

Summary:
Adherent-Invasive E. coli (AIEC) are enriched in the intestinal microbiota of patients with Crohn’s disease (CD) and promote intestinal inflammation. Yet, how AIEC metabolism of nutrients impacts intestinal homeostasis is poorly defined. Here, we show that AIEC encoding the large subunit of propanediol dehydratase (PduC), which facilitates utilization of fucose fermentation product 1,2-propanediol, are increased in the microbiome of CD patients and drive AIEC-induced intestinal T cell inflammation. In murine models, CX3CR1+ mononuclear phagocytes (MNPs) are required for PduC-dependent induction of Th17 cells and IL-1β production that leads to AIEC-induced inflammatory colitis. Activation of this inflammatory cascade requires catalytic activity of PduC to generate propionate, which synergizes with LPS to induce IL-1β by MNPs. Disrupting fucose availability limits AIEC-induced propionate production and intestinal inflammation. These findings identify MNPs as metabolic sensors linking AIEC metabolism with intestinal inflammation and identify microbial metabolism as a potential therapeutic target in Crohn’s disease treatment.
In Brief:
Viladomiu and Metz et al. show that propanediol metabolism by Crohn’s disease Adherent-Invasive E. coli is critical for promoting intestinal T cell inflammation. They identify a central role for CX3CR1+ mononuclear phagocytes in sensing AIEC-derived propionate to trigger IL-1β and establish a link between AIEC metabolism and tissue inflammation.
Graphical Abstract
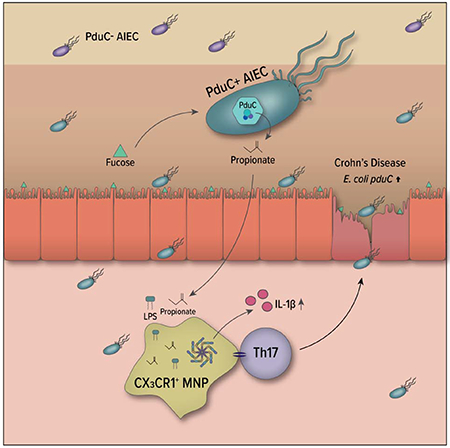
Introduction:
Crohn’s disease (CD) is a chronic inflammatory disease of the intestine that affects over 4 million people worldwide. There is no cure for CD and, despite medical treatment, nearly half of the patients who require therapy develop refractory disease. CD is associated with characteristic changes in the intestinal microbiome including the expansion of the pathobiont Adherent-Invasive E.coli (AIEC) (Darfeuille-Michaud et al., 2004; Dogan et al., 2014). Expansion of AIEC correlates with intestinal and systemic inflammatory burden in CD (Darfeuille-Michaud et al., 2004; Gevers et al., 2014; Viladomiu et al., 2017), and AIEC are sufficient to promote intestinal and systemic T cell inflammation in pre-clinical models (Eun et al., 2014; Viladomiu et al., 2017). The AIEC pathovar is defined by the ability to adhere to and invade epithelial cells, but comparative genomics have not identified any exclusive genetic signature for AIEC (O’Brien et al., 2017). As such, understanding AIEC-host interactions and the mechanisms by which microbial factors influence host immunity is needed to define therapeutic targets.
One key to understanding this interaction is the ability of AIEC to adhere to and invade the epithelial surface. While AIEC proteases facilitate mucin degradation and long polar fimbriae enable binding to M cells (Chassaing et al., 2011), the ability of AIEC to use degradation and fermentation products not accessible to commensals in the lumen promotes their fitness in the mucosal layer. These products enriched in the mucosal layer of the intestine include myo-inositol, ethanolamine cleaved from mucosal phospholipids, fucose cleaved from mucosal glycoconjugates, and 1,2-propanediol as the fermentation product of fucose or rhamnose (Delmas et al., 2019). Intestinal physiology and pH support AIEC growth (Zhang et al., 2020) and physiologic levels of bile salts induce these metabolic pathways in AIEC during homeostasis (Delmas et al., 2019); however, in the context of inflammation, this metabolic adaptation by AIEC allow them to competitively thrive in an inflamed gut (Kitamoto et al., 2020; Winter et al., 2013).
Under conditions of inflammation, enteropathogens benefit from their ability to utilize alternate carbon sources to fuel respiration (Faber et al., 2017). Salmonella serovars associated with gastroenteritis encode genes required for metabolic utilization of the fucose fermentation product, 1,2-propanediol, as a carbon source. The propanediol utilization (pdu) genes provide a mechanism for the competitive fitness of S. Typhimurium in the inflamed intestine (Faber et al., 2017). The large subunit of the propanediol dehydratase complex, which contains the active site that converts 1,2-propanediol to propionaldehyde, is encoded by pduC. AIEC encoding pduC show increased epithelial invasion and survival in macrophages (Dogan et al., 2014), but unlike Salmonella and other enteropathogens, AIEC lack the pathogenic effectors of the type III secretion systems. We propose that the metabolic fitness of AIEC allow it to act as an opportunistic pathobiont in conditions of intestinal inflammation or genetically susceptible individuals predisposed to inflammation, and that metabolic function plays a direct role in regulating AIEC-host immunity in Crohn’s disease.
Here, we show that utilization of the alternative carbon source, 1,2-propanediol, by AIEC is critical for promoting T cell-driven intestinal inflammation. Our work reveals a central role for CX3CR1+ mononuclear phagocytes (MNP) in sensing microbial metabolites to shape the balance between homeostasis and inflammation. Moreover, our results highlight the potential role for regulating nutrient availability to restore this balance and limit disease. These findings highlight a targetable link between AIEC metabolism and intestinal inflammation with therapeutic potential.
Results:
pduC-encoding AIEC are expanded in Crohn’s disease and drive inflammatory T cell-mediated colitis
Given the increased prevalence of pduC in AIEC compared to commensal E. coli (Dogan et al., 2014), we evaluated the association of pduC gene abundance with Crohn’s disease. We determined read count abundance of pduC using metagenomic data from 24 healthy and 22 CD participants. Defining pduC at 90% sequence homology (Suzek et al., 2015b), participants with CD had significantly higher reads for pduC which mapped primarily to the Escherichia genus compared to healthy controls (Fig. 1A, B). To validate these findings in an additional cohort, we compared pduC gene abundance in a publicly available cohort (McGovern et al., 2010). pduC abundance was 10-fold higher in CD compared to healthy controls while no difference was observed between ulcerative colitis participants and healthy controls (Fig. S1A). 16S rRNA sequencing composition analysis of our internal cohort revealed that Enterobacteriaceae and Escherichia/Shigella taxa are expanded in Crohn’s disease (Fig. S1B, C). Broader comparison of pduC at 50% homology to encompass the glycerol dehydratase enzyme family revealed no difference in the total read abundance, but a shift in the genera that encode these enzymes from commensals including Lachnospiraceae and Blautia to expanded Enterobacteriaceae in Crohn’s disease including Escherichia (Fig. 1C and Fig. S1C, D).
FIGURE 1. CD-derived AIEC induces intestinal Th17 cells and inflammation in a pduC-dependent manner.
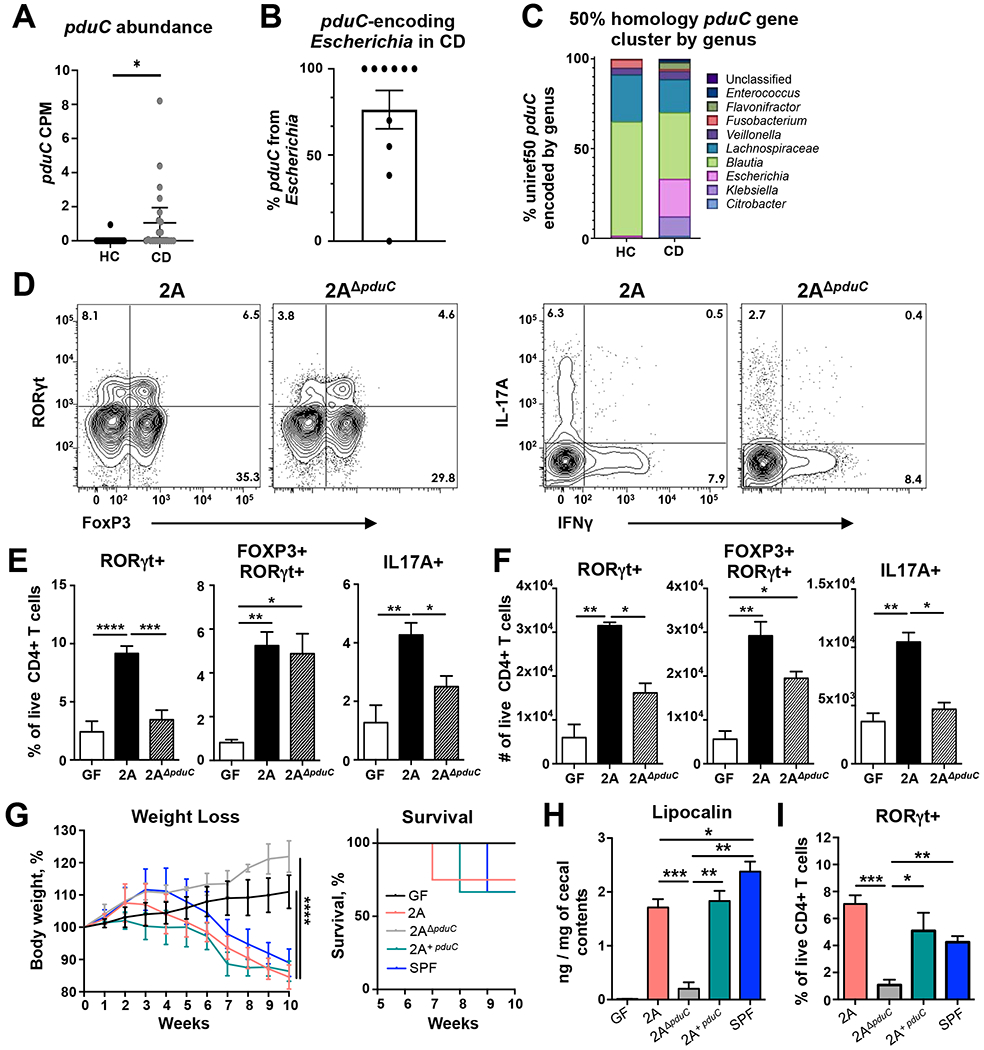
A. Read counts per million (CPM) base pairs of pduC in metagenomic sequencing from healthy controls (HC, N=24) compared to Crohn’s disease (CD, N=22). Error bars represent SEM. *p<0.05, unpaired t-test. B. Percentage of pduC reads encoded by the Escherichia genus. C. Average percentage of pduC reads by genus based on 50% homology for HC and CD patients. D-F. Germ-free C57BL/6 mice were colonized with 2×109 CFU of CD-derived, AIEC 2A or pduC-deficient isogenic mutant (2AΔpduC) and analyzed after 15 days. Representative flow cytometry of live, CD4+ T cells was used to evaluate RORγt/FoxP3 and IL17/IFNγ expression following a 4h stimulation with PMA/ionomycin and Brefeldin-A (D). Percentage (E) and total numbers (F) of colonic RORγt+, RORγt+FOXP3+ and IL17A+ producing CD4+ T cells was evaluated. Bar graphs represent geometric mean of 6-8 mice per group from two of four total experiments. Error bars represent SEM. *p<0.05, **p<0.01, ***p<0.005, ****p<0.001 ANOVA. G-I. Germ-free Rag1-deficient mice were colonized with 2×109 CFU of AIEC 2A, 2AΔpduC or complement 2AΔpduC+pduC. One week after colonization, mice received 500,000 FACS-sorted, naïve CD4+ T cells intraperitoneally. Mice were monitored for weight loss and survival (G) for 10 weeks. Levels of lipocalin in cecal contents was measured by ELISA at 10 weeks (H). Percentage of colonic RORγt+ CD4+ T cells was evaluated by flow cytometry (I). Graphs represent geometric mean of at least 3 mice per group from one of three experiments. Error bars represent SEM. *p<0.05, **p<0.01, ***p<0.005 ANOVA. See also Figures S1–S3.
Our previous work identified the ability of AIEC to induce inflammatory T cell immunity (Viladomiu et al., 2017). To test the contribution of the propanediol metabolic pathway to intestinal inflammation using a human-derived AIEC from a patient with CD, we generated a pduC-deficient mutant of AIEC 2A (Fig. S2A). pduC deletion was confirmed by PCR (Fig. S2B). Despite robust and equal mono-colonization in gnotobiotic C57BL/6 mice (Fig. S2C), WT AIEC 2A maintained a competitive fitness over 2AΔpduC during co-colonization in vivo (Fig. S2D,E). Mono-colonization with pduC-deficient mutant resulted in decreased number and frequency of RORγt+ CD4+ T cells, as well as, reduced IL-17A and inflammatory IL-17A/IFNγ-producing CD4+ T cells in the colonic lamina propria (Fig. 1D–F). No significant differences in the numbers or frequency of TBET+, GATA3+ or FOXP3+ CD4+ T cells were observed between AIEC 2A and isogenic mutant 2AΔpduC (Fig. S2F). Consistent with our previous work, AIEC does induce IL-22 production from intestinal ILC3s; however, no significant difference in ILC3 cytokine production was observed in mice colonized with 2AΔpduC at steady state (Fig. S2G).
To define the contribution of pduC to AIEC pathogenicity in a T cell-dependent model of colitis, the T cell transfer colitis model was evaluated in gnotobiotic mice. Briefly, Rag1-deficient, germ-free mice were monocolonized with AIEC 2A or 2AΔpduC for one week, followed by I.P. injection of 500,000 naïve CD4+ T cells to induce colitis. Mice colonized with 2AΔpduC showed significant reduction in weight loss, mortality, histology score, and fecal lipocalin compared to 2A and SPF controls (Fig. 1G–H, Fig. S2H–J). Cis-complementation of the pduC-deficient strain with pduC (called 2A+pduC) was sufficient to restore the more severe colitis phenotype. Mice colonized with AIEC 2A receiving IL-23R-deficient T cells lost less weight, showed improved survival, and had significantly lower fecal lipocalin than mice receiving WT T cells, revealing a primary role for Th17 inflammation (Fig. S3A–C). Consistent with these results, RORγt+ CD4+ T cells were significantly lower in mice colonized with 2AΔpduC compared to 2A and SPF controls (Fig. 1I). Collectively, these data highlight the critical contribution of pduC to T cell-mediated intestinal inflammation induced by CD-derived AIEC.
AIEC-encoded pduC promotes CX3CR1+ mononuclear phagocyte production of IL-1β driving intestinal inflammation
Our previous data (Viladomiu et al., 2017) as well as others (Ellermann et al., 2019) have shown that AIEC induction of IL-10 and IL-23 are important in regulating AIEC-induced immunity. To test the role for IL-10 in limiting pduC-dependent pathogenicity during colitis, wildtype or Il10-deficient mice mono-colonized with AIEC 2A or 2AΔpduC were subjected to dextran sodium sulfate (DSS)-induced colitis. Consistent with our previous data, IL-10 limited AIEC induced weight loss and mortality; however, in the absence of IL-10, mice colonized with 2ADpduC showed significantly less weight loss and mortality compared to AIEC 2A (Fig. 2A and 2B). Furthermore, qPCR of intestinal lamina propria mononuclear cells (LPMCs) from mice colonized with 2AΔpduC revealed no significant change in il10 and il23p19 compared to mice colonized with AIEC 2A (Fig. 2C). In contrast, LPMCs from mice colonized with 2AΔpduC showed a significant decrease in il1b. To determine regulation of secreted IL-1β protein, colon explants were incubated overnight. IL-1β ELISA of secreted protein in supernatant revealed that pduC was required for IL-1β protein secretion and that the induction of IL-1β was not elicited by another Gram-negative commensal, Bacteroides thetaiotamicron (Fig. 2D).
FIGURE 2. pduC-induced IL-1β mediates AIEC-dependent colitis.
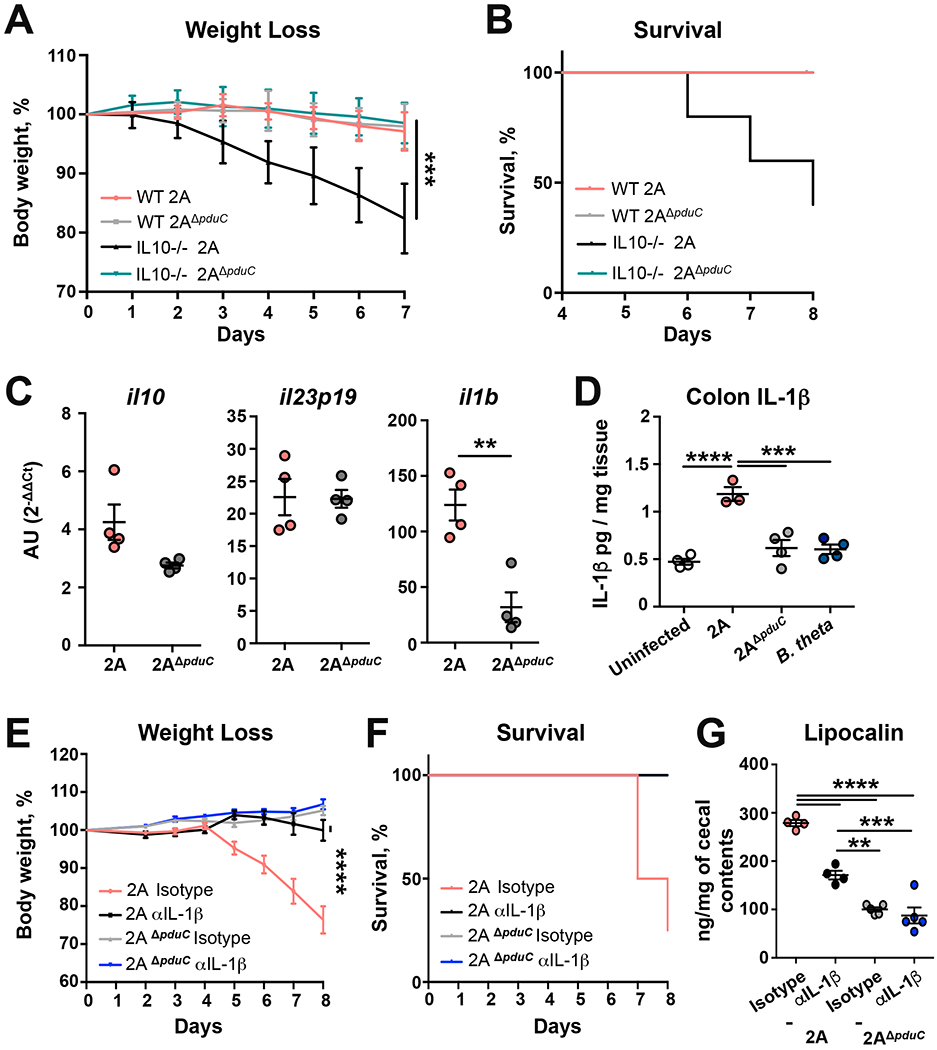
A-B. Germ-free C57BL/6 WT and Il10-deficient mice were colonized with 2×109 CFU of AIEC 2A or pduC-deficient isogenic mutant (2AΔpduC). Ten days after colonization, mice were exposed to 2% dextran sodium sulfate ad libitum for 7 days. Percent weight loss (A) and survival (B) is shown. C,D. C57BL/6 B6 SPF mice were treated with broad spectrum antibiotics for 2 weeks prior to colonization with 1010 CFU of AIEC 2A or 2AΔpduC. Il10, Il23p19 and Il1b expression in the colonic lamina propria mononuclear cells (LPMCs) was measured 5 days after colonization (C). IL-1β production was measured by ELISA in colon explants cultured overnight (D). Graphs show at least 4 mice from one of two experiments. Error bars represent SEM. **p<0.01, ***p<0.005, ****p<0.001 ANOVA. E-G. Germ-free C57BL/6 Il10-deficient mice were colonized with 2×109 CFU of AIEC 2A or 2AΔpduC. Ten days after colonization, mice were exposed to 2% dextran sodium sulfate ad libitum for 7 days. Mice were treated intraperitoneally with anti-IL1β or isotype control on days −2, 0, 2, 4, and 6 of DSS treatment. Weight loss (E), percent survival (F) and levels of lipocalin in cecal contents (G) are shown. Graphs show at least 4 mice from one of two experiments. Error bars represent SEM. **p<0.01, ***p<0.005, ****p<0.001 ANOVA. See also Figure S3.
To determine if IL-1β contributed to AIEC-induced intestinal inflammation, we used a neutralizing antibody to block IL-1β in mono-colonized, IL10-deficient, DSS-treated mice. Blockade of IL-1β reduced weight loss, improved survival, and reduced fecal lipocalin levels compared to mice treated with isotype control (Fig. 2E–G and Fig. S3D). Blockade of RORγt with the chemical inhibitor GSK805 confirmed a critical downstream role for pathogenic Th17 effectors in this model (Fig. S3E–I). In contrast to mice colonized with AIEC 2A, IL-1β blockade in mice colonized with 2AΔpduC revealed no significant difference in inflammation, weight loss, or fecal lipocalin (Fig. 2E–G). Collectively, these findings highlight a coordinated regulation of IL-10, IL-23, and pduC-dependent induction of IL-1β in AIEC-induced intestinal inflammation.
Intestinal mononuclear phagocytes (MNPs) and conventional dendritic cells (DCs) play a central role in mediating mucosal Th17 immunity to gut microbiota and regulating the production of IL-10, IL-23, and IL-1b (Kim et al., 2018; Kinnebrew et al., 2012; Panea et al., 2015; Welty et al., 2013). To evaluate the relative impact of these subsets in the induction of Th17 cells by AIEC 2A, we utilized genetic mouse models to perform selective depletion of these subsets in vivo (Fig. S4). Injection of diptheria toxin (DT) was used to selectively deplete CX3CR1+ MNPs expressing the diptheria toxin receptor (DTR) using Itgax-cre Cx3cr1-LSL-DTR mice (Diehl et al., 2013) or conventional DCs using Lang-DTREGFP mice (Welty et al., 2013). After 10 days of colonization with AIEC 2A, depletion of MNPs resulted in a significant reduction of RORγt+ and IL17A-producing CD4+ T cells as well as CD4+ Foxp3/RORγt+ Tregs compared to Cre-littermate controls (Fig. 3A–C). In contrast to CX3CR1+ MNPs, depletion of CD103+ DCs did not significantly impact RORγt+ CD4+ T cells and IL-17A production (Fig. 3A–C). Depletion of CX3CR1+ MNPs did not impact Th17 cells in mice colonized with 2AΔpduC (Fig. 3D). To test if CX3CR1+ MNPs were the major source of AIEC 2A-induced IL-1β in vivo, MNPs were depleted and LPMCs were assessed for IL-1β secretion. Mice with depleted MNPs showed significantly reduced IL-1β production compared to littermate controls, which was not observed in CD103+ DC-depleted mice or mice colonized with 2AΔpduC (Fig. 3E). These findings build a model in which CX3CR1+ MNPs regulate AIEC induction of inflammatory T cells through pduC-dependent induction of IL-1β.
FIGURE 3. CX3CR1+ mononuclear phagocytes regulate AIEC induction of intestinal Th17 cells.
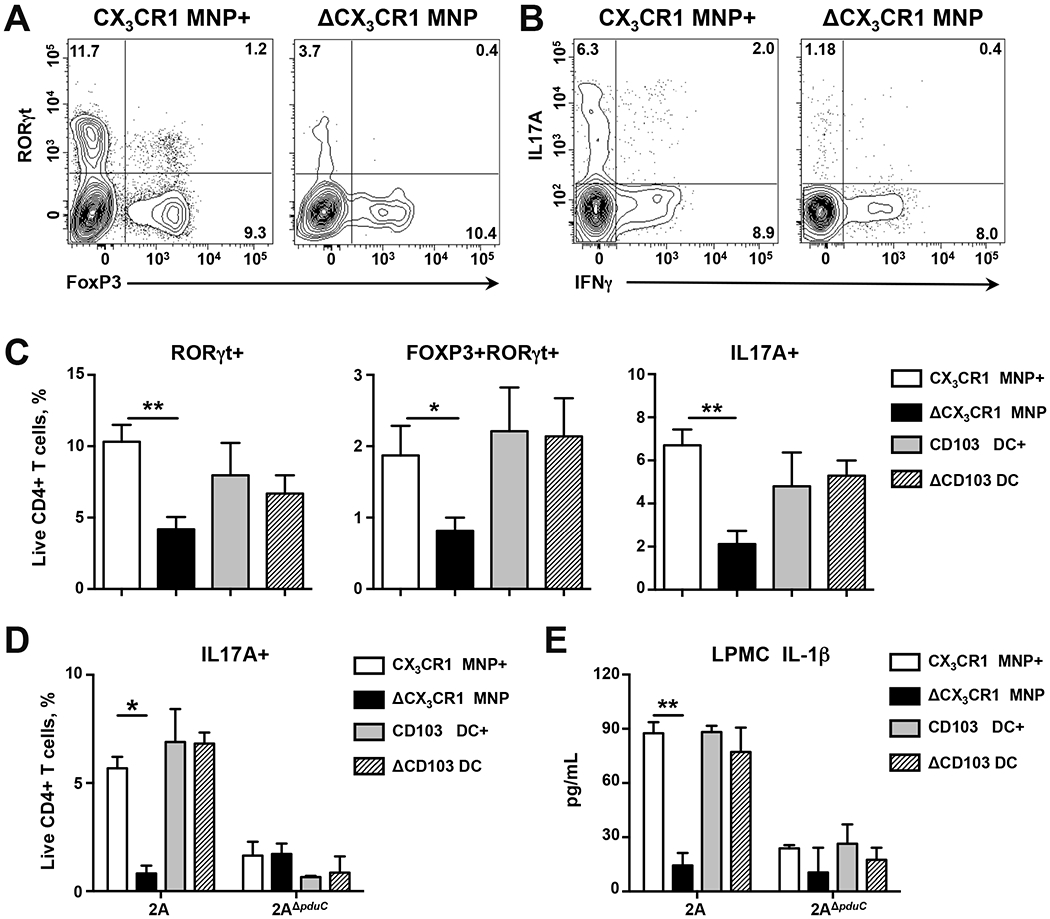
A-C. 4-week-old littermate Cx3cr1-LSL-DTR (labeled CX3CR1 MNP+) and Itgax-cre Cx3cr1-LSL-DTR (labeled ΔCX3CR1 MNP) mice were treated with diphtheria toxin (DT) before and during colonization with 1010 CFU AIEC 2A. 4-week-old littermate Lang-DTREGFP mice were treated with DT (labeled ΔCD103 DC) or PBS (labeled CD103 DC+) before and during colonization with AIEC 2A. Flow cytometry of live, CD4+ T cells was used to evaluate RORγt/FoxP3 and IL17/IFNγ expression following a 4h stimulation with PMA/ionomycin and Brefeldin-A (A, B). Percentage of colonic RORγt+, RORγt+FoxP3+ and IL-17A+ producing CD4+ T cells was evaluated (C). Bar graphs represent geometric mean of 12-15 mice per group from three total experiments. Error bars represent SEM. *p<0.05, **p<0.01 ANOVA. D, E. 4-week-old littermate Cx3cr1-LSL-DTR (labeled CX3CR1 MNP+) and Itgax-cre Cx3cr1-LSL-DTR (labeled ΔCX3CR1 MNP) mice were treated with diphtheria toxin (DT) before and during colonization with 1010 CFU AIEC 2A or 2AΔpduC. 4-week-old littermate Lang-DTREGFP mice were treated with DT (labeled ΔCD103 DC) or PBS (labeled CD103 DC+) before and during colonization with AIEC 2A or 2AΔpduC. Percentage of colonic IL-17A+ producing CD4+ T cells was evaluated following a 4h stimulation with PMA/ionomycin and Brefeldin-A (D). IL-1β production by colonic lamina propria mononuclear cells (LPMCs) was measured by ELISA (E). Bar graphs represent geometric mean of 4 mice per group. Error bars represent SEM. *p<0.05, **p<0.01 ANOVA. See also Figure S4.
Metabolic activity of PduC is required for AIEC-mediated intestinal inflammation
Propanediol dehydratase regulates the conversion of 1,2-propanediol to propionaldehyde, which is ultimately converted to propionate (Fig. S5A). In order to assess the importance of the catalytic activity of the dehydratase in AIEC-mediated immunity, we generated an AIEC 2A with a catalytically-inactive PduC (Fig. S5B). The active site of PduC was assessed by homology modeling and three residues were identified for site-directed mutagenesis and recombined into the locus for pduC (Fig. S5C). Biochemical assessment of the recombinant propanediol dehydratase complex, PduCDE, revealed a titratable and coenzyme B12-dependent conversion of 1,2-propanediol to propionaldehyde, which was lost in the enzyme complex containing the active site mutations (called PduC*) (Fig. 4A). Growth assays confirmed that while the active site disruption did not impact growth in glucose, a selective growth impairment was observed in 2AΔpduC and the catalytically-inactive mutant 2ApduC* compared to AIEC 2A when forced to use 1,2-propanediol as the sole carbon source (Fig. 4B).
FIGURE 4. PduC metabolic activity is required for T cell-dependent inflammatory colitis.
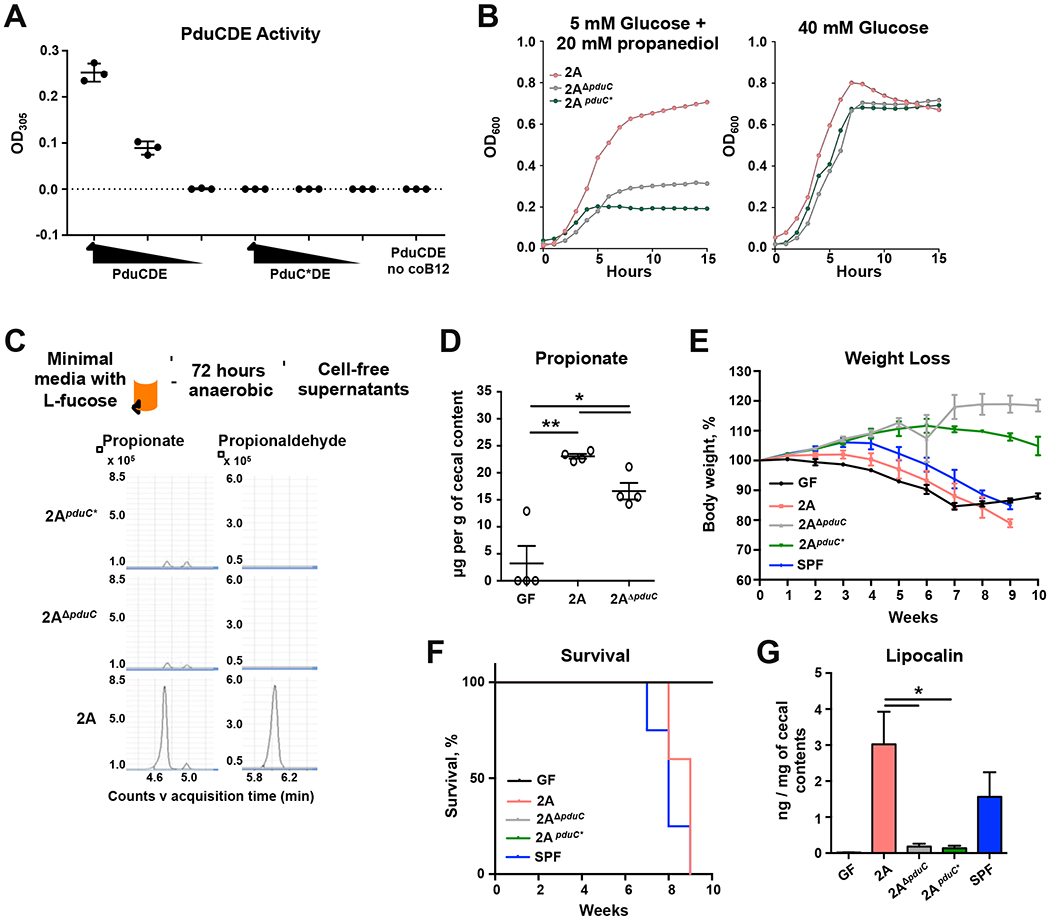
A. Purified, wild-type PduCDE or catalytically-inactive PduC*DE protein complexes (66 μL/mL, 33 μg/mL, 6.6 μg/mL, respectively) were incubated with 1,2-propanediol in the presence or absence of essential cofactor, vitamin B12. Catalytic activity was determined by absorbance after incubation with MBTH. B. Growth curves for AIEC 2A, 2AΔpduC or 2ApduC* grown anaerobically in M9-minimal media containing 40 mM D-glucose or 5 mM D-glucose with 20mM 1,2-propanediol as the sole carbon source for 15 hours. C. AIEC 2A, 2AΔpduC or 2ApduC* were grown anaerobically in M9-minimal media containing 20 mM L-fucose as the sole carbon source for 72 hours. Cell-free supernatants from these cultures were analyzed by derivatized LC-MS for targeted detection of propionate and propionaldehyde. Error bars represent SEM. *p<0.05, **p<0.01 Welch’s t-test. D. Germ-free C57BL/6 WT mice were colonized with 2×109 CFU of AIEC 2A or 2AΔpduC. Propionate levels in cecal contents were assessed five days after colonization. Graph shows 4 mice from one experiment. Error bars represent SEM. *p<0.05, **p<0.01 t-test, Welch’s test. E-G. Germ-free Rag1-deficient mice were colonized with 2×109 CFU of AIEC 2A, 2AΔpduC or 2ApduC*, or fecal contents from SPF mice. One week after colonization, mice received 500,000 FACS-sorted, naïve CD4+ T cells intraperitoneally. Mice were monitored for weight loss (E) and survival (F) for 10 weeks. Levels of lipocalin in colon contents was measured by ELISA (G). Graphs represent geometric mean of at least 3 mice per group from one of three experiments. Error bars represent SEM. *p<0.05 ANOVA. See also Figure S5.
In addition to providing a competitive advantage to scavenge for nutrients, we hypothesized that 1,2-propanediol utilization by pduC-encoding AIEC produces a metabolic signal that regulates the mucosal immune response. Mass spectrometry analysis of cell-free supernatants from AIEC cultures grown in minimal media with L-fucose revealed a specific reduction in the downstream product propionate and the aldehyde intermediate, propionaldehyde, from 2AΔpduC and 2ApduC* strains compared to WT AIEC 2A (Fig. 4C). To determine the impact of PduC metabolism from AIEC in vivo, short chain fatty acids (SFCAs) were quantitated in cecal contents of germ-free and mono-colonized mice with 2A or 2AΔpduC. Colonization with AIEC resulted in a detectable increase in propionate levels compared to germ-free controls, while 2AΔpduC-colonized mice showed significantly reduced propionate compared to AIEC 2A (Fig. 4D). Acetate levels were not affected by AIEC colonization and butyrate was not detected (Fig. S5D).
Next, to determine if this catalytic activity of PduC was required for AIEC-mediated, T cell-driven intestinal inflammation, gnotobiotic Rag1-deficient mice were mono-colonized with AIEC 2A, 2AΔpduC, or 2ApduC* and subjected to the T cell transfer colitis model. Consistent with our findings for pduC-deficient 2A, mice colonized with 2ApduC* had attenuated weight loss (Fig. 4C), improved survival (Fig. 4D), and a reduction in fecal lipocalin levels (Fig. 4E) despite equal colonization (Fig. S5E). These results demonstrate that metabolic activity of PduC is required for AIEC pathogenesis in T cell-dependent colitis.
AIEC-derived propionate regulates MNP production of IL1b
To evaluate if pduC-dependent metabolites mechanistically regulated mucosal immunity, cell-free supernatants from AIEC 2A or 2AΔpduC cultures were used to stimulate bone marrow-derived macrophages. Consistent with our observations in vivo (Fig. 2C), 2AΔpduC supernatants induced significantly less IL-1β compared to AIEC 2A (Fig. 5A) but no difference in the induction of IL-10 (Fig. S5F). To test if the product of the Pdu pathway, propionate, was sufficient to rescue 2AΔpduC supernatant induction of IL-1β, BM-derived macrophages were exposed to culture supernatants from 2AΔpduC supplemented with 100nM propionate (consistent with the physiologic concentration of propionate contributed by AIEC in vivo as detected in Fig. 4D) (Fig. 5A). Propionate, in synergy with 2AΔpduC supernatants, but not alone, increased IL-1β production. Induction of IL-1β was not observed with supernatants from another propionate-producing commensal B. thetaiotamicron. LPS depletion from the supernatants confirmed AIEC LPS is required for IL-1β secretion and that propionate alone does not synergize with another factor in the supernatant (Fig. 5A, Fig. S5G). While high levels of LPS (>100ng) can trigger detectable levels of IL-1β in our system, propionate synergized with lower levels of LPS, which were not sufficient to trigger detectable levels of IL-1β (Fig. 5B). Quantification of LPS in the cell-free supernatants revealed comparable levels of LPS from both AIEC 2A and 2AΔpduC (Fig. S5G), and this concentration (~1 ng/mL) was within the dynamic range in which propionate synergistically amplified induction of IL-1β. To further test the physiologic relevance of AIEC-dependent propionate, BM-derived macrophages were stimulated with cecal filtrates. Filtrates from 2AΔpduC mice stimulated significantly less IL-1β compared to filtrates from 2A colonized mice (Fig. 5C). Collectively these data demonstrate that under physiologic conditions, PduC-dependent propionate from AIEC synergizes with LPS to induce IL-1β production by macrophages.
FIGURE 5. AIEC-derived propionate regulates MNP production IL-1β production.
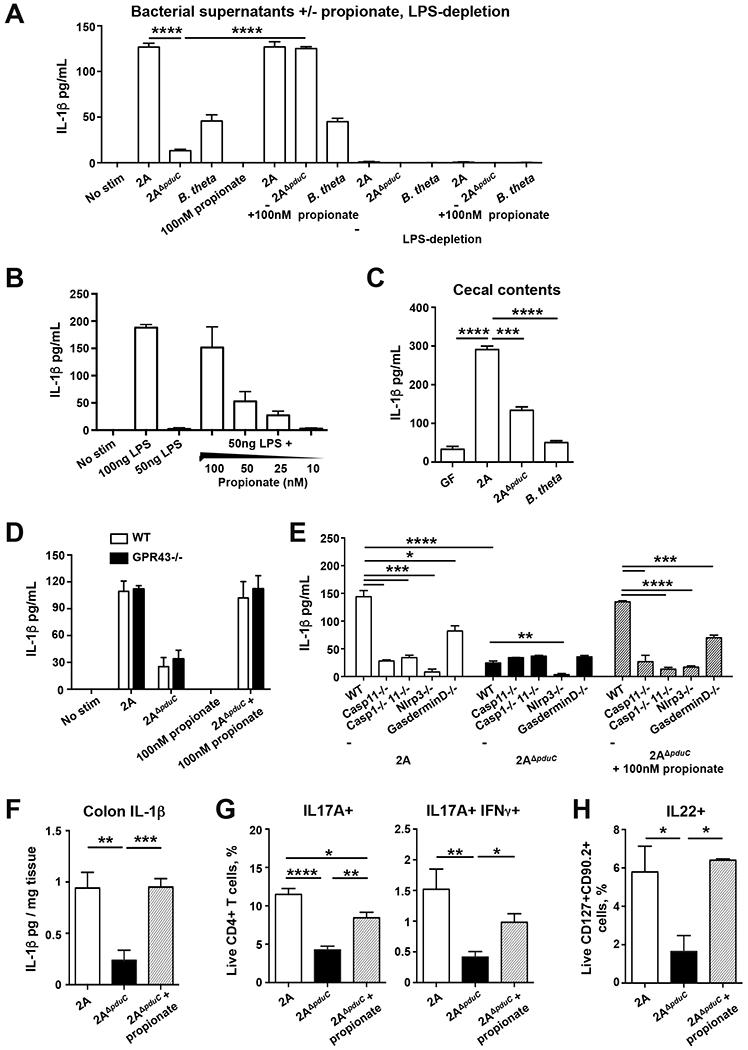
A-B. AIEC 2A or 2AΔpduC were grown anaerobically in M9-minimal media containing 20 mM L-fucose as the sole carbon source for 72 hours. BMDM were stimulated for 16h with cell-free supernatants and LPS-depleted supernatants in the presence or absence of 100nM propionate and IL-1β production was measured by ELISA (A). BMDM were stimulated with 50ng LPS along with decreasing concentrations of propionate to measure IL-1β induction (B). Graphs show at least 3 samples per group from one of four total experiments. Error bars represent SEM. *p<0.05, **p<0.01, ***p<0.005, ****p<0.001 ANOVA. C. BMDM were stimulated with cecal filtrates of monocolonized mice for 16h to measure IL-1β induction. Graphs show at 4 samples per group from one of two total experiments. Error bars represent SEM. ***p<0.005, ****p<0.001 ANOVA. D, E. Cell-free supernatants in the presence or absence of 100nM propionate were used to stimulate bone marrow-derived wild-type and GPR43-deficient macrophages (D) or immortalized WT, Caspase 11-, Caspase1/11-, Nlrp3- and Gasdermin D-deficient macrophages (E) for 16h. IL-1β production was measured by ELISA. Graphs show at least 4 samples from one of three total experiments. Error bars represent SEM. *p<0.05, **p<0.01, ***p=0.005, ****p=0.001, ANOVA. F-H. Germ-free Il10-deficient mice were colonized with 2×109 CFU AIEC 2A or pduC-deficient isogenic mutant (2AΔpduC) for 7 days. A subset of 2AΔpduC-colonized mice were treated with 150nM of propionate jn the drinking water starting one day before colonization. IL-1β production was measured in colon explants cultured overnight (F). Percentage of colonic IL17A+ and IL-17+/IFNγ+ producing CD4+ T cells (G) and IL-22-producing ILC3s (H) was evaluated following a 4h stimulation with PMA/ionomycin with Brefeldin-A. Graphs show 7-8 samples from two pooled experiments. Error bars represent SEM. *p<0.05, **p<0.01, ***p=0.005, ****p=0.001, ANOVA. See also Figure S5.
To determine if this synergy between AIEC-derived LPS and propionate required the short chain fatty acid (SCFA) receptor GPR43, BM-derived macrophages from WT and Gpr43-deficient mice were stimulated with AIEC 2A and 2AΔpduC supernatants with or without propionate. The effect of pduC-sufficient supernatants and the synergistic ability of propionate to trigger MNP production of IL-1β was independent of GPR43 (Fig. 5D). To assess if propionate regulation of MNP IL-1β production required conventional inflammasome machinery, immortalized wild-type, NLRP3-, Caspase11-, Caspase1/11-, and GasderminD-deficient macrophages were stimulated with cell-free supernatants with or without propionate. Induction of IL-1β by supernatants from WT AIEC 2A and propionate-supplemented 2AΔpduC supernatants required both NLRP3 and Caspase 11 and was partially dependent on Gasdermin D (Fig. 5E). These results show that pduC-dependent production of propionate by AIEC drives IL-1β secretion through NLRP3-and Caspase 11-dependent inflammasome activation.
Finally, to determine the sufficiency of propionate to regulate AIEC induction of inflammatory T cell immunity, Il10-deficient mice colonized with AIEC 2A or 2AΔpduC were treated with sodium propionate in the drinking water ad lib. In mice colonized with 2AΔpduC, propionate was sufficient to increase IL-1β production by colonic explants (Fig. 5F) as well as lamina propria Th17 (Fig. 5G). While no differences were seen in ILC production of IL-22 at steady state (Fig. S2G), 2AΔpduC-colonized mice showed reduced ILC production of IL-22 compared to AIEC 2A colonized mice following DSS-induced colitis and propionate increased IL-22 production consistent with previous reports (Fig. 5H) (Chun et al., 2019). Collectively, these data reveal a critical role for AIEC-derived propionate in regulating intestinal immunity.
Mucosal fucosylation promotes AIEC pathogenesis in colitis
In addition to revealing a link between AIEC metabolism and tissue MNP immunity, these results raise the possibility that regulating nutrient availability could shape AIEC-mediated intestinal inflammation. To functionally test the possibility that limiting mucosal fucosylation, and subsequent fermentation to 1,2-propanediol, could reduce AIEC-induced inflammation, we treated Il10-deficient mice mono-colonized with AIEC 2A with a (1,2)-fucosylation inhibitor 2-Deoxy-D-galactose (2-D-gal) (Li et al., 2014). As previously described, IL-10-deficient mice have enhanced epithelial fucosylation (Goto et al., 2015) (Fig. S6A). 2-D-gal is converted via the Leloir pathway to the activated UDP-2-deoxy-galactose which competes with UDP-galactose and blocks Fuca(1,2)-gal linkage. As a result, injection of 2-D-gal significantly reduced luminal propionate in mono-colonized mice (Fig. 6A). Consistent with the requirement for PduC metabolic activity and propionate in driving inflammation, limiting fucosylation during inflammation with 2-D-gal attenuated weight loss, improved survival and reduced fecal lipocalin levels following DSS-induced colitis (Fig. 6B–D). Bacterial colonization was not affected by 2-D-gal (Fig. S6B,C). Similarly, tissue immune phenotyping specifically revealed a reduction in RORγt+ and IFNγ/IL-17+ inflammatory CD4+ T cells (Fig. 6E). Transcriptional analysis of intestinal LPMCs revealed a significant reduction in il1b following 2-D-gal treatment (Fig. 6F). No effect of 2-D-gal treatment was seen in mice colonized with 2AΔpduC (Fig. S6D–E). These findings highlight the potential to regulate nutrient availability to modify AIEC’s metabolic contribution to inflammatory bowel disease.
FIGURE 6. Inhibition of mucosal fucosylation limits PduC-dependent AIEC-induced colitis.
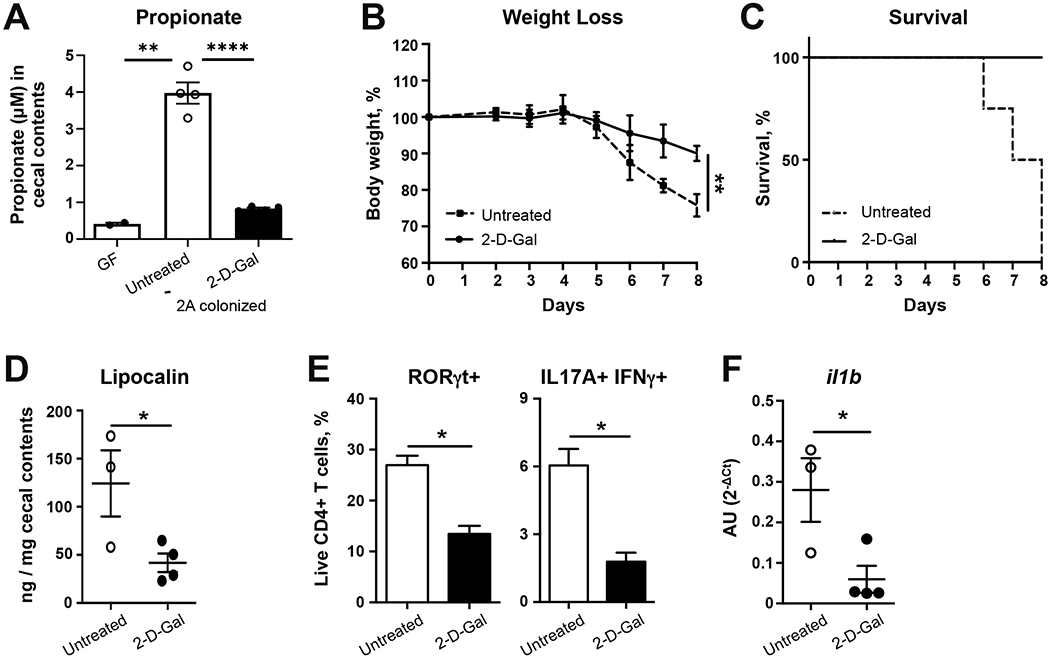
A-E. Germ-free C57BL/6 Il10-deficient mice were colonized with 2×109 CFU of AIEC 2A. Ten days after colonization, mice were exposed to 2% dextran sodium sulfate ad libitum for 7 days. Mice were treated intraperitoneally with 2-Deoxy-D-galactose (2-D-gal) or PBS control on days −2, 0, 2, 4, and 6 of DSS treatment. Propionate levels were measured in the cecal contents (A). Percent weight loss (B) and survival (C) is shown. Levels of lipocalin in cecal contents were measured by ELISA (D). Flow cytometry of live, CD4+ T cells was used to evaluate RORγt and IL17A/IFNγ (E) expression following a 4h stimulation with PMA/ionomycin and Brefeldin-A. Bar graphs represent geometric mean of at least 4 mice per group from one of three total experiments. Error bars represent SEM. *p<0.05, **p<0.01, ***p<0.005, ****p<0.001 t-test. F. C57BL/6 B6 SPF mice were treated with broad spectrum antibiotics for 2 weeks prior to infection with 1010 CFU of AIEC 2A. Mice were treated intraperitoneally with 2-D-gal or PBS control on days −2, 0, 2, and 4 after infection. Il1b expression in the colonic lamina propria was measured 5 days after colonization by quantitative PCR. Graphs show at least 3 mice from one of two total experiments. Error bars represent SEM. *p<0.05, t-test. See also Figure S6.
Discussion:
AIEC are enriched in the mucosal microbiome of CD patients, where they adhere to the intestinal epithelium and are associated with an increase in mucosal and systemic immune cell activation. Despite this association with intestinal inflammation, AIEC play an important role in maintaining homeostatic immunity at steady state. We and others have previously shown that AIEC induction of IL-10 restrains autologous (Ellermann et al., 2019; Viladomiu et al., 2017) and heterologous (Kim et al., 2018) inflammatory T cell responses. However, under conditions of induced inflammation or genetic susceptibility to inflammation, as seen in CD, the inflammatory arm of the homeostatic AIEC response tips the balance (Viladomiu et al., 2017). A more complete understanding of the microbial-host mechanisms regulating intestinal AIEC immunity are therefore critical to enable selective therapeutic targeting for CD. Moreover, the inability of comparative genomics to define the pathogenic elements of AIEC highlights the need for functional characterization of marker genes.
In both our own cohort of patients with CD as well as an external metagenomics data set, we identified pduC gene enrichment in the microbiome of CD patients, encoded primarily by the expanded Escherichia. Although the healthy microbiome includes commensals with the capacity for propanediol/glycerol metabolism, compositional changes associated with CD shift this metabolic capability to opportunistic pathobionts. In the case of infectious pathogens such as Salmonella, this pathway promotes expansion and pathogenic intestinal infection (Faber et al., 2017). In the case of the pathobiont AIEC, the response is context dependent. By enabling AIEC to utilize 1,2-propanediol for respiration, the pdu metabolic pathway promotes several aspects of mucosal fitness including access to an additional carbon source and production of propionate which has been shown to promote growth and virulence (Ormsby et al., 2020). Under wild type conditions, pduC promotes homeostatic AIEC-induced Th17 immunity. Excessive propionate can provide negative feedback on growth (Zhang et al., 2020) and the induction of IL-10 keeps inflammation in check (Kim et al., 2018). However, in the context of susceptibility to colitis (modeled here by transfer T cell colitis or IL10-deficiency), the propanediol utilization pathway induces inflammatory Th17 cells and causes more severe disease. The absence of IL-10, in particular, results in increased mucosal fucosylation (Goto et al., 2015) and may potentiate the role for the Pdu metabolic pathway in the pathogenic function of AIEC at the mucosal surface during inflammatory conditions. While E. coli can produce propionate through other mechanisms including the succinate pathway and catabolism of threonine (Gonzalez-Garcia et al., 2017; Haller et al., 2000), the enrichment of fucose at the epithelial barrier may potentiate the impact of propionate originating from the Pdu pathway. Similarly, while exogenous propionate is sufficient to trigger IL-1β and Th17 cell activation in mice colonized with pduC-deficient AIEC, the spatial location of AIEC as well as the synergy of propionate with E. coli-derived LPS may confer the specificity of this effect. These findings functionally link a fundamental metabolic pathway encoded by pathobionts enriched in CD with inflammatory Th17 cell immunity.
Our study identifies mononuclear phagocytes as key regulators of the intestinal immune response to AIEC. Mucosal association of AIEC (regulated in part by the Ipf operon) can trigger MNP production of IL-10, which in turns limits inflammatory T cell responses (Kim et al., 2018). In the context of genetic predisposition (e.g. an IL-10-deficient colitis or K/BxN arthritis model), AIEC can trigger mucosal and systemic immunity, supported in part by IL-23 (Viladomiu et al., 2017). Here, using mouse models for genetic deletion, we identified MNPs as critical regulators of AIEC-induced T cell tissue immunity. These findings are consistent with reports describing an essential role for MNPs in SFB-induced Th17 immunity (Panea et al., 2015) and may underlie a central role for these cells in tissue immunity of mucosal-associated microbes. Despite their ability to produce IL-23 (Kinnebrew et al., 2012), conventional DCs were dispensable for the AIEC-induced T cell response. While ex vivo studies have shown pduC-encoding AIEC survive better in macrophages (Dogan et al., 2014), our data show that pduC is required for robust production of MNP-derived IL-1β which drives AIEC-induced Th17 cell inflammatory disease. Collectively, these results highlight a central role for MNPs in dynamically balancing IL-10, IL-23 and pduC-dependent IL-1β to regulate AIEC-mediated T cell tissue immunity.
In achieving this balance, our results identify AIEC production of propionate as the mechanistic link between AIEC metabolism and MNP inflammasome activation leading to IL-1β production. Seminal work has highlighted the role for G protein-coupled receptors (GPCRs) in sensing microbial-derived SCFAs to regulate gut homeostasis and inflammasome activation in non-hematopoietic cells (Fujiwara et al., 2018; Macia et al., 2015; Maslowski et al., 2009). In addition, GPR43-dependent sensing of microbial-derived SCFA can regulate colonic ILC3 production of IL-22 in pathogen defense (Chun et al., 2019). The impact of these microbial-derived SCFA are not limited to non-hematopoietic cells or GPR43-dependent signaling. Recent work demonstrated that microbial-derived SCFA butyrate can also impact intestinal inflammation by regulating the monocyte to macrophage transition and imprint an antimicrobial transcriptional program independent of GPCRs under homeostatic conditions (Schulthess et al., 2019). In the context of intestinal inflammation and the expansion of pduC-encoding AIEC in CD, our work further reveals a functional role for microbial-derived propionate independent of GPR43 in potentiating the inflammasome signal in MNPs. As sentinels of the mucosal environment, this growing evidence suggest that the function of these MNPs are shaped by these environmental signals to regulate their immune impact in colitis.
The link between AIEC metabolism and intestinal inflammation raises the potential for therapeutic strategies for Crohn’s disease. Several therapeutic approaches focus on feeding the microbiome to build microbial structure and metabolism that promotes mucosal health. The intestinal mucosa is a unique space for nutrients including the fucose fermentation product 1,2-propanediol. Given our experimental data suggesting that limiting intestinal fucosylation during inflammation may functionally regulate AIEC induced propionate and the subsequent impact on intestinal inflammation, we propose that metabolic targeting of AIEC and / or limiting the availability of particular nutrients during inflammation can serve as a therapeutic approach to reduce mucosal inflammation. In contrast to antimicrobial strategies that simply target AIEC, regulating AIEC metabolism by limiting nutrients may preserve homeostatic function of AIEC in situ. Genetic variants in FUT2, the main mucosal fucosyltransferase, are associated with CD (McGovern et al., 2010) and interaction of this genotype status with AIEC-induced inflammation may identify an important subgroup for therapeutic regulation. Collectively, this study links AIEC metabolism of 1,2-propanediol with the metabolic sensing of MNPs to mediate T cell-dependent intestinal inflammation and reveals the potential for alternative approaches of selective, therapeutic manipulation in Crohn’s disease.
STAR Methods:
Resource availability:
Lead Contact:
Further information and requests for reagents may be directed to and will be fulfilled by Lead Contact, Randy S. Longman (ral2006@med.cornell.edu)
Materials Availability:
Bacterial strains generated in this study are available with MTA as per guidelines of Weill Cornell Medicine.
Data and Code Availability:
16SrRNA sequencing (Bioproject ID: PRJNA687480) and metagenomics data (Bioproject ID: PRJNA687536) has been deposited in SRA.
Experimental Model and Subject Details:
Housing and Husbandry of Experimental Animals:
C57BL/6, Itgax-cre, Cx3cr1-LSL-DTR and Lang-DTREGFP mice were purchased from The Jackson Laboratory. Cx3cr1-LSL-DTR (Diehl et al., 2013) and Lang-DTREGFP (Welty et al., 2013) mice have been previously described. Gpr43-deficient mice were generously provided by Dr. van den Brink (Fujiwara et al., 2018). Il23r-eGFP mice were provided by Dr. Mohammed Oukka (Awasthi et al., 2009) Mice were bred and maintained under specific pathogen free (SPF) conditions at Weill Cornell Medicine (WCM).
Gnotobiotic wild-type C57BL/6, Il10-deficient (Viladomiu et al., 2017) and Rag1-deficient (Fung et al., 2016) mice were bred and maintained in flexible film gnotobiotic isolators where fecal samples are collected biweekly for culture-based methods to verify germ-free status. For experiments, gnotobiotic mice were removed from isolators and placed in sterile, HEPA filter-controlled, isolator cages (Allentown) in the Gnotobiotic Facility at WCM. All mice were group housed with a 12 h light/dark cycle and allowed ad libitum access to diet and water. All experiments were performed with 6-8 week old littermates unless specified. Both male and female mice were used with random and equal assignment of same sex to each experimental group. All animal studies were carried out in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) at WCM.
Growth and Isolation of Bacterial Strains:
AIEC 2A was isolated from stool of a human with Crohn’s disease and spondyloarthritis (Viladomiu et al., 2017). Technical strains were obtained from Dr. Don Court at NCI, Novagen, and NEB (Supplemental Table 1). Bacteroidies thetaiotaomicron (Benjdia et al., 2011), AIEC 2A and all isogenic mutants were grown anaerobically at 37 °C in Peptone Yeast Extract Broth with Glucose (PYG, Anaerobe Systems) or Difco™ Brewer Anaerobic Agar (BD Biosciences) with or without antibiotics. All technical strains were grown aerobically at 37 °C in Difco™ Luria-Bertani Broth or Agar (BD Biosciences) with or without antibiotics. Assays using minimal media utilized starter cultures in PYG subcultured into M9 minimal salts supplemented with 100 μM iron sulfate, 2 mM magnesium sulfate, 100 μM calcium chloride, 10 mM ammonium chloride, 100 nM zinc sulfate, 100 nM copper sulfate, 3 μM cobalt chloride, 6 μM vitamin B1, 15 nM coenzyme B12, 150 nM B12, 15 nM dicyanocobinamide, and a carbon source (D-glucose, L-fucose, 1,2-propanediol).
Colonization of Mice with Cultured Bacteria:
Gnotobiotic mice were colonized with a single dose of 2 × 109 CFU AIEC 2A or its isogenic mutants in 200 μL sterile PBS by sterile oral gavage. SPF mice were treated with ampicillin (1g/L), neomycin (1g/L), vancomycin (1g/L) and metronidazole (1g/L) for 10 days ad libitum in the drinking water. After a one-day washout, mice were then gavaged with a single dose of 1010 CFU AIEC 2A or its isogenic mutants in 200 μL sterile PBS.
Human subjects:
Jill Roberts Institute for Research in Inflammatory Bowel Disease (JRI) LiveCell Bank Consortium was used to collect fecal samples from 24 healthy and 22 Crohn’s disease patients for metagenomic analysis. Sample collection was approved by the Institutional Review Board at Weill Cornell Medicine (1501015812, PI Longman). Inclusion criteria for Crohn’s disease included active endoscopic ileal disease consistent with a clinical diagnosis of ileal or ileo-colonic Crohn’s disease and a Harvery-Bradshaw score of >4 indicative of active disease. Healthy controls were age-matched controls without evidence of intestinal inflammation or a history of IBD.
Method Details:
Metagenomic analysis:
To evaluate pduC gene abundance during Crohn’s Disease, we performed metagenomic sequencing from fecal samples of healthy and CD patients (JRI Cohort described above). Briefly, bacterial genomic DNA was extracted using PowerSoil DNA Isolation Kit (Qiagen). Metagenomic library was constructed with Illumina barcodes from the Nextera XT kit (Illumina, San Diego, CA) and then loaded into Illumina Hiseq 4000 platform using 2 × 150 nucleotide pair-ending sequencing protocol at Weill Cornell Microbiome Core Facility. Metagenomics data was processed using Huttenhower lab bioinformatics tools (http://huttenhower.sph.harvard.edu/). Raw data was processed by KneadData (https://bitbucket.org/biobakery/kneaddata) to remove human contaminant reads. To filter out low quality sequence reads, sequences shorter than 50 bp and Illumina adapters reads, Trimmomatic (Bolger et al., 2014) was used as implemented in KneadData. After quality control, samples averaged 8 million reads (mean = 7,997,225 reads, SEM ± 267,135). Microbial functional potential profiling was determined by HUMAnN2 (Abubucker et al., 2012) version 0.11.2. For gene family profiling (proteincoding genes), DIAMOND (Buchfink et al., 2015) was used to map metagenome reads against UniRef90 (Suzek et al., 2015a). Hits were counted for each gene family and normalized for length and alignment quality. For pathway profiling, gene family abundances were then combined into structured pathways from MetaCyc (Caspi et al., 2018) and then sum-normalized to copies per million (CPM). The breakdown of pathways total abundance (CPM) into individual bacteria contribution was also computed by HUMAnN2. Uniref90_P37450 was used to assess pduC abundance and taxonomic contributions. For broader analysis at 50% homology, uniref90 values (UniRef90_P37450, UniRef90_P45514, UniRef90_R5U755, UniRef90_D6KQ92, UniRef90_D6LIA2, UniRef90_G9YVN5, UniRef90_R2NU32) which align to Uniref50_P37450 were assessed.
16S rRNA gene sequencing and analysis:
16S rRNA gene sequencing was performed by the Functional Genomics and Microbiome core at Baylor Medical College. Methods were adapted from the methods developed for the NIH-Human Microbiome Project (Human Microbiome Project, 2012). Briefly, bacterial genomic DNA was extracted using PowerSoil DNA Isolation Kit (Qiagen). The 16S rDNA V4 region was amplified by PCR and sequenced in the MiSeq platform (Illumina) using the 2×250 bp paired-end protocol (Caporaso et al., 2012). The 16S rRNA gene pipeline data incorporates phylogenetic and alignment-based approaches to maximize data resolution (Buffington et al., 2016). The read pairs were demultiplexed based on the unique molecular barcodes, and reads were merged using USEARCH v7.0.1090 (Edgar, 2010), allowing zero mismatches and a minimum overlap of 50 bases. Merged reads were trimmed at the first base with Q5. In addition, a quality filter was applied to the resulting merged reads and reads with > 0.05 expected errors were discarded. 16S rRNA gene sequences were clustered into Operational Taxonomic Units (OTUs) at a similarity cutoff value of 97% using the UPARSE algorithm. OTUs were mapped to an optimized version of the SILVA Database (Edgar, 2013; Quast et al., 2013) containing only the 16S V4 region to determine taxonomies. Abundances were recovered by mapping the demultiplexed reads to the UPARSE OTUs. A rarefied OTU table from the output files generated in the previous two steps was used for downstream analyses of α-diversity, β-diversity (Lozupone and Knight, 2005), and phylogenetic trends.
Generation of isogenic mutants:
AIEC 2A deficient in pduC was generated by Lambda-red recombineering of AIEC 2A as previously described (Thomason et al., 2014) using primers listed in Supplemental table 1. Recombinant clones were selected on Brewer Anaerobic Agar containing kanamycin. AIEC 2A with a catalytically-inactive or complemented wildtype PduC was generated by first generated an AIEC 2A deficient in pduC by Lambda-red recombination as previously described, with an oligo encoding a dual-selection, tetracycline resistance-sucrose sensitivity cassette surrounded by upstream and downstream homology of pduC. Recombinant clones were selected on Brewer Anaerobic Agar containing tetracycline. pSIM6 was retransformed into AIEC 2A ΔpduCtetAsacB to recombine with an oligo encoding wildtype or catalytically-inactive PduC and its upstream and downstream homologous sequences generating a cis-complemented AIEC 2A. Recombinant clones were selected on tetAsacB counterselection agar (Thomason et al., 2014). To generate the catalytically-inactive PduC, pduC was cloned into pUC19 for site-directed mutagenesis of the codons encoding H143, E170, and S362 to alanine using the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent Technologies).
Intestinal immune cell isolation and flow cytometry:
Lamina propria mononuclear cells were isolated from colonic tissue as previously described (Diehl et al., 2013). LIVE/DEAD fixable aqua dead cell stain kit (Molecular Probes) was used to exclude dead cells. For transcription factor analysis, cells were stained for surface markers before fixation and permeabilization with Intracellular Fixation and Permeabilization kit as per manufacturer’s instructions (eBiosciences) for intracellular staining. For cytokine detection, cells were stimulated with phorbol myristate acetate (PMA) and ionomycin with BD GolgiPlug for 4 hours. Following surface-marker staining cells were prepared as per manufacturer’s instruction with Cytofix/Cytoperm buffer set (BD Biosciences) for intracellular cytokine evaluation. Data acquisition was computed with BD LSRFortessa flow cytometer and analysis performed with FlowJo software (Tree Star).
Colitis models:
T cell transfer colitis:
Germ-free Rag1−/− mice were colonized with 2×109 CFU of AIEC 2A or its isogenic mutants or a SPF fecal pellet resuspended in sterile PBS. One week after colonization, mice received 500,000 FACS-sorted, CD45RbhiCD25−CD44−CD62L+ CD4+ T cells from wild-type or Il23r-eGFP (lacking IL-23R) mice intraperitoneally. Mice were monitored for weight loss and survival for 10 weeks. Flow cytometry and fecal lipocalin were assessed at 10 weeks.
DSS-induced colitis:
Wild type and Il10-deficient mice were treated with 2% dextran sulfate sodium w/v (M.W. 40,000-50,000, Affymetrix) in drinking water ad libitum for 7 days to induce colitis starting 10 days after monocolonization. Mice were monitored for weight loss and disease activity and tissues were collected 2 days after DSS removal. For RORγt inhibition experiments, mice were treated daily with 10mg/kg of the RORγt inverse agonist II, GSK805 (Calbiochem) or corn oil vehicle control throughtout the duration of the experiment. For IL-1β blockade experiments, functional grade anti-IL1β (B122) antibody or control rat IgG (Biolegend) was injected intravenously at 100 μg per mouse on days −2, 0, 2, 4 and 6 of treatment. For fucose availability disruption experiments, mice were injected intraperitoneally with 100mg of 2-Deoxy-D-galactose (2-D-gal, Sigma-Aldrich) on days −2, 0, 2, 4 and 6 of DSS treatment.
Propionate rescue model:
Germ-free, Il10-deficient mice were monocolonized with AIEC or its isogenic mutant 2AΔpduC for 7 days. A subset of 2AΔpduC colonized mice were given 150 nM sodium propionate in their drinking water starting one day before colonization. IL-1β detection and immune cell profiling was conducted on day 7 post-colonization.
Quantification of fecal lipocalin:
Fecal lipocalin-2 (Lcn-2) analyses were conducted using the mouse Lipocalin-2/NGAL DuoSet ELISA kit (R&D Systems).
Histopathology:
Segments of colon (3cm of the anatomic middle of the colon) were fixed in 4% buffered neutral formalin. Samples were embedded in paraffin, section (5μm), the stained with hematoxylin and eosin for histological examination. Tissue segments were blindly graded with a compouded histological score as previously described (Ostanin et al., 2009), including the extent of: 1) degree of inflammation in lamina propria (score 0–3); 2) goblet cell loss (score 0–2); 3) abnormal crypts (score 0–3); 4) presence of crypt abscesses (score 0–1); 5) mucosal erosion and ulceration (score 0–1); 6) submucosal spread to transmural involvement (score 0–3); 7) number of neutrophils counted at ×40 magnification (score 0–4). Total histopathological score was calculated by combining the scores for each of the seven parameters for a maximum score of 17.
Bacterial reisolation from mice:
For colonization counts, cecal content was resuspended in sterile PBS, serially diluted, and plated on Difco™ MacConkey agar (BD Biosciences) plates for calculation of CFU.
Conditional depletion of intestinal antigen-presenting cells:
Mice were treated intraperitoneally every other day with 200ng diphtheria toxin (DT, Sigma-Aldrich) during colonization for selective depletion of CX3CR1+ MNPs.
Gene expression by Quantitative RT-PCR:
Colonic lamina propria mononuclear cells were isolated from colonic tissue as previously described (Diehl et al., 2013) and stored in RLT buffer (Qiagen). RNA was extracted and purified using RNeasy Plus Mini Kit (Qiagen) and quantified by Nanodrop prior to reverse transcription with iScript cDNA synthesis kit (Bio-Rad). qPCR was performed on an Applied BioSciences Quant Studio 6 Flex Real-time PCR (Applied Biosystems) using PerfeCTa SYBR Green Fast mix, Low ROX (Quanta Biosciences). The following primers were used: for il10 IL10-F 5’-GAGAGCTGCAGGGCCCTTTGC-3’ and IL10-R 5’-CTCCCTGGTTTCTCTTCCCAAGACC-3’ (Viladomiu et al., 2017); for il1b, IL-1β-F 5’-TCAACTGTGAAATGCCACCT-3’ and IL-1β-R 5’-TCCACAGCCACAATGAGTGA-3’ (Pires and Parker, 2018); for il23p19, IL-23p19-F 5’-CCAGCAGCTCTCTCGGAATC-3’ and IL-23p19-R; 5’-GATTCATATGTCCCGCTGGTG-3’ (Liu et al., 2009); and for Hprt, HPRT-F 5’-GAGGAGTCCTGTTGATGTTGCCAG3’ and HPRT-R 5’-GGCTGGCCTATAGGCTCATAGTGC3’ (Stephens et al., 2011). The thermocycler program was as follows: initial cycle of 95°C for 60 s, followed by 40 PCR cycles at 95°C for 5 s, 60°C for 15 s, 72°C for 15 s. Relative levels of the target genes were determined by calculating the ΔCt to housekeeping gene Hprt expression.
In vivo IL-1β detection
To evaluate IL-1β production by LPMCs, cells were isolated as described above and stimulated with phorbol myristate acetate (PMA) and ionomycin for 5 hours. For tissue explants, a 1-cm piece of the colon was collected, throughly washed, and cultured overnight. IL-1β production was measured in the supernatants using Mouse IL-1 beta/IL-1F2 DuoSet ELISA as per manufacturer’s instructions (R&D Systems).
Isolation and purification of recombinant propanediol dehydratase:
Genes encoding wildtype and catalytically-inactive PduC were cloned with pduDE into the pET30b expression system (Novagen) and transformed into E. coli BL21 containing the λDE3 lysogen. A starter culture in LB with kanamycin was subcultured into LB with kanamycin and 0.1% 1,2-propanediol and incubated at 37°C with shaking until OD600 = 0.9 then 1 mM IPTG was added to induce expression. The culture was incubated an additional 6 hours. Protein was purified as previously described (Levin and Balskus, 2018). Briefly, bacterial cells were spun down and washed with buffer (50 mM potassium phosphate, pH 8, 2% 1,2-propanediol) then resuspended in lysis buffer (50 mM potassium phosphate, pH 8, 2 mM EDTA, 2 mM PMSF, 2% 1,2-propanediol), sonicated, and ultra-centrifuged for 30 minutes at 100,000 × g. Supernatants were transferred to a 10 kDa Slide-A-Lyzer cassette (Thermo Fisher) and dialyzed against buffer (50 mM potassium phosphate, pH 8, 0.1% Brij 35). Protein purity was assessed by SDS-PAGE and concentration was determined by Pierce BCA Protein Assay (Thermo Fisher).
Propanediol dehydratase activity assay:
Enzyme activity of purified 1,2-propanediol dehydratase was assessed using 3-methyl-2-benzothiazolinone hydrazone hydrochloride hydrate (MBTH) in the presence of 1.5% 1,2-propanediol and 15 μM coenzyme B12 under anaerobic conditions as previously described (Levin and Balskus, 2018; Toraya et al., 1977).
Ex vivo AIEC growth assay:
Starter cultures of AIEC in PYG were subcultured in reduced media specified by experiment in a sterile, clear, flat-bottom 96 well plate (Falcon®), overlayed with sterile mineral oil, and sealed with an optically clear plate seal (Microseal® ‘B’, BioRad). Growth was measured every 15 minutes by absorbance, OD600, on a Tecan Infinite F50 microplate reader with shaking at 7.8 Hz between readings. Growth assays were conducted at 37 °C in the anaerobic chamber.
Mass spectrometry of bacterial supernatants and 2-D-gal treated cecal content:
AIEC isolates were grown anaerobically for 72 hours in M9 minimal media (specified above) containing 20 mM L-fucose at 37 °C. Bacteria were then pelleted at 4000 rpm (2361 × g) for 7 minutes at 4 °C. Culture supernatants were filtered through a sterile, 0.22 μM filter. Cecal content was collected on day 8 after DSS from IL-10-deficient mice monocolonized with AIEC 2A and 10 mg aliquots were homogenized and derivatized as previously described (Lu et al., 2013). Propionate and propionaldehyde were detected and measured in the bacterial supernatants and cecal contents by liquid chromatography–quadrupole–time of flight mass spectrometry (LC–Q–TOF) as previously described (Lu et al., 2013).
SCFA measurement in cecal contents:
Cecal contents were collected from wild-type mice monocolonized with AIEC 2A or 2AΔpduC for 5 days and stored at −80°C. Samples were sent to Metabolon for short chain fatty acid (acetic acid, propionic acid and butyric acid) quantification by LCMS/MS Briefly, stool samples were spiked with stable labeled internal standards and were homogenized and subjected to protein precipitation with an organic solvent. After centrifugation, an aliquot of the supernatant was derivatized. The reaction mixture was diluted, and an aliquot was injected onto an Agilent 1290/AB Sciex QTrap 5500 LC MS/MS system equipped with a C18 reversed phase UHPLC column. The mass spectrometer was operated in negative mode using electrospray ionization (ESI). The peak area of the individual analyte product ions was measured against the peak area of the product ions of the corresponding internal standards. Quantitation was performed using a weighted linear least squares regression analysis generated from fortified calibration standards prepared immediately prior to each run. LC-MS/MS raw data were collected and processed using SCIEX OS-MQ software v1.7. Data reduction was performed using Microsoft Excel for Office 365 v.16.
LPS depletion and quantification from bacterial supernatants
Endotoxin was depleted from cell-free supernatants using the Pierce™ High Capacity Endotoxin Removal Spin Columns according to manufacturer protocol. Briefly, endotoxin removal resin was regenerated in sodium hydroxide overnight and equilibrated using sterile, endotoxin-free PBS. Supernatants were added and incubated with resin for 1 hour at 4°C with gentle rocking. Endotoxin levels were quantified from whole, and LPS-depleted supernatants using the Pierce™ Chromogenic Endotoxin Quant kit according to the manufacturer protocol. Endotoxin concentration was extrapolated based on a standard curve of the kit provided LPS standard and measured as Endotoxin Units (EU)/mL. 1 EU/mL is between 0.1 and 0.2 ng/mL of LPS.
Generation of BMDM:
Bone marrow (BM) was flushed out from sterilized femur and tibia of euthanized mice. Red blood cells were lysed using ACK Lysis buffer (Gibco). Cells were differentiated in cRPMI (RPMI 1640, 10% Fetal bovine serum, 2.5% Hepes, 1% Sodium pyruvate, 1% L-glutamine, 1% Penicillin/Streptomycin and 50 uM β-mercaptoethanol) supplemented with 25 ng/mL of recombinant mouse macrophage colony-stimulating factor (M-CSF, Peprotech) at 37 °C, 5% CO2 and 95% humidity. At day 3, fresh M-CSF-supplemented media was added. On day 6, BM-derived macrophages (BMDM) were harvested. Cells were re-suspended in cRPMI and left to adhere overnight at 37 °C, 5% CO2 and 95% humidity.
Immortalized macrophage cell lines:
Immortalized wild-type, NLRP3-, Caspase11-, Caspase1/11- and GasderminD-deficient macrophages were generously provided by Dr. Gretchen Diehl (Evavold et al., 2018). Cells were grown in complete DMEM (DMEM, 10% Fetal bovine serum, 2.5% Hepes, 1% non-essential aminoacids, 1% L-glutamine, 1% Penicillin/Streptomycin and 50 μM β-mercaptoethanol).
Macrophage stimulation:
BMDM were stimulated with 100ng/mL LPS (positive control) or 1:50 bacterial supernatants (as previously described) with or without 100nM propionate for 16h. Cell culture supernatants were collected and IL-1β production was measured using Mouse IL-1 beta/IL-1F2 DuoSet ELISA as per manufacturer’s instructions (R&D Systems). Cells were harvested and stored in RLT buffer (Qiagen) and qPCR for il10 quantification was performed as described above.
Epithelial cell isolation and flow cytometry:
Ileal tissue was collected and, after removal of Peyer’s patches, the intestine was opened longitudinally and washed extensively with ice-cold phosphate-buffered saline. The tissue was then cut into 1-cm pieces, which were incubated with 1mM DTT at room temperature with gentle shaking for mucus removal. Tissue was then incubated twice with 30mM EDTA at 37°C for 10min with gentle shaking. The suspension was passed through a 70μm strainer. UEA-1 was used to assess fucosylation in isolated epithelial cells. LIVE/DEAD fixable aqua dead cell stain kit (Molecular Probes) was used to exclude dead cells, and non-stained samples served as controls. Data acquisition was computed with BD LSRFortessa flow cytometer and analysis performed with FlowJo software (Tree Star).
Quantification And Statistical Analysis:
N indicated in the figure legends represents the number of mice or technical replicates per group in each experiment. Data are representative of at least two or more independent experiments as indicated, and error bars represent standard error of the mean (SEM). Significance of categorical variables such as gene expression, cytokine or lipocalin levels quantification, cell populations, or bacterial loads, were calculated using unpaired t-test for comparison of two groups or two-way ANOVA for comparison of more than two groups. Significance of survival curves was determined by log rank test. Statistics were calculated in GraphPad Prism (GraphPad software) and a p-value < 0.05 was considered as statistically significant.
Supplementary Material
| Reagent/Resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Live/Dead cell stain kit | ThermoFisher | Cat# L34962 |
| CD3-eFluor780 (clone ID UCHT1) | ThermoFisher | Cat# 47-0038-42; RRID: AB_1272042 |
| CD3-FITC (clone ID 145-2C11) | ThermoFisher | Cat# 11-0031-63; RRID: AB_464880 |
| CD4-AlexaFluor700 (clone ID GK1.5) | ThermoFisher | Cat# 54-0041-80; RRID: AB_494001 |
| CD4-eFluor780 (clone ID RM4-5) | ThermoFisher | Cat# 47-0042-80; RRID: AB_1272219 |
| CD44-APC (clone ID IM7) | ThermoFisher | Cat# 17-0441-82; RRID: AB_469390 |
| CD62L(L-Selectin)-PE-Cy7 (clone ID MEL-14) | ThermoFisher | Cat# 25-0621-81; RRID: AB_469632 |
| CD25-eFluor660 (clone ID eBio7D4) | ThermoFisher | Cat# ; RRID: 50-0252-82; AB_11149355 |
| IL-17-PE (clone ID B2D) | ThermoFisher | Cat# 12-7177-81; RRID: AB_763582 |
| IFNγ-PE-Cy7 (clone ID XMG1.2) | ThermoFisher | Cat# 25-7311-41; RRID: AB_1257211 |
| T-bet-e660 (clone ID eBio4B10) | ThermoFisher | Cat# 50-5825-82; RRID: AB_10596655 |
| RORγt-PE (clone ID B2D) |
ThermoFisher | Cat# 12-6981-80; RRID: AB_10805392 |
| GATA3-BUV395 (clone ID L50-823) | BD Biosciences | Cat# 565448; RRID: AB_2813884 |
| Foxp3-e450 (clone ID FJK-16 s) | ThermoFisher | Cat# 48-5773-80; RRID: AB_1518813 |
| MHCII-Alexa700 (clone ID M5/114.15.2) | ThermoFisher | Cat# 56-5321-80; RRID: AB_494010 |
| CD11b-eFluor780 (clone ID M1/70) | ThermoFisher | Cat# 47-0112-80; RRID: AB_1603195 |
| CD11c-PE-Cy7 (clone ID N418) | ThermoFisher | Cat# 25-0114-81; RRID: AB_469589 |
| CD103-APC (clone ID 2E7) | ThermoFisher | Cat# 17-1031-80; RRID: AB_1106993 |
| F4/80-PE (clone ID BM8) | ThermoFisher | Cat# 12-4801-80; RRID: AB_465922 |
| Ly6C-eFluor450 (clone ID HK1.4) | ThermoFisher | Cat# 48-5932-80; RRID: AB_10805518 |
| EpCAM-PE (clone ID G8.8) | ThermoFisher | Cat# 12-5791-82; RRID: AB_953615 |
| CD45-BUV395 (clone ID 30-F11) | BD Biosciences | Cat# 564279; RRID: AB_2651134 |
| UEA-1-Fluorescein | Vector Laboratories | Cat# FL-1061 |
| Ultra-LEAF Purified anti-mouse / rat IL-1β (Clone ID B112) | Biolegend | Cat# 503514; RRID: AB_2814400 |
| Ultra-LEAF Purified Armenian Hamster Isotype Control (Clone ID HTK888) | Biolegend | Cat# 400940 |
| Bacterial Strains | ||
| AIEC 2A | (Viladomiu et al., 2017) | See table S1 |
| AIEC 2AΔpduC | This work | See table S1 |
| AIEC 2AΔpduC::tetAsacB | This work | See table S1 |
| AIEC 2AΔpduC::tetAsacB (pSIM6) | This work | See table S1 |
| AIEC 2ApduC* | This work | See table S1 |
| AIEC 2A+pduC | This work | See table S1 |
| BL21(DE3) | Novagen | Cat #70781-3 |
| T-SACK | Court lab, NCI | See table S1 |
| Bacteroides thetaiotaomicron | (Benjdia et al., 2011) | |
| Biological samples | ||
| Human stool samples | Weill Cornell Medicine | IRB #1501015812 |
| Chemicals and Recombinant proteins | ||
| PduCDE | This work | See table S1 |
| PduC*DE | This work | See table S1 |
| Phorbol myristate acetate | Sigma-Aldrich | Cat# P1585 |
| Ionomycin calcium salt | Sigma-Aldrich | Cat# I0634 |
| GolgiPlug | BD Bioscience | Cat# 51-2301KZ |
| SYBR Green Supermix | Roche | Cat# 4887352001 |
| 0.5M EDTA pH8.0 | Invitrogen | Cat# AM9261 |
| DL-Dithiothreitol | Sigma-Aldrich | Cat# D9779 |
| RPMI without L-glutamine | GE Healthcare | Cat# SH30096.01 |
| DMEM, High Glucose, GlutaMAX Supplement | Gibco | Cat# 10-566-016 |
| Sodium pyruvate | Sigma-Aldrich | Cat# S8636 |
| L-glutamine | Corning Life Sciences | Cat# 25-005-Cl |
| Penicillin/Streptomycin | HyClone | Cat# SV30010 |
| 2-mercaptoethanol | Gibco | Cat# 21985-023 |
| HEPES | HyClone | Cat# SH30237.01 |
| Collagenase 8 | Sigma-Aldrich | Cat# C2139 |
| Deoxyribonuclease I from bovine pancreas | Sigma-Aldrich | Cat# DN25 |
| Percoll | GE Health | Cat# 17-0891-01 |
| Ampicillin | Sigma-Aldrich | Cat# A0166 |
| Neomycin | Sigma-Aldrich | Cat# N1876 |
| Metronidazole | Sigma Aldrich | Cat# M1547 |
| Vancomycin | VWR Scientific | Cat# 97062 |
| Kanamycin | Sigma Aldrich | Cat# K1377 |
| Tetracycline | Sigma Aldrich | Cat# T7660 |
| Dextran sodium sulfate | Affymetrix/USB | Cat# J14489 |
| ACK Lysis Buffer | Gibco | Cat# A1049201 |
| Diphtheria toxin | Sigma Aldrich | Cat# D0564 |
| 2-Deoxy-D-Galactose | Sigma Aldrich | Cat# 1949-89-9 |
| Recombinant Murine M-CSF Protein | Peprotech | Cat# 315-02 |
| TMB Substrate Reagent pack | R&D Systems | Cat# DY999 |
| Stop Solution 2N Sulfuric Acid | R&D Systems | Cat# DY994 |
| IPTG | Sigma Aldrich | Cat# 11411446001 |
| MBTH | Sigma Aldrich | Cat# 65875 |
| PMSF | Sigma Aldrich | Cat# P7626 |
| Potassium Phosphate Dibasic | Sigma Aldrich | Cat# P8281 |
| Potassium Phosphate Monobasic | Sigma Aldrich | Cat# P9791 |
| Potassium Chloride | Sigma Aldrich | Cat# P9541 |
| Brij 35 | Thermo Fisher Scientific | Cat# 20150 |
| Potassium Citrate | Sigma Aldrich | Cat# 60215 |
| Sodium propionate | Sigma Aldrich | Cat# P5436 |
| 1,2-propanediol | Sigma Aldrich | Cat# 82280 |
| D-glucose | Sigma Aldrich | Cat# G7021 |
| L-fucose | ApexBio | Cat# C3589 |
| Vitamin B12 | Sigma Aldrich | Cat# V2876 |
| Dicyanocobinamide | Sigma Aldrich | Cat# C3021 |
| Coenzyme B12 | Sigma Aldrich | Cat# C0884 |
| Vitamin B1 | Sigma Aldrich | Cat# T1270 |
| Magnesium Sulfate | Sigma Aldrich | Cat# M2643 |
| Calcium Chloride | Sigma Aldrich | Cat# C5670 |
| Ammonium Chloride | Sigma Aldrich | Cat# 254134 |
| Zinc Sulfate | Sigma Aldrich | Cat# Z0251 |
| Copper Sulfate | Sigma Aldrich | Cat# C8027 |
| Cobalt Chloride | Sigma Aldrich | Cat# C8661 |
| Iron Sulfate | Sigma Aldrich | Cat# F7002 |
| Peptone Yeast Glucose Broth | Anaerobe Systems | Cat# AS-822 |
| MacConkey Agar | Sigma-Aldrich | Cat# M7408 |
| Difco LB Agar | BD | Cat# 244520 |
| Difco Brewer Anaerobic Agar | BD | Cat# 227920 |
| RORγt inverse agonist II, GSK805 | Sigma-Aldrich | Cat# 5313690001 |
| Corn oil | Sigma-Aldrich | Cat# C8267 |
| Critical Commercial Assays | ||
| Mouse Lipocalin-2/NGAL DuoSet ELISA | R&D Systems | Cat# DY1857 |
| Mouse IL-1 beta/IL-1F2 DuoSet ELISA | R&D Systems | Cat# DY401 |
| RNeasy Plus Mini Kit | QIAGEN | Cat# 74134 |
| iScript cDNA synthesis kit | Bio-Rad | Cat# 1708891 |
| FoxP3 /Transcription Factor Staining Buffer set | eBioscience | Cat# 00-5523-00 |
| Fixation/Permeabilization Concentrate | ThermoFisher | Cat# 00-5123-43 |
| Fixation/Permeabilization Diluent | ThermoFisher | Cat# 00-5223-56 |
| QuikChange Lightning Multi Site-Directed Mutagenesis Kit | Agilent Technologies | Cat# 210515 |
| pET Expression System 30 | Novagen | Cat# 70781-3 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23227 |
| DNeasy PowerLyzer PowerSoil Kit | Qiagen | Cat# 12855 |
| Pierce High Capacity Endotoxin Removal Spin Columns | Sigma-Aldrich | Cat# 88275 |
| Pierce Chromogenic Endotoxin quantification kit | Thermofisher | Cat# A39552 |
| Lipopolysaccharides from Escherchia coli O111:B4 (LPS) | Sigma-Aldrich | Cat# L3024 |
| Deposited Data | ||
| Crohn’s Disease 16S sequencing | SRA | PRJNA687480 |
| Crohn’s Disease metagenomics sequencing | SRA | PRJNA687536 |
| Experimental Models: cell lines | ||
| Immortalized bone marrow-derived macrophages (iBMDM) | (Evavold et al., 2018) | N/A |
| Experimental Models: organism/strain | ||
| Mouse: C57BL/6 | Jackson Laboratories | Cat# 000664 |
| Mouse: Itgax-Cre | Jackson Laboratories | Cat# 008068 |
| Mouse: Lang-DTREGFP | Jackson Laboratories | Cat# 016940 |
| Mouse: Cx3cr1-LSL-DTR | Jackson Laboratories | Cat# 025629 |
| Mouse: il23r-GFP | (Awasthi et al., 2009) | N/A |
| Mouse: Gpr43-deficient | (Fujiwara et al., 2018) | N/A |
| Il10-deficient | (Viladomiu et al., 2017) | N/A |
| Rag1-deficient | (Fung et al., 2016) | N/A |
| Oligonucleotides | See table S2 | |
| il10: 5’-GAGAGCTGCAGGGCCCTTTGC-3’ 5’-CTCCCTGGTTTCTCTTCCCAAGACC-3’ | (Viladomiu et al., 2017) | N/A |
| il1b: 5’-TCAACTGTGAAATGCCACCT-3’ 5’-TCCACAGCCACAATGAGTGA-3’ | (Pires and Parker, 2018) | N/A |
| il23p19: 5’-CCAGCAGCTCTCTCGGAATC-3’ 5’-GATTCATATGTCCCGCTGGTG-3’ | (Liu et al., 2009) | N/A |
| Hprt: 5′-GAGGAGTCCTGTTGATGTTGCCAG-3′ 5′-GGCTGGCCTATAGGCTCATAGTGC-3′ | (Stephens et al., 2011) | N/A |
| Software and algorithms | ||
| R | R Core Team | N/A |
| GraphPad Prism 7 | GraphPad Software | N/A |
| FlowJo LLC v8.7 | Becton Dickenson | N/A |
Highlights:
pduC is enriched in Crohn’s disease-associated E. coli
AIEC induction of IL-1β and T cell-dependent colitis requires PduC
PduC-dependent propionate triggers IL-1β from CX3CR1+ MNPs and promotes Th17 cells
Limiting fucose availability reduces AIEC propionate production and inflammation
Acknowledgements:
We would like to thank Dr. David Eliezer for his insights on homology modeling and SDM site prediction, Dr. Ben Levin from Dr. Emily Balskus’ lab for his help with the isolation of PduCDE protein complex, and Dr. Silvia Pires for her input on bone marrow-derived macrophage experiments. Bioinformatics support was provided by the Jill Roberts Institute for Research in IBD and microbiome sequencing was performed by the Microbiome Core Lab at Weill Cornell Medicine. Fecal samples were provided by the Jill Roberts Institute Live Cell Bank. We would like to thank Dr. Gregory Putzel for helping with the bioinformatics analysis. Lastly, we would like to acknowledge Rielmer Pinedor for technical assistance with gnotobiotic husbandry.
JRI Live Cell Bank consortium members include David Artis, Randy Longman, Greg Sonnenberg, Ellen Scherl, Dana Lukin, Robert Battat, Jasmin Williams, Shaira Khan, Peik Sean Chong, Samah Mozumder, Lance Chou, Wenqing Zhou, Mohd Ahmed, Connie Zhong, Ann Joseph.
This work was supported by grants from the NIH R01DK114252-01A1 (R.S.L), Kenneth Rainin Foundation, and the Charina Foundation.
M.V.P. is supported by postdoctoral fellowships from Crohn’s Colitis Foundation and Weill Cornell Department of Medicine.
M.L.M is supported by graduate student fellowships from NIH-NIAMS and NSF.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests:
None
References:
- Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, Rodriguez-Mueller B, Zucker J, Thiagarajan M, Henrissat B, et al. (2012). Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol 8, e1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasthi A, Riol-Blanco L, Jager A, Korn T, Pot C, Galileos G, Bettelli E, Kuchroo VK, and Oukka M (2009). Cutting edge: IL-23 receptor gfp reporter mice reveal distinct populations of IL-17-producing cells. J Immunol 182, 5904–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjdia A, Martens EC, Gordon JI, and Berteau O (2011). Sulfatases and a radical S-adenosyl-L-methionine (AdoMet) enzyme are key for mucosal foraging and fitness of the prominent human gut symbiont, Bacteroides thetaiotaomicron. J Biol Chem 286, 25973–25982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B, Xie C, and Huson DH (2015). Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59–60. [DOI] [PubMed] [Google Scholar]
- Buffington SA, Di Prisco GV, Auchtung TA, Ajami NJ, Petrosino JF, and Costa-Mattioli M (2016). Microbial Reconstitution Reverses Maternal Diet-Induced Social and Synaptic Deficits in Offspring. Cell 165, 1762–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6, 1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R, Billington R, Fulcher CA, Keseler IM, Kothari A, Krummenacker M, Latendresse M, Midford PE, Ong Q, Ong WK, et al. (2018). The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res 46, D633–D639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing B, Rolhion N, de Vallee A, Salim SY, Prorok-Hamon M, Neut C, Campbell BJ, Soderholm JD, Hugot JP, Colombel JF, et al. (2011). Crohn disease--associated adherent-invasive E. coli bacteria target mouse and human Peyer’s patches via long polar fimbriae. J Clin Invest 121, 966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun E, Lavoie S, Fonseca-Pereira D, Bae S, Michaud M, Hoveyda HR, Fraser GL, Gallini Comeau CA, Glickman JN, Fuller MH, et al. (2019). Metabolite-Sensing Receptor Ffar2 Regulates Colonic Group 3 Innate Lymphoid Cells and Gut Immunity. Immunity 51, 871–884 e876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, Bringer MA, Swidsinski A, Beaugerie L, and Colombel JF (2004). High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 127, 412–421. [DOI] [PubMed] [Google Scholar]
- Delmas J, Gibold L, Fais T, Batista S, Leremboure M, Sinel C, Vazeille E, Cattoir V, Buisson A, Barnich N, et al. (2019). Metabolic adaptation of adherent-invasive Escherichia coli to exposure to bile salts. Sci Rep 9, 2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl GE, Longman RS, Zhang JX, Breart B, Galan C, Cuesta A, Schwab SR, and Littman DR (2013). Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX(3)CR1(hi) cells. Nature 494, 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan B, Suzuki H, Herlekar D, Sartor RB, Campbell BJ, Roberts CL, Stewart K, Scherl EJ, Araz Y, Bitar PP, et al. (2014). Inflammation-associated adherent-invasive Escherichia coli are enriched in pathways for use of propanediol and iron and M-cell translocation. Inflamm Bowel Dis 20, 1919–1932. [DOI] [PubMed] [Google Scholar]
- Edgar RC (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. [DOI] [PubMed] [Google Scholar]
- Edgar RC (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10, 996–998. [DOI] [PubMed] [Google Scholar]
- Ellermann M, Gharaibeh RZ, Fulbright L, Dogan B, Moore LN, Broberg CA, Lopez LR, Rothemich AM, Herzog JW, Rogala A, et al. (2019). Yersiniabactin-Producing Adherent/Invasive Escherichia coli Promotes Inflammation-Associated Fibrosis in Gnotobiotic Il10(−/−) Mice. Infect Immun 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eun CS, Mishima Y, Wohlgemuth S, Liu B, Bower M, Carroll IM, and Sartor RB (2014). Induction of bacterial antigen-specific colitis by a simplified human microbiota consortium in gnotobiotic interleukin-10−/− mice. Infect Immun 82, 2239–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evavold CL, Ruan J, Tan Y, Xia S, Wu H, and Kagan JC (2018). The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 48, 35–44 e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber F, Thiennimitr P, Spiga L, Byndloss MX, Litvak Y, Lawhon S, Andrews-Polymenis HL, Winter SE, and Baumler AJ (2017). Respiration of Microbiota-Derived 1,2-propanediol Drives Salmonella Expansion during Colitis. PLoS Pathog 13, e1006129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara H, Docampo MD, Riwes M, Peltier D, Toubai T, Henig I, Wu SJ, Kim S, Taylor A, Brabbs S, et al. (2018). Microbial metabolite sensor GPR43 controls severity of experimental GVHD. Nat Commun 9, 3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung TC, Bessman NJ, Hepworth MR, Kumar N, Shibata N, Kobuley D, Wang K, Ziegler CGK, Goc J, Shima T, et al. (2016). Lymphoid-Tissue-Resident Commensal Bacteria Promote Members of the IL-10 Cytokine Family to Establish Mutualism. Immunity 44, 634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M, et al. (2014). The treatment-naive microbiome in new-onset Crohn’s disease. Cell host & microbe 15, 382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Garcia RA, McCubbin T, Wille A, Plan M, Nielsen LK, and Marcellin E (2017). Awakening sleeping beauty: production of propionic acid in Escherichia coli through the sbm operon requires the activity of a methylmalonyl-CoA epimerase. Microb Cell Fact 16, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Lamichhane A, Kamioka M, Sato S, Honda K, Kunisawa J, and Kiyono H (2015). IL-10-producing CD4(+) T cells negatively regulate fucosylation of epithelial cells in the gut. Sci Rep 5, 15918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller T, Buckel T, Retey J, and Gerlt JA (2000). Discovering new enzymes and metabolic pathways: conversion of succinate to propionate by Escherichia coli. Biochemistry 39, 4622–4629. [DOI] [PubMed] [Google Scholar]
- Human Microbiome Project C (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Galan C, Hill AA, Wu WJ, Fehlner-Peach H, Song HW, Schady D, Bettini ML, Simpson KW, Longman RS, et al. (2018). Critical Role for the Microbiota in CX3CR1(+) Intestinal Mononuclear Phagocyte Regulation of Intestinal T Cell Responses. Immunity 49, 151–163 e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnebrew MA, Buffie CG, Diehl GE, Zenewicz LA, Leiner I, Hohl TM, Flavell RA, Littman DR, and Pamer EG (2012). Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity 36, 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamoto S, Alteri CJ, Rodrigues M, Nagao-Kitamoto H, Sugihara K, Himpsl SD, Bazzi M, Miyoshi M, Nishioka T, Hayashi A, et al. (2020). Dietary L-serine confers a competitive fitness advantage to Enterobacteriaceae in the inflamed gut. Nat Microbiol 5, 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin BJ, and Balskus EP (2018). Characterization of 1,2-Propanediol Dehydratases Reveals Distinct Mechanisms for B12-Dependent and Glycyl Radical Enzymes. Biochemistry 57, 3222–3226. [DOI] [PubMed] [Google Scholar]
- Li J, Hsu HC, Ding Y, Li H, Wu Q, Yang P, Luo B, Rowse AL, Spalding DM, Bridges SL Jr., et al. (2014). Inhibition of fucosylation reshapes inflammatory macrophages and suppresses type II collagen-induced arthritis. Arthritis Rheumatol 66, 2368–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Ouyang X, Yang J, Liu J, Li Q, Gu Y, Fukata M, Lin T, He JC, Abreu M, et al. (2009). AP-1 activated by toll-like receptors regulates expression of IL-23 p19. J Biol Chem 284, 24006–24016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C, and Knight R (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71, 8228–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Yao D, and Chen C (2013). 2-Hydrazinoquinoline as a Derivatization Agent for LC-MS-Based Metabolomic Investigation of Diabetic Ketoacidosis. Metabolites 3, 993–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, Maruya M, Ian McKenzie C, Hijikata A, Wong C, et al. (2015). Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat Commun 6, 6734. [DOI] [PubMed] [Google Scholar]
- Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, et al. (2009). Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461, 1282–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGovern DP, Jones MR, Taylor KD, Marciante K, Yan X, Dubinsky M, Ippoliti A, Vasiliauskas E, Berel D, Derkowski C, et al. (2010). Fucosyltransferase 2 (FUT2) non-secretor status is associated with Crohn’s disease. Hum Mol Genet 19, 3468–3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien CL, Bringer MA, Holt KE, Gordon DM, Dubois AL, Barnich N, Darfeuille-Michaud A, and Pavli P (2017). Comparative genomics of Crohn’s disease-associated adherent-invasive Escherichia coli. Gut 66, 1382–1389. [DOI] [PubMed] [Google Scholar]
- Ormsby MJ, Johnson SA, Carpena N, Meikle LM, Goldstone RJ, McIntosh A, Wessel HM, Hulme HE, McConnachie CC, Connolly JPR, et al. (2020). Propionic Acid Promotes the Virulent Phenotype of Crohn’s Disease-Associated Adherent-Invasive Escherichia coli. Cell Rep 30, 2297–2305 e2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostanin DV, Bao J, Koboziev I, Gray L, Robinson-Jackson SA, Kosloski-Davidson M, Price VH, and Grisham MB (2009). T cell transfer model of chronic colitis: concepts, considerations, and tricks of the trade. Am J Physiol Gastrointest Liver Physiol 296, G135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panea C, Farkas AM, Goto Y, Abdollahi-Roodsaz S, Lee C, Koscso B, Gowda K, Hohl TM, Bogunovic M, and Ivanov II (2015). Intestinal Monocyte-Derived Macrophages Control Commensal-Specific Th17 Responses. Cell Rep 12, 1314–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires S, and Parker D (2018). IL-1beta activation in response to Staphylococcus aureus lung infection requires inflammasome-dependent and independent mechanisms. Eur J Immunol 48, 1707–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, and Glockner FO (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41, D590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulthess J, Pandey S, Capitani M, Rue-Albrecht KC, Arnold I, Franchini F, Chomka A, Ilott NE, Johnston DGW, Pires E, et al. (2019). The Short Chain Fatty Acid Butyrate Imprints an Antimicrobial Program in Macrophages. Immunity 50, 432–445 e437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens AS, Stephens SR, and Morrison NA (2011). Internal control genes for quantitative RT-PCR expression analysis in mouse osteoblasts, osteoclasts and macrophages. BMC Res Notes 4, 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH, and Consortium U (2015a). UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH, and UniProt C (2015b). UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason LC, Sawitzke JA, Li X, Costantino N, and Court DL (2014). Recombineering: genetic engineering in bacteria using homologous recombination. Curr Protoc Mol Biol 106, 1 16, 11–39. [DOI] [PubMed] [Google Scholar]
- Toraya T, Ushio K, Fukui S, and Hogenkamp PC (1977). Studies on the mechanism of the adenosylcobalamin-dependent diol dehydrase reaction by the use of analogs of the coenzyme. J Biol Chem 252, 963–970. [PubMed] [Google Scholar]
- Viladomiu M, Kivolowitz C, Abdulhamid A, Dogan B, Victorio D, Castellanos JG, Woo V, Teng F, Tran NL, Sczesnak A, et al. (2017). IgA-coated E. coli enriched in Crohn’s disease spondyloarthritis promote TH17-dependent inflammation. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welty NE, Staley C, Ghilardi N, Sadowsky MJ, Igyarto BZ, and Kaplan DH (2013). Intestinal lamina propria dendritic cells maintain T cell homeostasis but do not affect commensalism. J Exp Med 210, 2011–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, Laughlin RC, Gomez G, Wu J, Lawhon SD, et al. (2013). Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science 339, 708–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Dogan B, Guo C, Herlekar D, Stewart K, Scherl EJ, and Simpson KW (2020). Short Chain Fatty Acids Modulate the Growth and Virulence of Pathosymbiont Escherichia coli and Host Response. Antibiotics (Basel) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
16SrRNA sequencing (Bioproject ID: PRJNA687480) and metagenomics data (Bioproject ID: PRJNA687536) has been deposited in SRA.