

Abstract
Background:
RRx-001, a clinical macrophage-stimulating anti-cancer agent that also produces nitric oxide (NO) was studied in a model of ischemia-reperfusion injury.
Methods:
The production of NO is dependent on the oxygen tension because nitric oxide synthases convert l-arginine to NO and l-citrulline in the presence of O2. Since the P450 enzymes, which metabolize nitrate esters such as nitroglycerin are dependent on oxygen, the generation of ‘exogenous’ NO is also sensitive to alterations in tissue PO2. I/R injury was studied in a hamster chamber window, with compression of the periphery of the window for 1 h to induce ischemia. Animals received RRx-001 (5 mg/kg) 24 h before ischemia and sodium nitrite (10 nmols/kg) was supplemented 10 min after the start of reperfusion. Vessel diameter, blood flow, adherent leukocytes, and functional capillary density were assessed by intravital microscopy at 0.5, 2, and 24 h following the release of the ischemia.
Results:
The results demonstrated that, compared to control, RRx-001 preconditioning increased blood flow and functional capillary density, and preserved tissue viability in the absence of side effects over a sustained time period.
Conclusion:
Thus, RRx-001 may serve as a long-lived protective agent during postsurgical restoration of flow and other ischemia-reperfusion associated conditions, increasing blood flow and functional capillary density as well as preserving tissue viability in the absence of side effects.
Keywords: Ischemia-reperfusion injury, RRx-001, oncology, nitric oxide, functional capillary density
1. Introduction
Ischemia-reperfusion (I/R) is a complex and multifactorial phenomenon encountered in diverse clinical conditions such as stroke, MI, hemorrhage, peripheral vascular disease and surgical interventions such as cardiac bypass, organ transplantation and aneurysm repair. I/R involves restricted blood supply and insufficient delivery of oxygen to cells (ischemia) followed by reinstatement of circulation (reperfusion) during which blood flow overshoots to a peak value (hyperemia) before returning to baseline. The hyperemia is accompanied by a burst in the generation of reactive oxygen species (ROS) as proinflammatory neutrophils infiltrate ischemic tissues to exacerbate the injury [1–4].
Within the first 30 min of reperfusion, the percentage of adherent neutrophils increases, which express pseudopods and adhere to the endothelial wall, thus elevating flow resistance (Figure 1), microvascular protein permeability is enhanced, and the formation of edema occurs [5].
Figure 1.
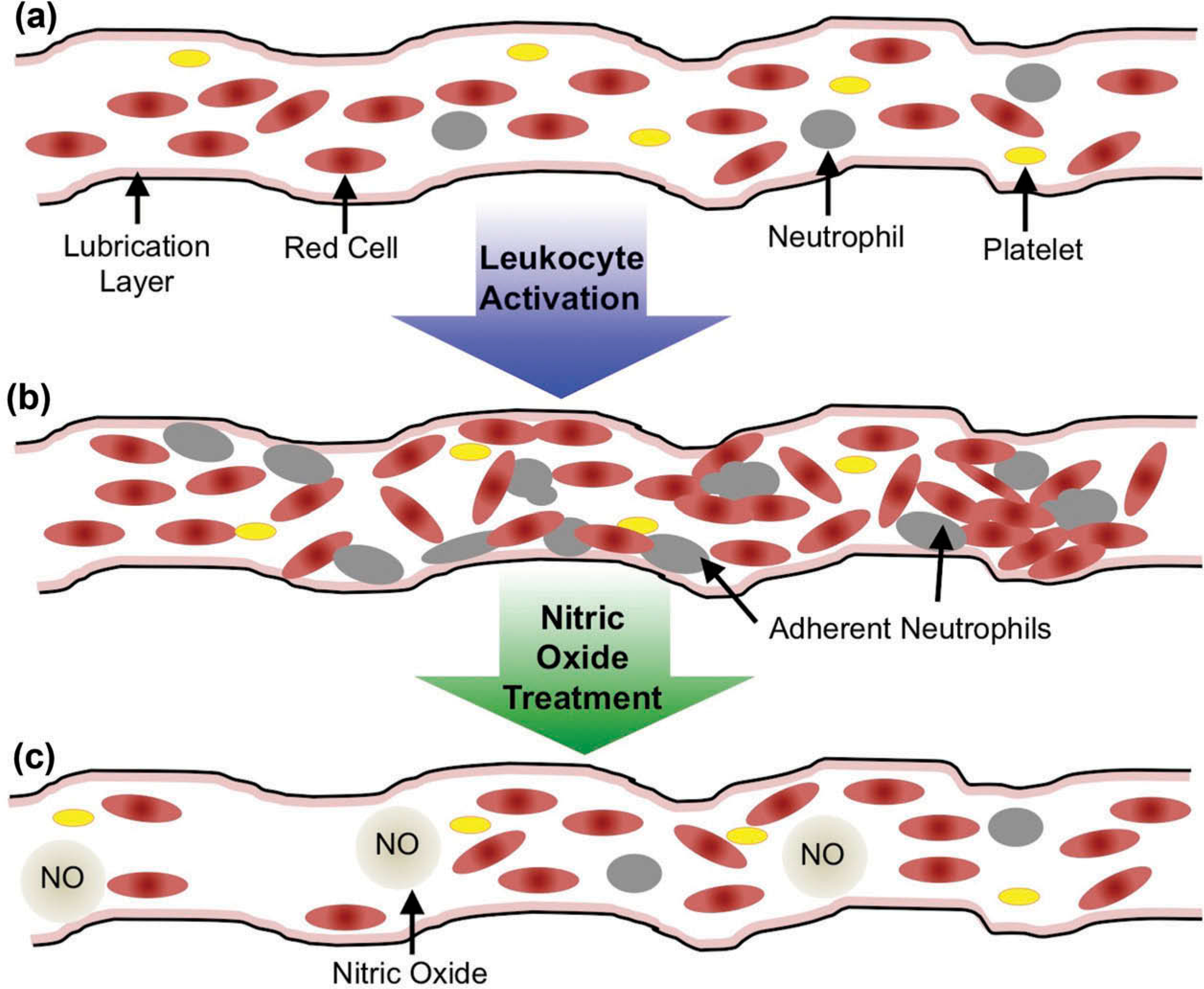
Impact of leukocyte activation on blood flow and rheology in the microcirculation. In ischemia-reperfusion, peripheral resistance is elevated due to leukocyte obstruction due to pseudopod projections or cell adhesion to the vascular wall. (a) Normal Blood Flow. (b) Vessel during ischemia-reperfusion. (c) Vessel during ischemia reperfusion, treated with nitric oxide.
The severity of changes that occur during reperfusion, such as accumulation of reactive oxygen products and leukocytes in postcapillary venules, appears to partially depend on the extent of the ischemic period. These effects have been substantiated in animal models of I/R by intravital fluorescence microscopy [6,7].
Nitric oxide (NO), which typically functions as a paracrine signaling molecule and subserves a central role in vascular biology, including vasodilation, suppression of neutrophil accumulation, reduction of inflammatory cytokines and inhibition of platelet aggregation and adhesion is predominately formed during conversion of L-arginine to L-citrulline by endothelial NO synthase (eNOS) in the presence of molecular O2 [8]. Nitrite, a dietary constituent, serves as an endogenous source of NO [9]. Nitrite reduction to NO may be catalyzed by hemoglobin, myoglobin or other metal-containing enzymes and occurs at increasing rates under conditions of physiologic hypoxia or ischemia [10]. A number of studies have demonstrated nitrite and NO-mediated cytoprotection against focal IR injury of the heart, liver, muscle, brain, and kidney [11–19].
The term preconditioning involves the application of a preceding noninjurious inflammatory stimulus that, overall, transiently results in protection from inflammation [20]. Oxidative stress has emerged as a sine qua non for preconditioning to occur [21].
RRx-001 represents the prototype of a new class of multidrug resistance reversing agents with preferential activity in glioblastoma and refractory small cell lung cancer (SCLC) [22] that substantially increases endogenous nitric oxide only in ischemic tissue [23,24]. RRx-001 binds to hemoglobin (Hb), where it catalyzes the nitrite reductase action of deoxygenated hemoglobin, which metabolizes nitrite to NO at low oxygen concentrations [20,21]. Since reduced NO availability is a hallmark of several hematological diseases, and RRx-001 generates nitric oxide under hypoxia, it has demonstrated beneficial effects on microvascular blood flow and vasoreactivity in pathologies of impaired endothelial function such as sickle cell disease, cerebral malaria and hemorrhagic shock [25–28].
The purpose of this study was to examine the therapeutic potential of RRx-001, as a hypoxic NO donor [29,30], in an I/R model [29]. Our hypothesis was that pretreatment with RRx-001 would protect against post-ischemia vasoconstriction and inflammation through nitric oxide preconditioning and thereby reduce tissue injury.
2. Methods
2.1. Animal preparation
Investigations were performed in 55–65-g male Syrian Golden hamsters (Charles River Laboratories) fitted with a dorsal chamber window. Animal handling and care followed the NIH Guide for the Care and Use of Laboratory Animals and the local animal care committee approved the experimental protocol. The hamster chamber window model is widely used for microvascular studies in non-anesthetized animals, and the complete surgical technique is described in detail elsewhere [31–33]. Briefly, the animal was prepared for chamber implantation with a 50 mg/kg intraperitoneal injection of pentobarbital sodium anesthesia. After hair removal, sutures were used to lift the dorsal skin away from the animal, and one frame of the chamber was positioned on the animal’s back. The chamber consists of two identical titanium frames with a 15-mm circular window (12-mm diameter circular visible field). With the aid of back- lighting and a stereomicroscope, one side of the skinfold was removed, following the outline of the window, until only a thin layer of retractor muscle and the intact subcutaneous skin of the opposing side remained. The cover glass was placed on the exposed skin and held in place by the other frame of the chamber. The intact skin of the other side was exposed to the ambient environment. The animal was allowed at least 2 days for recovery before the preparation was assessed under the microscope for any signs of edema, bleeding, or unusual neovascularization. Barring these complications, the animal was anesthetized again with sodium pentobarbital. Arterial and venous catheters were implanted in the carotid artery (PE-50) and jugular vein (PE-50), respectively, and filled with a heparinized saline solution (30 IU/mL) to ensure patency at the time of experiment. Catheters were tunneled under the skin and exteriorized at the dorsal side of the neck, where they were attached to the chamber frame with tape. 3–4 days after the initial operation, the microvasculature was examined, and only animals meeting established systemic and microcirculatory inclusion criteria, which included having tissue void of low perfusion, inflammation, and edema, were entered into the study [33,34].
2.2. Inclusion criteria
Animals were suitable for the experiments if: (1) systemic parameters were within normal range, namely, heart rate (HR) >340 beat/min, mean arterial blood pressure (MAP) >80 mmHg, systemic hematocrit >45%, and arterial oxygen partial pressure (paO2) >50 mmHg; and, (2) microscopic examination of the tissue in the chamber observed under ×650 magnification did not reveal signs of edema or bleeding.
2.3. I/R protocol
Ischemia was induced for 1 h in the window chamber model by a thin, flat rubber ring (circular clamp) that compressed the periphery of the window (Figure 2) with the awake and non-anesthetized animal restrained in a Plexiglass (Rohm & Haas) tube [35]. Flow cessation was induced by slowly tightening a precision threaded screw fixed to the intact skin side of the window chamber, progressively pressing the rubber ring against the intact skin toward the cover glass. Microvascular flow was continuously monitored under transillumination until it ceased in all feeding and draining microvessels leading in and out of the clamped area without compression injury. The chamber was checked during the clamping period to ensure that ischemia (no flow) was maintained. Animals received RRx-001 (5 mg/kg) 24 h before ischemia with and without sodium nitrite (10 nmols/kg), which was supplemented 10 min after the start of reperfusion.
Figure 2.
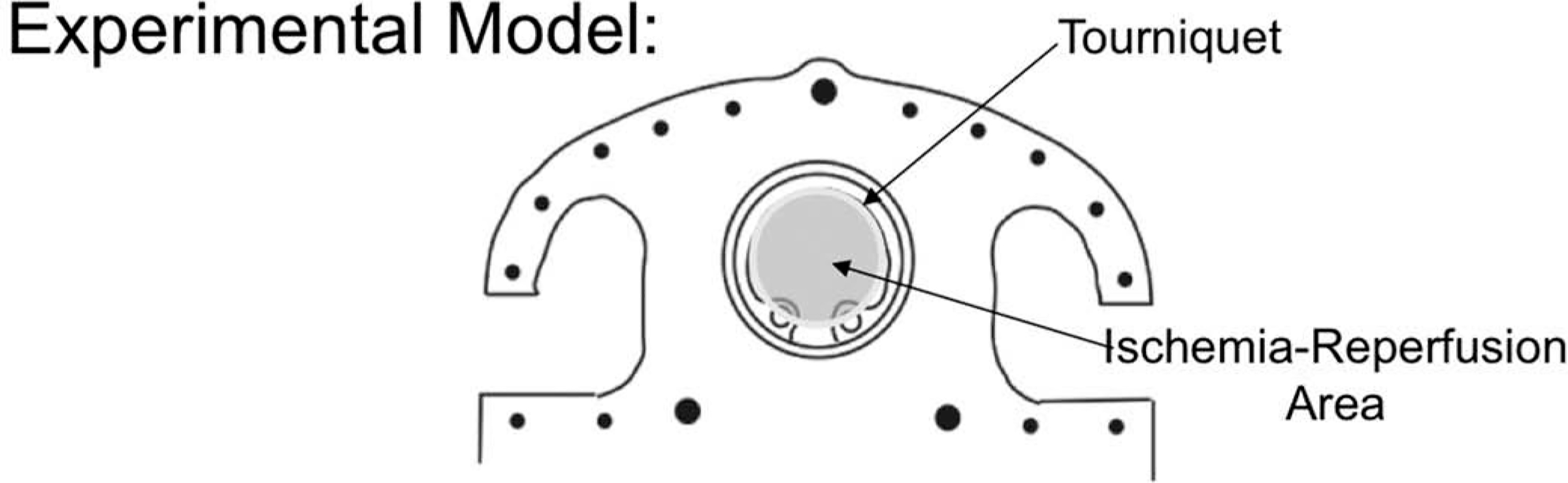
Experimental Model.
2.4. Experimental groups and protocol
Animals were randomly divided into three experimental groups (1) sham (no ischemia) 2) control (saline) and (3) RRx-001. Twelve animals were included in the I/R study, RRx-001 (5 mg/kg; n = 6); and Control (n = 6). Awake hamsters were placed in a restraining tube with a longitudinal slit from which the window chamber protruded. The animals were given 30 min to adjust to the tube environment before baseline systemic parameters (MAP, HR, blood gases, and Hct) were measured. The conscious animal in the tube was then fixed to the microscopic stage of a transillumination intravital microscope (BX51WI, Olympus, New Hyde Park, NY). The tissue image was projected onto a charge-coupled device camera (COHU 4815, San Diego, CA) connected to a videocassette recorder (AG-7355; JVC, Tokyo, Japan) and viewed on a monitor. Measurements were carried out using a 40× (LUMPFL-WIR, numerical aperture 0.8, Olympus) water immersion objective. Detection of RBC passage was enhanced by increasing contrast between RBCs and tissue using a BG12 (420 nm) band pass filter. Systemic and microvascular parameters were analyzed before hemorrhage (baseline), after hemorrhage (shock), and up to 90 min after volume replacement (resuscitation). Tissue viability was measured at 24 after the release of the ischemia. Fields of observation and vessels were chosen for study at locations in the tissue where the vessels were in sharp focus. Leukocyte-endothelium interaction was studied in all vessels included in the study. Detailed mappings were made of the chamber vasculature to record the vessel location and ensure that the same microvessels were studied throughout the experiment.
2.5. Systemic parameters
MAP and HR were recorded continuously (MP 150, Biopac System; Santa Barbara, CA) except during the actual blood exchange. Hematocrit (Hct) was measured from centrifuged arterial blood samples taken in heparinized capillary tubes (Readacrit; Becton-Dickinson, Parsippany, NJ). Hemoglobin (Hb) content was determined spectrophotometrically from a single drop of blood (B-Hemoglobin, Hemocue, Stockholm, Sweden). Arterial blood was collected in heparinized glass capillaries (0.05 ml) and immediately analyzed for PaO2, PaCO2, and pH (Blood Chemistry Analyzer 248, Bayer, Norwood, MA). The comparatively low PaO2 and high PaCO2 of these animals are a consequence of their adaptation to a fossorial or burrowing environment.
2.6. Microvascular measurements
Arteriolar and venular blood flow velocities were measured online by using the photodiode cross-correlation method (Photo Diode/Velocity Tracker Model 102B, Vista Electronics, San Diego, CA) [36]. The measured centerline velocity (V) was corrected according to vessel size to obtain the mean RBC velocity. A video image-shearing method was used to measure vessel diameter (D). Blood flow (Q) was calculated from the measured values as Q = π × V(D/2) [37]. Changes in arteriolar and venular diameter from baseline were used as indicators of a change in vascular tone. This calculation assumes a parabolic velocity profile and has been found to be applicable to tubes of 15–80 μm internal diameters and for Hcts in the range of 6–60%.
2.7. Functional capillary density (FCD)
Functional capillaries, defined as those capillary segments that have RBC transit of at least one RBC in a 60-s period in 10 successive microscopic fields were assessed, totaling a region of 0.46 mm2. Each field had between two and five capillary segments with RBC flow. FCD (cm−1), i.e. total length of RBC perfused capillaries divided by the area of the microscopic field of view, was evaluated by measuring and adding the length of capillaries that had RBC transit in the field of view.
2.8. Leukocyte-endothelium interaction
Intravenous injection of acridine orange (5 mg/kg solution in saline) was used to quantify microvascular leukocytes (adherence and rolling) in postcapillary venules (25–50 μm). Despite numerous examples of light-dye-induced toxicity in the microvasculature, little is known about the relative phototoxicity of commonly used fluorescent conjugates. So, low light fluorescent microscopy was used during leukocyte-endothelium assessment (ORCA 9247, Hamamatsu). Briefly, the straight portion of venule was exposed to low intensity epiillumination for 60 s and digitally video recorded (10 frames/sec). Leukocytes were counted during video playback in a 100-μm length segment and categorized according to their flow behavior as ‘passers,’ including ‘free-flowing’ leukocytes, ‘flowing with endothelial contact,’ and ‘immobilized’ cells [38].
2.9. Tissue viability
Equal volumes of Annexin V (Alexafluor 488 conjugate; Molecular Probes, Eugene, OR) and propidium iodide (PI, 0.2 mg/mL, Molecular Probes) were mixed, and 140 μl of the mixture was injected 30 min before visualization by intravital microscopy. Imaging of labeled cells was performed 8 h after the exchange transfusion. Microscopic images were obtained with a low light video camera (ORCA 9247, Hamamatsu, Tokyo, Japan) and recorded at 5 frames per second and 1344×1024 pixels per frame. Single and double-labeled cells were counted in the skinfold window, and the percentages of cells labeled with annexin V and/or propidium iodide were calculated at different time points. Data is given as the average of fluorescent cells counted in 40 selected visual fields (210 × 160 μm) for the tissue and the endothelial vessel wall separately. Sebaceous glands and hair follicles were identified and excluded from the cell counts due to their consistently high necrosis and apoptosis rate.
3. Results
Twelve animals were included in the I/R study, RRx-001 (5 mg/kg; n = 6); and Control (n = 6).All animals tolerated the experimental protocol without signs of stress or discomfort. Systemic and microvascular parameters of all animals at baseline passed the Grubbs’ test ensuring that all parameters at baseline were within a similar population (P > 0.3).
3.1. Microvascular measurements
We measured the critical microvascular parameters involved in I/R injury (i.e. arteriolar diameter, blood flow, functional capillary density (FCD), leukocyte adhesion).
Changes in arteriolar diameter and blood flow during reperfusion for control and RRx-001 treatment groups are presented in Figure 3. Compared to control, the RRx-001 group displayed an increased arteriolar diameter at 0.5 and 2 h during reperfusion. Similarly, the RRx-001 group displayed increased arteriolar blood flow all time points (e.g. 0.5, 2, and 24 h) during reperfusion compared to control. Arteriolar diameter and blood flow were significantly increased at 30 min (0.5 h) following the release of the ischemia (i.e. the start of reperfusion) compared to control. Supplementation of nitrite (NaNO2, 10ηmols/kg) 10 min after the start of reperfusion did not result in significant differences regarding the microvascular diameter (P = 0.59) and blood flow (P = 0.73). Other parameters were not studied for the nitrite supplementation group.
Figure 3.
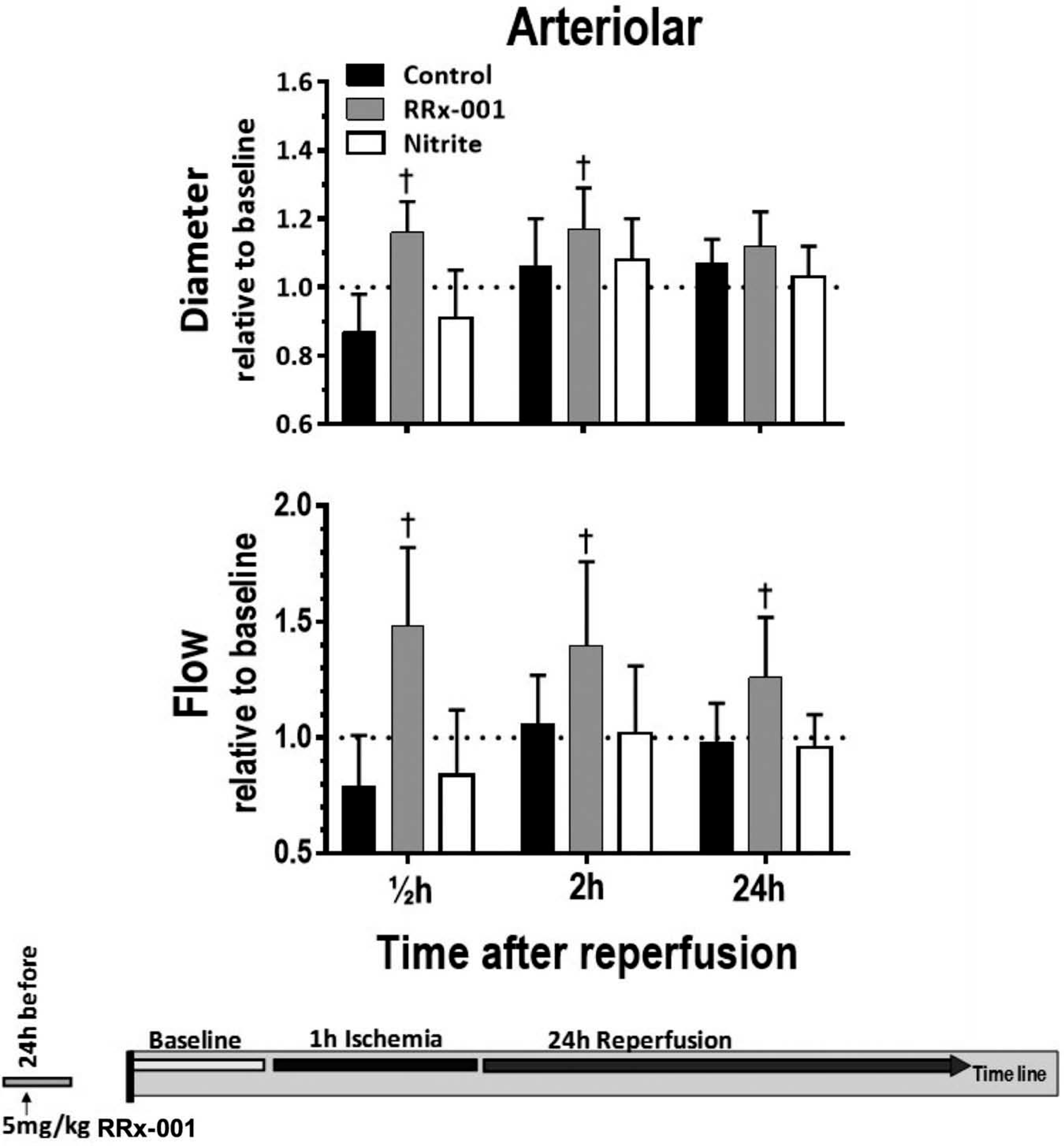
Arteriolar diameter and blood flow following the release of the ischemia. *p < 0.05 compared to control.
Changes in the number of capillaries perfused with RBCs during the protocol are presented in Figure 4. Again, compared to control, FCD was increased at all time points during resuscitation. FCD was significantly increased at 0.5 and 2 h following ischemia compared to control.
Figure 4.
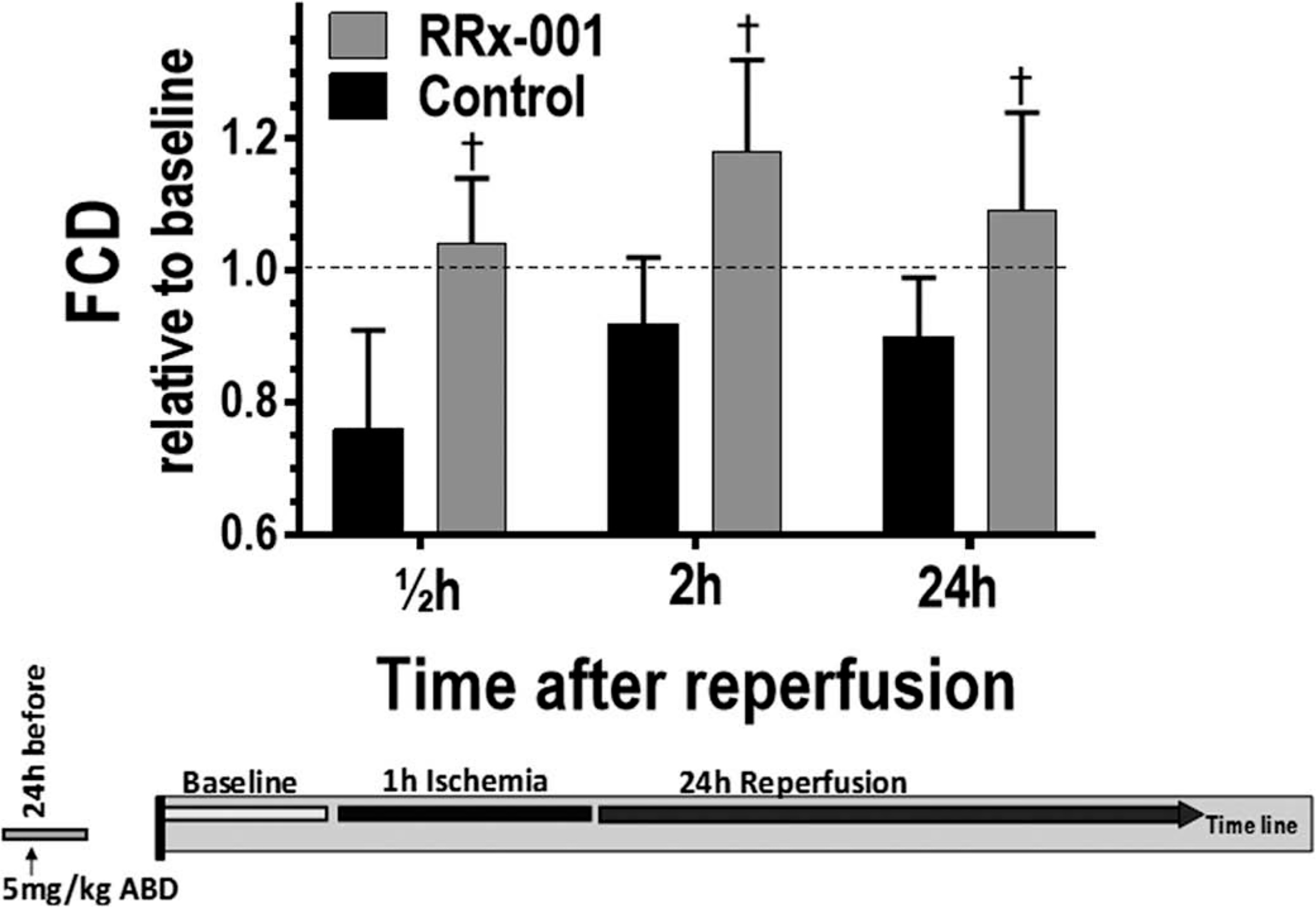
Functional capillary density (FCD) and immobilized leukocytes following the release of the ischemia. †, p < 0.05 compared to control.
3.2. Leukocyte adhesion
Changes in the number of immobilized leukocytes (leukocyte adhesion) following the release of the ischemia are presented in Figure 5. Preconditioning with RRx-001 resulted in decreased leukocyte adhesion at all time points following ischemia. Compared to control, leukocyte adhesion in the RRx-001 group was significantly decreased at 0.5, 2, and 24 h following ischemia.
Figure 5.
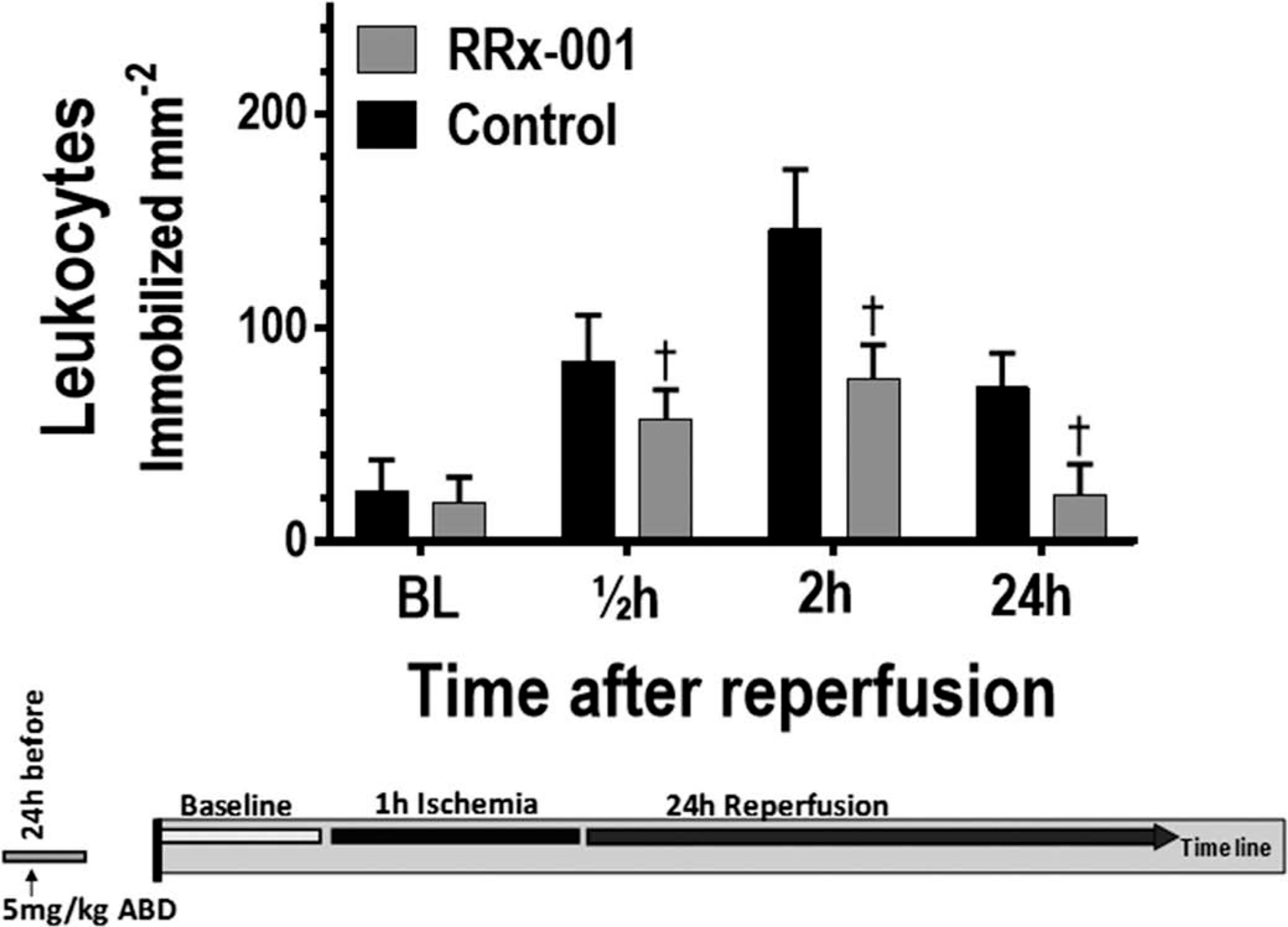
Immobilized leukocytes following the release of the ischemia. †, p < 0.05 compared to control.
3.3. Tissue viability
Tissue viability (the number of apoptotic and necrotic cells in 40 fields) for control and RRx-001 treatment groups at 24 following ischemia is presented in Figure 6. Treatment with RRx-001 resulted a significant decrease in both apoptotic and necrotic cells at 24 following the release of the ischemia.
Figure 6.
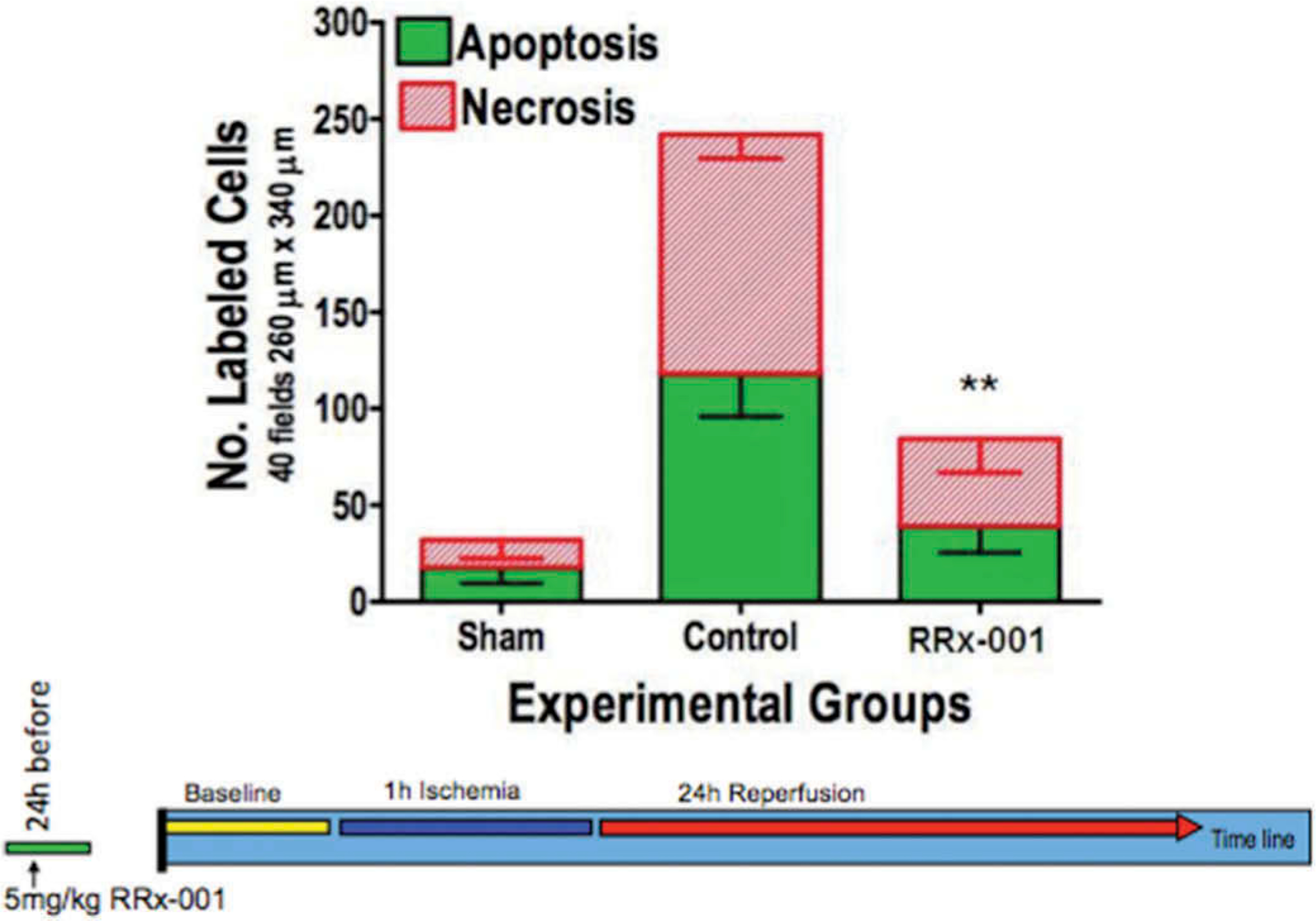
Tissue viability 24 following the release of the ischemia. **p < 0.05 for both the number of apoptotic and necrotic in the RRx-001 treatment group compared to control.
4. Discussion
The principal finding of the study was that preconditioning with RRx-001 significantly enhanced perfusion and preserved tissue viability during the reperfusion phase of I/R. RRx-001 also reduced leukocyte adhesion for up to 24 h following the onset of reperfusion. These RRx-001-mediated benefits on systemic, microvascular, and inflammatory reperfusion parameters are likely related to the modification of βeta-Cys-93, which enhances nitrite reductase activity and hypoxia-mediated nitrite reduction to NO by deoxyhemoglobin. Furthermore, RRx-001 modification of Hb cys-93 in blood has proven not to affect blood circulation half-life, as well as 80% of the RRx-001 modified hemoglobin in the blood remains in circulation 7 days after intravenous administration [39] (Figure 7). Hamsters have a lower arterial PO2 compared to other rodents; however the intravascular PO2s of their microvessels are similar to the PO2s found in other rodents (e.g. mice) instrumented with dorsal window chamber [40]. Furthermore, the intravascular PO2s of hamsters have a lower gradient between the systemic and the microvascular circulation compared to this PO2 gradient in other rodents (e.g. mice).
Figure 7.
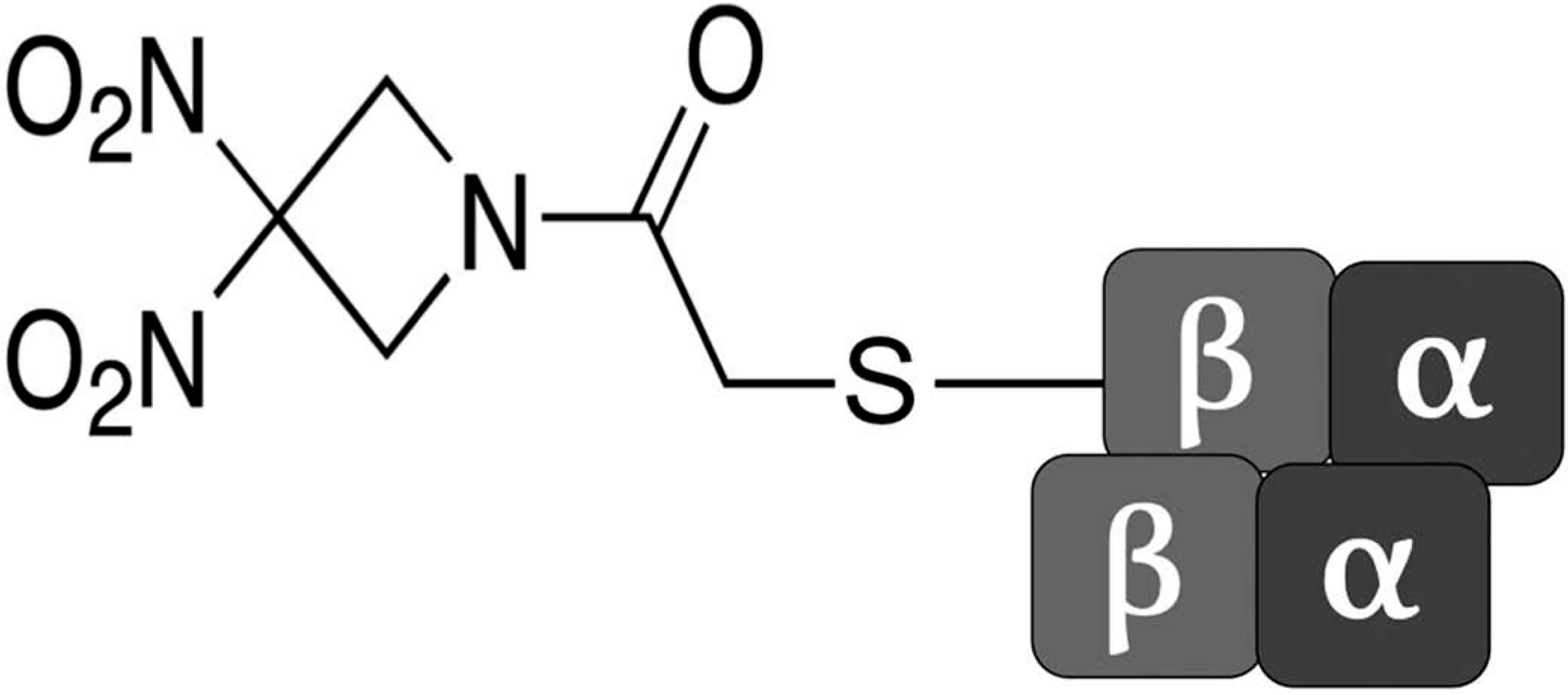
RRx-001 binds to a thiol residue on βeta-Cys-93.
The hemoglobin in hamsters transitions from partially saturated with oxygen in the systemic circulation to deoxygenated in the microvascular circulation, in contrast to the arterial circulation of humans; human arterial blood hemoglobin is rapidly converted to deoxyhemoglobin in the microcirculation. A faster and greater oxy to deoxy transition of the hemoglobin will result in a more efficient reduction of nitrite to NO in hypoxic zones. This allows for a more abundant production of NO as red cells deoxygenate along the arteriolar vascular tree. Additionally, experimental systems have proven that the more rapidly red cells are deoxygenated in the presence of nitrite, the faster vasodilation is observed [41]. Therefore, we speculate that the protective mechanism of hypoxia-mediated nitrite reduction to NO by deoxyhemoglobin enhanced by RRx-001, should translate into other species. If any, the nitrite reduction to NO mediated benefits on systemic, microvascular, and inflammatory reperfusion parameters are likely to increase as the intravascular gradients form systemic to microvascular pO2s are greater in other species (e.g. humans).
I/R injury, present in many medical conditions including shock, trauma, and cardiovascular related diseases is directly related to the development of inflammation; neutrophils, which produce oxygen-free radicals, are centrally involved [35,42,43].
A variety of treatment strategies including the use of nitrite and NO have been used to mitigate IR injury. NO, produced from endothelial NO synthase (eNOS), participates in the regulation of basal blood vessel tone, inhibits antiplatelet activity and aggregation, prevents neutrophil activation, and modulates oxidative/nitrosative stress and inflammation [9,44]. NO binds avidly to the heme of soluble guanylyl cyclase (sGC), produces (cGMP), activates cGMP-dependent protein kinases, and ultimately induces smooth muscle relaxation [9]. During ischemia and early during reperfusion, eNOS activity is impaired due to endothelial dysfunction, leading to loss of NO bioavailability. A decrease in NO availability may also result from its rapid reaction with superoxide to produce peroxynitrite [45].
The results of this study suggest that during hypoxia when NO synthase is inhibited and the bioavailability of NO is reduced, nitric oxide generation from RRx-001-modified RBCs restores the intravascular NO concentration and relaxes arteriolar smooth muscle, which increases tissue perfusion and attenuates leukocyte adhesion to the endothelium for up to 24 h after release of the ischemic insult. At the same time, FCD actually improved likely in part due to the decreased leukocyte trafficking, which would have otherwise driven endothelial cell disruption and capillary narrowing. Since RRx-001 pretreatment induced a decrease in both leukocyte adhesion and the number of apoptotic and necrotic cells, as well as an increase in blood flow and FCD in the first 24 h following ischemia, it is potentially a promising pharmacological strategy to prevent IR injury.
Nitrite, a dietary constituent, is a vascular endocrine nitric oxide reserve that contributes to hypoxic signaling and vasodilation [9], which is speculated to mediate cytoprotection after ischemia/reperfusion via NO release through mechanisms that involve the post-translational S-nitrosation of mitochondrial complex I [46,47]. Modification of complex I dampens electron transfer, effectively reducing reactive oxygen species generation during reperfusion, which, in turn, prevents cytochrome c release and apoptosis [47].
These experiments, which demonstrated that RRx-001 provided significant benefit in the critical parameters (microvascular arteriolar diameter, blood flow, FCD, tissue viability) that were examined compared to control suggest that preconditioning with RRx-001 is a useful strategy for treating IR-associated medical conditions.
A number of studies have demonstrated that nitrite and NO confer cytoprotection following focal I/R injury [11–13]. In hepatic I/R, nitrite exerted profound dose-dependent protective effects on cellular necrosis and apoptosis. In myocardial I/R injury, nitrite reduced cardiac infarct size by 67% [13]. In skeletal I/R, Wang et al. and Murata et al. demonstrated that low-dose nitrite significantly attenuated vasoconstriction, arteriole stagnation, and capillary no-reflow [14,15]. In brain I/R, nitrite was neuroprotective [16] and Campelo et al. demonstrated that NO release improved total brain infarction area and hippocampal neuronal viability [17].
However, contrary to these examples where nitrite provided benefit, Basireddy et al.[18] demonstrated that nitrite did not protect against renal I/R injury. As a systemic rather than local, on-demand treatment nitrite supplementation does not reach target tissues. In comparison, the hemoglobin-based reduction of nitrite to NO is an endogenous process that uses small circulating amounts of nitrite to promote NO production in hypoxic zones. Milsom et al. suggested that eNOS activity, compromised during ischemia as a result of its essential dependence on oxygen, is a critical component of renal I/R injury and that nitrite levels are not sustained for a long enough duration to provide a rescue effect [19]. These findings support the concept that naturally occurring nitrite is not present in sufficient quantities in the renal circulation to mediate a protective effect even with the RRx-001 hyper stimulation of nitrite reductase. The binding of RRx-001 to Hb Cys-93 is rapid and irreversible. These modified red blood cells are present for the duration of the lifetime of the RBC, rendering the NO pharmacodynamic effect long-lived [39]. Thus, RRx-001, which catalyzes a physiologic mechanism for generating nitric oxide under hypoxic conditions, i.e. the reduction of nitrite to NO by deoxyhemoglobin, thereby providing extended release of ‘on demand’ NO for the lifetime of the red blood cell, has the potential to significantly alleviate ischemic reperfusion injury in many organs. Future studies with RRx-001 will explore ischemia, myocardial infarction and secondary heart failure, liver ischemia/reperfusion and organ transplantation. Treatment with RRx-001 could reduce ischemic injury via an improved NO production pathway to increase blood flow by relaxing vascular smooth muscle in arterioles supplying the site of injury.
5. Conclusion
Herein, we have demonstrated that RRx-001, a macrophage-stimulating agent that repolarizes protumorigenic M2 tumor-associated macrophages (TAMs) to an antitumor M1 phenotype in phase II clinical trials, with activity in multiple additional disease states including sickle cell disease, cerebral malaria, leishmaniasis, and hemorrhagic shock, increased blood flow and FCD, and decreased leukocyte adhesion as well as the number of apoptotic and necrotic cells over 24 h following ischemia. Unlike nitrite, which historically only results in a transient effect on blood flow parameters and is associated with significant side effects, RRx-001 is systemically nontoxic, as evidenced by the absence of dose-limiting toxicities or serious drug-related adverse events in phase I [48], and the duration of nitric oxide release is theoretically as long-lived as the red blood cell lifespan. These results potentially have direct clinical applications since, for example, patients subjected to surgeries that involve inflow occlusion such as coronary artery bypass or organ transplantation may benefit from pharmacological preconditioning with RRx-001 to minimize the tissue damage associated with I/R.
Key issues.
RRx-001, a clinical macrophage-stimulating anti-cancer agent that also produces nitric oxide (NO) was studied in a model of ischemia-reperfusion injury.
I/R injury was studied in a hamster chamber window, with compression of the periphery of the window for 1 h to induce ischemia. Animals received RRx-001 (5 mg/kg) 24 h before ischemia and sodium nitrite (10 nmols/kg) was supplemented 10 min after the start of reperfusion. Vessel diameter, blood flow, adherent leukocytes, and functional capillary density were assessed by intravital microscopy at 0.5, 2, and 24 h following the release of the ischemia.
The results demonstrated that, compared to control, RRx-001 preconditioning increased blood flow and functional capillary density, and preserved tissue viability in the absence of side effects over a sustained time period.
The results suggest that RRx-001 may serve as a protective agent during postsurgical restoration of flow and other ischemia-reperfusion associated conditions, increasing blood flow and functional capillary density as well as preserving tissue viability in the absence of side effects.
Funding
This manuscript was not funded.
Footnotes
Declaration of interest
B Oronsky is employed by EpicentRx. S Caroen is employed by EpicentRx. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Toledo-Pereyra LH, Toledo AH, Walsh J, et al. Molecular signaling pathways in ischemia/reperfusion. Exp Clin Transplant. 2004;2:174–177. [PubMed] [Google Scholar]
- 2.Carden DL, Granger DN. Pathophysiology of ischaemia reperfusion injury. J Pathol. 2000;190:255–266. [DOI] [PubMed] [Google Scholar]
- 3.Granger DN, Benoit JN, Suzuki M, et al. Leukocyte adherence to venular endothelium during ischemia-reperfusion. Am J Physiol. 1989;257:G683–688. [DOI] [PubMed] [Google Scholar]
- 4.Schmid-Schonbein GW. Capillary plugging by granulocytes and the no-reflow phenomenon in the microcirculation. Fed Proc. 1987;46:2397–2401. [PubMed] [Google Scholar]
- 5.Inoue N, Ramasamy S, Fukai T, et al. Shear stress modulates expression of Cu/Zn superoxide dismutase in human aortic endothelial cells. Circ Res. 1996;79:32–37. [DOI] [PubMed] [Google Scholar]
- 6.Menger MD, Laschke MW, Amon M, et al. Experimental models to study microcirculatory dysfunction in muscle ischemia-reperfusion and osteomyocutaneous flap transfer. Langenbecks Arch Surg. 2003;388:281–290. [DOI] [PubMed] [Google Scholar]
- 7.Nolte D, Menger MD, Messmer K. Microcirculatory models of ischaemia-reperfusion in skin and striated muscle. Int J Microcirc Clin Exp. 1995;15(Suppl 1):9–16. [DOI] [PubMed] [Google Scholar]
- 8.Szabó C, Thiemermann C. Invited opinion: role of nitric oxide in hemorrhagic, traumatic, and anaphylactic shock and thermal injury. Shock. 1994;2:145–155. [PubMed] [Google Scholar]
- 9.Gladwin MT, Raat NJ, Shiva S, et al. Nitrite as a vascular endocrine nitric oxide reservoir that contributes to hypoxic signaling, cytoprotection, and vasodilation. Am J Physiol Heart Circ Physiol. 2006. November;291(5):H2026–35. [DOI] [PubMed] [Google Scholar]
- 10.Dezfulian C, Raat N, Shiva S, et al. Role of the anion nitrite in ischemia-reperfusion cytoprotection and therapeutics. Cardiovasc Res. 2007. July 15;75(2):327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lefer DJ. Emerging role of nitrite in myocardial protection. Arch Pharm Res. 2009. August;32(8):1127–1138. [DOI] [PubMed] [Google Scholar]
- 12.Abe Y, Hines I, Zibari G, et al. Hepatocellular protection by nitric oxide or nitrite in ischemia and reperfusion injury. Arch Biochem Biophys. 2009. April 15;484(2):232–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duranski MR, Greer JJ, Dejam A, et al. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J Clin Invest. 2005. May;115(5):1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang WZ, Fang XH, Stephenson LL, et al. Nitrite attenuates ischemia-reperfusion-induced microcirculatory alterations and mitochondrial dysfunction in the microvasculature of skeletal muscle. Plast Reconstr Surg. 2011. October;128(4):279e–287e. [DOI] [PubMed] [Google Scholar]
- 15.Murata I, Nozaki R, Ooi K, et al. Nitrite reduces ischemia/reperfusion-induced muscle damage and improves survival rates in rat crush injury model. J Trauma Acute Care Surg. 2012. June;72 (6):1548–1554. [DOI] [PubMed] [Google Scholar]
- 16.Jung KH, Chu K, Ko SY, et al. Early intravenous infusion of sodium nitrite protects brain against in vivo ischemia-reperfusion injury. Stroke. 2006. November;37(11):2744–2750. [DOI] [PubMed] [Google Scholar]
- 17.Campelo MW, Oriá RB, Lopes LG, et al. Preconditioning with a novel metallopharmaceutical NO donor in anesthetized rats subjected to brain ischemia/reperfusion. Neurochem Res. 2012. April;37(4):749–758. [DOI] [PubMed] [Google Scholar]
- 18.Basireddy M, Isbell TS, Teng X, et al. Effects of sodium nitrite on ischemia-reperfusion injury in the rat kidney. Am J Physiol Renal Physiol. 2006. April;290(4):F779–86. [DOI] [PubMed] [Google Scholar]
- 19.Milsom AB, Patel NS, Mazzon E, et al. Role for endothelial nitric oxide synthase in nitrite-induced protection against renal ischemia-reperfusion injury in mice. Nitric Oxide. 2010. February 15;22 (2):141–148. [DOI] [PubMed] [Google Scholar]
- 20.Oronsky B, Fanger GR, Oronsky N, et al. The implications of hyponitroxia in cancer. Transl Oncol. 2014. April;7(2):167–173.• Background on the role of nitric oxide in cancer.
- 21.Oronsky BT, Knox SJ, Scicinski JJ. Is Nitric Oxide (NO) the last word in radiosensitization? A review. Transl Oncol. 2012. April;5(2):66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carter CA, Oronsky BT, Caroen SZ, et al. RRx-001 in refractory small-cell lung carcinoma: a case report of a partial response after a third reintroduction of platinum doublets. Case Rep Oncol. 2016. March 11;9(1):171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scicinski J, Oronsky B, Ning S, et al. NO to cancer: the complex and multifaceted role of nitric oxide and the epigenetic nitric oxide donor, RRx-001. Redox Biol. 2015. December;6:1–8.• Background on the interaction between nitric oxide and RRx-001.
- 24.Fens MH, Larkin SK, Oronsky B, et al. The capacity of red blood cells to reduce nitrite determines nitric oxide generation under hypoxic conditions. PLoS One. 2014. July 9; 9(7):e101626.• Background on the interaction between nitric oxide and red blood cells.
- 25.Yalcin O, Oronsky B, Carvalho LJ, et al. From METS to malaria: RRx-001, a multi-faceted anticancer agent with activity in cerebral malaria. Malar J. 2015. May 28;14:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brouse C, Ortiz D, Su Y, et al. Impact of hemoglobin nitrite to nitric oxide reductase on blood transfusion for resuscitation from hemorrhagic shock. Asian J Transfus Sci. 2015. January-June;9(1):55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oronsky B, Scicinski J, Ning S, et al. RRx-001, A novel dinitroazetidine radiosensitizer. Invest New Drugs. 2016. February 3;34:371–377. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oronsky B, Scicinski J, Ning S, et al. RRx-001, a novel clinical-stage chemosensitizer, radiosensitizer and immunosensitizer that inhibits glucose 6-phosphate dehydrogenase in human tumor cells. Discov Med. 2016. April;21(116):251–265. [PubMed] [Google Scholar]
- 29.Ning S, Bednarski M, Oronsky B, et al. Dintroazetidines are a novel class of anticancer agents and hypoxia-activated radiation sensitizers developed from highly energetic materials. Cancer Res. 2012;15(72):2600–2608. [DOI] [PubMed] [Google Scholar]
- 30.Fens MHAM, Larkin SK, Morris CR, et al. NO or No NO, increased reduction of nitrite to nitric oxide by modified red blood cells. Blood (ASH Annual Meeting Abstracts). 2011;118:2125.• Background on the interaction between nitric oxide and red blood cells.
- 31.Colantuoni A, Bertuglia S, Intaglietta M. Quantitation of rhythmic diameter changes in arterial microcirculation. Am J Physiol. 1984;246:H508–H517. [DOI] [PubMed] [Google Scholar]
- 32.Endrich B, Asaishi K, Götz A, et al. Technical report: A new chamber technique for microvascular studies in unanaes-thetized hamsters. Res Exp Med. 1980;177:125–134. [DOI] [PubMed] [Google Scholar]
- 33.Cabrales P, Nacharaju P, Manjula BN, et al. Early difference in tissue pH and microvascular hemodynamics in hemorrhagic shock resuscitation using peg-albumin and hydroxyethyl starch based plasma expanders. Shock. 2005;24:66–73.• Background on the hemorrhagic shock and the circulatory system.
- 34.Tsai AG, Friesenecker B, McCarthy M, et al. Plasma viscosity regulates capillary perfusion during extreme hemodilution in hamster skin fold model. Am J Physiol. 1998;275:H2170–H2180. [DOI] [PubMed] [Google Scholar]
- 35.Friesenecker B, Tsai AG, Instaglietta M. Capillary perfusion during ischemia-reperfusion in subcutaneous connective tis- sue and skin muscle. Am J Physiol. 1994;267:H2204–2212. [DOI] [PubMed] [Google Scholar]
- 36.Intaglietta M, Silverman NR, Tompkins WR. Capillary flow velocity measure- ments in vivo and in situ by television methods. Microvasc Res. 1975;10:165–179. [DOI] [PubMed] [Google Scholar]
- 37.Cabrales P, Tsai AG, Intaglietta M. Exogenous nitric oxide induces protection during hemorrhagic shock. Resuscitation. 2009;80:707–712.• Background on nitric in hemorrhagic shock.
- 38.Childs EW, Udobi KF, Wood JG, et al. In vivo visualization of reactive oxidants and leukocyte-endothelial adherence following hemorrhagic shock. Shock. 2002;18:423–427. [DOI] [PubMed] [Google Scholar]
- 39.Scicinski J, Oronsky B, Taylor M, et al. Preclinical evaluation of the metabolism and disposition of RRx-001, a novel investigative anticancer agent. Drug Metab Dispos. 2012;40:1810–1816. [DOI] [PubMed] [Google Scholar]
- 40.Cabrales P, Tsai AG, Frangos JA, et al. Role of endothelial nitric oxide in microvascular oxygen delivery and consumption. Free Radic Biol Med. 2005;39:1229–1237.• Background on the interaction between nitric oxide and the circulatory system.
- 41.Crawford JH, Isbell TS, Huang Z, et al. Hypoxia, red blood cells, and nitrite regulate NO-dependent hypoxic vasodilation. Blood. 2006;107:566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paxian M, Keller SA, Huynh TT, et al. Perflubron emulsion improves hepatic microvascular integrity and mito- chondrial redox state after hemorrhagic shock. Shock. 2003;20:449–457. [DOI] [PubMed] [Google Scholar]
- 43.Baker JE, Boerboom LE, Olinger GN. Age and protection of the ischemic myocardium: is alkaline cardioplegia appropriate? Ann Thorac Surg. 1993;55:747–755. [DOI] [PubMed] [Google Scholar]
- 44.Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci U S A. 1991. June 1;88(11):4651–4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laude K, Thuillez C, Richard V, Reperfusion-induced coronary endothelial injury: A new target for ischemic preconditioning. Exp Clin Cardiol. 2001. Fall;6(3):149–152. [PMC free article] [PubMed] [Google Scholar]
- 46.Shiva S, Gladwin MT. Nitrite mediates cytoprotection after ischemia/reperfusion by modulating mitochondrial function. Basic Res Cardiol. 2009. March;104(2):113–119. [DOI] [PubMed] [Google Scholar]
- 47.Shiva S, Sack MN, Greer JJ, et al. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med. 2007. September 3;204(9):2089–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reid T, Oronsky B, Scicinski J, et al. Safety and activity of RRx-001 in patients with advanced cancer: a first-in-human, open-label, dose-escalation phase 1 study. Lancet Oncol. 2015. September;16 (9):1133–1142. [DOI] [PubMed] [Google Scholar]