

Abstract
Rationale:
In heart failure (HF), impaired sarcoplasmic reticulum (SR) Ca2+ release and cytosolic Na+ overload depress mitochondrial Ca2+ (mCa2+) signaling, resulting in a diminished ability to maintain matrix NAD(P)H redox potential, leading to increased oxidative stress when workload increases. Enhancing mCa2+ can reverse this defect but could potentially increase the likelihood of mitochondrial Ca2+ overload.
Objective:
To determine if moderate mitochondrial Ca2+ uniporter (MCU) overexpression has beneficial or detrimental effects on the development of HF and incident arrythmias in a guinea pig model (ACi) of HF and sudden cardiac death.
Methods and Results:
In vivo viral gene transfer was used to increase MCU levels by ~30% in ACi hearts. Left ventricular myocytes from hearts with MCU overexpression (ACi+MCU) displayed enhanced mCa2+ uptake, decreased oxidative stress, and increased β-adrenergic- and frequency-dependent augmentation of Ca2+ transients and contractions, compared to myocytes from ACi hearts. MCU overexpression decreased SR Ca2+ leak in the ACi group and mitigated the elevated ryanodine receptor disulfide crosslinks in HF. β-adrenergic responses were blunted in isolated perfused ACi hearts and these deficiencies were normalized in ACI+MCU hearts. To examine the in vivo effects of MCU overexpression, ACi hearts were transduced with the MCU virus 2–3w after aortic constriction, at the onset of cardiac decompensation. Two weeks later, cardiac function worsened in the untreated ACi group (fractional shortening: 39±1% at 2w and 32±1% at 4w), whereas MCU overexpression significantly improved cardiac function (36±1% at 2w and 42±2% at 4w). MCU overexpression in the decompensating ACi heart also attenuated pulmonary edema and interstitial fibrosis and prevented triggered arrhythmias.
Conclusions:
Moderate MCU overexpression in failing hearts enhances contractility and responses to β-adrenergic stimulation in isolated myocytes and perfused hearts by inhibiting mitochondrial oxidative stress-induced SR Ca2+ leak. MCU overexpression also reversed HF and inhibited ectopic ventricular arrhythmias.
Keywords: heart failure, sudden cardiac death, Mitochondrial Calcium Uniporter, Overexpression, ROS, guinea pig, ryanodine receptor, arrhythmia, gene transfer
Subject Terms: Arrhythmias, Calcium Cycling/Excitation-Contraction Coupling, Heart Failure, Ion Channels/Membrane Transport, Sudden Cardiac Death
INTRODUCTION
Heart failure (HF) is a progressive disease with considerable morbidity and mortality, with ventricular arrhythmia leading to sudden cardiac death (SCD) contributing significantly to death in the early stages 1, 2. Mitochondrial dysfunction, including impaired electron transport chain activity, altered fission-fusion dynamics, reduced high energy phosphate transfer, and increased reactive oxygen species (ROS) accumulation, is increasingly recognized as a major contributor to the pathophysiology of HF3–7.
Ca2+ is an important regulator of all of these mitochondrial functions. Mitochondrial Ca2+ (mCa2+) is responsible for sustaining respiration rate and ATP production during the constantly varying workloads of the heart by stimulating oxidative phosphorylation and by increasing NADH production through activation of Ca2+-sensitive enzymes in the Krebs cycle8, 9. In parallel, matrix NADPH/NADP+ redox potential drives the regeneration of reduced glutathione, thioredoxin, and peroxiredoxin, which are involved in detoxifying H2O210, 11. Mitochondria take up and extrude Ca2+ on a beat-to-beat basis, and with increased workload, or under β-adrenergic stimulation, a higher steady state mCa2+ level is reached in the matrix due to the relatively slower efflux rate compared to influx rate12. Previous work demonstrated that insufficient mCa2+ accumulation results in net oxidation of NAD(P)H and diminished ROS scavenging capacity when workload increases13, and that normalization of mCa2+ improves function in Na+-overloaded12, 14, 15 or failing cardiomyocytes16. On the other hand, excessive mCa2+ loading can induce the opening of the mitochondrial permeability transition pore (mPTP), potentially leading to myocyte dysfunction or cell death, hence, it is unclear whether enhancing mitochondrial Ca2+ uptake would be beneficial or detrimental to cardiac function in the context of HF.
The mitochondrial Ca2+ uniporter (MCU) is the major pathway for mitochondrial Ca2+ uptake and its recent molecular identification spurred intense interest in genetic manipulation of MCU to investigate the physiological and pathological roles of MCU in cells and animals17. In a mouse model overexpressing a dominant-negative form of MCU (DN-MCU), the “fight-or-flight” response to catecholamine stimulation was abrogated and NADH/NAD+ redox potential was decreased without a change in basal cardiac function.18 With a model of cardiac-specific MCU knockout, Luongo et al. also demonstrated that hearts without MCU failed to response to acute β-adrenergic stimulation.19 Surprisingly, hearts from DN-MCU18 or germline MCU knockout mice20 were not protected from ischemia-reperfusion injury, nevertheless, studies of cardiac-specific MCU knockouts showed decreased infarct size and improved function after left anterior descending artery ligation19. With respect to pressure overload HF, cardiac-specific MCU knockout had no effect on the development of HF induced by transverse aortic constriction (TAC)19, 20.
Our previous studies demonstrated that either enhancement of mCa2+ by partial inhibition of mitochondrial Na+/Ca2+ exchange21, or targeted mitochondrial antioxidant treatment22, can prevent cardiac decompensation and SCD in the guinea pig HF model. Because the effects of increasing MCU levels in HF are unknown, here, we use a novel strategy to moderately overexpress MCU to correct the mCa2+ problem, to determine if it benefits or exacerbates HF and the incidence of arrythmias. We find that MCU overexpression restores the fight-or-flight response in myocytes and hearts from ACi animals, improving oxidative stress, cardiac contractility, and SR Ca2+ load. Importantly, MCU treatment could reverse the course of cardiac decompensation and suppress arrhythmias when applied weeks after HF had begun to develop.
METHODS
Detailed Materials & Methods and Major Resources Table are available in the Supplemental Materials.
Data Availability.
The authors declare that all supporting data are available within the article. Constructs used will be made available upon request from the authors.
RESULTS
MCU overexpression in adult guinea pig heart.
An adenoviral vector expressing MCU and nuclear-targeted cyan fluorescent protein (nCFP) was directly injected into left ventricle (LV) wall of adult guinea pig hearts and MCU-flag expression was examined at 1, 2, and 4 weeks after injection. The distribution of adenovirus transduced myocytes in the heart was visualized by fluorescence imaging of nCFP in frozen sections of the heart (Figure 1A). nCFP positive cells were found to be widely distributed throughout the LV wall. MCU protein level was compared between hearts injected with control virus (Ad-nCFP) and Ad-MCU-flag (Figures 1B and 1C). While the endogenous MCU level was similar in both groups, the total MCU level (endogenous MCU+MCU-flag) in Ad-MCU-flag injected group was about 57% higher than the endogenous MCU in Ad-nCFP injected controls. We then investigated the time course of Adeno-viral vector-mediated MCU overexpression. MCU-flag level was measured at 1, 2, and 4 weeks. The ratio of MCU-flag/MCU was 0.32±0.04, 0.31±0.02, and 0.26±0.05 at 1w, 2w, and 4w after injection, respectively (Online figure I). The expression level did not significantly decline over 4 weeks. We also examined levels of EMRE, another key transmembrane component of the MCU complex 23. A trend towards increased EMRE was noted in Ad-MCU-flag transduced hearts compared to control Ad-nCFP hearts, but the increase did not reach statistical significance (Figure 1C). To quantitatively assess the efficiency of gene delivery to the mitochondria, an adenoviral vector expressing a MCU-GFP fusion protein was injected into the LV wall and mitochondria were isolated and then analyzed with flow cytometry. The results indicated that approximately 40% of the energized mitochondria were GFP-positive (Online Figure II).
Figure 1. Adenoviral vector-mediated MCU overexpression in adult guinea pig heart.
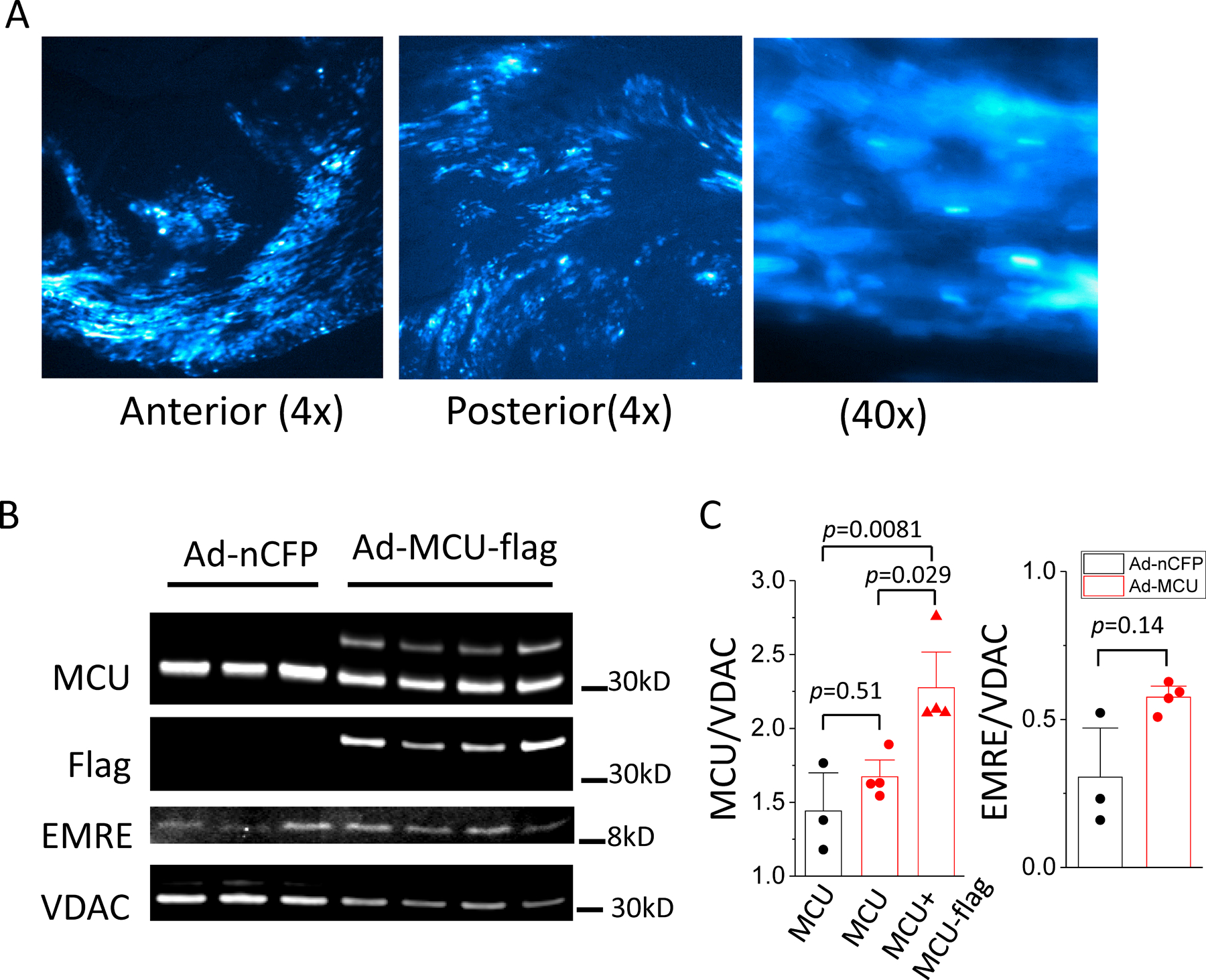
Adenoviral vector expressing MCU-3xflag with an independent reporter, nuclear-targeted CFP (nCFP), was administered by intramuscular injection into the LV wall of guinea pig hearts. A) Representative images of nCFP from frozen sections of a heart 1 week after injection. Left and middle panels show expression of nCFP across LV anterior and posterior walls; Right panel shows 40x image of nCFP positive and negative cells. B) images of western blot for MCU, EMRE, VDAC, and Flag in samples from heart injected control virus, Ad-nCFP (n=3) and Ad-MCU-flag (n=4); C) Left: average intensity of endogenous MCU in Ad-nCFP (black) and Ad-MCU-flag (red) injected heart and total MCU (endogenous MCU+MCU-flag) in Ad-MCU-flag injected heart (red), Data were analyzed with 1-way ANOVA followed with post-hoc Tukey test; Right: average intensity of EMRE in Ad-nCFP (black) and Ad-MCU-flag (red) injected heart, mean was compared with t-test.
MCU overexpression increases the rate of mCa2+ uptake and restores mCa2+ accumulation in myocytes from ACi hearts.
To examine the effect of MCU overexpression directly on mCa2+ uptake, we recorded mCa2+ uptake in permeabilized myocytes isolated from Normal and ACi hearts injected with Ad-MCU-flag-nCFP or Ad-nCFP using Rhod-2 as a mitochondrial Ca2+ indicator. Upon switching the perfusion buffer from Ca2+-free to 2 µM Ca2+, Rhod-2 fluorescence increased sigmoidally and this increase could be inhibited by 1 µM Ru360 (Online Figure III). To control for experimental variability in solution exchange rates or other factors, recordings were made from dishes including both transduced (nCFP positive) and non-transduced (nCFP negative) cells (Online Figure IV). The initial linear rapid mCa2+ uptake rate was then analyzed by pairwise comparisons between vector-transduced and non-transduced cells in each group. The initial linear rate of mCa2+ uptake was significantly increased by MCU overexpression in both normal (Normal, 0.11±0.01, n=56 vs Normal+MCU, 0.16±0.02, n=35, p<0.0001) and ACi myocytes (ACi, 0.08±0.01, n=57 vs ACi+MCU, 0.12±0.01, n=26, p<0.0001), compared to corresponding non-transduced controls (Figures 2A and 2B). There was no difference in mCa2+ uptake rate in myocytes transduced with control viruses versus non-transduced controls (ACi, 0.11±0.01, n=73 vs ACi+nCFP, 0.10±0.01, n=25, n.s.) (Figure 2C).
Figure 2. MCU overexpression increases the rate of mCa2+ uptake and restores mCa2+ accumulation and inhibits oxidative stress in failing cardiomyocytes.
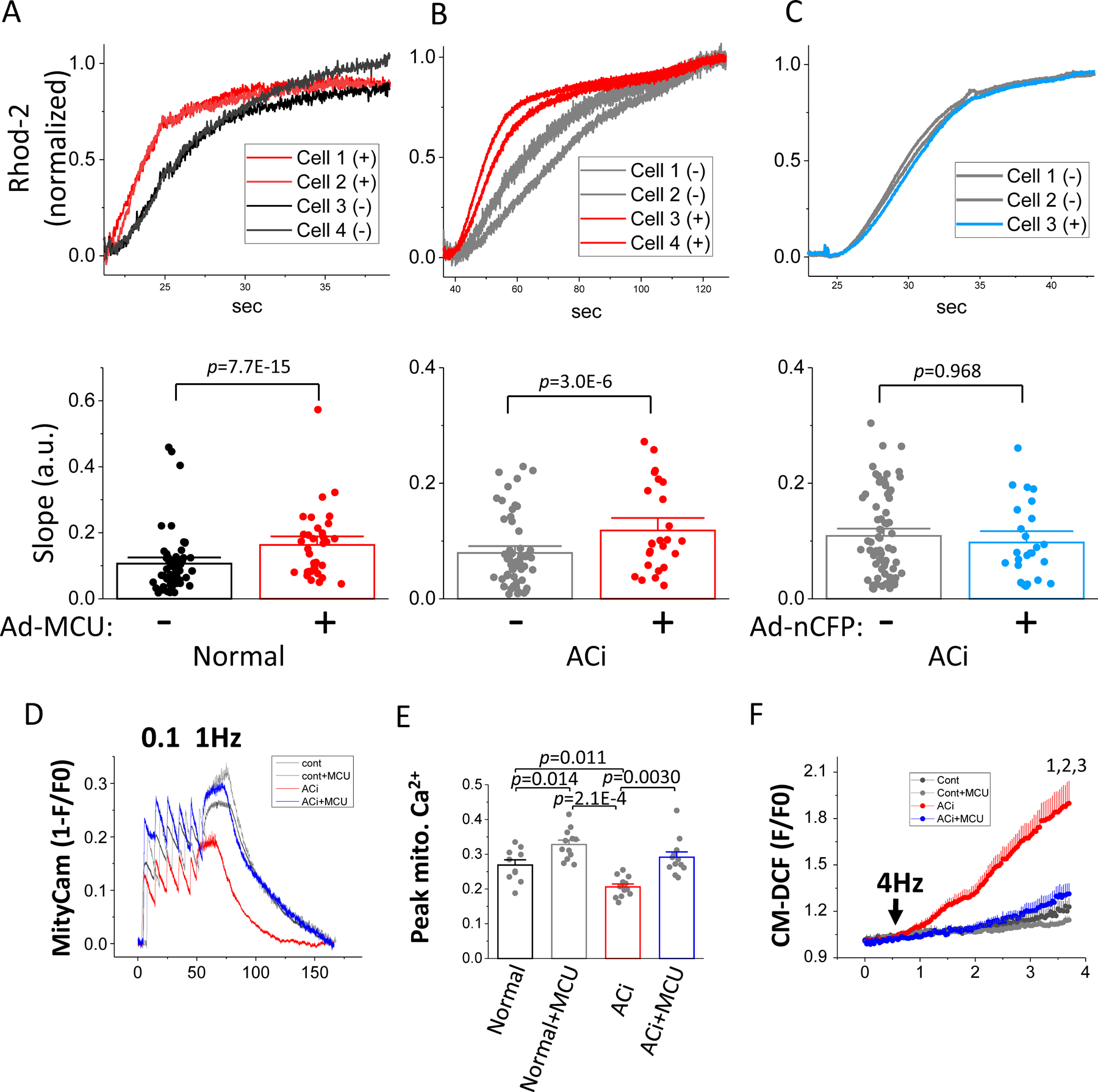
A-C) mCa2+ uptake was recorded in permeabilized myocytes from normal (A) and ACi (B and C) hearts injected with Ad-MCU-flag-nCFP (A and B) or control virus (Ad-nCFP) (C). Upper panels: representative trace of rhod-2 signals simultaneously recorded in 4 cells (transduced, (+) and non-transduced (−)) from each group. The raw signal was normalized to a scale of 0 to 1. The transmitted light and nCFP images and rhod-2 time-lapse recordings of these representative cells are shown in Online Figure III. Lower panels: slope of the initial linear increase in rhod-2 fluorescence measured in both transduced and non-transduced cells was pairwise compared using nested random effects model (Normal: n=56 for non-transduced cells and 35 transduced cells; ACi+MCU: n=57 non-transduced cells and 26 transduced cells; ACi+nCFP: n= 73 for non-transduced cells and 25 for transduced cells; cells in each group were isolated from 3 hearts). D) mCa2+ accumulation was measured in freshly isolated myocytes after in vivo viral transduction with MityCam. Representative MityCam signals (1-F/F0) were recorded from resting state to 0.1 Hz and 1Hz stimulation in the presence of 100nM ISO in myocytes from Normal, Normal+MCU, ACi, and ACi+MCU groups. E) Averaged steady state MityCam signal at 1Hz stimulation in myocyte from Normal (n=10 from 3 hearts), Normal+MCU (n=12, from 4 hearts), ACi (n=12 from 4 hearts), and ACi+MCU (n=12, from 4 hearts) groups showing increased mCa2+ accumulation with MCU overexpression in Normal+MCU and ACi+MCU (right). Data were analyzed with nested 2-way ANOVA followed with post-hoc Tukey test. F) Myocytes isolated from Normal, Normal+MCU, ACi, and ACi+MCU groups (n=8 from 3 hearts in each group) were loaded with dihydrodichlorofluorescein (DCF) and stimulated at 4 Hz (arrow) in the presence of 100nM ISO for 3 min. Data were analyzed with nested 2 way-ANOVA with repeated measure followed with post-hoc Tukey test. 1, p=0.020; 2, p=0.016; 3, p=0.032 compared to Normal, Normal+MCU, and ACi+MCU groups, respectively.
To assess the effects increased mCa2+ uptake rate on mCa2+ accumulation in electrically-paced intact myocytes, we used MityCam24 as the mCa2+ indicator. Ad-MityCam was injected into the LV wall at week 3 and cardiomyocytes were isolated from non-failing control hearts (Normal), Normal+MCU, ACi, and ACi+MCU hearts at week 4 after aortic banding. Myocytes were field-stimulated at 0.1 and 1Hz in the presence of 100nM isoproterenol (ISO) in the perfusion medium (Figure 2D). Mean mCa2+ accumulation was assessed at steady state during 1Hz stimulation. MCU overexpression significantly increased mCa2+ accumulation in both normal and failing hearts. The MityCam signal (1-F/F0) during pacing was increased by 22% in myocytes from Normal+MCU hearts compared to those from Normal hearts (Normal: 0.27±0.02, n=10; Normal+MCU: 0.33±0.01, n=12, p<0.02) and increased 40% in myocytes from ACi+MCU hearts compared to those from ACi hearts (ACi: 0.21±0.01, n=12; ACi+MCU: 0.29±0.02, n=12, p<0.001) (Figure 2). The MityCam signal (1-F/F0) in myocytes from ACi hearts was significantly decreased compared to those from control hearts (Normal vs. ACi; p<0.01) and was restored to normal in myocytes from the ACi group with MCU overexpression (ACi+MCU vs. ACi, p<0.005; Normal+MCU vs. Normal, p= NS) (Figure 2E).
MCU overexpression inhibits ROS accumulation during increased workload in myocytes from ACi hearts.
To investigate the effect of MCU overexpression on intracellular ROS accumulation, cardiomyocytes were loaded with carboxymethyl dihydrodichlorofluorescein diacetate (CM-H2DCFDA) and were field-stimulated at 4Hz in the presence of ISO for 3min after recording baseline fluorescence for 0.5min (Figure 2F). 4Hz stimulation in the presence of ISO induced an increase in oxidized DCF signal. MCU overexpression in Normal myocytes had no effect on the rate of DCF oxidation (Normal vs. Normal+MCU, p= NS). At the end of 3-minutes of stimulation, the oxidized DCF signal, normalized to baseline fluorescence (F/F0), was 1.22±0.06 and 1.12±0.02 in Normal and Normal+MCU groups, respectively. Compared to Normal myocytes, myocytes from the ACi group showed a significantly higher rate of DCF oxidation (ACi vs Normal or Normal+MCU, p<0.0001). F/F0 at the end of 3-min of stimulation in ACi myocyte was 1.84±0.13. The rate of DCF oxidation in ACi myocytes was significantly decreased by MCU overexpression in ACi+MCU myocyte (ACi vs. ACi+MCU, p<0.0001; ACi+MCU vs. Normal or Normal+MCU, NS.) with a F/F0 of 1.29±0.07 at the end of 3-min stimulation.
MCU overexpression restores the frequency-dependence of contractility and the Ca2+ transient in failing myocytes.
Sarcomere shortening and cytosolic Ca2+ transients were measured in isolated myocytes during field stimulation at 1, 2, and 4Hz in the absence or presence of ISO. The frequency- and ®-adrenergic effects were analyzed in myocytes from Normal, ACi, ACi+nCFP (empty virus with marker but no MCU), and ACi+MCU hearts, respectively. Myocytes of the Normal group displayed significant frequency-dependent augmentation of contractility and Ca2+ transient amplitude both in the absence (Baseline) or presence of ISO (Figure 3). Sarcomere shortening (SS) and Ca2+ transient amplitude (ΔR340/380) were increased at 4Hz, compared to 1Hz, by 218% and 106%, respectively (SS, 2.33%±0.67 at 1hz vs. 7.40%±0.80 at 4hz, p<0.0001; ΔR, 1.26±0.29 at 1hz vs. 2.59±0.39 at 4hz, p<0.0005) in the absence of ISO and 56% and 39%, respectively (SS, 6.56%±0.71 at 1hz vs. 10.21%±1.26 at 4hz, p<0.005; ΔR, 2.88±0.37 at 1hz vs. 4.00±0.52 at 4hz, p<0.05), in the presence of ISO. Normal myocytes also displayed strong ®-adrenergic responses. SS and ΔR were significantly increased by ISO at all stimulation-frequencies (Baseline vs. ISO: SS, p<0.0005 at 1hz; p<0.05 at 2hz; p<0.05 at 4hz; ΔR, p<0.0005 at 1hz, p<0.05 at 2hz, p<0.05 at 4hz) (Figure 3). The frequency- and β-adrenergic effects on contractility and Ca2+ were greatly diminished in myocytes from ACi hearts. SS and Ca2+ transient amplitude (ΔR340/380) at 4hz were not significantly different from those at 1hz in the absence of ISO (SS, 1.02%±0.22 at 1hz vs. 2.46%±0.68 at 4hz, p=0.41; ΔR, 0.99±0.10 at 1hz vs. 1.75±0.28 at 4hz, p=0.22) or with ISO (SS, 2.54%±0.58 at 1hz vs. 4.00%±0.61 at 4hz, p=0.54; ΔR, 2.10±0.28 at 1hz vs. 1.97±0.39 at 4hz, p=0.99). β-adrenergic responses of myocytes from ACi hearts were also blunted (p-value of baseline vs. ISO: SS, p=0.46 at 1hz, p<0.05 at 2hz, p=0.60 at 4hz; ΔR, p<0.05 at 1hz, p<0.05 at 2hz, p=0.99 at 4hz). Treatment with the control virus (nCFP marker virus with no MCU insert) had no effects on the contractility and peak Ca2+ in the ACi+nCFP group versus ACi, but the frequency- and β-adrenergic effects were restored to normal levels by MCU overexpression. SS and Ca2+ transient amplitude (ΔR340/380) at 4hz in myocytes from ACi+MCU hearts were increased by 269% and 178%, respectively, compared to 1Hz stimulation, in the absence of ISO (SS, 1.00%±0.50 at 1hz vs. 3.70%±0.84, p<0.005; ΔR, 0.84±0.21 at 1hz vs. 2.34±0.61 at 4hz, p<0.005) and 89% and 88% (SS, 4.48%±0.77 at 1hz vs. 8.44%±1.28 at 4hz, p<0.0001; ΔR, 2.07±0.43 at 1hz vs. 3.90±0.64 at 4hz, p<0.001) in the presence of ISO. Myocytes from ACi+MCU hearts also showed significant increases in SS and ΔR in response to ISO at all stimulation frequencies (p-value of baseline vs. ISO: SS, p<0.0001 at 1hz, p<0.0005 at 2hz, p<0.0001 at 4hz; ΔR, p<0.05 at 1hz, p<0.01 at 2hz, p<0.001 at 4hz). In addition to the impaired frequency- and β-adrenergic responses, myocytes from ACi hearts also displayed significantly reduced sarcomere shortening at baseline and with ISO. Although MCU overexpression restored frequency responses in myocytes from ACi hearts, the effects on sarcomere shortening were not statistically significant in the absence of ISO. Sarcomere shortening of myocytes from ACi+MCU heart were still lower than that of normal group at both 2hz and 4hz under basal conditions. When workload was further increased by ISO application, the improvement of sarcomere shortening in ACi+MCU myocytes became evident, and the ACi+MCU group was not significantly different from Normals at 1hz, 2hz, or 4hz (Figure 3). The largest benefit of MCU overexpression on sarcomere shortening in myocytes from failing hearts was thus evident at high workloads, i.e., when cells were stimulated at 4Hz in the presence of ISO, with fractional sarcomere shortening more than 2-fold higher in the ACi+MCU group (8.44%±1.28) as compared to ACi (4.00%±0.61) or ACi+nCFP (4.14%±0.94) groups, nearly matching the performance of non-failing myocytes from the Normal group (10.21%±1.27) (at 4hz with ISO, Normal vs. ACi+MCU: p=0.65; ACi vs. ACi+MCU: p<0.05; ACi+nCFP vs. MCU: p<0.05).
Figure 3. Blunted frequency-dependent augmentation of contractility in ACi myocytes was improved by MCU overexpression.
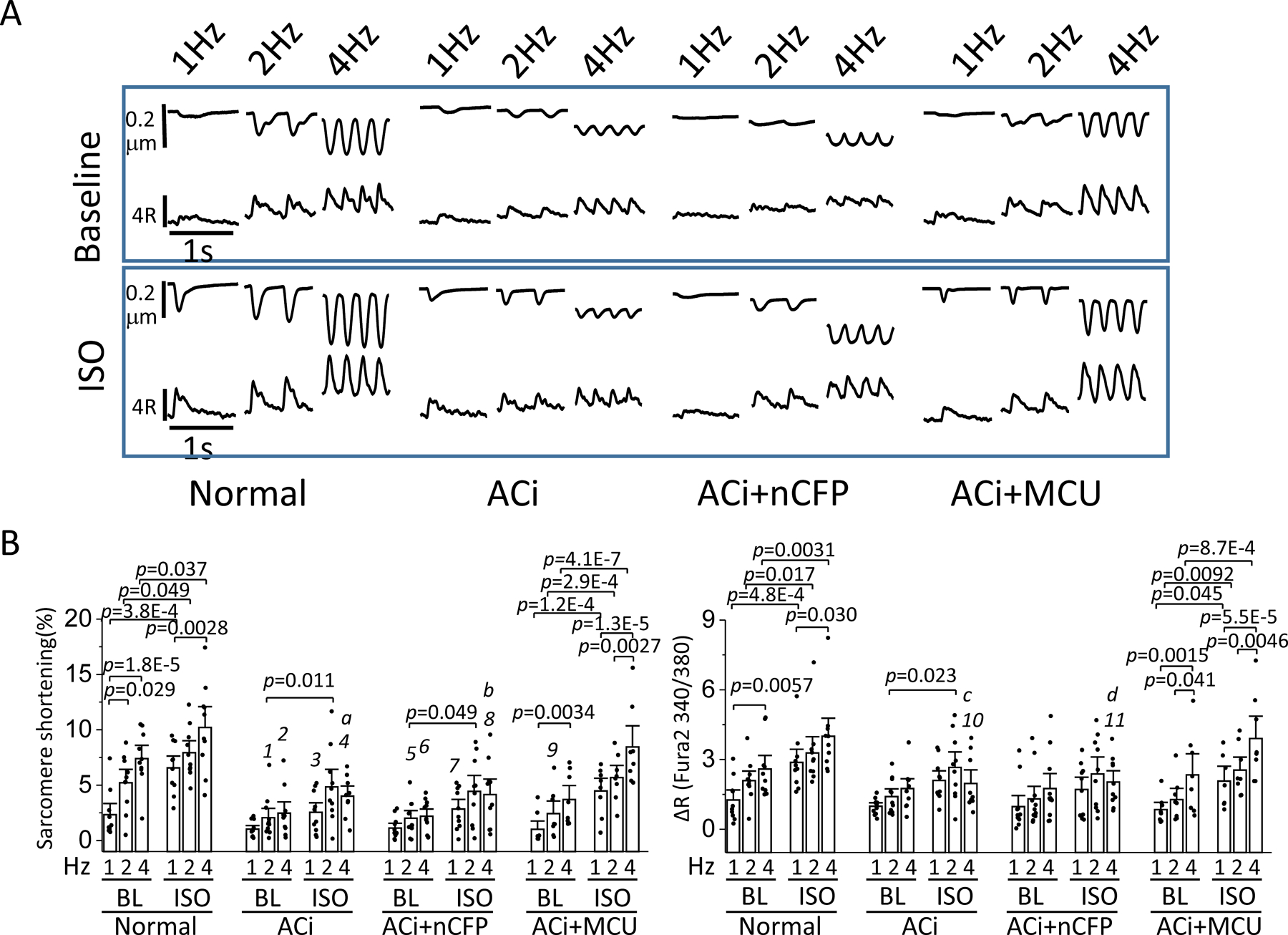
A) Representative recordings of sarcomere length and cytosolic Ca2+ transients with 1, 2, or 4hz stimulation in the absence (Baseline, upper panels) or presence of 100 nM ISO (lower panels) in myocytes of Normal, ACi, ACi+nCFP, and ACi+MCU groups. B) Average sarcomere shortening (%) and ΔR of Fura-2 (340/380nm) measured at 1, 2, or 4hz stimulation with and without ISO in myocytes of Normal (n=10 from 4 hearts), ACi (n=11 from 5 hearts), ACi+nCFP (n=11 from 4 hearts), and Aci+MCU (n=8 from 3 hearts) groups. Effects of stimulation frequency and ISO were analyzed within each group with nested 2-way ANOVA followed with post-hoc Tukey test. Comparisons across groups were performed with nested 2-way ANOVA followed with post-hoc Tukey test under each condition. 1, p=0.0065; 2, p=0.0069; 3, p=0.0070; 4, p=0.0062; 5, p=0.0037; 6, p=0.0064; 7, p=0.012; 8, p=0.0062; 9, p=0.022; 10, p=0.027; 11, p=0.026 compared to Normal group. a, p=0.040; b, p=0.040; c, p=0.044; d, p=0.045 compared to ACi+MCU group.
MCU Overexpression reduces SR Ca2+ leak and increases SR Ca2+ content in myocytes from failing hearts.
It has been shown previously that mitochondrial ROS release can affect diastolic Ca2+ spark frequency by redox modification of the ryanodine receptor (RyR)25,26; therefore, we hypothesized that SR Ca2+ leak may be increased in failing heart cells as a result of the altered ROS balance described above, which was abrogated by MCU overexpression. Hence, we measured SR Ca2+ load by caffeine exposure with and without tetracaine to inhibit RyR-mediated SR Ca2+ leak27. SR Ca2+ load was estimated as the peak change in cytosolic Ca2+ level upon caffeine-induced SR Ca2+ release (Figure 4A). The RyR leak was estimated by the difference in the caffeine response with and without TTC (though it should be recognized that the amplitude of the caffeine response is not a simple function of the RyR leak27). Myocytes from ACi hearts demonstrated a significant reduction in SR Ca2+ load compared to control cells (Figure 4B and 4C). In the absence of tetracaine, the amplitude of caffeine-induced Ca2+ release (ΔR of Fura-2) in ACi myocytes was only 27.6% of that in normal myocytes (ΔR: 2.06±0.48 in ACi vs. 7.46±0.47 in Normal, p<0.0001). Inhibition of RyR-dependent leak by tetracaine had no effect on SR Ca2+ load in normal cells (ΔR: −TTC, 7.46±0.47 vs. +TTC, 7.98±0.60 p=0.99), but significantly increased the amplitude of caffeine-induced Ca2+ release in ACi cells (ΔR: 2.06±0.48 −TTC vs 5.30±0.46 +TTC, p<0.001), suggesting significant RyR-dependent Ca2+ leak in ACi myocytes. Nevertheless, even with tetracaine, the SR Ca2+ load in ACi myocytes was still only 65% of that in control myocytes. MCU overexpression in myocytes from ACi+MCU hearts increased SR Ca2+ load and attenuated RyR-dependent Ca2+ leak. Without tetracaine, the amplitude of caffeine-induced Ca2+ release in ACi+MCU cells was 2.6-fold higher than that in ACi cells. Application of tetracaine showed no significant effect on SR Ca2+ load in ACi+MCU myocyte (ΔR: −TTC, 5.42±0.52 vs. +TTC, 6.92±0.37, p=0.41). Moreover, compared to the Normal group, there was no difference in the amplitude of caffeine-induced Ca2+ release in ACi+MCU myocytes with or without tetracaine (Figure 4C).
Figure 4. MCU overexpression restores SR Ca2+ loading and inhibits RyR-mediated Ca2+ leak.
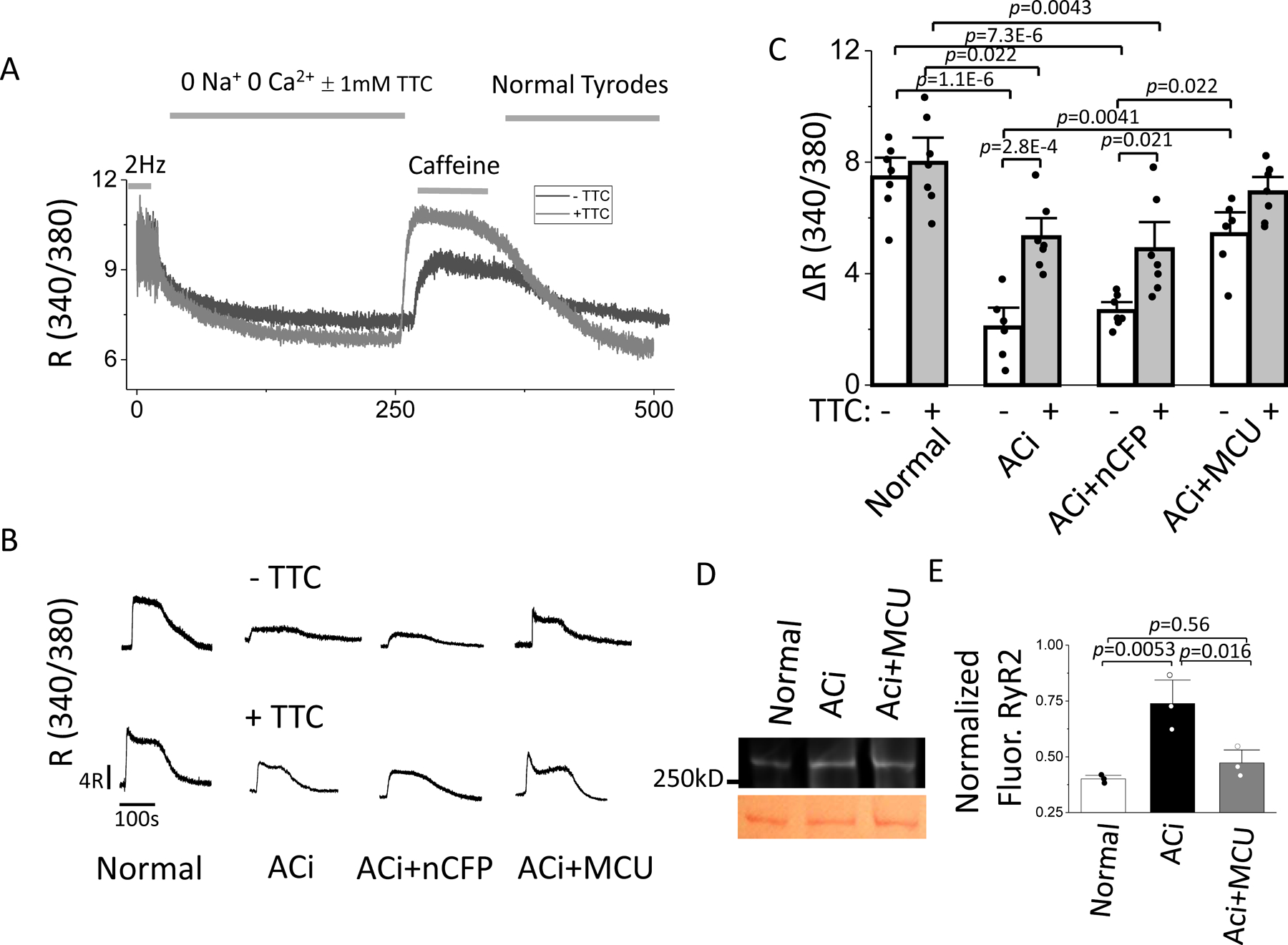
A) SR Ca2+ loading protocol and measurement of Ca2+ load (10 mM Caffeine) after a 200 sec rest subsequent to pacing with or without TTC. B) Representative recordings of caffeine-induced SR Ca2+ release in the 4 experimental groups in the absence (upper panels) or presence (lower panels) of TTC. C) Average SR Ca2+ release with or without TTC in myocytes from Normal, ACi, ACi+nCFP, and ACi+MCU groups. In each subgroup, n=7 from 3 hearts. Data of 8 groups were analyzed with nested 2-way ANOVA followed with Tukey test for multiple comparisons. D) Representative image of fluorescently-labelled disulfide bonds of RyR2 (above) and Ponceau S staining (below). E) Average levels of fluorescently-labelled disulfide bonds of RyR2 from Normal, ACi, and ACi+MCU (n=3) groups. Data were analyzed with 1-way ANOVA followed with post-hoc Tukey test.
MCU overexpression reversed RyR redox modification in ACi hearts.
To further investigate whether the effect of MCU overexpression on RyR leak in the ACi heart was associated with RyR2 redox modification, we compared the levels of disulfide bond crosslinks of RyR2 in Normal, ACi, and ACi+MCU groups. The intensity of fluorescently-labelled disulfide bonds on RyR2 in ACi hearts was 84% higher than that in normal hearts, whereas MCU overexpression decreased the fluorescent intensity in ACi+MCU to a level similar to that in normal hearts (Figure 4E and Online Figure V). The results indicate that RyR2 in the ACi heart is redox modified due to chronic oxidative stress, likely contributing to the increased SR Ca2+ leak, and that MCU overexpression inhibits RyR leak by preventing RyR2 redox modification.
MCU overexpression potentiates the β-adrenergic response of isolated perfused ACi hearts.
We next examined the effect of MCU overexpression on increased workload following β-adrenergic stimulation in Langendorff perfused hearts. Administration of 25nM ISO induced a significant increase in cardiac contractility in Normal hearts (Figure 5A). At steady state after ISO application, pressure development rate (+dP/dt), relaxation rate (−dP/dt), LVDP, HR, and oxygen consumption rate (OCR) were increased by 261%, 123%, 49%, 37%, and 45% compared to baseline, respectively (Figures 5B–5G). However, the positive inotropic and chronotropic effects of ISO were severely blunted in ACi hearts; none of the parameters (+dP/dt, −dP/dt, LVDP, HR, or OCR) measured after ISO application were statistically different from those under basal conditions. MCU overexpression strongly enhanced the positive inotropic, chronotropic and lusitropic effects of ISO in ACi hearts. In ACi+MCU hearts, ISO application significantly increased +dP/dt, −dP/dt, LVDP, HR, and OCR by 145%, 88%, 68%, 47%, and 54%, respectively, compared to baseline (Figure 5).
Figure 5. MCU overexpression potentiates the inotropic and chronotropic responses of perfused ACi hearts during β-adrenergic stimulation.
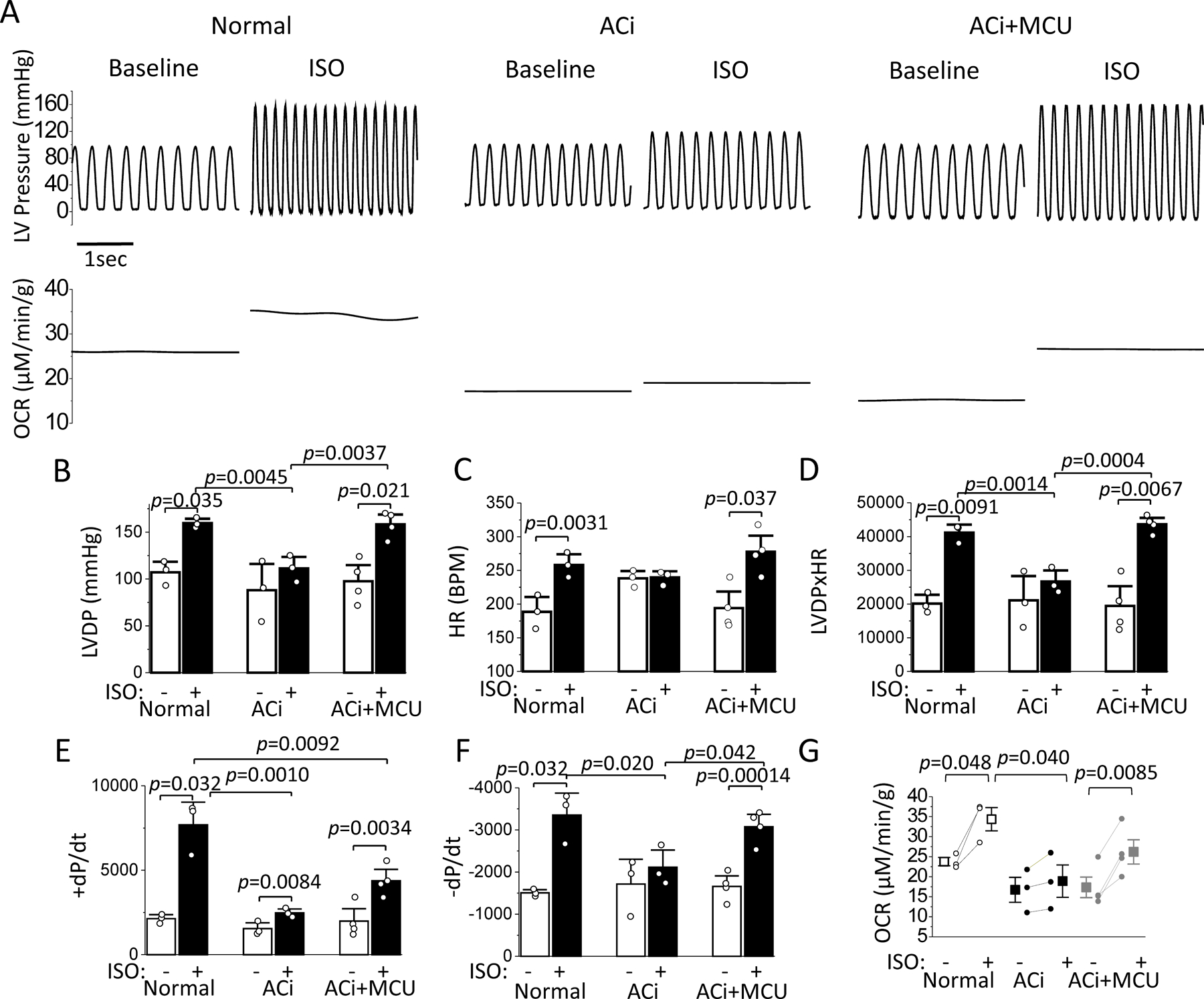
Normal and ACi hearts with or without MCU overexpression were retrogradely perfused and contractile and oxygen consumption parameters were measured at baseline and after 25nM ISO application. A) Representative recordings of LV pressure (upper panels) and oxygen consumption rate (OCR) (lower panels) of Normal, ACi, and ACi+MCU hearts at baseline and in the presence of ISO. B-G) Average LVDP (B), HR (C), rate pressure product (D), +dP/dtmax (E), -dP/dtmax (F), and OCR (G) at baseline and ISO in Normal (n=3), ACi (n=3), ACi+MCU (n=4) hearts. ISO effect was analyzed with paired t test in each group. Cross group comparisons were performed with 1-way ANOVA followed with post-hoc Tukey test at baseline or ISO.
MCU overexpression reverses the development of heart failure and improves cardiac function.
We next examined the effect of MCU overexpression on the development of HF in vivo. At 2 to 3 weeks after aortic banding or sham-operated surgery, a sham group was treated with control virus (Normal+nCFP), and ACi animals were randomly assigned to 2 groups for administration of MCU adenovirus (ACi+MCU) or vehicle (ACi) treatment. Echocardiographic studies were performed before adenoviral vector administration at baseline and 2 weeks following adenoviral vector administration. There was no significant difference in fractioning shortening (FS) between ACi and ACi+MCU groups before vector administration (FS: 44.1±1.8% in Normal+nCFP, 39±1% in ACi, and 36±1% in ACi+MCU; Figure 6A and 6B). 2 weeks after adenovirus or vehicle administration (4–5 weeks post-banding), FS of the ACi group was decreased from 39±1% to 32±1%, FS of the ACi+MCU group was increased from 36±1% to 42±2%, whereas FS of the Normal+nCFP group was not significantly changed (44.1±1.8% pre vs 43.7±0.8% post) (Figure 6A and 6B). These results show that MCU overexpression fully recovered cardiac function of the ACi+MCU group to normal levels. The ACi group displayed evident lung edema with a significant increase in lung weight-to-tibia length (LW/TL) compared to Normal+nCFP, whereas MCU overexpression prevented lung edema; LW/TL in ACi+MCU was not significantly different from that in Normal+nCFP (LW/TL: 0.69±0.03g/cm in Normal+nCFP, 1.62±0.17 g/cm in ACi, and 0.98±0.17 g/cm in ACi+MCU) (Figure 6D). Heart weight was slightly, but not significantly, decreased in the ACi+MCU group compared to ACi; both groups had significantly higher HW/TL than the Normal+nCFP group (HW/TL: 0.57±0.01g/cm in Normal+nCFP, 0.86±0.05g/cm in ACi, and 0.72±0.04 in ACi+MCU) (Figure 6C).
Figure 6. MCU overexpression reverses HF, attenuates LV fibrosis, and decreases the incidence of PVB of ACi model.
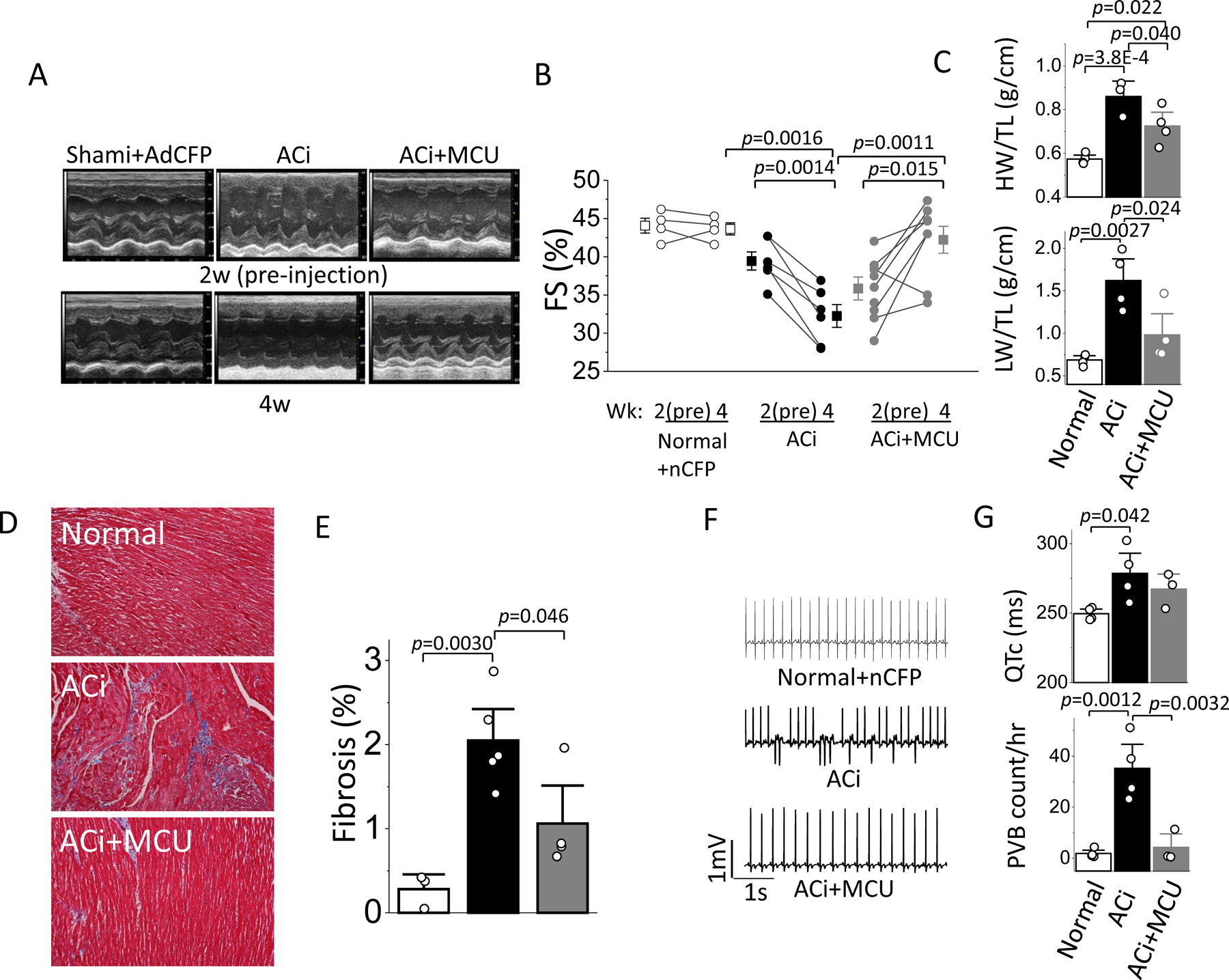
Hearts of a sham group were injected with Ad-nCFP and the ACi group was divided for injection of either Ad-MCU-3xFLAG or vehicle at week2. Echocardiography was performed at week 2 (before injection) and at week 4. A) Representative M-mode echocardiographic images of Normal+nCFP, ACi and ACi+MCU guinea pigs before and 2 weeks after injection. B) Fractional shortening measured in Normal+nCFP (n=4), ACi (n=6), and ACi+MCU (n=8) groups before and 2 weeks after injection showing reversal of HF in the MCU overexpression group. FS at 2w and 4w were compared with paired t-test within each group, FS at 4w cross groups was analyzed with 1-way ANOVA followed with post-hoc Tukey test. C-D) Average heart and lung weights normalized to tibia length in Normal+nCFP (n=4), ACi (n=3), and ACi+MCU (n=4) groups. E) Representative image of Masson’s trichrome staining in Normal (n=3), ACi (n=5), and ACi+MCU (n=4) hearts. F) Averaged fibrotic areas. G) Representative ECG recordings in Normal+nCFP (upper), ACi (middle), and ACi+MCU (lower) animals. H) Average HR (upper) and PVC frequency (counts/hr) at a time point 2–5 hr after the daily ISO injection. Data in C-G were analyzed with 1-way ANOVA followed with post-hoc Tukey test.
MCU overexpression on fibrosis.
Histological analysis indicated the development of interstitial fibrosis in ACi hearts (Figure 6E and F). The area of the fibrotic tissue increased from 0.28%±0.12 in Normal hearts to 2.05%±0.25 in ACi hearts (Figure 6E). Fibrosis was significantly reduced in the ACi+MCU hearts and was not statistically different from that in Normal hearts (Figure 6E).
MCU overexpression on LV arrhythmias.
ECGs were recorded with a telemetry system in Normal+nCFP, ACi, and ACi+MCU groups after administration of the Ad-nCFP in Normal animals, and Ad-MCU or vehicle in ACi animals. HR-corrected QT interval (QTc) in ACi group was significantly elongated compared to that in Normal+nCFP. The prolonged QTc in ACi was not significantly altered by MCU overexpression (250±2 in Normal+nCFP, 276±14 in ACi control, and 267±7 in ACi+MCU); however, the incidence of Premature Ventricular beats (PVB; Figure 6G) was suppressed in the ACi+MCU group, which was similar to that in Normal+nCFP group (2±1/hour in Normal+nCFP, 34±9/hour in ACi control, and 4±4/hour in ACi+MCU) (Figure 6H).
DISCUSSION
The present results show, for the first time, that moderate overexpression of MCU can reverse the course of HF when applied at the onset of cardiac decompensation and suppress HF-associated arrhythmias. These effects were correlated with improved mCa2+ accumulation, diminished mitochondrial oxidative stress, and decreased SR Ca2+ leak. MCU overexpression restored frequency- and β-adrenergic-dependent contractile and Ca2+ responses in HF myocytes and enhanced inotropic and metabolic responses to β-adrenergic stimulation in isolated perfused hearts. The findings are consistent with the general hypothesis that insufficient mCa2+ in the failing heart contributes to impaired energy and redox metabolism.
Previously, we reported that restoration of mCa2+ by partial inhibition of the mitochondrial Na+/Ca2+ exchanger could prevent HF and SCD in the ACi model21 by mitigating oxidative stress induced by increased work and concomitant cytosolic Na+ overload15. Using a genetically engineered mitochondrial Ca2+ reporter, we showed that mCa2+ accumulation upon electrical stimulation is significantly lower in ACi cardiomyocytes compared to non-failing controls, and this is attributable to several factors present in heart failure. First, mCa2+ uptake via MCU is Ca2+-concentration dependent, with a K0.5 for Ca2+ in the range of 20 µM, driven by the electrochemical gradient across the mitochondrial inner membrane. This is above the bulk concentration of cytosolic Ca2+ typically reached during systole in a cardiomyocyte (~1μM). Hence, mitochondrial Ca2+ signaling depends on close proximity to the SR Ca2+ release microdomain28, 29. The present findings show that the SR Ca2+ load in ACi myocytes is approximately one-third of that in controls, similar to previous reports in canine tachypacing-induced30 and human HF31, which severely impairs the peak of the Ca2+ transient and limits microdomain Ca2+. This deficiency would be further exacerbated by disruption of the diadic ultrastructure32 and disorganization of the mitochondrial network33 known to occur in the failing heart. Theoretically, increased cytosolic Na+ could offset this problem by altering the sarcolemmal Na+/Ca2+ exchange reversal potential to increase SR Ca2+ load; however, the Na+ dependence of Ca2+ efflux in heart mitochondria is sigmoidal with a K0.5 of ~5–10mM34, 35, directly in the range over which cytosolic Na+ increases in the HF model (up to 17mM15), resulting in a tripling of the efflux rate and further depletion of mCa2+36. In fact, as shown previously, lowering cytosolic Na+ to 5mM (by intracellular dialysis) in failing myocytes can prevent ROS accumulation and oxidation of the pyridine nucleotide (NAD(P)H) pool during rapid pacing16. The present work illustrates an additional component of the vicious cycle contributing to depletion of both SR Ca2+ load and mCa2+, which is that mitochondrial oxidative stress increases Ca2+ leak through the RyR, further impairing mitochondrial Ca2+ signaling during excitation-contraction coupling.
SR Ca2+ load is controlled by multiple Ca2+ handling components. During diastole, cytosolic Ca2+ is transported back into the SR by the sarco-endoplasmic reticulum Ca2+ ATPase (SERCA2a) and extruded from the myocyte by the sarcolemmal Na+/Ca2+ exchanger (NCX). In HF, reduced SERCA2a activity contributes to slowed relaxation and thus more Ca2+ is removed from the cell via NCX, leading to a reduced SR Ca2+ content. Increased RyR leak in HF further exacerbates the reduction in SR Ca2+ content and plays a critical role in triggering arrhythmias. RyR-mediated Ca2+ leak accounted for ~55% of the decrease in SR Ca2+ content in ACi in this study. Remarkably, MCU overexpression increased SR Ca2+ content by 2.6-fold and decreased RyR-dependent leak by 67% in ACi. In addition, MCU overexpression decreased the TTC-insensitive decline in SR Ca2+ load during rest by 55% in the ACi myocytes. This component reflects Ca2+ recycling into the SR mediated by SERCA2a, indicating that MCU overexpression improved deficient Ca2+ pump activity in HF. These effects can also be explained by the attenuated oxidative stress by MCU overexpression in ACi myocytes. By oxidative modification of −SH groups of RyR and SERCA2a, ROS can directly increase the open probability of RyR and decrease the activity of SERCA2a, thus depleting SR Ca2+.37
Similar changes in Ca2+ handling and Na+ level have been consistently observed in human HF and animal models of HF. Reduced SR loading and Ca2+-induced Ca2+ release during EC coupling is considered to be the major cause for contractile dysfunction in systolic HF.38 Elevated Na+ could be an adaptive response to contractile failure with positive inotropic effects39, 40, yet the negative impact on mCa2+ can be detrimental to function, as is true for cardiac glycoside toxicity14. Considering the common abnormalities of Ca2+ and Na+ homeostasis in HF, it is plausible to assume that insufficient mCa2+ accumulation is a general symptomatic feature of systolic HF and enhancing mCa2+ accumulation could thus be a therapeutic strategy for HF with similar abnormal ion homeostasis. However, mCa2+ overload is also suggested to contribute to HF and arrhythmias in some murine models41, 42. For example, Santulli and colleagues reported that mCa2+ overload played a role in HF in a murine model with leaky RyRs; however, the mCa2+ measurements in that study did not take precautions to separate the mitochondrial versus cytosolic components of the Rhod2 signal41, the latter of which can be substantial12. In another study using a hypertension-induced non-ischemic HF murine model, Xie and colleagues demonstrated that increased mCa2+ cycling due to increased MCU activity contributed to extreme action potential duration elongation and early afterdepolarizations, which were reversed by inhibition of MCU or NCLX, suggesting a model in which enhanced mitochondrial Ca2+ influx and efflux contribute to diastolic loading of SR Ca2+, accounting for the triggered arrhythmias.42
Despite the discrepant conclusions of these studies, it is clear that an optimal level of mCa2+ is required to prevent mitochondrial dysfunction and cardiac pathology during cardiac stress. To date, murine models of MCU ablation have not adequately resolved this issue, showing no differences in cardiac function under the stressors of chronic β-adrenergic stimulation or pressure overload.19, 20, 43 The lack of observable phenotype may be due to adaptive mechanisms that regulate mCa2+ or to energetic adaptation developing over time in knockout models. Kwong et al showed that although mitochondria without MCU are incapable of rapid Ca2+ loading, total Ca2+ loading eventually reaches similar levels as wild type mitochondria.44 Given the major differences in action potential morphology and Ca2+ handling properties between mice and guinea pigs, it is difficult to reconcile the differences observed between species and HF models employed; however, it is plausible to speculate that species with a high resting SR Ca2+ load (e.g., mice and rats) may also be closer to the threshold for mCa2+ overload than guinea pigs or large mammals with systolic HF, where SR Ca2+ load is severely depleted. The beneficial effects of increasing mCa2+ may therefore be more relevant for the latter. Compared to guinea pigs and larger mammals, mice have much higher heart rates and cytosolic Na+ levels, and always operate close to the maximum for mitochondrial ATP production, 45 which may result in different adaptations for regulating oxidative phosphorylation that are less dependent on mCa2+ dynamics.46, 47 Nevertheless, a consistent finding in mouse MCU knockout models is that “fight or flight” responses are impaired. We observed blunted force frequency response (FFR) in ACi hearts, which is a typical feature of contractile failure. Reduced SR Ca2+ loading underlies this defect and targeting SR Ca2+ uptake has been shown to improve FFR. Enhancement of SERCA2a activity restores impaired FFR in HF, 48, 49 whereas decreased SERCA activity leads to a negative FFR in normal hearts.50 Evidence also suggested that increased RyR activity by oxidation contributes to impaired FFR.51 Our results show that frequency-dependent augmentation of contractility and Ca2+ in myocytes from ACi animals depends on mCa2+ signaling via MCU. MCU overexpression also reversed deficient chronotropic, inotropic and lusitropic responses to ISO in perfused ACi hearts, while having no significant effect on cardiac output and the maximal rates of pressure development and relaxation at baseline. Oxygen consumption was also significantly increased in ACi+MCU hearts in parallel with contractility following ISO administration, while no significant increase was observed in ACi hearts, indicating that MCU overexpression improved not only SR Ca2+ loading but also mitochondrial energetics.
In summary, insufficient mCa2+ accumulation plays an essential role in the development of HF and arrhythmias in the ACi heart failure model. The present study provides evidence that overexpression of MCU restores mCa2+ accumulation, improves inotropic responses in myocytes, reverses HF development and prevents arrhythmias (Figure 7). The underlying mechanism of these beneficial effects is attenuated oxidative stress, which contributes to enhanced SR Ca2+ loading by inhibiting RyR leak and improving SERCA2a activity, while improving mitochondrial respiration.
Figure 7. Summary of the role of mCa2+ in the chain of events underlying cellular contractile/bioenergetic/redox dysfunction in HF.
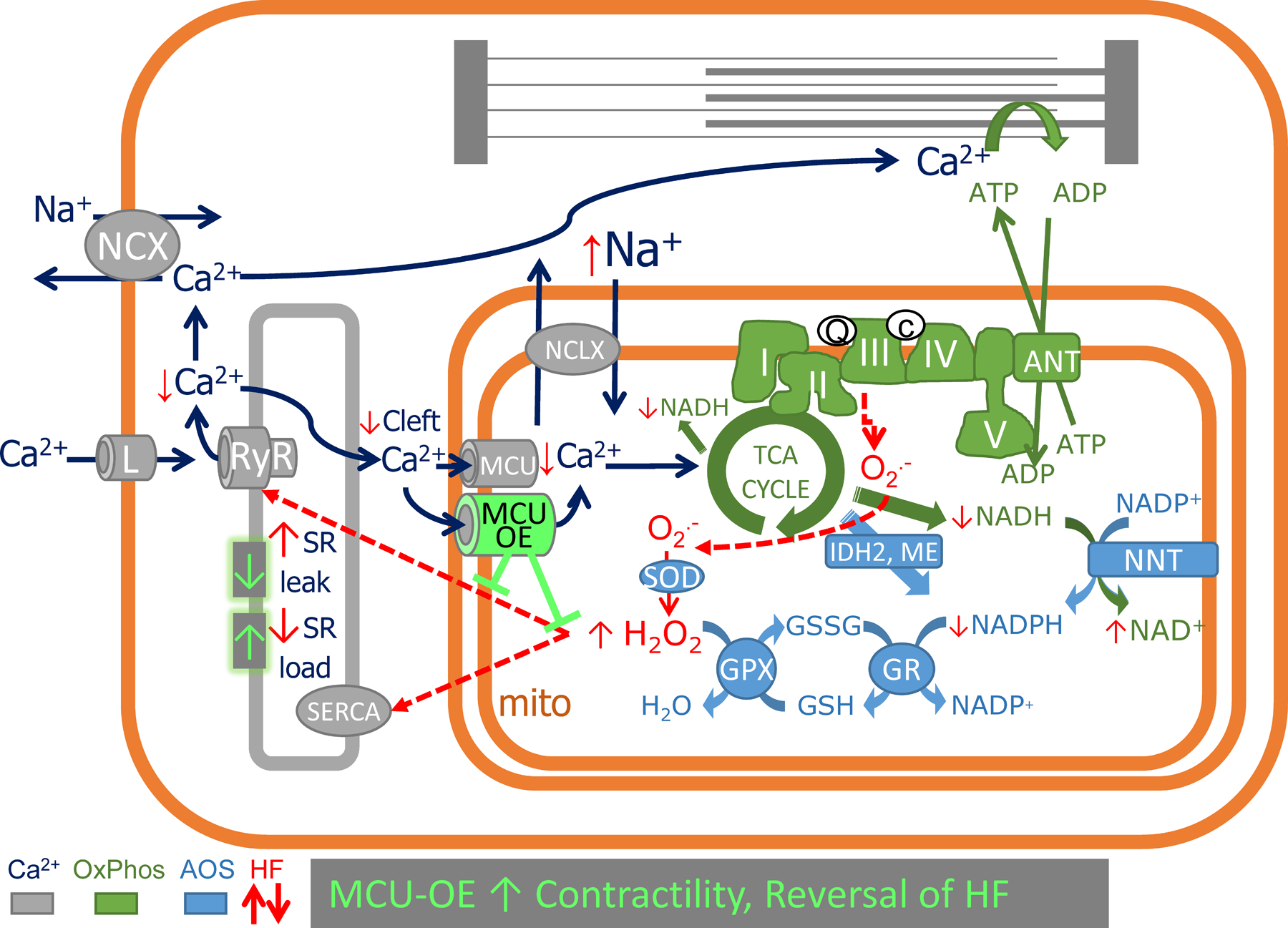
Insufficient mCa2+ accumulation in HF occurs due to impaired Ca2+ release into the diadic/mitochondrial microdomain and acceleration of Ca2+ extrusion via NCLX as a result of cytosolic Na+ overload. Impaired mCa2+ signaling renders the HF cell unable to upregulate NAD(P)H production to meet cellular energy demands, compromising mitochondrial energy production (via NADH) and antioxidant capacity (via NADPH). Increased ROS emission increases RyR-mediated SR Ca2+ leak, resulting in cellular Ca2+ depletion via sarcolemmal NCX. The inability to maintain SR Ca2+ stores further exacerbates the mCa2+ insufficiency in a vicious cycle. MCU overexpression (MCU-OE) restores mitochondrial redox balance, decreases SR Ca2+ leak, increases SR Ca2+ load, and improves frequency- or b-adrenergic- dependent inotropy to prevent or reverse heart failure. AOS, anti-oxidant system; OxPhos, oxidative phosphorylation system; TCA, tricarboxylic acid; RyR, ryanodine receptor; SERCA, SR Ca2+ ATPase pump; L, L-type Ca2+ channel; ANT, adenine nucleotide translocator; NNT, nicotinamide nucleotide transhydrogenase; NCLX, mitochondrial Na+/Ca2+ exchanger; IDH2, isocitrate dehydrogenase 2; ME, malic enzyme; SOD, super oxide dismutase; GPX, glutathione peroxidase; GR, glutathione reductase.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Mitochondrial Ca2+ (mCa2+) is important for the physiological regulation of Krebs cycle dehydrogenases, but excess mCa2+ can trigger the permeability transition pore and cell death.
In animal models of heart failure (HF), the balance between mCa2+ uptake, via the mitochondrial Ca2+ uniporter (MCU), and efflux, through the mitochondrial Na+/Ca2+ exchanger (NCLX) is altered, impacting redox homeostasis.
The benefits of increasing or decreasing mCa2+ have been debated.
What New Information Does This Article Contribute?
Here, we show that moderate overexpression of MCU by in vivo viral gene transfer, restores frequency- and β-adrenergic inotropic responses in isolated myocytes and hearts from a guinea model of HF.
Normalization of mCa2+ in HF mitigated oxidative stress secondary to increased work, which corrected defective sarcoplasmic reticular Ca2+ loading by decreasing ryanodine receptor-mediated Ca2+ leak and RyR2 oxidation and improving Ca2+ pump function.
In vivo MCU overexpression reversed cardiac decompensation and decreased the incidence of premature ventricular complexes (PVC), indicating the potential therapeutic benefit of modest increases in mCa2+ in HF with reduced ejection fraction.
Mitochondrial Ca2+ (mCa2+) plays a dual role in the cardiac cell, at physiological signaling levels, it contributes to energy supply and demand matching by activating Krebs cycle dehydrogenases, but in excess, mCa2+ can trigger the permeability transition pore and cell death. The balance between Ca2+ uptake, through the mitochondrial Ca2+ uniporter (MCU), and efflux, through the mitochondrial Na+/Ca2+ exchanger (NCLX), determines the total mCa2+ load, and in animal models of heart failure (HF), both of these processes are altered. Disparate conclusions have been reported regarding the benefits of increasing or decreasing mCa2+ in HF, leaving the question open as to which direction to modulate this important control node for bioenergetic and redox functions. Here, we show that moderate overexpression of the MCU by viral gene transfer restores frequency- and β-adrenergic- inotropic responses in isolated myocytes and hearts from a guinea model of HF, while reducing sarcoplasmic reticular Ca2+ leak. Interestingly, in vivo MCU overexpression also reversed HF and decreased arrhythmias, suggesting a novel strategy to improve function in HF with reduced ejection fraction.
SOURCES OF FUNDING
This work was supported by National Institutes of Health grants R01HL137259 and R01HL134821 (to B. O’Rourke) and American Heart Association SDG 15SDG25710017 (to T. Liu).
Nonstandard Abbreviations and Acronyms
- EMRE
essential MCU regulator
- FFR
force frequency response
- FS
fractioning shortening
- HF
Heart failure
- HW
heart weight
- ISO
isoprotereno
- LV
left ventricle
- LW
lung weight
- mCa2+
mitochondrial Calcium
- MCU
mitochondrial Calcium Uniporter
- mPTP
mitochondrial permeability transition pore
- NCLX
sodium calcium lithium exchanger
- NCX
sodium calcium exchanger
- PVB
premature Ventricular beat
- RyR
ryanodine receptor
- SCD
sudden cardiac death
- SERCA2a
sarco-endoplasmic reticulum Ca2+ ATPase
- SR
sarcoplasmic reticulum
- SS
sarcomere shortening
- TL
tibia length
- TTC
tetracaine
- VDAC
Voltage-dependent anion channel
Footnotes
Publisher's Disclaimer: This article is published in its accepted form. It has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation Research involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final, published version.
DISCLOSURES
None.
SUPPLEMENTAL MATERIALS
Expanded Materials & Methods (including references 52–56)
Previous data for test of normality
Major Resources Table
Online Figures I–V
REFERENCES
- 1.Bigger JT. Why Patients with Congestive-Heart-Failure Die - Arrhythmias and Sudden Cardiac Death. Circulation. 1987;75:28–35. [PubMed] [Google Scholar]
- 2.Goldman S, Johnson G, Cohn JN, Cintron G, Smith R and Francis G. Mechanism of Death in Heart-Failure - the Vasodilator-Heart Failure Trials. Circulation. 1993;87:24–31. [PubMed] [Google Scholar]
- 3.Kiyuna LA, Albuquerque RPE, Chen CH, Mochly-Rosen D and Ferreira JCB. Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities. Free Radic Biol Med. 2018;129:155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knowlton AA, Chen L and Malik ZA. Heart failure and mitochondrial dysfunction: the role of mitochondrial fission/fusion abnormalities and new therapeutic strategies. J Cardiovasc Pharmacol. 2014;63:196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okonko DO and Shah AM. Heart failure: mitochondrial dysfunction and oxidative stress in CHF. Nat Rev Cardiol. 2015;12:6–8. [DOI] [PubMed] [Google Scholar]
- 6.Rosca MG and Hoppel CL. Mitochondrial dysfunction in heart failure. Heart Fail Rev. 2013;18:607–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou B and Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest. 2018;128:3716–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu T and O’Rourke B. Regulation of mitochondrial Ca2+ and its effects on energetics and redox balance in normal and failing heart. J Bioenerg Biomembr. 2009;41:127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rizzuto R, De Stefani D, Raffaello A and Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nature Reviews Molecular Cell Biology. 2012;13:566–578. [DOI] [PubMed] [Google Scholar]
- 10.Hempel N and Trebak M. Crosstalk between calcium and reactive oxygen species signaling in cancer. Cell Calcium. 2017;63:70–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aon MA, Cortassa S and O’Rourke B. Redox-optimized ROS balance: A unifying hypothesis. Biochimica Et Biophysica Acta-Bioenergetics. 2010;1797:865–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T and O’Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. 2006;99:172–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Marchi U, Santo-Domingo J, Castelbou C, Sekler I, Wiederkehr A and Demaurex N. NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J Biol Chem. 2014;289:20377–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu T, Brown DA and O’Rourke B. Role of mitochondrial dysfunction in cardiac glycoside toxicity. J Mol Cell Cardiol. 2010;49:728–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu T and O’Rourke B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res. 2008;103:279–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Bohm M, O’Rourke B and Maack C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. 2010;121:1606–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pallafacchina G, Zanin S and Rizzuto R. Recent advances in the molecular mechanism of mitochondrial calcium uptake. F1000Res. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu Y, Rasmussen TP, Koval OM, Joiner ML, Hall DD, Chen B, Luczak ED, Wang Q, Rokita AG, Wehrens XH, Song LS and Anderson ME. The mitochondrial uniporter controls fight or flight heart rate increases. Nat Commun. 2015;6:6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, Koch WJ, Cheung JY, Madesh M and Elrod JW. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015;12:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E and Finkel T. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1464–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu T, Takimoto E, Dimaano VL, DeMazumder D, Kettlewell S, Smith G, Sidor A, Abraham TP and O’Rourke B. Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ Res. 2014;115:44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dey S, DeMazumder D, Sidor A, Foster DB and O’Rourke B. Mitochondrial ROS Drive Sudden Cardiac Death and Chronic Proteome Remodeling in Heart Failure. Circ Res. 2018;123:356–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sancak Y, Markhard AL, Kitami T, Kovacs-Bogdan E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, Calvo SE, Goldberger O and Mootha VK. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science. 2013;342:1379–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kettlewell S, Cabrero P, Nicklin SA, Dow JA, Davies S and Smith GL. Changes of intra-mitochondrial Ca2+ in adult ventricular cardiomyocytes examined using a novel fluorescent Ca2+ indicator targeted to mitochondria. Journal of molecular and cellular cardiology. 2009;46:891–901. [DOI] [PubMed] [Google Scholar]
- 25.Cooper LL, Li W, Lu Y, Centracchio J, Terentyeva R, Koren G and Terentyev D. Redox modification of ryanodine receptors by mitochondria-derived reactive oxygen species contributes to aberrant Ca2+ handling in ageing rabbit hearts. J Physiol. 2013;591:5895–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan Y, Liu J, Wei C, Li K, Xie W, Wang Y and Cheng H. Bidirectional regulation of Ca2+ sparks by mitochondria-derived reactive oxygen species in cardiac myocytes. Cardiovasc Res. 2008;77:432–41. [DOI] [PubMed] [Google Scholar]
- 27.Shannon TR, Ginsburg KS and Bers DM. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ Res. 2002;91:594–600. [DOI] [PubMed] [Google Scholar]
- 28.Rizzuto R, Brini M, Murgia M and Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science. 1993;262:744–7. [DOI] [PubMed] [Google Scholar]
- 29.Szalai G, Csordas G, Hantash BM, Thomas AP and Hajnoczky G. Calcium signal transmission between ryanodine receptors and mitochondria. J Biol Chem. 2000;275:15305–13. [DOI] [PubMed] [Google Scholar]
- 30.Hobai IA and O’Rourke B. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation. 2001;103:1577–84. [DOI] [PubMed] [Google Scholar]
- 31.Lindner M, Erdmann E and Beuckelmann DJ. Calcium content of the sarcoplasmic reticulum in isolated ventricular myocytes from patients with terminal heart failure. Journal of molecular and cellular cardiology. 1998;30:743–9. [DOI] [PubMed] [Google Scholar]
- 32.Heinzel FR, MacQuaide N, Biesmans L and Sipido K. Dyssynchrony of Ca2+ release from the sarcoplasmic reticulum as subcellular mechanism of cardiac contractile dysfunction. Journal of molecular and cellular cardiology. 2011;50:390–400. [DOI] [PubMed] [Google Scholar]
- 33.Goh KY, Qu J, Hong H, Liu T, Dell’Italia LJ, Wu Y, O’Rourke B and Zhou L. Impaired mitochondrial network excitability in failing guinea-pig cardiomyocytes. Cardiovasc Res. 2016;109:79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bers DM, Barry WH and Despa S. Intracellular Na+ regulation in cardiac myocytes. Cardiovasc Res. 2003;57:897–912. [DOI] [PubMed] [Google Scholar]
- 35.Cox DA and Matlib MA. A role for the mitochondrial Na(+)-Ca2+ exchanger in the regulation of oxidative phosphorylation in isolated heart mitochondria. J Biol Chem. 1993;268:938–47. [PubMed] [Google Scholar]
- 36.Wei AC, Liu T, Cortassa S, Winslow RL and O’Rourke B. Mitochondrial Ca2+ influx and efflux rates in guinea pig cardiac mitochondria: low and high affinity effects of cyclosporine A. Biochim Biophys Acta. 2011;1813:1373–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zima AV and Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res. 2006;71:310–21. [DOI] [PubMed] [Google Scholar]
- 38.Piacentino V 3rd, Weber CR, Chen X, Weisser-Thomas J, Margulies KB, Bers DM and Houser SR. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res. 2003;92:651–8. [DOI] [PubMed] [Google Scholar]
- 39.Pieske B and Houser SR. [Na+]i handling in the failing human heart. Cardiovasc Res. 2003;57:874–86. [DOI] [PubMed] [Google Scholar]
- 40.Pogwizd SM, Sipido KR, Verdonck F and Bers DM. Intracellular Na in animal models of hypertrophy and heart failure: contractile function and arrhythmogenesis. Cardiovasc Res. 2003;57:887–96. [DOI] [PubMed] [Google Scholar]
- 41.Santulli G, Xie W, Reiken SR and Marks AR. Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci U S A. 2015;112:11389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xie A, Song Z, Liu H, Zhou A, Shi G, Wang Q, Gu L, Liu M, Xie LH, Qu Z and Dudley SC Jr. Mitochondrial Ca(2+) Influx Contributes to Arrhythmic Risk in Nonischemic Cardiomyopathy. J Am Heart Assoc. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holmstrom KM, Pan X, Liu JC, Menazza S, Liu J, Nguyen TT, Pan H, Parks RJ, Anderson S, Noguchi A, Springer D, Murphy E and Finkel T. Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J Mol Cell Cardiol. 2015;85:178–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM and Molkentin JD. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep. 2015;12:15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phillips D, Covian R, Aponte AM, Glancy B, Taylor JF, Chess D and Balaban RS. Regulation of oxidative phosphorylation complex activity: effects of tissue-specific metabolic stress within an allometric series and acute changes in workload. Am J Physiol Regul Integr Comp Physiol. 2012;302:R1034–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Griffiths EJ. Species dependence of mitochondrial calcium transients during excitation-contraction coupling in isolated cardiomyocytes. Biochem Biophys Res Commun. 1999;263:554–9. [DOI] [PubMed] [Google Scholar]
- 47.Bertero E and Maack C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ Res. 2018;122:1460–1478. [DOI] [PubMed] [Google Scholar]
- 48.Maier LS, Wahl-Schott C, Horn W, Weichert S, Pagel C, Wagner S, Dybkova N, Muller OJ, Nabauer M, Franz WM and Pieske B. Increased SR Ca2+ cycling contributes to improved contractile performance in SERCA2a-overexpressing transgenic rats. Cardiovasc Res. 2005;67:636–46. [DOI] [PubMed] [Google Scholar]
- 49.Ziolo MT, Martin JL, Bossuyt J, Bers DM and Pogwizd SM. Adenoviral gene transfer of mutant phospholamban rescues contractile dysfunction in failing rabbit myocytes with relatively preserved SERCA function. Circ Res. 2005;96:815–7. [DOI] [PubMed] [Google Scholar]
- 50.Huke S, Liu LH, Biniakiewicz D, Abraham WT and Periasamy M. Altered force-frequency response in non-failing hearts with decreased SERCA pump-level. Cardiovasc Res. 2003;59:668–77. [DOI] [PubMed] [Google Scholar]
- 51.Bovo E, Mazurek SR and Zima AV. The role of RyR2 oxidation in the blunted frequency-dependent facilitation of Ca(2+) transient amplitude in rabbit failing myocytes. Pflugers Arch. 2018;470:959–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wei AC, Liu T, Cortassa S, Winslow RL and O’Rourke B. Mitochondrial Ca2+ influx and efflux rates in guinea pig cardiac mitochondria: low and high affinity effects of cyclosporine A. Biochim Biophys Acta. 2011;1813:1373–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu T, Takimoto E, Dimaano VL, DeMazumder D, Kettlewell S, Smith G, Sidor A, Abraham TP and O’Rourke B. Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ Res. 2014;115:44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chiang DY, Li N, Wang Q, Alsina KM, Quick AP, Reynolds JO, Wang G, Skapura D, Voigt N, Dobrev D and Wehrens XH. Impaired local regulation of ryanodine receptor type 2 by protein phosphatase 1 promotes atrial fibrillation. Cardiovasc Res. 2014;103:178–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu T, Brown DA and O’Rourke B. Role of mitochondrial dysfunction in cardiac glycoside toxicity. J Mol Cell Cardiol. 2010;49:728–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sikkel MB, Francis DP, Howard J, Gordon F, Rowlands C, Peters NS, Lyon AR, Harding SE and MacLeod KT. Hierarchical statistical techniques are necessary to draw reliable conclusions from analysis of isolated cardiomyocyte studies. Cardiovasc Res. 2017;113:1743–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all supporting data are available within the article. Constructs used will be made available upon request from the authors.