

Abstract
To maintain an optimal proteome, both codon choice of each mRNA and supply of aminoacyl-tRNAs are two principal factors in translation. Recent reports have revealed that the amounts of tRNAs in cells are more dynamic than we had expected. High-throughput methods such as RNA-seq and microarrays are versatile for comprehensive detection of changes in individual tRNA amounts, but they suffer from inability to assess signal production efficiencies of individual tRNA species. Thus, they are not the perfect choice to measure absolute amounts of tRNAs. Here, we introduce a novel method for this purpose, termed oligonucleotide-directed three-prime terminal extension of RNA (OTTER), which uses fluorescence-labeling at the 3′ terminus of a tRNA by optimized reverse primer extension and an assessment step of each labeling efficiency by northern blotting. Using this method, we quantified the absolute amounts of the 34 individual and four pairs of isoacceptor tRNAs out of the total 42 nuclear-encoded isoacceptors in the yeast Saccharomyces cerevisiae. We found that the amounts of tRNAs in log phase yeast cells grown in a rich glucose medium range from 0.030 to 0.73 pmol/µg RNA. The tRNA amounts seem to be altered at the isoacceptor level by a few folds in response to physiological growing conditions. The data obtained by OTTER are poorly correlated with those by simple RNA-seq, marginally with those by microarrays and by microscale thermophoresis. However, the OTTER data showed good agreement with the data obtained by 2D-gel analysis of in vivo radiolabeled RNAs. Thus, OTTER is a suitable method for quantifying absolute amounts of tRNAs at the level of isoacceptor resolution.
Keywords: tRNA, quantification, absolute amount
INTRODUCTION
Building of appropriate proteomes under various conditions is an essential task for living organisms. Although a repertoire of mRNAs is crucial for building proteomes, the supply of aminoacyl-tRNAs determined by a tRNA repertoire and replenishment speed of aminoacyl-tRNAs also have pivotal roles. Thus, selective usage of a particular codon among synonymous ones, in other words, selection of a certain isoacceptor tRNA for peptide bond formation, is crucial for optimal translation (Shi and Barna 2015; Brar 2016). Indeed, recent advances in translatome analyses, especially those via ribosome profiling (Ingolia et al. 2009), have revealed a global view that codon selection affects both the efficiency (Shah and Gilchrist 2011; Qian et al. 2012; Goodarzi et al. 2016) and accuracy (Drummond and Wilke 2008; Krisko et al. 2014) of translation. Real-time monitoring of protein folding of nascent chains also has demonstrated that protein folding of a polypeptide encoded by the mRNA is strongly affected by codon selection (Kim et al. 2015; Yu et al. 2015; Brar 2016). Furthermore, recent reports have shown that codon selection determines the stability of mRNAs under certain conditions (Presnyak et al. 2015; Mishima and Tomari 2016; Hia et al. 2019).
Codon optimality has been usually discussed on codon usage of a gene, and preference for a particular codon within the genome of an organism has been thought to parallel the amount of the cognate tRNA (Hanson and Coller 2018; Rak et al. 2018). In eukaryotes, tRNAs are usually encoded by multiple genes to produce sufficient amounts of individual tRNA species. Indeed, tRNAs are the most abundant class of RNAs in terms of molecular number (Kirchner and Ignatova 2015), and it has been thought that the amounts of isodecoder tRNAs, a class of tRNAs with the same anticodon sequence, are mostly determined by the numbers of corresponding genes in the genome, indicating that the tRNA repertoire remains constant throughout the life of an organism.
However, several recent reports have cast a doubt on this assumption. For example, over-expression of initiator or elongator tRNA-Met differently affects tRNA repertoires in human cell lines (Pavon-Eternod et al. 2013). Pilpel's group compared tRNA abundance in B cell lymphoma with that in normal B lymphocytes and found significant variations in the expression level (>10-fold) of certain tRNA species (Gingold et al. 2014). Especially, a pair of isoacceptor tRNAs, tRNA-LysCUU and tRNA-LysUUU, showed opposite expression patterns in the two cell types despite the fact that they decode codons for the same amino acid. Reports of a similar change of tRNA repertoires along with transformation and development have been accumulating in recent years (Schmitt et al. 2014; Goodarzi et al. 2016; Turowski et al. 2016). Overall, these facts indicate that the tRNA repertoire is not a fixed parameter within an organism but is variable according to environmental and developmental conditions.
To understand effects of global and local translational efficiency on various biological aspects, it is essential to know concentrations of individual tRNA species in the cytosol (or in genome-containing organelles) and to obtain a total view of the tRNA repertoire in a cell under specific conditions at the certain time points. The first choice for tRNA repertoire analysis is RNA-sequencing (RNA-seq). However, several steps in the RNA-seq process are strongly affected by the nature of each target. One of such steps is reverse transcription (RT) in the preparation of templates for RNA-seq. Especially in quantification of tRNAs, their heavy modifications and tight secondary structures hamper efficient and even full-length RT despite the fact that tRNA species are only 75–90 nt in length (Pang et al. 2014; Zheng et al. 2015). Thus, precise estimation of tRNA amounts by simple RNA-seq is difficult. It is also true to another RT-based quantification, qRT-PCR (Torrent et al. 2018). Nonetheless, the blockades of RT by base modifications can be positively utilized to identify modified bases in RNA species (Arimbasseri et al. 2015; Hauenschild et al. 2015; Wulff et al. 2017). Several methods have been invented to circumvent the problems associated with RT. One simple way is to utilize only the 3′ region of a tRNA sequence for quantification (Chen and Tanaka 2018). This method can be applied to organisms with simple genomes, such as Saccharomyces cerevisiae. The second choice is usage of RTase with a high tolerance to nucleotide modification (Nottingham et al. 2016). Usage of de-modification enzymes to reverse the modification blockades is the third way (Cozen et al. 2015). In particular, demethylases can be used to remove the methyl moieties on amino groups of nucleobases with certain success (Zheng et al. 2015). A different approach to avoid modification-related problems is to introduce additional sequencing start sites by randomly digesting tRNAs via alkaline treatment, enabling sequencing of more distal regions to the 3′-end of a tRNA molecule (Karaca et al. 2014; Arimbasseri et al. 2015). Another improvement has been introduced into the adaptor ligation step required for RT. Kirino's group reported several approaches that improve the adaptor ligation step, including the use of Y-shaped double-stranded primers instead of stem–loop primers (Honda et al. 2015; Shigematsu et al. 2017).
Another way to measure tRNA quantity is to adopt hybridization. Microarrays have been used to measure differences in the levels of tRNAs between samples (Dittmar et al. 2004, 2006; Gingold et al. 2014). If compared with RNA-seq-based methods, the resolution of tRNA species by microarray analyses is rather low because near-cognate tRNA species with a single nucleotide difference in the region used for probe design can cross-hybridize. On the other hand, there is no need of RT. As an alternative, the stint ligation-based method also utilizes two oligo DNAs that hybridize with adjacent regions on a tRNA molecule followed by ligation of these oligo DNAs. Then, the ligated oligo DNAs, hallmarks of the tRNAs, are subjected to next-generation sequencing for quantification (Goodarzi et al. 2016). In either case, base modifications can affect the hybridization efficiencies of the probes; therefore, in some cases, effective oligonucleotide probes for certain tRNAs cannot be designed based on the sequences of the tRNA genes only.
A more fundamental problem associated with RT-based and hybridization-based high-throughput techniques for tRNA quantification is that it is difficult to estimate the signal generation efficiency of each tRNA species. For example, it is difficult to estimate the individual efficiency of hybridization between a tRNA species and the corresponding probe on a microarray, and the same problem occurs with the RT step in RNA-seq. Therefore, the present data on tRNA quantification can be used to compare expression difference of each tRNA species under different conditions, such as physiological environments, developmental stages, genetical backgrounds, etc. On the other hand, they harbor inherent limitations to measure and compare the absolute levels of individual tRNA species in a strict sense. Thus, we still stay one step behind knowing an absolute amount of each tRNA species in a biological sample with high precision, which is an important parameter to understand biochemistry of translation in vivo.
Here, we describe a novel method, termed Oligonucleotide-directed Three-prime Terminal Extension of RNA, or OTTER, to measure the absolute amount of a tRNA species at an isoacceptor level. Although this method relies on hybridization between the 3′-terminal region of a tRNA species and an oligo DNA that acts as a DNA template, the reaction extends the tRNA molecule and labels it with one fluorescent nucleotide. Thus, by analyzing urea-PAGE and fluorescence scanning, the absolute amount of a tRNA species can be measured, while the modification efficiency can be monitored by northern blotting of the same sample. Using this technique, we determined the levels of 34 individual isoacceptors and four pairs of two isoacceptors out of the 42 isoacceptors in the yeast Saccharomyces cerevisiae.
RESULTS
Development and experimental design of a modern method to quantify tRNAs
For quantification of an absolute amount of a specific tRNA species, we applied reverse primer extension, which was originally reported by Huang and Szostak (1996), to introduce a fluorescent nucleotide instead of a radiolabeled nucleotide directly to the tRNA species (Fig. 1). This method consists of two steps: first, we hybridize an oligo DNA specific to an isodecoder/isoacceptor tRNA with a 5′-extension of 5′-dAdTdTdTdT-3′ (Type 1 oligo DNA; Fig. 1A) to generate a 5′-overhang. As shown in Supplemental Figure S1, the 3′-terminal regions of yeast tRNAs have enough diversity to enable the design of isoacceptor-specific oligo DNAs for almost all the tRNAs. One exception is tRNA-SerCGA and tRNA-SerUGA, whose last 33 nt sequences, including CCA, are the same. When a certain isoacceptor tRNA consists of multiple isodecoders with the different 3′-terminal sequences, we measured the total amount of the isoacceptor tRNA because there are several pairs of isodecoders for an isoacceptor with identical 3′-terminal sequences. In the second step, the tRNA/oligo DNA hybrid is subjected to DNA polymerization reaction using the specific Klenow enzyme that lacks the 3′–5′ exonuclease activity with the tRNA as an RNA primer and the oligo DNA as a template (Fig. 1A). Because the reaction mixture contains only dATP and tetramethylrhodamine-dUTP (TMR-dUTP), the polymerization reaction yields a 5-nt-extended tRNA derivative with a 3′ terminal TMR-dU. The fluorescent tRNA derivative is then detected and quantified by fluoro-scanning with a TMR-labeled synthetic oligo DNA as a standard (Fig. 1B), giving absolute measurement of the amount of the tRNA in the sample. Since the labeled tRNA was elongated by 5 nt, the labeled tRNA and the original tRNA can be separated by urea-PAGE, and the labeling efficiency of each reaction is then determined by northern blotting for the target tRNA (Fig. 1C). Northern blotting is also used to evaluate cross-reactions of a template oligo DNA with noncognate tRNA species harboring homologous 3′-terminal sequences (see below). By using these values for compensation, this procedure can quantify the absolute level of an isoacceptor tRNA, which is difficult when using microarray and RNA-seq analyses. We confirmed linearity and quantitativeness of this procedure using tRNA-PheGAA and tRNA-TrpCCA transcribed in vitro as pure model substrates. Especially, we wanted to verify whether predetermined amounts of the model tRNAs produce fluorescence signals that can be converted into the corresponding RNA amounts from a calibration curve with a TMR-labeled oligo DNA as a standard. As expected, the amounts of tRNAs estimated from the fluorescence signals of TMR-dU that were incorporated into the tRNAs agreed with the amounts of tRNA-PheGAA or tRNA-TrpCCA used in these assays (Fig. 1D). Thus, it was validated that a molecular quantity of a tRNA can be estimated using the standard TMR-oligo DNA in this type of reactions.
FIGURE 1.
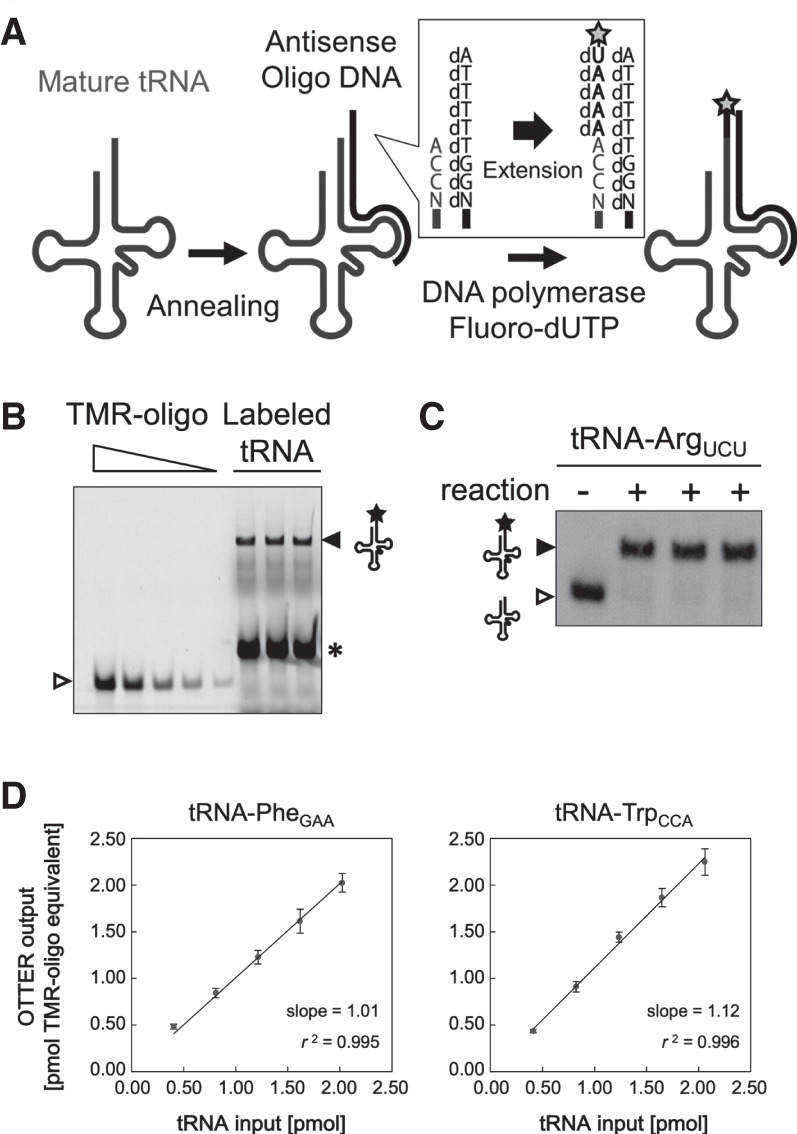
tRNA quantification using OTTER. (A) Schematic drawing of the tRNA fluorescence-labeling reaction in OTTER. A target tRNA is first hybridized with a specifically designed antisense oligo DNA with a 5′-extension of dAdTdTdTdT (Type 1 oligo DNA). The overhang in the tRNA/DNA hybrid is then filled by Klenow fragment (3′–5′ exo−) with dATP and fluorescence-labeled dUTP (such as TMR-dUTP; marked by a star) as substrates. (B) An example for tRNA-ArgUCU quantification using OTTER. Three replicates of a typical reaction were analyzed by urea-PAGE and fluorescence scanning. The fluorescence-labeled tRNA-ArgUCU (closed triangle) and the fluorescence-labeled template oligo DNA generated by a weak RT activity of the Klenow fragment (asterisk) were detected. Since the 3′ end of the oligo DNA hybridized to the TΨC region, which is rather conserved among different tRNA species, unrelated tRNAs can also act as templates to produce the strong RT signal. The standard TMR-oligo DNA (open triangle) was loaded onto the gel at 0.500, 0.250, 0.100, 0.050, and 0.020 pmol/lane. (C) The three OTTER reaction products for tRNA-ArgUCU shown in B (“+” lanes) and a reaction without the template oligo DNA (“−” lane) were subjected to northern blotting. (D) The linearity and quantitativeness of the OTTER procedure were confirmed using in vitro-transcribed tRNAs as pure model substrates. Indicated molecular quantities of tRNA-PheGAA (left) or tRNA-TrpCCA (right) subjected to the OTTER reactions (tRNA input; 0.40, 0.81, 1.21, 1.62, or 2.02 pmol for tRNA-PheGAA, and 0.41, 0.82, 1.24, 1.65, or 2.06 pmol for tRNA-TrpCCA) were analyzed on polyacrylamide gels. The amounts of fluorescence-labeled tRNAs on the gels were determined from a calibration curve of known amounts of the standard TMR-oligo DNA on the same gels and were expressed in “pmol TMR-oligo equivalent.” Each data point is an average of three measurements of dilution series from three independent stocks of the in vitro transcripts, and an error bar indicates the standard deviation.
Since the labeling efficiencies of individual tRNAs under the initial assay conditions with Type 1 oligo DNA templates varied significantly among tRNA species (Fig. 2A), we attempted to refine the assay conditions for each tRNA isoacceptor. During this process, we mainly encountered three problems in certain tRNAs: the first problem is incomplete extension where a part of the tRNAs ceased elongation without receiving the final TMR-dU (Fig. 2A, tRNA-ArgACG, asterisk). The second one is low extension efficiency where a considerable portion of certain tRNAs did not receive any extension (Fig. 2A, tRNA-LysCUU, open triangle in the “+” lane). The third is cross-hybridization of certain templates to near-cognate isoacceptor tRNAs (Fig. 2D). The incomplete extension was solved by redesigning the template oligo DNA to include a dA nucleotide that accepted TMR-dU in the middle part but not at the 5′-end of the template (Type 2 oligo DNA; Fig. 2B). Indeed, the previous study demonstrated that DNA polymerase I interacts with a template strand beyond the site of synthesis (Turner et al. 2003), which may affect the efficiency of incorporation of unnatural deoxynucleotides. Thereafter, the standard template choice was the Type 2 oligo DNA. The low extension efficiency seemed to be caused by inefficient hybridization between the target tRNA and the corresponding template oligo DNA, resulting from the structural rigidity of the tRNA. To deal with this problem, we used an “unfolder” oligo DNA that hybridizes with the anticodon stem–loop and variable loop regions of the target tRNA and destabilizes its overall structure (Buvoli et al. 2000). As shown in Figure 2C, introduction of the corresponding unfolder improved the extension efficiency for tRNA-LysCUU. To solve the cross-hybridization problem, the extent of cross-reactions with near-cognate tRNAs was monitored by northern blotting as described previously, and the results were used to optimize the annealing temperature to reduce cross-hybridization (Fig. 2D). In the case of tRNA-ValCAC, elevating the annealing temperature from 30°C to 52°C suppressed the cross-reaction of the oligo DNA to tRNA-ValUAC. Although the suppression was not complete, the individual amounts of these isoacceptors could be calculated based on the fluorescence signals and labeling efficiencies of the cognate and near-cognate isoacceptors (see Materials and Methods for more details). On the other hand, we could not resolve the quantitative data for three pairs of isoacceptors, tRNA-GlnCUG/tRNA-GlnUUG, tRNA-GluCUC/tRNA-GluUUC, and tRNA-ProAGG/tRNA-ProUGG. In addition, as described above, tRNA-SerCGA and tRNA-SerUAG cannot be resolved from their homology using our method. Therefore, we had no choice but to measure the tRNA amounts of these four pairs of tRNAs as the sum of the two isoacceptors. Overall, we were able to develop a set of optimized measurement conditions for 34 individual and four pairs of isoacceptor tRNAs out of the total 42 isoacceptors of S. cerevisiae. The final assay conditions are summarized in Supplemental Figure S3 and Supplemental Table S2. As shown in Supplemental Figure S3B, almost satisfactory, if not complete, reverse primer extensions were demonstrated by northern analyses under the optimized conditions for individual tRNAs summarized in Supplemental Figure S3A. We named this method as oligonucleotide-directed three-prime terminal extension of RNA, OTTER in short.
FIGURE 2.
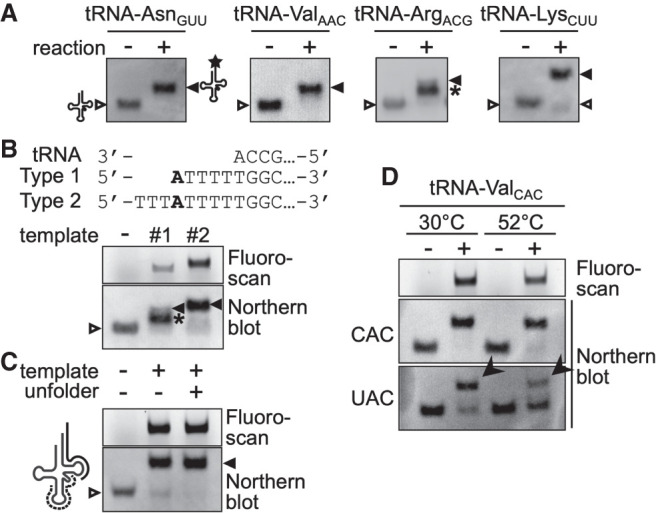
Optimization of OTTER reactions. (A) Northern blotting analyses of OTTER reactions of tRNA-AsnGUU, tRNA-ValAAC, tRNA-ArgACG, and tRNA-LysCUU to elucidate their labeling efficiencies. OTTER reactions were performed in the absence (−) or presence (+) of the corresponding Type 1 oligo DNA templates. Unextended tRNAs (open triangle), fully labeled species (closed triangle), and reaction intermediates (asterisk) are indicated. (B) A representative OTTER reaction with internal labeling. The full sequence of each oligo DNA is shown in Supplemental Table S1. OTTER reactions of tRNA-ArgACG with a Type1 template (#1), a Type 2 template (#2), or no template (−) were separated in an 8% polyacrylamide gel and were detected by fluorescence scanning (upper) and by northern blotting (lower). (C) The effect of the unfolder oligo DNA on OTTER reactions. An unfolder oligo DNA (dashed line in the schematic drawing on the left; Supplemental Table S1) was designed to be complementary to the anticodon stem–loop and variable loop regions of tRNA-LysCUU. OTTER reactions in the absence (−) or presence (+) of the unfolder were subjected to northern blotting with a probe for tRNA-LysCUU. (D) The effect of different annealing temperatures on OTTER reactions. OTTER reactions with a template oligo DNA for tRNA-ValCAC were subjected to fluorescence scanning (top) and to northern blotting with probes for tRNA-ValCAC (middle) and for tRNA-ValUAC (bottom). The bands marked by arrowheads correspond to tRNA-ValUAC cross-reacted to the tRNA-ValCAC template.
The following description is the standard procedure of OTTER after optimization (see also Supplemental Fig. S2 for the timeline of the OTTER assay and Supplemental Table S2 for the individual conditions): For the reaction preparation (the phase P1 in Supplemental Fig. S2B), a 2.0 µg aliquot of RNA is mixed with a specific Type 2 oligo DNA for a tRNA species that is to be analyzed and a corresponding unfolder oligo DNA if necessary in the assay buffer on ice. The mixtures are incubated at 94°C for 3 min and then cooled gradually to 30°C for 90 min to allow annealing. Some tRNA species require different annealing conditions, such as gradual cooling to 52°C. After transferring the reactions on ice, final 10 µM TMR-dUTP, 250 µM dATP and 10 mM MgCl2, and 2 units of Klenow enzyme (exo−) are added (the phase P2 in Supplemental Fig. S2B). Subsequently, the mixtures are incubated at 37°C for 90 min and then quickly mixed with EDTA solution to stop the reaction. Completion of a set of OTTER reactions including mixture preparation takes ∼220 min. After the reaction, the samples are subjected to gel electrophoresis and signal detection with a fluoro-scanner, which takes approximately 1.5 h. Therefore, the whole OTTER procedure can be completed in 5 h.
Quantification of tRNAs in yeast cells under several growth conditions
We applied OTTER to measure the abundances of tRNAs in our standard wild-type yeast strain BY418, grown under several laboratory conditions. First, we measured the amounts of tRNAs in RNA samples prepared from cells in the logarithmic growth phase in a rich glucose medium, YPD. As shown in Figure 3 and Table 1, the amounts of tRNAs in the log phase cells range from 0.030 ± 0.002 pmol/µg RNA to 0.728 ± 0.044 pmol/µg RNA (24-fold difference). The least abundant isoacceptor was tRNA-LeuGAG, which is encoded by a single gene, whereas the most abundant one was tRNA-AspGUC encoded by 16 synonymous genes. The total amount of the yeast tRNAs was 9.11 ± 0.56 pmol/µg RNA, which accounts for ∼20% of the total RNA in weight.
FIGURE 3.
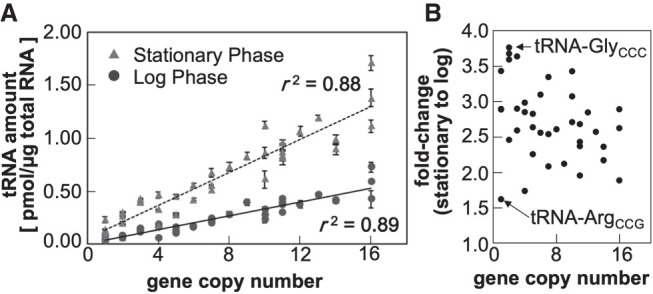
The tRNA repertoires of yeast cells grown in the rich glucose medium. (A) The absolute amounts of tRNAs in RNA samples extracted from yeast cells growing to the log phase (circles) or stationary phase (triangles) were measured by OTTER. Each data set represents the average of three biological replicates, and the error bars indicate the standard deviations. The tRNA gene copy numbers were obtained from Genomic tRNA Database (GtRNAdb) (Chan and Lowe 2016). (B) Fold-changes in tRNA abundances between the stationary phase and log phase cells. The tRNA species with the smallest change (tRNA-ArgCCG) and the largest change (tRNA-GlyCCC) are indicated.
TABLE 1.
Summary of tRNA measurements in S. cerevisiae strain BY418 using OTTER

The absolute amounts of tRNAs in the log phase yeast cells determined using OTTER were far more closely related to the number of synonymous genes than we had expected, with a correlation coefficient (r2) of 0.89 (Fig. 3A). On the other hand, the tRNA amounts per gene varied between 0.016 pmol/µg RNA/gene for tRNA-CysGCA and 0.080 pmol/µg RNA/gene for tRNA-ThrCGU, with an average of 0.035 pmol/µg RNA/gene in the log phase yeast, indicating an approximately fivefold difference in tRNA expression per gene.
Then, we compared the tRNA amounts of yeast cells under different growth conditions. RNA samples were prepared from yeast cells growing in the rich medium with a nonfermentable carbon source, glycerol (YPGly), and those in the synthetic glucose medium (SD). We took RNA samples from cells both in the logarithmic growth and stationary phases to see the effect of growth phase progression. The results are summarized in Table 1. First, we looked closer at effects of the growth phase in the YPD culture. As shown in Figure 3A, the tRNA amounts per µg of total RNA were substantially higher in the stationary phase than in the log phase. As seen in the log phase cells, the amount and the gene copy number of a tRNA are well-correlated in the stationary phase cells (Fig. 3A; r2 = 0.88). The increases in tRNA abundances from the log phase to the stationary phase varied from 1.6-fold (tRNA-ArgCCG) to 3.8-fold increase (tRNA-GlyCCC) with an average increase of 2.7-fold (Table 1; Fig. 3B). This observation directly shows that the amounts of tRNAs change according to the cellular proliferative states and that the amount of each isoacceptor tRNA is somehow altered individually within the range of a few folds. Similar increases in the tRNA amounts in the stationary phase were also observed when the cells were cultured in YPGly; 2.3-fold increase on average (ranging from 1.4-fold for tRNA-Metelongator to 3.3-fold for tRNA-HisGUG). Notably, this growth phase-related increase in the tRNA abundance was not obvious when the yeast cells were grown in the SD medium; however, the absolute amounts of tRNAs in log-phase cells cultured in SD were comparable to those cultured in YPD or YPGly (Table 1).
Next, we investigated differences among tRNA abundances in log-phase cells cultured in the three different media. As shown in Figure 4A, no extraordinary difference was seen among these three medium conditions including the total amounts of tRNAs (9.11, 8.37, and 9.02 pmol/µg RNA in YPD, YPGly, and SD, respectively). However, when investigated in detail, some characteristic differences among the tRNA repertoires of the three groups were revealed. First, we compared the total amounts of tRNAs for a certain amino acid. The amount of tRNA-Asp, which consists of a single isoacceptor tRNA-AspGUC, was approximately twofold higher in cells grown in the YPD medium than in those grown in YPGly or SD medium, whereas the abundances of most other tRNA species differed by approximately ±15% between the groups (Table 1). A similar, although milder, variation was seen for tRNA-Glu, consisting of tRNA-GluCUC and tRNA-GluUUC isoacceptors. On the other hand, tRNA-Trp, with a single tRNA-TrpCCA isoacceptor, behaved oppositely; specifically, the level of tRNA-TrpCCA in cells grown in the YPGly medium was approximately 1.8-fold and 1.4-fold higher than that in cells grown in the YPD and SD media, respectively. Even in the isoacceptor level, there were some interesting differences. For example, tRNA-Leu comprises one minor isoacceptor, tRNA-LeuGAG, and three more abundant isoacceptors, tRNA-LeuCAA, tRNA-LeuUAA and tRNA-LeuUAG (Fig. 4B, left). When comparing the tRNA mounts in the YPD-grown cells with those in the YPGly-grown cells, the levels of the three abundant isoacceptors were slightly lower in the latter cells (72%–92% of those in the YPD-grown cells), as was the total amount of tRNA-Leu (0.82 pmol/µg RNA in YPD vs. 0.69 pmol/µg RNA in YPGly). In contrast, the level of the minor isoacceptor tRNA-LeuGAG was almost twofold higher in cells grown in YPGly than that in cells grown in YPD. Thus, the isoacceptor ratio of tRNA-LeuGAG in the Leu-charged tRNAs increased from 3.6% in the YPD cells to 8.6% in the YPGly cells. Similar counteractive differences between major and minor isoacceptors were also observed for other tRNAs including tRNA-Thr (tRNA-ThrAGU and tRNA-ThrUGU vs tRNA-ThrCGU) and tRNA-Val (tRNA-ValAAC vs tRNA-ValUAC) (Fig. 4B, center and right). And any of these cases allow an approximately twofold increase in the contribution of the minor isoacceptor tRNAs to decoding of the particular amino acid under the respiratory conditions.
FIGURE 4.

Comparisons of tRNA amounts in the yeast cells growing in different media. (A) Comparison of tRNA amounts in the RNA samples prepared from log phase cells grown in YPD (top), YPGly (middle), and SD (bottom). The width of each bar represents the absolute amount of the indicated isoacceptor tRNA. (B) Alteration of isoacceptor contents in yeast cells grown in different carbon sources. The tRNA amounts of individual isoacceptors for tRNA-Leu (left), tRNA-Thr (center), and tRNA-Val (right) in yeast cells grown either in YPD or in YPGly are shown as bar graphs.
This was also examined by northern blotting, a gold standard of RNA quantification. As shown in Figure 5A, northern blotting also confirmed an approximately twofold increase of tRNA-LeuGAG between the YPGly-grown cells and the YPD-grown cells. Figure 5B summarizes variation of each tRNA-Leu isoacceptor level between the two medium conditions measured by northern blotting (light gray bars) and OTTER (dark gray bars). Both techniques demonstrated an apparent decrease of tRNA-LeuCAA in YPGly and a slight decrease of tRNA-LeuUAA in addition to the significant increase of tRNA-LeuGAG.
FIGURE 5.
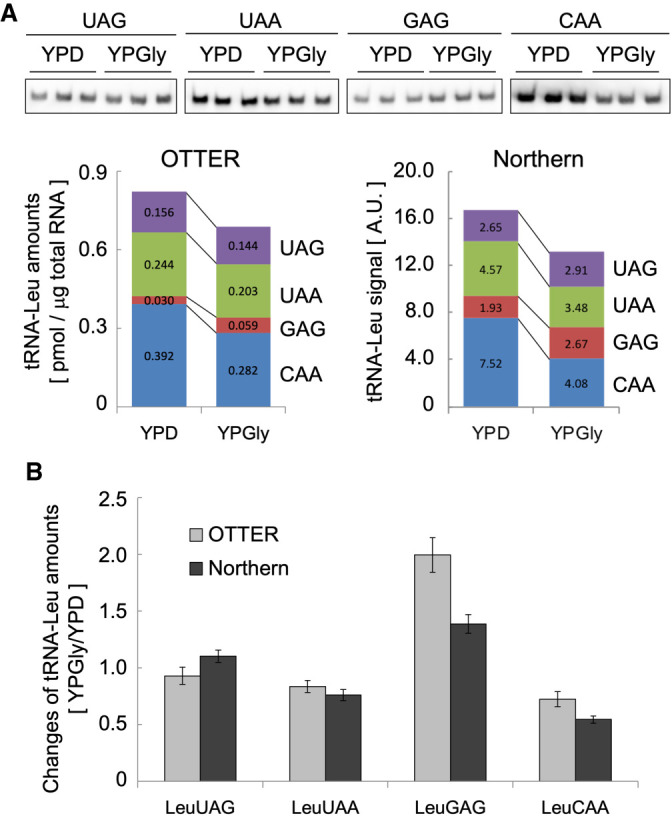
Comparison of tRNA quantification in yeast using OTTER and northern blotting. (A) The upper panel shows northern blotting analyses of tRNA-Leu isoacceptors in yeast cells grown in YPD or YPGly. Three replicates (0.50 µg of total RNA per lane) were hybridized with the probe shown on the top. The sequences of the probes are shown in Supplemental Table S1. The lower panel shows a quantification analysis of the northern blotting and a comparison of the tRNA-Leu isoacceptor amounts obtained from northern blotting and OTTER. (B) Changes in the amounts of the indicated tRNA-Leu isoacceptors in the YPGly-grown cells normalized by those in the YPD-grown cells. Error bar: standard deviation.
In summary, the data presented above demonstrated that tRNA repertoires are varied both in the amino acid isoform level and in the isoacceptor level in response to carbon sources. The range of variation in tRNA amounts was ∼40%–200% of the tRNA amounts in the YPD-grown cells.
Comparison of OTTER and other tRNA quantification methods
Finally, we compared our tRNA quantitation results with other tRNA quantification data obtained by different methods (Supplemental Table S3; Fig. 6). First, we took two early cases, simple RNA-seq analysis by Dedon's group (Pang et al. 2014) and a microarray analysis by Pilpel's group (Tuller et al. 2010) as high-throughput tRNA quantification studies. Essentially, there was no correlation between the tRNA amounts determined by OTTER and the RNA-seq reads (Fig. 6A; r2 = 0.03), and only a moderate correlation between the OTTER data and the microarray data (Fig. 6B; r2 = 0.44). Since both OTTER and microarray analyses rely on hybridization but not on RT, the big difference between the OTTER and the RNA-seq analyses probably comes from uneven cDNA synthesis among different tRNA species caused by nucleotide modifications and/or the tight secondary structures of tRNAs. Indeed, the correlation between the RNA-seq data and the microarray data was also very low (r2 = 0.002). Various methods have been integrated into the RNA-seq technique, such as demethylation of modified nucleobases, to improve the read counts of heavily modified RNAs. Indeed, the RNA-seq data reported by Cozen et al. (2015), which introduced improvements including the ARM-seq procedure using AlkB demethylase before RT, showed a higher correlation (r2 = 0.29) with the OTTER data than the RNA-seq data from Dedon's group (Fig. 6C). However, no significant differences were observed after demethylation of the tRNAs by AlkB, suggesting that other modifications may perturb uniform RT of tRNAs more strongly, although the improvement of the read counts enhanced comparison accuracy of tRNA levels in different samples. Recently, a unique approach to quantify tRNAs using microscale thermophoresis (MST) was reported (Jacob et al. 2019). MST also relies on hybridization between a tRNA and a corresponding DNA probe but measures the thermal motion of the probe under the microscope to yield a binding curve of the tRNA–DNA probe hybrid. While the correlation coefficient between the OTTER data and those obtained by MST was 0.49 (Fig. 6D), this value seemed to be strongly affected by a few outliners, such as tRNA-AspGUC, tRNA-ValAAC, and tRNA-ThrAGU. When these three data were omitted, a higher correlation coefficient was obtained (r2 = 0.74). Thus, the MST seems to produce results that are most consistent with our OTTER results among the techniques for tRNA quantification discussed above.
FIGURE 6.
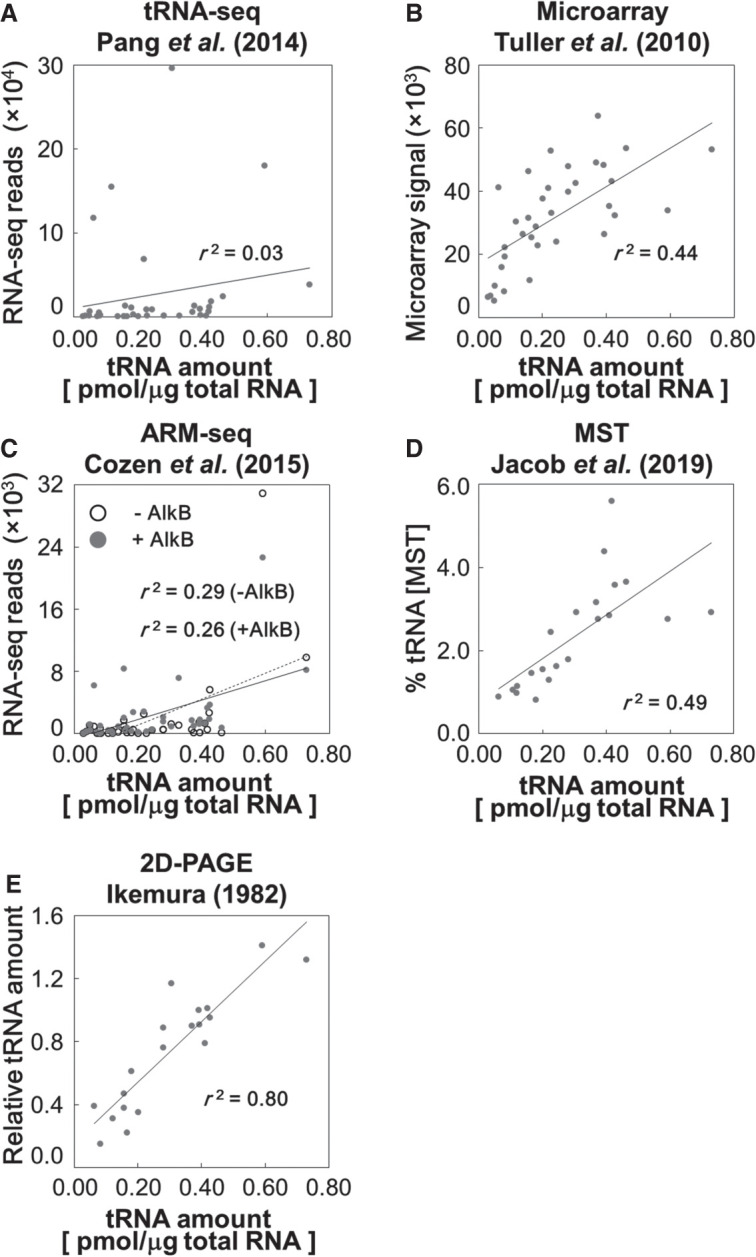
Comparisons of OTTER and other tRNA quantification methods. The absolute amounts of isoacceptor tRNAs in the RNA samples from log phase cells cultured in the glucose medium were measured by OTTER, and the results were compared with the relative tRNA levels in the previously reported simple tRNA-seq data (Pang et al. 2014) (A), the microarray data (Tuller et al. 2010) (B), the ARM-seq data (Cozen et al. 2015) (C), the MST data (Jacob et al. 2019) (D), and the 2D-PAGE data (Ikemura 1982) (E).
We did one more comparison of the OTTER data with those in the report by Ikemura (1982), where RNA samples prepared from 32P-labeled yeast were separated using 2D-urea PAGE and measured radioactivities of identified tRNA spots. Although this method only resolved 21 out of the 42 total tRNA species (one of the 20 tRNA spots on the 2D gel was a mixture of tRNA-SerCGA and tRNA-SerUGA), the quantification principle is quite simple, and has the least compromises and complexities in its experimental/data processing procedures among the methods analyzed here, including OTTER. As shown in Figure 6E, the results of the OTTER analysis showed high correlation (r2 = 0.80) to those of the 2D-PAGE method. Correlations of 2D gel's data to those of simple RNA-seq, ARM-seq, microarray, and MST analyses are 0.19, 0.22, 0.23, and 0.36, respectively, and the results obtained by OTTER show the best correlation with those obtained using the 2D-PAGE data among the five. Thus, these results indicate that OTTER is the most suitable method to compare the amounts of different tRNA species.
DISCUSSION
A modern method for tRNA quantification, OTTER; its pros and cons
Here, we described a method for quantifying absolute amounts of tRNAs, OTTER (Fig. 1). This method directly fluorescence-labels a specific tRNA species according to sequence diversity of the 3′ region of tRNAs. Because the OTTER procedure includes the assessment of signal production efficiency and cross-reactivity through northern blotting, it circumvents the problem associated with variations in signal generation among different tRNA species/sample conditions. In our experience, the efficiency of fluorescence-labeling of a certain isoacceptor tRNA did not differ much among RNA samples from yeast cells grown under different physiological conditions. Therefore, a correction coefficient determined using a standard sample, such as an RNA sample from log phase yeast cells grown in the YPD medium, can be applied to other measurements when high precision is not required for estimation of the tRNA repertoire. In this case, northern blotting analysis of each sample can be omitted, as far as cross-reactivity is not a problem. In addition, OTTER can be used to measure the levels of a subset of tRNA species; hence it is also a versatile method for tRNA quantification in various analytical scenarios.
As shown in Figure 6, the quantification data of OTTER showed a good correlation with those obtained by 32P in vivo-labeling and quantitative 2D-gel analysis (r2 = 0.80) (Ikemura 1982), the most straightforward method for tRNA quantification. On the other hand, the OTTER data were poorly correlated with those from the simple RNA-seq analysis (r2 = 0.03) (Pang et al. 2014), partly because of uneven RT caused by variable nucleotide modifications and/or the secondary structural stability of individual tRNAs. This was improved more recent RNA-seq data (r2 = 0.29) (Cozen et al. 2015). The moderate correlation between the OTTER data and the tRNA microarray data (r2 = 0.44) (Tuller et al. 2010) also supports this notion. Although the probes used in the microarray analysis are carefully designed, their hybridization efficiency and specificity cannot be evaluated one-by-one on the array. As shown in Figure 2, the DNA probes for the OTTER analyses described here hybridized to their cognate tRNAs with different efficiencies. In addition, certain tRNA pairs with similar 3′ sequences cannot be resolved (see below for more details) using the OTTER technique despite the fact that such pairs harbor at least 2 nt mismatches in the middle part of the region that hybridizes with the OTTER probes for the noncognate tRNAs. We speculate that microarrays may also suffer from similar problems. Thus, OTTER is superior to RNA-seq and microarrays in terms of its precision and ability to measure the absolute amounts of tRNA species. Another method of tRNA quantification, MST, also relies on hybridization between a target tRNA and the corresponding DNA probe and monitors how much of the probe is in the free or tRNA-bound form based on their thermal motion. The MST data showed a marginal correlation with the OTTER data (r2 = 0.49) with a similar extent to the microarray data. However, this value was increased to 0.74 when three outliners (tRNA-AspGUC, tRNA-ValAAC, and tRNA-ThrAGU) were removed from analysis. In contrast, in the case of the microarray data, we could not identify a limited number of outliners that would affect the correlation coefficient in a similar manner. We need to investigate the reason behind the discrepancy between the MST and OTTER data in these tRNAs. Since both techniques have ways to monitor hybridization efficiency between each tRNA and its probe, we suppose that these two techniques are the primary choices for measuring absolute amounts of tRNAs.
There are several drawbacks to the OTTER method. The first is limitation of tRNA resolution because our method fully relies on the diversity of the 3′ region of tRNAs. Indeed, we cannot resolve yeast tRNA-SerCGA and tRNA-SerUGA in principle. In addition, we could not resolve the GlnCUG/tRNA-GlnUUG, tRNA-GluCUC/tRNA-GluUUC, and tRNA-ProAGG/tRNA-ProUGG pairs in practice, despite the fact that all these tRNA pairs have some sequence differences within this region hybridized with the oligo DNA templates. Thus, RNA-seq and even microarray analyses are superior to the OTTER method in terms of tRNA resolution. The second drawback comes from the fact that no amplification step is integrated into the OTTER method although it is required for precise determination of absolute amounts of tRNAs. Thus, we need a relatively large amount of RNA samples for quantification of a full set of tRNAs. Finally, OTTER is a low throughput method for accurate analysis: it requires one assay per single tRNA species. However, OTTER is not such a time-consuming method if compared to some other methods. It takes about 5 h per a set of assays (Supplemental Fig. S2A) if calibration by northern blotting is not required, whereas RNA-seq analyses take hours to days to obtain data.
Quantification results of yeast tRNAs
Our OTTER analyses revealed that the quantities of tRNA species in log phase yeast cells range from 0.030 to 0.73 pmol/µg RNA, indicating that yeast cells have ∼9 pmol/µg RNA of tRNAs in total (∼20% of total RNA in weight). One yeast cell is estimated to have ∼0.5 pg RNA (Boehlke and Friesen 1975); therefore, the molecular numbers of individual tRNA species can be calculated as 8000–220,000 molecules/cell. Supposing that most of the yeast tRNAs are localized in the cytosol (Sarkar and Hopper 1998; Grosshans et al. 2000), their cytosolic concentrations can be estimated as 0.55–13 µM, based on the average haploid cell volume and the average cytoplasm/cell ratio (Jorgensen et al. 2002; Yamaguchi et al. 2011). These in vivo parameters estimated from our data can be compared with the translation-related numbers that were acquired previously through various biochemical and cell biological studies of yeast. For example, a yeast cell contains approximately 190,000 ribosomes (von der Haar 2008) and more than 500,000 molecules of eEF1A (Norbeck and Blomberg 1997). This means that, for low abundant tRNA species such as tRNA-LeuGAG, the tRNA/ribosome ratio is less than 1/20 while molecular numbers of major tRNAs, such as tRNA-AspGUC, are comparable to that of ribosomes. Therefore, these in vivo estimations support the notion that there is competition between ribosomes for recruiting minor tRNAs to their A site while such competition is quite mild, or perhaps nonexisting, for major tRNAs. Each tRNA species must be charged with its cognate amino acid by aminoacyl-tRNA synthetase (ARS). The affinities of yeast ARSs for the cognate tRNAs have been biochemically determined as ranging from a few 10 to a few 100 nM (for example, 12 nM, 130 nM, and 220 nM for AspRS, TyrRS, and SerRS, respectively) (Eriani et al. 1991; Soma and Himeno 1998). The range of tRNA concentrations estimated from our OTTER data is higher than these numbers, and it is consistent with the fact that most of the tRNA species are almost fully aminoacylated under the normal growth conditions (Zaborske et al. 2009).
The absolute amounts of tRNAs in yeast cells measured using the OTTER method correlated well with the numbers of synonymous genes in the yeast genome. Thus, it is feasible that the amount of a certain tRNA can be estimated roughly from its gene copy number as adopted by many researchers. On the other hand, we noticed that the tRNA amounts can change in response to different physiological conditions. First, the amounts of tRNAs per µg of total RNA increased by two- to threefold when yeast cells entered the stationary phase. This effect was seen in cells grown in YPD or YPGly but not in those grown in SD. Although the exact substance(s) that induces this phenomenon is unknown, we speculate that a shortage of some nutrients, such as nitrogen sources but not carbon sources, causes this increase. This level of increase in the amount of tRNA per µg of total RNA is unlikely to come from up-regulation of tRNA production because both RNA polymerases I and III are repressed in the stationary phase. However, repression of rRNA production precedes that of RNA polymerase III (Clarke et al. 1996), and this effect may contribute to the relative accumulation of tRNAs. In addition, ribosomal RNAs may be degraded under these conditions more efficiently than tRNAs partly via autophagy (Iwama and Ohsumi 2019). Because autophagy in the stationary phase seems to degrade the bulk of the cytosol, tRNAs might somehow avoid autophagy. It is also possible that ribosomes are selectively degraded by so-called ribophagy in the stationary phase (Wyant et al. 2018).
Another important point to note is that individual tRNA species responded differently to the environmental changes. As shown in Figure 3 and Table 1, increases in the amounts of tRNAs during the progression of the growth phase varied from 1.6-fold to 3.8-fold in cells grown in YPD and from 1.4-fold to 2.3-fold in those grown in YPGly, indicating that the amounts of individual tRNA species seem to be differently affected in the range of a few folds. In addition, the major and minor isoacceptors of tRNA-Leu, tRNA-Thr, etc. behave oppositely on change in the growth conditions (YPD vs. YPGly) (Fig. 4), implying a possibility that the major/minor isoacceptor ratio is regulated accordingly to minimize changes in the total amount of isoacceptor tRNAs for one amino acid. Alternatively, these changes may merely come from differences in modifications that might affect the stability or turnover rates of individual tRNAs. Although we still do not know whether such alterations are achieved at the transcriptional level or in the post-transcriptional (degradation) level, the amount of individual tRNAs may be under more complex and sophisticated regulation or on a more minute balance of synthesis and degradation than we expected.
MATERIALS AND METHODS
Yeast cell culture
Saccharomyces cerevisiae BY418 (MATα ade2Δ::hisG his3Δ200 leu2Δ1 lys2Δ202 met15Δ0 trp1Δ63 ura3-52) was grown at 30°C in liquid media YPD (1.0% [w/v] yeast extract, 2.0% [w/v] polypeptone, and 2.0% [w/v] d-glucose), YPGly (1.0% [w/v] yeast extract, 2.0% [w/v] polypeptone, and 2.0% [w/v] glycerol), or SD (0.67% [w/v] yeast nitrogen base without amino acids and 2.0% [w/v] d-glucose) with 20 µg/mL appropriate amino acid and nucleobase supplements. Cell growth was monitored by measuring OD660 using a Miniphoto518R spectrophotometer (Taitec). The cells were harvested at the log phase (OD660 ≈ 0.3) or at the stationary phase by centrifugation at 1610g for 3 min. The cell pellets were then frozen quickly in liquid nitrogen.
Total RNA isolation
Total RNA was extracted from yeast cells with SDS under acidic conditions (Hayashi et al. 2019). Frozen yeast cells were resuspended in AES Buffer (50 mM sodium acetate, pH 5.2, 10 mM EDTA, and 1.0% [w/v] SDS), and was then extracted with acidic phenol chloroform (phenol:chloroform = 5:1, pH 4.5) at 65°C for 10 min with occasional mixing. The water phase was separated by cooling in ice water and subsequent centrifugation. After extraction of the aqueous phase with phenol chloroform (phenol:chloroform = 1:1 with 0. 050% [w/v] 8-oxyquinoline) followed by chloroform extraction, RNAs were precipitated with 2-propanol, and the final pellet was dissolved in TE (10 mM Tris-HCl, pH 7.5, and 1.0 mM EDTA).
Fluorescence-labeling of tRNA
The template oligo DNAs shown in Supplemental Table S1 were synthesized by Eurofins Genomics. Two micrograms of yeast total RNA was incubated with an appropriate oligo DNA template (12.5 pmol) in 8 µL of assay buffer (10 mM Tris-HCl, pH 8.0, 50 mM NaCl, 0.50 mM EDTA, and 1.0 mM DTT) at 94°C for 3 min. The mixture was cooled gradually to 30°C and 52°C in 90 min and 59 min, respectively, and then chilled quickly at 4°C. After adding a final concentration of 10 µM TMR-dUTP (Roche Diagnostics), 250 µM dATP, 10 mM MgCl2, and 2.0 units of Klenow fragment (3′–5′ exo−) (New England Biolabs), a total of 10 µL of the mixture was incubated at 37°C for 90 min. The labeling reaction was stopped by mixing with 0.40 µL of 0.50 M EDTA. Details of the template oligo DNAs, annealing temperatures, and data processing for individual tRNA species are summarized in Supplemental Table S2.
To prepare pure model substrate tRNAs for the OTTER reactions, the mature forms of tRNA-PheGAA and tRNA-TrpCCA were transcribed in vitro from template plasmids pTYE341 harboring T7p::tF(GAA)M and pTYE349 harboring T7p::tW(CCA)G2 (Takano et al. 2015), respectively, using MEGAshortscript T7 Transcription Kit (Thermo Fisher Scientific) according to the manufacturer's instruction. The amounts of the tRNAs after urea-PAGE purification were quantified with Qubit RNA BR Assay Kit (Thermo Fisher Scientific) according to the manufacturer's instruction.
Quantification of fluorescence-labeled tRNAs
RNAs in the assay mixtures were separated by electrophoresis on an 8.0% (w/v) polyacrylamide gel containing 7.0 M urea in TBE buffer. The fluorescence signals of the labeled tRNAs in the gel were detected with a laser scanner, Typhoon FLA-7000 (GE Healthcare), and the data were processed by Image Quant TL software (GE Healthcare). The absolute amount of each tRNA species was determined by using a known concentration of TMR-labeled oligo DNA (Sigma Genosys) as a standard substance.
Northern blotting
For accurate measurements of tRNA abundances, the labeling efficiencies and cross-reactivities to homologous tRNAs of each target tRNA species were assessed by northern blotting. RNAs in the urea-polyacrylamide gel were electrophoretically transferred to a charged nylon membrane, Hybond-N+ (GE Healthcare). A particular tRNA was hybridized with the corresponding antisense oligo DNA probe shown in Supplemental Table S1 labeled with digoxigenin (DIG) using DIG Oligonucleotide Tailing Kit (Roche Diagnostics) in hybridization solution (0.50 M Na2HPO4, 0.34% [v/v] H3PO4, 7.0% [w/v] SDS, and 1.0 mM EDTA, pH 7.0) at 37°C. After stringent washing in 4× SSC with 0.10% (w/v) SDS at 37°C, the hybridized DIG probe was decorated with alkaline phosphatase-conjugated anti-DIG antibodies (Roche Diagnostics) and was detected via CDP-Star chemiluminescence. The signal was captured and analyzed using a cooled CCD camera system, Ez-Capture and imaging analysis software, CS Analyzer 3 (ATTO).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank our current and previous laboratory members for their support and useful discussions. This work was supported by Grants-in-Aid (KAKENHI) for Scientific Research (C) (grant numbers JP17K07289, JP17KT0113, and JP20K06490) from the Japan Society for the Promotion of Science, and by KAKENHI for Scientific Research on Innovative Areas (grant numbers JP17H05672 and JP20H05338) from the Japanese Ministry of Education, Culture, Sports, Science and Technology Japan.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.076489.120.
REFERENCES
- Arimbasseri AG, Blewett NH, Iben JR, Lamichhane TN, Cherkasova V, Hafner M, Maraia RJ. 2015. RNA polymerase III output is functionally linked to tRNA dimethyl-G26 modification. PLoS Genet 11: e1005671. 10.1371/journal.pgen.1005671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehlke KW, Friesen JD. 1975. Cellular content of ribonucleic acid and protein in Saccharomyces cerevisiae as a function of exponential growth rate: calculation of the apparent peptide chain elongation rate. J Bacteriol 121: 429–433. 10.1128/JB.121.2.429-433.1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar GA. 2016. Beyond the triplet code: context cues transform translation. Cell 167: 1681–1692. 10.1016/j.cell.2016.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buvoli A, Buvoli M, Leinwand LA. 2000. Enhanced detection of tRNA isoacceptors by combinatorial oligonucleotide hybridization. RNA 6: 912–918. 10.1017/S1355838200000339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PP, Lowe TM. 2016. GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res 44: D184–D189. 10.1093/nar/gkv1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C-W, Tanaka M. 2018. Genome-wide translation profiling by ribosome-bound tRNA capture. Cell Rep 23: 608–621. 10.1016/j.celrep.2018.03.035 [DOI] [PubMed] [Google Scholar]
- Clarke EM, Peterson CL, Brainard AV, Riggs DL. 1996. Regulation of the RNA polymerase I and III transcription systems in response to growth conditions. J Biol Chem 271: 22189–22195. 10.1074/jbc.271.36.22189 [DOI] [PubMed] [Google Scholar]
- Cozen AE, Quartley E, Holmes AD, Hrabeta-Robinson E, Phizicky EM, Lowe TM. 2015. ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat Methods 12: 879–884. 10.1038/nmeth.3508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmar KA, Mobley EM, Radek AJ, Pan T. 2004. Exploring the regulation of tRNA distribution on the genomic scale. J Mol Biol 337: 31–47. 10.1016/j.jmb.2004.01.024 [DOI] [PubMed] [Google Scholar]
- Dittmar KA, Goodenbour JM, Pan T. 2006. Tissue-specific differences in human transfer RNA expression. PLoS Genet 2: 2107–2115. 10.1371/journal.pgen.0020221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO. 2008. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134: 341–352. 10.1016/j.cell.2008.05.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriani G, Prevost G, Kern D, Vincendon P, Dirheimer G, Gangloff J. 1991. Cytoplasmic aspartyl-tRNA synthetase from Saccharomyces cerevisiae. Study of its functional organisation by deletion analysis. Eur J Biochem 200: 337–343. 10.1111/j.1432-1033.1991.tb16190.x [DOI] [PubMed] [Google Scholar]
- Gingold H, Tehler D, Christoffersen NR, Nielsen MM, Asmar F, Kooistra SM, Christophersen NS, Christensen LL, Borre M, Sørensen KD, et al. 2014. A dual program for translation regulation in cellular proliferation and differentiation. Cell 158: 1281–1292. 10.1016/j.cell.2014.08.011 [DOI] [PubMed] [Google Scholar]
- Goodarzi H, Nguyen HCB, Zhang S, Dill BD, Molina H, Tavazoie SF. 2016. Modulated expression of specific tRNAs drives gene expression and cancer progression. Cell 165: 1416–1427. 10.1016/j.cell.2016.05.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosshans H, Hurt E, Simos G. 2000. An aminoacylation-dependent nuclear tRNA export pathway in yeast. Genes Dev 14: 830–840. [PMC free article] [PubMed] [Google Scholar]
- Hanson G, Coller J. 2018. Codon optimality, bias and usage in translation and mRNA decay. Nat Rev Mol Cell Biol 19: 20–30. 10.1038/nrm.2017.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauenschild R, Tserovski L, Schmid K, Thüring K, Winz M-L, Sharma S, Entian K-D, Wacheul L, Lafontaine DLJ, Anderson J, et al. 2015. The reverse transcription signature of N-1-methyladenosine in RNA-Seq is sequence dependent. Nucleic Acids Res 43: 9950–9964. 10.1093/nar/gkv895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, Mori S, Suzuki T, Suzuki T, Yoshihisa T. 2019. Impact of intron removal from tRNA genes on Saccharomyces cerevisiae. Nucleic Acids Res 47: 5936–5949. 10.1093/nar/gkz270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hia F, Yang SF, Shichino Y, Yoshinaga M, Murakawa Y, Vandenbon A, Fukao A, Fujiwara T, Landthaler M, Natsume T, et al. 2019. Codon bias confers stability to human mRNAs. EMBO Rep 20: e48220. 10.15252/embr.201948220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda S, Shigematsu M, Morichika K, Telonis AG, Kirino Y. 2015. Four-leaf clover qRT-PCR: a convenient method for selective quantification of mature tRNA. RNA Biol 12: 501–508. 10.1080/15476286.2015.1031951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Szostack JW. 1996. A simple method for 3′-labeling of RNA. Nucleic Acids Res 24: 4360–4361. 10.1093/nar/24.21.4360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemura T. 1982. Correlation between the abundance of yeast transfer RNAs and the occurrence of the respective codons in protein genes. Differences in synonymous codon choice patterns of yeast and Escherichia coli with reference to the abundance of isoaccepting transfer RNAs. J Mol Biol 158: 573–597. 10.1016/0022-2836(82)90250-9 [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS. 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223. 10.1126/science.1168978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwama R, Ohsumi Y. 2019. Analysis of autophagy activated during changes in carbon source availability in yeast cells. J Biol Chem 294: 5590–5603. 10.1074/jbc.RA118.005698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob D, Thüring K, Galliot A, Marchand V, Galvanin A, Ciftci A, Scharmann K, Stock M, Roignant J-Y, Leidel SA, et al. 2019. Absolute quantification of noncoding RNA by microscale thermophoresis. Angew Chem Int Ed Engl 58: 9565–9569. 10.1002/anie.201814377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen P, Nishikawa JL, Breitkreutz B-J, Tyers M. 2002. Systematic identification of pathways that couple cell growth and division in yeast. Science 297: 395–400. 10.1126/science.1070850 [DOI] [PubMed] [Google Scholar]
- Karaca E, Weitzer S, Pehlivan D, Shiraishi H, Gogakos T, Hanada T, Jhangiani SN, Wiszniewski W, Withers M, Campbell IM, et al. 2014. Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function. Cell 157: 636–650. 10.1016/j.cell.2014.02.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Yoon JS, Shishido H, Yang Z, Rooney LAA, Barral JM, Skach WR. 2015. Translational tuning optimizes nascent protein folding in cells. Science 348: 444–448. 10.1126/science.aaa3974 [DOI] [PubMed] [Google Scholar]
- Kirchner S, Ignatova Z. 2015. Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat Rev Genet 16: 98–112. 10.1038/nrg3861 [DOI] [PubMed] [Google Scholar]
- Krisko A, Copic T, Gabaldón T, Lehner B, Supek F. 2014. Inferring gene function from evolutionary change in signatures of translation efficiency. Genome Biol 15: R44. 10.1186/gb-2014-15-3-r44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishima Y, Tomari Y. 2016. Codon usage and 3′ UTR length determine maternal mRNA stability in zebrafish. Mol Cell 61: 874–885. 10.1016/j.molcel.2016.02.027 [DOI] [PubMed] [Google Scholar]
- Norbeck J, Blomberg A. 1997. Two-dimensional electrophoretic separation of yeast proteins using a non-linear wide range (pH 3-10) immobilized pH gradient in the first dimension; reproducibility and evidence for isoelectric focusing of alkaline (pI>7) proteins. Yeast 13: 1519–1534. [DOI] [PubMed] [Google Scholar]
- Nottingham RM, Wu DC, Qin Y, Yao J, Hunicke-Smith S, Lambowitz AM. 2016. RNA-seq of human reference RNA samples using a thermostable group II intron reverse transcriptase. RNA 22: 597–613. 10.1261/rna.055558.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang YLJ, Abo R, Levine SS, Dedon PC. 2014. Diverse cell stresses induce unique patterns of tRNA up- and down-regulation: tRNA-seq for quantifying changes in tRNA copy number. Nucleic Acids Res 42: e170. 10.1093/nar/gku945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavon-Eternod M, Gomes S, Rosner MR, Pan T. 2013. Overexpression of initiator methionine tRNA leads to global reprogramming of tRNA expression and increased proliferation in human epithelial cells. RNA 19: 461–466. 10.1261/rna.037507.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presnyak V, Alhusaini N, Chen Y-H, Martin S, Morris N, Kline N, Olson S, Weinberg D, Baker KE, Graveley BR, et al. 2015. Codon optimality is a major determinant of mRNA stability. Cell 160: 1111–1124. 10.1016/j.cell.2015.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W, Yang J-R, Pearson NM, Maclean C, Zhang J. 2012. Balanced codon usage optimizes eukaryotic translational efficiency. PLoS Genet 8: e1002603. 10.1371/journal.pgen.1002603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rak R, Dahan O, Pilpel Y. 2018. Repertoires of tRNAs: the couplers of genomics and proteomics. Annu Rev Cell Dev Biol 34: 239–264. 10.1146/annurev-cellbio-100617-062754 [DOI] [PubMed] [Google Scholar]
- Sarkar S, Hopper AK. 1998. tRNA nuclear export in Saccharomyces cerevisiae: in situ hybridization analysis. Mol Biol Cell 9: 3041–3055. 10.1091/mbc.9.11.3041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt BM, Rudolph KLM, Karagianni P, Fonseca NA, White RJ, Talianidis I, Odom DT, Marioni JC, Kutter C. 2014. High-resolution mapping of transcriptional dynamics across tissue development reveals a stable mRNA-tRNA interface. Genome Res 24: 1797–1807. 10.1101/gr.176784.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah P, Gilchrist MA. 2011. Explaining complex codon usage patterns with selection for translational efficiency, mutation bias, and genetic drift. Proc Natl Acad Sci 108: 10231–10236. 10.1073/pnas.1016719108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Barna M. 2015. Translating the genome in time and apace: specialized ribosomes, RNA regulons, and RNA-binding proteins. Annu Rev Cell Dev Biol 31: 31–54. 10.1146/annurev-cellbio-100814-125346 [DOI] [PubMed] [Google Scholar]
- Shigematsu M, Honda S, Loher P, Telonis AG, Rigoutsos I, Kirino Y. 2017. YAMAT-seq: an efficient method for high-throughput sequencing of mature transfer RNAs. Nucleic Acids Res 45: e70. 10.1093/nar/gkx005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soma A, Himeno H. 1998. Cross-species aminoacylation of tRNA with a long variable arm between Escherichia coli and Saccharomyces cerevisiae. Nucleic Acids Res 26: 4374–4381. 10.1093/nar/26.19.4374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano A, Kajita T, Mochizuki M, Endo T, Yoshihisa T. 2015. Cytosolic Hsp70 and co-chaperones constitute a novel system for tRNA import into the nucleus. eLife 4: e04659. 10.7554/eLife.04659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrent M, Chalancon G, de Groot NS, Wuster A, Madan Babu M. 2018. Cells alter their tRNA abundance to selectively regulate protein synthesis during stress conditions. Sci Signal 11: eaat6409. 10.1126/scisignal.aat6409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuller T, Carmi A, Vestsigian K, Navon S, Dorfan Y, Zaborske J, Pan T, Dahan O, Furman I, Pilpel Y. 2010. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 141: 344–354. 10.1016/j.cell.2010.03.031 [DOI] [PubMed] [Google Scholar]
- Turner RM, Grindley NDF, Joyce CM. 2003. Interaction of DNA polymerase I (Klenow fragment) with the single-stranded template beyond the site of synthesis. Biochemistry 42: 2373–2385. 10.1021/bi026566c [DOI] [PubMed] [Google Scholar]
- Turowski TW, Leśniewska E, Delan-Forino C, Sayou C, Boguta M, Tollervey D. 2016. Global analysis of transcriptionally engaged yeast RNA polymerase III reveals extended tRNA transcripts. Genome Res 26: 933–944. 10.1101/gr.205492.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Haar T. 2008. A quantitative estimation of the global translational activity in logarithmically growing yeast cells. BMC Syst Biol 2: 87. 10.1186/1752-0509-2-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff TF, Argüello RJ, Jordàn MM, Frigolé HR, Hauquier G, Filonava L, Camacho N, Gatti E, Pierre P, Ribas de Pouplana L, et al. 2017. Detection of a subset of posttranscriptional transfer RNA modifications in vivo with a restriction fragment length polymorphism-based method. Biochemistry 56: 4029–4038. 10.1021/acs.biochem.7b00324 [DOI] [PubMed] [Google Scholar]
- Wyant GA, Abu-Remaileh M, Frenkel EM, Laqtom NN, Dharamdasani V, Lewis CA, Chan SH, Heinze I, Ori A, Sabatini DM. 2018. NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science 360: 751–758. 10.1126/science.aar2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Namiki Y, Okada H, Mori Y, Furukawa H, Wang J, Ohkusu M, Kawamoto S. 2011. Structome of Saccharomyces cerevisiae determined by freeze-substitution and serial ultrathin-sectioning electron microscopy. J Electron Microsc 60: 321–335. 10.1093/jmicro/dfr052 [DOI] [PubMed] [Google Scholar]
- Yu C-H, Dang Y, Zhou Z, Wu C, Zhao F, Sachs MS, Liu Y. 2015. Codon usage influences the local rate of translation elongation to regulate co-translational protein folding. Mol Cell 59: 744–754. 10.1016/j.molcel.2015.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborske JM, Narasimhan J, Jiang L, Wek SA, Dittmar KA, Freimoser F, Pan T, Wek RC. 2009. Genome-wide analysis of tRNA charging and activation of the eIF2 kinase Gcn2p. J Biol Chem 284: 25254–25267. 10.1074/jbc.M109.000877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Qin Y, Clark WC, Dai Q, Yi C, He C, Lambowitz AM, Pan T. 2015. Efficient and quantitative high-throughput tRNA sequencing. Nat Methods 12: 835–837. 10.1038/nmeth.3478 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.