

Abstract
Rationale:
Increased myocardial activity of Ca/calmodulin-dependent kinase II (CaMKII) leads to heart failure (HF) and arrhythmias. In Drosophila neurons, interaction of CaMKII with Ca/CaM-dependent serine protein kinase (CASK) has been shown to inhibit CaMKII activity, but the consequences of this regulation for HF and ventricular arrhythmias are unknown.
Objective:
We hypothesize that CASK associates with CaMKII in human and mouse hearts thereby limiting CaMKII activity, and that altering CASK expression in mice changes CaMKII activity accordingly, with functional consequences for contractile function and arrhythmias.
Methods and Results:
Immunoprecipitation revealed that CASK associates with CaMKII in human hearts. CASK expression is unaltered in HF but increased in patients with aortic stenosis. In mice, cardiomyocyte-specific knockout of CASK (CASK-KO) increased CaMKII auto-phosphorylation at the stimulatory T287 site, but reduced phosphorylation at the inhibitory T305/306 site. CASK-KO mice showed increased CaMKII-dependent sarcoplasmic reticulum (SR) Ca leak, reduced SR Ca-content, increased susceptibility to ventricular arrhythmias, greater loss of ejection fraction, and increased mortality after transverse aortic constriction.
Intriguingly, stimulation of the cardiac glucagon-like peptide 1-receptor with exenatide increased CASK expression resulting in increased inhibitory CaMKII T305 phosphorylation, reduced CaMKII activity, and reduced SR Ca leak in WT but not CASK KO.
Conclusions:
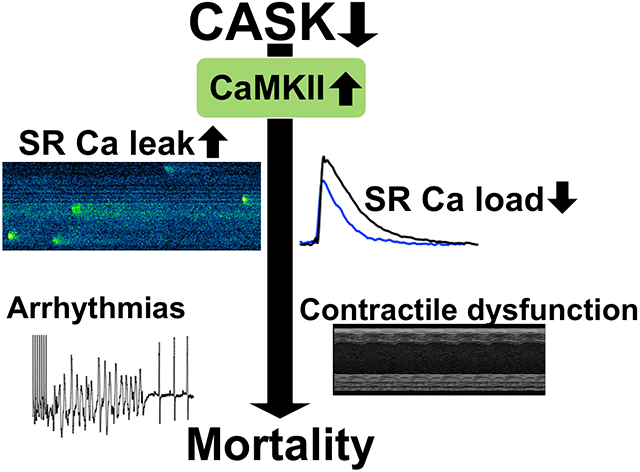
CASK associates with CaMKII in the human heart. CASK-KO in mice increases CaMKII activity, leading to contractile dysfunction and arrhythmias. Increasing CASK expression reduces CaMKII activity, improves Ca handling and contractile function.
Keywords: Ca, calmodulin, CaMKII, CaMKII inhibition, heart failure, SR Ca leak, calcium sparks, calcium transients, basic science
Subject Terms: Calcium cycling/excitation-contraction coupling, Cell Signaling/Signal Transduction, Contractile function, Ion Channels/Membrane Transport, Mechanisms
Graphical Abstract
INTRODUCTION
Despite therapeutic advancements, heart failure (HF) mortality and morbidity remain high. Ca/calmodulin-dependent kinase II (CaMKII) expression and activity is increased in HF, contributing to contractile dysfunction and arrhythmias.1-3
Increased CaMKII-activity results in action potential (AP) prolongation, disturbed Na- and Ca-handling,2,4 and increased diastolic Ca leak from the sarcoplasmic reticulum (SR) contributing to arrhythmias5 and reduced contractility in HF.6
Despite the growing body of evidence about involvement of CaMKII in HF, information is scarce on how CaMKII-activation is regulated. CaMKII-activation requires first the binding of Ca/calmodulin (Ca/CaM) to the regulatory domain,7 resulting in conformational changes that enable access to catalytic and regulatory domain. Consequent intersubunit autophosphorylation at T286 further stimulates CaMKII-activity and renders CaMKII Ca/CaM-autonomous.7 Alternatively, oxidation (at M281/282), O-glycnacylation (at S280) and S-nitrosylation (at C290) can also confer autonomous activity.8-10 Prevention of Ca/CaM-binding to CaMKII, has not only been shown to inhibit CaMKII activity but also prevents these stimulatory posttranslational CaMKII modifications.4 Importantly, the Ca/CaM-CaMKII interaction itself is subject to regulation. It has been shown that phosphorylation at threonine 305/306 (T305/306) in the CaM-binding domain prevents Ca/CaM-binding,11 thereby inhibiting CaMKII-activity. Intriguingly, in neurons, CaMKII T305/306-phosphorylation was shown to be specifically regulated by the membrane associated guanylate kinase (MAGUK) Ca/CaM-dependent serine protein kinase (CASK).12 Unlike other MAGUK-proteins that have primarily scaffolding functions, CASK possesses a CaMKII-homolog domain in the N-terminal region.13 This domain has an unusual catalytic activity, and can only phosphorylate substrates that are recruited by the scaffolding activity.13 In Drosophila neurons, lack of CASK was shown to shift the balance towards increased CaMKII T286-phosphorylation and less inhibitory T305/306-phosphorylation, resulting in enhanced CaMKII-activity.12 Despite previous data reporting the expression of CASK in myocardium,14,15 neither its role for CaMKII-activity, nor its consequences for HF and ventricular arrhythmias have previously been investigated.
We show here that myocardial CASK associates with CaMKII in human myocardium. Moreover, lack of CASK results in increased CaMKII-activity, arrhythmias and contractile dysfunction in mice. In contrast, stimulation of CASK expression by exenatide increased inhibitory CaMKII T305 phosphorylation, reduced CaMKII activity, and reduced SR Ca leak suggesting that CASK and its interaction with CaMKII may be a potential drug target.
METHODS
Data availability.
Data can be made available by the authors upon request.
Study approval.
All experiments were approved by local committees and are in accordance with the Helsinki Declaration. Written consent by patients had been given prior to tissue donation.
All animal procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals and approved by institutional committees.
Human cardiac tissue.
Left ventricular (LV) myocardium was acquired from explanted hearts of heart transplant recipients with end-stage heart failure after informed consent. LV samples of donor hearts that were not eligible for transplantation due to technical limitations served as controls. Additionally, left ventricular samples were acquired from patients with severe aortic stenosis and preserved ejection fraction who underwent cardiac surgery and valve replacement. All samples were from patients with a caucasian background. After explantation, hearts were stored in a cardioprotective solution containing (in mmol/L): 152 Na+, 3.6 K+, 135 Cl−, 25 HCO3−, 0.6 Mg2+, 1.3 H2PO4−, 0.6 SO42−, 2.5 Ca2+, 11.2 glucose, 10 2,3-butanedione monoxime (BDM), oxygenated with 95% O2 and 5% CO2 at 4°C.
CASK knock-out mice.
The knock-out of cardiac CASK was achieved in transgenic mice using the previously described Cre-Lox-recombination method 16,17 and the α-myosin heavy chain promotor (α-MHC).18 We used mutant mice, in which the first coding exon of the CASK gene is flanked by loxP sites (CASKtm1sud). These mice were kindly provided by the Südhof lab.19 The cardiomyocyte-specific knockout of CASK (CASK-KO) was accomplished by crossing these CASK-FLOX mice with mutant mice expressing Cre-recombinase (heterozygous) under control of the α-MCH promotor18 (The Jackson Laboratory). Atasoy et al. suggested that changes in the CASK gene introduced by insertion of loxP sites and/or the neomycin resistance gene cassette may impair neuronal CASK expression. They found a reduction in CASK expression to 33% in brain homogenates from newborn CASK-Flox mice.19 On the other hand, off-target effects of Cre recombinase 20,21 are also an important consideration. Therefore, mice with heterozygous Cre-expression but wild-type CASK were used as control (CASK-CTRL), but for key experiments we also tested mice with homozygously floxed CASK, but without expression of Cre-recombinase (CASK-Flox). To obtain a homogenous background, mice homozygous for the floxed CASK gene were bred with mice heterozygous for Cre recombinase (controlled by the α-MCH promotor), and litters were back-crossed with CASK flox mice for four generations before being used for experiments. The following mating scheme was implemented to optimize the number of littermates used for experiments: Breeding pairs consisted of a mate with heterozygous expression of floxed CASK and heterozygous expression of Cre recombinase and a mate with heterozygous expression of floxed CASK and WT (null) expression of Cre recombinase. However, since such a breeding pair generates only a limited the number of the required littermates CASK-KO (homozygous for floxed CASK, heterozygous for Cre), CASK-CTRL (WT for CASK, heterozygous for Cre), and CASK-Flox (homozygous for floxed CASK and null for Cre), we have also used the litter from multiple pairs of mice bred in parallel. Genotyping was done by PCR. Animals were on standard chow and bedding. Since female CASK-KO mice have been shown not to thrive well,19 only male mice were used for experiments. Moreover, the use of both sexes may have caused huge variation in jugular vein diameters compromising the in vivo catheter studies. Baseline survival was monitored for all mice. Some mice were randomly sacrificed for cell isolation or protein analysis studies. As such, those mice cannot be included attributed to having died spontaneously but do explain the decreasing census at older ages (figure 2B). All animal procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee.
Figure 2: CASK-KO mice have no cardiac structural or functional abnormalities.

A) Original M-mode acquisitions (left panel, parasternal long axis view) from anesthetized CASK-CTRL (CTRL), CASK-Flox (Flox) and CASK-KO (KO) mice. Upper right panel shows mean data of left ventricular ejection fraction and left ventricular end diastolic diameter. Data follows normal distribution (Shapiro-Wilk-test). No significant differences could be detected (One way ANOVA with Tukey’s post-test). EF: ANOVA p=0.8668. LVEDD: ANOVA p=0.9447. B) Kaplan-Meier survival analysis for CASK-CTRL, CASK-Flox, and CASK-KO mice shows no increased mortality. Log-rank (Mantel-Cox) p=0.6462. n=mice at risk. C) Western blots and mean data of CaMKII T286-phosphorylation (normalized to CaMKII-expression; data are normally distributed [Kolmogorov-Smirnov-test], one way ANOVA *p=0.0293), phospholamban T17-phosphorylation (normal distribution [D’Agostino-Pearson test], one way ANOVA *p=0.0052), and CaMKII T305-phosphorylation (normal distribution [Shapiro-Wilk-test], one way ANOVA p=0.2758) in ventricular homogenates. For multiple comparisons within each data set, Holm-Sidak post-tests were applied (p in figures). For pT286/CaMKII ratio, one extreme statistical outlier was removed from the CASK-Flox group after statistical outlier analysis.
Animal model of heart failure.
Heart failure was induced in 12±2 week-old mice by transverse aortic constriction. Mice were anesthetized using intraperitoneal injections of medetomidine (0.5 mg/kg), midazolam (5 mg/kg) and fentanyl 0.05 mg/kg body weight). A horizontal incision (1-1.5 cm) at the suprasternal notch was used to display the transversal aorta. A 27 gauge needle was tied against the aorta using a 6-0 non-absorbable suture. After removal of the 27 gauge needle, the skin was closed and the mice were kept on a heating plate (37° C) until recovery from anesthesia. Sham animals underwent the same procedure except for banding of the transversal aorta. Allocation to experimental groups (TAC vs. Sham) was done on a random basis; littermates of equal age and sex were used to avoid selection bias; investigator was not aware of the genotypes. At the end of the surgery, anesthesia was antagonized using intraperitoneal injections of atipamezol (2.5 mg/kg), flumazenil (0.5 mg/kg) and buprenorphine (0.1 mg/kg body weight). For analgesia, metamizole (1.33 mg/ml) was added to the drinking water 2 days before surgery and supplied for 7 days after operation. In addition, buprenorphine (60 μg/kg body weight) was administered s.c. 1 hour before surgery. Animals that died during surgery or within 48 h were excluded from the study (not reported; on average 15% of all operated mice for TAC and below 5% for Sham procedure). In case of disturbed wound-healing, animals were killed at the discretion of the veterinarian and excluded from the study. At the end of the experiments, mice were sacrificed under isoflurane anesthesia (5%) by cervical dislocation.
For the in vivo experiments with GLP1-receptor agonist treatment, all experiments were approved by the government of Nordrhein-Westfalen (Germany). Male C57Bl/6J mice, 5 weeks of age were obtained from Charles River Laboratory and placed in a 12 h day-night cycle with unlimited supply of food and water in the animal facility of the University Hospital of the RWTH Aachen University. After a one-week adaptation to our facility, mice received 5×1012 particles of AAV-CMV-GLP-1 (7-37) or AAV-CMV-LacZ as control. AAV were generated as previously reported22. Two weeks after vector injection mice underwent transverse aortic constriction (TAC) surgery to induce cardiac hypertrophy. Mice were anesthetized with Ketamin/Xylazin and temgesic was used for analgesia. After intubation mice were ventilated and chest was opened at second intercostal space at the left upper sternal border through a small incision and aortic constriction was performed by tying a ligature against a 27G needle. Control mice underwent a sham operation in which the thread was placed, but not tied. Mice were sacrificed 5 weeks after TAC surgery.
Mouse echocardiography.
Transthoracic echocardiography was performed by blinded investigators using a Vevo2100 or 3100 (VisualSonics, Toronto, Canada) system with a 30 MHz center frequency transducer as described previously.23 The animals were anesthetized with isoflurane (induction with 3%, maintenance with 1.5%), while temperature, respiration, and ECG were continuously monitored. Two-dimensional cine loops with frame rates of >200 frames/s of a long axis view and a short axis view at mid-level of the papillary muscles as well as M-mode loops of the short axis view were recorded. Thicknesses of the septum, the anterior and posterior myocardial wall, the inner diameter of the left ventricle (LVEDD) and the area of the left ventricular cavity were measured in systole (sys) and diastole (dia) from the short axis view according to standard procedures. Maximal left ventricular length was measured from the long axis view. Systolic and diastolic left ventricular volumes were calculated using the area-length method 2 and the ejection fraction was derived.
Electrophysiological studies (EP) in mice.
Mice were anesthetized using intraperitoneal injections of medetomidine (0.5 mg/kg), midazolam (5 mg/kg) and fentanyl 0.05 mg/kg body weight) by blinded investigators. During EP studies, body temperature was monitored by a rectal probe and controlled using a mousepad circuit board equipped with a heating element (Mousepad, THM 100, Indus Instruments, USA). All studies were performed at 37±0.5°C. We used a Millar 1.1F octapolar EP catheter (EPR-800; Millar Instruments) inserted via the right jugular vein, as previously described 24. A computer-based data acquisition system (Powerlab 16/35; ADI Instruments) was used to record body surface ECG (lead II) and 4 intra-cardiac bipolar electrograms (Labchart Pro software, version 7; AD Instruments). QT interval was corrected for heart rate (QTc) using Mitchell’s formula.25 Right ventricular pacing was performed using 2 ms current pulses delivered by an external stimulator (STG-3008FA; Multi Channel Systems). Ventricular capture was confirmed by ventricular pacing prior to the arrhythmia protocol. Mice without ventricular capture were excluded from the study. For some experiments, mice were treated with the novel CaMKII-inhibitor RA608.26 For oral administration of RA608, a stock solution of 15 mg/ml was generated once every day and kept at 4°C until used. 15 mg of the compound was solubilized in 800 μl of an acidic cyclodextrin (captisol) and HCl mixture (10% in HCl 0.1 N). Thereafter, the pH was adjusted to 4 with NaOH and the final volume of 1 ml was adjusted by addition of neutral cyclodextrin (10% in H2O). The gavage mass for in vivo experiments was 30 mg/kg body weight and the inhibitor was applied once <4h prior to the experiment.
Inducibility of ventricular arrhythmias was tested by decremental burst pacing. Burst pacing started at a 40 ms cycle length, decreasing by 2 ms every 2 s to a cycle length of 20 ms. Burst pacing was repeated one min after the previous burst concluded or the termination of arrhythmias. Pacing was performed five times in each mouse. Ventricular arrhythmias were defined as rapid ventricular potentials that occurred independently from atrial potentials and displayed altered QRS morphology. Ventricular arrhythmias were considered significant if their duration was longer than one sec. A second train of burst pacing was started 3 min after intraperitoneal injection of isoproterenol (2 mg/kg body weight). At the end of the experiments, mice were killed by cervical dislocation under anesthesia.
Murine ventricular cardiomyocyte isolation.
Murine ventricular cardiomyocyte isolation was performed as described previously27. In brief, mice were anaesthetized with isoflurane. After death by cervical dislocation, hearts were quickly excised, mounted on a Langendorff perfusion apparatus and retrogradely perfused with nominally Ca-free solution containing (in mmol/l) NaCl 113, KCl 4.7, KH2PO4 0.6, Na2HPO4 x 2 H2O 0.6, MgSO4 x 7 H2O 1.2, NaHCO3 12, KHCO3 10, HEPES 10, taurine 30, BDM 10, glucose 5.5, phenol-red 0.032 for 4 min at 37°C (pH 7.4). Then, 7.5 mg/ml liberase TM (Roche diagnostics, Mannheim, Germany), trypsin 0.6%, and 0.125 mmol/l CaCl2 were added to the perfusion solution. Perfusion was continued for 3-4 min until the heart became flaccid. Ventricular tissue was collected in perfusion buffer supplemented with 5% bovine calf serum, cut into small pieces, and dispersed by repeatedly pipetting until no solid cardiac tissue was left. Ca-reintroduction was performed by stepwise increasing [Ca] from 0.1 to 1.0 mmol/l.
Measurement of Ca sparks via confocal microscopy.
Ca sparks were assessed via confocal microscopy. Isolated ventricular myocytes were loaded with 10 μmol/L fluo-4 acetoxymethylester (Molecular Probes; for 12 min at room temperature) and mounted on an inverted laser scanning confocal microscope (Zeiss Pascal 5). Regular myocyte contraction was elicited by electrical field stimulation (1 Hz). For measurements of Ca sparks, line scans (512 pixel of 0.075 μm size, 1309 lines per second, 10000 lines per scan, 488 nm excitation, 505 nm long pass emission filter) were acquired immediately after stop of electrical field stimulation. Ca spark characteristics were analyzed using Image J (Sparkmaster plugin).28 Ca spark frequency (CaSpF) was measured as number of sparks per cell volume and time (100 μm−1s−1). Ca spark width and duration were taken from the full-width-half-maximum (FWHM) and full-duration-half-maximum (FDHM), respectively. The diastolic Ca leak was calculated as follows: CaSpF x Ca spark amplitude x FWHM x FDHM.28
Epifluorescence experiments.
Intracellular Ca ([Ca]i) concentrations were measured as described previously.2,29 Briefly, myocytes on laminin-coated recording chambers were loaded with 10 μmol/L Fluo-4-AM in the presence of 0.02% (w/v) pluronic acid (Molecular Probes, Eugene, OR) for 12 min, , at room temperature in the dark. The chambers were mounted on the stage of an inverted microscope (Nikon Eclipse TE2000-U) and superfused with Tyrode solution (37°C) containing (in mmol/L) 140 NaCl, 4 KCl, 1 MgCl2, 5 HEPES, 10 glucose, 1 CaCl2, pH 7.4. After 10 min of washing out the external dye, myocytes were electrical field-stimulated (voltage 25% above threshold) at various basic cycle lengths. Intracellular Fluo-4 was excited at 480±15 nm, emitted fluorescence was detected at 535±20 nm.30 All fluorescence emission was recorded using IonWizard software (IonOptix Corporation, Boston, MA). Background fluorescence was subtracted, the ratio F/F0 was calculated. A CCD camera was also used to simultaneously record sarcomere length as a function of time. For each pacing frequency, both Ca transients and cell contractions were averaged (typically 10 traces) during steady-state and analyzed using IonWizard software. For some experiments, caffeine (10 mmol/L) was applied. To investigate the effect of β-receptor stimulation, myocytes were exposed to isoproterenol (1 μmol/L) for 5 min. Cellular arrhythmias were categorized according to a scoring system developed by Wu et al 31, which was modified by our group as described previously4. In short, early non-stimulated Ca release events (ENSE), delayed non-stimulated Ca release events (DNSE), doublets or triplets, runs or alternans, and sustained premature contractions were counted and the cell was assigned a point value for each of these arrhythmia categories (1 for no arrhythmia of this category, 2 for ENSE or DNSE, 3 for doublets, triplets or runs or alternans, and 4 for sustained premature contractions). The point value from each of these arrhythmia categories was then multiplied, and the final score subtracted by 1 to remove non-arrhythmogenic cells (as these would have a total point value of 1). The resulting score was then normalized to observation time.
Co-Immunoprecipitation.
LV myocardium was mechanically homogenized (using a stainless-steel pestle) in Tris buffer containing (in mmol/L): 50 Tris-HCl, 200 NaCl, 20 NaF, 1 Na3VO4, 1 DTT [pH 7.4] and protease inhibitor cocktail. Protein concentration was determined by BCA assay. The homogenate (1 mg protein) was suspended in dilution medium containing (in mmol/L): 50 Tris-HCl, 154 NaCl, 1% CHAPS, 1 NaF, 1 Na3VO4 [pH 7.4] and protease inhibitor cocktail. CASK, NaV1.5 or CaMKII, respectively, were immunoprecipitated with rabbit polyclonal anti-CASK (1.4 mg/ml, Abcam, discontinued, AB11343), rabbit polyclonal anti-NaV1.5 (1 mg/ml, Alomone, ASC-013) or rabbit polyclonal anti-CaMKII antibody (1 mg/ml32, Don Bers' lab) by preincubation at 4°C overnight in protein G–sepharose Fast Flow (prewashed; 2 hours, 4°C; Amersham Biosciences). As control, rabbit polyclonal anti-calsequestrin (1 mg/ml, Thermo Scientific) was used. After centrifugation, pellets were washed with Tris buffer containing (in mmol/L): 50 Tris-HCl, 154 NaCl [pH 7.4], and immunoprecipitated proteins were eluted in 2X Laemmli sample buffer containing 4% β-mercaptoethanol (30 minutes, 37°C) followed by centrifugation. Supernatants were subjected to Western blotting.
Western blot analysis.
Cell lysates or whole-heart homogenates were used. After denaturation (for analysis of CaMKII for 5 min at 95°C, all other proteins for 30 min at 37°C in 2% β-mercaptoethanol), proteins were separated on 5% (RyR2 [Sigma, HPA020028], S2814 [Badrilla, A010-31AP]), 8% (CaMKII [Don Bers' lab], ox-CaMKII [Mark Anderson's lab], pT286 [Thermo Scientific, MA1-047) pT305 [Antibodies online, discontinued], SERCA [Thermo Scientific, MA3-919], CASK [StressMarq, discontinued, S56A-50] Nav1.5 [Alomone, ASC-013]), or 13% (phospholamban [Milipore, discontinued, 05205], pT17 [Badrilla, A010-13AP]) SDS-polyacrylamide gels, then transferred to a nitrocellulose membrane (or PDVF membrane) and incubated with primary antibodies: mouse monoclonal anti-CASK (1:2000), rabbit polyclonal anti-CaMKII (1:1200033), mouse monoclonal anti-phospho-threonin-286-CaMKII (pT286, 1:1000), polyclonal rabbit anti-phospho-threonin305-CaMKII (1:1000), rabbit polyclonal anti-phospho-threonin17-PLN (1:10000), mouse monoclonal anti-PLN (1:10000), rabbit polyclonal anti-RyR2 (1:10000), rabbit polyclonal anti-phospho-serin2814-RyR (1:10000), mouse monoclonal anti-SERCA (1:20000), and mouse monoclonal anti-GAPDH (1:50000, BIOTREND [discontinued, BT46-9995-55]) at 4°C overnight. Secondary antibodies were HRP-conjugated donkey anti-rabbit and sheep anti-mouse IgG (1:10000, GE Healthcare, NA934 and NA931) that were incubated for 1 h at room temperature. For chemiluminescent detection, Immobilon™ Western Chemiluminescent HRP Substrate (Millipore) was used.
For detection of oxidized CaMKII, an immune serum directed against oxizided M281/M282 (ox-CaMKII) was used (polyclonal rabbit, 1:1500034, Mark Anderson's lab). To avoid unspecific reduction of oxidized CaMKII, β-mercaptoethanol was omitted during the denaturation process (5 min at 95°C). For some experiments, cardiomyocytes were exposed to isoproterenol at 1 μmol/L for 10 min.
Statistics.
All data were analyzed by blinded investigators. Data are expressed as mean or mean ± s.e.m. Normality of data was tested by hierarchical application of D'Agostino-Pearson, Shapiro-Wilk, or Kolmogorov-Smirnov test, respectively, depending on the number of experiments. For statistical analysis of the arrhythmia score in figure 4A, a Box-Cox transformation was applied prior to normality and statistical testing. A power analysis was performed for the TAC experiments with the primary outcome left ventricular ejection fraction. For this, the significance level α was taken to p<0.05 (two-tailed) with a power of 0.8. We assumed an EF of 45±12 (SD) in the CASK-CTRL and 30±8 in the CASK-KO group two weeks after TAC (Cohen's d 1.78). This yielded a group size of 9 animals per group.
Figure 4: CASK-KO mice show disturbed Ca handling and ventricular arrhythmias in vivo.

A) Original trace of Fluo-4 fluorescence in an isolated ventricular myocyte of a CASK KO mouse. Arrows indicate electrical field-stimulation. The left panel displays a burst of Ca release events followed by a single delayed non-stimulated event (DNSE). In the right panel, data for scored arrhythmias normalized to observation time are shown. Data were normally distributed (D’Agostino-Pearson) and analyzed after Box-Cox transformation. Two-way repeated measured mixed-effects analysis with two-stage linear step-up post-test procedure of Benjamini, Krieger and Yekutieli was performed (n=cells). In the lower left panel, the proportions of cells showing DNSE upon isoproterenol are shown. Lower mid panel shows the proportions of cells with Ca alternans or runs upon isoproterenol. Lower right panel shows the proportions of cells with severe arrhythmias upon isoproterenol (defined as either doublets, triplets, runs, alternans or sustained premature contractions, Fisher’s exact tests, n=cells). B) Original surface ECG acquisitions (lead II) in mice subjected to burst stimulation at the right ventricular apex in vivo. C) Mean data of the proportion of mice showing arrhythmias (Fisher’s exact test, n=mice). Label information: CTRL=CASK-CTRL, Flox=CASK-Flox, KO=CASK-KO
For longitudinal data, 2-way repeated-measures ANOVA or mixed-effects analysis was run; where appropriate, one-way ANOVA with multiple comparison test was used. For categorical data, Fisher's exact test was used. Otherwise, Student’s unpaired t-test was applied. Two-sided P<0.05 was considered significant. The respective post-tests are mentioned in the figure legends to improve legibility. Statistics were performed using Graph Pad Prism 9.
RESULTS
CASK is expressed in human heart and associates with CaMKII.
To test if CASK is expressed in human ventricular myocardium, we collected samples of left ventricular myocardium from explanted human hearts of patients with end-stage HF or non-failing (NF) donors not eligible for transplantation, as well as patients with aortic stenosis (AS) undergoing valve replacement. Clinical data can be found in table 1 (no data is available for the NF donors).
Table 1: Clinical data of patients:
LVAD=left ventricular assist device, ACE/AT1-RA/ARNI= Angiotensin-converting enzyme inhibitor/angiotensin receptor antagonist/angiotensin receptor neprilysin inhibitor, MRA=mineralocorticoid receptor antagonist, N/A=not available/not applicable
| HF (n=18) |
AS (n=5) |
|
|---|---|---|
| Male gender, n (%) | 14 (78) | 1 (20) |
| Age (years), mean±SD | 49.9±10.7 | 73.8±9.7 |
| Body mass index (kg/m2), mean±SD | 24.3±4.8 | N/A |
| Left ventricular ejection fraction (%),mean±SD | 18.6±5.8 | 57.0±6.7 |
| Dilative cardiomyopathy, n (%) | 17 (94) | N/A |
| Ischemic cardiomyopathy, n (%) | 1 (6) | N/A |
| LVAD, n (%) | 5 (28) | 0 (0) |
| Atrial fibrillation, n (%) | 9 (56)* | N/A |
| Diabetes mellitus, n (%) | 2 (11) | N/A |
| Creatinine (mg/dl), mean±SD | 1.3±0.5X | N/A |
| Pharmacological Therapy: | ||
| ACE/AT1-RA/ARNI, n (%) | 12 (67) | 4 (80) |
| MRA, n (%) | 12 (67) | 0 (0) |
| Loop diuretics, n (%) | 17 (94) | 3 (60) |
| Beta blocker, n (%) | 14 (78) | 4 (80) |
| Ca antagonist, n (%) | 4 (22) | 0 (0) |
| Statin, n (%) | 3 (17) | N/A |
data from n=16 available,
data from n=12 available
Figure 1A shows that CASK is strongly expressed in human ventricle with no difference between NF and HF (fig. 1A). In contrast, a significant increase in CASK expression was found in hypertrophied LV-myocardium of patients with AS and preserved ejection fraction (EF, fig. 1A, table 1). To further investigate the interaction of CASK with CaMKII in human ventricle, we immunoprecipitated CaMKII and measured CASK-expression (and vice versa). Co-immunoprecipitation analysis revealed a strong interaction of CaMKII with CASK in both NF and HF (fig. 1B). Since the magnitude of sepharose bead binding by a specific antibody cannot be exactly controlled, these results should only be interpreted in a qualitative manner. We also found an association of CASK with the cardiac isoform of voltage-gated Na-channels (NaV1.5, fig. 1B), which we have shown to be regulated by CaMKII.2
Figure 1: CASK expression is regulated in hypertrophy and HF.
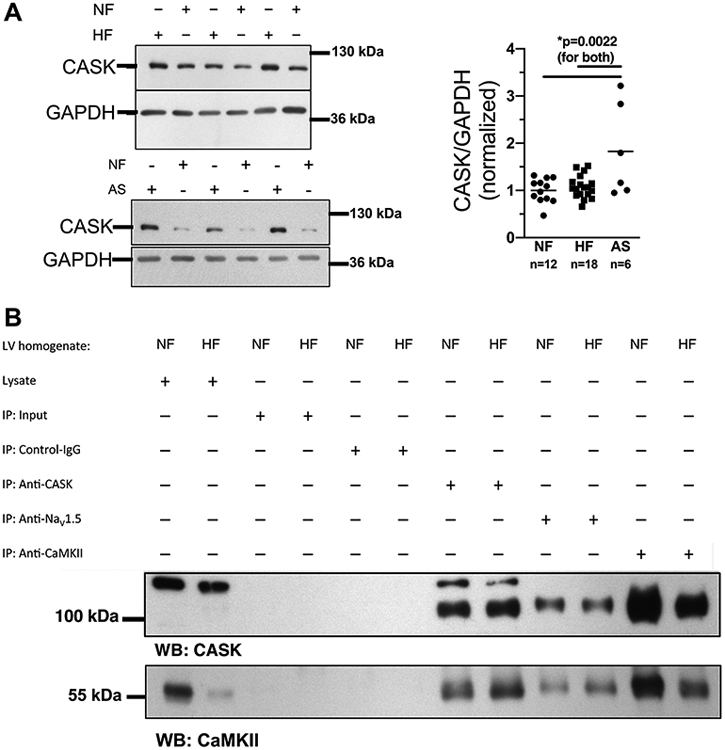
A) Western blots (left panels) and mean analysis (right panel) of CASK expression in human left ventricular (LV) myocardium from patients with end-stage heart failure (HF) or patients with aortic stenosis (AS) in comparison to LV samples from healthy organ donor hearts (NF). CASK expression is increased in AS compared to HF and NF. Data are normally distributed (Shapiro-Wilk-test). *One way ANOVA p=0.0017, Holm-Sidak post-test (p in figure, n=mice). B) Original blots analyzing CASK (upper panel) and CaMKII (lower panel) expression in immunoprecipitated proteins CASK (anti-CASK), NaV1.5 (anti-NaV1.5) and CaMKII (anti-CaMKII) from homogenates of HF vs. NF.
CASK-KO mice display an unaltered ejection fraction at rest.
To identify the specific role of CASK for CaMKII regulation in the heart, we employed a cardiomyocyte-specific CASK-knockout mouse model (CASK-KO). As control, we used mice with wild-type CASK-alleles and cardiomyocyte-specific expression of Cre-recombinase (CASK-CTRL), and for key experiments also homozygous floxed mice without expression of Cre-recombinase (CASK-Flox). CASK-expression was similar in ventricular cardiomyocytes of CASK-Flox and CASK-CTRL mice but abolished in CASK-KO (Online Fig. I A). We have no evidence that the floxed allele affects the heart per se, since the slightly smaller heart weight found in these mice (HW, Online Fig. I B) was proportionate and HW/BW-ratios were not different (Online Fig. I D).
Echocardiography was performed in 12-week old mice to investigate contractile function at rest (fig. 2A, Table 1). Compared to CASK-CTRL and CASK-Flox, there was no difference in left ventricular ejection fraction (EF) or left ventricular end diastolic internal diameter (LVEDD) in CASK-KO mice (fig. 2A). Behavior and motor activity of CASK-KO mice were not discernable from their littermate controls and long-term survival of CASK-KO mice was also not different (fig. 2B).
To test, if lack of CASK-expression affects CaMKII-activity, stimulatory CaMKII-autophosphorylation at threonine 286 (pT286-CaMKII) was assessed. CASK-KO mice showed an increase in myocardial T286-phosphorylation compared to CASK-CTRL and CASK-Flox (Fig. 2C) while CaMKII-expression was not different (Online Fig. I E).
We assessed phospholamban (PLN)-phosphorylation at threonine 17 (pT17), which is an established marker of CaMKII-activity35. In accordance with T286-phosphorylation, CaMKII-dependent PLN-phosphorylation was significantly increased in CASK-KO compared to both CASK-CTRL and CASK-Flox (Fig. 2C).
To test if counterregulatory pathways limiting CaMKII-activity are already activated at baseline, we measured inhibitory T305-autophosphorylation (pT305-CaMKII), which has been shown to be mediated by CASK in Drosophila neurons 12, thereby limiting CaMKII-activity. However, as CaMKII-dependent dysregulation of Ca-handling is most relevant under pathophysiological conditions like increased afterload36 and increased β-adrenergic stimulation,37,38 we were not surprised to find no significant difference in inhibitory T305 CaMKII-phosphorylation (Fig. 2C), suggesting that CaMKII-inhibitory pathways may not play a significant role at rest, i.e. under normal physiological conditions without catecholaminergic or afterload stress. We thus proceeded with experiments investigating cellular response to β-adrenergic stimulation with isoproterenol (ISO).
CASK-KO mice show disturbed Ca handling upon β-adrenergic stimulation.
Acute exposure of isolated ventricular myocytes to ISO increased CaMKII autophosphorylation at T286 and reduced inhibitory CaMKII-autophosphorylation at T305. Importantly, ISO-dependent stimulation of CaMKII-activity was significantly greater in CASK KO compared to CASK-CTRL (Fig. 3A). CaMKII-expression, again, was unaltered by ISO (Online Fig. I F). Frequency of Ca sparks are known to be increased by CaMKII39, and in line with the increased CaMKII-activity, CaMKII-dependent RyR2-phosphorylation was strongly increased in CASK-KO (Fig. 3B). Consistently, compared to CASK-CTRL (or CASK-Flox), diastolic SR Ca spark frequency was significantly more increased in CASK-KO myocytes upon ISO (Fig. 3C). As increased Ca sparks can limit SR Ca content1, we measured SR Ca content in isolated cardiomyocytes using caffeine-induced Ca transients. At baseline, SR Ca content was already significantly decreased in CASK-KO vs. CASK-CTRL (or Flox, Fig. 3C, right panel). Exposure to ISO, clearly augmented SR Ca content in CASK-KO, but it was still significantly lower than that for CASK-CTRL with ISO. Consequently, systolic Ca-transient amplitudes, which are critical for systolic contraction, were smaller in CASK-KO myocytes and this reduction was maintained upon ISO (Fig. 3D).
Figure 3: CASK-KO mice show increased Ca leak.

A) Western blots and mean data of CaMKII T286 (pT286) phosphorylation (normalized to CaMKII-expression), and CaMKII T305-phosphorylation in ventricular homogenates from mice that were harvested 30 min after intraperitoneal injection of ISO (2 mg/kg body weight). Left-panel: data are not normally distributed (D’Agostino-Pearson test) and Mann-Whitney-Test was applied; right panel: data are normally distributed (Shapiro-Wilk-test) and Student’s unpaired t-test was applied. n=mice. B) Western blots and mean data of RyR2 serine 2814 (pS2814) phosphorylation (normalized to RyR2-expression) in ventricular homogenates from mice that were harvested 30 min after intraperitoneal injection of ISO (2 mg/kg body weight). Data are normally distributed (Shapiro-Wilk test) and unpaired t-test applied, n=mice. C) Left panel: original line scans analyzing elementary SR Ca release events (Ca sparks) in resting Fluo-4-loaded ventricular myocytes. Center panel: violin plot of SR Ca spark frequency. n=cells. Data are not normally distributed (D’Agostino-Pearson test), and Kruskal-Wallis-test (p<0.0001) with Dunn’s post-test (p in graph) was applied. Right panel: mean data for Caffeine transient amplitude. Data are normally distributed (Shapiro-Wilk-test) and two-way ANOVA was applied (p for substance p<0.0001 and for genotype p<0.0243). Multiple comparisons were done by Newman-Keuls post-test (p in graph). n=cells. D) Ca transient amplitude at 1 Hz before and after exposure to ISO (10-7 mol/l) in Fluo-4 –loaded isolated ventricular myocytes. Data are not normally distributed (D’Agostino-Pearson test) and *Kruskal-Wallis-test (p<0.0001) was applied. Multiple comparison by Dunn’s post-test (p in graph). n=cells.
Ca export pathways (SERCA and NCX) may partly compensate for the higher SR Ca leak, which is consistent with our finding of unaltered SERCA expression levels (Online Fig. II B), but enhanced T17 phosphorylation of PLN (Fig. 2C). Nevertheless, since SR Ca content was significantly reduced, this compensation is incomplete.
Disturbed Ca-handling may also lead to arrhythmias in vitro and in vivo. Thus, we assessed the propensity for non-stimulated Ca transients, which was normalized to observation time. At baseline, the severity and frequency of non-stimulated cellular arrhythmias was not different between CASK-KO and CASK-CTRL (or CASK-Flox; Figure 4A), but the incidence of arrhythmic events was generally low. Exposure to ISO increased the propensity for arrhythmias in CASK-CTRL (and CASK-Flox) significantly compared to baseline, but even more in CASK-KO myocytes (Figure 4A, upper right panel). Upon ISO, the proportions of CASK-KO myocytes that showed delayed non-stimulated events, alternans or runs, and severe overall arrhythmias (defined as either doublets, triplets, alternans, runs or sustained premature ventricular contractions) were significantly increased (Figure 4A, lower panels).
Figure 4B shows an original ECG of a ventricular tachycardia induced by right ventricular burst stimulation in CASK-KO in vivo. Compared to CASK-CTRL (and also CASK-Flox), the propensity for these life-threatening ventricular arrhythmias was strongly increased in CASK-KO mice (Figure 4C), consistent with arrhythmogenic SR Ca release (fig 3 and 4A). Intriguingly, CASK KO mice also showed an increased propensity for spontaneous unprovoked arrhythmias (premature ventricular contractions or runs) at baseline (Online Fig. III B). CASK-KO mice also displayed prolonged heart rate-corrected QT intervals compared to CASK-CTRL and CASK-Flox (Online Fig. III A+C, Online table II) consistent with work showing CaMKII-dependent APD prolongation.2 Interestingly, oral gavage of CASK-KO mice with the novel orally applicable CaMKII-inhibitor RA608 (30 mg/kg body weight) completely abolished inducibility of ventricular arrhythmias in these mice, further underscoring the dysregulation of CaMKII in CASK-KO (Figure 4C). RA608 also significantly reduced QTc in CASK-KO upon ISO to values comparable with CASK-CTRL upon ISO (Online Fig. III A+C).
CASK-KO accelerates afterload-induced heart failure development.
To test if CASK-dependent CaMKII-regulation is important for HF development, we subjected mice to pressure-overload by transverse aortic constriction (TAC, Fig. 5, Online table III). Two weeks after TAC, control mice (CASK-CTRL/CASK-Flox) developed left ventricular hypertrophy, but showed unaltered EF (Fig. 5A and B, Online table III). In contrast, left ventricular function was significantly reduced in mice lacking CASK two weeks after TAC (fig. 5A and B, Online table III). Importantly, this HF-development was clearly enhanced in CASK-KO mice.
Figure 5: CASK-KO accelerates heart failure progression and increases mortality upon TAC.

A) Original traces of echocardiographic M-mode acquisitions (parasternal long axis view) in CASK-CTRL or CASK-KO mice before TAC, and at two weeks after TAC operation. B) Mean data of left ventricular ejection fraction (EF). Data follows normally distribution (Shapiro-Wilk-test, *two-way repeated measured mixed-effects analysis p for TAC=0.0459 with Holm-Sidak post-test (p values in graph, n=mice). C) Kaplan-Meier survival analysis for CASK-CTRL, CASK-Flox and CASK-KO mice upon TAC. Data were tested by Log-rank (Mantel-Cox) with p in graph. n=mice at risk. D) Mean densitometric expression of CASK (normalized to GAPDH, western blot) in left ventricular homogenates of mice after TAC (vs. Sham-operated mice). Data are normally distributed (Shapiro-Wilk-test) and tested by unpaired t-test (p in graph, n=mice. E) Original registrations (western blot) and mean densitometric data for CaMKII expression and T286 autophosphorylation in ventricular homogenates from CASK-CTRL and KO mice 2 weeks (2w) after TAC. Left panel: Data are not normally distributed (Shapiro-Wilk) and tested by *Kruskall-Wallis p=0.0089. Multiple comparison were done by two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli (p in graph, n=mice. Right panel: Data are normally distributed (Shapiro-Wilk-test) and tested by *ANOVA p=0.0132 with Holm-Sidak post-test (p in graph, n=mice). F) Mean densitometric data for T305-autophosphorylation shows that compared to CASK-CTRL, T305 autophosphorylation was significantly reduced in CASK KO upon TAC. Data are normally distributed (Shapiro-Wilk-test) and compared by unpaired t-test (p in graph, n=mice). G) Mean densitometric analysis of CaMKII-oxidation relative to CaMKII expression indicates enhanced CaMKII oxidation in CASK KO upon TAC. Data are normally distributed (Shapiro-Wilk-test) and compared by unpaired t-test (p in graph, n=mice. Label information: CASK-CTRL = CTRL, CASK-Flox = Flox, CASK-KO = KO
Also, CASK-KO mice showed an signficantly impaired survival after TAC (Fig. 5C). This suggests that CASK may be revelant in limiting CaMKII-activity during early pressure overload.
Accordingly, we found a two-fold increase in CASK-expression 2 weeks after TAC (Fig. 5D) in CASK-CTRL compared to Sham-operated CASK-CTRL. The strong TAC-dependent increase in CASK-expression strikingly resembles the increased CASK-expression found in patients with aortic stenosis and preserved EF (Fig. 1B). In accordance with the regulatory role of CASK for CaMKII-autophosphorylation, stimulatory CaMKII T286-phosphorylation was significantly increased (Fig. 5E), and inhibitory CaMKII T305-phosphorylation was significantly reduced in left ventricular myocardium of CASK-KO mice 2 weeks after TAC (vs. CASK-CTRL, fig. 5F). Consistent with that, CaMKII-oxidation was also increased in CASK-KO 2 weeks after TAC (Fig. 5G).
GLP1-receptor agonists induce a protective overexpression of CASK in cardiomyocytes.
In the pancreas, CASK has been shown to be upregulated by exposure to the GLP1-receptor agonist exenatide.40 Thus, we exposed isolated murine ventricular cardiomyocytes from CASK-CTRL mice to 100 nmol/L40 exenatide (GLP1-RA) or vehicle control for 24h before harvesting for cellular experiments or Western blotting (cell culture performed as described previously41,42). Interestingly, exenatide exposure significantly increased CASK expression (Fig 6A). In accordance, inhibitory T305 phosphorylation was significantly increased (Fig. 6B) upon GLP1-RA exposure in CASK-CTRL. While GLP1-RA increases CASK expression and T305 phosphorylation, CASK expression also correlates significantly with T305 phosphorylation(Fig. 6B). Further, stimulatory T286 phosphorylation upon GLP1-RA was reduced, but only in CASK CTRL (Fig. 6C).
Figure 6: CASK expression is increased upon GLP1-receptor agonist treatment.

A) Original Western blots and mean densitometric data of CASK-expression in isolated ventricular cardiomyocytes from CASK-CTRL or CASK-KO mice that were harvested 24 h after culture with either vehicle control (DMSO) or 100 nmol/L of the GLP1-receptor agonist (GLP1-RA) exenatide. Data follows normal distribution (D’Agostino-Pearson-test) and was compared by paired t-test (p in graph, n=mice). B) Left panel: original Western blots and mean densitometric data of T305-phosphorylation (original registration contrast-enhanced equally across all lanes). Data are normally distributed (D’Agostino-Pearson test) and tested by two-way mixed-effects analysis (substance *p=0.0278, genotype *p=0.0115, interaction *p=0.0295) with Sidak post-tests (p in graph). Right panel: CASK-expression correlates strongly with T305-phosphorylation (linear regression p<0.05, R2=0.45).
C) Original Western blots and mean densitometric data of CaMKII T286-phosphorylation. Data are normally distributed (D’Agostino-Pearson test) and tested by two-way ANOVA (genotype *p=0.0274, substance p=0.8033, interaction *p=0.0339). n=mice)
D) Mean data of SR Ca spark frequency (CaSpF) as a marker of SR Ca leak in isolated ventricular cardiomyocytes from CASK-CTRL (left panel) or CASK-KO (right panel) mice that were harvested 24 h after culture with either vehicle control (DMSO) or 100 nmol/L of GLP1-RA and exposed to 10−7 mol/l isoproterenol. Data are normally distributed (D’Agostino-Pearson test) and compated by paired t-test (p in graph, n=mice)
E) Mean data of Caffeine transient amplitude in Fluo-4 –loaded isolated ventricular myocytes that were harvested 24 h after culture with either vehicle control (DMSO) or 100 nmol/L of GLP1-RA and exposed to 10-7 mol/l isoproterenol. Left panel: CASK-CTRL. Data are normally distributed (Shapiro-Wilk-test) and compared by paired t-test (p in graph, n=mice). Right panel: CASK-KO. Data are not normally distributed (Shapiro-Wilk-test) and tested by Wilcoxon test (p in graph, n=mice).
To test, if this GLP1-RA-dependent regulation of CaMKII autophosphorylation would translate into reduced SR Ca leak, we measured SR Ca sparks as a measure of Ca leak in CASK-CTRL or CASK-KO cardiomyocytes exposed to GLP1-RA for 24h. As mentioned above, Ca leak at baseline is low in both CASK-CTRL and CASK-KO, as such we exposed cardiomyocytes to 10−7 mol/l isoproterenol to stimulate CaMKII. Interestingly, in CASK-CTRL, Ca spark frequency was significantly blunted by the exposure to GLP1-RA, while no effect could be observed in CASK-KO (Fig. 6D). In parallel, SR Ca content was significantly increased in CASK-CTRL cardiomyocytes exposed to isoproterenol after 24h GLP1-RA pretreatment, while no such effect was observed in CASK-KO (Fig. 6E).
To test, if the protective effect GLP1-RA can also be observed in failing ventricular myocytes, we subjected WT mice to TAC. Ventricular cardiomyocytes were isolated 5 weeks after TAC and exposed to GLP1-RA or vehicle control for 24h. Interestingly, exposure to GLP1-RA significantly reduced CaSpF, while SR Ca content was significantly increased, leading to increased Ca transient amplitude (Fig. 7A). In accordance, in vivo treatment of TAC mice with GLP-1RA resulted in a significant reduction of CaMKII T286 autophosphorylation 5 weeks after TAC (figure 7B).
Figure 7: GLP-1 RA inhibits CaMKII activation upon TAC.

A) Original confocal line scans (upper panel) and mean data of SR Ca2+ spark frequency (CaSpF) as a marker of SR Ca2+−leak (lower left panel), Caffeine transient amplitude (lower mid panel), and Ca2+ transient amplitude (lower right panel) in isolated ventricular cardiomyocytes from mice exposed to 5 weeks of transverse aortic constriction (TAC) that were harvested 24 h after culture with either vehicle control (DMSO) or 100 μmol/l of the GLP1-receptor agonist (GLP1-RA) exenatide. Lower left: Data are not normally distributed (D’Agostino-Pearson test) and tested by Mann-Whitney-test (p in graph, n=cells/mice). Lower mid panel: Data are normally distributed (D’Agostino-Pearson) and tested by unpaired t-test (p in graph, n=cells/mice). Lower right panel: Data are normally distributed (D’Agostino-Pearson) and tested by two-way-ANOVA (p in graph, n=cells/mice).
B) Original Western blots and mean densitometric data of CaMKII T287-phosphorylation and CaMKII expression in hearts at 5 weeks after TAC and in vivo treatment with either vehicle or the GLP1-RA peptide 7-36. Data are normally distributed (Shapiro-Wilk-test) and tested by unpaired t-test for both panels (p in graph, n=mice).
DISCUSSION
Here we show that the MAGUK protein CASK associates with CaMKII in human ventricular myocardium and that modulation of CASK expression affects CaMKII activity. Lack of CASK in mice enhances CaMKII-activity by increased T286- and reduced T305-phosphorylation, leads to contractile dysfunction and increased mortality upon pressure overload thus increasing the propensity for ventricular arrhythmias. In contrast, stimulation of CASK expression by GLP1 receptor agonism inhibits CaMKII activity and improves Ca handling. This suggests that CASK is a crucial regulator of cardiac excitation-contraction coupling by modulation of CaMKII-activity.
CASK associates with CaMKII.
We and others have shown that CaMKII is crucially involved in arrhythmias and contractile dysfunction in HF5,32,43,44, where CaMKII-expression and CaMKII-dependent phosphorylation of Ca regulatory proteins are enhanced.45-47
While various pathways of CaMKII-activation have been described,2,8-10,48 the mechanism of CaMKII inactivation has been less well studied. In neurons, the MAGUK-protein CASK interacts with CaMKII, which leads to increased CaMKII-autophosphorylation at T305/306.11 The phosphorylation of these residues at the calmodulin-binding site leads to inhibition of calmodulin-binding, thereby limiting CaMKII-activity,12,49 which alters memory function.12,50 CASK contains a CaMKII-homologue domain in the N-terminal region. This domain was initially considered as catalytically inactive,13,51 however, it was shown that the domain can phosphorylate substrates if they had been recruited by the CASK scaffolding-activity.13 This may explain, why direct CASK-CaMKII interaction has been shown to promote CaMKII-autophosphorylation at T305/306 and inhibit CaMKII-activity in neurons.12
Despite previous data reporting CASK-expression in the myocardium,14,15 its role for CaMKII-activity in the heart has not been investigated.
CASK regulates CaMKII-activity and excitation-contraction coupling.
To investigate the role of CASK for CaMKII activity, we made use of a novel cardiomyocyte-specific CASK-KO mouse model. We showed that CASK KO mice display increased CaMKII T286-phosphorylation but reduced T305-phosphorylation upon stimulation with ISO. This suggests that CASK indeed regulates CaMKII-activity in the myocardium which is in accordance with data from neuronal synapses in Drosophila.12
Despite these CASK effects on CaMKII-function, CASK-KO mice show normal cardiac-morphology and -function in vivo at rest without altered mortality. The basal increase in CaMKII-activity in CASK-KO is thus not sufficient to induce HF alone, but may be a sentinel for a preclinical stage, as implied by the fact that ISO-induced arrhythmogenic effects are already apparent in the CASK-KO even at baseline. Indeed, different levels of CaMKII overexpression in mice have different rates of heart failure and mortality.43 The lack of a basal cardiac phenotype may seem surprising given the multiple functional roles that have been ascribed to CASK, but is in accordance with previously published data.14 CASK may be dispensable for heart development and cardiac function at rest, similar to CaMKII.35
Using CaMKII knockout mouse models, we and others have shown that CaMKII is also not essential for cardiac function at rest in vivo.29,35,52 In sharp contrast, exposure of mice to stressors like increased afterload not only activates CaMKII, but also leads to CaMKII-dependent disturbance of Ca handling, ion channels, contractile dysfunction and arrhythmias.23,36,53 Similarly, increased β-adrenergic stimulation is known to result in profound CaMKII-activation, and CaMKII-dependent changes in excitation-contraction coupling.37 Consistent with this fact, we show here that isoproterenol enhances CaMKII-dependent RyR2-phosphorylation in CASK-KO and diastolic SR Ca sparks (as a measure of Ca leak). This increase in SR Ca leak in CASK KO is not compensated by isoproterenol-dependent stimulation of SERCA activity, since SR Ca content and Ca transients are also diminished.
Increased SR Ca leak is not only important for SR Ca content but also a major trigger of arrhythmias.5,6,54 Right ventricular burst stimulation together with intraperitoneal isoproterenol injection resulted in life-threatening ventricular tachyarrhythmias in CASK-KO mice in vivo, but not in CASK-CTRL. This is in accordance with animal models of enhanced CaMKII activity showing increased propensity for ventricular arrhythmias.2,6 The role of elevated CaMKII activity in CASK-KO is underscored by the fact that oral gavage of CASK-KO mice with a novel CaMKII inhibitor completely prevented the induction of ventricular arrhythmias.
Lack of CASK accelerates HF upon pressure overload.
In order to test, if CASK-dependent regulation of CaMKII activity is important for HF development, we induced pressure overload by TAC in mice. Interestingly, echocardiographic analysis revealed that CASK-KO mice displayed a reduction in left ventricular EF already two weeks after TAC, while CASK-CTRL mice were still at the stage of compensated hypertrophy. This suggests that CASK may be most important at the early stages of cardiac remodeling. In WT mice CASK-expression was increased more than 2-fold at 2 weeks (compared to Sham). Similarly, our human data showing more than 2-fold-increased CASK-expression in hearts of patients with aortic stenosis and preserved EF but no significant increase in CASK-expression in the hearts of patients with end-stage HF. This suggests that increased afterload may be involved in the regulation of CASK-expression. In contrast to CASK-CTRL, lack of CASK (in CASK KO) and especially lack of increased CASK expression after two weeks of TAC resulted in an impairment of EF and increased mortality. This suggests that CASK may be important for the heart to adapt to increased afterload possibly by limiting excessive CaMKII activation.
Increased CASK expression by GLP1-receptor stimulation protects the heart.
In contrast, stimulation of CASK expression appeared to confer cardioprotection. It was shown that the GLP1-receptor agonist exenatide increased CASK expression in the pancreas.40 Similarly, 24h exposure of cardiomyocytes to exenatide resulted in increased cardiomyocyte CASK-expression. Moreover, this led to both decreased stimulatory T286-CaMKII phosphorylation and increased inhibitory T305-CaMKII phosphorylation, neither of which occurred in the CASK-KO. This suggests that the modulation of CaMKII activity by exenatide requires CASK expression. Functionally, the exenatide-effect on CaMKII activity translated into decreased isoproterenol-induced SR Ca sparks and increased SR Ca content in CASK-CTRL, which could not be seen in CASK-KO. Moreover, exposure to GLP-1-RA inhibited CaMKII activity, decreased SR Ca leak and improved systolic Ca handling upon TAC. This CASK-dependent regulation of CaMKII activity may, thus, partly explain the protective effect that has been observed in large clinical trials using GLP-1 receptor agonists.55,56
In conclusion, we propose here a novel regulation of CaMKII-activity in the heart by CASK. CASK increases inhibitory CaMKII T305-phosphorylation, which limits pathological CaMKII-activation. This regulation appears to be important under conditions that are known to stimulate CaMKII-activity like β-adrenergic stimulation or increased afterload and may lead to the development of novel treatment options for HF.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
The Calcium/Calmodulin-dependent kinase II (CaMKII) regulates excitation and contraction in the heart.
Increased expression and activity of CaMKII contributes to heart failure (HF) development and arrhythmias
What New Information Does This Article Contribute?
We describe the role of Ca/CaM-dependent serine protein kinase (CASK) in the heart, a scaffolding protein that can regulate CaMKII activity.
Knock-out of CASK (KO) results in increased CaMKII activity leading to development of arrhythmias and enhancement of HF progression.
Increasing CASK expression, on the other hand, reduces CaMKII activity and improves contractile function.
Increased CaMKII expression and activity contributes to contractile dysfunction and arrhythmias. However, clinical application of pharmacological CaMKII inhibition may be limited by organ-selectivity and off-target toxicity. While many studies have focused on stimulatory pathways of CaMKII, few is known about proteins limiting CaMKII activity. We describe here cardiac CASK, which can limit CaMKII activation by promoting inhibitory CaMKII T305 phosphorylation. In cardiomyocytes, loss of CASK (KO) resulted in increased CaMKII activity with cellular arrhythmias and disturbed Ca handling. In vivo, CASK KO led to enhanced CaMKII-dependent HF progression and ventricular arrhythmias. In contrast, pharmacological stimulation of CASK expression enhanced inhibitory CaMKII T305-phosphorylation and limited pathological CaMKII-activation thereby improving Ca handling and contractile performance. Thus, the CASK regulation of CaMKII may be a promising novel treatment option for patients with HF.
ACKNOWLEDGEMENT
We acknowledge the help of Felicia Radtke, Thomas Sowa, and Lukas Schöberl. We thank Prof. Thomas Südhof, Stanford University, for graciously providing the CASK mouse. The authors declare no competing financial interests.
SOURCES OF FUNDING
This work was supported by grants from Deutsche Forschungsgemeinschaft [WA 2539/5-1 to SW, MA 1982/ 7-1 to LSM, as well as SFB 1350 project number 387509280], faculty grants from University of Regensburg [ReForM C program, LSM, STS and SW]. JM is funded by a faculty grant from University of Regensburg (ReForM A program) and by the German Cardiac Society Clinician Scientist program. SW and LSM were funded by German Centre for Heart Research (DZHK). This work was also supported by US National Institutes of Health grants [R01-HL114383, R01-HL134824, and R35 HL-135754 to PJM, R01-HL142282 to DMB, and R35-HL140034 to MEA].
Nonstandard Abbreviations and Acronyms:
- AP
action potential
- AS
aortic stenosis
- BW
body weight
- CaMKII
Ca/calmodulin-dependent kinase II
- CASK
Ca/CaM-dependent serine protein kinase
- CaSpF
Calcium spark frequency
- ECG
electrocardiogram
- ENSE
early non-stimulated Ca release event(s)
- DNSE
delayed non-stimulated Ca release event(s)
- GPL1-RA
GLP1-receptor agonist
- HF
heart failure
- HW
heart weight
- ISO
isoproterenol
- KO
knock out
- LV
left ventricular / left ventricle
- LVEDD
left ventricular end-diastolic diameter
- MAGUK
membrane associated guanylate kinase
- Nav1.5
voltage-gated sodium channel 1.5
- NCX
sodium calcium exchanger
- NF
non-failing
- PCR
polymerase chain reaction
- pT17
phosphorylated threonine 17 (on phospholamban)
- PLN
phospholamban
- QTc
heart rate corrected QT interval
- RyR2
ryanodine receptor type 2
- pS2814
phosphorylated serine 2814 (on RyR2)
- SDS
sodium dodecyl sulfate
- SERCA
sarcoplasmic reticulum calcium ATPase
- SR
sarcoplasmic reticulum
- TAC
transverse aortic constriction
- WT
wild type
Footnotes
DISCLOSURES
The authors declare that no conflict of interest exists.
Publisher's Disclaimer: This article is published in its accepted form. It has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation Research involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final, published version.
REFERENCES
- 1.MAIER LS, BERS DM. CALCIUM, CALMODULIN, AND CALCIUM-CALMODULIN KINASE II: HEARTBEAT TO HEARTBEAT AND BEYOND. J MOL CELL CARDIOL 2002;34:919–939. [DOI] [PubMed] [Google Scholar]
- 2.WAGNER et al. CA/CALMODULIN-DEPENDENT PROTEIN KINASE II REGULATES CARDIAC NA CHANNELS. J CLIN INVEST 2006; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.LUO M, ANDERSON ME. MECHANISMS OF ALTERED CA2+ HANDLING IN HEART FAILURE. CIRC RES 2013;113:690–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.WAGNER S, RUFF HM, WEBER SL, BELLMANN S, SOWA T, SCHULTE T, ANDERSON ME, GRANDI E, BERS DM, BACKS J, BELARDINELLI L, MAIER LS. REACTIVE OXYGEN SPECIES–ACTIVATED CA/CALMODULIN KINASE IIΔ IS REQUIRED FOR LATE INA AUGMENTATION LEADING TO CELLULAR NA AND CA OVERLOAD. CIRC RES 2011;108:555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.NEEF S, DYBKOVA N, SOSSALLA S, ORT KR, FLUSCHNIK N, NEUMANN K, SEIPELT R, SCHÖNDUBE FA, HASENFUSS G, MAIER LS. CAMKII-DEPENDENT DIASTOLIC SR CA2+ LEAK AND ELEVATED DIASTOLIC CA2+ LEVELS IN RIGHT ATRIAL MYOCARDIUM OF PATIENTS WITH ATRIAL FIBRILLATION. CIRC RES 2010;106:1134. [DOI] [PubMed] [Google Scholar]
- 6.SAG CM, WADSACK DP, KHABBAZZADEH S, ABESSER M, GREFE C, NEUMANN K, OPIELA M-K, BACKS J, OLSON EN, BROWN JH, NEEF S, MAIER SK, MAIER LS. CALCIUM/CALMODULIN-DEPENDENT PROTEIN KINASE II CONTRIBUTES TO CARDIAC ARRHYTHMOGENESIS IN HEART FAILURE. CIRC HEART FAIL 2009;2:664–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.HUDMON A, SCHULMAN H. STRUCTURE-FUNCTION OF THE MULTIFUNCTIONAL CA2+/CALMODULIN-DEPENDENT PROTEIN KINASE II. BIOCHEM J 2002;364:593–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.ERICKSON JR, JOINER MA, GUAN X, KUTSCHKE W, YANG J, ODDIS CV, BARTLETT RK, LOWE JS, O’DONNELL SE, AYKIN-BURNS N, ZIMMERMAN MC, ZIMMERMAN K, HAM A-JL, WEISS RM, SPITZ DR, SHEA MA, COLBRAN RJ, MOHLER PJ, ANDERSON ME. A DYNAMIC PATHWAY FOR CALCIUM-independent ACTIVATION OF CAMKII BY METHIONINE OXIDATION. CELL 2008;133:462–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.ERICKSON JR, PEREIRA L, WANG L, HAN G, FERGUSON A, DAO K, COPELAND RJ, DESPA F, HART GW, RIPPLINGER CM, BERS DM. DIABETIC HYPERGLYCAEMIA ACTIVATES CAMKII AND ARRHYTHMIAS BY O-LINKED GLYCOSYLATION. NATURE 2013;502:372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.ERICKSON JR, NICHOLS CB, UCHINOUMI H, STEIN ML, BOSSUYT J, BERS DM. S-NITROSYLATION INDUCES BOTH AUTONOMOUS ACTIVATION AND INHIBITION OF CALCIUM/CALMODULIN-DEPENDENT PROTEIN KINASE II Δ. J BIOL CHEM 2015;290:25646–25656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.HANSON PI, SCHULMAN H. INHIBITORY AUTOPHOSPHORYLATION OF MULTIFUNCTIONAL CA2+/CALMODULIN-DEPENDENT PROTEIN KINASE ANALYZED BY SITE-DIRECTED MUTAGENESIS. J BIOL CHEM 1992;267:17216–17224. [PubMed] [Google Scholar]
- 12.HODGE JJL, MULLASSERIL P, GRIFFITH LC. ACTIVITY-DEPENDENT GATING OF CAMKII AUTONOMOUS ACTIVITY BY DROSOPHILA CASK. NEURON 2006;51:327–337. [DOI] [PubMed] [Google Scholar]
- 13.MUKHERJEE K, SHARMA M, URLAUB H, BOURENKOV GP, JAHN R, SÜDHOF TC, WAHL MC. CASK FUNCTIONS AS A MG 2+-INDEPENDENT NEUREXIN KINASE. CELL 2008;133:328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.EICHEL CA, BEURIOT A, CHEVALIER MYE, ROUGIER J-S, LOUAULT F, DILANIAN G, AMOUR J, COULOMBE A, ABRIEL H, HATEM SN, BALSE E. LATERAL MEMBRANE-SPECIFIC MAGUK CASK DOWN-REGULATES NA V 1.5 CHANNEL IN CARDIAC MYOCYTESNOVELTY AND SIGNIFICANCE. CIRC RES 2016;119:544–556. [DOI] [PubMed] [Google Scholar]
- 15.LEONOUDAKIS D, CONTI LR, RADEKE CM, MCGUIRE LMM, VANDENBERG CA. A MULTIPROTEIN TRAFFICKING COMPLEX COMPOSED OF SAP97, CASK, VELI, AND MINT1 IS ASSOCIATED WITH INWARD RECTIFIER KIR2 POTASSIUM CHANNELS. J BIOL CHEM 2004;279:19051–19063. [DOI] [PubMed] [Google Scholar]
- 16.ORBAN PC, CHUI D, MARTH JD. TISSUE-AND SITE-SPECIFIC DNA RECOMBINATION IN TRANSGENIC MICE. PROC NATL ACAD SCI 1992;89:6861–6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.SAUER B FUNCTIONAL EXPRESSION OF THE CRE-LOX SITE-SPECIFIC RECOMBINATION SYSTEM IN THE YEAST SACCHAROMYCES CEREVISIAE. MOL CELL BIOL 1987;7:2087–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.SUBRAMANIAM A, JONES WK, GULICK J, WERT S, NEUMANN J, ROBBINS J. TISSUE-SPECIFIC REGULATION OF THE ALPHA-MYOSIN HEAVY CHAIN GENE PROMOTER IN TRANSGENIC MICE. J BIOL CHEM 1991;266:24613–24620. [PubMed] [Google Scholar]
- 19.ATASOY D, SCHOCH S, HO A, NADASY KA, LIU X, ZHANG W, MUKHERJEE K, NOSYREVA ED, FERNANDEZ-CHACON R, MISSLER M. DELETION OF CASK IN MICE IS LETHAL AND IMPAIRS SYNAPTIC FUNCTION. PROC NATL ACAD SCI 2007;104:2525–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.NAGY A CRE RECOMBINASE: THE UNIVERSAL REAGENT FOR GENOME TAILORING. GENES N Y N 2000 2000;26:99–109. [PubMed] [Google Scholar]
- 21.HEFFNER CS, HERBERT PRATT C, BABIUK RP, SHARMA Y, ROCKWOOD SF, DONAHUE LR, EPPIG JT, MURRAY SA. SUPPORTING CONDITIONAL MOUSE MUTAGENESIS WITH A COMPREHENSIVE CRE CHARACTERIZATION RESOURCE. NAT COMMUN 2012;3:1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.LEBHERZ C, GAO G, LOUBOUTIN J-P, MILLAR J, RADER D, WILSON JM. GENE THERAPY WITH NOVEL ADENO-ASSOCIATED VIRUS VECTORS SUBSTANTIALLY DIMINISHES ATHEROSCLEROSIS IN A MURINE MODEL OF FAMILIAL HYPERCHOLESTEROLEMIA. J GENE MED JOHN WILEY & SONS, LTD; 2004;6:663–672. [DOI] [PubMed] [Google Scholar]
- 23.TOISCHER K, HARTMANN N, WAGNER S, FISCHER TH, HERTING J, DANNER BC, SAG CM, HUND TJ, MOHLER PJ, BELARDINELLI L, HASENFUSS G, MAIER LS, SOSSALLA S. ROLE OF LATE SODIUM CURRENT AS A POTENTIAL ARRHYTHMOGENIC MECHANISM IN THE PROGRESSION OF PRESSURE-INDUCED HEART DISEASE. J MOL CELL CARDIOL 2013;61:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.PUROHIT A, ROKITA AG, GUAN X, CHEN B, KOVAL OM, VOIGT N, NEEF S, SOWA T, GAO Z, LUCZAK ED, STEFANSDOTTIR H, BEHUNIN AC, LI N, EL-ACCAOUI RN, YANG B, SWAMINATHAN PD, WEISS RM, WEHRENS XHT, SONG L-S, DOBREV D, MAIER LS, ANDERSON ME. OXIDIZED CA2+/CALMODULIN-DEPENDENT PROTEIN KINASE II TRIGGERS ATRIAL FIBRILLATION. CIRCULATION 2013;128:1748–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MITCHELL GF, JERON A, KOREN G. MEASUREMENT OF HEART RATE AND Q-T INTERVAL IN THE CONSCIOUS MOUSE. AM J PHYSIOL 1998;274:H747–751. [DOI] [PubMed] [Google Scholar]
- 26.MUSTROPH J, DRZYMALSKI M, BAIER M, PABEL S, BIEDERMANN A, MEMMEL B, DURCZOK M, NEEF S, SAG CM, FLOERCHINGER B, RUPPRECHT L, SCHMID C, ZAUSIG Y, BÉGIS G, BRIAND V, OZOUX M, TAMARELLE D, BALLET V, JANIAK P, BEAUVERGER P, MAIER LS, WAGNER S. THE ORAL CA/CALMODULIN - DEPENDENT KINASE II INHIBITOR RA608 IMPROVES CONTRACTILE FUNCTION AND PREVENTS ARRHYTHMIAS IN HEART FAILURE. ESC HEART FAIL 2020;EHF2.12895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.SAG CM, MALLWITZ A, WAGNER S, HARTMANN N, SCHOTOLA H, FISCHER TH, UNGEHEUER N, HERTING J, SHAH AM, MAIER LS, SOSSALLA S, UNSÖLD B. ENHANCED LATE INA INDUCES PROARRHYTHMOGENIC SR CA LEAK IN A CAMKII-DEPENDENT MANNER. J MOL CELL CARDIOL 2014;76:94–105. [DOI] [PubMed] [Google Scholar]
- 28.PICHT E, ZIMA AV, BLATTER LA, BERS DM. SPARKMASTER: AUTOMATED CALCIUM SPARK ANALYSIS WITH IMAGEJ. AM J PHYSIOL - CELL PHYSIOL 2007;293:C1073. [DOI] [PubMed] [Google Scholar]
- 29.NEEF S, SAG CM, DAUT M, BÄUMER H, GREFE C, EL-ARMOUCHE A, DESANTIAGO J, PEREIRA L, BERS DM, BACKS J, MAIER LS. WHILE SYSTOLIC CARDIOMYOCYTE FUNCTION IS PRESERVED, DIASTOLIC MYOCYTE FUNCTION AND RECOVERY FROM ACIDOSIS ARE IMPAIRED IN CAMKIIΔ-KO MICE. J MOL CELL CARDIOL 2013;59:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.GEE KR, BROWN KA, CHEN W-NU, BISHOP-STEWART J, GRAY D, JOHNSON I. CHEMICAL AND PHYSIOLOGICAL CHARACTERIZATION OF FLUO-4 CA2+-INDICATOR DYES. CELL CALCIUM 2000;27:97–106. [DOI] [PubMed] [Google Scholar]
- 31.WU Y, TEMPLE J, ZHANG R, DZHURA I, ZHANG W, TRIMBLE R, RODEN DM, PASSIER R, OLSON ERICN, COLBRAN RJ. CALMODULIN KINASE II AND ARRHYTHMIAS IN A MOUSE MODEL OF CARDIAC HYPERTROPHY. CIRCULATION 2002;106:1288–1293. [DOI] [PubMed] [Google Scholar]
- 32.MAIER LS. TRANSGENIC CAMKIIDELTAC OVEREXPRESSION UNIQUELY ALTERS CARDIAC MYOCYTE CA2+ HANDLING: REDUCED SR CA2+ LOAD AND ACTIVATED SR CA2+ RELEASE. CIRC RES 2003;92:904–911. [DOI] [PubMed] [Google Scholar]
- 33.HUKE S, BERS DM. TEMPORAL DISSOCIATION OF FREQUENCY-DEPENDENT ACCELERATION OF RELAXATION AND PROTEIN PHOSPHORYLATION BY CAMKII. J MOL CELL CARDIOL 2007;42:590–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.LUO M, GUAN X, LUCZAK ED, LANG D, KUTSCHKE W, GAO Z, YANG J, GLYNN P, SOSSALLA S, SWAMINATHAN PD, WEISS RM, YANG B, ROKITA AG, MAIER LS, EFIMOV IR, HUND TJ, ANDERSON ME. DIABETES INCREASES MORTALITY AFTER MYOCARDIAL INFARCTION BY OXIDIZING CAMKII. J CLIN INVEST 2013;123:1262–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.BACKS J, BACKS T, NEEF S, KREUSSER MM, LEHMANN LH, PATRICK DM, GRUETER CE, QI X, RICHARDSON JA, HILL JA, KATUS HA, BASSEL-DUBY R, MAIER LS, OLSON EN. THE Δ ISOFORM OF CAM KINASE II IS REQUIRED FOR PATHOLOGICAL CARDIAC HYPERTROPHY AND REMODELING AFTER PRESSURE OVERLOAD. PROC NATL ACAD SCI 2009;106:2342–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.TOISCHER K, ROKITA AG, UNSÖLD B, ZHU W, KARARIGAS G, SOSSALLA S, REUTER SP, BECKER A, TEUCHER N, SEIDLER T, GREBE C, PREUß L, GUPTA SN, SCHMIDT K, LEHNART SE, KRÜGER M, LINKE WA, BACKS J, REGITZ-ZAGROSEK V, SCHÄFER K, FIELD LJ, MAIER LS, HASENFUSS G. DIFFERENTIAL CARDIAC REMODELING IN PRELOAD VERSUS AFTERLOAD. CIRCULATION 2010;122:993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DYBKOVA N, WAGNER S, BACKS J, HUND TJ, MOHLER PJ, SOWA T, NIKOLAEV VO, MAIER LS. TUBULIN POLYMERIZATION DISRUPTS CARDIAC B-ADRENERGIC REGULATION OF LATE I(NA). CARDIOVASC RES 2014;103:168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.SAG CM, WOLFF HA, NEUMANN K, OPIELA M-K, ZHANG J, STEUER F, SOWA T, GUPTA S, SCHIRMER M, HÜNLICH M, RAVE-FRÄNK M, HESS CF, ANDERSON ME, SHAH AM, CHRISTIANSEN H, MAIER LS. IONIZING RADIATION REGULATES CARDIAC CA HANDLING VIA INCREASED ROS AND ACTIVATED CAMKII. BASIC RES CARDIOL 2013;108:385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.GUO T, ZHANG T, MESTRIL R, BERS DM. CA2+/CALMODULIN-DEPENDENT PROTEIN KINASE II PHOSPHORYLATION OF RYANODINE RECEPTOR DOES AFFECT CALCIUM SPARKS IN MOUSE VENTRICULAR MYOCYTES. CIRC RES 2006;99:398–406. [DOI] [PubMed] [Google Scholar]
- 40.ZHU Z-Q, WANG D, XIANG D, YUAN Y-X, WANG Y. CALCIUM/CALMODULIN-DEPENDENT SERINE PROTEIN KINASE IS INVOLVED IN EXENDIN-4-INDUCED INSULIN SECRETION IN INS-1 CELLS. METABOLISM 2014;63:120–126. [DOI] [PubMed] [Google Scholar]
- 41.MUSTROPH J, WAGEMANN O, LÜCHT CM, TRUM M, HAMMER KP, SAG CM, LEBEK S, TARNOWSKI D, REINDERS J, PERBELLINI F, TERRACCIANO C, SCHMID C, SCHOPKA S, HILKER M, ZAUSIG Y, PABEL S, SOSSALLA ST, SCHWEDA F, MAIER LS, WAGNER S. EMPAGLIFLOZIN REDUCES CA/CALMODULIN-DEPENDENT KINASE II ACTIVITY IN ISOLATED VENTRICULAR CARDIOMYOCYTES. ESC HEART FAIL 2018;5:642–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.HOFHUIS J, BERSCH K, BÜSSENSCHÜTT R, DRZYMALSKI M, LIEBETANZ D, NIKOLAEV VO, WAGNER S, MAIER LS, GÄRTNER J, KLINGE L, THOMS S. DYSFERLIN MEDIATES MEMBRANE TUBULATION AND LINKS T-TUBULE BIOGENESIS TO MUSCULAR DYSTROPHY. J CELL SCI 2017;130:841. [DOI] [PubMed] [Google Scholar]
- 43.ZHANG T THE DELTAC ISOFORM OF CAMKII IS ACTIVATED IN CARDIAC HYPERTROPHY AND INDUCES DILATED CARDIOMYOPATHY AND HEART FAILURE. CIRC RES 2003;92:912–919. [DOI] [PubMed] [Google Scholar]
- 44.ANDERSON ME. OXIDANT STRESS PROMOTES DISEASE BY ACTIVATING CAMKII. J MOL CELL CARDIOL 2015;89:160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.HOCH B, MEYER R, HETZER R, KRAUSE E-G, KARCZEWSKI P. IDENTIFICATION AND EXPRESSION OF -ISOFORMS OF THE MULTIFUNCTIONAL CA2+/CALMODULIN-DEPENDENT PROTEIN KINASE IN FAILING AND NONFAILING HUMAN MYOCARDIUM. CIRC RES 1999;84:713–721. [DOI] [PubMed] [Google Scholar]
- 46.FISCHER TH, HERTING J, TIRILOMIS T, RENNER A, NEEF S, TOISCHER K, ELLENBERGER D, FÖRSTER A, SCHMITTO JD, GUMMERT J, SCHÖNDUBE FA, HASENFUSS G, MAIER LS, SOSSALLA S. CA2+/CALMODULIN-DEPENDENT PROTEIN KINASE II AND PROTEIN KINASE A DIFFERENTIALLY REGULATE SARCOPLASMIC RETICULUM CA2+ LEAK IN HUMAN CARDIAC PATHOLOGY. CIRCULATION 2013;128:970. [DOI] [PubMed] [Google Scholar]
- 47.KOVAL OM, GUAN X, WU Y, JOINER M-L, GAO Z, CHEN B, GRUMBACH IM, LUCZAK ED, COLBRAN RJ, SONG L-S, HUND TJ, MOHLER PJ, ANDERSON ME. CAV1.2 BETA-SUBUNIT COORDINATES CAMKII-TRIGGERED CARDIOMYOCYTE DEATH AND AFTERDEPOLARIZATIONS. PROC NATL ACAD SCI U S A 2010;107:4996–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.ZHANG T, ZHANG Y, CUI M, JIN L, WANG Y, LV F, LIU Y, ZHENG W, SHANG H, ZHANG J, ZHANG M, WU H, GUO J, ZHANG X, HU X, CAO C-M, XIAO R-P. CAMKII IS A RIP3 SUBSTRATE MEDIATING ISCHEMIA- AND OXIDATIVE STRESS-INDUCED MYOCARDIAL NECROPTOSIS. NAT MED 2016;22:175–182. [DOI] [PubMed] [Google Scholar]
- 49.GILLESPIE JM, HODGE JJL. CASK REGULATES CAMKII AUTOPHOSPHORYLATION IN NEURONAL GROWTH, CALCIUM SIGNALING, AND LEARNING. FRONT MOL NEUROSCI 2013;6:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.MALIK BR, GILLESPIE JM, HODGE JJL. CASK AND CAMKII FUNCTION IN THE MUSHROOM BODY A′/B′ NEURONS DURING DROSOPHILA MEMORY FORMATION. FRONT NEURAL CIRCUITS 2013;7:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.BOUDEAU J, MIRANDA-SAAVEDRA D, BARTON GJ, ALESSI DR. EMERGING ROLES OF PSEUDOKINASES. TRENDS CELL BIOL 2006;16:443–452. [DOI] [PubMed] [Google Scholar]
- 52.KREUSSER MM, LEHMANN LH, KERANOV S, HOTING M-O, OEHL U, KOHLHAAS M, REIL J-C, NEUMANN K, SCHNEIDER MD, HILL JA, DOBREV D, MAACK C, MAIER LS, GRÖNE H-J, KATUS HA, OLSON EN, BACKS J. CARDIAC CAM KINASE II GENES Δ AND Γ CONTRIBUTE TO ADVERSE REMODELING BUT REDUNDANTLY INHIBIT CALCINEURIN-INDUCED MYOCARDIAL HYPERTROPHY. CIRCULATION 2014;130:1262–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.LING H, ZHANG T, PEREIRA L, MEANS CK, CHENG H, GU Y, DALTON ND, PETERSON KL, CHEN J, BERS D, HELLER BROWNJ. REQUIREMENT FOR CA2+/CALMODULIN–DEPENDENT KINASE II IN THE TRANSITION FROM PRESSURE OVERLOAD–INDUCED CARDIAC HYPERTROPHY TO HEART FAILURE IN MICE. J CLIN INVEST 2009;119:1230–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.VOIGT N, LI N, WANG Q, WANG W, TRAFFORD AW, ABU-TAHA I, SUN Q, WIELAND T, RAVENS U, NATTEL S, WEHRENS XH, DOBREV D. ENHANCED SARCOPLASMIC RETICULUM CA(2+)-LEAK AND INCREASED NA(+)-CA(2+)-EXCHANGER FUNCTION UNDERLIE DELAYED AFTERDEPOLARIZATIONS IN PATIENTS WITH CHRONIC ATRIAL FIBRILLATION. CIRCULATION 2012;125:2059–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.MARSO SP, DANIELS GH, BROWN-FRANDSEN K, KRISTENSEN P, MANN JFE, NAUCK MA, NISSEN SE, POCOCK S, POULTER NR, RAVN LS, STEINBERG WM, STOCKNER M, ZINMAN B, BERGENSTAL RM, BUSE JB. LIRAGLUTIDE AND CARDIOVASCULAR OUTCOMES IN TYPE 2 DIABETES. N ENGL J MED MASSACHUSETTS MEDICAL SOCIETY; 2016;375:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.GERSTEIN HC, COLHOUN HM, DAGENAIS GR, DIAZ R, LAKSHMANAN M, PAIS P, PROBSTFIELD J, RIESMEYER JS, RIDDLE MC, RYDÉN L, XAVIER D, ATISSO CM, DYAL L, HALL S, RAO-MELACINI P, WONG G, AVEZUM A, BASILE J, CHUNG N, CONGET I, CUSHMAN WC, FRANEK E, HANCU N, HANEFELD M, HOLT S, JANSKY P, KELTAI M, LANAS F, LEITER LA, LOPEZ-JARAMILLO P, et al. DULAGLUTIDE AND CARDIOVASCULAR OUTCOMES IN TYPE 2 DIABETES (REWIND): A DOUBLE-BLIND, RANDOMISED PLACEBO-CONTROLLED TRIAL. THE LANCET ELSEVIER; 2019;394:121–130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data can be made available by the authors upon request.