Cat# 344090). Seven additional iodixanol layers (18% to 0%, 80 μL each) were carefully placed on top of one another to form a quasi-linear gradient. The tube, loaded with the liposome sample in the iodixanol gradient, was spun in an SW55-Ti rotor at 48,000 rpm and RT for 4.5 hr. For large scale preparations (e.g., 10× and 20× in Supplementary Table 3), linear 0–18% iodixanol gradients (4.2 mL each) were formed in 5-mL tubes (Beckman Coulter, Cat# 344057) using a Gradient Master (BioComp Instruments). Seven-hundred microliters of DNA-coated liposomes in 22.5% iodixanol were carefully layered at the bottom of the gradient using a syringe and a needle. The tubes were spun at 50,000 rpm and RT for 4.5 hours. Proteoliposomes (see Supplementary Figure 21), were sorted in the same way, except using gradients made in Buffer Y.
3c. Post-centrifugation recovery
After ultracentrifugation, the content of a tube was collected from top to bottom with 52 μL (800 μL tube) or 200 μL (5 mL tube) per fraction. We used caution to minimize disturbance to the gradient when pipetting. The recovered fractions were transferred to a 96-well plate, sealed with aluminum film, and stored at RT in the dark. To remove iodixanol and concentrate sorted liposomes, selected fractions were combined and concentrated to 50–100 μL by centrifugation (8 min at 10,000 rcf) on Amicon filtration units with 30 kD NMWL. The concentrated liposomes were diluted in Buffer X or Y to 500 μL and concentrated again for a total of 4–5 times. Optionally, to remove DNA bricks, sorted liposomes were treated with DNase I (Thermo Fisher Scientific) following the manufacturer’s recommendation (see Supplementary Figure 22).
4. Characterization of sorted liposomes
4a. Agarose gel electrophoresis
Recovered fractions of a post-centrifugation gradient (5 μL each) were electrophoresed in a 3.5% agarose gel (casted with 0.05% sodium dodecyl sulfate, SDS) at 10 V/cm for 1.5 hr in a 0.5×TBE buffer containing 10 mM MgCl2 and 0.05% SDS. The gels were imaged on a multi-color laser scanner (Typhoon FLA 9500).
4b. Negative stain TEM study
A drop of sample (~5 μL) was deposited on a glow discharged formvar/carbon-coated copper grid (Electron Microscopy Sciences), incubated for 1–3 minutes and blotted away. The grid was then washed briefly and stained for 1 minute with 2% (w/v) uranyl formate. Images were acquired on a JEOL JEM-1400 Plus microscope (acceleration voltage: 80 kV) with a bottom-mount 4k×3k CCD camera (Advanced Microscopy Technologies), using AMT Image Capture Engine. Liposome sizes were measured from electron micrographs by ImageJ (National Institutes of Health). The image analysis workflow is summarized in Supplementary Figure 5.
In our experience, sorting the same liposome mixtures following an established protocol (Method 1–3) yielded consistent liposome size distributions. In Supplementary Table 4, we summarize the gradient fractions where liposomes of particular sizes reside after sorting as a guide for others who want to reproduce our experiments or develop new protocols.
4c. Cryo-EM imaging
A drop (3.5 μL) of liposome sample was loaded onto a glow-discharged lacey carbon film, copper, 300 mesh grids, and plunge frozen in liquid ethane using an FEI Mark III Vitrobot operating at 100% humidity, 22°C temperature, 5 s blot time and −4 force.
The grids were imaged on an FEI Talos L120C TEM equipped with a Ceta CCD camera. The images were collected at magnifications of 36K/45K/57K/92K (with the pixel size of 4.01/3.21/2.53/1.57 Å) and a dose of 50 e/Å2, using a defocus range of −2 to −4 μm.
5. Leakage assay (deoxyribozyme self-cleavage)
As described in Method 2a, deoxyribozyme I-R1a (10 μM, with 5’-FAM label) in Buffer X was first loaded into liposomes through a rehydration process. After sequential extrusions (Method 2b), the liposomes were coated with 6HB bricks and sorted as described in Method 3. Fractions from density ultracentrifugation, as well as a control sample containing unsorted liposomes (free I-R1a pre-removal through a separate isopycnic centrifugation without DNA-brick coating), were normalized to 0.75 mM lipid concentration. A deoxyribozyme reaction buffer (DRB+) was prepared to contain 25 mM HEPES, 400 mM KCl, 6 mM MgCl2 and 4 mM ZnCl2, which provides the same osmotic pressure as Buffer X but with 2 mM Zn2+ for I-R1a cleavage once mixed with the sample in a 1:1 ratio.
Three microliters of Buffer X containing 0% (for permeability test) or 4% n-octyl-β-D-glucopyranoside (OG, for liposome lysis) were added to 9 μL of each sample (fraction 6, 8, 10, 12, 14, 16, 18, and unsorted), then mixed with 12 μL DRB+ and incubated at 37°C for 12 hr. After incubation, samples were mixed with 16 μL denaturing loading buffer (90% formamide, 10 mM NaOH, 1 mM EDTA, 0.1% Xylene Cyanole FF) and boiled for 3 min. Samples (10 μL each) were electrophoresed in a 12% urea polyacrylamide gel containing 0.1% SDS in 1×TBE buffer with 0.1% SDS at 10 V/cm for 1.5 hours (Figure 2). The gels were imaged on a multi-color laser scanner (Typhoon FLA 9500).
6. ATG7/ATG3 catalyzed GL1 lipidation assay
6a. Lipidation reaction
Protein expression and membrane-curvature dependent lipidation reactions were performed as described previously with minor changes noted below47–49. Sorted liposomes were concentrated to a lipid concentration ≥1.2 mM in Buffer X, pretreated with DNaseI (Method 3c), and mixed with ATG7, ATG3, and human GABARAP L1 (GL1) proteins purified in SN buffer (20 mM Tris at pH 7.6, 100 mM NaCl and 5 mM MgCl2). The ~20-μL reaction mixture (~1:4 protein:liposome, v/v ratio), which contained purified ATG7 (1.5 μM), ATG3 (2.5 μM), and GL1 (8 μM) proteins, unsorted or sorted liposomes (1 mM total lipid), as well as 1 mM dithiothreitol (DTT) and 1 mM ATP, was incubated at 30 °C for 90 min. The reaction was stopped by 4× SDS-PAGE loading buffer and boiled at 90°C for 5 min. Electrophoresis was performed in precast 10% Bis-Tris gels (Novex, Thermo Fisher Scientific) running in 1×MES SDS Running Buffer (NuPAGE, Thermo Fisher Scientific) at 180 V (18V/cm) for 60 min. The proteins were visualized with Coomassie blue stain following the manufacturer’s instruction (Imperial Protein Stain, Thermo Fisher Scientific).
6b. Immunoblotting
After electrophoresis, samples were transferred onto a PVDF membrane (Amersham, GE Healthcare), blocked with 5% BSA and probed with anti-GL1 (1:1000, Cell Signaling Technology clone D5R9Y) antibody in 2.5% BSA (Sigma). HRP-conjugated anti-mouse (NA931) and anti-rabbit (NA934) secondary antibodies were purchased from Amersham, GE Healthcare. Both secondary antibodies were diluted 1:10,000 in 5% nonfat dry milk in 1× phosphate-buffered saline with Tween detergent (PBST, from Sigma).
7. SNARE-mediated liposome fusion assay
7a. Plasmid constructs and protein purification
The vectors encoding full-length t-SNARE complex including rat Stx1A and mouse 6×His-SNAP25 (plasmid pTW34) and 6×His-SUMO-VAMP2 (plasmid pET-SUMO-VAMP2), were transfected into the BL21-Gold (DE3) E. coli strain (Agilent Technologies; Cat# 230132) and purified as previously described50. Briefly, bacteria carrying SNARE plasmids were cultured in 2 L LB media at 37 °C until OD600 reached 0.7, induced by 1 mM isopropyl β-D-thiogalactoside, and cultured for additional 3 hr at 37 °C. The pelleted cells were resuspended in breaking buffer (25 mM HEPES pH 7.4, 400 mM KCl, 10% glycerol, 4% Triton X-100, 1 mM TCEP, protease cocktail inhibitors) and lysed by cell disruptor (Avestin) with 3–5 passages at ~15,000 psi. The cell lysate was clarified by centrifugation at 40,000 rpm for 30 min; the supernatant was collected and incubated with nickel-NTA agarose (Qiagen) for 4 hr to overnight at 4°C. t-SNARE bound beads were rinsed with 25 mM HEPES pH 7.4, 400 mM KCl, 10% glycerol, 1% (w/v) OG, 1 mM TCEP. t-SNARE proteins were eluted off the beads by elution buffer (25 mM HEPES pH 7.4, 400 mM KCl, 10% glycerol, 1% OG, 1 mM TCEP, 400 mM imidazole). 6×His-SUMO tags on VAMP2 were cleaved by SUMO protease.
7b. Proteoliposome preparation
SNARE proteins were reconstituted into liposomes at physiologically relevant densities, with protein:lipid ratio at 1:200 or 1:400 for v-SNARE liposome and 1:400 for t-SNARE liposomes. A vacuum-dried lipid film was dissolved in the reconstitution buffer (25 mM HEPES pH 7.4, 140 mM KCl, 0.2 mM TCEP, 10% glycerol, 1% OG) and mixed with SNARE proteins. OG-free reconstitution buffer was added to reach a final OG concentration of 0.33%. Detergent was then removed in a Slide-A-Lyzer dialysis cassette (Thermo Fisher Scientific) against 4 L of OG-free reconstitution buffer at 4°C overnight. Proteoliposomes were separated in a Nycodenz (Progen Biotechnik) density gradient via centrifugation30. For t-SNARE liposomes, centrifugation was done in an SW60-Ti rotor (Beckman Coulter) at 55,000 rpm for 3hr 40min at 4 °C; for v-SNARE liposomes, centrifugation was done in an SW55-Ti rotor (Beckman Coulter) at 48,000 rpm for 4 hr at 4 °C. The enriched proteoliposomes were collected from 0/30% Nycodenz interface. These proteoliposomes were sorted as described in Method 3 and analyzed by negative-stain TEM (Method 4b) and SDS-PAGE (Supplementary Figure 21). The v-SNARE concentrations of proteoliposomes were determined using VAMP2 concentration standards by densitometry (ImageJ). Lipid concentrations of v-SNARE liposomes were determined by rhodamine absorbance at 574 nm.
7c. Lipid mixing assay
A typical fusion reaction was initiated by mixing 5 μL of v-SNARE liposomes (3 mM lipid concentration) labeled with a pair of FRET dyes (donor: NBD-DOPE, acceptor: Rhodamine-DOPE) and 45 μL (3 mM lipid concentration) of unlabeled t-SNARE liposomes30 (Supplementary Table 2), with a total lipid amount of 150 nmol. For sorted v-SNARE liposomes, every two fractions (F3+F4, F5+F6, …, F17+F18) were combined and concentrated by Amicon filtration units with 30 kD NMWL. Lipid concentration in each sample were measured based on the Rhodamine absorption at 574 nm. Sorted v-SNARE liposomes (5 μL, 3 mM lipid concentration) were then mixed with 45 μL (3 mM lipid concentration) unlabeled t-SNARE liposomes for fusion as previously described. These mixtures were pre-incubated at 4 °C for 2 hr for trans-SNARE complex assembly, before being transferred to a Falcon 96-well plate with a black skirt and clear glass bottom and heated to 37 °C. NBD fluorescence was monitored at excitation/emission of ~460/535 nm every 1 min for 2 hr by Synergy H1 Hybrid Multi-Mode Reader (BioTek Instruments). At the end of the 2-hr reaction, 10 μL of 20% Triton X-100 was added and fluorescence was recorded for another 30 min to obtain the maximum fluorescence. As a negative control in addition to mixing protein-free liposomes, we performed a competition assay using the cytosolic domain of VAMP (CDV; amino acids 1–94). The CDV was recombinantly expressed in E.coli and purified via a C-terminal His6-tag. For each competition assay, CDV (10 μM) was incubated with t-liposomes to pre-inhibit t-SNAREs and block membrane fusion.
7d. Rounds of fusion assay
Rounds of fusion were calculated through a standard curve which was determined by the fold of NBD fluorescent dilution39. Colored (PC : PS : NBD-DOPE : Rhodamine-DOPE = 82: 15: 1.5: 1.5 at molar ratio) and clear (POPC : DOPS : POPE : PIP2 = 58 : 25 : 15 : 2 at molar ratio) lipid stocks were made to resemble the lipid compositions of v-SNARE and t-SNARE liposomes, respectively. Vesicles were made from mixtures of colored and clear lipid stock in 1: 0, 1: 0.5, 1: 1, 1: 2, 1: 5, and 1: 8 ratios. For each sample, 1.5 μmol of total phospholipid were dried by nitrogen for 15 min and then vacuum-dried for at least 2 hrs. These lipidic films were rehydrated by 0.5 mL of reconstitution buffer, agitated for 30 min, and diluted to 1.5 mL using OG-free reconstitution buffer. The OG was removed by dialysis and reconstituted liposomes were separated through gradient density ultra-centrifugation as described in 7b. The NBD fluorescence of these vesicles were measured before and after detergent treatment. The liposomes’ NBD fluorescence measured before detergent treatment were background corrected (by subtracting the 1: 0 group NBD fluorescence before detergent) and normalized to the maximal fluorescence after detergent treatment. This data (Supplementary Table 5) was used to generate a standard curve and fitted with an empirical function (Supplementary Figure 25) that describes the relationship between lipid dilution factor and normalized NBD signal.
For a SNARE-mediated liposome fusion event, the relationship between dilution factor F(x) and rounds of fusion (x) that a v-SNARE liposome undergo can be written as:
where Dv is the measured mean diameter of sorted v-SNARE liposomes, and Dt (52 nm, cf. Ref.36) is the mean diameter of t-SNARE liposomes.
Therefore,
Using this function on every time point of kinetic measurement, the NBD fluorescence traces (Figure 4b) can be replotted as rounds of fusion versus time. The fusion rates (slope values) were calculated from such replotted lipid mixing curves (between 30 and 60 min) for 4 trials of fusion assays, normalized by the surface area of v-SNARE liposomes and the corresponding v-SNARE copy number, and plotted against mean diameters of v-SNARE liposomes (Figure 4i). Total rounds of fusion in 2 hours were calculated for 4 trials of fusion assays and plotted against mean diameters of v-SNARE liposomes (Figure 4f). Data were plotted using Origin-2018 (OriginLab).
Supplementary Material
Acknowledgements
This work is supported by a National Institutes of Health (NIH) Director’s New Innovator Award (GM114830), an NIH grant (GM132114), and a Yale University faculty startup fund to C.L., NIH grants to E.R.C. (MH061876 and NS097362), to T.M. (GM100930 and GM109466) and to E.K. (NS113236), and a National Key Research and Development Program of China grant (2020YFA0908901) and National Natural Science Foundation of China grants (21673050, 91859104, and 81861138004) to H.G.. E.R.C. is an Investigator of the Howard Hughes Medical Institute. Q.X. is supported by a Graduate Scholarship from Agency for Science, Technology and Research (Singapore).
Footnotes
Competing Interests
Yale University has filed a provisional patent (U.S. Application No. 62/968,683; inventors: C.L. and Y.Y.) on the DNA-assisted liposome sorting method.
Data Availability
The data (TEM images, gel and blot images, fluorescence traces, and statistical data) supporting the findings of this study are available within the paper and its supplementary information files. Source data for Figures 1–4 are provided with the paper.
References
- 1.McMahon HT & Gallop JL Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature 438, 590–596 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Jarsch IK, Daste F & Gallop JL Membrane curvature in cell biology: An integration of molecular mechanisms. J Cell Biol 214, 375–387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woodle MC & Papahadjopoulos D Liposome preparation and size characterization. Methods Enzymol 171, 193–217 (1989). [DOI] [PubMed] [Google Scholar]
- 4.Schubert R Liposome preparation by detergent removal. Methods Enzymol 367, 46–70 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Patil YP & Jadhav S Novel methods for liposome preparation. Chem Phys Lipids 177, 8–18 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Berger N, Sachse A, Bender J, Schubert R & Brandl M Filter extrusion of liposomes using different devices: comparison of liposome size, encapsulation efficiency, and process characteristics. Int J Pharm 223, 55–68 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Szoka F et al. Preparation of unilamellar liposomes of intermediate size (0.1–0.2 mumol) by a combination of reverse phase evaporation and extrusion through polycarbonate membranes. Biochim Biophys Acta 601, 559–571 (1980). [DOI] [PubMed] [Google Scholar]
- 8.Hinna A et al. Filter-extruded liposomes revisited: a study into size distributions and morphologies in relation to lipid-composition and process parameters. J Liposome Res 26, 11–20 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Silva R, Ferreira H, Little C & Cavaco-Paulo A Effect of ultrasound parameters for unilamellar liposome preparation. Ultrason Sonochem 17, 628–632 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Goormaghtigh E & Scarborough GA Density-based separation of liposomes by glycerol gradient centrifugation. Anal Biochem 159, 122–131 (1986). [DOI] [PubMed] [Google Scholar]
- 11.Lundahl P, Zeng CM, Lagerquist Hagglund C, Gottschalk I & Greijer E Chromatographic approaches to liposomes, proteoliposomes and biomembrane vesicles. J Chromatogr B Biomed Sci Appl 722, 103–120 (1999). [DOI] [PubMed] [Google Scholar]
- 12.van Swaay D & deMello A Microfluidic methods for forming liposomes. Lab Chip 13, 752–767 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Jahn A, Vreeland WN, Gaitan M & Locascio LE Controlled vesicle self-assembly in microfluidic channels with hydrodynamic focusing. J Am Chem Soc 126, 2674–2675 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Jahn A, Vreeland WN, DeVoe DL, Locascio LE & Gaitan M Microfluidic directed formation of liposomes of controlled size. Langmuir 23, 6289–6293 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Yang Y et al. Self-assembly of size-controlled liposomes on DNA nanotemplates. Nat Chem 8, 476–483 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Z, Yang Y, Pincet F, Llaguno MC & Lin CX Placing and shaping liposomes with reconfigurable DNA nanocages. Nat Chem 9, 653–659 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perrault SD & Shih WM Virus-Inspired Membrane Encapsulation of DNA Nanostructures To Achieve In Vivo Stability. Acs Nano 8, 5132–5140 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daniel E Equilibrium sedimentation of a polyelectrolyte in a density gradient of a low-molecular weight electrolyte. I. DNA in CsCl. Biopolymers 7, 359–377 (1969). [DOI] [PubMed] [Google Scholar]
- 19.Seeman NC & Sleiman HF DNA nanotechnology. Nat Rev Mater 3 (2018). [Google Scholar]
- 20.Kwak M & Herrmann A Nucleic acid amphiphiles: synthesis and self-assembled nanostructures. Chem Soc Rev 40, 5745–5755 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Langecker M, Arnaut V, List J & Simmel FC DNA nanostructures interacting with lipid bilayer membranes. Acc Chem Res 47, 1807–1815 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Howorka S NANOTECHNOLOGY. Changing of the guard. Science 352, 890–891 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Shen Q, Grome MW, Yang Y & Lin C Engineering Lipid Membranes with Programmable DNA Nanostructures. Advanced Biosystems 4, 1900215 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He Y, Chen Y, Liu H, Ribbe AE & Mao C Self-assembly of hexagonal DNA two-dimensional (2D) arrays. J Am Chem Soc 127, 12202–12203 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Mathieu F et al. Six-helix bundles designed from DNA. Nano Lett 5, 661–665 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hill HD, Millstone JE, Banholzer MJ & Mirkin CA The role radius of curvature plays in thiolated oligonucleotide loading on gold nanoparticles. ACS Nano 3, 418–424 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du XY, Zhong X, Li W, Li H & Gu HZ Retraining and Optimizing DNA-Hydrolyzing Deoxyribozymes for Robust Single- and Multiple-Turnover Activities. Acs Catal 8, 5996–6005 (2018). [Google Scholar]
- 28.Nguyen N, Shteyn V & Melia TJ Sensing Membrane Curvature in Macroautophagy. J Mol Biol 429, 457–472 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nath S et al. Lipidation of the LC3/GABARAP family of autophagy proteins relies on a membrane-curvature-sensing domain in Atg3. Nat Cell Biol 16, 415–424 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weber T et al. SNAREpins: minimal machinery for membrane fusion. Cell 92, 759–772 (1998). [DOI] [PubMed] [Google Scholar]
- 31.Jahn R & Scheller RH SNAREs--engines for membrane fusion. Nat Rev Mol Cell Biol 7, 631–643 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Sudhof TC & Rothman JE Membrane fusion: grappling with SNARE and SM proteins. Science 323, 474–477 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hernandez JM et al. Membrane fusion intermediates via directional and full assembly of the SNARE complex. Science 336, 1581–1584 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hernandez JM, Kreutzberger AJ, Kiessling V, Tamm LK & Jahn R Variable cooperativity in SNARE-mediated membrane fusion. Proc Natl Acad Sci U S A 111, 12037–12042 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mostafavi H et al. Entropic forces drive self-organization and membrane fusion by SNARE proteins. Proc Natl Acad Sci U S A 114, 5455–5460 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ji H et al. Protein determinants of SNARE-mediated lipid mixing. Biophys J 99, 553–560 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stratton BS et al. Cholesterol Increases the Openness of SNARE-Mediated Flickering Fusion Pores. Biophys J 110, 1538–1550 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu WM et al. A Programmable DNA Origami Platform to Organize SNAREs for Membrane Fusion. J Am Chem Soc 138, 4439–4447 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parlati F et al. Rapid and efficient fusion of phospholipid vesicles by the alpha-helical core of a SNARE complex in the absence of an N-terminal regulatory domain. Proc Natl Acad Sci U S A 96, 12565–12570 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malinin VS & Lentz BR Energetics of vesicle fusion intermediates: comparison of calculations with observed effects of osmotic and curvature stresses. Biophys J 86, 2951–2964 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang B et al. Synaptic vesicle size and number are regulated by a clathrin adaptor protein required for endocytosis. Neuron 21, 1465–1475 (1998). [DOI] [PubMed] [Google Scholar]
- 42.Qu L, Akbergenova Y, Hu Y & Schikorski T Synapse-to-synapse variation in mean synaptic vesicle size and its relationship with synaptic morphology and function. J Comp Neurol 514, 343–352 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Czogalla A et al. Amphipathic DNA Origami Nanoparticles to Scaffold and Deform Lipid Membrane Vesicles. Angew Chem Int Edit 54, 6501–6505 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Grome MW, Zhang Z, Pincet F & Lin CX Vesicle Tubulation with Self-Assembling DNA Nanosprings. Angew Chem Int Edit 57, 5330–5334 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Franquelim HG, Khmelinskaia A, Sobczak JP, Dietz H & Schwille P Membrane sculpting by curved DNA origami scaffolds. Nat Commun 9, 811 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams S, Lund K, Lin C, Wonka P, Lindsay S, Yan H (2009) Tiamat: A Three-Dimensional Editing Tool for Complex DNA Structures. In: Goel A, Simmel FC, Sosík P (eds) DNA Computing. DNA 2008. Lecture Notes in Computer Science, vol 5347. Springer, Berlin, Heidelberg. 10.1007/978-3-642-03076-5_8 [DOI] [Google Scholar]
- 47.Nair U et al. SNARE proteins are required for macroautophagy. Cell 146, 290–302 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choy A et al. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science 338, 1072–1076 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jotwani A, Richerson DN, Motta I, Julca-Zevallos O & Melia TJ Approaches to the study of Atg8-mediated membrane dynamics in vitro. Methods Cell Biol 108, 93–116 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Wu Z et al. Dilation of fusion pores by crowding of SNARE proteins. Elife 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.