

Abstract
Cryogenic electron microscopy (cryo-EM) methods began to be used in the mid-1970s to study thin and periodic arrays of proteins. Following a half-century of development in cryo-specimen preparation, instrumentation, data collection, data processing, and modeling software, cryo-EM has become a routine method for solving structures from large biological assemblies to small biomolecules at near to true atomic resolution. This review explores the critical roles played by the Protein Data Bank (PDB) and Electron Microscopy Data Bank (EMDB) in partnership with the community to develop the necessary infrastructure to archive cryo-EM maps and associated models. Public access to cryo-EM structure data has in turn facilitated better understanding of structure–function relationships and advancement of image processing and modeling tool development. The partnership between the global cryo-EM community and PDB and EMDB leadership has synergistically shaped the standards for metadata, one-stop deposition of maps and models, and validation metrics to assess the quality of cryo-EM structures. The advent of cryo-electron tomography (cryo-ET) for in situ molecular cell structures at a broad resolution range and their correlations with other imaging data introduce new data archival challenges in terms of data size and complexity in the years to come.
Keywords: cryo-electron microscopy, imaging, single particle analysis, cryo-electron tomography, electron crystallography, protein structure, data archiving
Abbreviations: cryo-EM, cryogenic electron microscopy; cryo-ET, cryo-electron tomography; EMDB, Electron Microscopy Data Bank; EMPIAR, Electron Microscopy Public Image Archive; PDB, Protein Data Bank; TEM, transmission electron microscope
Cryogenic electron microscopy (cryo-EM) refers to an imaging method using a transmission electron microscope (TEM) operated with an electron energy typically between 100 and 300 kV to collect images of frozen specimens. This corresponds to a wavelength range of 0.02 to 0.037 Å, which is not a limiting factor to resolve atomic details. However, achieving atomic resolution imaging of biological specimens required developing workarounds for many roadblocks, most fundamentally radiation damage and specimen dehydration in the microscope’s vacuum, which were circumvented by cryo-EM.
A penalty cost of this energy range of electrons is their high probability to cause radiation damage in biological molecules, initially by chemical bond breakage and followed by release of damaged fragments (1, 2). A low-dose method coupled with computational averaging of repeating units was proposed to minimize specimen damage and to restore the image contrast (3, 4). Bacteriorhodopsin was the first demonstration of this approach yielding a 7 Å projection density map from invisible low-dose images (5). The adoption of maintaining the specimen at low temperature was the very beginning of cryo-EM, enabling reduction of radiation damage in biological specimens in order to obtain near atomic-resolution electron microscopic data (6, 7).
Another constraint of high-energy electrons is the need for biological specimens to be in a high vacuum, which is incompatible with their need to be in a hydrated state to be functional. The pioneering work of Taylor and Glaeser in the mid 1970s demonstrated the feasibility of immersing a thin protein crystal directly in liquid nitrogen and recording electron diffraction data beyond 3 Å resolution with the specimen kept at liquid nitrogen temperature (8). This initial breakthrough was further extended by the innovative approach of Dubochet and colleagues to freeze biological samples very quickly and effectively in liquid ethane with its high heat transfer coefficient so that the water is frozen into a vitrified rather than a crystalline state (9, 10, 11). This game-changing protocol has become the standard for cryo-EM structure determination since the mid-1980s.
In this review, we provide a brief history of the evolution of cryo-EM from nanometer to atomic resolution over a period of 40 years and the synergistic relationship between Protein Data Bank (PDB), Electron Microscopy Data Bank (EMDB), and cryo-EM community in the last 20 years in shaping public sharing of data and evaluation metrics, as was done for X-ray crystallography over 50 years ago. Currently, cryo-EM is a highly sought method for determining near atomic-resolution structures of small (30 kDa) and large (tens of MDa) macromolecular complexes, including those that may exist in multiple conformations or compositions in a single sample.
Early efforts (1980–2005): Quarter century progress in cryo-EM
The idea of using low temperature to reduce radiation damage and push toward atomic resolution imaging and structure determination was pursued successfully in 1980 to 1990 with thin protein crystals preserved in glucose instead of ice, examined in the electron microscope at low temperatures. The advantage of utilizing thin crystals was the ease of performing image averaging as in crystallography. The detection of 3.5 Å diffraction intensities in the Fourier power spectrum of the image of glucose-embedded crotoxin complex crystal kept at low temperature (12) pointed to the possibility of retrieving the phase information directly from electron images for structure determination. A series of two-dimensional protein crystal structures (where crystal is one unit cell thick in one direction) were subsequently solved with diffraction and images to near atomic-resolution including bacteriorhodopsin (13), light harvesting complex (14), Zn-induced tubulin sheet (15), and aquaporin-1 (16) (Fig. 1). The data collection for each of these projects was laborious because of low yield of high-resolution images later found to be attributable to electron-beam-induced movement (17). These experiments led to the first few cryo-EM structures with models deposited to PDB.
Figure 1.

Early de novo models from 2D protein crystals embedded in glucose. All were derived from 3D maps computed from electron diffraction intensities and images recorded from specimens embedded in glucose and kept at either liquid nitrogen or liquid helium temperatures. A, bacteriorhodopsin: PDB:1BRD, (B) light harvesting complex II: PDB:2BHW (shown: X-ray model that built upon the original electron diffraction analysis), (C) tubulin: PDB:1TUB, (D) aquaporin-1 PDB:1FQY.
Paralleling the excitement of these electron crystallographic structure determinations at near atomic resolution, single-particle cryo-EM began its independent path using Dubochet’s ice-embedding protocol to study single particles of spherical viruses, membrane channels, and ribosomes at nanometer resolution, as exemplified by some early structures (18, 19, 20, 21, 22). In some cases, reconstructions reached subnanometer resolutions where α-helices were clearly visible (23, 24, 25, 26, 27). These early structures were unfortunately not archived in PDB because there was no associated model. In 2003, a de novo model derived from a 4 Å map of a helical filament of membrane protein embedded in vitreous ice was deposited to PDB (Fig. 2) (28). All these developments inspired more enthusiasm for the potential of single-particle cryo-EM to reach atomic resolution (29, 30).
Figure 2.
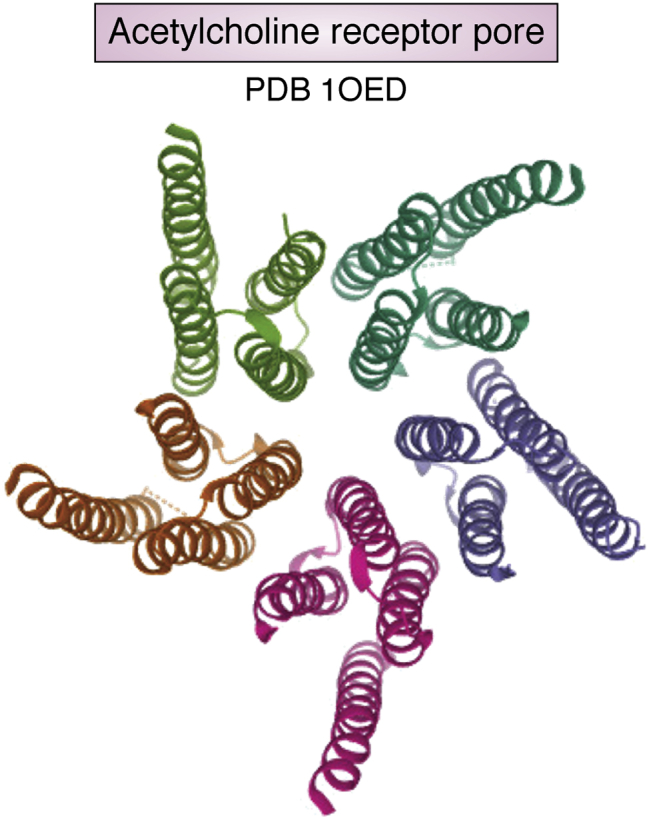
De novo model (PDB:1OED) of helical acetylcholine receptor core embedded in vitreous ice.
Toward the end of this period, some protein crystallographers began to integrate cryo-EM and X-ray crystallography in two ways. One was to fit the crystal structure of a molecular component into a low-resolution cryo-EM map of a large molecular complex in order to shed light on biological assembly principles. This approach began attracting the modeling community to develop tools that combine both crystallographic and cryo-EM data to generate structural models (31). Notably, several biologically important protein structures were reported with this approach such as actin-myosin filament (32), foot and mouth virus-Fab complex (33), Rhinovirus-receptor complex (34), actin bundle (35). Some of them were indeed deposited to the PDB (Fig. 3). The second use of cryo-EM maps by crystallographers was to build an initial low-resolution model of a large complex and then apply phase extension to solve its high-resolution crystal structure. This approach was successful to solve challenging crystallographic projects at that time such as ribosome subunit and large icosahedral virus structures (36, 37).
Figure 3.

Early depositions of all-atom (A and C) and Cα (B) models of macromolecular complexes with crystal structures of their molecular components fitted into low and moderate-resolution cryo-EM maps (A) Foot-and-mouth virus with Fab (PDB 1QGC) (B) Rhinovirus with its receptor (PDB 1D3I), (C) Actin filament in the acrosomal bundle (PDB 3B5U).
Archiving cryo-EM structures: EMDB, PDB, and EMDataResource
With the aforementioned achievements and strong potential for use of cryo-EM as a supplementary source of structural data for large biological complexes, Kim Henrick, former director of the European PDB group at the European Bioinformatics Institute (EBI), recognized the need for a public archive of cryo-EM maps, enabling reuse of a growing body of novel structural results that could not be archived in PDB. In 2002, he and his colleagues launched the EMDB for archiving single-particle reconstructions with any symmetry, subtomogram averages of subcellular tomograms, and electron crystallography maps of thin crystals, along with metadata describing the full experimental workflow (38). Henrick, in collaboration with Helen Berman, former director of the USA PDB group at the Research Collaboratory for Structural Bioinformatics (RCSB), led efforts to integrate the EMDB data model for cryo-EM maps with PDB’s data dictionary, significantly extending it beyond its original context of crystallography. In a 2004 meeting co-organized with Michael Rossmann (Purdue), input was sought from an international group of cryo-EM practitioners (Fig. 4, A and B).
Figure 4.

Workshops held at EBI and RCSB to derive metadata dictionaries, map, and model validation standards.A and B, cryo-EM Structure Deposition Workshop 2004 (Rutgers USA), (C) 3DEM Validation Task Force 2010 (Rutgers USA), (D) Cryo-EM Structure Joint Map and Model Challenges Workshop 2017 (SLAC, Stanford, USA), and (E) Frontiers in cryo-EM Validation Meeting 2019 (EBI UK).
In 2006, Henrick and Berman recruited Wah Chiu, former director of National Center for Macromolecular Imaging, an NIH-supported Cryo-EM Resource Center, to form the Unified Data Resource for 3D Electron Microscopy collaborative project (EMDataResource (EMDR); emdataresource.org, previously called EMDataBank). The project was targeted partly to reach out to the broader cryo-EM community and partly to leverage the long-standing experience of the protein crystallography community in developing and maintaining public archives for structure coordinates and experimental metadata (39). This partnership has continuously sought community consensus to refine the meta data dictionary, data formats, and validation standards through 19 in-person workshops held mainly at EBI and RCSB (40). In 2008, the first map plus model joint deposition system (“one stop shop”) was developed by the EMDataResource project team in collaboration with the worldwide PDB (wwPDB) (41). The first gathering of 28 cryo-EM and modeling experts (Fig. 4C) laid out the need to (i) define what is meant by the resolution of a cryo-EM map in terms of Fourier Shell Correlation and (ii) refine models that comply with protein stereochemistry (42). Significantly, the participants overwhelmingly recommended that all published cryo-EM maps should be deposited to EMDB and all models to PDB prior to publication and that journals should develop policies to encourage this practice. Through numerous communications with editors of prominent journals, it has gradually become the rule rather than an exception that a manuscript cannot be published without accession codes for EMDB and PDB if applicable.
Steady increase in cryo-EM resolution between 2005 and 2020
Owing to advances in software development, cryo-EM structure resolution improved steadily from subnanometer to near atomic resolution by the late 2000s. In 2008, several single-particle structures were reported to reach ∼4 Å resolution where polypeptide backbone tracing was possible (43, 44, 45, 46) (Fig. 5). The inherent high symmetry of the specimens meant that fewer particles were needed to reach this technology milestone.
Figure 5.

Cryo-EM maps and Cα atom model derived from 4.5 to 3.88 Å resolution cryo-EM density maps (A) GroEL, (B) epsilon15 bacteriophage, (C) rotavirus, and (D) cytoplasmic polyhedrosis virus.
As shown in Figure 6, the number of structures reaching a resolution of 3 to 4 Å has continued to rise since 2008. A further technological breakthrough was made in the mid-2010s in cryo-EM data recording devices different from photographic films or charge-coupled devices. The direct electron detector enabled cryo-EM scientists to overcome two major barriers, resulting in more effective recording of high-resolution images (47). Image blurring attributable to the beam-induced specimen motion was reduced by digitally realigning and averaging multiple frames of the same specimen area, and quantum detective efficiency was also significantly improved (48, 49, 50). This detector advance was regarded as leading to so-called cryo-EM’s resolution revolution (51). In parallel, the emergence of new image processing software made structure studies possible even for conformationally heterogeneous particles (52). The commercially available TEM with automated data collection software makes large data set collection more tractable in round-the-clock data collection style. Many near atomic-resolution structures began to emerge unexpectedly and rapidly (Fig. 6).
Figure 6.

Growth of archived electron microscopy density maps at different resolution ranges between 2002 and 2020 since the inception of EMDB with the early deposition statistics shown in the inset. Each color denotes a different resolution range of deposited structures. Because of the early adoption of a community-agreed resolution definition for map resolution, this measure is self-reported by authors for nearly every deposited cryo-EM map. The shift in resolution from subnanometer to near atomic resolution is evident since 2016.
Even though the electron optics of all the high-end instruments should be good enough for atomic-resolution imaging, many projects were still hindered by specimen quality: either from denaturation during cryo-specimen preparation or from the presence of inherently flexible domains. Various experimental and computational approaches have been introduced to resolve some but not all of these difficulties (53, 54, 55). Nevertheless, the number of cryo-EM structures better than 4 Å has been rising exponentially since 2016 (Fig. 6). In 2020, three independent laboratories using different electron optics and cameras reported true atomic-resolution cryo-EM structures of octahedral apoferritin, resolving protein atoms, water molecules, and ions (56, 57, 58) (Fig. 7).
Figure 7.

Selected side-chain densities and atoms in apoferritin taken from one X-ray crystal structure (PDB: 6B8F) and multiple cryo-EM maps at atomic resolution obtained with different electron microscopes.A, Q-scores are computed for each of the residues with higher Q-scores seen in higher-resolution maps. B, Apoferritin model (PDB: 7K3V) based on one of the cryo-EM maps (EMD:22657) shows protein ribbons in white, water oxygens in red, and ions in green and blue.
Accelerated activities in cryo-EM data archiving and structure validation since 2010
In 2016, cryo-EM map and model deposition was integrated into the wwPDB OneDep system, which also collects structures determined using X-ray crystallography and NMR (59). In addition, a validation report was developed to inform depositors how their structure is ranked with respect to all other cryo-EM structures at equivalent resolution in the EMDB and PDB (60).
Also in 2016, EBI established the Electron Microscopy Public Image Archive (EMPIAR) (61), which enables cryo-EM scientists to archive and share raw images and intermediate data files associated with their maps deposited to EMDB. Making raw image data broadly available has multiple benefits, including accelerating development of reconstruction software and enriching resources for cryo-EM scientists in training.
In this rapid growth phase of cryo-EM, it is necessary to establish rigorous methods to validate the maps and associated models. EMDR has taken an initiative to develop validation tools and simultaneously promote the public awareness of this necessary step in the structure determination as is done in crystallography (Table 1).
Table 1.
EMDataResource challenges
| Activity | Participants’ goals | Outcomes |
|---|---|---|
| 2010 Model Challenge (62) | Produce best models against 13 selected maps (2.5–24 Å) |
|
| 2016/17 Map Challenge (63) | Produce best maps from seven raw image datasets (2.5–5 Å), compare reconstruction practices |
|
| 2016/17 Model Challenge (63) | Produce best models from 12 selected maps (2.5–5 Å), compare modeling practices |
|
| 2019 Model Challenge (64) | Produce best models from four selected maps (1.8–3.1 Å), evaluate model metrics |
|
The first EMDR Challenge project in 2010 (Fig. 4D) was targeted to attract modeling experts to engage with the cryo-EM investigators and data that were mostly at subnanometer resolution at that time. The outcomes were reported in a series of articles (summarized in (62)). Additional challenges were organized in 2016 with two separate groups (63), making use of benchmark raw images drawn from EMPIAR and density maps from EMDB. The first group was charged with evaluating whether some recently published reconstructions could be improved. The challengers computed maps using their own choice of image processing pipeline. Submitted maps were then assessed by a group of image processing software developers. The analysis of 66 submitted maps for seven specimen targets with different reported resolutions from 2.7 to 6.8 Å showed that some participants could do better while others did worse than the published maps even with the same image reconstruction software package. In parallel, a second group was charged with evaluating whether improved models could be built from several recently published reconstructions. Interestingly, many of the submitted models were better than the published ones (65, 66). During these two challenge activities, many participants were stimulated to improve data processing algorithms, modeling pipelines, and to generate novel validation procedures; outcomes included the need in particular for careful review of metrics that evaluate the fit of the model to its cryo-EM map (63).
In 2019, we used our prior experience in setting up challenge events and aimed at modeling maps of two targets determined between 1.8 and 3.1 Å resolutions (64). This challenge was aimed at evaluating the quality of models produced by current modeling tools, the reproducibility of modeling results from different software developers and users, and the performance of current metrics used for evaluation of models. It was found that no single metric commonly used is sufficient to assess the quality of the model. An interesting outcome of this challenge was the finding that CaBLAM (67) and Q-scores (68) can respectively assist to generate a more accurate backbone trace and to assess the resolvability of the atoms, residues, or backbone atoms. CaBLAM considers pseudo-dihedrals between Cα atoms and compares them to those of high-resolution PDB structures, while Q-scores consider the map values around each atom and compare them to what the values would be if the atom was well resolved. These local and quantitative assessments are important new tools for investigators to build better models and to assess the density reliability at the local regions of structural interest.
Cryo-electron tomography (cryo-ET)
Cryo-ET is a method to collect images of the same specimen tilted at different angles, and the 3D map is reconstructed from the tilted images. Multiple reconstructions can be merged to obtain a final structure as exemplified by vitrified insect flight muscle (69) (Fig. 8). For cellular materials, cryo-ET is emerging as a very exciting means to extract structural information that is beyond the reach of fluorescence optical microscopy (70). Correlative study of vitrified cell samples containing targets of interest tagged via fluorescence microscopy is beginning to be used routinely (71). Focused ion beam milling of thick specimens has become a standard protocol to prepare thin lamellae with thickness suited for 200 to 300 kV transmission electron microscope collection of tilted specimen images and computation of a 3D map (72, 73). Tomographic data is more complex to archive partly because of its massive data size (e.g., 150 GB per tilt series with a 8k x 8k camera). If the aim of the study is to computationally extract, classify, and average tomogram subvolumes of the object of interest (74), the archiving task is less complex because one can follow the validation principles as used for single particle cryo-EM (75). However, if the aim of the study is to make morphological observations of subcellular structure arrangements under different conditions, it becomes more challenging as to what to archive and how to validate the conclusions based on the morphological observations. Feature extraction using convolutional neural networks (76) is a promising method that will likely be further developed to enable reproducible and statistically meaningful conclusions.
Figure 8.
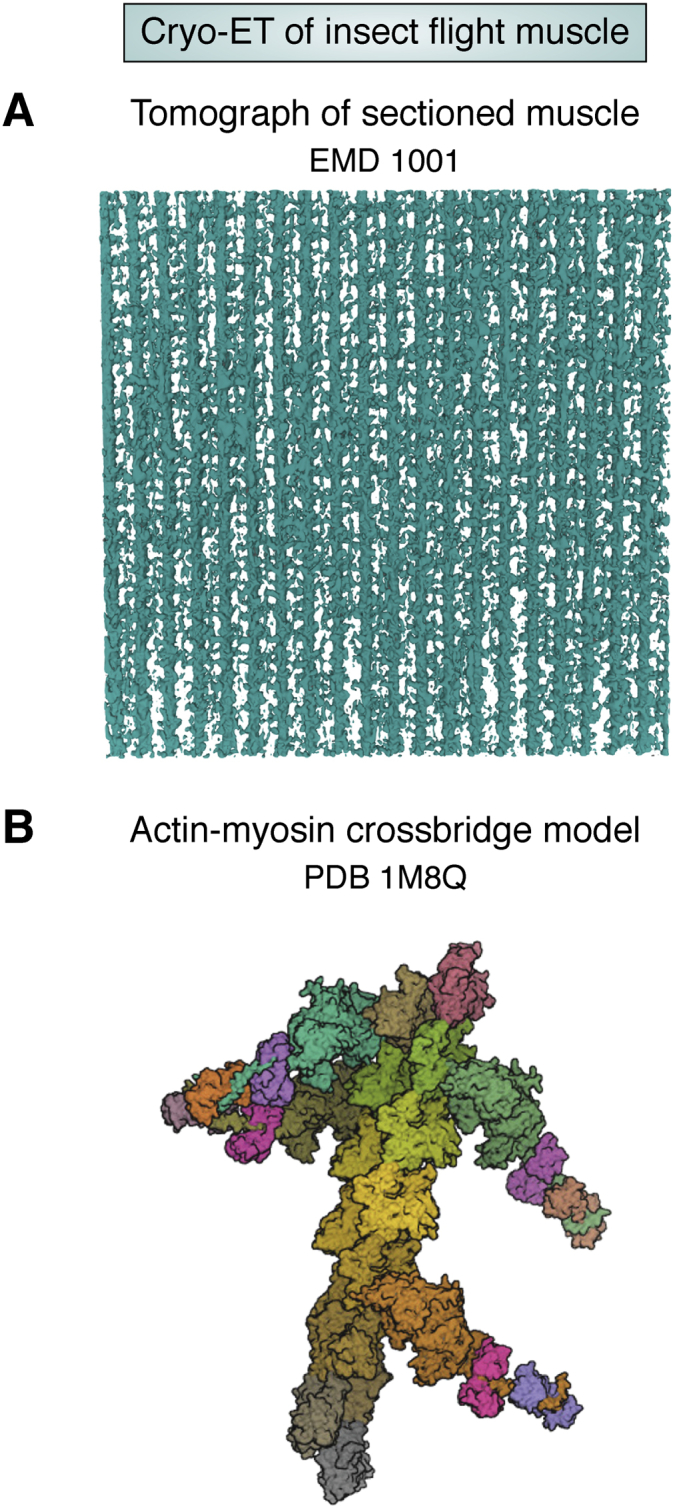
Early Cryo-ET investigation of Flight Insect Muscle.A, tomogram of sectioned muscle (EMD-1001), B, representative actin-myosin crossbridge model (PDB: 1M8Q) based on subtomogram averaging of the sectioned muscle.
Future trends
The examples cited above are not exhaustive but demonstrate some early and some more recent cryo-EM structures at different resolution scales in the EMDB and PDB inspiring the development of public data archiving and validation standards. Facilitating interactions between data scientists and cryo-EM scientists accelerates both platforms’ developments. As shown in Figure 9A, the growth of cryo-EM maps and models has followed an exponential trend in the past decade. We therefore anticipate an increasing need for robust validation methods going forward. If current trends continue, cryo-EM will become the dominant structure determination method within the next two decades (Fig. 9B). The development and implementation of rigorous validation methods require the efforts of individual investigators as well as community consensus. For instance, using sophisticated classification of single-particle images, it is not uncommon to have multiple structure models per specimen that one can derive an atomic model per particle (77). The validation report from wwPDB and EMDB has raised the quality of final structures because it raises concerns when structure quality is flagged as below average. Additional work remains to develop more quantitative tools to assess maps at a broad range of resolutions from atomic to nanometer particularly in view of the anticipated improvements in resolution from subtomogram average maps (Fig. 4E). The recent advances in artificial intelligence provide a great opportunity for data scientists using this branch of computational methods for data processing, segmentation, and structure validation. Though cryo-EM/ET are relatively young fields relative to X-ray crystallography, close collaboration between the structural biology and data science community will bring them forward to enable new discoveries in molecular and cellular structure biology. This integrative approach will ultimately benefit human health, as evidenced by the rapid accumulation of structures of coronavirus spike and other related proteins during the SARS-CoV-2/COVID-19 pandemic.
Figure 9.

Current and predicted growth of cryo-EM in the EMDB and PDB archives.A, cumulative statistics for maps in EMDB and EM coordinate entries in PDB, by year, (B) predicted growth of EMDB plus EM, X-ray, and NMR methods in PDB through 2040, based on simple exponential fits of 2010 to 2020 growth statistics.
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgments
This research has been supported by National Institutes of Health (R01GM079429). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Drs Helen Berman, Ardan Patwardhan, Gerard Kleywegt, and Kim Henrick for their continuous support, collaboration, and advice.
Author contributions
W. C. conceived the initial manuscript content and collaborated with C. L. L. to produce a draft which was refined by all authors. M. F. S., G. P., and C. L. L. prepared the figures and tables.
Biography

Wah Chiu, Wallenberg-Bienenstock Chair Professor in the Department of Bioengineering, Department of Microbiology and Immunology, and the Division of CryoEM and Bioimaging, Stanford Synchrotron Radiation Lightsource of SLAC National Accelerator Laboratory at Stanford University, is a pioneer in the use of cryo-electron microscopy to determine the structures of biological molecules.
Edited by Wolfgang Peti
References
- 1.Glaeser R.M. Limitations to significant information in biological electron microscopy as a result of radiation damage. J. Ultrastruct. Res. 1971;36:466–482. doi: 10.1016/s0022-5320(71)80118-1. [DOI] [PubMed] [Google Scholar]
- 2.Baumeister W., Hahn M., Seredynski J., Herbertz L.M. Radiation damage of proteins in the solid state: Changes of amino acid composition in catalase. Ultramicroscopy. 1976;1:377–382. doi: 10.1016/0304-3991(76)90052-8. [DOI] [PubMed] [Google Scholar]
- 3.Kuo I.A., Glaeser R.M. Development of methodology for low exposure, high resolution electron microscopy of biological specimens. Ultramicroscopy. 1975;1:53–66. doi: 10.1016/s0304-3991(75)80007-6. [DOI] [PubMed] [Google Scholar]
- 4.Saxton W.O., Frank J. Motif detection in quantum noise-limited electron micrographs by cross-correlation. Ultramicroscopy. 1977;2:219–227. doi: 10.1016/s0304-3991(76)91385-1. [DOI] [PubMed] [Google Scholar]
- 5.Unwin P.N.T., Henderson R. Molecular structure determination by electron microscopy of unstained crystalline specimens. J. Mol. Biol. 1975;94:425–440. doi: 10.1016/0022-2836(75)90212-0. [DOI] [PubMed] [Google Scholar]
- 6.Hayward S.B., Stroud R.M. Projected structure of purple membrane determined to 3.7 A resolution by low temperature electron microscopy. J. Mol. Biol. 1981;151:491–517. doi: 10.1016/0022-2836(81)90007-3. [DOI] [PubMed] [Google Scholar]
- 7.Jeng T.W., Chiu W. Low dose electron microscopy of the crotoxin complex thin crystal. J. Mol. Biol. 1983;164:329–346. doi: 10.1016/0022-2836(83)90080-3. [DOI] [PubMed] [Google Scholar]
- 8.Taylor K.A., Glaeser R.M. Electron diffraction of frozen, hydrated protein crystals. Science. 1974;186:1036–1037. doi: 10.1126/science.186.4168.1036. [DOI] [PubMed] [Google Scholar]
- 9.Dubochet J., McDowall A.W. Vitrification of pure water for electron microscopy. J. Microsc. 1981;124:3–4. [Google Scholar]
- 10.Dubochet J., Lepault J., Freeman R., Berriman J.A., Homo J.-C. Electron microscopy of frozen water and aqueous solutions. J. Microsc. 1982;128:219–237. [Google Scholar]
- 11.Lepault J., Booy F.P., Dubochet J. Electron microscopy of frozen biological suspensions. J. Microsc. 1983;129:89–102. doi: 10.1111/j.1365-2818.1983.tb04163.x. [DOI] [PubMed] [Google Scholar]
- 12.Jeng T.-W., Chiu W., Zemlin F., Zeitler E. Electron imaging of crotoxin complex thin crystal at 3.5 Å. J. Mol. Biol. 1984;175:93–97. doi: 10.1016/0022-2836(84)90449-2. [DOI] [PubMed] [Google Scholar]
- 13.Henderson R., Baldwin J.M., Downing K.H., Lepault J., Zemlin F. Structure of purple membrane from halobacterium halobium: Recording, measurement and evaluation of electron micrographs at 3.5 Å resolution. Ultramicroscopy. 1986;19:147–178. [Google Scholar]
- 14.Kühlbrandt W., Wang D.N., Fujiyoshi Y. Atomic model of plant light-harvesting complex by electron crystallography. Nature. 1994;367:614–621. doi: 10.1038/367614a0. [DOI] [PubMed] [Google Scholar]
- 15.Nogales E., Wolf S.G., Downing K.H. Structure of the alpha beta tubulin dimer by electron crystallography. Nature. 1998;391:199–203. doi: 10.1038/34465. [DOI] [PubMed] [Google Scholar]
- 16.Murata K., Mitsuoka K., Hirai T., Walz T., Agre P., Heymann J.B., Engel A., Fujiyoshi Y. Structural determinants of water permeation through aquaporin-1. Nature. 2000;407:599–605. doi: 10.1038/35036519. [DOI] [PubMed] [Google Scholar]
- 17.Henderson R., Glaeser R.M. Quantitative analysis of image contrast in electron micrographs of beam-sensitive crystals. Ultramicroscopy. 1985;16:139–150. [Google Scholar]
- 18.Adrian M., Dubochet J., Lepault J., McDowall A.W. Cryo-electron microscopy of viruses. Nature. 1984;308:32–36. doi: 10.1038/308032a0. [DOI] [PubMed] [Google Scholar]
- 19.Talmon Y., Prasad B.V., Clerx J.P., Wang G.J., Chiu W., Hewlett M.J. Electron microscopy of vitrified-hydrated La Crosse virus. J. Virol. 1987;61:2319–2321. doi: 10.1128/jvi.61.7.2319-2321.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prasad B.V.V., Venkataram Prasad B.V., Wang G.J., Clerx J.P.M., Chiu W. Three-dimensional structure of rotavirus. J. Mol. Biol. 1988;199:269–275. doi: 10.1016/0022-2836(88)90313-0. [DOI] [PubMed] [Google Scholar]
- 21.Orlova E.V., Serysheva I.I., van Heel M., Hamilton S.L., Chiu W. Two structural configurations of the skeletal muscle calcium release channel. Nat. Struct. Mol. Biol. 1996;3:547–552. doi: 10.1038/nsb0696-547. [DOI] [PubMed] [Google Scholar]
- 22.Frank J., Zhu J., Penczek P., Li Y., Srivastava S., Verschoor A., Radermacher M., Grassucci R., Lata R.K., Agrawal R.K. A model of protein synthesis based on cryo-electron microscopy of the E. coli ribosome. Nature. 1995;376:441–444. doi: 10.1038/376441a0. [DOI] [PubMed] [Google Scholar]
- 23.Jeng T.-W., Crowther R.A., Stubbs G., Chiu W. Visualization of alpha-helices in tobacco mosaic virus by cryo-electron microscopy. J. Mol. Biol. 1989;205:251–257. doi: 10.1016/0022-2836(89)90379-3. [DOI] [PubMed] [Google Scholar]
- 24.Unwin N. Nicotinic acetylcholine receptor an 9 Å resolution. J. Mol. Biol. 1993;229:1101–1124. doi: 10.1006/jmbi.1993.1107. [DOI] [PubMed] [Google Scholar]
- 25.Zhou Z.H., Dougherty M., Jakana J., He J., Rixon F.J., Chiu W. Seeing the Herpesvirus capsid at 8.5 Å. Science. 2000;288:877–880. doi: 10.1126/science.288.5467.877. [DOI] [PubMed] [Google Scholar]
- 26.Böttcher B., Wynne S.A., Crowther R.A. Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy. Nature. 1997;386:88–91. doi: 10.1038/386088a0. [DOI] [PubMed] [Google Scholar]
- 27.Conway J.F., Cheng N., Zlotnick A., Wingfield P.T., Stahl S.J., Steven A.C. Visualization of a 4-helix bundle in the hepatitis B virus capsid by cryo-electron microscopy. Nature. 1997;386:91–94. doi: 10.1038/386091a0. [DOI] [PubMed] [Google Scholar]
- 28.Miyazawa A., Fujiyoshi Y., Unwin N. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 2003;423:949–955. doi: 10.1038/nature01748. [DOI] [PubMed] [Google Scholar]
- 29.Henderson R. The potential and limitations of neutrons, electrons and X-rays for atomic resolution microscopy of unstained biological molecules. Q. Rev. Biophys. 1995;28:171–193. doi: 10.1017/s003358350000305x. [DOI] [PubMed] [Google Scholar]
- 30.Henderson R. Realizing the potential of electron cryo-microscopy. Q. Rev. Biophys. 2004;37:3–13. doi: 10.1017/s0033583504003920. [DOI] [PubMed] [Google Scholar]
- 31.Wriggers W., Milligan R.A., Andrew McCammon J. Situs: A package for docking crystal structures into low-resolution maps from electron microscopy. J. Struct. Biol. 1999;125:185–195. doi: 10.1006/jsbi.1998.4080. [DOI] [PubMed] [Google Scholar]
- 32.Rayment I., Holden H.M., Whittaker M., Yohn C.B., Lorenz M., Holmes K.C., Milligan R.A. Structure of the actin-myosin complex and its implications for muscle contraction. Science. 1993;261:58–65. doi: 10.1126/science.8316858. [DOI] [PubMed] [Google Scholar]
- 33.Hewat E.A., Verdaguer N., Fita I., Blakemore W., Brookes S., King A., Newman J., Domingo E., Mateu M.G., Stuart D.I. Structure of the complex of an Fab fragment of a neutralizing antibody with foot-and-mouth disease virus: Positioning of a highly mobile antigenic loop. EMBO J. 1997;16:1492–1500. doi: 10.1093/emboj/16.7.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kolatkar P.R., Bella J., Olson N.H., Bator C.M., Baker T.S., Rossmann M.G. Structural studies of two rhinovirus serotypes complexed with fragments of their cellular receptor. EMBO J. 1999;18:6249–6259. doi: 10.1093/emboj/18.22.6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmid M.F., Sherman M.B., Matsudaira P., Chiu W. Structure of the acrosomal bundle. Nature. 2004;431:104–107. doi: 10.1038/nature02881. [DOI] [PubMed] [Google Scholar]
- 36.Ban N., Freeborn B., Nissen P., Penczek P., Grassucci R.A., Sweet R., Frank J., Moore P.B., Steitz T.A. A 9 Å resolution X-ray crystallographic map of the large ribosomal subunit. Cell. 1998;93:1105–1115. doi: 10.1016/s0092-8674(00)81455-5. [DOI] [PubMed] [Google Scholar]
- 37.Grimes J.M., Jakana J., Ghosh M., Basak A.K., Roy P., Chiu W., Stuart D.I., Prasad B.V.V. An atomic model of the outer layer of the bluetongue virus core derived from X-ray crystallography and electron cryomicroscopy. Structure. 1997;5:885–893. doi: 10.1016/s0969-2126(97)00243-8. [DOI] [PubMed] [Google Scholar]
- 38.Henrick K., Newman R., Tagari M., Chagoyen M. EMDep: A web-based system for the deposition and validation of high-resolution electron microscopy macromolecular structural information. J. Struct. Biol. 2003;144:228–237. doi: 10.1016/j.jsb.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 39.Berman H.M., Lawson C.L., Vallat B., Gabanyi M.J. Anticipating innovations in structural biology. Q. Rev. Biophys. 2018;51 doi: 10.1017/S0033583518000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lawson C.L., Berman H.M., Chiu W. Evolving data standards for cryo-EM structures. Struct. Dyn. 2020;7 doi: 10.1063/1.5138589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lawson C.L., Baker M.L., Best C., Bi C., Dougherty M., Feng P., van Ginkel G., Devkota B., Lagerstedt I., Ludtke S.J., Newman R.H., Oldfield T.J., Rees I., Sahni G., Sala R. EMDataBank.org: Unified data resource for CryoEM. Nucleic Acids Res. 2010;39:D456–D464. doi: 10.1093/nar/gkq880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Henderson R., Sali A., Baker M.L., Carragher B., Devkota B., Downing K.H., Egelman E.H., Feng Z., Frank J., Grigorieff N., Jiang W., Ludtke S.J., Medalia O., Penczek P.A., Rosenthal P.B. Outcome of the first electron microscopy validation task force meeting. Structure. 2012;20:205–214. doi: 10.1016/j.str.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ludtke S.J., Baker M.L., Chen D.-H., Song J.-L., Chuang D.T., Chiu W. De novo backbone trace of GroEL from single particle electron cryomicroscopy. Structure. 2008;16:441–448. doi: 10.1016/j.str.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 44.Jiang W., Baker M.L., Jakana J., Weigele P.R., King J., Chiu W. Backbone structure of the infectious ε15 virus capsid revealed by electron cryomicroscopy. Nature. 2008;451:1130–1134. doi: 10.1038/nature06665. [DOI] [PubMed] [Google Scholar]
- 45.Yu X., Jin L., Zhou Z.H. 3.88 Å structure of cytoplasmic polyhedrosis virus by cryo-electron microscopy. Nature. 2008;453:415–419. doi: 10.1038/nature06893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang X., Settembre E., Xu C., Dormitzer P.R., Bellamy R., Harrison S.C., Grigorieff N. Near-atomic resolution using electron cryomicroscopy and single-particle reconstruction. Proc. Natl. Acad. Sci. U. S. A. 2008;105:1867–1872. doi: 10.1073/pnas.0711623105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McMullan G., Faruqi A.R., Clare D., Henderson R. Comparison of optimal performance at 300keV of three direct electron detectors for use in low dose electron microscopy. Ultramicroscopy. 2014;147:156–163. doi: 10.1016/j.ultramic.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Campbell M.G., Cheng A., Brilot A.F., Moeller A., Lyumkis D., Veesler D., Pan J., Harrison S.C., Potter C.S., Carragher B., Grigorieff N. Movies of ice-embedded particles enhance resolution in electron cryo-microscopy. Structure. 2012;20:1823–1828. doi: 10.1016/j.str.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li X., Mooney P., Zheng S., Booth C.R., Braunfeld M.B., Gubbens S., Agard D.A., Cheng Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Z., Hryc C.F., Bammes B., Afonine P.V., Jakana J., Chen D.-H., Liu X., Baker M.L., Kao C., Ludtke S.J., Schmid M.F., Adams P.D., Chiu W. An atomic model of brome mosaic virus using direct electron detection and real-space optimization. Nat. Commun. 2014;5:4808. doi: 10.1038/ncomms5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuhlbrandt W. The resolution revolution. Science. 2014;343:1443–1444. doi: 10.1126/science.1251652. [DOI] [PubMed] [Google Scholar]
- 52.Scheres S.H.W. Classification of structural heterogeneity by maximum-likelihood methods. Methods Enzymol. 2010;482:295–320. doi: 10.1016/S0076-6879(10)82012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Glaeser R.M. How good can single-particle cryo-EM become? What remains before it approaches its physical limits? Annu. Rev. Biophys. 2019;48:45–61. doi: 10.1146/annurev-biophys-070317-032828. [DOI] [PubMed] [Google Scholar]
- 54.Scheres S.H.W. Processing of structurally heterogeneous cryo-EM data in RELION. Methods Enzymol. 2016;579:125–157. doi: 10.1016/bs.mie.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 55.Punjani A., Rubinstein J.L., Fleet D.J., Brubaker M.A. cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods. 2017;14:290–296. doi: 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 56.Zhang K., Pintilie G.D., Li S., Schmid M.F., Chiu W. Resolving individual atoms of protein complex by cryo-electron microscopy. Cell Res. 2020;30:1136–1139. doi: 10.1038/s41422-020-00432-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakane T., Kotecha A., Sente A., McMullan G., Masiulis S., Brown P.M.G.E., Grigoras I.T., Malinauskaite L., Malinauskas T., Miehling J., Uchański T., Yu L., Karia D., Pechnikova E.V., de Jong E. Single-particle cryo-EM at atomic resolution. Nature. 2020;587:152–156. doi: 10.1038/s41586-020-2829-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yip K.M., Fischer N., Paknia E., Chari A., Stark H. Atomic-resolution protein structure determination by cryo-EM. Nature. 2020;587:157–161. doi: 10.1038/s41586-020-2833-4. [DOI] [PubMed] [Google Scholar]
- 59.Young J.Y., Westbrook J.D., Feng Z., Sala R., Peisach E., Oldfield T.J., Sen S., Gutmanas A., Armstrong D.R., Berrisford J.M., Chen L., Chen M., Di Costanzo L., Dimitropoulos D., Gao G. OneDep: Unified wwPDB system for deposition, biocuration, and validation of macromolecular structures in the PDB archive. Structure. 2017;25:536–545. doi: 10.1016/j.str.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gore S., Sanz García E., Hendrickx P.M.S., Gutmanas A., Westbrook J.D., Yang H., Feng Z., Baskaran K., Berrisford J.M., Hudson B.P., Ikegawa Y., Kobayashi N., Lawson C.L., Mading S., Mak L. Validation of structures in the Protein Data Bank. Structure. 2017;25:1916–1927. doi: 10.1016/j.str.2017.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Iudin A., Korir P.K., Salavert-Torres J., Kleywegt G.J., Patwardhan A. Empiar: A public archive for raw electron microscopy image data. Nat. Methods. 2016;13:387–388. doi: 10.1038/nmeth.3806. [DOI] [PubMed] [Google Scholar]
- 62.Ludtke S.J., Lawson C.L., Kleywegt G.J., Berman H., Chiu W. The 2010 cryo-EM modeling challenge. Biopolymers. 2012;97:651–654. doi: 10.1002/bip.22081. [DOI] [PubMed] [Google Scholar]
- 63.Lawson C.L., Chiu W. Comparing cryo-EM structures. J. Struct. Biol. 2018;204:523–526. doi: 10.1016/j.jsb.2018.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lawson C.L., Kryshtafovych A., Adams P.D., Afonine P.V., Baker M.L., Barad B.A., Bond P., Burnley T., Cao R., Cheng J., Chojnowski G., Cowtan K., Dill K., DiMaio F., Farrell D.P. Outcomes of the 2019 EMDataResource model challenge: Validation of cryo-EM models at near-atomic resolution. Nat. Methods. 2021;18:156–164. doi: 10.1038/s41592-020-01051-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kryshtafovych A., Adams P.D., Lawson C.L., Chiu W. Evaluation system and web infrastructure for the second cryo-EM model challenge. J. Struct. Biol. 2018;204:96–108. doi: 10.1016/j.jsb.2018.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kryshtafovych A., Monastyrskyy B., Adams P.D., Lawson C.L., Chiu W. Distribution of evaluation scores for the models submitted to the second cryo-EM model challenge. Data Brief. 2018;20:1629–1638. doi: 10.1016/j.dib.2018.08.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Williams C.J., Headd J.J., Moriarty N.W., Prisant M.G., Videau L.L., Deis L.N., Verma V., Keedy D.A., Hintze B.J., Chen V.B., Jain S., Lewis S.M., Arendall W.B., 3rd, Snoeyink J., Adams P.D. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018;27:293–315. doi: 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pintilie G., Zhang K., Su Z., Li S., Schmid M.F., Chiu W. Measurement of atom resolvability in cryo-EM maps with Q-scores. Nat. Methods. 2020;17:328–334. doi: 10.1038/s41592-020-0731-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen L.F., Winkler H., Reedy M.K., Reedy M.C., Taylor K.A. Molecular modeling of averaged rigor crossbridges from tomograms of insect flight muscle. J. Struct. Biol. 2002;138:92–104. doi: 10.1016/s1047-8477(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 70.Mahamid J., Pfeffer S., Schaffer M., Villa E., Danev R., Kuhn Cuellar L., Forster F., Hyman A.A., Plitzko J.M., Baumeister W. Visualizing the molecular sociology at the HeLa cell nuclear periphery. Science. 2016;351:969–972. doi: 10.1126/science.aad8857. [DOI] [PubMed] [Google Scholar]
- 71.Dahlberg P.D., Saurabh S., Sartor A.M., Wang J., Mitchell P.G., Chiu W., Shapiro L., Moerner W.E. Cryogenic single-molecule fluorescence annotations for electron tomography reveal in situ organization of key proteins in. Proc. Natl. Acad. Sci. U. S. A. 2020;117:13937–13944. doi: 10.1073/pnas.2001849117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rigort A., Bauerlein F.J.B., Villa E., Eibauer M., Laugks T., Baumeister W., Plitzko J.M. Focused ion beam micromachining of eukaryotic cells for cryoelectron tomography. Proc. Natl. Acad. Sci. U. S. A. 2012;109:4449–4454. doi: 10.1073/pnas.1201333109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu G.-H., Mitchell P.G., Galaz-Montoya J.G., Hecksel C.W., Sontag E.M., Gangadharan V., Marshman J., Mankus D., Bisher M.E., Lytton-Jean A.K.R., Frydman J., Czymmek K., Chiu W. Multi-scale 3D cryo-correlative microscopy for vitrified cells. Structure. 2020;28:1231–1237.e3. doi: 10.1016/j.str.2020.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dai W., Fu C., Raytcheva D., Flanagan J., Khant H.A., Liu X., Rochat R.H., Haase-Pettingell C., Piret J., Ludtke S.J., Nagayama K., Schmid M.F., King J.A., Chiu W. Visualizing virus assembly intermediates inside marine cyanobacteria. Nature. 2013;502:707–710. doi: 10.1038/nature12604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen M., Bell J.M., Shi X., Sun S.Y., Wang Z., Ludtke S.J. A complete data processing workflow for cryo-ET and subtomogram averaging. Nat. Methods. 2019;16:1161–1168. doi: 10.1038/s41592-019-0591-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen M., Dai W., Sun S.Y., Jonasch D., He C.Y., Schmid M.F., Chiu W., Ludtke S.J. Convolutional neural networks for automated annotation of cellular cryo-electron tomograms. Nat. Methods. 2017;14:983–985. doi: 10.1038/nmeth.4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roh S.-H., Hryc C.F., Jeong H.-H., Fei X., Jakana J., Lorimer G.H., Chiu W. Subunit conformational variation within individual GroEL oligomers resolved by Cryo-EM. Proc. Natl. Acad. Sci. U. S. A. 2017;114:8259–8264. doi: 10.1073/pnas.1704725114. [DOI] [PMC free article] [PubMed] [Google Scholar]