

Abstract
Glioblastoma multiforme (GBM) is the most lethal primary brain tumor characterized by high cellular and molecular heterogeneity, hyper-vascularization, and innate drug resistance. Current treatment options include a combination of surgical resection, radiotherapy, and chemotherapy primarily with temozolomide, but the prognosis is poor with an average life expectancy of 15 months. Despite significant research and drug development efforts, therapeutic advances to treat glioblastoma remain stagnant. Cellular components and extracellular matrix (ECM) are the two primary sources of heterogeneity in GBM. One of the major roadblocks in understanding the genetic basis of the cancer and developing new therapies is the lack of physiologically relevant and patient-specific GBM tumor models. Here, we develop biomimetic tri-regional GBM models with a tumor region, an acellular ECM region, and an endothelial region – with regional stiffnesses patterned corresponding to the GBM stroma, pathological or normal brain parenchyma, and brain capillaries. Patient-derived GBM cells, human endothelial cells, and hyaluronic acid derivatives are used to generate a species-matched and biochemically relevant microenvironment. This in vitro study demonstrates that biophysical cues are involved in various tumor cell behaviors and angiogenic potentials and promote different molecular subtypes of GBM. The stiff models are enriched in the mesenchymal subtype, exhibit diffuse invasion of tumor cells, and induce protruding angiogenesis and higher drug resistance to temozolomide. Meanwhile, the soft models demonstrate enrichment in the classical subtype and support expansive cell growth. The 3D bioprinting technology utilized in our study enables rapid, flexible, and reproducible GBM modeling with biophysical heterogeneity that can be employed by future studies as a tunable system to interrogate GBM disease mechanisms and screen drug compounds.
Keywords: Glioblastoma, 3D printing, biophysical regulation, stiffness, angiogenesis
Graphical Abstract
1. Introduction
Glioblastoma (GBM) is the most lethal central nervous system (CNS) cancer that has a notable short median patient survival time of 14.6 months.[1] Therapeutic dilemma of GBM results from its genetic heterogeneity, diffusive infiltration, hyper-angiogenesis, and innate resistance to treatments. GBM is characterized by a unique extracellular matrix (ECM) composition, mainly composed of hyaluronic acid (HA), glycosaminoglycans, proteoglycans, and glycoproteins, and lack of collagens.[2] Proteases secreted by cells within the GBM tumor microenvironment (TME) constantly remodel the ECM, leading to altered expression of ECM components as well as changes in biophysical properties such as the stiffness.[2] Dynamic interactions of the ECM with cells regulate GBM initiation, progression, invasion, and treatment responses through both biochemical and biophysical cues. The role of biochemical cues of ECM on tumor cells have been widely studied owing to the availability of ECM-derived materials that can be used for cell culture.[3–5] Biophysical cues such as stiffness, geometry, and topography regulates gene expression and cell behaviors, and cells reciprocally exert forces on ECM and remodel the microenvironment.[6] Mechanoreciprocity involved in these dynamic interactions has been identified to cause tissue stiffening in several cancer types. However, precisely creating the biophysical properties in vitro and investigating their roles in GBM are relatively challenging using traditional tissue engineering techniques. Hyper-angiogenesis is another characteristic of GBM that promotes GBM growth and invasion.[7] Biophysical cues of the TME regulate the abnormal tumor angiogenesis, and anti-angiogenesis treatment leads to changes in GBM stiffness.[8,9] However, models for investigating the angiogenesis activity of endothelial cells and GBM cell responses to vascularization in a stiffness-matched model has not been developed.
In vitro 3D models have gained popularity in investigating the cellular crosstalk and cell-ECM interactions due to their improved biomimicry compared to conventional models. Organoids as a self-assembled 3D model system have been explored for various cancer types, including pancreatic cancer, bladder cancer, as well as GBM.[10–12] Organoids generally better retain the tumor heterogeneity and transcriptional signatures compared to the traditional 2D culture. However, organoids are limited by innate variations and limited control over the structures due to the self-assembly process. Among various biofabrication technologies, 3D bioprinting enables cell-encapsulation in native ECM-derived biomaterials with defined architectures and matrix properties, and thus is increasingly employed for modeling complex cellular tissues and investigating the role of ECM in cancer progression.[13] 3D-printed models have successfully modeled the cellular heterogeneity, a characteristic of many cancer types including GBM, by creating a stromal layer surrounding a tumor zone, for the investigation of stromal impacts on tumor development and treatment responses.[14–16] In addition to cellular components, ECM is another critical aspect of the tumor microenvironment. Previously, polyethylene glycol (PEG)-based or HA-based hydrogels with different stiffnesses have been developed to evaluate stiffness impact on GBM cell growth.[17,18] PEG-HA composite hydrogels with tunable stiffnesses independent of the HA concentration have been employed for 3D culture of GBM cells.[19] Most studies either investigated ECM biophysical impacts on GBM using hydrogels with a bulk stiffness or relied on synthetic materials to modulate the stiffnesses.[17] Use of synthetic materials reduces the biomimicry of models due to the lack of proper biochemical cues. Stiffnesses of the tumor stroma, brain parenchyma, and brain capillaries are not homogeneous in vivo, thus hydrogels with bulk mechanical properties may also be insufficient to recapitulate the TME.[17,20–23] Investigating the biophysical aspects of ECM requires 3D matrices that can faithfully recapitulate native architectures as well as mechanical properties. Digital light processing (DLP)-based bioprinting is a rapid biofabrication technique compatible with various light-sensitive biomaterials.[24] Several tissue models and cancer models have been developed using this technology and ECM-derived biomaterials.[15,24–26] Orthogonal control of biophysical properties and biochemical cues have also been achieved using DLP-based printing and ECM-derived materials, making the technology ideal for the investigation of biophysical impacts on GBM development within a biomimetic ECM-based model.[27,28]
We hereby developed the first species-matched in vitro models that recapitulate the biophysical heterogeneity of GBM – with regionally varied stiffnesses corresponding to GBM stroma, pathological or healthy brain parenchyma, and brain capillaries – based on patient-derived cells and HA derivatives via DLP-based bioprinting. HA-based biomaterial and patient-derived cells enable the evaluation of biophysical impacts on GBM in a biochemically relevant and species-matched microenvironment. By adjusting printing parameters and the concentrations of the biomaterials, we were able to modulate the stiffness of three distinct regions in 3D-printed GBM models with a constant HA concentration. Modeling the biophysical heterogeneity potentially enhanced the biomimicry of models. Changes associated with tumorigenesis, including cell morphologies, invasion behaviors, gene expressions, angiogenic potentials, and drug responses rapidly occurred within two weeks in the 3D-printed GBM models with different stiffness conditions. The stiff ECM microenvironment induced the mesenchymal phenotype associated with recurrence and poorest treatment outcomes in patients. The soft ECM microenvironment promoted rapid cell proliferation and supported the expansion of cells with the classical phenotype. Endothelial cells incorporated in the 3D-printed GBM models also demonstrated different modes of growth and interaction, such as protruding morphologies in the stiff models and expansive growth in the soft models. Endothelial co-culture also induced differential drug responses in the GBM cells in the 3D-printed models, suggesting their potential roles in GBM drug resistance.
2. Results
2.1. 3D bioprinted GBM models with regionally varied biophysical properties
GBM tumor cells respond to the complex ECM cues and constantly remodel the ECM as cancer progresses. The remodeled ECM affects both the tumor survival and progression, and other critical stromal cell events such as angiogenesis by the endothelial cells. To recapitulate the spatially inhomogeneous ECM microenvironment and interrogate how ECM heterogeneity impacts on GBM development and endothelial cell growth, we utilized our DLP-based 3D bioprinting system and brain tumor-specific ECM-derived bioinks consisted of glycidyl methacrylate hyaluronic acid (GMHA) and gelatin methacrylate (GelMA), to create four different models: the tumor-only stiff model, the tumor-only soft model, the coculture stiff model, and the coculture soft model (Figure 1A and Figure S1). The 3D bioprinting system utilized a digital micromirror device chip consisting of over a million independently controlled micromirrors and a light source to project predesigned patterns on the bioink, allowing rapid polymerization of each region with 20–30 seconds of light exposure. A complete multi-stiffness GBM model could be created within 2 minutes. Considering the diffusion limit of nutrition and oxygen, the models were designed with a thickness of 250 μm. For each model, four initial tumor regions were generated with GBM cells with a diameter of 500 μm, surrounded by a donut-shape acellular ECM region with the ring width of 500 μm. For coculture models, an additional endothelial region was printed with human umbilical vascular endothelial cells (HUVECs) to encompass the tumor and ECM regions (Figure 1B). Matrix stiffness of the tumor core and the endothelial regions was designed to mimic that of GBM patient tissues (7 kPa) and normal brain tissues (0.45 kPa).[21] Encapsulating the GBM cells in a pathologically stiffened matrix was to mimic their physiological conditions and promote cell growth. Both the tumor cells and endothelial cells demonstrated high viability in their corresponding hydrogel environment through one week of culture (Figure S2a).[20–23] Clinical measurements indicated that the matrix stiffness could increase up to 26 kPa in GBM tissues.[17,22,29,30] For the ECM regions, two different stiffnesses, 21 kPa (hereby referred to as stiff models) and 2 kPa (referred to as soft models), were designed to mimic the GBM tissue stiffness and healthy brain stiffness (Figure 1c). Leveraging the GelMA concentration and printing parameters, we were able to obtain desired stiffness for each region while keeping the HA content constant across all models. Keeping a constant HA concentration avoided potential impacts on the tumor cells due to the difference in the amount of biochemical cues provided by HA. For all three ranges, hydrogel stiffnesses remained stable through one week of incubation at 37 °C and 5% CO2, the same condition used for all samples (Figure S2b). Difference in the appearance of stiff and soft ECM hydrogels was observed using bright field imaging or scanning electron microscopy (SEM) (Figure 1D). The pore sizes of the stiff ECM were significantly smaller than the pore sizes of the soft ECM. We further used these 3D-printed GBM models with regionally varied mechanical properties to perform various assessments and studies, including gene expressions, drug responses, tumor cell migration behaviors, and angiogenesis activities, occurred in response to the biophysical cues and culture conditions (Figure 1E and Figure S2C).
Figure 1.
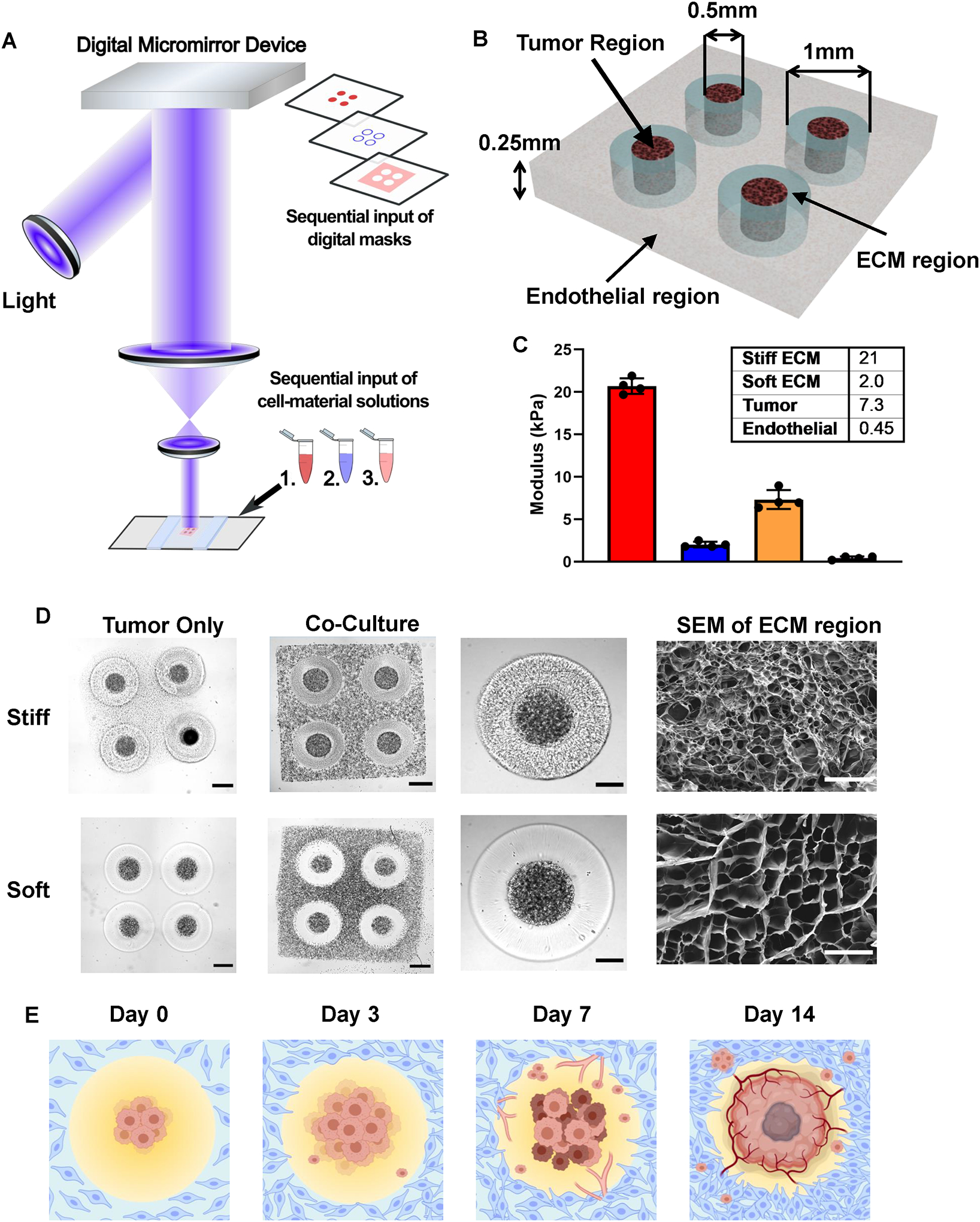
3D-bioprinted GBM models with regionally varied biophysical properties. (A) Schematic diagram of a multi-step digital light processing-based bioprinting approach. For each model, tumor cores with GBM cells were printed with the first digital mask, followed by acellular ECM regions patterned by the second mask. For co-culture models, endothelial cells were subsequently printed with the third mask. (B) Illustration of the model dimensions. (C) Stiffness of each region in the 3D-printed model. Bar charts represent mean compressive modulus of each region ± standard deviation. n = 4 technical replicates per group. (D) Representative bright field image and scanning electron microscopy of the 3D printed models with stiff (upper row) and soft conditions (lower row) on day 0. (Scale bars from left to right: 500 μm, 500 μm, 250 μm, 50 μm.) (E) Illustration of the timeline of tumor development and angiogenesis event in the 3D-bioprinted models, created with BioRender.com.
2.2. 3D models have distinct transcriptional profiles compared to sphere culture
Traditional cell culture methods including 2D culture and sphere culture have been extensively used for in vitro expansion and maintenance of GBM cells. Patient-derived cells cultured as spheres in serum-free conditions were enriched for GBM stem cells that better replicate the transcriptional signatures of the original tumor tissue than cell line-based 2D cultures. However, studies have demonstrated that cells maintained in traditional culture conditions still display distinct transcriptional profiles and cellular dependencies compared to primary tissue, 3D culture, or xenografts.[15,31]
Global transcriptome profiling was performed through RNA extraction and RNA sequencing (RNAseq) on TS576 GBM cells cultured as spheres and isolated from the tumor-only 3D soft and the tumor-only 3D stiff conditions. We first interrogated the difference among the sphere culture and the two 3D conditions to investigate the impact of 3D culture and ECM cues on the transcriptional signature of tumor cells. Principal component analysis revealed that the sphere culture had a drastically different transcriptional profile compared to either 3D condition, while the difference between the two 3D conditions was to a lesser extent (Figure 2a). A few protein coding cancer-related genes or prognostic genes were significantly upregulated over 16 folds in both 3D conditions, such as SLCO2A1, TCN1, and NTN4. Overexpression of the solute carrier organic anion transporter SLCO2A1 has been observed in GBM cells compared to normal brain tissues, promoting colon cancer tumorigenesis and mediating lung cancer invasion through the PI3K/AKT/mTOR pathway.[32–34] TCN1 encodes vitamin B12 binding proteins and is a prognostic marker in renal cancer. NTN4 promotes GBM proliferation and is associated with TMZ resistance.[35,36] Genes significantly downregulated in both 3D conditions but enriched in sphere cultures included CYP24A1, DIRAS2, KANK4, KRT75, and PRSS35.
Figure 2.
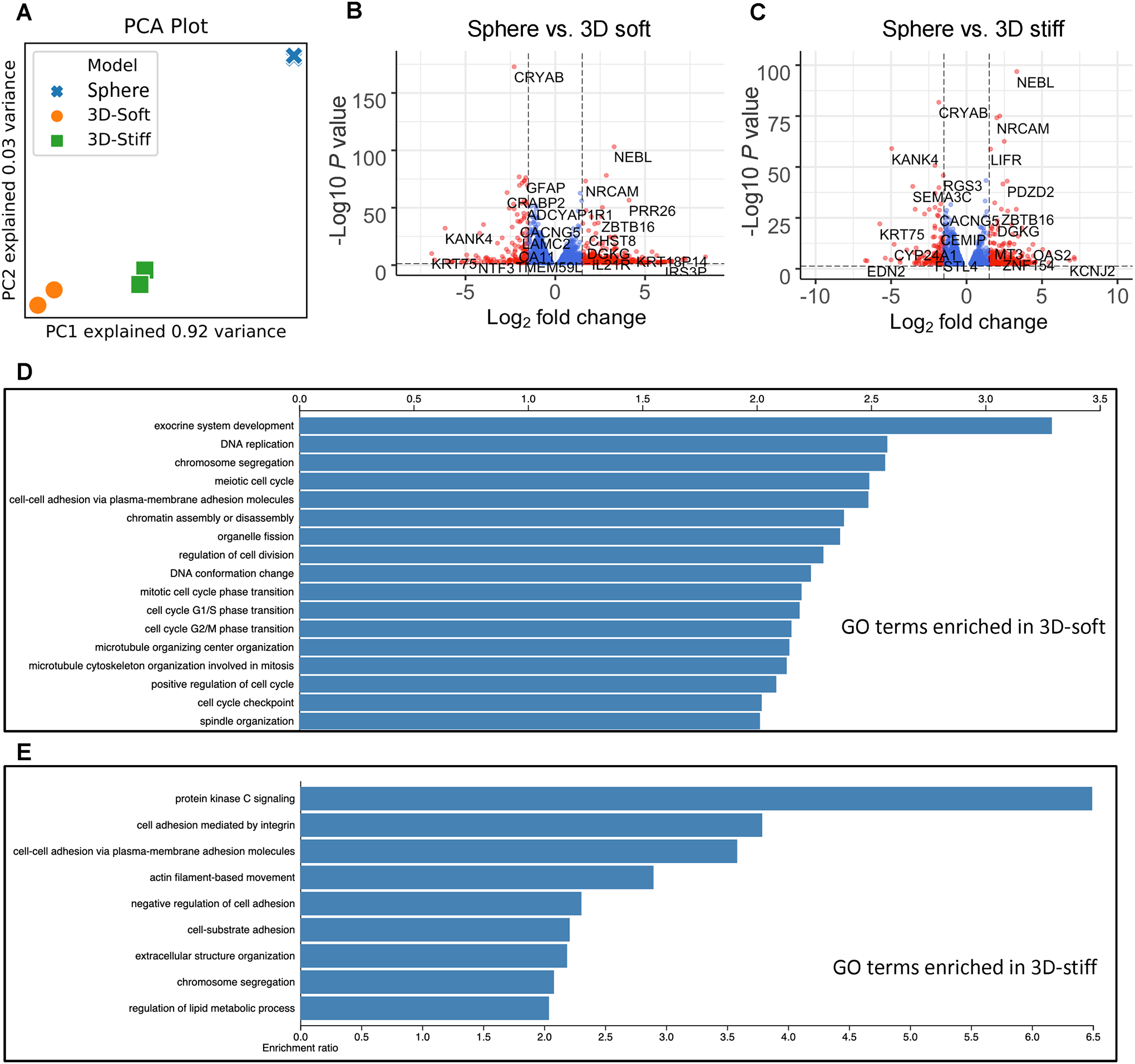
Distinct transcriptional profiles between the GBM sphere culture and 3D models. (A) Principal component analysis (PCA) of the global transcriptional landscape profiled by RNA sequencing of TS576 cells in (1) sphere culture, (2) tumor-only 3D stiff condition, and (3) tumor-only 3D soft. (B) Volcano plot of transcriptional landscape comparing TS576 in the sphere culture vs. TS576 in the 3D tumor-only soft model. The x-axis represents log2 transformed fold change, and the y-axis shows the log transformed p-value adjusted for multiple test correction. n=2 replicates per condition. (C) Volcano plot of transcriptional landscape comparing TS576 in the sphere culture vs. TS576 in the 3D tumor-only stiff model. The x-axis represents log2 transformed fold change, and the y-axis shows the log transformed p-value adjusted for multiple test correction. n=2 replicates per condition. (D) Gene ontology (GO) terms enriched in TS576 cells cultured in 3D soft condition vs. standard sphere culture. (E) GO terms enriched in TS576 cells cultured in 3D stiff condition vs. standard sphere culture.
We performed a gene ontology (GO) enrichment analysis with the RNAseq results of sphere culture and the 3D tumor-only models to investigate the changes in biological processes and molecular functions of the tumor cells in different culture conditions. Cell-cell adhesion via plasma-membrane adhesion molecules was over-represented in both 3D conditions compared to the sphere culture, demonstrating the biomimetic ECM materials used for printing promoted cell adhesion and cellular crosstalk. GO terms about DNA replication, cell cycle regulation, and cell division, were significantly over-represented in the 3D soft model compared to the sphere culture (Figure 2D). Enriched GO terms involved in cellular component organizations such as chromosome segregation, organelle fission, microtubule cytoskeleton organization, and spindle organization were identified in the 3D soft model. In addition, positive regulation of cell cycles, G1/S phase transition and G2/M phase transition, suggests an enhanced proliferation of tumor cells in the 3D soft model compared to the sphere culture. Highly enriched term of exocrine system development implied that cell differentiation probably occurred in the 3D soft condition. Gene set enrichment analysis (GSEA) revealed that compared to the 3D soft condition, sphere cultured cells express enriched gene sets involved in STAT pathway, chemotaxis, autophagosome, and cell differentiation (Figure S3A).
Cells in the 3D stiff condition also demonstrated distinctly enriched GO terms compared to sphere cultures (Figure 2E). The protein kinase C (PKC) activity is highly enriched in the 3D stiff condition compared to the sphere control, and previous studies have demonstrated that the PKC pathway is involved in the aggressive phenotype of GBM.[37] Cell adhesion mediated by integrin, cell-substrate adhesion, and extracellular structure organization were over-represented by the cells in the stiff condition. GSEA comparing the expression of cells cultured as spheres and in the 3D stiff condition revealed that sphere cultured cells expressed highly enriched cellular organization and modification activities including DNA repair and histone modifications (Figure S3B). Sphere culture were also enriched in pathways related to signal transduction and metabolism, including NGF-stimulated transcription, MAPK signaling pathways, oxidative phosphorylation, respiratory electron transport, complex I biogenesis, response of EIF2AK4 to amino acid deficiency, eukaryotic translation elongation, and KEGG ribosome related pathway.
The changes in gene expression and subsequent functional changes of cells cultured in the 3D models compared to the control are the combined results of the dimensionality, biochemical cues and mechanical cues of the ECM-like matrix, and cellular crosstalk enabled by 3D modeling. While in both 3D soft and 3D stiff conditions, biomimetic HA-based hydrogel were used to fabricate the models, genes related to cell-ECM interactions were more enriched in the stiff condition, suggesting that the stiff microenvironment might have enhanced the cellular response to the ECM-derived cues.
2.3. Stiff model promotes hypoxia and tumorigenicity signature in GBM
We further interrogated the transcriptional profiles of the GBM cells encapsulated in the 3D stiff and 3D soft models. The amount of HA in the two models was identical so that we could evaluate the effects of the biophysical cues from the ECM on the tumor development. Gene sets related to hypoxia conditions, cancer invasiveness, E-cadherin loss-induced metastasis, and responses to external stimulations including interferons, inflammation, ECM, and cell apoptosis were significantly enriched in the stiff model (Figure 3A–B). A cut-off of fold change greater than 2 and false discovery rate (FDR) less than 0.05 was used to identify the most differentially expressed genes (DEGs) in either 3D tumor-only condition.
Figure 3.
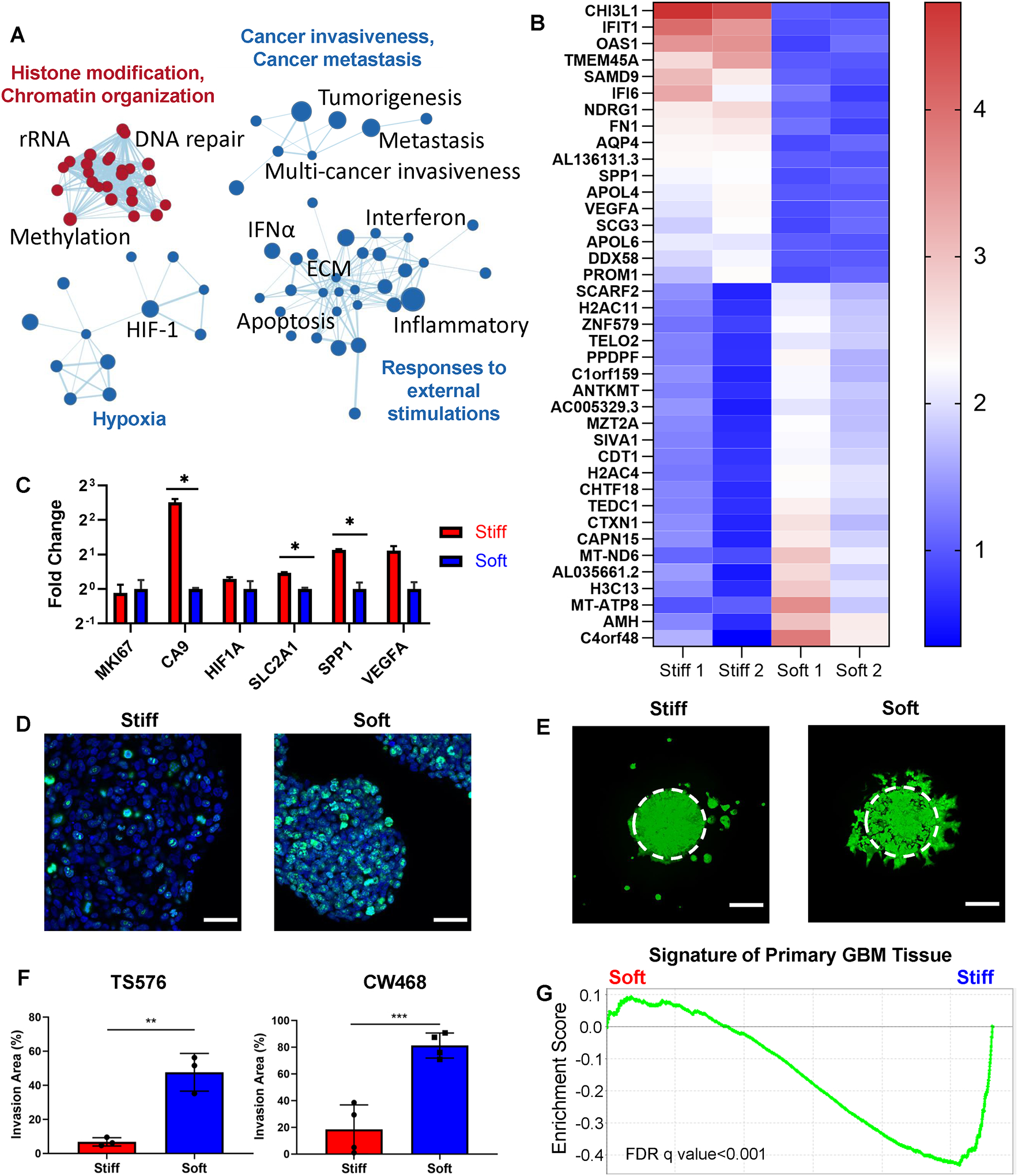
Biophysical patterning induced distinct transcriptional profiles and invasion patterns of GBM cells. (A) Gene set enrichment connectivity diagram displaying enriched pathways in the soft condition (red) and the stiff condition (blue) in TS576 in the 3D-printed tumor-only models. (B) Top differentially expressed genes in the 3D stiff condition vs. the 3D soft condition. Scale bar represents fold change. (C) mRNA expression of representative genes related to proliferation, hypoxia responses, and hypoxia-induced angiogenesis in TS576 in the 3D tumor-only conditions. P-values were calculated using unpaired Student’s t-test corrected for multiple comparisons using the Holm-Sidak method. *, p < 0.05. (D) Immunofluorescence staining of Ki67 in TS576 cells in the 3D tumor-only stiff condition vs. the 3D tumor-only soft condition. Scale bars: 50 μm. (E) Fluorescence imaging showing invasion patterns of fluorescently labeled TS576 cells in the 3D tumor-only conditions. Image taken on day 7. Scale bars: 250 μm. (F) Quantification of the invasion areas of TS576 or CW468 in the 3D tumor-only stiff and the 3D tumor-only soft conditions. n = 3 replicates for TS576 cells; n = 4 replicates for CW468 cells. P-values were calculated using unpaired Student’s t-test corrected for multiple comparisons using the Holm-Sidak method. (G) Gene set enrichment analysis of the primary GBM tissue signatures between the 3D tumor-only stiff and soft conditions. FDR q-value < 0.001.
Genes significantly elevated by the 3D stiff condition included CHI3L1, IFIT1, OAS1, TMEM45A, SAMD9, IFI6, NDRG1, FN1, AQP4, AL136131.3, SPP1, APOL4, VEGFA, SCG3, APOL6, DDX58, and PROM1. High expression of CHI3L1 and SAMD9 has been identified to be negatively correlated with GBM patient survival time.[38,39] TMEM45A and NDRG1 are involved in hypoxia-associated chemoresistance, and the knockdown of TMEM45A reduces glioma proliferation and invasion.[40–42] FN1 is highly upregulated in GBM and involved in the adhesion, growth, angiogenesis, and recurrence of GBM.[43] GBM expresses higher level of AQP4 compared to low-grade gliomas, and AQP4 with its highest water flux capacity in the CNS is potentially associated with tumor edema, migration, and proliferation.[44] Long non-coding RNA (lncRNA) genes AL136131.3, an antisense to VEGFA, was concordantly upregulated in the stiff condition with the angiogenesis markers VEGFA and SPP1. Genes encoding apolipoproteins including APOL4 and APOL6 were also significantly upregulated in the stiff condition. IFIT1, OAS1, IFI6, and DDX58 are associated with the interferon (IFN) signaling pathways which has been suggested to be involved in the immune escape of GBM.[45] CD133 (or RPOM1) upregulated in the stiff condition is essential for the maintenance of GBM stem cells.[46]
Gene sets related to cell cycle regulation, such as phase transition, DNA synthesis, chromosome organization, transcriptional regulation, and DNA repair were upregulated in the soft condition, more specifically, the most upregulated genes in the soft models included AMH, MT-ATP8, H3C13, MT-ND6, CAPN15, CTXN1, TEDC1, CHTF18, H2AC4, CDT1, SIVA1, MZT2A, ANTKMT, PPDPF, TELO2, ZNF579, H2AC11, SCARF2. AMH belongs to the transforming growth factor β (TGFβ) superfamily which plays a role in the initiation and progression of gliomas.[47] MT-ATP8 and MT-ND6 are mitochondrially encoded genes, and ANTKMT regulates mitochondrial respiration. Various cell cycle related genes were enriched. H3C13, H2AC4, and H2AC11 are histone genes whose upregulation often occur during the S phase of the cell cycle. Genes associated with biogenesis, such DNA replication, DNA synthesis, DNA repair, or spindle organization, were also upregulated, including TEDC1, CHTF18, CDT1, SIVA1, MZT2A, and TELO2. CAPN15, CTXN1, and ZNF579 are involved in transcriptional regulations or cell signaling. High expression of PPDPF correlates with cancer progression and tumor size in hepatocellular carcinoma.[48] SCARF2 upregulated in the soft model has been suggested as a risk gene for glioma.[49]
A hypoxic microenvironment has been shown to promote cell stemness, cancer invasiveness, endothelial-mesenchymal transition, and cell-cycle arrest.[50] Hypoxia related genes including CA IX, HIF1-α, SLC2A1 (encoding glucose transporter 1 protein), and hypoxia-associated angiogenesis markers VEGFA and SPP1 were upregulated in the stiff condition (Figure 3C). While mRNA expression of the proliferation marker MKI67 of cells from the two conditions showed no significant difference, immunofluorescence (IF) staining revealed that more KI67 positive cells were present in the soft model, suggesting that post translational regulations may be involved (Figure 3D). In addition, IF staining showed that the stiff model better maintained the overall stemness of the TS576 cells, while the SOX2+ tumor cells were mainly located on the invasive edge of the tumor core in the soft model (Figure S5). The astrocytic differentiation marker glial fibrillary acidic protein (GFAP) was observed in the soft model but not in the stiff mode.
2.4. 3D models induce different GBM invasion patterns and transcriptional subtypes
Using green fluorescent protein labeled tumor cells, the migration patterns of GBM cells were imaged on day 7. Distinct modes of invasion were observed (Figure 3E). GBM cells diffusively migrated out from the original region as single cells or small clusters and proliferated with a rounded morphology at new locations in the stiff model. Diffusive invasion is a characteristic of GBM making it difficult for complete surgical removal. The cells in the soft model expanded from their original location with protrusions, forming invasive margins as observed in a GBM mouse xenograft model.[51] Multiple invasion patterns have been observed in xenografts, in vitro cell culture, and biopsies, such as single cell invasion, cluster invasion, and expansive-growth, demonstrating the highly heterogeneous and dynamic behavior of GBM.[51,52] The area of invasion in the soft models was 7 folds and 4.4 folds higher than in the stiff models for TS576 cells and CW468 cells, respectively, consistent with the KI67 staining (Figure 3F).
We next generated a “primary GBM tissue” gene set with a core set of genes upregulated in GBM surgical specimens compared to sphere cultured GBM cells in vitro. Principle component analysis demonstrated that the sphere culture had a distinct transcriptome profile from primary GBM tissues (Figure S4A). TS576 cells isolated from the stiff model were highly enriched with the primary GBM tissue signatures when compared to TS576 cells isolated from the soft model (Figure 3G), indicating that the stiff model transformed the cells to a more clinically relevant state. Bulk transcriptional profiling of the primary GBM tissue from The Cancer Genome Atlas (TCGA) identified three major subtypes of GBM, the proneural, the mesenchymal, and the classical subtype, each enriched for different genetic alterations.[53] GSEA revealed that gene sets related to epithelial-mesenchymal transition (EMT) and mesenchymal signatures were enriched in the stiff condition (Figure S4B,C). Expression of genes associated with the mesenchymal subtype such as FN1 and CHI3L1 were 2-fold and 4-fold higher in the stiff model than in the soft model (Figure S4D). PDGFRA commonly altered in the proneural subtype had a higher expression level in the stiff model, while EGFR related genes commonly amplified in the classical subtype showed higher expressions in the soft model. These findings suggested that the stiff model could better model the mesenchymal subtype and proneural subtype, while the soft model was more suitable for modeling the classical subtype. The stiffness-patterned GBM model enables us to create different GBM situations in vitro.
2.5. Endothelial cells exhibit different growth patterns and angiogenic events in 3D-printed models
To further investigate the crosstalk between the GBM cells and endothelial cells, we incorporated HUVEC into the 3D-printed multi-stiffness models. The endothelial region and the tumor cores were separated by the ECM regions with either the stiff or the soft type hydrogels. The spatial separation allowed investigation of paracrine signaling-induced endothelial cell growth and migration towards the tumor core as well as the tumor cell migration. In both 3D co-culture models, migration of HUVEC towards GBM cells were observed. In the stiff model, the migrated CD31+ HUVECs exhibited a sprouted blood-vessel like morphology and were in close contact with the SOX2+ GBM cells (Figure 4A, Figure S6). In the soft model, the HUVECs exhibited expansive-growth morphology without visible sprouting. We performed a quantitative real-time PCR (qPCR) on tumor cells isolated from all our 3D printed models and sphere cultures. Angiogenic markers SPP1 and VEGFA were upregulated in all 3D models, and significantly enriched by the stiff culture condition or the co-culture condition (Figure 4B). More specifically, in the tumor-only 3D models, cells expressed significantly higher VEGFA in the stiff model than in the soft model; comparing the tumor-only models and the co-culture models, the co-culture condition significantly increased the expression of VEGFA in the tumor cells in both the soft and the stiff conditions. Tumor cells in the stiff co-culture condition expressed the highest level of VEGFA and SPP1. Prior studies showed that VEGFA and other signals generated by hypoxic tumor cells within the pseudo-palisades near necrotic tumor core could trigger sprouting angiogenesis events near the tumor-parenchyma interface, consistent with our observation of more active sprouting events in the 3D stiff co-culture model.[54,55]
Figure 4.
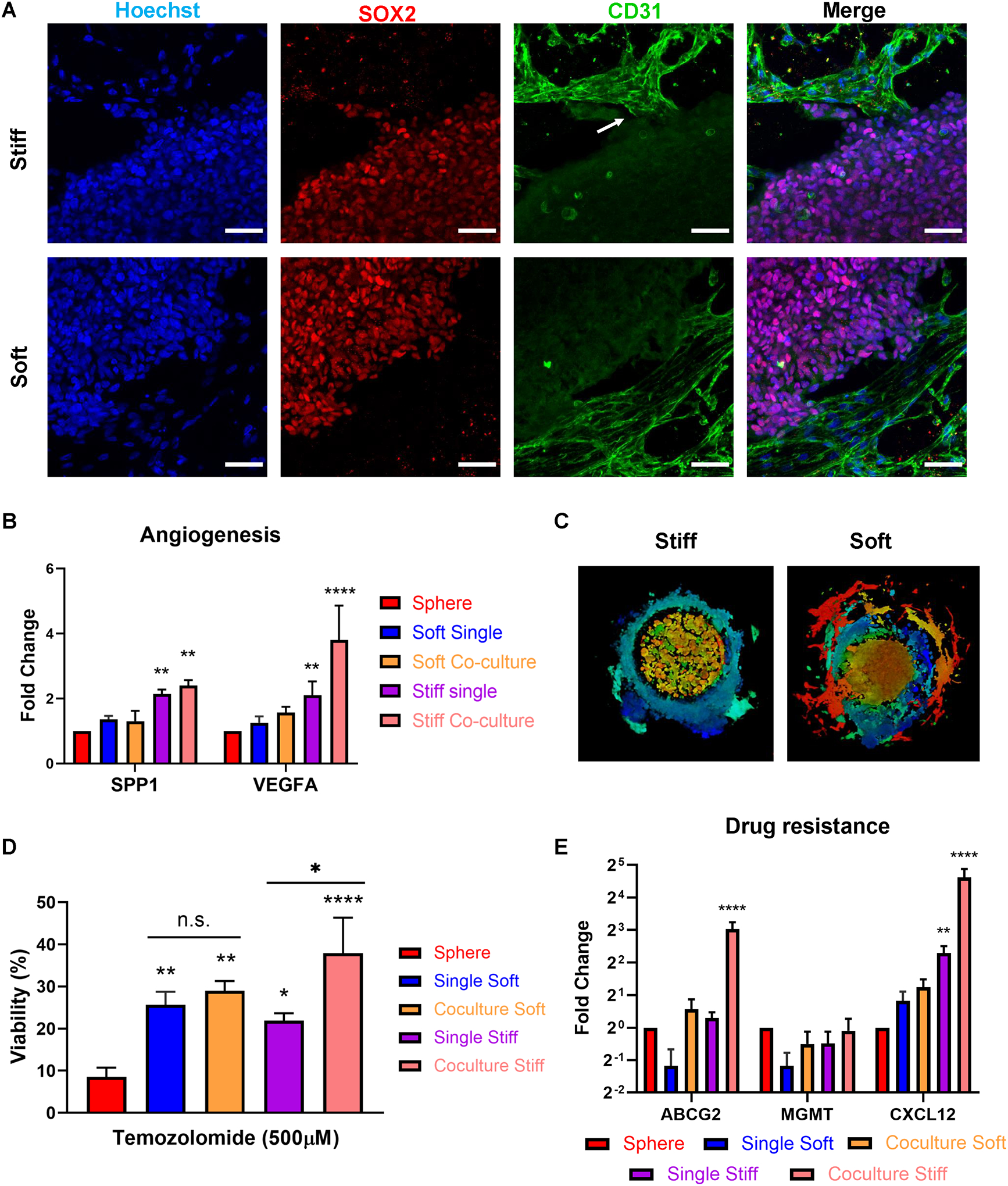
Stiff condition induced sprouting angiogenesis of endothelial cells and enhanced drug resistance of GBM cells. (A) Immunofluorescence staining of SOX2 (GBM cells) and CD31 (endothelial cells) in the stiff co-culture condition vs. the soft co-culture condition. White arrow: sprouting angiogenesis. Scale bars: 50 μm. (B) mRNA expressions of angiogenesis markers SPP1 and VEGFA in TS576 in (1) sphere culture, (2) tumor-only soft condition, (3) co-culture soft condition, (4) tumor-only stiff condition, and (5) co-culture stiff condition measured by real-time quantitative PCR (RT-qPCR). N=3 technical replicates for each condition. P-values were calculated using two-way ANOVA corrected for multiple comparisons using Dunnett test. **, p < 0.01; ****, p < 0.0001. (C) Invasion patterns of TS576 cells in 3D co-culture models. (D) TS576 viability after 6 days of Temozolomide treatment. n=3 technical replicates for each condition. P-values were calculated using ordinary one-way ANOVA corrected for multiple comparisons using Tukey test. *, p < 0.05; **, p < 0.01; ****, p < 0.0001. (E) mRNA expressions of drug resistance markers ABCG2, MGMT, and CXCL12 in TS576 measured by RT-qPCR. N=3 technical replicates for each condition. P-values were calculated using two-way ANOVA corrected for multiple comparisons using Turkey test. **, p < 0.01; ****, p < 0.0001.
2.6. GBM-endothelial crosstalk enhances tumor invasion and drug resistance
We then interrogated the impact of co-culture condition on GBM cell behaviors and functions. Similar to their tumor-only 3D counterparts, GBM cells in the co-culture models also demonstrated different invasion patterns. Tumor cells expanded with a fibroblastic morphology in the soft co-culture model, and more rounded morphology in the stiff model (Figure 4C). The co-culture condition promoted CW468 invasion into the ECM regions in both the soft and stiff models and promoted invasion of TS576 cells only in the stiff condition (Figure S7, S8). The observed difference may be resulted from the innate heterogeneity of GBM cells. SOX2 was expressed by most tumor cells in the stiff condition but mainly by the cells on the outer rim of the tumor region in the soft condition (Figure S9A). The expression of differentiation marker GFAP was significantly elevated in the soft condition compared to the stiff condition. The IF staining results suggested that the stiff co-culture condition promoted the stemness of tumor cells and the soft co-culture condition promoted cell differentiation.
All 3D-bioprinted models demonstrated enhanced drug resistance of tumor cells to TMZ compared to sphere cultured cells (Figure 4D). IC50 of TMZ on sphere cultured TS576 cells was measured to be 30 μM, but the dosage was ineffective in the 3D models (Figure S9B). For fair comparison, all conditions were treated with 500 μM of TMZ for 6 days. No significant difference in the viability of tumor cells were detected in the tumor-only stiff or soft models. The co-culture condition significantly increased the viability of TS576 cells to TMZ treatment in the stiff models but had no significant impact on cell viability in the soft models. We have previously observed sprouting events of endothelial cells in the stiff models. The sprouting endothelial cells were in close contact with a cluster of SOX2+ tumor cells, potentially forming a perivascular niche near the invading edge of the tumor zone. The perivascular niche has been reported to enrich cancer stem cells that are highly drug resistant.[56] It is possible that the interaction between endothelial cells and tumor cells in the stiff model have enriched the drug resistant population more than that in the soft model, leading to higher viability of tumor cells after TMZ treatment. To confirm that differential drug responses were not a result of diffusion kinetics of drugs into the 3D hydrogels, we simulated the diffusion of drugs with fluorescently labeled Dextran molecules. After 30 minutes of incubation, the fluorescence signals reached plateau in both the stiff hydrogel and the soft hydrogel and demonstrated no statistical significance in the signal intensities (Figure S10C). QPCR analysis revealed that drug resistance related genes such as ABCG2 and CXCL12 were upregulated 8-fold and 24-fold, respectively, in the stiff co-culture condition (Figure 4E), consistent with the TMZ treatment responses.
3. Conclusion
We have developed biochemically and biophysically relevant GBM models with stiffness patterning in HA-rich matrix for investigation of behaviors and interactions of tumor cell and endothelial cells in a heterogeneous and biomimetic microenvironment. Tumor regions and endothelial regions were designed to have stiffness resembling their native states. Two stiffness conditions of the ECM region specifically designed to mimic GBM-remodeled stroma or healthy brain parenchyma were printed between the tumor region and the endothelial region, resulting in differential tumor cell growth and behaviors. While cell proliferation and expansion occurred rapidly in the soft models, hypoxia, stemness, and angiogenic potentials related to malignant phenotypes were enhanced in the stiff models. Tumor cells invade the ECM regions with distinct morphologies and patterns in the two-stiffness microenvironment. The stiff condition demonstrated a single cell diffuse invasion pattern, a characteristic of GBM precluding complete surgical removal, while the soft condition exhibited an expansive growth pattern. Both invasion patterns have been previously observed for GBM cells, suggesting that our stiffness-patterned models may be suitable for modeling different states of GBM development. Gene set enrichment analysis suggested that the stiff condition recapitulated the primary GBM tissue signatures and was enriched in gene sets related to the mesenchymal and proneural subtypes. Meanwhile, GBM cells in the soft condition were more enriched in gene sets related to the classical subtype, indicating that the two types of stiffness are suitable for modeling different subtypes.
Vascularization is a characteristic feature of GBM that promotes tumor growth and facilitates tumor invasion. The incorporation of HUVECs into the stiffness-patterned model allowed us to observe different endothelial cell growth patterns and potential angiogenic events in two different stiffness conditions, as well as altered tumor invasion patterns and drug responses in the presence of endothelial cells. Sprouting and proliferation of endothelial cells coordinate to mediate blood vessel formation.[54,57] While proliferation of HUVECs was observed in the soft models, proliferation and sprouting were both observed in the stiff models. The gene expression revealed that tumor cells in the stiff model expressed high angiogenic markers, consistent with the observation of sprouting events in the stiff models. The soft condition in general promoted cell proliferation, demonstrated by the larger invasion area of tumor cells and the proliferation of endothelial cells. Moreover, TMZ treatment on all models and a sphere culture control demonstrated that the stiff co-culture model had the highest tumor cell viability, suggesting that the stiff condition as well as co-culture with endothelial cells enhanced the drug resistance of GBM. Many cancer drugs including TMZ induce cell cycle arrest and block cell division, thus are more effective on proliferating cells, consistent with the higher TMZ sensitivity of cells in the soft models compared to the stiff models.
To conclude, the tri-regional stiffness-patterned GBM models presented in this work are the first to incorporate physiologically relevant biophysical heterogeneity of GBM with biochemically relevant ECM materials. The regional stiffnesses better recapitulate the native environments and potentially favor more biomimetic cell-ECM and cellular interactions. Transcriptional profiling has demonstrated the potential of using these models to investigate different subtypes of GBM as well as different stages of GBM. Flexibility of the DLP-based bioprinting process and versatile material selection allow orthogonal modulation of biophysical properties and biochemical characteristics. The stiffness patterning can also be applied to future models to study the biophysical impacts on other tumor-stromal interactions, such as macrophages and astrocytes, abundant in the GBM microenvironment.
4. Experimental Section/Methods
All studies were conducted in accordance with approved IRB protocols by the University of California, San Diego. All animal work was approved by the Institutional Review Board at the University of California, San Diego and was performed in accordance with Institutional Animal Care and Use Committee guidelines.
4.1. GMHA and GelMA synthesis and characterization
GMHA was synthesized using 200 kDa hyaluronic acid (HA, Lifecore Biomedicals) and GelMA using gelatin from porcine skin (Sigma-Aldrich) with methods described previously.[58,59] Briefly, HA was dissolved in a 1:1 water and acetone solution at room temperature overnight. On the next day, triethylamine and glycidyl methacrylate was sequentially added to the mixture and stirred overnight. GMHA was precipitated with acetone and re-dissolve in de-ionized water. The resuspended GMHA solution was collected in 12–14 kDa rated dialyzer tubes and dialyzed at room temperature for 12 hours, with water replacement every 3 hours. For the synthesis of GelMA, gelatin was first dissolved in a 0.25M carbonate-bicarbonate buffer solution at 50 °C. For each gram of gelatin, 0.1ml methacrylic anhydride was added dropwise to the gelatin solution and stirred for 1 hour at 50 °C. The solution is then dialyzed at 42°C for a week. After dialysis, both GMHA and GelMA solutions were collected in 50 mL tubes to be frozen overnight at −80 °C and lyophilized. Freeze-dried GelMA and GMHA were stored at −80 °C before reconstitution. To prepare the printing biomaterials, GelMA was reconstituted to a stock solution of 20% (w/v) and GMHA to a stock solution of 4% (w/v). All stock solutions were sterilized using filters (Millipore) with 0.22 μm pore size and stored at 4°C before use. Proton nuclear magnetic resonance (1H NMR, Bruker) was used to characterize the degree of substitution (methacrylation) of GelMA and GMHA.
4.2. Cell culture
Human patient derived glioblastoma stem cells (TS576) were obtained from Dr. Frank Furnari Lab at the University of California, San Diego (UCSD) and cultured as described previously.[60,61] TS576 cells were cultured in DMEM/F12 medium containing 1% (v/v) B27 supplement without vitamin A, 20 ng/mL EGF, 10 ng/mL bFGF, and 100 IU/mL penicillin/streptomycin (P/S) at 37°C. CW468 cells obtained from Dr. Jeremy Rich Lab at UCSD were cultured in serum-free Neurobasal medium supplemented with 2% (v/v) B27 supplement without vitamin A, 10 ng/mL basic human fibroblast growth factor, 10 ng/mL human epidermal growth factor, and 1% P/S. HUVECs were cultured in EGM-2 (Lonza) complete medium supplemented with 1% P/S.
4.3. 3D printing of the GBM models
Before printing, TS576 cells were digested with Accutase (Stemcell Technology), and HUVECs were digested with 0.05% Trypsin-EDTA (Gibco). For all 3D samples, the cell suspension solution for the tumor region was resuspended to 10 × 107 cells/mL of TS576s. For the co-culture model, the cell suspension solution for the endothelial region was resuspended to 5 × 107 cells/mL of HUVECs. The cell suspension was then prepared as 10 μL aliquots and stored on ice before printing. Bioinks for each region was prepared with concentrations in Table 1. For tumor and endothelial regions, the bioinks were prepared with twice of their desired final concentrations (final biomaterial concentrations of these two regions are included in the parentheses).
Table 1.
Material composition of different regions.
| Region | GelMA (w/v) | GMHA (w/v) |
|---|---|---|
| Tumor region | 10% (5%) | 2% (1%) |
| Endothelial region | 5% (2.5%) | 1% (0.5%) |
| Stiff ECM | 10% | 1% |
| Soft ECM | 5% | 1% |
The bioinks were stored on heat block at 37 °C and covered with foil to avoid light exposure. Bioinks for the tumor and endothelial regions were mixed with the corresponding cell suspension solution at 1:1 ratio immediately before printing to maximize cell viability. Bioinks for the ECM region was directly used for printing with no further dilution.
A customized DLP-based 3D bioprinting system was used for the multicomponent printing process. The primary components of the 3D bioprinting system are a digital micromirror device (DMD) chip (Texas Instruments), optics for light control, a printing stage with motion controller (Newport), and a light source (Hamamatsu). Specialized computer software was developed to coordinate the loaded patterns, light exposure time, and stage movement. A specialized printing apparatus was used to precisely control the thickness of the printed structure. After loading the cell-biomaterial mixture onto the printing stage, light was switched on with a set of exposure times. The exposure time was 20 seconds for the soft ECM and 25 seconds for the endothelial region. The exposure time was 25 seconds for the tumor region and 30 seconds for the stiff ECM. Printed constructs were rinsed with DPBS supplemented with 1% P/S and 0.2% Normocin (Invitrogen) and cultured in maintenance medium at 37 °C. The maintenance medium composed of 50% of complete TS576 medium and 50% EGM-2.
4.4. Mechanical testing
MicroSquisher (CellScale) was used to measure the compressive modulus of the printed samples. For each prepolymer mixture, pillars with 250 μm in diameter and 250 μm in height were printed with the same printing setup used for the tumor models. Pillars were stored at 37 °C before measurement to mimic the culture condition. Mechanical testing was performed on Day 1, 3, and 7 to evaluate the stability of printed constructs. For measurement on the MicroSquisher, stainless steel beams and platens were used to consecutively compress the constructs at 10% displacement of their height for three times. The last measurement is used for analysis. Compressive modulus was generated using customized MATLAB scripts from the force and displacement data.
4.5. Scanning electron microscopy (SEM)
Micron-scale patterns of the printed constructs were imaged using Zeiss Sigma 500. Samples were prepared using a chemical dehydration protocol optimized for printed hydrogels. Briefly, samples were fixed with 2.5% glutaraldehyde for 1 hour at room temperature and overnight at 4°C. Then the samples were rinsed with DPBS and sequentially soaked in 70%, 90%, 95%, and 100% ethanol. After replacing the 100% ethanol solution for 3 times, the samples were transferred to hexamethyldisilazane (HDMS):EtOH (1:2) for 15 minutes, HDMS:EtOH (2:1) for another 15 minutes, and eventually 100% HDMS for 15 minutes. The samples were left in a chemical hood overnight. Right before SEM imaging, the chemically dried samples were coated with iridium using a sputter coater (Emitech).
4.6. Immunofluorescence staining and image acquisition
3D bioprinted constructs were rinsed with DPBS for three times and fixed with 4% paraformaldehyde for 1 hour at room temperature. The block/permeabilization solution was prepared by dissolving 5% (w/v) bovine serum albumin (BSA, Gemini Bio-Products) and 0.1% Triton X-100 (Promega) in PBS and filtered after fully dissolved. Fixed samples were blocked/permeabilized for 1 hour at room temperature on a shaker at 100 rpm.
Primary antibodies (Table 2) were diluted in PBS, and samples were incubated in primary antibody solution overnight at 4 °C. Samples were rinsed three times using DPBS with 0.05% Tween 20 (PBST) at 100 rpm at room temperature. Secondary Alexa Fluor-conjugated antibodies (1:200; Abcam) and Hoechst 33342 (1:1000; Life Technologies) were diluted in DPBS with 3% (w/v) BSA. Samples were incubated in secondary antibody and counterstain solutions in dark for 1 hour at room temperature. Samples were rinsed three times with PBST after secondary incubation. Before imaging, the samples were soaked in a 0.05% sodium azide (Alfa Aesar) solution and stored at 4 °C in dark. A confocal microscope (Leica SP8) was used for image acquisition with consistent settings for each primary antibody. Fluorescence images of EGFP labeled cells were also acquired using the confocal microscope.
Table 2.
Antibodies used for immunofluorescence staining.
| Primary Antibody | Species | Dilution | Manufacturer |
|---|---|---|---|
| SOX2 | rabbit anti human | 1:100 | Abcam 97959 |
| GFAP | mouse anti human | 1:100 | Millipore Sigma G3893 |
| CD31 | mouse anti human | 1:100 | Abcam 24590 |
| Ki67 | rabbit anti human | 1:100 | Abcam ab16667 |
4.7. RNA isolation and quantitative reverse transcription polymerase chain reaction (RT-qPCR)
For 3D single cell culture models, TS576 cells were retrieved from printed constructs by dissociating the hydrogel with collagenase type II (Sigma-Aldrich). For 3D co-culture models, endothelial layers were mechanically removed before using collagenase II to retrieve tumor cells. TRIzol reagent (Life Technologies) was mixed with tumor cells isolated from 3D models or their sphere culture counterparts to prepare cell lysates. Total RNA of each sample was extracted using the Direct-zol RNA MiniPrep Kit (Zymo). The RNA concentration from each sample was evaluated using a Tecan plate reader after resuspending RNAs in RNase free water. The RNA samples were immediately stored at −80 °C.
For RT-qPCR, cDNA was first synthesized from the RNA samples using the ProtoScript® First Strand cDNA Synthesis Kit (New England BioLabs). Input RNA was 200ng for each sample. The primers were designed using NCBI primer-BLAST and purchased from Integrated DNA Technologies. RT-qPCR was performed using PowerUp SYBR Green master mix (Applied Biosystems) and the Quantstudio 3 RT-PCR system. Expressions of specific genes were determined by normalizing the threshold cycle (Ct) values against the housekeeping gene. Primer sequences are listed in Table 3.
Table 3.
Primers for RT-qPCR.
| Gene | Accession Number | Forward Primer (5’->3’) | Reverse Primer (5’->3’) |
|---|---|---|---|
| ABCG2 | NM_004827.3 | AAGCCACAGAGATCATAGAGCC | TCTTCTTCTCACCCCCGGAA |
| CXCL12 | NM_199168.4 | AGATGCCCATGCCGATTCTT | AGGGCACAGTTTGGAGTGTT |
| GAPDH | NM_002046.7 | ACAACTTTGGTATCGTGGAAGG | GCCATCACGCCACAGTTTC |
| MGMT | NM_002412.5 | GCACCGTTTGCGACTTGG | GCTCACAACCAGACAGCTCC |
| SPP1 | NM_000582.3 | AGCTTTACAACAAATACCCAGATGC | GACTTACTTGGAAGGGTCTGTGG |
| VEGFA | NM_001025366.3 | ACGAAAGCGCAAGAAATCCC | CTCCAGGGCATTAGACAGCA |
4.8. RNA sequencing and data analysis
For RNA-seq analysis, RNA was extracted from sphere culture and 3D printed samples of TS576 cells using Direct-zol RNA MiniPrep Kit (Zymo, USA) and sequenced as described previously.[62] RNA was ribo-depleted and RNA-seq was performed using high-throughput Illumina sequencing system, Illumina NovaSeq 6000 (Illumina, San Diego, CA, USA) at the UC San Diego IGM Genomics Center. The single-end reads that passed Illumina filters were filtered for reads aligning to transfer RNA, ribosomal RNA, adapter sequences and spike-in controls.
We used Trim Galore (v0.6.5, https://github.com/FelixKrueger/TrimGalore) to trim and filter low-quality reads. After quality control, each FASTQ file was mapped to the human hg38 genome with gene annotation from GENCODE version 33 using STAR (v2.5.3a, https://github.com/alexdobin/STAR).[63] We then used htseq-count (v0.9.1, https://htseq.readthedocs.io/en/master/) to count exonic reads of each BAM file at the gene level and identify pairwise differentially expressed genes (DEG) between three experimental groups (sphere, 3D-stiff, 3D-soft) using R package DESeq2 (v1.24.0) with default settings.[64] Significant DEGs were determined by false discovery rate < 0.05. GO terms were identified using WebGestaltR (v0.4.4, http://www.webgestalt.org/).
Gene set enrichment analysis was performed with the GSEA desktop application (http://software.broadinstitute.org/gsea/downloads.jsp) and gene sets from molecular signatures database.[65] Processed data from primary patient GBM tissues and in vitro cultured GBM cells were derived from Mack et al.[31] and the list of upregulated expressed genes in primary GBM tissue were obtained using python package Scanpy (v1.6.0).[66] DEGs from the primary GBM tissue with adjusted p-value < 0.01 and log2 fold change >5 were selected to generate the input gene set for GSEA. Pathway enrichment bubble plots were generated using the Enrichment Map App of Cytoscape (v3.8.0).[67] Principal component analysis was performed using the top 5,000 DEGs.
4.9. Drug response assessment
Sphere cultured TS576 cells were seeded at a cell density of 1 × 105 cells per well and cultured for 5 days before treatment. Spheres were treated with different dosages of TMZ (Sigma-Aldrich) to generate the IC50 value of the TMZ on the sphere cultured TS576. For comparison of drug sensitivity on TS576 cells in different culture conditions, spheres and 3D-printed samples were cultured for 5 days and treated with 500 μM TMZ. Cell viability was evaluated using CellTiter-Glo 3D after 6 days of TMZ treatment.
4.10. Molecular diffusion assessment
3D constructs with the same material composition and stiffness as the stiff ECM and soft ECM were printed and stabilized overnight. FITC-dextran with a molecular weight 4.4 kDa (Sigma) was prepared at a concentration of 0.5 mg/ml. A FITC-dextran solution was added to 3D-printed samples and incubated at 37 °C. Samples were rinsed and imaged at several time points (5, 15, 30, 60, 120 minutes). Intensity quantification was performed using ImageJ.
4.11. Statistical analysis
The results were presented as mean ± standard deviations. The statistical significance was evaluated using unpaired Student’s t-test, ordinary one-way ANOVA, or two-way ANOVA with GraphPad Prism. *p < 0.05, ** p < 0.01, *** p < 0.001, **** p<0.0001.
4.12. Data deposition
All raw sequencing data reported in this paper has been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (GEO) database, https://www.ncbi.nlm.nih.gov/geo/, at the accession numbers GSE158097. There are no restrictions on data availability, and all data will be made available upon request directed to the corresponding authors.
Supplementary Material
Acknowledgement
We thank members of the Chen and Rana lab for helpful discussions and advices. M.Tang, S.K.T., K.A., M.Tan, J.D. contributed to the conceptualization; M.Tang, S.K.T, T.T., J.T., and X.W. performed the experiments; M.Tang, S.T., and K.A. analyzed data; J.S. performed material synthesis and characterization; S.Y. and Q.X. contributed to the printer and software optimization. M.Tang wrote the manuscript with help from S.K.T. and K.A.. T.R. and S.C. participated in the overall experimental design, data analyses, interpretation, manuscript writing, and obtained funding for the project. We thank Hanqing Liu for helpful suggestions on RNAseq analysis. This publication includes data generated at the UC San Diego IGM Genomics Center utilizing an Illumina NovaSeq 6000 that was purchased with funding from a National Institutes of Health SIG grant (#S10 OD026929). Confocal images were taken at the UCSD School of Medicine Microscopy Core, which is supported by a NINDS P30 grant (NS047101). This work was supported in part by grants from the National Institutes of Health (EB021857, CA253615, CA177322, DA039562, DA046171, and NS118250) and National Science Foundation (1937653). This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE-1650112.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Declaration of Interest. T.M.R. is a founder of ViRx Pharmaceuticals and has an equity interest in the company. S. C. is a Scientific Advisor of Allegro 3D, Inc. and has an equity interest in the company. The terms of these arrangements have been reviewed and approved by the University of California San Diego in accordance with its conflict of interest policies.
Contributor Information
Min Tang, Department of NanoEngineering, University of California San Diego, La Jolla, California 92093, USA..
Shashi Kant Tiwari, Division of Genetics, Department of Pediatrics, Institute for Genomic Medicine, Program in Immunology, University of California San Diego, La Jolla, California 92093, USA..
Kriti Agrawal, Division of Genetics, Department of Pediatrics, Institute for Genomic Medicine, Program in Immunology, University of California San Diego, La Jolla, California 92093, USA..
Matthew Tan, Division of Genetics, Department of Pediatrics, Institute for Genomic Medicine, Program in Immunology, University of California San Diego, La Jolla, California 92093, USA..
Jason Dang, Division of Genetics, Department of Pediatrics, Institute for Genomic Medicine, Program in Immunology, University of California San Diego, La Jolla, California 92093, USA..
Trevor Tam, Department of Bioengineering, University of California San Diego, La Jolla, California 92093, USA..
Jing Tian, Department of Bioengineering, University of California San Diego, La Jolla, California 92093, USA..
Xueyi Wan, Department of Bioengineering, University of California San Diego, La Jolla, California 92093, USA..
Jacob Schimelman, Department of NanoEngineering, University of California San Diego, La Jolla, California 92093, USA..
Shangting You, Department of NanoEngineering, University of California San Diego, La Jolla, California 92093, USA..
Qinghui Xia, Department of NanoEngineering, University of California San Diego, La Jolla, California 92093, USA..
Tariq M. Rana, Division of Genetics, Department of Pediatrics, Institute for Genomic Medicine, Program in Immunology, University of California San Diego, La Jolla, California 92093, USA..
Shaochen Chen, Department of NanoEngineering, University of California San Diego, La Jolla, California 92093, USA.; Department of Bioengineering, University of California San Diego, La Jolla, California 92093, USA.
References
- [1].Koshy M, Villano JL, Dolecek TA, Howard A, Mahmood U, Chmura SJ, Weichselbaum RR, McCarthy BJ, J Neurooncol 2012, 107, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bellail AC, Hunter SB, Brat DJ, Tan C, Van Meir EG, Int. J. Biochem. Cell Biol 2004, 36, 1046. [DOI] [PubMed] [Google Scholar]
- [3].Chen J-WE, Pedron S, Shyu P, Hu Y, Sarkaria JN, Harley BAC, Front. Mater 2018, 5, DOI 10.3389/fmats.2018.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gilg AG, Tye SL, Tolliver LB, Wheeler WG, Visconti RP, Duncan JD, Kostova FV, Bolds LN, Toole BP, Maria BL, Clin. Cancer Res 2008, 14, 1804. [DOI] [PubMed] [Google Scholar]
- [5].Koh I, Cha J, Park J, Choi J, Kang S-G, Kim P, Sci Rep 2018, 8, 4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Butcher DT, Alliston T, Weaver VM, Nature Reviews Cancer 2009, 9, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ahir BK, Engelhard HH, Lakka SS, Mol. Neurobiol 2020, 57, 2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zanotelli MR, Reinhart-King CA, Adv Exp Med Biol 2018, 1092, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Schregel K, Nowicki MO, Palotai M, Nazari N, Zane R, Sinkus R, Lawler SE, Patz S, Cancer Imaging 2020, 20, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tiriac H, Belleau P, Engle DD, Plenker D, Deschênes A, Somerville TDD, Froeling FEM, Burkhart RA, Denroche RE, Jang G-H, Miyabayashi K, Young CM, Patel H, Ma M, LaComb JF, Palmaira RLD, Javed AA, Huynh JC, Johnson M, Arora K, Robine N, Shah M, Sanghvi R, Goetz AB, Lowder CY, Martello L, Driehuis E, LeComte N, Askan G, Iacobuzio-Donahue CA, Clevers H, Wood LD, Hruban RH, Thompson E, Aguirre AJ, Wolpin BM, Sasson A, Kim J, Wu M, Bucobo JC, Allen P, Sejpal DV, Nealon W, Sullivan JD, Winter JM, Gimotty PA, Grem JL, DiMaio DJ, Buscaglia JM, Grandgenett PM, Brody JR, Hollingsworth MA, O’Kane GM, Notta F, Kim E, Crawford JM, Devoe C, Ocean A, Wolfgang CL, Yu KH, Li E, Vakoc CR, Hubert B, Fischer SE, Wilson JM, Moffitt R, Knox J, Krasnitz A, Gallinger S, Tuveson DA, Cancer Discov 2018, 8, 1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lee SH, Hu W, Matulay JT, Silva MV, Owczarek TB, Kim K, Chua CW, Barlow LJ, Kandoth C, Williams AB, Bergren SK, Pietzak EJ, Anderson CB, Benson MC, Coleman JA, Taylor BS, Abate-Shen C, McKiernan JM, Al-Ahmadie H, Solit DB, Shen MM, Cell 2018, 173, 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hubert CG, Rivera M, Spangler LC, Wu Q, Mack SC, Prager BC, Couce M, McLendon RE, Sloan AE, Rich JN, Cancer Res. 2016, 76, 2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Murphy SV, Atala A, Nature Biotechnology 2014, 32, 773. [DOI] [PubMed] [Google Scholar]
- [14].Langer EM, Allen-Petersen BL, King SM, Kendsersky ND, Turnidge MA, Kuziel GM, Riggers R, Samatham R, Amery TS, Jacques SL, Sheppard BC, Korkola JE, Muschler JL, Thibault G, Chang YH, Gray JW, Presnell SC, Nguyen DG, Sears RC, Cell Reports 2019, 26, 608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Tang M, Xie Q, Gimple RC, Zhong Z, Tam T, Tian J, Kidwell RL, Wu Q, Prager BC, Qiu Z, Yu A, Zhu Z, Mesci P, Jing H, Schimelman J, Wang P, Lee D, Lorenzini MH, Dixit D, Zhao L, Bhargava S, Miller TE, Wan X, Tang J, Sun B, Cravatt BF, Muotri AR, Chen S, Rich JN, Cell Res 2020, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yi H-G, Jeong YH, Kim Y, Choi Y-J, Moon HE, Park SH, Kang KS, Bae M, Jang J, Youn H, Paek SH, Cho D-W, Nat Biomed Eng 2019, 3, 509. [DOI] [PubMed] [Google Scholar]
- [17].Wang C, Tong X, Yang F, Molecular Pharmaceutics 2014, 11, 2115. [DOI] [PubMed] [Google Scholar]
- [18].Ananthanarayanan B, Kim Y, Kumar S, Biomaterials 2011, 32, 7913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Xiao W, Ehsanipour A, Sohrabi A, Seidlits SK, J Vis Exp 2018, DOI 10.3791/58176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang T-W, Spector M, Acta Biomaterialia 2009, 5, 2371. [DOI] [PubMed] [Google Scholar]
- [21].Engler AJ, Sen S, Sweeney HL, Discher DE, Cell 2006, 126, 677. [DOI] [PubMed] [Google Scholar]
- [22].Chauvet D, Imbault M, Capelle L, Demene C, Mossad M, Karachi C, Boch A-L, Gennisson J-L, Tanter M, Ultraschall in Med 2015, 37, 584. [DOI] [PubMed] [Google Scholar]
- [23].Ulrich TA, de Juan Pardo EM, Kumar S, Cancer Res. 2009, 69, 4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hribar KC, Finlay D, Ma X, Qu X, Ondeck MG, Chung PH, Zanella F, Engler AJ, Sheikh F, Vuori K, Chen SC, Lab Chip 2015, 15, 2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ma X, Qu X, Zhu W, Li Y-S, Yuan S, Zhang H, Liu J, Wang P, Lai CSE, Zanella F, Feng G-S, Sheikh F, Chien S, Chen S, Proc. Natl. Acad. Sci. U.S.A 2016, 113, 2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Soman P, Tobe BTD, Lee JW, Winquist AM, Singec I, Vecchio KS, Snyder EY, Chen S, Biomed Microdevices 2012, 14, 829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ma X, Yu C, Wang P, Xu W, Wan X, Lai CSE, Liu J, Koroleva-Maharajh A, Chen S, Biomaterials 2018, 185, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yu C, Ma X, Zhu W, Wang P, Miller KL, Stupin J, Koroleva-Maharajh A, Hairabedian A, Chen S, Biomaterials 2019, 194, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Polacheck WJ, Zervantonakis IK, Kamm RD, Cellular and molecular life sciences : CMLS 2013, 70, 1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Netti PA, Baxter LT, Boucher Y, Skalak R, Jain RK, Cancer research 1995, 55, 5451. [PubMed] [Google Scholar]
- [31].Mack SC, Singh I, Wang X, Hirsch R, Wu Q, Villagomez R, Bernatchez JA, Zhu Z, Gimple RC, Kim LJY, Morton A, Lai S, Qiu Z, Prager BC, Bertrand KC, Mah C, Zhou W, Lee C, Barnett GH, Vogelbaum MA, Sloan AE, Chavez L, Bao S, Scacheri PC, Siqueira-Neto JL, Lin CY, Rich JN, J. Exp. Med 2019, 216, 1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang Y, Ma S, Ruzzo WL, Scientific Reports 2020, 10, 3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Nakanishi T, Ohno Y, Aotani R, Maruyama S, Shimada H, Kamo S, Oshima H, Oshima M, Schuetz JD, Tamai I, Scientific Reports 2017, 7, 16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhu Q, Liang X, Dai J, Guan X, Int J Clin Exp Pathol 2015, 8, 9175. [PMC free article] [PubMed] [Google Scholar]
- [35].Hu Y, Ylivinkka I, Chen P, Li L, Hautaniemi S, Nyman TA, Keski-Oja J, Hyytiäinen M, Neoplasia 2012, 14, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Li L, Hu Y, Ylivinkka I, Li H, Chen P, Keski-Oja J, Hyytiäinen M, PLoS ONE 2013, 8, e80363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].do Carmo A, Balça-Silva J, Matias D, Lopes MC, Cancer Biol Ther 2013, 14, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Steponaitis G, Skiriutė D, Kazlauskas A, Golubickaitė I, Stakaitis R, Tamašauskas A, Vaitkienė P, Diagn Pathol 2016, 11, DOI 10.1186/s13000-016-0492-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Liang A, Zhou B, Sun W, Cancer Cell International 2017, 17, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Schmit K, Chen J-W, Ayama-Canden S, Fransolet M, Finet L, Demazy C, D’Hondt L, Graux C, Michiels C, Cell Death & Disease 2019, 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sun W, Qiu G, Zou Y, Cai Z, Wang P, Lin X, Huang J, Jiang L, Ding X, Hu G, Int J Clin Exp Pathol 2015, 8, 12657. [PMC free article] [PubMed] [Google Scholar]
- [42].Weiler M, Blaes J, Pusch S, Sahm F, Czabanka M, Luger S, Bunse L, Solecki G, Eichwald V, Jugold M, Hodecker S, Osswald M, Meisner C, Hielscher T, Rübmann P, Pfenning P-N, Ronellenfitsch M, Kempf T, Schnölzer M, Abdollahi A, Lang F, Bendszus M, von Deimling A, Winkler F, Weller M, Vajkoczy P, Platten M, Wick W, PNAS 2014, 111, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chen X, Pan C, Xu C, Sun Y, Geng Y, Kong L, Xiao X, Zhao Z, Zhou W, Huang L, Song Y, Zhang L, Int. J. Mol. Med 2019, 43, 1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lan Y-L, Wang X, Lou J-C, Ma X-C, Zhang B, Oncotarget 2017, 8, 32345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Silginer M, Nagy S, Happold C, Schneider H, Weller M, Roth P, Neuro Oncol 2017, 19, 1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Brescia P, Ortensi B, Fornasari L, Levi D, Broggi G, Pelicci G, Stem Cells 2013, 31, 857. [DOI] [PubMed] [Google Scholar]
- [47].Han J, Alvarez-Breckenridge CA, Wang Q-E, Yu J, Am J Cancer Res 2015, 5, 945. [PMC free article] [PubMed] [Google Scholar]
- [48].Mao Z, Li X, Ma X, Wang X, Zhang J, Fan X, Medicine (Baltimore) 2019, 98, DOI 10.1097/MD.0000000000014552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhang Y, Xu Y, Li F, Li X, Feng L, Shi X, Wang L, Li X, Oncotarget 2016, 7, 25769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhang K, Xu P, Sowers JL, Machuca DF, Mirfattah B, Herring J, Tang H, Chen Y, Tian B, Brasier AR, Sowers LC, Mol Cell Proteomics 2017, 16, 1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Alieva M, Leidgens V, Riemenschneider MJ, Klein CA, Hau P, van Rheenen J, Scientific Reports 2019, 9, 2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Diao W, Tong X, Yang C, Zhang F, Bao C, Chen H, Liu L, Li M, Ye F, Fan Q, Wang J, Ou-Yang Z-C, Sci Rep 2019, 9, DOI 10.1038/s41598-018-36347-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, Richman AR, Silverbush D, Shaw ML, Hebert CM, Dewitt J, Gritsch S, Perez EM, Gonzalez Castro LN, Lan X, Druck N, Rodman C, Dionne D, Kaplan A, Bertalan MS, Small J, Pelton K, Becker S, Bonal D, Nguyen Q-D, Servis RL, Fung JM, Mylvaganam R, Mayr L, Gojo J, Haberler C, Geyeregger R, Czech T, Slavc I, Nahed BV, Curry WT, Carter BS, Wakimoto H, Brastianos PK, Batchelor TT, Stemmer-Rachamimov A, Martinez-Lage M, Frosch MP, Stamenkovic I, Riggi N, Rheinbay E, Monje M, Rozenblatt-Rosen O, Cahill DP, Patel AP, Hunter T, Verma IM, Ligon KL, Louis DN, Regev A, Bernstein BE, Tirosh I, Suvà ML, Cell 2019, 178, 835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Pauty J, Usuba R, Cheng IG, Hespel L, Takahashi H, Kato K, Kobayashi M, Nakajima H, Lee E, Yger F, Soncin F, Matsunaga YT, EBioMedicine 2018, 27, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ngo MT, Harley BA, Adv. Healthcare Mater 2017, 6, 1700687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lathia JD, Heddleston JM, Venere M, Rich JN, Cell Stem Cell 2011, 8, 482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Pitulescu ME, Schmidt I, Giaimo BD, Antoine T, Berkenfeld F, Ferrante F, Park H, Ehling M, Biljes D, Rocha SF, Langen UH, Stehling M, Nagasawa T, Ferrara N, Borggrefe T, Adams RH, Nat Cell Biol 2017, 19, 915. [DOI] [PubMed] [Google Scholar]
- [58].Leach JB, Bivens KA, Jr CWP, Schmidt CE, Biotechnology and Bioengineering 2003, 82, 578. [DOI] [PubMed] [Google Scholar]
- [59].Shirahama H, Lee BH, Tan LP, Cho N-J, Scientific Reports 2016, 6, 31036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Benitez JA, Ma J, D’Antonio M, Boyer A, Camargo MF, Zanca C, Kelly S, Khodadadi-Jamayran A, Jameson NM, Andersen M, Miletic H, Saberi S, Frazer KA, Cavenee WK, Furnari FB, Nat Commun 2017, 8, 15223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wang S, Zhang Q, Tiwari SK, Lichinchi G, Yau EH, Hui H, Li W, Furnari F, Rana TM, Cell Rep 2020, 30, 969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Zhang Q, Chao T-C, Patil VS, Qin Y, Tiwari SK, Chiou J, Dobin A, Tsai C-M, Li Z, Dang J, Gupta S, Urdahl K, Nizet V, Gingeras TR, Gaulton KJ, Rana TM, The EMBO Journal 2019, 38, e100041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Frankish A, Diekhans M, Ferreira A-M, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, Barnes I, Berry A, Bignell A, Carbonell Sala S, Chrast J, Cunningham F, Di Domenico T, Donaldson S, Fiddes IT, García Girón C, Gonzalez JM, Grego T, Hardy M, Hourlier T, Hunt T, Izuogu OG, Lagarde J, Martin FJ, Martínez L, Mohanan S, Muir P, Navarro FCP, Parker A, Pei B, Pozo F, Ruffier M, Schmitt BM, Stapleton E, Suner M-M, Sycheva I, Uszczynska-Ratajczak B, Xu J, Yates A, Zerbino D, Zhang Y, Aken B, Choudhary JS, Gerstein M, Guigó R, Hubbard TJP, Kellis M, Paten B, Reymond A, Tress ML, Flicek P, Nucleic Acids Res. 2019, 47, D766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Love MI, Huber W, Anders S, Genome Biol. 2014, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Proc. Natl. Acad. Sci. U.S.A 2005, 102, 15545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wolf FA, Angerer P, Theis FJ, Genome Biology 2018, 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Merico D, Isserlin R, Stueker O, Emili A, Bader GD, PLoS ONE 2010, 5, e13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.