

Summary
Murine regulatory T (Treg) cells in tissues promote tissue homeostasis and regeneration. We sought to identify features that characterize human Treg cells with these functions in healthy tissues. Single-cell chromatin accessibility profiles of murine and human tissue Treg cells defined a conserved, microbiota-independent tissue-repair Treg signature with a prevailing footprint of the transcription factor BATF. This signature, combined with gene expression profiling and TCR fate mapping, identified a population of tissue-like Treg cells in human peripheral blood that expressed BATF, chemokine receptor CCR8 and HLA-DR. Human BATF+CCR8+ Treg cells from normal skin and adipose tissue shared features with nonlymphoid T follicular helper-like (Tfh-like) cells, and induction of a Tfh-like differentiation program in naive human Treg cells partially recapitulated tissue Treg regenerative characteristics, including wound healing potential. Human BATF+CCR8+ Treg cells from healthy tissue share features with tumor-resident Treg cells, highlighting the importance of understanding the context-specific functions of these cells.
Graphical abstract
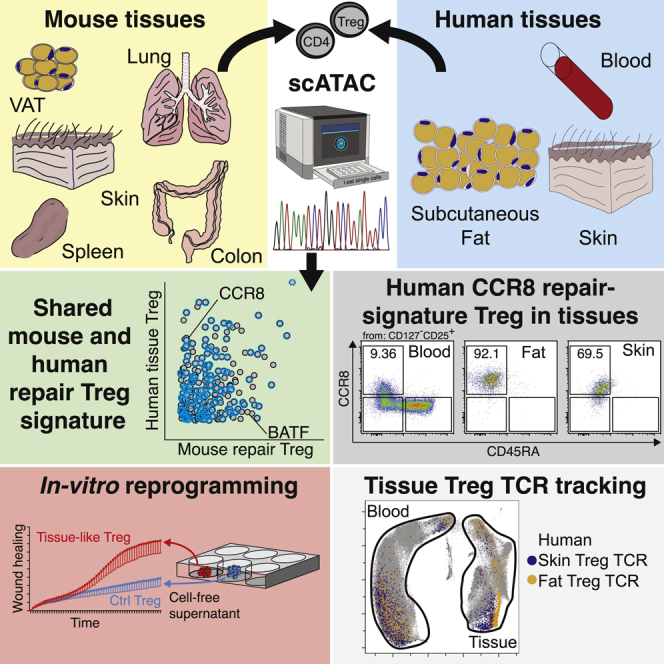
Highlights
-
•
Comparison of chromatin accessibility between murine und human tissue Treg cells
-
•
TCR tracking revealed clonal relationship between tissue and blood BATF+CCR8+ Tregs
-
•
Treg cells from healthy tissues were similar to CCR8+ Treg cells from tumors
-
•
Tfh-like differentiation program induces tissue Treg cell repair characteristics
Delacher et al. identify a conserved transcriptional and epigenetic signature that defines tissue regulatory T (Treg) cells in mice and humans. BATF+CCR8+ Treg cells from healthy human tissue share features with Treg cells found in tumors, suggesting that characteristics associated with tumor-residency may rather reflect the tissue repair functions of these cells.
Introduction
Regulatory T (Treg) cells exert various pivotal functions. In particular, they restrain self-reactivity and excessive inflammation, thereby promoting immune regulation (Sakaguchi et al., 2020). They can also promote tissue homeostasis and regeneration following tissue damage. The repair function of Treg cells in nonlymphoid tissues is seen in a variety of murine systems, including virus-infected lungs (Arpaia et al., 2015), damaged muscle (Burzyn et al., 2013), experimental brain ischemia, and autoimmune encephalitis (Dombrowski et al., 2017; Ito et al., 2019; Liesz et al., 2009). In visceral adipose tissue (VAT), Treg cells mediate metabolic functions (Cipolletta et al., 2012; Feuerer et al., 2009; Vasanthakumar et al., 2015). In addition, Treg cells facilitate regeneration of hair follicles in mice (Ali et al., 2017) and heart, retina, and spinal cord tissue in zebrafish (Hui et al., 2017).
A common epigenetic and transcriptional framework of murine tissue repair Treg cells classifies them as killer cell lectin-like receptor subfamily G1 (Klrg1)+ and ST2+, referred to as tisTregST2, herein (Delacher et al., 2017). Murine tisTregST2 cells are found in virtually all nonlymphoid tissues. They readily secrete amphiregulin (Areg) and IL-10 and express T-helper 2 (Th2)-associated molecules such as the transcription factors GATA binding protein 3 (Gata3), musculoaponeurotic fibrosarcoma (Maf), and basic leucine zipper ATF-like (Batf) (Delacher et al., 2017; DiSpirito et al., 2018). Murine tisTregST2 precursor cells develop in a two-stage process, where Batf is pivotal for the differentiation of tisTregST2 precursor cells in secondary lymphoid tissues (Delacher et al., 2020; Li et al., 2018; Miragaia et al., 2019).
Whereas the relevance of murine tissue-repair Treg cells has been well established and their development described on single-cell level, the characterization of a human tissue-repair Treg counterpart remains limited. Single-cell studies have addressed human Treg cells in the context of tumors (Azizi et al., 2018; Plitas et al., 2016; Sakaguchi et al., 2020; Szabo et al., 2019) and healthy tissues (James et al., 2020; Miragaia et al., 2019; Wu et al., 2019), but little is known about the human repair Treg program.
Here, we examined murine and human tissue-resident CD4+ T cells by single-cell chromatin accessibility and defined a shared tissue-repair Treg signature. This signature and TCR tracking identified human BATF+CCR8+ tissue-like Treg cells in peripheral blood, and molecular analysis revealed a Tfh-like differentiation program of this population in the tissue. Inducing a Tfh program in naive Treg cells promoted a tissue phenotype, including a repair function. A comparison of CCR8+ Treg cells from healthy tissues with CCR8+ Treg cells isolated from tumor tissues revealed multiple similarities, underlining the importance of understanding the contribution of these cells to the human tissue repair program in both health and disease.
Results
Single-cell chromatin landscape of murine CD4+ T cells establishes a tissue repair Treg cell signature
CD4+ T cells were purified from murine spleen, lung, colon, skin, and VAT using fluorescence-activated cell sorting (FACS) followed by the assay for transposase-accessible chromatin using sequencing on single-cell level (scATAC-seq). In total, scATAC-profiles were generated for more than 26,000 CD4+ T cells (Figure 1A), with ∼18,000 to ∼35,000 unique nuclear fragments per cell (Figures S1A–S1E). All data were group-normalized using Harmony (Korsunsky et al., 2019) and subjected to Uniform Manifold Approximation and Projection for Dimension Reduction (UMAP; Figure 1A). To identify populations, clusters were computed and color-coded (Figure 1A, right). The cellular compositions of the clusters were evaluated using the gene accessibility of lineage-defining genes. For example, Foxp3, the master transcription factor of Treg cells, was identified in clusters derived from spleen Treg cells (00, 03, 05, 11, 14, 18, 22) as well as several clusters from colon (10, 19), skin (16), and VAT (23; Figure 1B). In contrast to Foxp3, the Il2 locus was accessible only in tissue-derived CD4+Foxp3- T-conventional (Tconv) cells, e.g., in colon (06), skin (12, 20), and VAT (02, 17, 21). The locus encoding for CD62L (Sell) served to identify naive-like T cell clusters (e.g., 00, 07; Figure 1C). The Tbx21 locus, encoding for the T-helper 1 (Th1) transcription factor T-bet, and the Ifng locus were accessible only in effector CD4+ T cells from tissues (e.g., in skin [12] or colon [06], Figure 1D), but not in the Treg clusters. To evaluate whether our T cell scATAC-seq dataset was able to identify the Treg cell ‘regeneration and repair’ program, a bulk ATAC-seq derived signature of tisTregST2 from murine skin, VAT, and colon was used (Delacher et al., 2020). When plotting the skin-specific tisTregST2 signature (340 peaks; Table S1), the majority of signature-positive cells were detected in the skin Treg cluster (16), whereas the VAT tisTregST2 signature (417 peaks) identified the VAT-Treg cluster (23, Figure 1E). A core tissue repair signature based on 2,267 peaks derived from tisTregST2 of murine skin, VAT, and colon identified repair Treg cells in all those tissues (16, 23, 10). The core signature was comprised of loci encoding particular surface receptors (e.g., Klrg1), transcription factors (e.g., Pparg), and key effector molecules (e.g., Areg, Il10), many of which showed a very strong overlap with repair Treg cell clusters (Figure S1F).
Figure 1.

Single-cell ATAC landscape identifies repair Treg signature in murine tissues and spleen
(A) UMAP of scATAC-seq data derived from FACS-sorted CD4+ and CD25+ T cell populations of spleen, colon, lung, skin, and VAT. Sort layout and post-sort QC in Figure S1. Left, cells color-coded based on tissue of origin, batch-corrected using Harmony. Right, cells grouped in 26 clusters.
(B) Chromatin accessibility of Foxp3 and Il2 gene locus, with blue = low and red = high accessibility.
(C and D) Chromatin accessibility of the Sell, Tbx21 and Ifng gene locus.
(E and F) ATAC signature of skin tisTregST2 (340 peaks), VAT tisTregST2 (417 peaks), a core tisTregST2 signature (2,267 peaks) or early (3,323 peaks), and late (1,726 peaks) tisTregST2 precursor signature overlaid on UMAP, with blue = low and yellow = high overlap. Contribution of signature to clusters in pie chart. All signatures in Table S1.
(G) Pseudotime values and Batf chromVAR scores for selected clusters.
(H) UMAP of scATAC-data with colon T cells. Outlined fraction localization of tisTregST2 cells. Right, chromatin accessibility of the Foxp3, Klrg1, Rorc and Ikzf2 gene locus with blue = low and red = high accessibility. Data derived from four experiments with 18 C57BL/6J male mice bred under specified pathogen-free (SFP) conditions.
Tissue-repair Treg precursor cells differentiate in a two-step process starting in lymphoid organs (Delacher et al., 2020). When bulk ATAC-seq signatures of both early (PD1+Klrg1-, 3,323 peaks) and late (PD1+Klrg1+, 1,726 peaks) stages of tisTregST2 precursors were plotted on our scATAC-dataset, precursor cells were readily detected in spleen (05 and 11) but not in tissue clusters, highlighting the specificity of these signatures (Figure 1F).
A pseudotemporal ordering of the scATAC-seq data revealed a straight developmental path from naive Treg cells (00) via early (05) and late precursors (11) to tisTregST2 from skin, VAT and colon (16, 23, 10), with cells expressing the core tissue repair signature during the final step in the Treg differentiation trajectory (Figure 1G, top). This developmental path depends on the transcriptional regulator Batf (Delacher et al., 2020), and accordingly, an increased Batf chromVAR score was detected during pseudotemporal differentiation (Figure 1G, bottom). In summary, scATAC-seq provides the resolution to identify and characterize tissue repair Treg cells.
The tissue repair program in Treg cells is independent of the microbiome
The microbiota influence Treg cells in the colon (Atarashi et al., 2011; Geuking et al., 2011), but the impact of the microbiota on tisTregST2 cells is unclear. For this purpose, all colon-derived clusters were selected (06, 09, 10, 19, Figure 1H). Foxp3 and Klrg1 gene activity was overlaid and two Treg populations were identified: one cluster with high accessibility of both the Klrg1 and Foxp3 locus, indicating tisTregST2 (outlined fraction of cluster 10, Figure 1H). The other population was accessible at the Foxp3, but not at the Klrg1 locus (cluster 19). In the colon, two different populations of Treg cells exist: peripherally induced Treg cells (pTreg), converted locally from naive cells, and thymus-derived Treg cells (tTreg) (Sefik et al., 2015; Thornton et al., 2010). Plotting the accessibility of Rorc and Ikzf2 identified both populations: the circled fraction of cluster 10 represented thymus-derived Treg cells and constituted the tisTregST2 group (Foxp3openKlrg1openRorcclosedIkzf2open), cluster 19 represented pTreg cells (Foxp3openKlrg1closedRorcopenIkzf2closed) (Figure 1H).
In order to determine whether the generation of tisTregST2 cells is influenced by the microbiota, CD4+ T cells from spleen, skin, VAT, and colon of germ-free animals were sorted and subjected to scATAC-seq. More than 30,000 single nuclei were recovered (Figures S2A–S2D) and plotted in a UMAP (Figure 2A). As described in Figure 1, Foxp3, Il2, Sell, Tbx21, and Ifng were used to identify Treg, Tconv, effector and naive-like T cell populations (Figure 2B). As before, bulk-derived ATAC signatures of tisTregST2 cells and their precursors identified the populations on a single-cell level (Figures 2C and S2E). Bulk ATAC signatures of skin tisTregST2 cells identified a skin repair Treg cluster (13), the core tisTregST2 signature identified the skin (13) and colon (09) Treg repair clusters, while the precursor signature identified a spleen-derived cluster (06), verified by gene accessibility of Klrg1, Areg, Il10, and Pparg (Figure S2F). As before, the pseudotemporal ordering of scATAC-seq data revealed a straight developmental path from naive Treg cells (01) via late precursors (06) to tisTregST2 cells from skin, VAT, and colon (13, 09), again correlated with an increasing Batf chromVAR score (Figure 2D). These data indicate that the tissue-repair program in tisTregST2 cells can be induced in gnotobiotic mice. To verify this, flow cytometry was performed, and no differences were observed in overall Treg cell percentage, whereas the frequency of PD1+Klrg1+ tissue Treg precursors was slightly increased in spleens of gnotobiotic animals (Figure 2E). In addition, an increased percentage of tisTregST2 cells in colon and skin, but not VAT, of gnotobiotic mice was seen, whereas tisTregST2 cells from gnotobiotic mice had the same capacity to produce effector molecules, such as Areg or IL-10 (Figures 2E, S2G, and S2H). It is likely that the relative increase in tisTregST2 cells and their corresponding precursor cells could be due to a loss of pTreg cells in gnotobiotic animals. Therefore, the colons of these animals were analyzed in more detail: the colon-derived T cell clusters were plotted, the colon tisTregST2 repair signature (307 peaks) overlaid, and a high signal in cluster 09 identified (outlined fraction, Figures 2F and S2I). When plotting the gene accessibility of Foxp3, Klrg1, Ikzf2, and Rorc, tisTregST2 cells were detected in the tTreg population in this cluster (Foxp3openKlrg1openRorcclosedIkzf2open). In contrast, the pTreg population (Foxp3openKlrg1closedRorcopenIkzf2closed) was absent, confirming previous results (Atarashi et al., 2011; Geuking et al., 2011). These data indicate that the murine Treg repair program is established independently of microbiota-derived factors in gnotobiotic mice.
Figure 2.

Treg repair signature in single-cell ATAC landscape of T cells from germ-free animals
(A) UMAP of scATAC-seq data derived from FACS-sorted CD4+ T cell populations of spleen, colon, skin, and VAT of germ-free animals. Sort layout and post-sort QC in Figure S2. Left, cells color-coded based on tissue of origin. Right, cells grouped in 21 clusters.
(B) Chromatin accessibility of the Foxp3, Il2, Sell, Tbx21, and Ifng gene locus.
(C) Bulk ATAC signature of skin tisTregST2, a core tisTregST2 signature or a late tisTregST2 precursor signature overlaid on UMAP. Contribution of signature to clusters in pie chart.
(D) Pseudotime values and Batf chromVAR scores for selected clusters.
(E) Flow cytometry of T cells from SPF versus gnotobiotic mice. Top, percentage of Foxp3+CD25+ Treg cells of CD4+ T cells in spleen and mesenteric LN (mes LN). Below, Klrg1+PD1+ late tisTregST2 precursors of CD4+Foxp3+ Treg cells in spleen and tissues (2-way ANOVA with Sidak’s multiple comparison test, n = 4–13 with ∗∗∗p < 0.001 or ns = p > 0.05).
(F) UMAP of colon T cells. Outlined fraction localization of tisTregST2 cells. Right, chromatin accessibility of the Foxp3, Klrg1, Rorc, and Ikzf2 gene locus. Data derived from three experiments with 18 male mice bred under germ-free conditions or SPF conditions.
Single-cell chromatin landscape of human CD4+ T cells identifies tissue Treg cells
Given that murine tissue repair Treg cells develop independently of the microbiota, we asked whether such a repair Treg program was A) also present in human Treg cells and B) whether it can be directly compared between both species. scATAC-seq was performed on CD4+ T cells or CD4+CD25+CD127- Treg cells sorted from human blood, skin, and fat tissue of cosmetic surgery patients. Single-cell ATAC profiles of more than 20,000 CD4+ T cells from human skin, more than 20,000 CD4+ and CD25+ T cells from human fat and more than 35,000 CD4+ and CD25+ cells from human blood of several independent donors were obtained (Figures S3A–S3G). All data were donor-normalized using Harmony (Korsunsky et al., 2019) and a tissue-based as well as a cluster-based UMAP was computed (Figure 3A). Analogous to the data in the murine system, chromatin accessibility was used to identify T cell subpopulations; the human FOXP3 gene locus identified Treg cells in blood-based clusters (01, 07, 08, 09, 11) and in one cluster merged from skin and fat CD4+ T cells (03), therefore representing tissue Treg cells (Figure 3B). The human IKZF2 locus was open in tissue and blood-derived Treg cells, indicating thymic origin (Figure 3C). In blood, antigen-naive T cells were found in cluster 07 (Treg) and 06 (Tconv), whereas memory-type Treg cells reside in cluster 01 and 03 (Figure 3D). The human IL2 locus was open in tissue and blood-derived Tconv cells (00, 02, 04, 05, 10) and did not overlap with Treg cell clusters, comparable to the IFNG or the BHLHE40-accessible populations (Figures 3E and S3H). Other characteristic human Treg genes such as ENTPD1 (CD39), CTLA4, and TIGIT showed a clear overlap with the Treg clusters, whereas KLRG1, a key marker for murine tisTregST2, showed no accessibility in Treg clusters (Figure S3H). In contrast to this, the ZC3H12C locus encoding for Regnase-3 was accessible only in tissue Treg cells and a Treg subpopulation in the memory Treg cluster in blood (Figure S3H).
Figure 3.

Single-cell ATAC and gene expression landscape of human blood, fat and skin CD4+ T cells
(A) UMAP of scATAC-seq data derived from FACS-sorted CD4+ T cell populations of human peripheral blood, skin, and fat of individual donors. Sort layout and post-sort QC in Figure S3. Left, cells color-coded based on tissue of origin, batch-corrected for donor differences using Harmony. Right, cells grouped in 16 clusters.
(B–E) Chromatin accessibility of the FOXP3, IKZF2, SELL, IL2, and IFNG gene loci.
(F) PCA of DEG (padj < 0.001) in a comparison of bulk RNA-seq data from CD4+CD25+CD127- human fat and skin Treg cells and blood CD45RA+CD45RO- naive or blood CD45RA-CD45RO+ memory Treg cells. Sort layout and post-sort QC in Figure S3, Rpkm table and statistics in Table S2.
(G) DEG between blood and tissue Treg cells. Several genes and number of DEG labeled.
(H) Hierarchical clustering highlighting a common RNA tissue Treg signature with 2,779 DEG, selected by comparing memory Treg versus fat Treg cells (foldchange (fc) < 0.5 or > 2) and fat Treg versus skin Treg (fc < 0.5 or > 2).
(I) Left, DEG between skin and fat Treg cells. Right, UMAP of scATAC-seq data from skin and fat Treg cells with tissue of origin and clustering.
(J) ATAC-seq data for the GPR55, AHRR, and ITGA4 locus with two clusters (0, Skin Treg and 1, Fat Treg). All datasets group-normalized to maximum peak height indicated in brackets. scATAC-seq data are derived from four experiments with five female donors. Bulk RNA-seq data are derived from 11 experiments with 14 female donors.
To quantify the differences between human tissue Treg cells and their blood counterparts on a gene expression level, human CD4+CD25+CD127- Treg cells from fat and skin, as well as blood-derived naive (CD45RA+CD45RO-) and memory (CD45RA-CD45RO+) Treg cells were sorted and subjected to bulk RNA-seq (Figures 3F–3H; Figure S3I and Table S2). In total, more than 12,000 genes were differentially expressed (padj < 0.05) between fat Treg and blood naive Treg cells, while almost 11,000 genes were still differentially expressed between fat and blood memory Treg cells (Figure 3G). For skin, more than 7,000 and 5,500 differentially expressed genes (DEG) between skin Treg and blood naive Treg cells and between skin and blood memory Treg cells, respectively, were identified (Figure 3G). These numbers highlight a considerable difference between tissue Treg cells and circulating blood Treg cells, while about 2,500 DEG separate skin and fat-derived tissue Treg cells. Examples include GRP55 and AHRR expressed in skin and ITGA4 expressed in fat Treg cells (Figures 3H and 3I). To compare chromatin and gene expression data, fat and skin Treg cells were subclustered (Figure 3I, right panel) and aggregated ATAC reads for GPR55, AHRR, and ITGA4 displayed, showing differential chromatin accessibility (Figure 3J).
As in mice, the analysis of human skin, fat, and blood single-cell ATAC-seq data provided high resolution on functional and anatomical dimension, thereby allowing the characterization of human tissue Treg cells and their functional programs.
Species-conserved repair signature identifies BATF+CCR8+ Treg population in human blood and tissues
First, the murine skin and VAT repair Treg clusters (16, 23) versus spleen Treg clusters (00, 03, 14, from Figure 1A) were compared and a murine tissue repair Treg signature was computed (14,594 peaks, Figure 4A and Table S3). Next, human fat and skin Treg cells versus human blood naive Treg cells (clusters 03 versus 07 from Figure 3A) were compared, resulting in a human tissue Treg signature with 12,236 differential peaks (Figure 4B). To identify shared peaks between human and mouse tissue Treg cells, a liftover of peaks identified in the murine mm10 genomic annotation to the human hg19 genome was performed. Of the murine peaks that could be mapped on the human genome, candidates were restricted to regions inside the gene body plus 2,000 bp upstream of the transcription start site. In addition, only regions in genes which had the same direction of change in chromatin accessibility in mouse and human were selected. This resulted in a list of 643 peaks (Figure 4C). This species-conserved peakset of 643 loci contained several surface receptors (e.g., CCR2, CCR5, CCR6, CCR8), members of the TNF receptor superfamily (TNFRSF8, TNFRSF9) and transcription factors such as GATA3, TOX, BACH2, and BATF (Figure 4C). Gene expression data derived from bulk RNA-seq confirmed that, for several loci, open chromatin was linked to increased gene expression (Figure 4C, lower part, right panel).
Figure 4.

Species-conserved tissue Treg peakset identifies CCR8+ Treg cell population in human blood
(A) Murine tissue repair Treg signature. All peaks in a heatmap (left) and volcano plot (right). Some peaks highlighted and labeled by their closest gene. Peaks as hexagons p value < 10−300.
(B) Human tissue Treg signature.
(C) Shared peaks in murine (A) and human (B) tissue Treg datasets. Peaks as hexagons log FC > 2.0. Peaks overlapping with ChIP-confirmed BATF binding sites (B cells, GSM803538) blue and counted in pie charts. All peaks in heatmap with ATAC signal (left) and corresponding gene expression (right). All peaksets in Table S3.
(D) HOMER de novo motif results in the comparison of tissue Treg (03) versus blood naive Treg (07). Below, read coverage versus distance from BATF motif center.
(E) BATF chromVAR deviation against pseudotime for human Treg cell cluster 01, 03, and 07. Smoothing line fitted to the data.
(F) UMAP with scATAC-seq data (cluster 01, 03, 07) and chromatin accessibility of the BATF and CCR8 gene locus.
(G) Chromatin accessibility of the CCR8 locus in human Treg clusters 01, 03, and 07 with BATF ChIP-Seq data. Below, flow cytometry of BATF or FOXP3 of CCR8+ Treg cells (protein: n = 14, one-way ANOVA; RNA: n = 5, Deseq2).
(H) Chromatin accessibility of the Ccr8 locus in murine Treg cluster 00, 11 and 16 combined with publicly available Batf ChIP-Seq (CD8 T cells, GSE54191). Below, expression of Ccr8 in tisTregST2 and precursor cells (n = 5, Deseq2). Pseudocolor dot plots: co-staining of Nfil3(GFP) or Areg(GFP) versus Ccr8 in spleen, skin or VAT-derived CD4+CD25+Treg cells from Nfil3(GFP) or Areg(GFP) reporter mice. All data are derived from two to five independent experiments with five or more individual mice or donors.
In order to identify a key transcription factor responsible for the human tissue Treg signature, HOMER (Heinz et al., 2010) was used to search motifs in differential peaks between human tissue and blood Treg (accessible peaks cluster 03 versus 07). The de novo motif with the most significant enrichment (rank 1, p < 10−389) in the tissue Treg group closely matched a basic leucine zipper domain (bZIP) motif (Figure 4D). Of the bZIP family members, BATF was selectively expressed in both fat and skin Treg cells (Figure 3G). To demonstrate the impact of BATF, reads around the BATF motif center were displayed (Figure 4D) and illustrated increased activity in tissue Treg cells versus naive or memory Treg cells from blood. In addition, an increased BATF chromVAR score during pseudotemporal differentiation was observed (Figure 4E), also highlighted by chromatin accessibility of the BATF gene in a cluster-based UMAP with all three populations (Figure 4F). These results indicate a developmental trajectory from blood naive Treg cells via blood memory Treg cells to tissue Treg cells, making tissue Treg cells the final developmental and differential step, as seen in the mouse dataset (Figures 1G, 2D, and 4E).
We wondered whether BATF would have confirmed binding sites located inside the species-conserved repair Treg signature. To this end, human BATF chromatin immunoprecipitation following sequencing (ChIP-seq) data were extracted (Pope et al., 2014) and binding sites determined: as many as 145 out of the 278 peaks that gain accessibility in repair Treg cells showed BATF binding sites (Figure 4C), with one example being the CCR8 gene (Figure 4G). Upon closer examination of the CCR8 locus, differential chromatin accessibility between tissue and blood naive Treg cells in the promoter region, the gene body as well as a potential upstream enhancer site was detected (Figure 4G, upper panel, additional genes in Figures S4A–S4D). Blood memory Treg cells showed somewhat increased accessibility at the CCR8 enhancer locus, which could indicate a subpopulation of CCR8open Treg cells. To investigate this, human peripheral blood was analyzed by flow cytometry and a subpopulation of CCR8+ Treg cells in the memory, but not the naive Treg cell pool was identified (Figure 4G, lower part). This population was sorted and increased BATF expression on protein and RNA level was confirmed (Figures 4G and S4E). In addition, the population showed increased expression of FOXP3 and CD25, making them part of the highly differentiated effector Treg population (‘Fraction II’) described previously (Miyara et al., 2009; Tanaka and Sakaguchi, 2017).
Since CCR8 was also part of the murine repair Treg signature (Figure 4A), Ccr8 chromatin accessibility for tissue-repair Treg cells, their circulating precursor cells and naive Treg cells was displayed (Cluster 00, 11 and 16 from Figure 1A), and murine Batf ChIP data were overlaid (Hasan et al., 2017); Figure 4H). Again, differential peaks in the promoter region as well as an upstream potential enhancer site were detected, with Batf binding at the upstream enhancer of the murine Ccr8 gene (Figure 4H upper part, yellow box). Ccr8 gene expression was verified by RNA-seq of tisTregST2 cells from lung, liver, VAT and skin, as well as the precursor populations (Figure 4H lower part, left). To analyze the relationship of Ccr8-expressing Treg cells with the tisTregST2 repair phenotype, Nfil3(GFP) reporter animals, were analyzed. In these animals, Nfil3(GFP) labels the precursor and tissue repair program in Treg cells (Delacher et al., 2020). In addition, we generated an amphiregulin reporter mouse (Areg(GFP)), in which GFP reports the production of the tissue repair factor Areg. In murine skin and VAT, the majority of tisTregST2 cells were positive for both Ccr8 and Nfil3(GFP) or Areg(GFP), identifying Ccr8 as a good marker to recognize tisTregST2 in the murine system and potentially also in the human system (Figure 4G-H; Figures S4F–S4I).
In summary, open chromatin data of murine repair Treg cells were translated to human tissue Treg cell data and identified a conserved chromatin repair signature, associated with the transcription factor BATF.
Surface characterization of CCR8+ Treg cells reveals co-expressed proteins
To perform a deeper characterization of BATF+CCR8+ Treg cells in human blood, Treg cells from several donors were pre-enriched and co-stained with more than 350 individual surface antibodies labeled in PE, followed by flow cytometry (Figures 5A, S5A, and S5B). Gating was performed for naive Tconv cells, naive Treg cells, memory Tconv cells, CCR8- memory Treg cells, and CCR8+ memory Treg cells, and surface protein expression data were subjected to hierarchical clustering. Of the proteins analyzed with this screen, 287 were not expressed (group 9), while 47 were ubiquitously expressed (group 8). Group 2 identified proteins that were enriched in CCR8+ Treg cells (CD39, CD71, CD195, HLA-DR), whereas group 1 revealed proteins that were less expressed on CCR8+ Treg cells (CD26, CD49d, CD197; Figures 5B and 5C). To correlate protein expression to chromatin accessibility, aggregated scATAC-seq data for blood naive Treg, blood memory Treg (which include the CCR8+ population), and skin and fat Treg cells were plotted for HLADR, TFRC (CD71), and CCR5 (Figure 5D). Differential peaks between tissue and blood as well as BATF binding were detected in all three genes, including different HLA-DR loci (HLADRA, HLADRB1, HLADRB3); Figures 5D and S5C). For ITGA4, encoding CD49d, a protein not expressed in CCR8+ Treg cells, chromatin was less accessible in tissue Treg cells as compared to naive Treg cells from blood (Figure 5E).
Figure 5.

Surface protein, transcriptional and epigenetic analysis of human CCR8+ Treg cells in blood
(A) Human Treg cells from healthy donors were pre-enriched and surface-stained (CD3+CD4+CD25+CD127-CD45RO+CCR8+), followed by individual staining with 361 PE-conjugated anti-human surface antibodies and measurement by flow cytometry. Color code indicates expression with blue = low and red = high. Gating, isotype control staining and additional results are shown in Figure S5.
(B) Expression of HLA-DR in five subpopulations of human Treg cells.
(C) Co-expression of CCR8 (x axis) and PE-coupled antibody (y axis), pre-gated on memory Treg cells. Blue = negative correlation, red = positive correlation.
(D-E) Chromatin accessibility of the human HLA-DRB1, TFRC, NR_125405 and ITGA4 locus. Data are derived from antigen-naive Treg cells (07), memory Treg cells (01), and fat and skin Treg cells (03). BATF ChIP-Seq signal below (GSM803538).
(F) DEG between blood memory CCR8+ and blood naive Treg cells. Several genes and total number of DEG are labeled.
(G) Flow cytometry of Treg cells (CD4+CD25+CD127-) isolated from human blood, fat and skin tissue with CCR8 and HLA-DR expression (n = 4–5, one-way ANOVA with Tukey post-test).
(H) Flow cytometry and tSNE with 100,000 human blood Treg cells, 1,763 fat Treg cells and 5,546 skin Treg cells. tSNE grouping based on CD25, CD39, BATF, CD45RA, CD45RO, CD195, CCR8, CD49d, HLA-DR, CD71 and FOXP3 expression. All data are derived from one independent experiment with three donors (A-C) or five independent experiments with five human female donors (F) and (G) or an individual donor, (H).
To directly verify HLA-DR and CCR8 expression, bulk RNA-seq of blood CCR8+ Treg cells was performed and gene expression of several HLADR loci, CCR8, and BATF was verified (Figure 5F). In addition, human skin, fat, and blood Treg cells were analyzed via flow cytometry and high protein expression levels of CCR8 and HLA-DR in tissue Treg cells were detected (Figures 5G and S5D). Recently, a CD161+ human repair Treg population, induced from Tconv cells and found in the lamina propria of the intestine, has been identified (Povoleri et al., 2018). The CCR8+ tissue Treg population in human skin and fat was distinct from this CD161+ Treg population in chromatin accessibility and CD161 protein expression (Figure S5E).
Finally, several proteins with specificity for the CCR8+ Treg population (e.g., CD71, CCR8, HLA-DR, CD195) were selected, followed by multicolor flow cytometry with Treg cells from skin, fat, and blood. Unsupervised grouping of individual cells from all three tissues using T-distributed Stochastic Neighbor Embedding (t-SNE) clustered skin, fat, and blood CCR8+ Treg cells into a common CCR8+ cluster (Figure 5H). These data indicate a close relationship between CCR8+ Treg cells from fat and skin with CCR8+ Treg cells found in blood.
Single-cell TCR-seq of tissue and blood confirms clonal relationship of CCR8+ Treg cells
T cells have a natural barcoding feature, the TCR-sequence, which allows tracking their clonal relationship. Therefore, CD4+ T cells from fat and skin were sorted and combined scRNA and scTCR sequencing was performed. This technology allows both the identification of cells via gene expression programs and tracking of different clones via their TCR α and β chain sequences. Blood from the same individuals was processed and CD4+ T cells, Treg cells (CD25+CD127-), memory Treg cells (CD45RA-CD45RO+) and memory CCR8+ Treg cells were sorted, followed by scRNA/scTCR-seq (Figures S6A–S6D). All data from the same individual were combined, UMAP graphs computed and tissue origin (Figure 6A, left) and input sort population (Figure 6A, right) highlighted.
Figure 6.

Single-cell RNA and TCR landscape of donor-matched human blood, fat and skin CD4+ T cells
(A) UMAP of scRNA-seq data derived from FACS-sorted CD4+ T cell populations of human peripheral blood, skin and fat of one individual donor, second donor in Figure S6. Left, cells color-coded based on tissue of origin. Right, cells color-coded based on sort strategy.
(B and C) Gene expression of FOXP3, CCR8, and HLA-DRB1 plotted on UMAP. Color code indicates expression strength.
(D) TCRs derived from all skin Treg cells (blue) and all fat Treg cells (yellow) extracted and highlighted. TCRs listed in Table S4.
(E and F) Tracking of fat (E) or skin (F) Treg TCR clones in different sorted populations of peripheral blood of the same donor. Percentage indicates fraction of detected clones, total number of clones shown below.
(G and H) scATAC-seq based pseudotime trajectory and tissue Treg signature (2687 peaks) score (∗∗∗p < 0.001, Kruskal-Wallies test).
(I) UMAP of scATAC-seq data derived from FACS-sorted CCR8+ T cell population of human peripheral blood of female donors highlighting the skin Treg signature (1,030 peaks) and fat Treg signature (437 peaks). All data are derived from two independent experiments with one female donor (scRNA/scTCR) or two independent experiments with two female donors (scATAC).
To identify the Treg cluster in the tissue samples, among others, Foxp3 gene expression was used (Figure 6B). To highlight the location of CCR8+ Treg cells in tissue and blood, CCR8 and HLADRB1 gene expression were plotted (Figure 6C). Next, the TCR-sequences of fat- and skin-derived Treg cells were extracted and all T cells with identical TCRs found in either fat or skin tissue Treg cells were highlighted in the UMAP plot (Figure 6D and Table S4). Skin and fat Treg TCR clones accumulated in the CCR8+ and HLA-DR+ regions of blood Treg cells. To confirm this accumulation, the presence of tissue Treg TCR clones within the different FACS-sorted blood Treg input populations was evaluated: Fat Treg TCR clones constituted 0.52% of all TCRs detected in blood CD4+ T cells, 1.14% in blood Treg cells, 1.62% in blood memory Treg cells and 4.42% in sorted blood CCR8+ Treg cells (Figure 6E). Analogously, skin Treg TCR clones in blood constituted 1.21% in blood CD4+, 2.67% in blood Treg, 3.61% in blood memory Treg, and 7.18% in blood CCR8+ Treg cells (Figure 6F). Analysis of a second fat, skin, and blood donor confirmed our TCR tracking results and overall TCR clonalities (Figures S6E–S6H). Thus, these data indicate a close clonal relationship between tissue Treg cells and blood CCR8+ Treg cells.
To understand the complexity of the CCR8+ Treg population in human blood, scATAC-seq on sorted CCR8+ Treg cells was performed (Figure S6D), and the data were integrated into a pseudotime-based developmental trajectory (Figure 6G), where pseudotime values indicated a developmental path from blood naive Treg cells via blood memory Treg cells to CCR8+ tissue Treg cells. To assess the contribution of the tissue phenotype in blood CCR8+ Treg cells, a human tissue Treg signature was computed, tissue Tconv peaks were excluded, and its contribution (2,687 peaks) to all four populations described above was calculated. Based on this calculation, CCR8+ Tregs from blood were closest to tissue Treg cells from fat and skin (Figure 6H). Next, human skin- or fat-specific tissue Treg peaksets were computed (skin: 1,030 peaks; fat: 437 peaks) and two largely non-overlapping populations in CCR8+ Treg cells from blood were identified, possibly containing recirculating tissue Treg cells or committed tissue Treg precursor cells (Figure 6I).
In summary, TCR tracking, pseudotemporal projection and tissue signature-based tracing all imply a close relationship between CCR8+ Treg cells from human blood and tissue Treg cells from skin and fat tissue.
Human tissue Treg cells integrate a Tfh-like signature which can be induced in-vitro
Having established a link between blood CCR8+ Treg cells and tissue Treg cells from skin and fat, we were wondering which factors contribute to the differentiation into human tissue Treg cells. While mouse tisTregST2 are characterized by a Th2-like expression profile (Delacher et al., 2020; Delacher et al., 2017; Delacher et al., 2019; Schiering et al., 2014), human tissue Treg cells from fat and skin do not display such an obvious Th2-bias (Figure 3G). However, in both species, BATF seems to be a critical factor for the tissue Treg program. Since BATF is also important for T-follicular helper (Tfh) differentiation (Ise et al., 2011), we extracted a Tfh-like signature from a published scATAC-seq dataset characterizing tumor infiltrating T cell subtypes (Satpathy et al., 2019). The tissue-Tfh-signature (3,099 peaks) was plotted on our scATAC-seq UMAP of human blood and tissue T cells (Figure 7A). As indicated by the color code, increased Tfh-signature activity was detected in fat and skin Treg cells and, to a lesser degree, in a subpopulation of blood memory Treg cells (Figure 7A). To confirm this, a Tfh signature score was determined, confirming the increase of a Tfh-like signature in CCR8+ Treg cells from blood as well as tissue Treg cells from fat and skin (Figure 7B). On the RNA level, Tfh-associated genes such as the transcription factors BCL6, BATF and MAF were expressed in tissue Treg cells from fat and skin, and BATF and MAF transcripts were increased in CCR8+ Treg cells from blood. Other Tfh-associated genes such as PDCD1 (PD-1) and ICOS were also expressed in tissue Treg cells, whereas CXCR5, the defining marker for germinal center Tfh cells, was not expressed in tissue Treg cells (Figures 7B and S7A).
Figure 7.

Treg cells in wound healing and cancer
(A) UMAP with Tfh signature (3,099 peaks).
(B) Tfh signature score and RNA expression of Tfh-related molecules (n = 5, Deseq2).
(C and D) Human blood-derived naive Treg cells treated for six days with IL-2 or Tfh mix, followed by ATAC and RNA-seq (n = 4, Deseq2, Table S5). ATAC loci highlighted, peaks padj < 10−40 capped. Right, HOMER de-novo motif search and reads around BATF motif center.
(E) RNA-seq data (Tfh Treg versus IL-2 Treg) compared to RNA-seq of CCR8+ skin Treg versus blood naive Treg.
(F) Supernatants of Tfh-like Treg cells or IL-2 Treg cells from 5 donors used in wound healing assay (HaCaT, 1+7 dilution, n = 5, unpaired t testing with Holm-Sidak post-test).
(G) Tfh-like Treg or IL-2 Treg supernatants from 16 donors used in human epidermal reconstruction model. Left, electrical impedance (n = 3 models); right, surface area of Stratum corneum (n = 3, two scans of each model, ANOVA and Tukey post-test).
(H) UMAP of CD4+ T cell populations of human basal cell carcinoma (Satpathy et al., 2019) with cell type, tissue Treg signature, IKZF2 and CCR8 gene activity.
(I) CCR8 and CD45RA expression in Treg cells from liver tumor, blood, skin or fat (one-way ANOVA with Tukey post-test, n = 4–5).
(J) PCA with 500 most differential DEG for Treg cells from tumors or healthy donors (Table S5).
(K) DEG between liver tumor CCR8+ Treg versus blood CD45RA+ Treg (left) or liver tumor CCR8+ Treg versus NAT liver CCR8+ Treg.
(L) Top, DEG between skin and fat CCR8+ Treg versus blood CD45RA+ Treg with heatmap. Below, DEG between liver tumor CCR8+ Treg versus patient blood CD45RA+ Treg cells with heatmap. Data are derived from 11 experiments with 14 donors (B), 2 experiments 4 donors (C-D), one experiment with 5 (F) or 16 donors (G), or 9 experiments with 10 female and male donors (I–L).
To model whether human tissue-Treg differentiation is influenced by the Tfh-pathway, blood naive Treg cells were cultured with a cytokine cocktail described to induce Tfh differentiation in-vitro. This cocktail contained TGF-β and IL-12 as essential factors for human Tfh-induction, as well as IL-21, and IL-23 to stabilize the program (Qin et al., 2018). The Tfh mix induced MAF, BATF, and BCL6 in cytokine-treated Treg cells as compared to IL-2-only treated Treg cells, while FOXP3 expression and suppressive capacity remained high (Figures 7C, S7B, and S7C). Gene expression and bulk ATAC-seq profiles of Tfh-like Treg cells versus IL-2 Treg cells were generated and identified more than 4,500 DEG and more than 17,000 differentially accessible peaks between both groups, some of which were shared with tissue Treg and Tfh cells (Figure 7D, left, Figure S7D and Table S5). HOMER de novo analysis revealed closely matched bZIP and IRF:BATF motifs with low p value and rank, and transcription factor footprinting around the BATF motif center visualized the impact of BATF in the chromatin landscape of Tfh-like Treg cells (Figure 7D, right).
To identify key genes shared between the in-vitro induced Tfh-like Treg cells and the in situ BATF+CCR8+ tissue Treg cells, CCR8+ Treg cells from human fat and skin were sorted, followed by bulk RNA-seq and calculation of DEG. When comparing DEG between Tfh-like versus IL-2 Treg cells with DEG between skin and fat Treg cells versus naive blood Treg cells, we identified 228 genes that showed increased expression in both human CCR8+ tissue Treg cells and in-vitro induced Tfh-like Treg cells (Figure 7E), including Tfh-typical TFs such as BCL6, BATF and MAF. In addition, a number of remodeling enzymes from the matrix metalloproteinase (MMP) and the disintegrin/metalloproteinase (ADAM) family, such as MMP25 and ADAM19, were identified. But also tissue repair-related genes, such as platelet-derived growth factor subunit A (PDGFA), which acts as a mitogenic factor for fibroblasts, or GRN were detected. GRN encodes for the secreted protein Progranulin, which is important for normal development, wound healing, and tumorigenesis (Bateman et al., 2018).
In-vitro induced human tissue Treg cells promote tissue repair
To investigate the impact of the Tfh-program on Treg function, the tissue-like Treg program was induced in naive Treg cells, cells were washed vigorously to remove any traces of cytokines and re-stimulated for an additional 20 h to allow the secretion of factors into the supernatant. Cell-free supernatant was collected and evaluated in a wound healing assay with a keratinocyte cell line in a live cell imaging system (Figure 7F). For the first day after wounding, no differences between the groups were observed. After that, wounds treated with supernatant derived from Tfh-induced tissue-like Treg cells closed faster than controls and achieved complete wound closure after about 45 h, not observed with supernatant from IL-2-only treated Treg cells (Figures 7F, S7E, and S7F). To evaluate the effect on skin tissue repair, a reconstructed human epidermis model was used. In this model, primary human epidermal keratinocytes are grown on a polycarbonate membrane forming a well stratified epidermis with all physiological epidermal layers. Following the maturation, models were punched to generate a circular wound with a diameter of 2 mm. After wounding, supernatants were applied in the basolateral medium and wound healing was monitored via impedance spectroscopy and histological analysis of the Stratum corneum (Groeber et al., 2015) (Figures 7G and S7G). While electrical impedance was not different between groups 48 h after wounding, models treated with Tfh-like Treg supernatant significantly increased TEER 1000 Hz values 96 h and 168 h after treatment compared to IL-2 Treg supernatant or no supernatant controls. These results indicate enhanced reconstruction of the epidermal layer, thereby resulting in an enhanced barrier function. Quantitative histological analysis revealed that the wound healing-associated regeneration of the Stratum corneum was increased upon treatment with Tfh-like Treg supernatants compared to IL-2 Treg supernatants. Based on these data, one can conclude that installing a tissue Tfh-like signature in naive Treg cells induces tissue-reparative functions, which may be comparable with the in vivo situation.
Tumor-resident CCR8+ Treg share features with normal tissue CCR8+ Treg
Recently, CCR8+ Treg cells have also been described in tumor tissues such as lung cancer (Alvisi et al., 2020; De Simone et al., 2016) and breast cancer (Plitas et al., 2016; Wang et al., 2019). Since cancers can co-opt wound healing responses to promote tumor cell maintenance and growth (Dvorak, 2015; Sundaram et al., 2018), we wondered whether tumor-resident CCR8+ Treg cells had similarities to CCR8+ Treg cells found in normal tissues. Therefore, published scATAC-seq data from CD4+ T cells isolated from human skin cancer were extracted (Satpathy et al., 2019) and different CD4+ T cell subtype clusters were plotted as described by the authors. When plotting the human tissue-Treg cell signature (described in Figure 6H; 2687 peaks), derived from skin and fat Treg cells of healthy donors, on this dataset, a large fraction of tumor Treg cells was highlighted, along with Tfh-like CD4 T cells (Figure 7H). In addition, Treg cells in the tumor showed elevated chromatin accessibility for IKZF2, indicating thymic origin, and CCR8. These findings were recapitulated in scATAC-seq data of tumor-infiltrating CD4+ T cells from a spontaneous murine HER2-transgenic mammary carcinoma model (BALB-NeuT; (Hosseini et al., 2016); Figure S7H): A fraction of tumor infiltrating Treg cells were positive for the murine repair Treg signature and showed accessible chromatin for the Ikzf2 and Ccr8 locus.
To further investigate the overlap between CCR8+ Treg cells in the tumor microenvironment and CCR8+ tissue Treg cells found in healthy tissues, we obtained human liver tumor samples, normal liver adjacent tissue (NAT), and healthy skin and fat tissues. In liver tumors and normal skin and fat tissue, on average 60% to 80% of Treg cells were positive for CCR8 (Figures 7I and S7I). Next, CCR8+ Treg cells from all tissues were sorted together with different blood Treg cell populations followed by bulk RNA-seq. Principal component analysis of all groups revealed a high overlap between human fat, skin, liver NAT and liver tumor Treg cells on PC1 (Figure 7J). While the comparison of liver tumor CCR8+ Treg versus blood memory-type Treg cells from the same patients identified almost 2,000 DEGs, liver tumor-derived CCR8+ Treg versus liver NAT CCR8+ Treg cells from the same patients were almost identical (17 DEG with padj < 0.05, Figure 7K). To further investigate this, a tissue Treg signature using gene expression data from skin and fat CCR8+ Treg cells was calculated and plotted for all cell types (Figure 7L, top). While there was a substantial overlap between healthy tissue CCR8+ Treg cells and tumor CCR8+ Treg cells, much less similarity was observed with blood CCR8+, blood CCR8- memory or blood naive Treg cell populations. And vice versa, a calculated liver tumor Treg signature showed substantial gene expression overlap of tumor CCR8+ Treg cells with tissue CCR8+ Treg cells isolated from healthy skin and fat as well as from liver NAT (Figure 7L, bottom). Examples for this high degree of overlap are the transcription factors BCL6 and BATF (Figure S7J).
These findings could be recapitulated by looking at chromatin accessibility. A chromatin signature calculated from tumor-derived Treg cells (Satpathy et al., 2019) with 940 peaks also identified tissue Treg cells from normal fat and skin tissue (Figure S7K), indicating that tumor residing CCR8+ Treg cells and normal-tissue CCR8+ Treg cells are indeed very similar.
Discussion
In this study, we performed single-cell chromatin accessibility profiling and single-cell RNA and TCR-sequencing of human and mouse tissue-resident and circulatory Treg cells. A cross-species analysis enabled us to identify features of nonlymphoid Treg cells from human skin and fat tissue, which includes the identification of a tissue-repair program.
Currently, most studies about human Treg cells focus on circulating cells in the blood or Treg cells isolated from tumor tissue. Therefore, differences observed between tumor tissue and blood Treg cells are usually attributed to the tumor environment. Our data suggest that Treg cells isolated from healthy nonlymphoid tissues share many features with tumor-resident Treg cells. Several surface proteins are thought to characterize tumor-specific Treg cells, including CCR8, TIGIT, PD1, and ICOS (Azizi et al., 2018; Plitas et al., 2016). However, all of these markers are already constitutively expressed on Treg cells in normal nonlymphoid tissue, highlighting that they are not tumor-specific, but rather indicative of tissue residency.
Blood-based CCR8+ Treg cells are suggested to be circulating precursors of intratumoral Treg cells (Wang et al., 2019). We found that blood memory CCR8+ Treg cells constituted putative tissue Treg precursors and/or recirculating tissue Treg cells, thus acting as a physiological reservoir of tissue-committed Treg cells with tissue-specific TCRs, tissue-specific gene expression programs and potentially tissue-repair capacity.
The BATF+CCR8+ tissue Treg population is independent from the previously described CD161+ human repair Treg population found in blood and the lamina propria of the intestine (Povoleri et al., 2018). BATF+CCR8+ Treg cells do not express CD161 and have a distinct gene expression profile compared to CD161+ Treg cells including the expression of IKZF2, separating them from the retinoic acid-converted CD161+ Treg population.
CCR8+ Treg cells share many features with ‘Fraction II’ or ‘effector’ Treg cells, initially described as a population with elevated FOXP3 and CD25 expression and strong suppressive capacity (Sakaguchi et al., 2020). These features, together with the here proposed repair function of CCR8+ Treg cells, make them an undesirable cell type in the tumor microenvironment: while downregulating potential anti-tumor immune responses via their suppressive effect on anti-tumor immunity, they might contribute to tumor growth and matrix re-organization via the secretion of tissue-repair factors. Our data also imply that using strategies to eliminate CCR8+ Treg cells from tumors, via CCR8-depleting antibodies or other means, may have side effects because such interventions would likely affect the CCR8+ Treg populations in other normal, non-cancerous tissues.
We are just beginning to understand what the function of CCR8+ tissue Treg cells in normal human tissues might be. As in mouse, BATF seems to be a central factor to induce the tissue and presumably the repair program in human Treg cells, promoting considerable changes in the chromatin accessibility and gene expression landscape (Delacher et al., 2020). This change might induce yet-unknown properties of human repair Treg cells to aid in the remodeling of the extracellular matrix and to provide signals which stimulate keratinocytes and other parenchymal and stromal cell types to rebuild tissues. One could envision a highly relevant function of CCR8+ Treg cells for tissue regeneration after inflammatory responses. This feature could make this cell type a good candidate to be utilized in regenerative medicine.
Limitations of study
The human CCR8+ Treg cell repair function has not been evaluated in vivo, and whether CCR8+ Treg cells promote tumor growth in cancer patients has not been determined. Whether tissue Treg differentiation in the human context is also a multistep process as described for mouse Treg cells needs further analysis. Circulating CCR8+ Treg cells could be a good starting point to address this question. In the future, it will be important to find ways to efficiently expand tissue CCR8+ Treg cells in-vitro in order to further study their functional capacity in different assay systems. We identified a molecular program, parts of which were initially described in germinal center T-follicular helper cells, which is diverted by human Treg cells located in tissues to induce repair capacities. Whether T-follicular helper-like cells residing in inflamed or tumor tissues have a similar ability to promote wound healing needs to be further analyzed.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies anti-mouse | ||
| AF488 anti-mouse CD3 antibody | Biolegend | AB_312671 |
| APC anti-mouse CD4 antibody | Biolegend | AB_312719 |
| APC/Cy7 anti-mouse CD4 antibody | Biolegend | AB_312699 |
| Biotin anti-mouse CD4 antibody | Biolegend | AB_312711 |
| BV421 anti-mouse CD4 antibody | Biolegend | AB_11219790 |
| BV510 anti-mouse CD4 antibody | Biolegend | AB_2562608 |
| BV605 anti-mouse CD4 antibody | Biolegend | AB_2563054 |
| BV711 anti-mouse CD4 antibody | Biolegend | AB_2562099 |
| BV786 anti-mouse CD4 antibody | Biolegend | AB_2563053 |
| FITC anti-mouse CD4 antibody | Biolegend | AB_312713 |
| PE anti-mouse CD4 antibody | Biolegend | AB_312715 |
| PE/Cy7 anti-mouse CD4 antibody | Biolegend | AB_312729 |
| PerCP/Cy5.5 anti-mouse CD4 antibody | Biolegend | AB_893326 |
| PE-Dazzle 594 anti-mouse CD4 antibody | Biolegend | AB_2563684 |
| BUV395 anti-mouse CD4 antibody | BD Biosciences | AB_2738426 |
| BUV737 anti-mouse CD4 antibody | BD Biosciences | AB_2732918 |
| Biotin anti-mouse CD8a antibody | Biolegend | AB_312743 |
| BV605 anti-mouse CD8a antibody | Biolegend | AB_2562609 |
| PE/Cy7 anti-mouse CD8a antibody | Biolegend | AB_312761 |
| PerCP/Cy5.5 anti-mouse CD8a antibody | Biolegend | AB_2075238 |
| APC anti-mouse CD8a antibody | Biolegend | AB_312751 |
| FITC anti-mouse CD8a antibody | Biolegend | AB_312745 |
| BV421 anti-mouse CD8a antibody | Biolegend | AB_11204079 |
| APC/Cy7 anti-mouse CD8a antibody | Biolegend | AB_312753 |
| APC/Cy7 anti-mouse/human CD11b antibody | Biolegend | AB_830641 |
| APC/Cy7 anti-mouse CD19 antibody | Biolegend | AB_830707 |
| Pacific Blue anti-mouse CD19 antibody | Biolegend | AB_439718 |
| APC anti-mouse CD25 antibody | Biolegend | AB_312861 |
| APC anti-mouse CD25 antibody | Miltenyi | AB_2752169 |
| Biotin anti-mouse CD25 antibody | Biolegend | AB_312853 |
| PE anti-mouse CD25 antibody | Biolegend | AB_312857 |
| PE anti-mouse CD25 antibody | Miltenyi | AB_2656655 |
| PE/Cy7 anti-mouse CD25 antibody | Biolegend | AB_312865 |
| BV711 anti-mouse CD25 antibody | Biolegend | AB_2564130 |
| BV421 anti-mouse CD25 antibody | Biolegend | AB_11203373 |
| AF488 anti-mouse CD25 antibody | Biolegend | AB_493333 |
| PerCP/Cy5.5 anti-mouse CD25 antibody | Biolegend | AB_893288 |
| Pacific Blue anti-mouse/human CD44 antibody | Biolegend | AB_493683 |
| BV421 anti-mouse/human CD44 antibody | Biolegend | AB_10895752 |
| BV605 anti-mouse/human CD44 antibody | Biolegend | AB_2562451 |
| APC/Cy7 anti-mouse/human CD44 antibody | Biolegend | AB_830785 |
| BV421 anti-mouse CD45 antibody | Biolegend | AB_10899570 |
| BUV395 anti-mouse CD45 antibody | BD Biosciences | AB_2651134 |
| APC/Cy7 anti-mouse CD45 antibody | Biolegend | AB_312981 |
| Pacific Blue anti-mouse CD45 antibody | Biolegend | AB_493535 |
| APC anti-mouse CD45 antibody | Biolegend | AB_312977 |
| FITC anti-mouse CD45.1 antibody | Biolegend | AB_313495 |
| PerCP/Cy5.5 anti-mouse CD45.1 antibody | Biolegend | AB_893346 |
| PE/Cy7 anti-mouse CD45.1 antibody | Biolegend | AB_1134168 |
| BV605 anti-mouse CD45.1 antibody | Biolegend | AB_2562565 |
| PerCP/Cy5.5 anti-mouse CD45.2 antibody | Biolegend | AB_893350 |
| AF488 anti-mouse CD45.2 antibody | Biolegend | AB_492869 |
| AF647 anti-mouse CD45.2 antibody | Biolegend | AB_492870 |
| FITC anti-mouse CD45.2 antibody | BD Biosciences | AB_395041 |
| APC/Cy7 anti-mouse CD45.2 antibody | Biolegend | AB_830789 |
| Pacific Blue anti-mouse CD45.2 antibody | Biolegend | AB_492872 |
| APC anti-mouse CD62L antibody | Biolegend | AB_313099 |
| APC/Cy7 anti-mouse CD62L antibody | Biolegend | AB_830799 |
| PerCP/Cy5.5 anti-mouse CD62L antibody | Biolegend | AB_2285839 |
| PE anti-human/mouse/rat CD278 (ICOS) antibody | Biolegend | AB_416332 |
| FITC anti-mouse/human KLRG1 antibody | Biolegend | AB_10643998 |
| PE anti-mouse/human KLRG1 antibody | Biolegend | AB_10574005 |
| PE/Dazzle 594 anti-mouse/human KLRG1 | Biolegend | AB_2564050 |
| BV711 anti-mouse/human KLRG1 antibody | Biolegend | AB_2629721 |
| BV421 anti-mouse/human KLRG1 antibody | Biolegend | AB_10918627 |
| FITC anti-mouse/human KLRG1 antibody | Biolegend | AB_10643582 |
| BV421 anti-mouse/human KLRG1 antibody | Biolegend | AB_2565613 |
| BV605 anti-mouse/human KLRG1 antibody | Biolegend | AB_2563357 |
| BV510 anti-mouse TCR-β chain antibody | Biolegend | AB_2562350 |
| APC/Cy7 anti-mouse TCR-β chain antibody | Biolegend | AB_893624 |
| PE anti-mouse TCR-β chain antibody | Biolegend | AB_313431 |
| PerCP/Cy5.5 anti-mouse TCR-β chain antibody | Biolegend | AB_1575173 |
| BV605 anti-mouse TCR-β chain antibody | Biolegend | AB_2629563 |
| BV711 anti-mouse TCR-β chain antibody | Biolegend | AB_2629564 |
| PE/Cy7 anti-mouse TCR-β chain antibody | Biolegend | AB_893625 |
| Biotin anti-mouse IL-33Ra (ST2) antibody | eBioscience | AB_2572809 |
| PE anti-mouse IL-33Ra (ST2) antibody | Biolegend | AB_2728176 |
| PerCP/Cy5.5 anti-mouse IL-33Ra (ST2) antibody | Biolegend | AB_2565636 |
| APC anti-mouse IL-33Ra (ST2) antibody | Biolegend | AB_2561917 |
| PE/Cy7 anti-mouse IL-33Ra (ST2) antibody | Biolegend | AB_2687367 |
| BV421 anti-mouse IL-33Ra (ST2) antibody | Biolegend | AB_2565634 |
| PE anti-mouse IL-33Ra (ST2) antibody | Biolegend | AB_2561915 |
| Biotin anti-mouse Amphiregulin antibody | R&D Systems | AB_2060662 |
| PE/Cy7 anti-mouse CD279 (PD-1) antibody | Biolegend | AB_10689635 |
| PE anti-mouse CD279 (PD-1) antibody | Biolegend | AB_1877231 |
| BV421 anti-mouse CD279 (PD-1) antibody | Biolegend | AB_2561447 |
| BV711 anti-mouse CD279 (PD-1) antibody | Biolegend | AB_2566158 |
| Biotin anti-mouse FoxP3 antibody | eBioscience | AB_763540 |
| PE anti-mouse FoxP3 antibody | eBioscience | AB_465936 |
| AF488 anti-mouse FoxP3 antibody | eBioscience | AB_763537 |
| APC anti-mouse Foxp3 antibody | eBioscience | AB_469457 |
| Anti-Mouse/Rat Foxp3 AF647 antibody | Biolegend | AB_763538 |
| Anti-Mouse/Rat Foxp3 Biotin antibody | Biolegend | AB_763540 |
| Anti-Mouse/Rat Foxp3 PE antibody | Biolegend | AB_465936 |
| PE anti-mouse/human c-MAF antibody | eBioscience | AB_2572747 |
| Purified anti-mouse/human BATF antibody | Cell Signaling | AB_11141425 |
| Goat Anti-Rabbit IgG (H+L) antibody, AF 647 | Cell Signaling | AB_10562581 |
| PE anti-mouse/human BATF antibody | Cell Signaling | AB_2798938 |
| BV605 anti-mouse CD127 (IL-7Ra) ab | Biolegend | AB_2562114 |
| BV421 anti-mouse CD127 (IL-7Ra) ab | Biolegend | AB_11218800 |
| AF647 anti-mouse CD279 (PD-1) antibody | Biolegend | AB_2566008 |
| BV421 anti-mouse CCR8 antibody | Biolegend | AB_2616650 |
| BUV737 anti-mouse IL-33R antibody | BD Biosciences | AB_2873697 |
| BV711 anti-mouse CD3 antibody | Biolegend | AB_2563945 |
| PE/Cy7 anti-mouse CD195 antibody | Biolegend | AB_2617012 |
| BV421 Rat IgG2a, К Isotype control | BD Biosciences | AB_11153860 |
| PE anti-mouse CD127 antibody | BD Biosciences | AB_394417 |
| AF700 anti-mouse CD8 antibody | BD Biosciences | AB_396959 |
| APC anti-mouse CD3 antibody | Biolegend | AB_312677 |
| BV510 anti-mouse CD3 antibody | Biolegend | AB_2562555 |
| PE anti-mouse IL10 antibody | Biolegend | AB_315362 |
| Biotin anti-mouse Amphiregulin antibody | R&D systems | AB_2060662 |
| BUV496 anti-mouse CD4 antibody | BD Biosciences | AB_2870665 |
| Antibodies anti-human | ||
| Biotin anti-human CD25 | Biolegend | AB_830747 |
| Biotin anti-human CD4 | Biolegend | AB_571949 |
| PE anti-human CD4 | Biolegend | AB_571955 |
| PerCP/cy5.5 anti-human TCRbeta chain | Biolegend | AB_2563002 |
| BV510 anti-human CD3 | Biolegend | AB_2561943 |
| BV421 anti-human CD8 | Biolegend | AB_10960142 |
| APC/Cy7 anti-human CD19 | Biolegend | AB_314248 |
| PE/Cy7 anti-human CD127 | Biolegend | AB_10897098 |
| AF488 anti-human CD25 | Biolegend | AB_493043 |
| PE anti-human CD25 | Biolegend | AB_314276 |
| PE anti-human CD25 | BD | AB_2783790 |
| BV711 anti-human CD4 | Biolegend | AB_2562912 |
| BUV737 anti-human CD45 | BD Biosciences | AB_2873123 |
| APC anti-human CD127 | Biolegend | AB_2564137 |
| BV605 anti-human CD19 | BD Biosciences | AB_2740124 |
| BUV395 anti-human CD14 | BD Biosciences | AB_2744288 |
| PE/Cy7 anti-human TCR beta chain | Biolegend | AB_10639947 |
| BV786 anti-human CD3 | BD Biosciences | AB_2869863 |
| BV421 anti-human CD45RO | Biolegend | AB_2563817 |
| BV605 anti-human CD45RA | Biolegend | AB_2563814 |
| BV510 anti-human CD45RA | Biolegend | AB_2561947 |
| AF488 anti-human FoxP3 | Biolegend | AB_430883 |
| BUV395 anti-human CCR8 | BD Biosciences | AB_2744144 |
| BV421 anti-human CCR8 | BD Biosciences | AB_2744265 |
| BB515 anti-human CD39 | BD Biosciences | AB_2722754 |
| PerCP/Cy5.5 anti-human CD45RO | Biolegend | AB_2174124 |
| Goat Anti-Rabbit IgG (H+L) antibody, AF488 | Cell Signaling | AB_1904025 |
| PE/Dazzle™ 594 anti-human FoxP3 | Biolegend | AB_2564025 |
| BUV395 anti-human CD4 | BD Biosciences | AB_2738596 |
| BUV737 anti-human CD25 | BD Biosciences | AB_2870132 |
| PE anti-human FoxP3 | Biolegend | AB_492986 |
| BUV496 anti-human CD45RA | BD Biosciences | AB_2874456 |
| BUV661 anti-human CD195 | BD Biosciences | AB_2874490 |
| BUV805 anti-human CD8 | BD Biosciences | AB_2871326 |
| BV510 anti-human CD49d | Biolegend | AB_2563820 |
| BV605 anti-human HLA-DR | Biolegend | AB_2561913 |
| PE anti-human HLA-DR | Biolegend | AB_314684 |
| BV711 anti-human CD45RO | Biolegend | AB_2562107 |
| BB515 anti-human CD25 | BD Biosciences | AB_2744340 |
| BB700 anti-human CD39 | BD Biosciences | AB_2743331 |
| PE anti-human CD71 | Biolegend | AB_2201481 |
| PE/Cy7 anti-human CD200R | Biolegend | AB_2783197 |
| PE/Cy7 anti-human CD45RO | Biolegend | AB_11203900 |
| APC/Cy7 anti-human CD8 | Biolegend | AB_10613636 |
| BV605 anti-human CD8 | Biolegend | AB_2563185 |
| BV711 anti-human CD8 | Biolegend | AB_2562906 |
| APC anti-human CD2 | Biolegend | AB_10900259 |
| VioBright 515 anti-human PD-1 (CD279) | Miltenyi | AB_2752077 |
| BUV395 anti-human CD206 | BD Biosciences | AB_2740047 |
| PE/Cy7 anti-human CD4 | Biolegend | AB_571959 |
| BV711 anti-human CD25 | Biolegend | AB_2562910 |
| APC anti-mouse anti-human Bcl-6 | Biolegend | AB_2562472 |
| PE anti-mouse anti-human Helios | Biolegend | AB_10660749 |
| BV421 anti-human FOXP3 | Biolegend | AB_2565972 |
| Fixable Viability Dyes | ||
| Fixable Viability Dye eFluor 450 | eBioscience | Cat# 65-0863-18 |
| Fixable Viability Dye eFluor 506 | eBioscience | Cat# 65-0866-18 |
| Fixable Viability Dye eFluor 780 | eBioscience | Cat# 65-0865-18 |
| Streptavidin conjugates | ||
| AF488 Streptavidin | Biolegend | Cat# 405235 |
| APC/Cy7 Streptavidin | Biolegend | Cat# 405208 |
| E450 Streptavidin | eBioscience | Cat# 48-4317-82 |
| FITC Streptavidin | Biolegend | Cat# 405201 |
| PE Streptavidin | Biolegend | Cat# 405204 |
| PE/Cy7 Streptavidin | Biolegend | Cat# 405206 |
| PerCP/Cy5.5 Streptavidin | Biolegend | Cat# 405214 |
| PE-Dazzle 594 Streptavidin | Biolegend | Cat# 405248 |
| BUV395 Streptavidin | BD Biosciences | Cat# 564176 |
| BUV737 Streptavidin | BD Biosciences | Cat# 564293 |
| BV711 Streptavidin | Biolegend | Cat# 405241 |
| BV605 Streptavidin | Biolegend | Cat# 405229 |
| APC Streptavidin | Biolegend | Cat# 405207 |
| BV421 Streptavidin | Biolegend | Cat# 405225 |
| Bacterial and Virus Strains | ||
|---|---|---|
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant human IL-2 | Novartis | Proleukin S® 18 Mio U |
| Pancoll human, Density: 1.077 g/mL | PAN Biotech | P04-601000 |
| HBSS | GIBCO | 14025092 |
| PBS | GIBCO | 10010023 |
| DMEM | GIBCO | 41965 |
| TexMACS medium | Miltenyi Biotec | 130-097-196 |
| Collagenase Type II | Sigma-Aldrich | C6885 |
| Collagenase Type IV | Sigma-Aldrich | C5138 |
| DNase I | Roche | 11284932001 |
| Bovine Serum Albumin | Sigma | A4503 |
| Fetal Bovine Serum | N/A | |
| Percoll | GE Healthcare | 17-0891-01 |
| Power SYBR Green Master Mix | Thermo Fisher | Cat# 4367659 |
| Taqman Gene Expression Master Mix | Thermo Fisher | Cat# 4359016 |
| SuperScript II Reverse Transcriptase | Thermo Fisher | Cat# 18064071 |
| SYBR-Green | Thermo Fisher | S7563 |
| Oligo d(T) 12-18 Primer | Thermo Fisher | Cat# 18418012 |
| Compensation Beads | eBiosciences | 01-1111-41 |
| Hair removal creme | Reckitt Benckiser (Veet) | NA |
| Critical Commercial Assays | ||
| ChromiumTM Single Cell Controller & Accessory Kit | 10X | 120212 |
| ChromiumTM Chip E Single Cell ATAC Kit v1.0 | 10X | 1000082 |
| ChromiumTM Single Cell ATAC Library & Gel Bead Kit v1.0 | 10X | 1000110 |
| ChromiumTM i7 Multiplex Kit N, Set A | 10X | 1000084 |
| ChromiumTM Next GEM Single Cell ATAC Library & Gel Bead Kit v1.1 | 10X | 1000175 |
| ChromiumTM Next GEM Chip H Single Cell Kit v1.1 | 10X | 1000161 |
| ChromiumTM Single Index Kit N Set A | 10X | 1000212 |
| ChromiumTM Next GEM Single Cell 5¢ Library and Gel Bead Kit v1.1 | 10X | 1000165 |
| ChromiumTM Next GEM Chip G Single Cell Kit | 10X | 1000120 |
| ChromiumTM Single Cell 5¢ Library Construction Kit | 10X | 1000020 |
| ChromiumTM Single Cell V(D)J Enrichment Kit, Mouse T Cell | 10X | 1000071 |
| ChromiumTM Single Cell V(D)J Enrichment Kit, Human T Cell | 10X | 1000005 |
| ChromiumTM Single Index Kit T Set A | 10X | 1000213 |
| GentleMACS C tube | Miltenyi | 130-096-334 |
| Lamina Propria Dissociation Kit | Miltenyi Biotec | 130-097-410 |
| Dead cell removal Kit | Miltenyi Biotec | 130-090-101 |
| Anti-biotin microbeads, ultrapure | Miltenyi Biotec | 130-105-637 |
| Anti-PE microbeads, ultrapure | Miltenyi Biotec | 130-105-639 |
| Anti-APC microbeads | Miltenyi Biotec | 130-090-855 |
| Anti-mouse CD45 Microbeads | Miltenyi Biotec | 130-052-301 |
| LS column | Miltenyi Biotec | 130-042-401 |
| MS column | Miltenyi Biotec | 130-042-201 |
| T cell TransAct, Human | Miltenyi Biotec | 130-111-160 |
| ACK lysis buffer | GIBCO | A1049201 |
| Foxp3 / Transcription Factor Buffer Set | eBiosciences | 00-5523-00 |
| RNEasy Plus Mini Kit | QIAGEN | 74134 |
| InnuPREP RNA Mini Kit 2.0 Analytik Jena | Analytik Jena | 845-KS-2040250 |
| SMARTer Ultra Low Input RNA kit | Takara | 634894 |
| SMART-Seq Stranded Kit | Takara | 634455 |
| NEXT CHiP-Seq Library Prep Master Mix | NEB | E6240L |
| NEBNext Multiplex Oligos | NEB | E7335L |
| NEBNext High Fidelity PCR Master mix | NEB | M0541S |
| Foxp3 / Transcription Factor Staining Buffer Set | eBioscience | Cat# 00-5523-00 |
| Agencourt AMPure XP beads | Beckman Coulter | A63880 |
| Illumina Nextera DNA preparation Kit | Illumina | FC1211030 |
| Qubit dsDNA HS Kit | Thermo Fisher Scientific | Q32851 |
| Qubit Protein Assay Kit | Thermo Fisher Scientific | Q33211 |
| High Sensitivity D1000 ScreenTape | Agilent | 50675584 |
| High Sensitivity D1000 Reagents | Agilent | 50675585 |
| RNA ScreenTape | Agilent | 50675576 |
| RNA ScreenTape Sample Buffer | Agilent | 50675577 |
| Zymo clean & concentrator kit | Zymo | D4013 |
| LEGENDScreen™ Human PE Kit | Biolegend | cat # 700007 |
| CellTrace™ CFSE Cell Proliferation Kit, for flow cytometry | Invitrogen | C34554 |
| Deposited Data | ||
| Murine scATAC-Seq data | This paper | GEO GSE156112 |
| Human scATAC-Seq data | This paper | EGA S00001004900 |
| Human scRNA-Seq data | This paper | EGA S00001004900 |
| Human scTCR-Seq data | This paper | EGA S00001004900 |
| Human bulk RNA-Seq data | This paper | EGA S00001004900 |
| Human bulk ATAC-Seq data | This paper | EGA S00001004900 |
| Experimental Models: Cell Lines | ||
| HaCaT | CVCL_0038 | |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Markus Feuerer (Markus.Feuerer@ukr.de).
Materials availability
The Areg(GFP) and Nfil3(GFP) reporter mouse lines used in this study are available upon request.
Data and code availability
The accession numbers for murine scATAC-Seq data reported in this paper are: Gene Expression Omnibus (GEO) GSE156112. The accession numbers for human scATAC-Seq, scRNA-Seq, scTCR-Seq, bulk ATAC-seq and bulk RNA-seq data reported in this paper are: European Genome-phenome Archive (EGA) S00001004900.
Experimental model and subject details
Ethics statement
Human skin and adipose tissue used for scATAC-seq and scRNA/scTCR-seq were obtained from five healthy female donors with an average age of 44.6 years (±14; range from 26 to 56) undergoing abdominoplasty procedures after weight loss (4 donors) and epigastric hernia repair (1 donor). The average BMI of all patients was 31.0 (±8.9).
Human skin and adipose tissue used for bulk RNA-seq of skin and fat tissue Treg cells (CD4+CD127-CD25+) were obtained from 7 donors undergoing abdominoplasty procedures after weight loss (5 donors) and epigastric hernia repair (2). The average BMI of all patients was 33.2 (±6.3).
Human skin and adipose tissue used for bulk RNA-seq of skin and fat tissue CCR8+ Treg cells (CD4+CD127-CD25+CD45RA-CCR8+) were obtained from 5 patients undergoing abdominoplasty procedures after weight loss (4 donors) and epigastric hernia repair (1 donor). The average BMI of all patients was 26.1 (±2.8).
Human primary liver tumors (4 hepatocellular carcinomas and 2 cholangiocellular carcinomas) and surrounding healthy liver tissue as well as PBMCs used for bulk RNA-seq of CD45RA-CCR8+ Treg cells, CD45RA-CCR8- Treg cells and CD45RA+ Treg cells were obtained from six patients (3 male and 3 female) with an average age of 74.5 years (±7; range from 62 to 82) undergoing major liver surgery.
Collection of skin, fat and blood samples from donors was performed after ethical approval by the local ethical committee (Regensburg University, reference number 19-1453-101) and signed informed consent. Collection of primary liver tumors, surrounding liver tissue and blood samples from tumor patients was performed after ethical approval of the local ethical committee (Regensburg University, reference number 18-1075-101) and signed informed consent.
Human primary keratinocytes for the generation of epidermal models were isolated from foreskin biopsies obtained from juvenile donors under informed consent, according to ethical approval granted by the local ethic committee (Würzburg University, reference numbers 182/10 and 280/18sc) and the written consent of guardians.
Peripheral blood mononuclear cells for CD4 T cell enrichment were isolated from leukocyte reduction chambers from healthy female donors donating thrombocytes. Collection of immune cells from those donors was performed in compliance with the Helsinki Declaration after ethical approval by the local ethical committee (Regensburg University, reference number 13-0240-101 and 19-1414-101) and signed informed consent.
Mice
To generate scATAC-seq data of SPF animals, we used adult male C57BL/6 mice of 10 or more weeks of age. To generate scATAC-seq data of germ-free animals, we used adult male wild type C57BL/6 mice of 9 or more weeks of age housed under germ-free conditions at the Helmholtz Centre for Infection Research (Braunschweig). Proof of germ-free hygiene status was performed bi-weekly through Sytox cell staining and 16S rRNA gene analysis of stool samples. Nfil3(GFP) mice were housed under SPF conditions and described previously (Delacher et al., 2020). Foxp3(huCD2) mice (Foxp3tm1(CD2/CD52)Shori) were a gift from S. Hori (Komatsu et al., 2009). For generation of Areg(GFP) BAC-transgenic mice, a fusion construct composed of the DNAs for iCre and eGFP, separated by a 2A sequence, was generated and inserted at the start codon of the Areg gene in the BAC RP23-190I9 (BioScience) using Escherichia coli DH10B (Copeland et al., 2001). The final construct was linearized and injected into the pronuclei of fertilized C57BL/6N mouse eggs at the animal facility of the German Cancer Research Center (DKFZ). Genotyping was carried out by PCR from genomic DNA of tail biopsies. Animals were housed under specific pathogen-free conditions at the DKFZ animal care facility or the Regensburg University Clinics animal care facility, and the governmental committee for animal experimentation (Regierungspräsidium Karlsruhe, Germany for DKFZ Heidelberg or Regierungspräsidium Unterfranken, Würzburg for Regensburg) approved these animal experiments.
Tumor bearing BALB-NeuT transgenic mice (Hosseini et al., 2016) were a gift from Christoph Klein. Mice were screened at 3–4 weeks of age for hemizygosity (neuT+/neuT−). Mammary glands of BALB-NeuT female mice were inspected twice a week and CD4+ T cells from growing tumors were analyzed by scATAC-seq. Experimental animal procedures regarding BALB-NeuT were approved and conducted according to German federal and state regulations (Regierungspräsidium Unterfranken, Würzburg).
Method details
Tissue digestion for flow cytometry and FACS sorting of murine T cells
To isolate T cells from VAT tissue, gonadal fat pads of male mice were excised, cut into small pieces and digested for 45 min at 37°C (base medium DMEM (GIBCO #41965), 1 mg/mL collagenase type II (Sigma-Aldrich #C6885), 20 μg/mL DNase I (Roche #11284932001), 20 mg/mL bovine serum albumin (Sigma-Aldrich #A4503)) on a MACSmix tube rotator (Miltenyi Biotec 130-090-753), followed by incubation with 2 mM EDTA-PBS for 2 min and centrifugation and filtration steps.
To isolate T cells from skin tissue, hair and hair follicles from the back of the animal were removed with an electric shaver and depilatory cream. Skin was separated from the dorsal surface, cut into small pieces and digested (base medium DMEM (GIBCO #41965), 4 mg/mL collagenase type IV (Sigma-Aldrich #C5138), 10 μg/mL DNase I (Roche #11284932001), 2% fetal bovine serum). Digestion was performed directly in a GentleMACS C tube (Miltenyi Biotec #130-096-334) and the program “37_C_Multi_H” for 90 min, followed by centrifugation and filtration steps. Dead cells were removed using a dead cell removal kit (Miltenyi Biotec # 130-090-101).
To isolate T cells from lung tissue, animals were perfused by opening the inferior vena cava and flushing the left ventricle with 10ml PBS to clear the body circulation. Lungs were excised, cut in small pieces and digested (base medium DMEM (GIBCO #41965), 1 mg/mL collagenase type IV (Sigma-Aldrich #C5138), 20 μg/mL DNase I (Roche #11284932001), 5 mg/mL bovine serum albumin (Sigma-Aldrich #A4503)) for 30-45 min at 37°C on a MACSmix tube rotator, followed by centrifugation and filtration steps. Lung samples were pre-enriched using biotinylated antibodies (Clone PC61, Biolegend) and anti-biotin ultrapure, anti-PE ultrapure or anti-APC microbeads (Miltenyi Biotec #130-105-637, #130-105-639, #130-090-855).
To isolate T cells from colon tissue, colons were isolated, cleared of feces and prepared according to manufacturer’s instructions with a lamina propria dissociation kit (Miltenyi #130-097-410). Samples were either pre-enriched with CD4 (Clone RM4-5, Biolegend) or CD25 (Clone REA568, Miltenyi Biotec) antibody followed by bead-based magnetic purification or measured directly. More detailed protocols about T cell isolation from murine tissues are published (Cossarizza et al., 2019).
To isolate T cells from murine spleen and lymph nodes, tissues were mechanically dissociated on a 70 μm filter unit and red blood cells lysed using ACK lysis buffer (Thermo Fisher # A1049201). Samples were either pre-enriched with CD4 (Clone RM4-5, Biolegend) or CD25 (Clone PC61, Biolegend) antibody followed by column-based magnetic purification or measured directly.
To isolate T cells from tumor tissue of HER2-transgenic tumor-bearing animals, mammary carcinoma depots were surgically excised, cut into small pieces and digested for 45 min at 37°C (base medium DMEM (GIBCO #41965), 1 mg/mL collagenase type II (Sigma-Aldrich #C6885), 20 μg/mL DNase I (Roche #11284932001), 20 mg/mL bovine serum albumin (Sigma-Aldrich #A4503) on a MACSmix tube rotator, followed by incubation with 2 mM EDTA-PBS for 2 min and centrifugation and filtration steps. Cells were pre-enriched with anti-mouse CD45 microbeads (Miltenyi #130-052-301), followed by column-based magnetic enrichment and staining for FACS-based sorting.
Tissue digestion for flow cytometry and FACS sorting of human T cells
To isolate T cells from human skin and subcutaneous fat tissue, skin and underlying fat were first mechanically separated, followed by tissue-individual preparation steps. Fat was cut in into small pieces and digested for 90 min at 37°C (base medium DMEM (GIBCO #41965), 1 mg/mL collagenase type II (Sigma-Aldrich #C6885), 20 μg/mL DNase I (Roche #11284932001), 20 mg/mL bovine serum albumin (Sigma-Aldrich #A4503), 10 mM HEPES) on a MACSmix tube rotator, followed by filtration and centrifugation steps as well as red blood cell lysis using ACK lysis buffer (GIBCO # A1049201).
To isolate T cells from human skin tissue, skin was cut into small pieces and digested (base medium DMEM (GIBCO #41965), 4 mg/mL collagenase type IV (Sigma-Aldrich #C5138), 10 μg/mL DNase I (Roche #11284932001), 2% fetal bovine serum, 10 mM HEPES). Digestion was performed directly in a GentleMACS C tube and the program “37_C_Multi_H” for 90 min, followed by centrifugation and filtration steps. Dead cell removal was performed with a dead cell removal kit.
To isolate T cells from human liver and liver tumor tissue, samples were cut into small pieces and digested (base medium DMEM (GIBCO #41965), 1 mg/mL collagenase type IV (Sigma-Aldrich #C5138), 25 μg/mL DNase I (Roche #11284932001), 10% fetal bovine serum, 10 mM HEPES). Digestion was performed directly in a GentleMACS C tube. Tissues were minced using the program “h.tumor” on the GentleMACS, and were subsequently attached to a MACSmix tube rotator and placed at 37°C for 60 min. Digestion was stopped and cells were washed with PBS + 2mM EDTA. Single cell solution was obtained by centrifugation and filtration steps as well as red blood cell lysis using ACK lysis buffer (GIBCO # A1049201).
Peripheral blood mononuclear cell isolation and pre-enrichment of blood lymphocytes
To isolate T cells from human blood, leukocyte reduction chambers (provided by Transfusion Medicine, University Clinics Regensburg) were used. Leukocytes were first diluted 3 times with DPBS (GIBCO #14190-094), and the resulting blood and PBS mixture was split in two fractions and underlayed with an equal amount of Pancoll (PAN biotech #P04-601000). Samples were centrifuged at 1,000xg for 20 min at RT, with acceleration set to 4 and brake to 0. The PBMC layer was isolated and washed twice by centrifugation steps. Cells were pre-enriched with either biotinylated anti-human CD4 (clone OKT4, Biolegend), biotinylated anti-human CD8 (clone HIT8a, Biolegend) or PE-labeled or biotinylated anti-human CD25 (Clone BC96, Biolegend; Clone 2A3, BD), followed by column-based magnetic separation with anti-biotin or anti-PE ultrapure microbeads (Miltenyi Biotec #130-105-637 or #130-105-639) following manufacturer’s protocol.
Preparation of samples for FACS sorting or flow cytometry
T cells were isolated and pre-enriched as described in the previous sections. Cells were stained in 1.5 mL Eppendorf tubes or 96-well plates in FACS buffer (2%FCS in PBS). Surface staining was performed at 4°C for 20 min in 50-100μl staining volume. Antibodies were used, if not indicated otherwise, as recommended by the manufacturer. The following anti-human antibodies were used for surface staining: CD4 (OKT4/L200 SK3), CD3 (OKT3), TCR-β chain (IP26), CD8 (RPA-T8/HIT8a), CD19 (HIB19), CD25 (BC96/2A3), CD45RA (HI100), CD45RO (UCHL1), CD127 (A019D5), CD45 (HI30/2D1/REA747), CD14 (MφP9), CCR8 (433H), CD39 (TU66), HLA-DR (L243), CD71 (CY1G4), CD195 (3A9), CD49d (9F10) CD279 (REA1165), CD206 (19.2). The following anti-mouse antibodies were used for surface staining: CD4 (RM4-5), CD3 (17A2), TCR-β chain (H57-597), CD8 (53-6.7), CD19 (6D6), CD45 (30-F11), CCR8 (SA214G2), CD25 (PC61), Klrg1 (2F1/KLRG1), Pd1 (29F.1A12), St2 (DIH9).
Intracellular staining was performed with the Foxp3/Transcription Factor Buffer Set (eBioscience 00-5521-00) according to manufacturer’s protocol with the following adaptations: intracellular staining steps were performed for 60 min at room temperature. The following antibodies were used for intracellular staining of mouse and human proteins: FOXP3 (206D) at a dilution of 1:20, BCL-6 (7D1) at a dilution of 1:20, Helios (22F6) at a dilution of 1:20, IL10 (JES5-16E3) at a dilution of 1:25, Amphiregulin (IPO0314111) at a dilution of 1:50 and BATF (D7C4) at a dilution of 1:400. For BATF staining, secondary intracellular staining was performed with AF647 (Cell Signaling Cat#4414) or AF488 (Cell Signaling Cat#4412) coupled anti-rabbit antibody at 1:400. Dead cells were excluded with a fixable live/dead dye (Fixable Viability Dye eFlour780, eBioscience Cat# 65-0865-14).
Flow cytometry and FACS sorting of T cells from blood and tissues
T cells were isolated, pre-enriched and stained as described previously. Afterward, samples were filtered with a 40μM filter unit and acquired on a BD LSRII, BD FACSymphony or BD FACSFusion flow cytometer. BD CS&T beads were used to validate machine functionality. Fluorescence spillover compensation was performed with lymphocytes stained with CD4 (OKT4) in the respective colors. Flow cytometry data were analyzed using BD FlowJo (Version 10.6.2). Sorting was performed with a BD FACSAriaII or BD FACSFusion cell sorter with 85μm nozzle and 45 psi of pressure. Post-sort quality controls were performed as applicable. For scATAC-seq, target cells were sorted into 500 μl 10%FCS-PBS, followed by nuclei preparation. For scRNA-seq, target cells were sorted into 500 μl 10%FCS-PBS. For bulk RNA-seq, cells were sorted directly into 500 μL RLT+ lysis buffer (QIAGEN RNEasy Plus Micro Kit #74034). For bulk ATAC-seq, target cells were sorted into 500 μl 10%FCS-PBS. For cultivation experiments, cells were sorted directly into cell culture medium. All procedures were performed in DNA low-bind tubes (Eppendorf #0030108051). For cytokine re-stimulation experiments, cells were incubated with either 1X cell stimulation cocktail with transport inhibitors or transport inhibitor only (both eBiosciences) in cell culture medium for 4 h at 37°C. Afterward, cells were surface-stained, followed by intracellular staining with the Foxp3 Fix/perm kit.
Single-cell ATAC-seq of blood and tissue T cells from mice
Murine T cells were isolated, pre-enriched, stained and sorted as described previously. Sort gates and post-sort QC are shown in Figure S1 (SPF animals) and Figure S2 (Gnotobiotic animals). From spleens of SPF animals, we sorted 100,000 CD45+Dead-CD19-CD3+TCRβ+CD8-CD4+ and two independent runs of 100,000 CD45+Dead-CD19-CD3+TCRβ+CD8-CD4+CD25+ T cells. From colons of SPF animals, we sorted 79,000 CD45+Dead-CD19-CD3+TCRβ+CD8-CD4+ T cells. From skin of SPF animals, we sorted 73,000 CD45+Dead-CD19-CD3+TCRβ+CD8-CD4+ T cells. From lung of SPF animals, we sorted 100,000 CD45+Dead-CD19-CD3+TCRβ+CD8-CD4+ T cells. From VAT of SPF animals, we sorted 70,000 and 100,000 CD45+Dead-CD19-CD3+TCRβ+CD8-CD4+ T cells in two independent runs (Figure S1).
From spleens of germ-free animals, we sorted 100,000 CD45+Dead-CD19-CD3+TCRβ+CD8-CD4+ and 100,000 CD45+Dead-CD19-CD3+TCRβ+CD8-CD4+CD25+ T cells. From colons of germ-free animals, we sorted 77,000 CD45+Dead-CD19-CD3+TCRβ+CD8-CD4+ T cells. From VAT of germ-free animals, we sorted 37,000 CD45+Dead-CD19-CD3+TCRβ+CD8-CD4+ T cells. From skin of germ-free animals, we sorted 17,500 CD45+Dead-CD19-CD3+TCRβ+CD8-CD4+ T cells (Figure S2).
From tumors of HER2-transgenic tumor-bearing animals, we sorted two independent runs of 100,000 CD45+Dead-CD19-CD3+TCRβ+CD8-CD4+ and 25,000 CD45+Dead-CD19-CD3+TCRβ+CD8-CD4+CD25+ T cells (Figure S7).
Sorted cells were pelleted by centrifugation (300xg, 5min 4°C). Supernatant was removed and cells were resuspended in 50 μl - 100 μl 0.04%BSA-PBS buffer. Cells were centrifuged (300xg, 5min 4°C), supernatant was completely removed and chilled nuclei lysis buffer (10mM TRIS-HCL pH7.4, 10mM NaCl, 3mM MgCl2, 0.1% Tween-20, 0.1% NP-40, 0.01% Digitonin, 1% BSA in nuclease-free water) was added. Lysis was performed for 2 min at 4°C, followed by addition of 50 μl washing buffer (10mM TRIS-HCl ph7.4, 10mM NaCl, 3mM MgCL2, 1%BSA, 0.1% Tween-20 in nuclease-free water). After centrifugation (300xg, 5min 4°C), supernatant was removed and 45 μl chilled diluted nuclei buffer (10X Genomics) was added. After centrifugation (300xg, 5min 4°C), supernatant was removed and 7μl chilled diluted nuclei buffer (10X Genomics) was added. Nuclei were counted (where applicable) and transposition mix was prepared with 5 μl of nuclei suspension based on the manufacturer’s protocol. Transposition was performed for one h at 37°C, followed by supplementation of master mix and beads (Single Cell ATAC Gel Beads V1.0 or V1.1 and reagents, 10X Genomics #1000175), loading on 10X Chromium Next GEM Chip H (10X Genomics #1000161) and processing on 10X Chromium Controller (10X Genomics #120212). GEM incubation was performed with 12 cycles of PCR. Library was prepared according to the protocol with cycle numbers dependent on input nuclei concentration. scATAC libraries were sequenced on an Illumina NextSeq™ 550 with NextSeq™ 500/550 High Output Kit v2.5 (75 cycles, Illumina #20024906).
Single-cell ATAC-seq of blood and tissue T cells from human donors
Human T cells were isolated, pre-enriched, stained and sorted as described previously. Sort gates and post-sort QC are shown in Figure S3.
From peripheral blood of healthy donor 1, we sorted 100,000 CD3+Dead-CD19-TCRβ+CD8-CD4+ and two independent runs of 100,000 CD3+Dead-CD19-TCRβ+CD8-CD4+CD25+CD127- T cells. From peripheral blood of healthy donor 2, we sorted 100,000 CD3+Dead-CD19-TCRβ+CD8-CD4+ and two independent runs of 100,000 CD3+Dead-CD19-TCRβ+CD8-CD4+CD25+CD127- T cells. From peripheral blood of healthy donor 3, we sorted two independent runs of 100,000 CD3+Dead-CD19-TCRβ+CD8-CD4+CD25+CD127-CD45RA-CD45RO+CCR8+ T cells.
From fat tissue of donor 3, we sorted 100,000 CD45+Dead-CD14-CD19-CD3+TCRβ+CD8-CD4+ and 15,000 CD45+Dead-CD14-CD19-CD3+TCRβ+CD8-CD4+CD25+CD127- T cells. From fat tissue of donor 4, we sorted 100,000 CD45+Dead-CD14-CD19-CD3+TCRβ+CD8-CD4+ T cells. From skin tissue of donor 4, we sorted 70,000 CD45+Dead-CD14-CD19-CD3+TCRβ+CD8-CD4+ T cells. From fat tissue of donor 5, we sorted 100,000 CD45+Dead-CD14-CD19-CD3+TCRβ+CD8-CD4+ and 25,000 CD45+Dead-CD14-CD19-CD3+TCRβ+CD8-CD4+CD25+CD127- T cells. From skin tissue of donor 5, we sorted 90,000 CD45+Dead-CD14-CD19-CD3+TCRβ+CD8-CD4+ T cells.
Sorted cells were pelleted by centrifugation (300xg, 5min 4°C) and processed as described above (Single Cell ATAC Gel Beads V1.1). scATAC libraries were sequenced on an Illumina NextSeq™ 550 with NextSeq™ 500/550 High Output Kit v2.5 (75 cycles).
Preprocessing of scATAC-seq data
All preprocessing steps were performed using “Cell Ranger ATAC version 1.1.0” (10X Genomics). Read filtering, alignment, peak calling and count matrix generation from fastq files were done per sample using ‘cellranger-atac count’. Reference genome assemblies mm10 (refdata-cellranger-atac-mm10-1.1.0) and hg19 (refdata-cellranger-atac-hg19-1.1.0) provided by 10xGenomics were used for murine and human samples, respectively. Gel bead and barcode multiplets were detected with a custom script provided by 10xGenomics (clean_barcode_multiplets_1.1.py), which leverages the frequency of adjacent fragment ends within and between barcodes. Subsequently, barcodes were annotated as cells if they met the following criteria: at least 1,000 (scATAC-data derived from HER2-transgenic tumor samples) or at least 5,000 read-pairs (scATAC-data derived from murine and human tissues) passed read filters defined by Cell Ranger ATAC version 1.1.0; less than 20% read-pairs with low mapping quality (< 0.2 fraction of unmapped + low mapping quality (mapq < 30) + chimeric read-pairs); less than 90% read pair duplicates; less than 10% read pairs from mitochondrial DNA; no annotation as gel bead or barcode multiplets. Then, CD4+ T cell samples were aggregated using the command ‘cellranger-atac aggr’ on barcodes annotated as cells without normalization for library size (–normalize = none). To reduce the influence of cell count depth on the downstream analysis, fragments from barcodes with more than 50,000 unique fragments which passed filters were randomly subsampled to 50,000 fragments.
All further analysis steps were performed in R (Version 3.6.0). Fragments were loaded into R using the package Seurat (version 3.1.2 (Stuart et al., 2019)). Transcription start site (TSS) scores were calculated for each barcode as previously described (Satpathy et al., 2019). Briefly, the per-base coverage was obtained in a 1,000 bp flanking region around all transcription start sites from the R packages ‘TxDb.Mmusculus.UCSC.mm10.knownGene’ (version 3.4.7, murine samples) or ‘TxDb.Hsapiens.UCSC.hg19.knownGene’ (version 3.2.2, human samples). These values were divided by the mean coverage at the 100 bp end regions of the flanking regions for normalization. Finally, the normalized coverage values were smoothed within a window of 50 bp and the highest value was maintained as TSS score. Barcodes with a TSS score smaller than 8 were excluded from further analysis. Peaks were filtered out if they met any of the following criteria: less than 10 fragments across all cells; overlap with chromosomes chrY, chrM or unplaced sequences; overlap with blacklisted regions provided by the ENCODE consortium using the blacklist assembly provided in R package Signac (version 0.1.3, blacklist_mm10 for murine cells and blacklist_hg19 for human cells).
For normalization and dimensionality reduction, the R package Signac (version 0.1.3, https://github.com/timoast/signac) was used. The peak-barcode matrix was then binarized and normalized using the implementation of the TF-IDF transformation described in ((Stuart et al., 2019), RunTFIDF(method = 1)). Subsequently, singular value decomposition was run (RunSVD) on the upper quartile of accessible peaks (FindTopFeatures(min.cutoff = ‘q75’)). Batch correction for donors in the human and pooled samples in the murine CD4+ T cell data were done with Harmony (Korsunsky et al., 2019). Harmony was run on the previously calculated SVD embeddings with the command ‘HarmonyMatrix’ with default parameters for lambda and theta. Sigma was set to 1 for human data and to 0.3 for murine data, respectively. The first 20 components from the SVD or Harmony reduction were used for secondary dimensionality reduction with UMAP (RunUMAP(metric = ‘euclidean’)).
Gene activity scores of scATAC-seq data
To calculate gene activity scores we first obtained gene body coordinates by using the command genes (TxDb.Mmusculus.UCSC.mm10.knownGene) for murine samples and genes (TxDb.Hsapiens.UCSC.hg19.knownGene) for human samples from the package GenomicFeatures in R. The coordinates were filtered for standard chromosomes (keepStandardChromosomes(pruning.mode = ‘coarse’)) and extended by 2,000 bp upstream of the transcription start sites to include promoter regions (Extend(upstream = 2000)). Then, the command ‘FeatureMatrix’ from the Signac package was used with the ‘features’ parameter set to the extended gene coordinates to sum up the number of unique reads within gene regions for each cell. Eventually, these gene activity scores were log-normalized and multiplied by the median read counts per cell (nCount_Reads) with the command NormalizeData(normalization.method = ’LogNormalize’,scale.factor = median(nCount_Reads). Normalized gene activities were capped at the 95th quantile for plotting.
For the tumor T cell dataset from (Satpathy et al., 2019), the provided Cicero gene activity scores (Pliner et al., 2018) were used to visualize gene activities.
Signature scores of scATAC-seq data
To calculate an accessibility score for a set of regions for each cell, we first filtered the dataset for peaks overlapping with any of the signature regions (subsequently referred to as signature peaks). For each cell in the dataset of n cells, the number of peaks (n_peaks) was counted. The relative frequency for each peak in the dataset was calculated by dividing the number of cells with a signal for this peak through the total peak count of the binarized peak-cell matrix. For a peak x in the dataset:
We then simulated cells with (1000, 2000, 3000, .., max(n_peaks)) peaks for our datasets. Due to the higher number of peaks, we used (1000, 6000, 11000, .., max(n_peaks)) for the human tumor T cells from (Satpathy et al., 2019). For each of these cells, we sampled the respective number of peaks from the dataset with probabilities given by the previously calculated frequencies for each peak. We then counted the number of signature peaks among these samples. For each simulated cell, sampling was repeated 48 times and the average sum of signature peaks + 1 was assigned as final value. These values represent the number of signature peaks overlapping by chance with random peaks for cells with increasing count depth. Finally, we calculated the enrichment of signature peaks for each cell by dividing the sum of observed signature peaks + 1 for the cell by the simulated cell with the closest number of observed peaks.
Estimation of transcription factor activity with chromVAR
We measured transcription factor (TF) activities for each cell using chromVAR (Schep et al., 2017). TF position weight matrices were downloaded from the Homer website (http://homer.ucsd.edu/homer/custom.motifs). We then used Signac to build a motif-peak matrix for all peaks in the murine and human datasets (CreateMotifMatrix) using reference genomes from the packages BSgenome.Mmusculus.UCSC.mm10 and BSgenome.Hsapiens.UCSC.hg19, respectively. After assembling and adding the Motif object to the Seurat object (CreateMotifObject, AddMotifObject), information on the base composition was calculated for each peak (RegionStats). Eventually, the wrapper function ‘RunChromVAR’ was called to obtain chromVAR deviation z-scores. For plotting, chromVAR deviation z-scores below the 5th and above the 95th quantile was capped.
Cell clustering of scATAC-seq data
We identified cell clusters using graph-based clustering implemented in the Seurat package. The shared nearest neighbor graph was constructed based on the Harmony or SVD embeddings as indicated in the table below (FindNeighbors(dims = 1:20)). Subsequently, clusters were determined using the function ‘FindClusters’ with the indicated resolutions: Mouse SPF tissue and spleen CD4+ with 1.7 (Harmony); Mouse gnotobiotic tissue and spleen CD4+ with 1.7 (SVD); Mouse tumor CD4+ with 0.5 (SVD); Human tissue and blood CD4+ with 1.0 (Harmony).
Identification of differential chromatin accessibility between scATAC clusters
To identify differentially accessible regions between cell clusters, we built a logistic regression model for each region and used a likelihood ratio test to compare it to a null model with the number of peaks per cell as latent variable. Regions were filtered for Bonferroni-corrected p value below 0.05 and average log-fold change above 0.25 (FindMarkers(test.use = ‘LR’, latent.vars = ’n_peaks’, min.pct = 0.1, logfc.threshold = 0.25) from Seurat).
Usage of bigWig tracks
For the generation of per-cluster BigWig tracks, we first identified all barcodes within the clusters. We then used Sinto (https://github.com/timoast/sinto) to extract reads associated with these barcodes from the sample bam files and combined them into one bam file per cluster (samtools merge). Eventually ‘bamCoverage’ was called to generate BigWig coverage tracks from the bam file. BigWig and broadPeak files from BATF-ChIP-seq data of human B cells (replicate 2) were downloaded from GSM803538 for comparisons with the chromatin accessibility BigWig data.
Homer transcription factor analysis of scATAC-seq data
We used Homer (Heinz et al., 2010) for the identification of known transcription factor motifs and de novo motif discovery in sets of cluster-specific peaks (findMotifsGenome.pl peaks.bed hg19 -mask -size given -len 8,10,12,14). We used the motif position weight matrices provided on the Homer website as reference for known transcription factors (http://homer.ucsd.edu/homer/custom.motifs). To generate BATF transcription factor footprints for human CD4+ T cells from different clusters, we first converted the per-cluster bam files into tag directories (makeTagDirectory). We then centered the peaks from the human CD4+ T cell dataset around the BATF (Th17-BATF-ChIP-Seq(GSE39756)/Homer) transcription factor binding motif (annotatePeaks.pl -center -mask -size −1000,1000). Finally, we quantified the tags within 2,000 bp around the BATF motif using the centered peaks (annotatePeaks.pl -fragLength 1 -size −1000,1000 -hist 1).
Cross-species regions liftover of murine and human scATAC-seq data
We used the liftOver tool from UCSC (Kuhn et al., 2013) to transfer mouse signature regions (original peaks) to the human genome (liftover regions). First, liftOver was called with the mm10 to hg19 chain file on the original peaks (http://hgdownload.cse.ucsc.edu/goldenpath/mm10/liftOver/mm10ToHg19.over.chain.gz). Subsequently the liftover regions were lifted back to the mouse genome with the hg19 to mm10 chain file (http://hgdownload.cse.ucsc.edu/goldenpath/hg19/liftOver/hg19ToMm10.over.chain.gz). We used `minMatch = 0.2` to require a substantial sequence conservation between the corresponding murine and human DNA sequences. We filtered out all liftover regions with an overlap smaller than 90% or a width difference greater than 40% with the original peaks after transfer back to the mm10 genome. To include information on the localization with respect to genes, we obtained gene regions and extended these by 2,000 bp upstream to include promoter regions (Signac::Extend(upstream = 2000)). We then annotated both the original peaks and liftover regions with the gene symbols of overlapping genes as well as the closest gene. We used biomart (Durinck et al., 2009) to translate MGI symbols to HGNC symbols and filtered out all liftover regions without any matching gene symbol to the original peak.
To derive a repair Treg signature that is conserved between mouse and human, we searched peaks from the human tisTregST2 signature which overlap by more than 40% with the liftover regions or vice versa. This set of peaks was further filtered for peaks within gene bodies or promoter regions and same direction of chromatin accessibility difference in mouse and human.
Signatures displayed on scATAC-seq data
The following previously derived signatures (Delacher et al., 2020) were used to visualize signature enrichment in murine cells: Core tisTregST2 (Figures 1E, 2C, and S7H, 2267 peaks), Skin tisTregST2 (Figure 1E and Figure 2C, 340 peaks), VAT tisTregST2 (Figures 1E and S2E, 417 peaks), Colon tisTregST2 (Figure S2I, 307 peaks). In addition, two tisTregST2 precursor signatures derived from the aforementioned study were used: early tisTregST2 precursors (Klrg1-Nfil3(GFP)+) and late tisTregST2 precursors (Klrg1+Nfil3(GFP)+) were compared against a combined in silico generated tisTregST2 sample containing all tisTregST2 ATAC-seq samples from all nonlymphoid tissues. Differential peaks with mean accessibility > 10, log2 fc > 2 and padj < 0.01 were selected; early tisTregST2 precursor signature (Figures 1F and S2E): 3323 peaks, late tisTregST2 precursor signature (Figures 1F and 2C): 1726 peaks.
A human tissue Treg signature was calculated from differential peaks between tissue Treg cluster 03 and blood naive Treg cluster 07 of the human CD4 T cell dataset (padj < 0.05, average log fc > 0.25, 4416 peaks). In a second comparison, differential peaks between Tconv clusters 00, 02, 04, 10, 12 and 14 and blood naive Treg cluster 07 were identified (Tconv signature, padj < 0.05, average log fc > 0.25, 3588 peaks). To obtain a Tconv-corrected tissue Treg signature (Figures 6H and 7H), peaks from the human tissue Treg signature also present in the Tconv signature were removed resulting in 2687 peaks.
To derive tissue-specific Treg signatures (Figure 6I), cluster 03 tissue Treg cells were selected from the human CD4+ T cell dataset. Subsequently, tissue Treg cells isolated from fat were compared against those isolated from skin (human fat Treg signature: padj < 0.05, average log fc > 0.25, 437 peaks, human skin Treg signature: padj < 0.05, average log fc < −0.25, 1030 peaks).
A previously published dataset of tumor infiltrating T cells (Satpathy et al., 2019) was used to derive a human Tfh signature (Figure 7A and Figure 7B). Based on provided cell annotations, Tfh cells were compared against Th17 cells, naive CD4+ T cells, activated CD4+ T cells and Th1 cells (padj < 0.05, average log fc > 0.25, 3099 peaks). The dataset was also used to derive a human tumor Treg signature (Figure S7K). In detail, Treg cells were compared against Th17 cells, naive CD4+ T cells, activated CD4+ T cells, memory CD4+ T cells, Tfh cells and Th1 cells (padj < 0.05, average log fc > 0.25, 940 peaks).
To assess similarities between the tisTreg and Tfh differentiation programs (Figure S7D), we annotated peaks from the tissue Treg, Tfh-like Treg and tumor Tfh signatures with their closest gene. Subsequently, overlaps between unique genes in these signatures were counted.
All signatures are provided in Table S1.
Trajectory analysis of scATAC-seq data
To construct a possible developmental trajectory for human or murine Treg cells, we used Monocle (Trapnell et al., 2014). We extracted the Treg clusters (human CD4+ T cell dataset: 01, 03, 07; mouse CD4+ T cell dataset: 00, 05, 10, 11, 16, 23; gnotobiotic mouse CD4+ T cell dataset: 01, 06, 09, 13) for the analysis and rerun the normalization and dimensionality reduction steps described before. To reduce the size of the input matrix from the human dataset, we sampled 5,000 cells from each cluster. We subsequently kept the 3,000 most accessible peaks in the matrix. We then counted the number of sites per cell (detectGenes) and performed size factor estimation (estimateSizeFactors) before reducing dimensions with DDRTree using all features while accounting for the total accessible sites per cell (reduceDimensions(reduction_method = “DDRTree,” residualModelFormulaStr = “∼num_genes_expressed”)). Finally, cells were ordered along pseudotime with the function ‘orderCells’.
Bulk RNA-seq of human tissue Treg cells
Human T cells were isolated, pre-enriched, stained and sorted as described previously. Sort gates are shown in Figure S3. From human fat samples of five individual donors, we sorted 2,500 to 10,000 Treg cells (CD45+CD3+TCRb+CD4+CD8-CD25+CD127-). From human skin samples of five individual donors, we sorted 3,500 to 10,000 Treg cells (CD45+CD3+TCRb+CD4+CD8-CD25+CD127-). From human peripheral blood samples of five individual donors, we sorted 10,000 naive Treg cells (CD45+CD3+TCRb+CD4+CD8-CD25+CD127-CD45RA+CD45RO-), 10,000 memory Treg cells (CD45+CD3+TCRb+CD4+CD8-CD25+CD127-CD45RA-CD45RO+), and 10,000 CCR8 Treg cells (CD45+CD3+TCRb+CD4+CD8-CD25+CD127-CD45RA-CD45RO+ CCR8+). Total RNA was isolated using a DNA/RNA micro kit (Quiagen Cat#80284) and RNA was eluted in 14 μL RNase-free water. RNA quality was assessed using Tapestation 4200 and High-Sensitivity RNA Screentape (Agilent Cat# 5067-5579). 8 μl of the RNA was used for generating RNA-seq libraries using the SMART-seq Stranded Kit (Takara Cat# 634444). Indexed libraries were pooled in an equimolar ratio and sequenced on an Illumina NextSeq 550 machine with NextSeq 500/550 High Output Kit v2.5 (75 cycles).
To isolate and compare CCR8+ Treg cells from healthy and tumor patients, we sorted 800 to 5,500 CCR8+ Treg cells (CD45+Dead-CD206-CD3+CD4+CD8-CD25+CD127-CD45RA-CCR8+) from human fat, 850 to 2,500 CCR8+ Treg from human skin, 1,000 to 10,000 CCR8+ Treg or Treg from human NAT liver tissue, and 2,000 to 10,000 CCR8+ Treg or Treg from human liver tumor tissue. From blood of skin and fat donors, we sorted 750 to 2,500 CCR8+ Treg cells, 2,000 to 2,500 CCR8- Treg cells and 2,000 to 2,500 CD45RA+ Treg cells. From blood of tumor patients, we sorted 2,000 CCR8+ Treg cells, 2,000 CCR8- Treg cells and 2,000 CD45RA+ Treg cells. RNA was isolated, reversely transcribed, indexed and sequenced as described above.
Mapping of RNA-seq data, statistical evaluation and plotting
For all samples, low quality bases were removed with Fastq_quality_filter from the FASTX Toolkit 0.0.13 (http://hannonlab.cshl.edu/fastx_toolkit/index.html) with 90% of the reads needing a quality phred score > 20. Homertools 4.7 (Heinz et al., 2010) were used for PolyA-tail trimming, and reads with a length < 17 were removed. PicardTools 1.78 (https://broadinstitute.github.io/picard/) were used to compute the quality metrics with CollectRNASeqMetrics. With STAR 2.3 (Dobin et al., 2013), the filtered reads were mapped against human genome 38 using default parameters. Count data and RPKM tables were generated by mapping filtered reads against union transcripts using a custom pipeline. Mapping was carried out with bowtie2 version 2.2.4 (Langmead and Salzberg, 2012) against union human genes: every gene is represented by a union of all its transcripts (exons). The count values (RPKM and raw counts) were calculated by running CoverageBed from Bedtools v2.26.0 (Quinlan and Hall, 2010) of the mapped reads together with the human annotation file (Ensembl 90) in gtf format and parsing the output with custom perl scripts. The input tables containing the replicates for groups to compare were created by a custom perl script. For the pairwise comparisons DESeq2 (Love et al., 2014), DESeqDataSetFromMatrix was applied, followed by estimateSizeFactors, estimateDispersions, and nbinomWald testing. The MA plots (Figures 3G, 3I, 5F, 7D, 7K, and 7L) were generated using the plotMA function of DESeq2 using all data. The PCA plots were generated by DESeq2’s plotPCA after transforming the counts using varianceStabilizingTransformation and selecting the genes from the DESeq2 result according to the adjusted p value (< 0.05, < 0.01, < 0.001). Dendrogram (Figure 3H) was generated using R. First, tables were integrated (read.csv) followed by conversion (as.matrix.data.frame). Heatmap was generated using library (pheatmap) and pheatmap function (cutree_rows = 2, cellwidth = 50, cellheight = 1, scale = ”row,” color = greenred(500)). As source data, all genes identified in bulk RNA-seq were first sorted in a comparison of skin – or fat Treg cells versus naive Treg cells. All values unequal to zero in either comparison were allowed. Next, correlated genes were identified by dividing skin Treg/fat Treg (0.5 < x > 2.0) and top 1000 genes with increased expression in fat Treg versus naive Treg and bottom 1000 genes in the comparison fat Treg versus naive Treg were selected and displayed as dendrogram. Rpkm table and statistical results are provided in Table S2.
Single-cell RNA/TCR-seq of human blood and tissue T cells
Human T cells were isolated, pre-enriched, stained and sorted as described previously. Sort gates are shown in Figure S6.
From fat tissue of donor 6, we sorted 40,000 CD45+Dead-CD14-CD19-CD3+TCRβ+CD8-CD4+ T cells. From skin tissue of donor 6, we sorted 40,000 CD45+Dead-CD14-CD19-CD3+TCRβ+CD8-CD4+ T cells. From peripheral blood of donor 6, we sorted 50,000 CD45+Dead-CD14-CD19-CD3+TCRβ+CD8-CD4+ T cells, 50,000 CD45+Dead-CD14-CD19-CD3+TCRβ+CD8-CD4+CD25+CD127- Treg cells, 50,000 CD45+Dead-CD14-CD19-CD3+TCRβ+CD8-CD4+CD25+CD127-CD45RA-CD45RO+ memory Treg cells and 27,000 CD45+Dead-CD14-CD19-CD3+TCRβ+CD8-CD4+CD25+CD127-CD45RA-CD45RO+CCR8+ Treg cells.
From fat tissue of donor 7, we sorted 40,000 CD45+Dead-CD14-CD19-CD3+TCRβ+CD8-CD4+ T cells. From skin tissue of donor 7, we sorted 40,000 CD45+Dead-CD14-CD19-CD3+TCRβ+CD8-CD4+ T cells. From peripheral blood of tissue donor 7, we sorted 50,000 CD45+Dead-CD14-CD19-CD3+TCRβ+CD8-CD4+ T cells, 50,000 CD45+Dead-CD14-CD19-CD3+TCRβ+CD8-CD4+CD25+CD127- Treg cells and 50,000 CD45+Dead-CD14-CD19-CD3+TCRβ+CD8-CD4+CD25+CD127-CD45RA-CD45RO+ memory Treg cells.
After sorting, cells were centrifuged (1000xg, 5min, 4°C) and reconstituted in 38 μl 10%FCS-PBS buffer. Master Mix (Chromium Next GEM Single Cell 5¢ Gel bead V1.1, 10X Genomics #1000165) was added to the cells and samples were loaded on 10X Chromium Next GEM Chip G (10X Genomics #1000120). GEM incubation was performed for 45 min at 53°C according to manufacturer’s protocol. Single-cell libraries were prepared as by manufacturer’s protocol (Chromium Next GEM Single Cell 5¢ Library and Gel Bead Kit v1.1). scRNA libraries were sequenced on an Illumina NextSeq™ 550 with NextSeq™ 500/550 High Output Kit v2.5 (75 cycles). scTCR libraries were sequenced on an Illumina NextSeq™ 550 with NextSeq™ 500/550 Mid Output Kit v2.5 (300 cycles, Illumina Cat# 20024905).
Analysis of scTCR-seq data
Fastq files were processed using Cell Ranger (version 3.1.0) based on 10xGenomics provided VDJ reference (version 3.1.0). Clones from different samples were matched by TRA and TRB amino acid sequences. Clonal abundance pie charts were generated using ggplot2 (version 2.3.3) and R (version 3.6.0). Different TCR numbers between samples and respective pie charts based on removal of duplets or certain clusters.
Analysis of scRNA-seq data
Fastq files were processed using Cell Ranger (version 3.1.0) based on 10xGenomics provided hg19 reference genome (version 3.0.0). Cell Ranger was run per sample (using cellranger count). For downstream analysis, the R package Seurat (Butler et al., 2018) (version 3.4.1) together with R (version 3.6.0) was used. Cells with fewer than 500 were discarded as well as cells exceeding a 5% threshold of mitochondrial transcripts. The data was log normalized (using NormalizeData) and scaled (using ScaleData). Highly variable genes were identified (using FindVariableFeatures) with default parameter settings and principle components calculated (using RunPCA(npcs = 40)). UMAP dimensionality reduction was performed (using RunUMAP).
LegendSCREEN with human blood T cells
Human peripheral blood T cells from three donors isolated and pre-enriched using CD25-biotinylalted antibody and anti-biotin ultrapure microbeads. Samples were surface-stained with antibodies against CCR8, CD45, CD3, CD4, CD8, CD19, Dead cells, CD45RO, and CD127 for 20 min at 4°C. CD25 was detected by Streptavidin staining (detailed gating in Figure S5). Pre-enriched and pre-stained cells were distributed among LegendSCREEN™ plates and stained, washed and fixed according to manufacturer’s protocol. Samples were acquired on a BD FACSSymphony™ using a high-throughput sampling unit (BD HTS™) with following settings: Sample flow rate 3.0 μL/sec, sample volume 100 μL, mixing volume 50 μL/sec, mixing speed 200 μL/sec, 5 number of mixes per well, wash volume 400 μL and BLR period of 5 s, throughput mode standard. Analysis was performed using BD FlowJo™. Clustermap (Figure 5A) was generated using R. First, tables were integrated (read.csv) followed by conversion (as.matrix.data.frame). Clustermap was generated using library (pheatmap) and pheatmap function (cutree_rows = 9, cellwidth = 10, cellheight = 8, scale = ”row”).
Induction of Tfh-like Treg in-vitro followed by bulk ATAC-seq and RNA-seq
Human peripheral blood was separated by Ficoll gradient centrifugation and pre-enriched with anti-human CD25 biotinylated antibody and anti-biotin beads or anti-human CD25 PE-conjugated antibody and anti-PE ultrapure beads. Cells were purified using magnetic columns followed by fluorescence-activated cell sorting of CD45+Dead-CD19-CD3+TCRβ+CD8-CD4+CD25+CD127-CD45RA+ antigen-naive Treg cells. 100,000 cells per well were activated with anti-human TransAct and either treated with IL-2 only (500 units/mL, “IL-2 Treg”) or with IL-2 (500 units/mL), IL-12, IL-21, IL-23 and TGF-b (50 ng/mL, “Tfh-like Treg”) in TexMACS medium (Miltenyi130-097-196) for 3 days, followed by re-supplementation of cytokines and additional cultivation for 3 days. After five days, a QC staining was performed: samples meeting quality requirements (> 85% FOXP3, induction of BCL-6 and BATF) were FACS-sorted on day 6 (CD45+Dead-CD19-CD3+TCRβ+CD8-CD4+) and 100,000 cells were subjected to bulk ATAC-seq and 50,000 cells were subjected to bulk RNA-seq.
RNA was isolated using the RNEasy Plus Micro Kit (QIAGEN #74034), and samples meeting quality criteria (RIN > 9.5) were used to generate libraries with the Truseq total RNA kit (100 ng total RNA as input, Ilumina Cat# 20020596). Indexed samples were pooled in an equimolar ratio and sequenced on an Illumina NextSeq™ 550 with NextSeq™ 500/550 High Output Kit v2.5 (75 cycles).
Chromatin accessibility mapping was performed using the ATAC-seq method as previously described (Buenrostro et al., 2013; Corces et al., 2017), with minor adaptations. Briefly, in each experiment 30,000 – 100,000 sorted cells were pelleted by centrifuging for 10 min at 4°C at 500 x g. After centrifugation, the pellet was carefully lysed in 50 μl resuspension buffer supplemented with NP-40 (Sigma), Tween-20 and Digitonin (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40, 0.1% Tween-20, 0.01% Digitonin) and incubated for 3 min on ice. Then, 1 mL of ice-cold resuspension buffer supplemented with Tween-20 was added, and the sample was centrifuged at 4°C at 500 x g for 10 min. The supernatant was discarded, and the cell pellet was carefully resuspended in the transposition reaction (25 μl 2 x TD buffer (20 mM Tris-HCL pH 7.6, 10 mM MgCl2 20% Dimethyl Formamide in Ambion water), 2.5 μl TDE1 (Illumina), 16.5 μl PBS, 5 μl nuclease-free water, 0.5 μl 1% Digitonin (Promega), 0.5 μl 10% Tween-20 (Sigma)) for 30 min at 37°C on a shaker at 1000 rpm. Following DNA purification with the Clean and Concentrator-5 kit (Zymo) eluting in 23 μl, 2 μl of the eluted DNA was used in a quantitative 10 μL PCR reaction (1.25 μM forward and reverse custom Nextera primers (Corces et al., 2017), 1x SYBR green final concentration) to estimate the optimum number of amplification cycles with the following program 72°C 5 min; 98°C 30 s; 25 cycles: 98°C 10 s, 63°C 30 s, 72°C 1 min; the final amplification of the library was carried out using the same PCR program and the number of cycles according to the Cq value of the qPCR. Library amplification using custom Nextera primers was followed by SPRI size selection with AmpureXP beads to exclude fragments larger than 1,200 bp and smaller than 150 bp (e.g., primer dimers). DNA concentration was measured with a Qubit fluorometer (Life Technologies). The libraries were sequenced using the Illumina NextSeq 550 system.
Preprocessing and analysis of ATAC-seq data
ATAC-seq reads were trimmed using Skewer (Jiang et al., 2014) and aligned to the hg19 assembly of the human genome using Bowtie2 (Langmead and Salzberg, 2012) with the ‘-very-sensitive’ parameter and a maximum fragment length of 2,000 bp. Duplicate and unpaired reads were removed using the sambamba (Tarasov et al., 2015) ‘markdup’ command, and reads with mapping quality > 30 and alignment to the nuclear genome were kept. All downstream analyses were performed on these filtered reads. For visualization purposes bigwig files were created using the bamCoverage function from deeptools with the “RPGC” normalization option.
Bioinformatic analysis of chromatin accessibility data
Peak calling for each sample was performed using MACS2 with the parameters ‘–nomodel–extsize 147 -g 2700000000’ and a qvalue cutoff of 5.00e-02. Peaks overlapping blacklisted features as defined by the ENCODE project were discarded. For the analysis of sample sets we always created a consensus region set by merging the called peaks from all involved samples, and we quantified the accessibility of each region in each sample by counting the number of reads from the filtered BAM file that overlapped each region.
To normalize the chromatin accessibility signal across samples, we performed quantile normalization followed by GC content normalization by regression using DESeq2’s “varianceStabilizingTransformation” approach (Love et al., 2014). DESeq2 was used to identify differential chromatin accessibility between samples. Significant regions were defined as having an FDR-corrected p value below 0.05, an absolute log2 fold change above xxx, and a mean accessibility equal or greater than 10.
Motif enrichment analysis was done using HOMER (Heinz et al., 2010) with the function findMotifsGenome using “-size 500 -len 8,10,12 -h” parameters. Statistical significances of motif enrichment using HOMER were calculated using hypergeometric testing indicated by the “-h” parameter. Histograms of reads around transcription factor binding motifs were generated using HOMER by centering the peaks of interest on the investigated motifs using the annotatePeaks function, followed by counting reads from individual experiments at single base pair resolution in a radius of 1,000 bp (or 150 bp) around the peak centers using the annotatePeaks function with the parameters “-hist -fragLength 1.” Tn5 background was calculated by processing publicly available sequencing data of tagmented “naked” DNA fragments (ERR2213810_1, ERR2213810_2, ERR2213806_1, and ERR2213806_2) (Henriksson et al., 2019) similar to all other ATAC-seq data.
In-vitro suppression assay with Tfh-like Tregs
Tfh-like Tregs were generated as described previously, and responder cells from the same donor were frozen in FCS with 10% DMSO. After 6 days of culture, Tregs were pooled into low bind Eppendorf tubes, stained and 100.000 live cells were sorted. Sorted cells were cultured overnight in TexMACS medium supplemented with 1% Penicillin/Streptomycin and IL-2 (50u/mL). Responder cells were thawed and cultured overnight in the same conditions. After overnight culture, responder cells were labeled with CFSE (Invitrogen), and cultured with Tregs of the same donor, where Tregs were diluted 1+1, 1+3, and 1+15. Cells were stimulated with anti-CD3/anti-CD8 beads (Miltenyi) in a ratio of 1 bead to 5 responder cells. Cells were taken out of culture, surface stained and acquired on a BD FACSymphony.
Generation of supernatant for in-vitro wound-healing assay
Human peripheral blood was separated by Ficoll gradient centrifugation and pre-enriched with anti-human CD25 biotinylated antibody and anti-biotin beads or anti-human CD25 PE-conjugated antibody and anti-PE ultrapure beads. Cells were purified using magnetic columns followed by fluorescence-activated cell sorting of CD45+Dead-CD19-CD3+TCRβ+CD8-CD4+CD25+CD127-CD45RA+ antigen-naive Treg cells. 100,000 cells per well were activated with anti-human TransAct and either treated with IL-2 only (500 units/mL, “IL-2 Treg”) or with IL-2 (500 units/mL), IL-12, IL-21, IL-23 and TGF-b (50 ng/mL, “Tfh-like Treg”) in TexMACS medium (Miltenyi 130-097-196) for 3 days, followed by re-supplementation of cytokines and additional cultivation for 2-3 days. After five to six days, cells were washed vigorously to remove all remaining cytokines. Cells were resuspended in fresh medium with IL-2 and TransAct and allowed to produce metabolites for 20 h. After 20 h, supernatant was collected and cells were used for flow cytometry-based quality control, where FOXP3, HELIOS, BATF and BCL-6 expression were evaluated.
In-vitro wound healing assay with HaCaT cells
The human keratinocyte cell line HaCaT (RRID: CVCL_0038) was grown in TexMACS medium supplemented with 1% Penicillin/Streptomycin. 25.000 Cells/ well were seeded in an ImageLock plate (Essen Bioscience Cat# 4379) and were allowed to settle and expand for 18 h at 37°C. Wounds were made with a WoundMaker™ (Essen Bioscience). Supernatant was diluted at various ratios (1+1, 1+3, 1+7, 1+15) and added to HACAT cells, which were then placed at 37 degrees. Culture plate was placed in the IncuCyte Zoom (Essen Bioscience) and scans of the wells were scheduled for every 60 min for 48 h. Relative wound density was calculated win IncuCyte® Scratch Wound Cell Migration and Invasion System software (Essen Bioscience). Relative wound density values were analyzed, and statistical significance was established with GraphPad Prism (GraphPad software).
In-vitro wound healing assay with reconstructed epidermis
The reconstructed human epidermis wound healing assay was performed using models generated with primary human epidermal keratinocytes (hEK) isolated from juvenile foreskin samples, according to the open source method described in literature (Groeber et al., 2016). In short, hEKs were seeded on a polycarbonate membrane and cultivated using supplemented EpiLife medium. At day 12 of airlift culture, models were wounded using a 2 mm biopsy punch. After wounding, models were supplemented with fresh cell culture supernatant of either IL-2 Treg cells or Tfh-like Treg cells at a 1+5 dilution on day 0. On day 2 after wounding, IL-2 or Tfh-like Treg supernatant was supplemented again. On day 4 after wounding, cell culture supernatant was washed off and growth medium was used. Impedance spectroscopy measurement was performed before wounding, 3 h after wounding, and 2 days, 4 days and 7 days after wounding. Seven days after wounding, models were fixed in Roti Histofix (Carl Roth GmbH, Germany) and embedded in paraffin. Cross sections of 5 μm were cut and hematoxylin & eosin staining (H&E; Morphisto, Germany) was performed, followed by computer-assisted histological studies. In this context, slides were scanned at 40-fold magnification with the high-resolution microscope system TissueFAXSiPLUS (TissueGnostics, Vienna, Austria) operated by TissueFAXS software (TissueGnostics). This technique allows both image processing and automated stitching of single images to one dataset automatically (Schmid et al., 2015). The StrataQuest digital image analysis software was used to quantify the progress of Stratum corneum reconstruction, known to be associated with wound healing in the applied assay (Kiesewetter et al., 2019). Based on the tissue detection algorithm, the area of the Stratum corneum including the adjoining transition zone to the Stratum granulosum was determined.
Calculation of correlation between RNA-seq data of Tfh-like Treg and skin CCR8+ Treg cells
DEG between skin CD4+CD25+CD127-CD45RA-CCR8+ Treg cells and blood CD4+CD25+CD127-CD45RA+ naive Treg cells (Figures 7J and 7K) were selected (padj < 0.05, 4,136 genes). DEG between in-vitro IL-2 Treg cells and Tfh-like Treg cells (Figure 7D) were selected (padj < 0.05, log2 fc > 1 or < -1, 2,222 genes). In the comparison of DEG between IL-2 Treg and Tfh-like Treg cells with skin CCR8-specific DEG, 499 genes were identified. Of those 499 genes, 228 showed increased expression in Tfh-like Treg cells and skin CCR8+ Treg cells (Figure 7E).
Calculation of tissue Treg and tumor Treg cell signature
DEG between skin and fat CD4+CD25+CD127-CD45RA-CCR8+ Treg cells and blood CD4+CD25+CD127-CD45RA+ naive Treg cells (Figures 7J–7L) were selected (padj < 0.05, log2 fc > 2 or < -2, 1,393 genes). For those genes, expression data for all groups were extracted and shown in a heatmap (Figure 7L). DEG between HCC CD4+CD25+CD127-CD45RA-CCR8+ Treg cells and blood CD4+CD25+CD127-CD45RA+ naive Treg cells (Figure 7J–7L) were selected (padj < 0.05, log2 fc > 2 or < -2, 1,408 genes). For those genes, expression data for all groups were extracted and shown in a heatmap (Figure 7L). All signatures are listed in Table S6.
Quantification and statistical analysis
Data were analyzed with Prism software or algorithm. Statistical details are indicated in the figure legend. Population size is described in the figure legend.
Acknowledgments
We thank the RCI NGS core facility and the RCI flow cytometry core for technical support. We thank B. Ruhland, V. Hofmann, H. Stanewsky, J. Raitel, L. Schwarzfischer, U. Ackermann, R. Eder, and I. Fink for technical support and G. Küblbeck (DKFZ) for help with the BAC construct. We thank C. Klein and C. Mulas for providing BALB-NeuT transgenic mice and S. Hori for Foxp3(huCD2) mice. We thank C. Albert and K.K. Tiltmann (TLZ-RT) for technical support. This work was supported by grants from the European Research Council (ERC-CoG, #648145 REGiREG) to M.F. and by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - Projektnummer 324392634 - TRR 221 to M.F., and under Germany’s Excellence Strategy – EXC 2155 – Projektnummer 390874280 – to T.S. and J.H.
Author contributions
Conceptualization, M.D. and M.F.; Methodology, M.D., M.S., L.S. and M.F.; Software, M.D., M.S., A.H.-W., U.R., C.S., N.S., B.B. and C.I.; Investigation, M.D., L.S., M.W., K.S., L.S., S.B., A.P., T.H., D.R., V.S., F.K.G.B., C.G. and A.F.; Resources, A.E., M.R., P.H., M.E., T.S., J.H., L.P. and J.W.; Writing – Original Draft, M.D. and M.F.; Writing – Review & Editing, M.D., M.S., L.S. and M.F.; Visualization, M.D., M.S. and C.I.; Supervision: M.D. and M.F.; Project Administration, M.D. and M.F.; Funding Acquisition, M.F.
Declaration of interest
The authors declare no competing financial interests.
Published: March 30, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.immuni.2021.03.007.
Supplemental information
This is an extension of Figure 1, 2, 6, and 7. This table contains peak data for the skin tisTregST2 signature (Figure 1E and 2C), VAT tisTregST2 signature (Figure 1E and S2E), the core tisTregST2 signature (Figure 1E, 2C and S7H), early tisTregST2 precursor signature (Figure 1F and S2E), late tisTregST2 precursor signature (Figure 1F and 2C), colon tisTregST2 signature (Figure S2I), human tissue Treg signature (Figures 6H and 7H), human skin Treg signature (Figure 6I), human fat Treg signature (Figure 6I), human tissue Tfh signature (Figure 7A and 7B) and human tumor Treg signature (Figure S7K). Comparison of Tfh versus tissue Treg signature (Figure S7D).
This is an extension of Figures 3 and 7. RPKM values for five replicates of blood naive Treg cells, blood memory Treg cells, blood CCR8 Treg cells, fat Treg cells and skin Treg cells along with Ensembl ID and gene symbol (Figures 3F–3I and Figure 7B); Deseq2 results for fat Treg versus blood naive Treg, skin Treg versus blood naive Treg, fat Treg versus blood memory Treg, skin Treg versus blood memory Treg and skin Treg versus fat Treg as well as blood CCR8+ Treg versus blood naive Treg (Figures 3G and 3I); Input data for PCA (Figure 3F); Common RNA tissue Treg signature with 2779 genes used in clustermap (Figure 3H).
This is an extension of Figure 4. Murine tisTregST2 peakset (14,594 peaks; Figure 4A), human tissue Treg peakset (12,236 peaks, Figure 4B) and species-conserved shared peakset (643 peaks, Figure 4C).
This is an extension of Figure 6. Sample overview (with sample name, source name, tissue, donor and organism) and clonotype ID, frequency of the clonotype, proportion of the clonotype, and the CDR3 sequence of TCRα and/or TCRβ as amino acid (aa) information.
This is an extension of Figure 7. Rpkm table with expression values for four replicates of blood naive Treg cells treated for six days with IL-2 or a Tfh mix. Ensembl ID and gene symbol (Figure 7C); Deseq2 results for the comparison of Treg IL-2 versus Treg Tfh (Figure 7D); Peak table with 17,577 peaks identified in the comparison of four replicates of blood naive Treg cells treated for six days with IL-2 or Tfh mix in-vitro, followed by bulk ATAC-seq (Figure 7D).
This is an extension of Figure 7. RPKM values for five replicates of blood naive Treg along with Ensembl ID and gene symbol; Deseq2 results for skin CCR8+ Treg cells versus blood CD45RA+ naive Treg; Deseq2 results for fat CCR8+ Treg cells versus blood CD45RA+ naive Treg; Overlap between in-vitro Tfh signature and skin CCR8 Treg versus blood naive Treg signature (Figure 7E); Deseq2 results for fat and skin CCR8+ Treg cells versus blood CD45RA+ naive Treg; Input data for PCA in Figure 7J; common fat and skin CCR8+ tissue Treg cell signature with 3,801 genes; Input data with 1,393 genes for heatmap in Figure 7L; Deseq2 results for HCC CCR8+ Treg cells versus blood CD45RA+ naive Treg; HCC CCR8+ versus blood naive Treg cell signature with 3,792 genes; Input data with 1,408 genes for heatmap in Figure 7L.
References
- Ali N., Zirak B., Rodriguez R.S., Pauli M.L., Truong H.A., Lai K., Ahn R., Corbin K., Lowe M.M., Scharschmidt T.C. Regulatory T Cells in Skin Facilitate Epithelial Stem Cell Differentiation. Cell. 2017;169:1119–1129. doi: 10.1016/j.cell.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvisi G., Brummelman J., Puccio S., Mazza E.M., Tomada E.P., Losurdo A., Zanon V., Peano C., Colombo F.S., Scarpa A. IRF4 instructs effector Treg differentiation and immune suppression in human cancer. J. Clin. Invest. 2020;130:3137–3150. doi: 10.1172/JCI130426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpaia N., Green J.A., Moltedo B., Arvey A., Hemmers S., Yuan S., Treuting P.M., Rudensky A.Y. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell. 2015;162:1078–1089. doi: 10.1016/j.cell.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi K., Tanoue T., Shima T., Imaoka A., Kuwahara T., Momose Y., Cheng G., Yamasaki S., Saito T., Ohba Y. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:337–341. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizi E., Carr A.J., Plitas G., Cornish A.E., Konopacki C., Prabhakaran S., Nainys J., Wu K., Kiseliovas V., Setty M. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell. 2018;174:1293–1308. doi: 10.1016/j.cell.2018.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A., Cheung S.T., Bennett H.P.J. A Brief Overview of Progranulin in Health and Disease. Methods Mol. Biol. 2018;1806:3–15. doi: 10.1007/978-1-4939-8559-3_1. [DOI] [PubMed] [Google Scholar]
- Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzyn D., Kuswanto W., Kolodin D., Shadrach J.L., Cerletti M., Jang Y., Sefik E., Tan T.G., Wagers A.J., Benoist C., Mathis D. A special population of regulatory T cells potentiates muscle repair. Cell. 2013;155:1282–1295. doi: 10.1016/j.cell.2013.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 1514. 2013 Apr;128 doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolletta D., Feuerer M., Li A., Kamei N., Lee J., Shoelson S.E., Benoist C., Mathis D. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. 2012;486:549–553. doi: 10.1038/nature11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland N.G., Jenkins N.A., Court D.L. Recombineering: a powerful new tool for mouse functional genomics. Nat. Rev. Genet. 2001;2:769–779. doi: 10.1038/35093556. [DOI] [PubMed] [Google Scholar]
- Corces M.R., Trevino A.E., Hamilton E.G., Greenside P.G., Sinnott-Armstrong N.A., Vesuna S., Satpathy A.T., Rubin A.J., Montine K.S., Wu B. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods. 2017;14:959–962. doi: 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossarizza A., Chang H.D., Radbruch A., Acs A., Adam D., Adam-Klages S., Agace W.W., Aghaeepour N., Akdis M., Allez M. Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition) Eur J Immunol. 2019;49:1457–1973. doi: 10.1002/eji.201970107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simone M., Arrigoni A., Rossetti G., Gruarin P., Ranzani V., Politano C., Bonnal R.J.P., Provasi E., Sarnicola M.L., Panzeri I. Transcriptional Landscape of Human Tissue Lymphocytes Unveils Uniqueness of Tumor-Infiltrating T Regulatory Cells. Immunity. 2016;45:1135–1147. doi: 10.1016/j.immuni.2016.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacher M., Imbusch C.D., Weichenhan D., Breiling A., Hotz-Wagenblatt A., Träger U., Hofer A.C., Kägebein D., Wang Q., Frauhammer F. Genome-wide DNA-methylation landscape defines specialization of regulatory T cells in tissues. Nat. Immunol. 2017;18:1160–1172. doi: 10.1038/ni.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacher M., Schmidl C., Herzig Y., Breloer M., Hartmann W., Brunk F., Kägebein D., Träger U., Hofer A.C., Bittner S. Rbpj expression in regulatory T cells is critical for restraining TH2 responses. Nat. Commun. 2019;10:1621. doi: 10.1038/s41467-019-09276-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacher M., Imbusch C.D., Hotz-Wagenblatt A., Mallm J.P., Bauer K., Simon M., Riegel D., Rendeiro A.F., Bittner S., Sanderink L. Precursors for Nonlymphoid-Tissue Treg Cells Reside in Secondary Lymphoid Organs and Are Programmed by the Transcription Factor BATF. Immunity. 2020;52:295–312. doi: 10.1016/j.immuni.2019.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiSpirito J.R., Zemmour D., Ramanan D., Cho J., Zilionis R., Klein A.M., Benoist C., Mathis D. Molecular diversification of regulatory T cells in nonlymphoid tissues. Sci. Immunol. 2018;3 doi: 10.1126/sciimmunol.aat5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombrowski Y., O’Hagan T., Dittmer M., Penalva R., Mayoral S.R., Bankhead P., Fleville S., Eleftheriadis G., Zhao C., Naughton M. Regulatory T cells promote myelin regeneration in the central nervous system. Nat. Neurosci. 2017;20:674–680. doi: 10.1038/nn.4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S., Spellman P.T., Birney E., Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009;4:1184–1191. doi: 10.1038/nprot.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak H.F. Tumors: wounds that do not heal-redux. Cancer Immunol. Res. 2015;3:1–11. doi: 10.1158/2326-6066.CIR-14-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerer M., Herrero L., Cipolletta D., Naaz A., Wong J., Nayer A., Lee J., Goldfine A.B., Benoist C., Shoelson S., Mathis D. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009;15:930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geuking M.B., Cahenzli J., Lawson M.A., Ng D.C., Slack E., Hapfelmeier S., McCoy K.D., Macpherson A.J. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity. 2011;34:794–806. doi: 10.1016/j.immuni.2011.03.021. [DOI] [PubMed] [Google Scholar]
- Groeber F., Engelhardt L., Egger S., Werthmann H., Monaghan M., Walles H., Hansmann J. Impedance spectroscopy for the non-destructive evaluation of in vitro epidermal models. Pharm. Res. 2015;32:1845–1854. doi: 10.1007/s11095-014-1580-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groeber F., Schober L., Schmid F.F., Traube A., Kolbus-Hernandez S., Daton K., Hoffmann S., Petersohn D., Schäfer-Korting M., Walles H., Mewes K.R. Catch-up validation study of an in vitro skin irritation test method based on an open source reconstructed epidermis (phase II) Toxicol. In Vitro. 2016;36:254–261. doi: 10.1016/j.tiv.2016.07.008. [DOI] [PubMed] [Google Scholar]
- Hasan Z., Koizumi S.I., Sasaki D., Yamada H., Arakaki N., Fujihara Y., Okitsu S., Shirahata H., Ishikawa H. JunB is essential for IL-23-dependent pathogenicity of Th17 cells. Nat. Commun. 2017;8:15628. doi: 10.1038/ncomms15628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksson J., Chen X., Gomes T., Ullah U., Meyer K.B., Miragaia R., Duddy G., Pramanik J., Yusa K., Lahesmaa R., Teichmann S.A. Genome-wide CRISPR Screens in T Helper Cells Reveal Pervasive Crosstalk between Activation and Differentiation. Cell. 2019;176:882–896. doi: 10.1016/j.cell.2018.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini H., Obradović M.M.S., Hoffmann M., Harper K.L., Sosa M.S., Werner-Klein M., Nanduri L.K., Werno C., Ehrl C., Maneck M. Early dissemination seeds metastasis in breast cancer. Nature. 2016;540:552–558. doi: 10.1038/nature20785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui S.P., Sheng D.Z., Sugimoto K., Gonzalez-Rajal A., Nakagawa S., Hesselson D., Kikuchi K. Zebrafish Regulatory T Cells Mediate Organ-Specific Regenerative Programs. Dev Cell. 2017;43:659–672. doi: 10.1016/j.devcel.2017.11.010. [DOI] [PubMed] [Google Scholar]
- Ise W., Kohyama M., Schraml B.U., Zhang T., Schwer B., Basu U., Alt F.W., Tang J., Oltz E.M., Murphy T.L., Murphy K.M. The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nat. Immunol. 2011;12:536–543. doi: 10.1038/ni.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M., Komai K., Mise-Omata S., Iizuka-Koga M., Noguchi Y., Kondo T., Sakai R., Matsuo K., Nakayama T., Yoshie O. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature. 2019;565:246–250. doi: 10.1038/s41586-018-0824-5. [DOI] [PubMed] [Google Scholar]
- James K.R., Gomes T., Elmentaite R., Kumar N., Gulliver E.L., King H.W., Stares M.D., Bareham B.R., Ferdinand J.R., Petrova V.N. Distinct microbial and immune niches of the human colon. Nat. Immunol. 2020;21:343–353. doi: 10.1038/s41590-020-0602-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H., Lei R., Ding S.W., Zhu S. Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics. 2014;15:182. doi: 10.1186/1471-2105-15-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002 Jun.;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiesewetter L., Littau L., Walles H., Boccaccini A.R., Groeber-Becker F. Reepithelialization in focus: Non-invasive monitoring of epidermal wound healing in vitro. Biosens. Bioelectron. 2019;142:111555. doi: 10.1016/j.bios.2019.111555. [DOI] [PubMed] [Google Scholar]
- Komatsu N., Mariotti-Ferrandiz M.E., Wang Y., Malissen B., Waldmann H., Hori S. Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc. Natl. Acad. Sci. USA. 2009;106:1903–1908. doi: 10.1073/pnas.0811556106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn R.M., Haussler D., Kent W.J. The UCSC genome browser and associated tools. Brief. Bioinform. 2013;14:144–161. doi: 10.1093/bib/bbs038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., McDermott M.G., Monteiro C.D., Gundersen G.W., Ma'ayan A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016 Jul 8;44 doi: 10.1093/nar/gkw377. W90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., DiSpirito J.R., Zemmour D., Spallanzani R.G., Kuswanto W., Benoist C., Mathis D. TCR Transgenic Mice Reveal Stepwise, Multi-site Acquisition of the Distinctive Fat-Treg Phenotype. Cell. 2018;174:285–299. doi: 10.1016/j.cell.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesz A., Suri-Payer E., Veltkamp C., Doerr H., Sommer C., Rivest S., Giese T., Veltkamp R. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat. Med. 2009;15:192–199. doi: 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miragaia R.J., Gomes T., Chomka A., Jardine L., Riedel A., Hegazy A.N., Whibley N., Tucci A., Chen X., Lindeman I. Single-Cell Transcriptomics of Regulatory T Cells Reveals Trajectories of Tissue Adaptation. Immunity. 2019;50:493–504. doi: 10.1016/j.immuni.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyara M., Yoshioka Y., Kitoh A., Shima T., Wing K., Niwa A., Parizot C., Taflin C., Heike T., Valeyre D. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009;30:899–911. doi: 10.1016/j.immuni.2009.03.019. [DOI] [PubMed] [Google Scholar]
- Pliner H.A., Packer J.S., McFaline-Figueroa J.L., Cusanovich D.A., Daza R.M., Aghamirzaie D., Srivatsan S., Qiu X., Jackson D., Minkina A. Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol Cell. 2018;71:858–871. doi: 10.1016/j.molcel.2018.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plitas G., Konopacki C., Wu K., Bos P.D., Morrow M., Putintseva E.V., Chudakov D.M., Rudensky A.Y. Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer. Immunity. 2016;45:1122–1134. doi: 10.1016/j.immuni.2016.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope B.D., Ryba T., Dileep V., Yue F., Wu W., Denas O., Vera D.L., Wang Y., Hansen R.S., Canfield T.K. Topologically associating domains are stable units of replication-timing regulation. Nature. 2014;515:402–405. doi: 10.1038/nature13986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povoleri G.A.M., Nova-Lamperti E., Scottà C., Fanelli G., Chen Y.C., Becker P.D., Boardman D., Costantini B., Romano M., Pavlidis P. Human retinoic acid-regulated CD161+ regulatory T cells support wound repair in intestinal mucosa. Nat. Immunol. 2018;19:1403–1414. doi: 10.1038/s41590-018-0230-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L., Waseem T.C., Sahoo A., Bieerkehazhi S., Zhou H., Galkina E.V., Nurieva R. Insights Into the Molecular Mechanisms of T Follicular Helper-Mediated Immunity and Pathology. Front. Immunol. 2018;9:1884. doi: 10.3389/fimmu.2018.01884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat Biotechnol. 2011 Jan;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S., Mikami N., Wing J.B., Tanaka A., Ichiyama K., Ohkura N. Regulatory T Cells and Human Disease. Annu. Rev. Immunol. 2020;38:541–566. doi: 10.1146/annurev-immunol-042718-041717. [DOI] [PubMed] [Google Scholar]
- Satpathy A.T., Granja J.M., Yost K.E., Qi Y., Meschi F., McDermott G.P., Olsen B.N., Mumbach M.R., Pierce S.E., Corces M.R. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 2019;37:925–936. doi: 10.1038/s41587-019-0206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep A.N., Wu B., Buenrostro J.D., Greenleaf W.J. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods. 2017;14:975–978. doi: 10.1038/nmeth.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiering C., Krausgruber T., Chomka A., Fröhlich A., Adelmann K., Wohlfert E.A., Pott J., Griseri T., Bollrath J., Hegazy A.N. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature. 2014;513:564–568. doi: 10.1038/nature13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M., Dufner B., Dürk J., Bedal K., Stricker K., Prokoph L.A., Koch C., Wege A.K., Zirpel H., van Zandbergen G. An Emerging Approach for Parallel Quantification of Intracellular Protozoan Parasites and Host Cell Characterization Using TissueFAXS Cytometry. PLoS ONE. 2015;10:e0139866. doi: 10.1371/journal.pone.0139866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sefik E., Geva-Zatorsky N., Oh S., Konnikova L., Zemmour D., McGuire A.M., Burzyn D., Ortiz-Lopez A., Lobera M., Yang J. MUCOSAL IMMUNOLOGY. Individual intestinal symbionts induce a distinct population of RORγ+ regulatory T cells. Science. 2015;349:993–997. doi: 10.1126/science.aaa9420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield N.C., Bock C. LOLA: enrichment analysis for genomic region sets and regulatory elements in R and Bioconductor. Bioinformatics. 2016;15:587–589. doi: 10.1093/bioinformatics/btv612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram G.M., Quah S., Sampath P. Cancer: the dark side of wound healing. FEBS J. 2018;285:4516–4534. doi: 10.1111/febs.14586. [DOI] [PubMed] [Google Scholar]
- Szabo P.A., Levitin H.M., Miron M., Snyder M.E., Senda T., Yuan J., Cheng Y.L., Bush E.C., Dogra P., Thapa P. Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nat. Commun. 2019;10:4706. doi: 10.1038/s41467-019-12464-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A., Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27:109–118. doi: 10.1038/cr.2016.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov A., Vilella A.J., Cuppen E., Nijman I.J., Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31:2032–2034. doi: 10.1093/bioinformatics/btv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton A.M., Korty P.E., Tran D.Q., Wohlfert E.A., Murray P.E., Belkaid Y., Shevach E.M. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J. Immunol. 2010;184:3433–3441. doi: 10.4049/jimmunol.0904028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N.J., Livak K.J., Mikkelsen T.S., Rinn J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasanthakumar A., Moro K., Xin A., Liao Y., Gloury R., Kawamoto S., Fagarasan S., Mielke L.A., Afshar-Sterle S., Masters S.L. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat. Immunol. 2015;16:276–285. doi: 10.1038/ni.3085. [DOI] [PubMed] [Google Scholar]
- Wang L., Simons D.L., Lu X., Tu T.Y., Solomon S., Wang R., Rosario A., Avalos C., Schmolze D., Yim J. Connecting blood and intratumoral Treg cell activity in predicting future relapse in breast cancer. Nat. Immunol. 2019;20:1220–1230. doi: 10.1038/s41590-019-0429-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D., Han J.M., Yu X., Lam A.J., Hoeppli R.E., Pesenacker A.M., Huang Q., Chen V., Speake C., Yorke E. Characterization of regulatory T cells in obese omental adipose tissue in humans. Eur. J. Immunol. 2019;49:336–347. doi: 10.1002/eji.201847570. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This is an extension of Figure 1, 2, 6, and 7. This table contains peak data for the skin tisTregST2 signature (Figure 1E and 2C), VAT tisTregST2 signature (Figure 1E and S2E), the core tisTregST2 signature (Figure 1E, 2C and S7H), early tisTregST2 precursor signature (Figure 1F and S2E), late tisTregST2 precursor signature (Figure 1F and 2C), colon tisTregST2 signature (Figure S2I), human tissue Treg signature (Figures 6H and 7H), human skin Treg signature (Figure 6I), human fat Treg signature (Figure 6I), human tissue Tfh signature (Figure 7A and 7B) and human tumor Treg signature (Figure S7K). Comparison of Tfh versus tissue Treg signature (Figure S7D).
This is an extension of Figures 3 and 7. RPKM values for five replicates of blood naive Treg cells, blood memory Treg cells, blood CCR8 Treg cells, fat Treg cells and skin Treg cells along with Ensembl ID and gene symbol (Figures 3F–3I and Figure 7B); Deseq2 results for fat Treg versus blood naive Treg, skin Treg versus blood naive Treg, fat Treg versus blood memory Treg, skin Treg versus blood memory Treg and skin Treg versus fat Treg as well as blood CCR8+ Treg versus blood naive Treg (Figures 3G and 3I); Input data for PCA (Figure 3F); Common RNA tissue Treg signature with 2779 genes used in clustermap (Figure 3H).
This is an extension of Figure 4. Murine tisTregST2 peakset (14,594 peaks; Figure 4A), human tissue Treg peakset (12,236 peaks, Figure 4B) and species-conserved shared peakset (643 peaks, Figure 4C).
This is an extension of Figure 6. Sample overview (with sample name, source name, tissue, donor and organism) and clonotype ID, frequency of the clonotype, proportion of the clonotype, and the CDR3 sequence of TCRα and/or TCRβ as amino acid (aa) information.
This is an extension of Figure 7. Rpkm table with expression values for four replicates of blood naive Treg cells treated for six days with IL-2 or a Tfh mix. Ensembl ID and gene symbol (Figure 7C); Deseq2 results for the comparison of Treg IL-2 versus Treg Tfh (Figure 7D); Peak table with 17,577 peaks identified in the comparison of four replicates of blood naive Treg cells treated for six days with IL-2 or Tfh mix in-vitro, followed by bulk ATAC-seq (Figure 7D).
This is an extension of Figure 7. RPKM values for five replicates of blood naive Treg along with Ensembl ID and gene symbol; Deseq2 results for skin CCR8+ Treg cells versus blood CD45RA+ naive Treg; Deseq2 results for fat CCR8+ Treg cells versus blood CD45RA+ naive Treg; Overlap between in-vitro Tfh signature and skin CCR8 Treg versus blood naive Treg signature (Figure 7E); Deseq2 results for fat and skin CCR8+ Treg cells versus blood CD45RA+ naive Treg; Input data for PCA in Figure 7J; common fat and skin CCR8+ tissue Treg cell signature with 3,801 genes; Input data with 1,393 genes for heatmap in Figure 7L; Deseq2 results for HCC CCR8+ Treg cells versus blood CD45RA+ naive Treg; HCC CCR8+ versus blood naive Treg cell signature with 3,792 genes; Input data with 1,408 genes for heatmap in Figure 7L.
Data Availability Statement
The accession numbers for murine scATAC-Seq data reported in this paper are: Gene Expression Omnibus (GEO) GSE156112. The accession numbers for human scATAC-Seq, scRNA-Seq, scTCR-Seq, bulk ATAC-seq and bulk RNA-seq data reported in this paper are: European Genome-phenome Archive (EGA) S00001004900.