

Abstract
Supramolecular self-assembly enables living organisms to form highly functional hierarchical structures with individual components self-organized across multiple length scales. This has inspired work on multicomponent supramolecular materials to understand factors behind co-assembly versus self-sorting of molecules. We report here on a supramolecular system comprised of negatively charged peptide amphiphile (PA) molecules, in which only a tiny fraction of the molecules (0.7 mol %) were covalently conjugated to one of two different fluorophores, half to fluorescein isothiocyanate (FTIC) and the other half to tetramethylrhodamine (TAMRA). Confocal microscopy of the system revealed self-sorting of the two different fluorescent PA molecules, where TAMRA PA is concentrated in micron-scale domains while FITC PA remains dispersed throughout the sample. From Förster resonance energy transfer and fluorescence recovery experiments, we conclude that conjugation of the negatively charged FITC to PA significantly disrupts its co-assembly with the 99.3 mol % of unlabeled molecules, which are responsible for formation of micron-scale domains. Conversely, conjugation of the zwitterionic TAMRA causes no such disruption. Interestingly, this dissimilar behavior between FITC and TAMRA PA causes them to self-sort at large length scales in the supramolecular system, mediated not by specific interactions among the individual fluorophores but instead by their different propensities to co-assemble with the majority component. We also found that greater ionic strength in the aqueous environment of the system promotes mixing by lowering the electrostatic barriers involved in self-sorting. Our results demonstrate great thermodynamic subtlety in the driving forces that mediate self-sorting versus co-assembly in supramolecular peptide assemblies.
Graphical Abstract
We report on self-sorting of two minority fluorescent components in a multicomponent supramolecular system. The self-sorting is mediated by interactions of the fluorescent components with a third non-fluorescent component that comprises 99 mol% of the system.
Introduction
Living organisms maintain a high degree of self-organization using non-covalent self-assembly, which allows formation of ordered structures from individual components. The DNA double helix is held together by hydrogen bonds between complementary base pairs while microtubule filaments in the cytosol of cells undergo dynamic assembly and disassembly mediated by hydrogen bonding among tubulin subunits.1, 2 Recent work has shown that formation of heterochromatin domains relies on phase-separation mediated by weak hydrophobic interactions.3 These highly functional biological structures have inspired research activity on synthetic supramolecular materials over the past decade.4–6 In the design of synthetic supramolecular systems, two or more components can be mixed to achieve properties not attainable with only a single molecule. When different molecules are mixed and allowed to self-assemble, they may either self-sort, that is phase separate, or co-assemble, and the factors that promote one versus the other are poorly understood. Previous work has shown that molecular recognition events, such as hydrogen bonding complementarity,7, 8 chirality,9 or sterics,10 can mediate self-sorting in supramolecular systems. Furthermore, kinetic control of self-assembly has been used to achieve self-sorting. This was achieved by slowly changing the pH to trigger self-assembly of molecular components with different pKa values, thereby providing pathways to segregated systems.11, 12 More recently, manipulation of thermodynamic and kinetic self-assembly pathways, generally through temperature-controlled protocols, has allowed control over self-sorting versus co-assembly in supramolecular polymers.13, 14 Self-sorted configuration outcomes can also be controlled by directing seed formation that initiates self-assembly.15
Peptide amphiphiles (PAs), peptides modified with hydrophobic segments such as alkyl segments, are an extensive class of molecules that self-assemble into nanostructures and supramolecular materials in aqueous solution through hydrophobic collapse and hydrogen bond formation.16–19 Our laboratory and others have previously shown that supramolecular assemblies are dynamic and can exchange monomers over time, as weak non-covalent interactions allow individual molecules to escape.20–24 This phenomenon could be used to create co-assemblies containing two or more different molecules, and was recently shown to be useful in the formation of superstructures driven by the formation of thermodynamically favorable interactions.22 Strong cohesive forces within supramolecular assemblies have been shown to suppress this dynamic exchange, thus maintaining mixtures of assemblies in a segregated state.24 In the specific case of PAs, our laboratory previously showed that electrostatic screening of charged PAs by adding salt increases their propensity for hydrogen bonding.18 Based on this previous observation, we hypothesized that increased cohesion due to screening with added salt could inhibit exchange of molecules among assemblies that would otherwise enable co-assembly rather than self-sorted states.
To investigate the hypothesis stated above, we synthesized in this work two different fluorescently labeled PAs and mixed a small mole percent of the fluorescent PAs with unlabeled molecules. We used Förster resonance energy transfer (FRET) to examine the spatial proximity of the two different fluorescent PAs, as a way to establish the extent of co-assembly. Furthermore, we performed fluorescence recovery after photobleaching (FRAP) experiments to determine the mobility of fluorescent PAs in solution, which we believed would reflect the rate of PA exchange among nanostructures. Using these techniques, we studied the behavior of the fluorescent PAs at various ionic strengths and after exposure to different thermal pathways, which provided insights on the factors that drive self-sorting versus co-assembly. The fluorophore-conjugated PA molecules can be considered a model for those containing biological or chemical signals in the design of bioactive biomaterials.25–27 PAs bearing these signals are often mixed with un-functionalized diluent PAs in similar fashion to the unlabeled non-fluorescent PA molecules used to create supramolecular assemblies in this work. In these multicomponent supramolecular biomaterials with important biomedical functions in regenerative medicine, the factors that lead to self-sorting or co-assembly of the diluent and signal-bearing PAs are not fully understood.
Results
Mixtures of FITC, TAMRA, and Diluent PAs
Our laboratory has worked extensively with the PA containing the peptide sequence V3A3E3, which promotes self-assembly of the molecules into high aspect ratio nanofibers.18, 28–30 In this work we synthesized fluorescent analogs of this PA with the goal of examining molecular exchange among the PA nanofibers.20, 22 We first prepared separate 2 wt% aqueous solutions of V3A3E3 PA (diluent PA, Fig. 1 and SI Appendix Fig. S1), V3A3E3 PA conjugated to fluorescein isothiocyanate (FITC PA, Fig. 1 and SI Appendix Fig. S2), and V3A3E3 PA conjugated to carboxytetramethylrhodamine (TAMRA PA, Fig. 1 and SI Appendix Fig. S3). In both fluorescent PAs, the fluorophore is covalently conjugated to the PA molecule and does not interrupt the V3A3E3 peptide sequence responsible for self-assembly (Fig. 1). All three PAs were dissolved to 17.32 mM with water containing 30 mM NaOH, which helps deprotonate the acidic residues and improve solubility. Since ionic strength is an important aspect of this system, we chose to add a known amount of NaOH instead of titrating to a target pH with concentrated base; however, we note that the final PA solutions were in a pH range from 7 to 8. Fluorescent PA solutions (either FITC or TAMRA PA) were then mixed with diluent PA at 1% (v/v) These 2 wt% PA mixtures (diluent PA with either FITC or TAMRA PA) were then diluted with equal volume of pure water or water containing NaCl, such that the resultant solutions contained 1 wt% (8.66 mM) total PA and 0, 26, or 150 mM NaCl. At each NaCl concentration, PA solutions containing diluent and FITC PA were mixed in equal volume with PA solutions containing diluent and TAMRA PA. Thus, the final composition in the PA mixture was 0.99 wt% diluent PA, 0.005 wt% FITC PA, and 0.005 wt% TAMRA PA, and the solution contained either 0, 26, or 150 mM NaCl. Previous work from our laboratory has shown that electrostatic screening of charged PAs can promote more cohesive assemblies,18 and we therefore expected that Na+ ions would screen repulsive interactions among glutamate residues in the V3A3E3 sequence. At a concentration of 8.66 mM PA molecules each bearing three glutamic acid residues, 26 mM Na+ ions should provide complete electrostatic screening. In the 150 mM NaCl sample, the salt is in excess of the PA and this condition happens to correspond to physiological ionic strength. As mentioned earlier, our laboratory has shown in previous work that PA assemblies are dynamic and can exchange monomers over time,20 and we therefore assumed NaCl would slow this exchange by charge screening PA molecules and allowing them to engage in more cohesive interactions within the assemblies.
Figure 1:

Chemical structures of PA molecules used in this work.
In order to investigate the spatial proximity of the two fluorescent PAs, we first carried out Förster resonance energy transfer (FRET) experiments on the solutions. Since FITC can act as a FRET donor for TAMRA, we irradiated the sample at 450 nm to directly excite FITC (excitation maximum: 490 nm, emission maximum: 525 nm) but not TAMRA (excitation maximum: 555 nm, emission maximum: 585 nm). TAMRA, the FRET acceptor, will emit only if it receives energy from FITC, which can only occur if the two fluorophores are within a few nanometers of each other. If this energy transfer occurs, donor (FITC PA) emission is quenched while acceptor (TAMRA PA) emission increases. When PA solutions were analyzed immediately after their preparation, the spectra showed a TAMRA emission peak in samples with and without NaCl (Fig. 2a left and SI Appendix Fig. S4). The fluorescent signal was found to be lower when NaCl was present, which is likely due to self-quenching by FITC residues (SI Appendix Section S9, Fig. S17). As mentioned above, counterion screening of the PA’s glutamic acid side chains promotes more cohesive assemblies,18 which should result in closer packing of FITC PAs and increase the probability of self-quenching interactions. Although charge screening can also cause different nanofibers to pack closer together (i.e. nanofiber bundling), the distance at which two FITC fluorophores can self-quench is around 5 nm31, making intra-fiber quenching events far more likely. This would reduce the amount of FITC fluorophores that can potentially interact with TAMRA, but the emission spectra still showed a TAMRA peak, indicating that FITC and TAMRA engage in FRET interactions when NaCl is present. Next, the PA mixtures were subjected to a thermal annealing cycle (80°C for 30 minutes and slowly cooled overnight), which causes elongation of short nanofibers into longer, more cohesive nanofibers.18, 30 We found that, in the absence of NaCl, the emission spectrum shifts to show noticeably less donor quenching and less acceptor excitation (Fig. 2a right and Fig. S5). In samples containing either 26 or 150 mM NaCl, the spectra do not show significant changes in FRET upon annealing (Figure 2a right, S5). We quantified the FRET behaviors shown in Fig. 2a by calculating a FRET ratio for each sample, with a higher ratio indicating more FRET behavior (Fig. 2b and SI Appendix Fig. S6). As suggested by the emission spectra, PA mixtures containing 0 mM NaCl have a lower FRET ratio than PA mixtures containing 26 or 150 mM NaCl, both before and after annealing. The annealing procedure causes a statistically significant decrease in the FRET ratio of the 0 mM NaCl sample, while the FRET ratios of the 26 and 150 mM NaCl samples remain unchanged (Fig. 2b). The data suggest that NaCl enables more mixing between FITC and TAMRA PAs, and also prevents the annealing-induced separation observed in the 0 mM NaCl sample. This result was somewhat surprising because, as mentioned earlier, counterion screening should promote more cohesive assemblies, which we believed would decrease the rate of exchange among different nanostructures. Since the annealing procedure should help equilibrate the self-assembling system to lower energy states in its energy landscape,18 we hypothesized that a phase-separated system must be more stable under salt-free conditions. Thus, we proceeded to further examine the annealed PA mixtures.
Figure 2.

(a) Fluorescence emission spectra of PA mixtures excited at 450 nm, while freshly dissolved (left) and after annealing (right), at the indicated NaCl concentrations. (b) Förster resonance energy transfer (FRET) ratios of fluorescence emission spectra shown in panel a (*FRET ratio of annealed 0 mM NaCl sample is significantly different than the freshly dissolved 0 mM NaCl sample, paired t-test p < 0.05; ns: FRET ratio of annealed 26 and 150 mM NaCl samples are not significantly different than their corresponding freshly dissolved samples; error bars represent the standard deviation). Confocal micrographs of annealed PA mixtures containing (c) 0, (d) 26, and (e) 150 mM NaCl (left, FITC channel, middle, TAMRA channel, right, merge FITC and TAMRA channels). Cryogenic transmission electron micrographs (cryoTEM) of PA mixtures containing (f) 0, (g) 26, and (h) 150 mM NaCl. Cross polarized light micrographs of PA mixtures containing (i) 0, (j) 26, and (k) 150 mM NaCl.
Since FRET efficiency can be affected by the relative orientation of the fluorophore transition dipoles,32 we wanted to examine the spatial distribution of dyes using a different technique. We imaged the annealed PA mixtures using confocal microscopy, which probes the samples at the microns scale. Conversely, since FRET interactions can only occur if fluorophores are within a few nanometers of each other, those experiments report on nanoscale features of the samples. In annealed PA mixtures not containing NaCl, the FITC and TAMRA channels appear different and the merged image shows poor overlap (Fig. 2c and SI Appendix Fig. S10). Conversely, PA mixtures containing either 26 or 150 mM NaCl show similar features in the FITC and TAMRA channels, and the merged image shows more overlap compared to the 0 mM NaCl sample (Fig. 2d–e and SI Appendix Figs. S11–12). Interestingly, the FITC channel shows qualitatively weaker features than the TAMRA channel when NaCl is not present. When NaCl is added, the FITC channel exhibits stronger micron-scale features, which also match the micron-scale features of the TAMRA channel. Thus, the FRET and confocal microscopy data are consistent, with both showing separation of FITC and TAMRA PA assemblies when NaCl is absent and mixing when NaCl is present.
We wanted to further investigate how NaCl affects the PA self-assembly, in order to gain insight on how it enables mixing of the fluorescent PAs. We imaged the annealed PA mixtures using cryogenic transmission electron microscopy (cryoTEM), which can reveal the individual PA nanofibers in their hydrated state, avoiding drying effects associated with drop casting samples. We also imaged the samples using polarized light microscopy, which can reveal organized micron-scale features formed by the nanoscale assemblies. While FRET and confocal microscopy can only interrogate the fluorescently labeled PAs, these techniques can assess the entire PA sample, including the 99% of diluent PA in the mixture. CryoTEM reveals long nanofibers in all samples (Fig. 2f–h), which are expected to form after the thermal annealing procedure.18, 30 Based on our previous studies, these long nanofibers are expected to form liquid crystalline domains,30 and indeed polarized light microscopy shows the presence of birefringent features (Fig. 2i–k). The cryoTEM and polarized light microscopy data show that NaCl does not cause any obvious differences in the morphology of supramolecular assemblies, and small angle x-ray scattering (SAXS) experiments also show little change with added NaCl (SI Appendix Fig. S13). Thus, morphology alone cannot explain how NaCl encourages mixing of the fluorescent PAs, but morphology does not report on molecular packing arrangement within the nanofibers. Charge screening by salts is known to promote closer association among PA molecules by decreasing repulsive forces between them,18 but may not necessarily change the filamentous structure. Indeed, we probed the β-sheet content of diluent PA at different NaCl concentrations using Thioflavin T dye, and found that it increased with NaCl (SI Appendix Fig. S14). We note that the FITC fluorophore attached to the PA bears two negative charges, while the TAMRA fluorophore is zwitterionic and bears zero net charge (Fig. 1). Thus, it is possible that charge screening by NaCl affects these two PAs differently, which may change how they interact with the negatively charged diluent PA matrix. Therefore, we wanted to examine the two fluorescent PAs separately when mixed with diluent PA but not with each other.
Mixtures of FITC and Diluent PAs
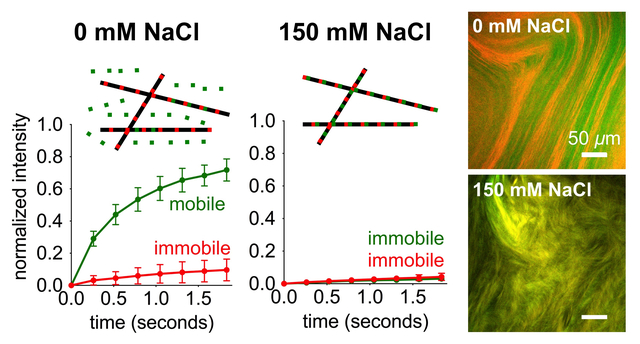
We mixed FITC PA with diluent PA at a range of salt concentrations to find out how these assemblies interact. Separate 2 wt% (17.32 mM) diluent PA and 2 wt% FITC PA solutions were prepared by dissolving PA with 30 mM NaOH, and then FITC PA was mixed with diluent PA at a volume ratio of 1:100. This 2 wt% PA mixture, containing both diluent PA and FITC PA, was then diluted to 1 wt% PA with either pure water or NaCl, such that the final PA solution contained 0.99 wt% diluent PA, 0.1 wt% FITC PA, and 0, 26, or 150 mM NaCl. We used confocal microscopy to image these solutions while freshly dissolved, and found no discernable micron-scale features with any amount of NaCl (Fig. 3a–c). After thermal annealing, the PA mixture containing 0 mM NaCl remains featureless, while the 26 and 150 mM NaCl samples exhibit micron-scale hierarchical structures (Fig. 3d–f and SI Appendix Fig. S15). These micron-scale features are stable enough to image while the sample is a bulk solution, which led to us to believe that FITC PA molecules may be less mobile when NaCl is present and these micron-scale structures appear. Therefore, we performed fluorescence recovery after photobleaching (FRAP) experiments to measure the diffusion of FITC PA. We photobleached 10 μm diameter circles and monitored fluorescence intensity recovery, which only occurs from diffusion of FITC PA into the bleached spot because photobleaching is irreversible. We monitored fluorescence recovery at short time points after photobleaching (< 2 seconds), during which the recovery profile should be dominated by the fastest diffusing species, which is likely to be PA molecules or small aggregates as opposed to entire nanofibers. In both freshly dissolved and annealed PA solutions, the presence of NaCl hinders diffusion of FITC PA, indicated by the decreased fluorescence recovery after ~1.8 seconds (Figure 3g–h, S16). Surprisingly, although the 0, 26, and 150 mM NaCl freshly dissolved samples look identical by confocal microscopy, their FRAP signatures are extremely different (Fig. 3g and SI Appendix S16). The lower mobile fractions in the 26 and 150 mM NaCl samples suggest that in those conditions, the FITC PA exists in relatively longer nanofibers than the FITC PA in the condition without NaCl, but before annealing these nanofibers are not long enough to form micron-scale bundles. Confocal microscopy cannot resolve individual nanofibers; it can only reveal micron-scale arrangements of several nanofibers. Thus, all three freshly dissolved conditions appear featureless regardless of nanofiber length. Upon annealing, the mobility of FITC PA in 150 mM NaCl is further decreased and is almost completely immobile, while the mobility of FITC PA in 0 mM and 26 mM NaCl remains similar (Fig. 3h and SI Appendix S16). Despite the elongation of nanofibers and obvious appearance of micron-scale bundles in the annealed 26 mM NaCl condition, the immobile fraction remains similar because the short timescale of our FRAP experiments (<2 seconds) cannot capture this elongation. In this short timeframe, only extremely short nanofibers would be expected to be mobile (~200 nm, Fig. 5b, 5d), so any changes at longer lengths would not appreciably affect the mobile fraction. Experiments at additional NaCl concentrations further demonstrate these trends, with more salt leading to less fluorescence recovery and more micron-scale features in a concentration-dependent manner (SI Appendix, Section S9). Although the confocal microscopy and FRAP experiments reveal information on the dynamics of FITC PA, we cannot draw any conclusions about the non-fluorescent diluent PA. Since confocal microscopy and FRAP can only interrogate FITC PA and not diluent PA, we interpret these data to be the behavior of FITC PA as it interacts with the diluent PA matrix.
Figure 3:

Confocal micrographs of freshly dissolved FITC PA mixed with diluent PA, containing (a) 0, (b) 26, and (c) 150 mM NaCl. Confocal micrographs of annealed FITC PA mixed with diluent PA, containing (d) 0, (e) 26, and (f) 150 mM NaCl. Fluorescence recovery after photobleaching (FRAP) experiments on (g) freshly dissolved and (h) annealed FITC PA mixed with diluent PA, at indicated NaCl concentrations (error bars represent the standard deviation).
Figure 5.

(a) Immobile fraction of FITC PA when mixed with diluent PA and (b) diffusion coefficient of mobile FITC PA, at indicated conditions (the dashed lines are hypothetical values). (c) Immobile fraction of TAMRA PA when mixed with diluent PA and (d) diffusion coefficient of mobile TAMRA PA at indicated conditions (the dashed lines are hypothetical values). To compare FITC PA with TAMRA PA, the same data in panels a-d are plotted again to group (e) fresh and (f) annealed samples together.
Mixtures of TAMRA and Diluent PAs
Next we repeated the confocal microscopy and FRAP experiments for TAMRA PA to determine if these assemblies behaved differently from FITC PA ones in the presence of diluent PA matrix. Using the same procedure as described for FITC PA mixtures, we prepared PA mixtures containing 0.99 wt % diluent PA and 0.01 wt % TAMRA PA and 0, 26, or 150 mM NaCl. Similar to the FITC PA mixtures, when the TAMRA PA samples were freshly dissolved, confocal micrographs did not reveal micron-scale features regardless of the NaCl concentration (Fig. 4a–c). After annealing, micron-scale features appear in all these samples, whether they contain 0, 26, or 150 mM NaCl (Fig. 4d–f). Thus, all confocal micrographs of TAMRA PA are similar to their FITC PA counterparts (Fig. 4a–f versus Fig. 3a–f), except for the sample that was annealed in 0 mM NaCl. In this condition, TAMRA PA exhibits micron-scale features (Fig. 4d) but FITC PA does not (Fig. 3d). We note that this was the condition where an apparent separation between the two fluorescent PAs occurred (Fig. 2c), and that confocal micrographs showed qualitatively weaker micron-scale features in the FITC channel.
Figure 4:

Confocal micrographs of freshly dissolved TAMRA PA mixed with diluent PA, containing (a) 0, (b) 26, and (c) 150 mM NaCl. Confocal micrographs of annealed TAMRA PA mixed with diluent PA, containing (d) 0, (e) 26, and (f) 150 mM NaCl. Fluorescence recovery after photobleaching (FRAP) experiments on (g) freshly dissolved and (h) annealed TAMRA PA mixed with diluent PA, at indicated NaCl concentrations. Error bars represent the standard deviation.
We performed FRAP experiments on TAMRA PA, to determine if its diffusion rate was different than FITC PA. In both experiments, the diluent PA matrix is exactly the same, and the fluorescent PA is only 1% of all PA in the system. While freshly dissolved, the 0 mM NaCl TAMRA PA sample appears slightly less mobile than its corresponding FITC PA sample, with ~40% (TAMRA PA) versus ~60% (FITC PA) recovery at ~1.8 seconds (Fig. 3g versus Fig. 4g). Upon annealing, this difference becomes far more drastic, as the TAMRA PA becomes almost completely immobile (Fig. 4h versus Fig. 3h). In the presence of either 26 or 150 mM NaCl, the FRAP curves of FITC and TAMRA PA are similar whether freshly dissolved or annealed, although the annealed 26 mM NaCl FITC PA sample is more mobile than its TAMRA PA counterpart (Fig. 3h versus Fig. 4h). Thus, similar to the confocal microscopy data, the most drastic difference between FITC and TAMRA PA is in the 0 mM NaCl annealed condition, where FITC PA was highly mobile while the TAMRA PA was almost completely immobile. Again, this is the condition under which apparent separation between the two fluorescent PAs occurred (Fig. 2c).
Modeling of FRAP Data
We modeled the diffusion observed by FRAP using a single exponential to describe the extent of fluorescence recovery (0–100%) at various time points after photobleaching.33 We fit the FRAP data to the equation:
| 1 |
where y is the normalized fluorescence intensity (on a scale of 0 to 1, 0 indicates no recovery after photobleaching and 1 indicates full recovery) and t is the time after photobleaching. By fitting the experimental data, we computed the constants in the model: y0, the mobile fraction (thus, 1−y0 is the immobile fraction), and τ, the characteristic diffusion time of the mobile species. We note that the mobile fraction y0 only reflects species that are mobile in the timeframe of the FRAP experiments, which is slightly less than 2 seconds (Figs. 3 and 4). Diffusion that occurs on longer time scales, which reflects slower diffusion, is not captured by our FRAP data. For mobile species that exhibit diffusion on this timescale, we calculated their diffusion coefficient using the Soumpasis equation. This equation relates the diffusion coefficient of mobile species to their half recovery time in FRAP experiments with a circular bleach area.34 The TAMRA PA FRAP experiments could not be performed with a circular bleach area due to experimental constraints with photobleaching (SI Appendix, Section S7), but we performed the same analysis for rough estimates. A diffusion coefficient for mobile species is correlated with their size, with larger objects exhibiting slower diffusion than smaller ones, so we calculated the theoretical diffusion coefficients for PA spherical micelles (10 nm in diameter) and PA short nanofibers (200 nm in length) for comparison with our data. These theoretical values are based on the Stokes–Einstein equation, which assumes non-interacting spherical objects, and therefore do not directly apply to anisotropic structures. We also note that a diffusion coefficient can only be measured for mobile components, and thus only reflects the behavior of the mobile component and not the average diffusion of the entire system. For a component to be considered “mobile,” it must diffuse in the specific timescale of our FRAP experiments (<2 seconds). Any species that diffuse on larger timescales would not be reflected in the calculated diffusion coefficients. Additionally, especially for systems with a smaller mobile fraction, the accuracy of the diffusion coefficient measurement will be limited by the fluorescence sensitivity of the experiment. Thus, we make these approximations to estimate the diffusing species’ size to an order of magnitude, not to obtain a precise size or fiber length.
Using this mathematical model, we analyzed the FRAP data (Fig. 3g–h) for the FITC PA when it is mixed with diluent PA. In freshly dissolved samples, approximately 30% of FITC PA is immobile if NaCl is not added, and this immobile fraction increases to 70% if 26 mM or more NaCl is added (Fig. 5a and SI Appendix Fig. S21). After annealing, the immobile fraction remains similar to the freshly dissolved state in the 0 and 26 mM NaCl samples, but increases to 97% in the 150 mM NaCl sample (Fig. 5a and SI Appendix Fig. S21). The similar order of magnitudes of the mobile species’ diffusion coefficients suggest that, regardless of the amount of mobile FITC PA diffusing in solution, the FITC PA that is mobile exists in structures of sizes consistent with micelles or short nanofibers (Fig. 5b and SI Appendix Fig. S21). Since FITC PA self-assembles into anisotropic nanofibers in the absence of any diluent PA (SI Appendix Fig. S22), we speculate that in the mobile fraction short nanofibers are more likely to be present than spherical micelles. We also suggest that the immobile fraction present in FITC PA contains structures significantly larger than 200 nm, which is consistent with highly anisotropic long nanofibers. As expected, the addition of NaCl to charge screen PAs allows a greater amount of FITC PA to exist in long nanofibers, thus increasing the immobile fraction (Fig. 5a).
Relating these calculations back to the confocal micrographs (Fig. 3), we note that in the 0 mM NaCl condition, the 30% immobile fraction does not increase with annealing (Fig. 5a) and hierarchical structures never develop (Fig. 3a vs. Fig. 3d). During annealing, the diluent PA rearranges into long nanofibers that organize into hierarchical structures (Fig. 2i), but the −2 charge on FITC PA prevents it from participating in this process. Instead, FITC PA remains in shorter nanofibers throughout annealing. Interestingly, in the 26 mM NaCl sample, annealing does cause formation of FITC PA hierarchical structures (Fig. 3b vs. Fig. 3e) but the immobile fraction remains at its pre-annealing level of 70% (Fig. 5a). Since the immobile fraction simply reflects nanofibers longer than 200 nm, annealing likely causes elongation that is not captured by the FRAP data. When we add 26 mM NaCl to screen the FITC’s −2 charge, the FITC PA can now exist in long nanofibers, which are necessary for the formation of hierarchical structures. Similar to the 26 mM NaCl condition, FITC PA in the 150 mM NaCl sample also forms hierarchical structures after annealing (Fig. 3c vs. Fig. 3f). Unlike the 26 mM NaCl condition, annealing increases the FITC PA’s immobile fraction from 70% to 97% (Fig. 5a). At the higher NaCl concentration, the negatively charged nanofibers are further charge screened, which may cause increased bundling behavior that further impedes FITC PA mobility. We note that the FITC PA’s micron-scale features in the annealed 150 mM NaCl condition are qualitatively more defined than in the annealed 26 mM NaCl condition (Fig. 3e vs. Fig. 3f).
Using the FRAP data shown in Fig. 4g–h, we carried out the same analysis for TAMRA PA mixed with diluent PA. In freshly dissolved samples, the immobile fraction is around 50% when either 0 or 26 mM NaCl is present, and increases to approximately 65% when 150 mM NaCl is added (Fig. 5c). Upon annealing, the immobile fraction increases to approximately 80% in both the 0 and 26 mM NaCl samples, and to approximately 95% in the 150 mM NaCl sample. In contrast to the FITC PA system, the immobile fraction of TAMRA PA is not strongly dependent on NaCl concentration but increases in response to annealing (Fig. 5a versus Fig. 5c). Similar to its FITC counterpart, the diffusion coefficients of mobile TAMRA PA suggests that it exists in small structures such as spherical micelles or short nanofibers (Fig. 5d), so immobile TAMRA PA must exist in nanofibers much longer than 200 nm. Also similar to FITC PA, we believe that short nanofibers are more likely than micelles in the mobile fraction, since TAMRA PA also forms long nanofibers in the absence of diluent PA (SI Appendix Fig. S22). Unlike negatively charged FITC fluorophores, TAMRA has a zwitterionic charge, so its conjugation to PA will not create electrostatic repulsion. Thus, unlike FITC PA, TAMRA PA assemblies remain as long nanofibers even in the absence of Na+ counterion screening. Elongation of PA nanofibers in response to annealing is well documented,18, 28, 30 and the annealing-induced, salt-independent increase in TAMRA PA’s immobile fraction simply reflects this elongation.
In both the 0 mM and 26 mM NaCl samples, annealing causes the appearance of TAMRA PA hierarchical structures (Fig. 4a–b vs. Fig. 4d–e) and this is accompanied by an increase in the TAMRA PA’s immobile fraction from 50% to 80% (Fig. 5c). Aided by diluent PA, the TAMRA PA rearranges into long nanofibers as annealing occurs, and this allows them to participate in the formation of hierarchical structures. Similar to the 0 and 26 mM NaCl samples, TAMRA PA in the 150 mM NaCl condition also forms hierarchical structures during annealing (Fig. 4c vs. Fig. 4f), and the immobile fractions increase from 65% to 95% during annealing (Fig. 5c). Similar to FITC PA, the extra charge screening provided by 150 mM NaCl may promote more nanofiber bundling and thus less TAMRA PA mobility, leading to more defined micron-scale hierarchical structures (Fig. 4f vs. Fig. 4d–e).
To compare FITC PA with TAMRA PA, we re-plotted the immobile fraction data (Fig. 5a, c) so that data for both PAs were in the same graph (Fig. 5e–f). Again, both fluorescent PAs are mixed with diluent PA in these experiments. In freshly dissolved samples, the immobile fractions of FITC PA and TAMRA PA do not appear drastically different from each other at any NaCl condition (Figure 5e), and likely reflect metastable nanofibers of highly heterogenous lengths. Freshly dissolved PA solutions are known to contain metastable assemblies that that are still rearranging towards their thermodynamic minimum, which is the formation of very long nanofibers for this particular diluent PA.18, 28 These metastable, relatively short nanofibers cannot efficiently align into hierarchical structures, so it is not surprising that the confocal micrographs of freshly dissolved FITC PA and TAMRA PA do not show micron-scale features, in the absence or presence of NaCl (see Fig. 3a–c and Fig. 4a–c). However, when the solutions are annealed to allow rearrangement into thermodynamically preferred states, the relationship between the immobile fractions of FITC PA and TAMRA PA changes (Fig. 5e vs. Fig. 5f). In annealed samples, the immobile fractions of FITC PA and TAMRA PA are very similar at 26 and 150 mM NaCl (Fig. 5f), and both fluorescent PAs showed micron-scale features at these salt conditions (Figs. 3e–f and 4–e–f). If the two fluorescent PAs are in the same solution, their micron-scale features even overlap (Fig. 2d–e). However, in the annealed 0 mM NaCl condition, the FITC PA’s immobile fraction is approximately 30% while the TAMRA PA’s immobile fraction is approximately 80% (Fig. 5f). This large difference (30% versus 80%) is accompanied by a clear visual difference in confocal micrographs of the two PAs, with TAMRA PA exhibiting micron-scale hierarchical structures (Fig. 4d) while FITC PA does not (Fig. 3d). Consistently, confocal micrographs of mixtures of the two fluorescent PAs in the absence of NaCl and annealed reveal weaker micron-scale features in the FITC channel than the TAMRA channel (Fig. 2c). This is the condition where self-sorting among the supramolecular assemblies is occurring.
Discussion
Previous work in our laboratory showed that molecular exchange among supramolecular co-assemblies can spatially concentrate certain components driven by thermodynamically favorable interactions.22 In this work, annealing PA mixtures (99% diluent PA, 0.05% FITC PA, 0.05% TAMRA PA) at 0 mM NaCl rearranges the system towards a segregated configuration, causing a statistically significant decrease in the FRET ratio between FITC PA and TAMRA PA (Fig. 2b). During annealing, the diluent PA rearranges into long nanofibers, which in turn align into hierarchical structures (Fig. 2i).18, 28, 30 Annealing of the TAMRA PA also lengthens the nanofibers (Fig. 5c), which in turn arrange into hierarchical structures (Fig. 4d). Since annealed pure TAMRA PA (without diluent) forms short nanofibers with heterogeneous lengths (SI Appendix Fig. S22), TAMRA PA is likely co-assembled with diluent PA to form long supramolecular nanofibers. Although both diluent and TAMRA PA exist in hierarchical structures and are likely co-assembled, we cannot ascertain if TAMRA PA is homogenously distributed among diluent PA assemblies. In fact, based on our previous work,22 we speculate that TAMRA PA concentrates in particular nanofiber bundles through electrostatic “self-pairing” of the zwitterionic TAMRA moieties in different nanofibers. In the previous work, a small amount of PA bearing complementary DNA strands (DNA-PA) was mixed with diluent PA, and the DNA-PA concentrated in nanofibers that became intertwined due to DNA hybridization. Although this proposed TAMRA PA “self-pairing” interaction is less specific than hybridization of complementary DNA strands, rhodamine dimers in water have been observed.35 In contrast to TAMRA PA, the majority of FITC PA exists in structures smaller than 200 nm (Fig. 5a, f) due to electrostatic repulsion from the fluorophore’s −2 charge. Since pure FITC PA (without diluent PA) and pure diluent PA both form anisotropic nanofibers, these small structures are likely nanofibers shorter than 200 nm as opposed to spherical aggregates. These short nanofibers are excluded from the hierarchical structures of aligned long nanofibers, and thus phase separate from TAMRA PA and do not reveal micron scale order (Fig. 3d). We suggest that electrostatic repulsion is the dominant force that limits the co-assembly of pure FITC PA and diluent PA, thus FITC PA forms only the smaller nanostructures. Obviously the driving force for some limited co-assembly of these two molecules is hydrophobic collapse of their C16 alkyl tails and their identical β-sheet forming peptide domain V3A3E3.
In summary, with TAMRA PA concentrated in long nanofiber bundles and FITC PA in short nanofibers, self-sorting is observed at 0 mM NaCl. Mechanical stirring does not disrupt self-sorting (SI Appendix, Section S12), suggesting that physical breakage of long nanofibers does not change the outcome. Interestingly, heating times shorter than the 30 minutes of our standard annealing procedure are also sufficient to induce the separation observed by FRET (SI Appendix Fig. S7), and this separation also occurs at lower PA concentrations (SI Appendix Fig. S8). However, in the presence of 26 or 150 mM NaCl, the FRET ratio of the fluorescent PAs does not change with annealing (Fig. 2b) and self-sorting is suppressed (Fig. 2d–e vs. Fig. 2c). We suggest that the salt ions electrostatically screen the FITC fluorophore’s −2 charge, allowing FITC PA to co-assemble into long nanofibers and form hierarchical structures during annealing. With salt counterion screening, both FITC PA and TAMRA PA can now be a part of long nanofibers and hierarchical structures. Overlap of the FITC and TAMRA channels in confocal micrographs suggests they are present in the same hierarchical structures (Fig. 2d–e). These micrographs demonstrate that FITC PA and TAMRA PA are mixed at the micron-scale, but cannot reveal if they are co-assembled on the nano-scale. However, FRET interactions can only occur if fluorophores are within a few nanometers of each other, which is much more probable if FITC PA and TAMRA PA are co-assembled in the same nanofiber. Thus, the FRET ratios of approximately 0.6 – 0.7 in annealed mixtures with 26 or 150 mM NaCl (Fig. 2b) suggest that at least a portion of FITC PA and TAMRA PA are co-assembled. Furthermore, as mentioned earlier, TAMRA’s zwitterionic charge may contribute to its ability to “self-pair” and Na+ screening should disrupt this. Thus, with less self-interactions, the TAMRA PA has more opportunities to engage in FRET interactions with FITC PA. Interestingly, when NaCl is added to an already annealed, salt-free, separated system, it appears to induce mixing almost immediately (SI Appendix Fig. S9). This suggests that as soon as FITC PA is charge screened, it leaves short nanofibers to join longer ones, putting it in closer proximity to TAMRA PA. Another possible factor is that Na+ instantly disrupts TAMRA self-interactions, allowing more FRET interactions between TAMRA PA and FITC PA.
While this work has focused on the behavior of the FITC PA and TAMRA PA, we emphasize the crucial role of diluent PA, which constitutes a 99% majority of PA in the mixtures in the observed self-sorting. Neither fluorescent PA forms infinitely long nanofibers on their own (SI Appendix Fig. S22), so it is only through co-assembly with diluent PA that they can exist in long nanofibers and hierarchical structures. When NaCl is not present, FITC PA cannot co-assemble with long diluent PA nanofibers while TAMRA PA can, leading to spatial separation between the two fluorophores. Although some diluent PA likely ends up co-assembled into FITC PA’s short nanofibers, the majority of the 99% of diluent PA still forms long nanofibers that can incorporate TAMRA PA. Also, our hypothesized TAMRA PA “self-pairing” phenomenon would not be possible if TAMRA moieties were not spaced out at the surface of nanofibers containing non-fluorescent diluent PA. The diluent PA matrix in this work is composed of a single non-fluorescent PA, which has a relatively strong propensity for hydrogen bonding due to its V3A3 peptide sequence. We also explored the possibility of forming two-component diluent PA matrices, using mixtures of two distinct PAs with different propensities for hydrogen bonding in different ratios (SI Appendix Section S13). When the amount of the weaker hydrogen bonding PA is increased relative to the stronger hydrogen bonding PA (SI Appendix Fig. S29), we observed enhanced FRET signal between the minority fluorescent PAs, suggesting a shift from self-sorting to mixing (SI Appendix Fig. S30). Thus, varying the non-fluorescent diluent PA composition causes a change in the self-sorting behavior of the minority fluorescent PA components.
We have investigated here supramolecular co-assembly in water of systems containing potentially three different molecules that include a majority component and two molecules present only in extremely small amounts as dopants. We found that differences in the nature of interactions between the majority component and the two different dopant molecules can either favor self-sorting of co-assemblies containing only one dopant or promote global co-assembly of all three components. These systems are of interest to create multi-functional systems in which two or more molecules present in very small amounts provide biological or chemical signals. In this work, PA molecules containing fluorophores served as a “model” for PAs bearing signals. We conclude that subtle electrostatic differences between these fluorophores, present only in extremely small concentrations, drove their self-sorting versus co-assembly with diluent PA. Since the driving force was electrostatic, self-sorting or co-assembly could be controlled by altering the ionic strength of the aqueous solvent. The principles that govern self-sorting or co-assembly of these fluorescent PAs are likely applicable to other multicomponent supramolecular systems.
Materials and Methods
Details on material synthesis and purification, and all materials characterization, can be found in the SI Appendix.
Supplementary Material
Acknowledgments
This work was primarily supported by the Center for Bio-Inspired Energy Science (CBES), an Energy Frontier Research Center (EFRC) funded by the US Department of Energy, Office of Science, Basic Energy Sciences under award number DE-SC0000989. Additional support was provided by the Northwestern University Center for Regenerative Nanomedicine (synthesis of PA molecules) and the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number R01AR072721 (fluorescence spectroscopy experiments). C.H.C. has received support from a National Defense Science and Engineering Graduate Fellowship and the National Institutes of Health Training Program in Pathophysiology and Rehabilitation of Neural Dysfunction (NIH T32 HD07418). The following research facilities at Northwestern University were used in this work: Peptide Synthesis and Analytical BioNano Technology Equipment Cores of the Simpson Querrey Institute (U.S. Army Research Office, U.S. Army Medical Research and Materiel Command, Northwestern University, Soft and Hybrid Nanotechnology Experimental [SHyNE] Resource [NSF NNCI-1542205]), Optical Microscopy and Metallography Facility (MRSEC Program [DMR-1121262]), Keck Biophysics Facility (NCI CCSG P30 CA060553 grant awarded to the Robert H Lurie Comprehensive Cancer Center), Biological Imaging Facility (Northwestern University Office for Research), Center for Advanced Microscopy (NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center). This work also made use of the DuPont-Northwestern-Dow Collaborative Access Team (Northwestern University, E.I. DuPont de Nemours & Co., and The Dow Chemical Company) located at Sector 5 of the Advanced Photon Source (U.S. Department of Energy [DOE] Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357).
References
- 1.Watson JD; Crick FH. Nature. 1953. 171(4356), 737–738. [DOI] [PubMed] [Google Scholar]
- 2.Nogales E; Wolf SG; Downing KH. Nature. 1998. 391(6663), 199–203. [DOI] [PubMed] [Google Scholar]
- 3.Strom AR; Emelyanov AV; Mir M; Fyodorov DV; Darzacq X; Karpen GH. Nature. 2017. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Versluis F; van Esch JH; Eelkema R. Adv. Mater. 2016. 28(23), 4576–4592. [DOI] [PubMed] [Google Scholar]
- 5.Aida T; Meijer EW; Stupp SI. Science. 2012. 335(6070), 813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dumele O; Chen J; Passarelli JV; Stupp SI. Adv. Mater. 2020. 32(17), 1907247. [DOI] [PubMed] [Google Scholar]
- 7.Ligthart G; Ohkawa H; Sijbesma RP; Meijer E. J. Am. Chem. Soc. 2005. 127(3), 810–811. [DOI] [PubMed] [Google Scholar]
- 8.Onogi S; Shigemitsu H; Yoshii T; Tanida T; Ikeda M; Kubota R; Hamachi I. Nat. Chem. 2016. 8(8), 743. [DOI] [PubMed] [Google Scholar]
- 9.Ishida Y; Aida T. J. Am. Chem. Soc. 2002. 124(47), 14017–14019. [DOI] [PubMed] [Google Scholar]
- 10.Ma Y; Kolotuchin SV; Zimmerman SC. J. Am. Chem. Soc. 2002. 124(46), 13757–13769. [DOI] [PubMed] [Google Scholar]
- 11.Morris KL; Chen L; Raeburn J; Sellick OR; Cotanda P; Paul A; Griffiths PC; King SM; O’Reilly RK; Serpell LC. Nat. Commun. 2013. 4, 1480. [DOI] [PubMed] [Google Scholar]
- 12.Ardoña HAM; Draper ER; Citossi F; Wallace M; Serpell LC; Adams DJ; Tovar JD. J. Am. Chem. Soc. 2017. 139(25), 8685–8692. [DOI] [PubMed] [Google Scholar]
- 13.Aratsu K; Takeya R; Pauw BR; Hollamby MJ; Kitamoto Y; Shimizu N; Takagi H; Haruki R; Adachi S.-i.; Yagai S. Nat. Commun. 2020. 11(1), 1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarkar A; Sasmal R; Empereur-mot C; Bochicchio D; Kompella SVK; Sharma K; Dhiman S; Sundaram B; Agasti SS; Pavan GM; George SJ. J. Am. Chem. Soc. 2020. 142(16), 7606–7617. [DOI] [PubMed] [Google Scholar]
- 15.Kubota R; Nagao K; Tanaka W; Matsumura R; Aoyama T; Urayama K; Hamachi I. Nat. Commun. 2020. 11(1), 4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartgerink JD; Beniash E; Stupp SI. Science. 2001. 294(5547), 1684–8. [DOI] [PubMed] [Google Scholar]
- 17.Hartgerink JD; Beniash E; Stupp SI. Proc. Natl. Acad. Sci. U. S. A. 2002. 99(8), 5133–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tantakitti F; Boekhoven J; Wang X; Kazantsev RV; Yu T; Li J; Zhuang E; Zandi R; Ortony JH; Newcomb CJ; Palmer LC; Shekhawat GS; Olvera de la Cruz M; Schatz GC; Stupp SI. Nat. Mater. 2016. 15(4), 469–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ortony JH; Newcomb CJ; Matson JB; Palmer LC; Doan PE; Hoffman BM; Stupp SI. Nat. Mater. 2014. 13(8), 812–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.da Silva RM; van der Zwaag D; Albertazzi L; Lee SS; Meijer E; Stupp SI. Nat. Commun. 2016. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hendrikse SI; Wijnands SP; Lafleur RP; Pouderoijen MJ; Janssen HM; Dankers PY; Meijer E. ChemComm. 2017. 53(14), 2279–2282. [DOI] [PubMed] [Google Scholar]
- 22.Freeman R; Han M; Álvarez Z; Lewis JA; Wester JR; Stephanopoulos N; McClendon MT; Lynsky C; Godbe JM; Sangji H. Science. 2018. 362(6416), 808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Albertazzi L; van der Zwaag D; Leenders CM; Fitzner R; van der Hofstad RW; Meijer E. Science. 2014. 344(6183), 491–495. [DOI] [PubMed] [Google Scholar]
- 24.Wester JR; Lewis JA; Freeman R; Sai H; Palmer LC; Henrich SE; Stupp SI. J. Am. Chem. Soc. 2020. 142(28), 12216–12225. [DOI] [PubMed] [Google Scholar]
- 25.Shah RN; Shah NA; Lim MMDR; Hsieh C; Nuber G; Stupp SI. Proc. Natl. Acad. Sci. U. S. A. 2010. 107(8), 3293–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sur S; Tantakitti F; Matson JB; Stupp SI. Biomater. Sci. 2015. 3(3), 520–532. [DOI] [PubMed] [Google Scholar]
- 27.Lee SS; Hsu EL; Mendoza M; Ghodasra J; Nickoli MS; Ashtekar A; Polavarapu M; Babu J; Riaz RM; Nicolas JD; Nelson D; Hashmi SZ; Kaltz SR; Earhart JS; Merk BR; McKee JS; Bairstow SF; Shah RN; Hsu WK; Stupp SI. Adv. Healthc. Mater. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen CH; Palmer LC; Stupp SI. Nano Lett. 2018. 18(11), 6832–6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pashuck ET; Cui H; Stupp SI. J. Am. Chem. Soc. 2010. 132(17), 6041–6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang S; Greenfield MA; Mata A; Palmer LC; Bitton R; Mantei JR; Aparicio C; Olvera de la Cruz M; Stupp SI. Nat. Mater. 2010. 9(7), 594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deka C; Lehnert B; Lehnert N; Jones G; Sklar L; Steinkamp J. Cytometry: The Journal of the International Society for Analytical Cytology. 1996. 25(3), 271–279. [DOI] [PubMed] [Google Scholar]
- 32.Iqbal A; Arslan S; Okumus B; Wilson TJ; Giraud G; Norman DG; Ha T; Lilley DM. Proc. Natl. Acad. Sci. U. S. A. 2008. 105(32), 11176–11181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kang M; Day CA; DiBenedetto E; Kenworthy AK. Biophys. J. 2010. 99(9), 2737–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soumpasis DM. Biophys. J. 1983. 41(1), 95–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ilich P; Mishra PK; Macura S; Burghardt TP. Spectrochim. Acta A. 1996. 52(10), 1323–1330. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.