

To build the brain, embryonic neural stem cells (NSCs) tightly regulate their cell divisions, undergoing a polarized form of cytokinesis that is poorly understood. Cytokinetic abscission is mediated by the midbody to sever the daughter cells at the apical membrane. In cell lines, the coiled-coil protein Cep55 was reported to be required for abscission. Mutations of Cep55 in humans cause a variety of cortical malformations. However, its role in the specialized divisions of NSCs is unclear.
Keywords: abscission, Cep55, cortical development, microcephaly, midbody, neural stem cell
Abstract
To build the brain, embryonic neural stem cells (NSCs) tightly regulate their cell divisions, undergoing a polarized form of cytokinesis that is poorly understood. Cytokinetic abscission is mediated by the midbody to sever the daughter cells at the apical membrane. In cell lines, the coiled-coil protein Cep55 was reported to be required for abscission. Mutations of Cep55 in humans cause a variety of cortical malformations. However, its role in the specialized divisions of NSCs is unclear. Here, we elucidate the roles of Cep55 in abscission and brain development. KO of Cep55 in mice causes abscission defects in neural and non-neural cell types, and postnatal lethality. The brain is disproportionately affected, with severe microcephaly at birth. Quantitative analyses of abscission in fixed and live cortical NSCs show that Cep55 acts to increase the speed and success rate of abscission, by facilitating ESCRT recruitment and timely microtubule disassembly. However, most NSCs complete abscission successfully in the absence of Cep55. Those that fail show a tissue-specific response: binucleate NSCs and neurons elevate p53, but binucleate fibroblasts do not. This leads to massive apoptosis in the brain, but not other tissues. Double KO of both p53 and Cep55 blocks apoptosis but only partially rescues Cep55−/− brain size. This may be because of the persistent NSC cell division defects and p53-independent premature cell cycle exit. This work adds to emerging evidence that abscission regulation and error tolerance vary by cell type and are especially crucial in neural stem cells as they build the brain.
SIGNIFICANCE STATEMENT During brain growth, embryonic neural stem cells (NSCs) must divide many times. In the last step of cell division, the daughter cell severs its connection to the mother stem cell, a process called abscission. The protein Cep55 is thought to be essential for recruiting proteins to the mother-daughter cell connection to complete abscission. We find that Cep55 mutants have very small brains with disturbed structure, but almost normal size bodies. NSC abscission can occur, but it is slower than normal, and failures are increased. Furthermore, NSCs that do fail abscission activate a signal for programmed cell death, whereas non-neural cells do not. Blocking this signal only partly restores brain growth, showing that regulation of abscission is crucial for brain development.
Introduction
Embryonic neural stem cells (NSCs) must undergo rapid divisions during development to produce sufficient daughter cells of various fates to build a brain of correct size and structure. They divide within a polarized epithelium, with mitosis and cytokinesis occurring at the apical membrane. The cleavage furrow ingresses asymmetrically, forming the midbody at the apical membrane. The midbody mediates severing of the intercellular bridge, in a process called abscission. Errors in cytokinesis are one etiology of microcephaly, in which the brain is disproportionately small relative to body size (Di Cunto et al., 2000; Li et al., 2016; Bondeson et al., 2017; Frosk et al., 2017; Moawia et al., 2017; Makrythanasis et al., 2018). We previously showed that mutation of the Kinesin-6 family member Kif20b disrupts normal cytokinetic abscission to cause microcephaly in mice (Dwyer et al., 2011; Janisch et al., 2013; Little and Dwyer, 2019). The unique constraints of NSCs may make them more vulnerable to defects in cytokinesis than other cell types. However, the cellular and developmental mechanisms that drive these tissue-specific requirements are just beginning to be identified.
Cytokinesis studies in cell lines and single-cell models have identified many required proteins, and generated a mechanistic model of abscission (Green et al., 2012; Mierzwa and Gerlich, 2014). The midbody has a thick central bulge flanked on either side by dense microtubule bundles. This complex structure consists of >450 proteins that mediate the abscission process: midbody formation, maturation, and the final scission event (Skop et al., 2004; Hu et al., 2012; Addi et al., 2020). Scission involves both microtubule disassembly and membrane severing, thought to be mediated by endosomal sorting complexes required for transport (ESCRT) filaments (Connell et al., 2009; Guizetti et al., 2011). After scission on each side, the central bulge domain becomes a midbody remnant (MBR).
Studies of abscission mechanisms and roles in tissues in vivo are just beginning (Addi et al., 2018).The duration of abscission can be regulated, and slower abscission has been linked to stemness (Lenhart and DiNardo, 2015; Chaigne et al., 2020; McNeely and Dwyer, 2020). In cell lines, the MBR may mediate cell signaling and fate by binding surface receptors, or through engulfment into daughter or neighbor cells (Crowell et al., 2014; Peterman et al., 2019). We previously showed that, in embryonic brains, cytokinesis of NSCs and MBR persistence is developmentally regulated (McNeely and Dwyer, 2020).
The coiled-coil scaffolding protein Cep55 is thought to be essential for abscission in cell lines (Fabbro et al., 2005; Zhao et al., 2006), with a key role in ESCRT recruitment in midbodies (Stoten and Carlton, 2018). Human mutations of Cep55 cause a variety of brain malformations (Bondeson et al., 2017; Frosk et al., 2017; Rawlins et al., 2019; Barrie et al., 2020). That human embryos with Cep55 mutation are able to develop at all is surprising, given that knockdown of CEP55 in human cell lines caused almost universal failure of cell division (Fabbro et al., 2005; Zhao et al., 2006).
Here, we use a mouse KO to elucidate the roles and requirements of Cep55 in abscission in vivo. We find that Cep55 is not absolutely required for abscission in NSCs or other embryonic cells, but its loss causes abnormalities and increases failures. NSCs are especially vulnerable to loss of Cep55 compared with other tissues, and brain size is disproportionally reduced. Surprisingly, we find that ESCRT recruitment is not eliminated in Cep55−/− cell midbodies, although it is impaired. We use fixed and live imaging of intact cortical epithelium to probe Cep55's role in abscission. Additionally, we find that cells' response to abscission failure varies: binucleate fibroblasts do not activate p53, but binucleate NSCs do, and this may underlie the severe effect on the nervous system compared with other tissues. We test this using double KO (dKO) of Cep55 and p53. Analyses reveal that, in addition to p53-dependent apoptosis, Cep55 loss in cortical NSCs causes p53-independent early cell cycle exit.
Materials and Methods
Mice
Mouse colonies were maintained in accordance with National Institutes of Health guidelines and policies approved by the Institutional Animal Care and Use Committee. Embryos were harvested by cesarean section, and the morning of the vaginal plug was considered E0.5. Littermate embryos served as controls for all experiments, and all parent and embryo genotypes were confirmed by PCR. The Cep55 allele (strain C57BL/6NCep55em1(IMPC)Tcp) was made as part of the KOMP2 Phase 2 project at the Toronto Center for PhenoGenomics for the Canadian Mouse Mutant Repository. It was maintained on C57BL/6 and 50%/50% C57BL/6 and FVB/N background embryos, which were used for experiments. We verified the correct mutation in our mutants with DNA sequencing and RT-PCR. p53 KO (Trp53tm1Tyj) mice on C57BL/6 background were obtained from The Jackson Laboratory (JAX stock #002101) (Jacks et al., 1994). These mice were bred with 50%/50% C57BL/6 and FVB/N Cep55 KO embryos for creation of the Cep55;p53 mouse line. Cep55 heterozygous mice were crossed with both the mT/mG reporter line (JAX stock #007576, The Jackson Laboratory) and Sox2-Cre mice (JAX stock #0085454, The Jackson Laboratory) (Hayashi et al., 2002; Muzumdar et al., 2007) to produce mice that express plasma membrane-localized GFP. Sex of embryonic mice was not noted as sex was not a relevant biological variable for these experiments. The specific ages of embryonic mice used for each experiment are noted in the figure legends.
For survival analyses, pups were kept with their mother and littermates. Milk was noted in the bellies of Cep55−/− and Cep55−/−;p53−/− mice until at least P2. Pups were weaned at 21-25 d, unless too small to be separated from mothers. In some situations, pups were killed because of severe failure to thrive for humane reasons, and that was recorded as the day of death; in most cases, pups died without intervention.
For in vivo cell fate mapping, E11.5 females were injected intraperitoneally with warmed BrdU (200 μl of 10 mg/ml, made in sterile PBS) 24 h before sacking.
Immunoblotting
Mouse embryonic fibroblast (MEF) cells were washed with ice-cold PBS, and lysed directly on the plate (6 cm) into lysis buffer (500 µl); 50 mm Tris HCl, 150 mm NaCl, 1% NP-40, 1 mm EDTA, 1 mm DTT, 1 mm PMSF, 1× HALT protease/phosphatase inhibitor cocktail. After incubating lysates on a rotisserie at 4°C for 30 min, lysate was cleared by centrifugation at 21,000 × g for 15 min at 4°C. Supernatants were diluted to 1× with 5× Laemmli buffer for Western blot. Equivalent amounts of cells within each experiment were run on a 4%-20% polyacrylamide gel. After transfer onto nitrocellulose with the Bio-Rad Transblot Turbo, membrane was blocked with LI-COR TBS blocking buffer for 1 h at room temperature. Primary and secondary antibodies were diluted in 50% blocking buffer, 50% TBS, final 0.1% Tween-20. Primary antibodies were incubated overnight at 4°C, and after washing, followed by appropriate species specific near-infrared (680 or 800 nm) secondary antibodies for 1 h at room temperature. Blots were imaged with the dual-color LI-COR Odyssey CLx imager. Densitometry of Western blots was performed with LI-COR Image Studio software. Primary antibodies used are as follows: rabbit monoclonal anti human β-actin (1:10,000, Clone13E5 4970, Cell Signaling Technology) and mouse monoclonal anti-mouse Cep55 raised against amino acids 163-462 mapping at the C-terminus (1:1000, sc-377018, Santa Cruz Biotechnology).
Immunostaining and H&E staining
To collect cryosections for immunohistochemistry in Figures 1–3, 9–12 and Extended Data Figure 11-1, age E12.5, E14.5, and P0 brains were removed from heads and fixed for 4, 6, and 24 h, respectively, in 4% PFA, followed by immersion in 30% sucrose in PBS overnight. Whole E10.5 embryos (used in Fig. 8) were fixed overnight. Next, whole brains or embryos were embedded in OCT compound (Tissue-Tek 4583, Sakuraus), and cryosections were cut at 20 μm thickness and collected on Superfrost Plus slides (Thermo Fisher Scientific, 12-550-15). Frozen sections were stored at −80°C. Before immunostaining, cryosections were warmed to room temperature, then if antigen retrieval was needed (Pax6, Tbr2, p53, BrdU, Ctip2, Satb2 antibodies), immersed in 10 mm citrate buffer at 95°C for 20 min. After cooling, sections were blocked in 2% normal goat serum for 1 h, followed by incubation with primary antibodies overnight at 4°C. The next day, after PBS washes, sections were incubated with AlexaFluor secondary antibodies at 1:200 and DAPI at 1:100 for 30 min followed by PBS washes and coverslipping with VectaShield fluorescent mounting medium.
Figure 1.
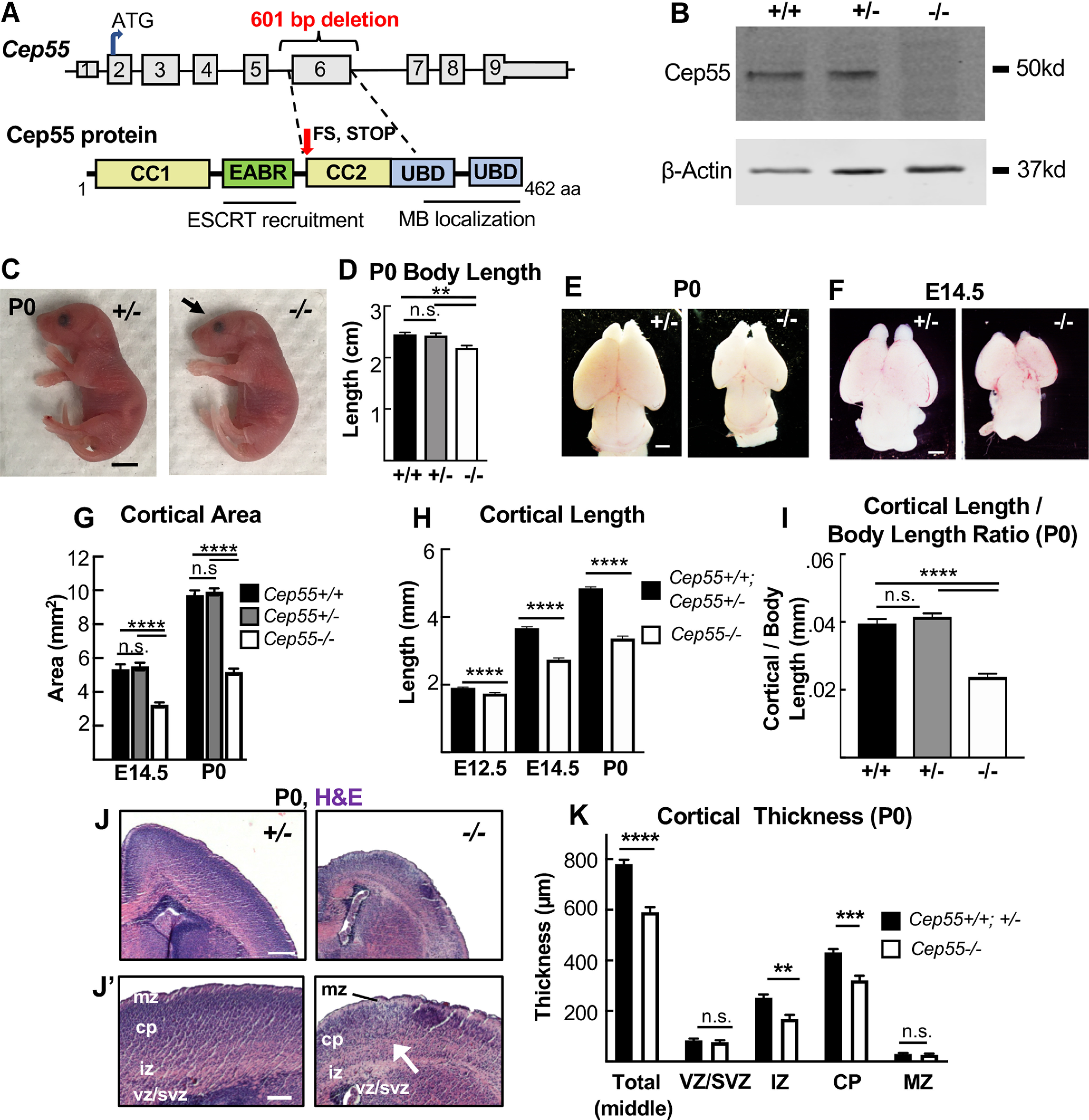
Cep55 KO causes microcephaly with severe thinning of neuronal and axon layers. A, Schematics of Cep55 mouse gene and protein (not to scale). The 601 bp deletion includes all of exon 6 (311 bp) and portions of flanking introns. Cep55 protein domains include two coiled-coil regions (CC1 and CC2) surrounding the ESCRT- and Alix-binding region (EABR), and two ubiquitin binding domains (UBD). The deletion of exon 6 is predicted to result in a frameshift (FS) at amino acid 226, which causes multiple premature stop codons starting 12 residues later. B, Immunoblots of E14.5 MEF lysates show the expected Cep55 protein product at ∼55 kDa in WT and heterozygote samples, but not in KO samples. C, D, Newborn Cep55−/− pups have 10% shorter body length and a flatter head (arrow) but otherwise appear morphologically similar to controls. E–G, Representative images and measurements show that mean cortical area is reduced in −/− brains at P0 and E14.5 compared with +/+ and +/− controls. H, Mean cortical length is significantly reduced in −/− mice at E12.5, E14.5, and P0. I, The cortical length in −/− P0 pups is disproportionately small relative to body size. J, Representative images of P0 +/− and −/− coronal sections at level of corpus callosum, stained with H&E. −/− mice have reduced cell density in neuronal layers (J′, arrow). K, Mean total cortical thickness is significantly reduced in −/− brains at P0. The vz/svz and mz thicknesses are unaltered, but the iz and cp are significantly thinner. P0 (C–I), n = 7 +/+, 14 +/−, and 8 −/− mice. E14.5 (E–H), n = 5 +/+, 13 +/−, and 10 −/− mice. E12.5 (H), n = 17 +/+;+/− and 9 −/− mice. K, n = 3 +/+, 5 +/−, and 5 −/− mice. Scale bars: C, 5 mm; E, F, 1 mm; J, 250 µm, J′, 100 µm. **p < 0.01. ***p < 0.001. ****p < 0.0001. D, G, I, One-way ANOVA; H, K, Student's t test.
Figure 3.

Cep55 KO cortex shows reduced and disorganized neuron and NSC layers by E14.5. A, Representative images of cortical sections from Cep55 +/− and −/− embryos immunostained for neuronal tubulin (Tubb3, gray) show cp and axons in the iz. In −/− images, the cp is disorganized. B, Mean thicknesses of total cortex and iz are significantly decreased in −/− brains. C, The −/− cp/iz occupies proportionally less of cortical width than normal, while the vz/svz occupies more. D, Representative images of E14.5 Cep55+/− and −/− coronal, middle sections stained with H&E. E, The iz and cp are ∼ 30% thinner in −/− cortices compared with controls; however, the mz is 20% thicker. F, Cortical sections stained for NSC marker Pax6 (red) and BP marker Tbr2 (green) show that NSCs in the vz of Cep55−/− brains are more disorganized, with some empty spaces (square), and some NSC nuclei mislocalized above the svz (arrows). G, H, NSCs (Pax6+) per cortical length are reduced in −/−, and some are mislocalized. BP numbers (Tbr2+) are not significantly changed. I, PH3 immunostaining marks cells in mitosis. J, −/− cortices show a normal number of mitotic cells at the apical membrane but an increased number of basal mitotic cells. K, The mitotic index of NSCs (Pax6+) is normal, but of BPs (Tbr2+) is increased in −/− cortices. L, Representative images of E12.5 control (+/+, +/−) and −/− coronal cortical sections immunostained for neuronal tubulin (Tubb3, white) to label preplate (pp) and BP marker Tbr2 (green). M, The mean cortical thickness is not different between control and −/− brains. N, O, Preplate of −/− brains is not thicker but is an increased proportion of total thickness. P, Cep55−/− cortices have decreased Tbr2 cell number per 100 µm of the VZ. A, F, I, L, Dashed line indicates apical membrane. B, Total thickness n = 7 +/− and 8 −/− brains. B, Layer thicknesses. C, F-K, n = 4 +/− and 4 −/− brains. E, n = 6 +/− and 5 −/− mice. M-O, n = 6 controls (4 +/+, 2 +/−), 6 −/− brains. P, n = 3 controls (2 +/+, 1 +/−), 3 −/− brains. pp, Preplate. Scale bars: A, L, 20 µm; D, 100 µm. *p < 0.05; **p < 0.01; ***p < 0.001; t test.
Figure 9.
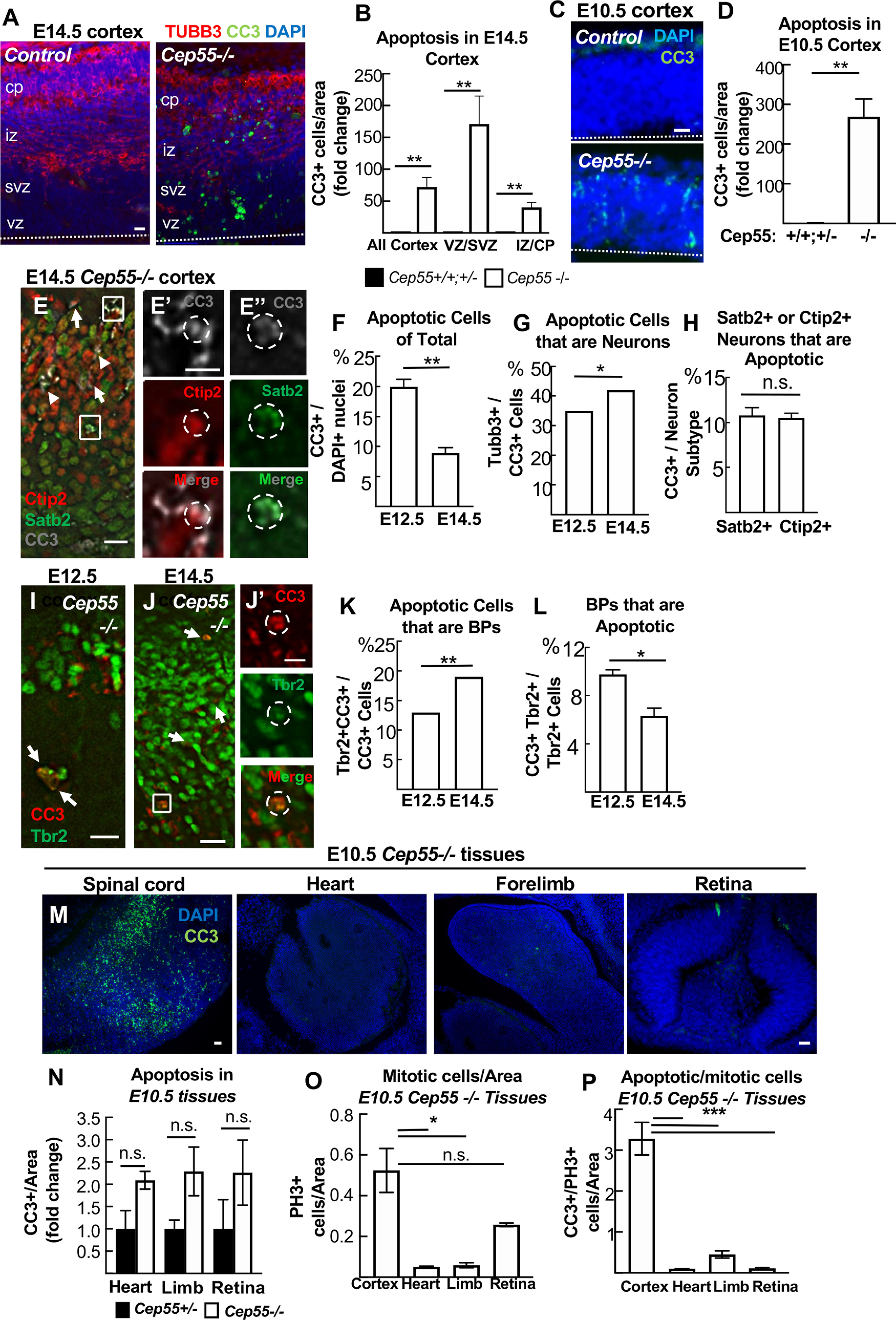
Apoptosis is greatly increased in embryonic neural tissue of Cep55 KO embryos. A, Cortical sections immunostained for apoptotic cell marker CC3 (green) show almost no CC3+ cells in control but many in Cep55−/− cortex. B, At E14.5, apoptotic cells were most increased in the vz/svz, containing nuclei of NSCs and BPs, but also increased in neurons in cp/iz of −/− brains. C, D, At E10.5, before neurogenesis, apoptosis is greatly increased in −/− forebrain NSCs. E-E′′, Representative images of coronal cortical sections from E14.5 Cep55−/− brains immunostained for neuron markers Satb2 (green) and Ctip2 (red) and apoptotic marker CC3 (gray). E′, Inset of top box in A shows a CC3+ Ctip2+ cell. E′′, Inset of bottom box in A shows a CC3+ Satb2+ cell. Arrows indicate additional CC3+ Ctip2+ cells. Arrowheads indicate additional CC3+ Satb2+ cells. F, There are increased numbers of CC3+ cells (cells defined as DAPI+) in Cep55−/− cortex at E12.5 compared with E14.5. G, The proportion of CC3+ cells that are copositive for neuron marker Tubb3 is slightly increased at E14.5 compared with E12.5. H, A similar percentage of Satb2+ and Ctip2+ cells are copositive for CC3 at E14.5. I-J′, Representative images of cortical sections from E12.5 (I) and E14.5 (J) −/− cortex immunostained for BPs (Tbr2) and CC3. Copositive cells are observed (arrows and box inset, J′). K, The proportion of CC3+ cells that are copositive for Tbr2 is increased at E14.5 compared with E12.5. L, Approximately 10% of Tbr2+ cells are copositive for CC3 at E12.5 compared with 8% at E14.5 in −/− cortices. M, N, Apoptosis is high in the E10.5 −/− spinal cord, (and midbrain and hindbrain, not shown), but not in heart, forelimb, or retina. O, P, The brain specificity of the apoptosis is not simply because of higher mitotic index. A, C, Dashed line indicates apical membrane. All experiments, n = 3 control and −/− brains or embryos at each age. Scale bars: A, C, E, 20 µm; E′, J′, 5 μm; I, 10 μm; M, left, 40 µm; retina, 20 µm. *p <0.05. **p < 0.01. ***p < 0.001. All experiments, t test.
Figure 12.
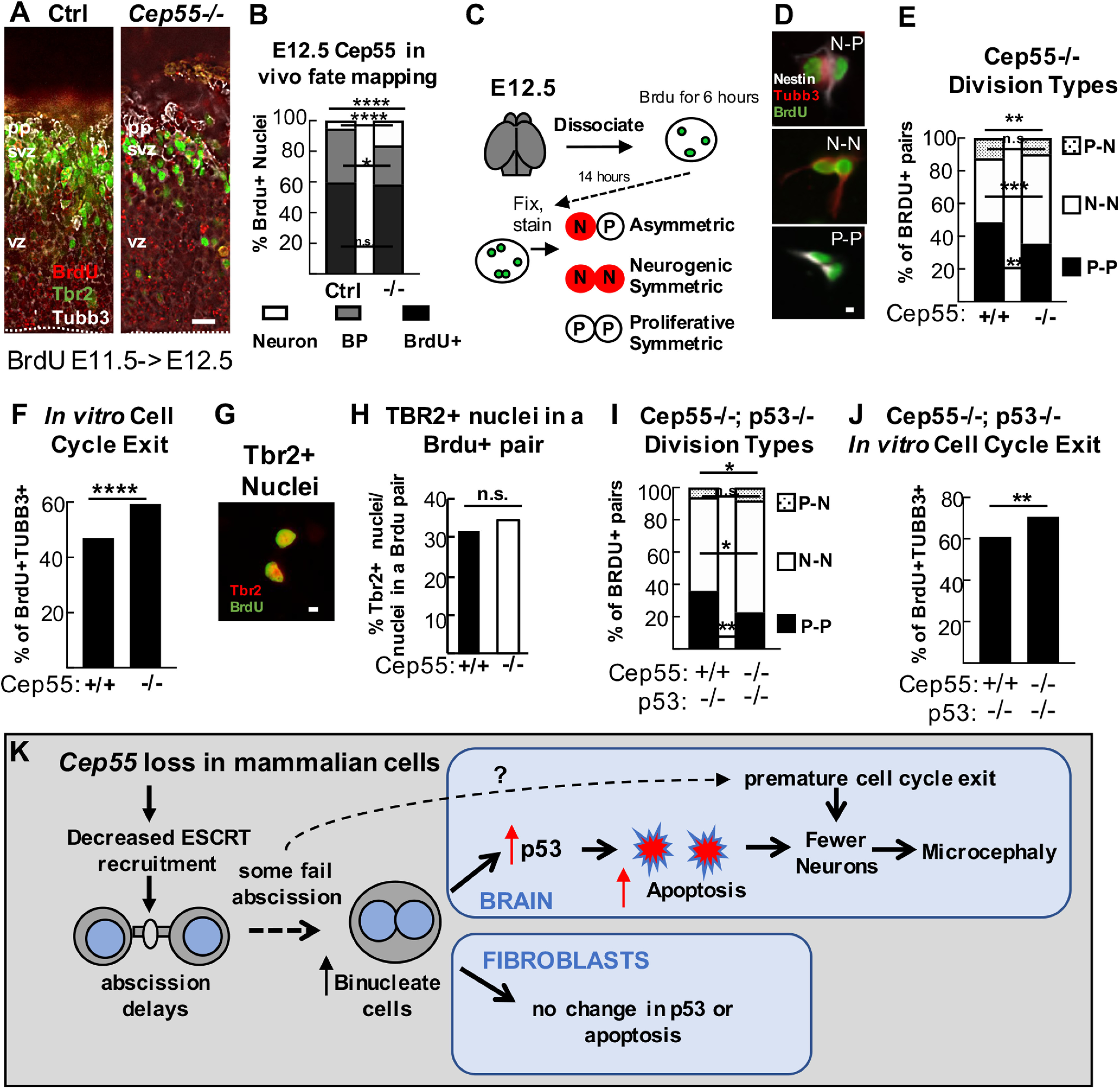
Cell cycle exit is increased in Cep55−/− cortices at E12.5 and is p53-independent. A, Representative images of coronal cortical sections from Cep55 control (+/+, +/−) and −/− embryos injected with BrdU at E11.5 and collected at 24 h, immunostained for neuron marker Tubb3 (gray), BP marker Tbr2 (green), and recently divided cells (BrdU+ only, red). B, Increased numbers of BrdU+ nuclei were copositive for Tubb3+ in Cep55−/− cortices. n = 308 control, 204 −/− nuclei from 3 control and 3 −/− brains. C, Schematic of modified BrdU pair cell assay. E12.5 progenitors were dissociated and plated with BrdU followed by washout, incubation, fixation, and immunostaining with BrdU, Tubb3, and Nestin (progenitors). Pairs of BrdU+ cells were identified and marked as containing progenitors (Nestin+, Tuj1–), or neurons (Tuj1+, Nestin+, or Nestin–). D, Representative images of BrdU+ pairs that are P-P, N-N, or N-P. E, Cep55−/− NSCs have reduced numbers of P-P and increased N-N divisions. n = 309 +/+, 256 −/− pairs; 3 coverslips/animals each. F, There are more BrdU+Tubb3+ nuclei indicating cell cycle exit in −/− cultures. G, Representative image of paired nuclei that are Tbr2+. n = 750 +/+ and 622 −/− nuclei; 3 coverslips/animals each. H, The percentage of Tbr2+ nuclei in a BrdU pair is not altered in −/− cultures. n = 566 +/+ and 528 −/− nuclei; 3 coverslips/animals each. I, dKO E12.5 NSCs have reduced proliferative symmetric and increased neurogenic symmetric division compared with controls. J, There are more BrdU+Tubb3+ nuclei in dKO cultures. K, L, n = 217 Cep55+/+;p53−/− pairs/528 nuclei; 3 coverslips/2 animals and 189 Cep55−/−;p53−/− pairs/481 nuclei; 4 coverslips/2 animals. Scale bars: A, C, 20 µm; F, I, 5 µm. *p < 0.05. **p < 0.01. ***p < 0.001. ****p < 0.0001. B, D, G, K, χ2; others, Fisher's exact. K, Working model for the consequences of Cep55 loss on abscission of mammalian cells. Cep55−/− mouse cells have impaired ESCRT recruitment and abscission delay. Most cells complete abscission, but at least some fail to complete cytokinesis resulting in increased binucleate cells. Binucleate NSCs and neurons have elevated p53 expression, but not binucleate MEFs. p53 activation in NSCs and neurons causes apoptosis in Cep55 KO cortices, which partially accounts for microcephaly. Precocious cell cycle exit occurs after some NSC divisions and is p53-independent.
Figure 8.
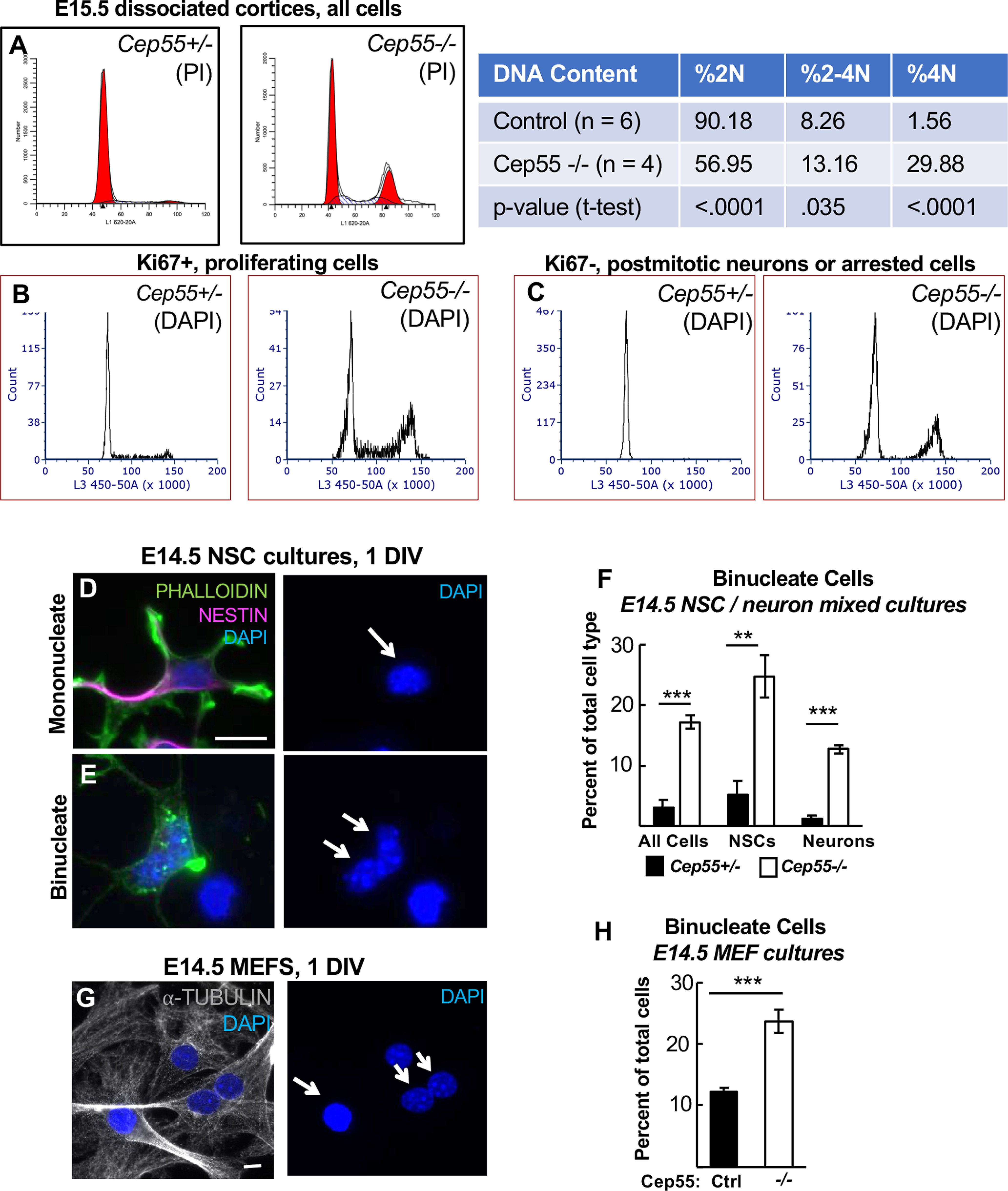
Cep55 KO mice have increased numbers of binucleate cortical cells and MEFs. A, Flow cytometric analysis of E15.5 dissociated cortices labeled with propidium iodide (PI) indicates an increase in cells with tetraploid (4N) DNA content in Cep55−/− brains, and concomitant decrease in cells with 2N DNA content. B, C, Both proliferating (Ki67+) and nonproliferating (Ki67–, presumed neurons) populations of −/− brains show increases in 4N DNA cells. D–F, Increased numbers of binucleate progenitors (Nestin+) and neurons (Nestin–) are seen in Cep55−/− dissociated cortical cultures at 1 DIV. D, Arrow indicates example of mononucleate progenitor. E, Arrow indicates example of binucleate neuron. G, H, Primary cultures of MEFs from −/− embryos have twice as many binucleate cells as control cultures at 1 DIV. Arrow indicates example of mononucleate cell. Double arrow indicates example of binucleate cell. A, n = 6 Cep55+/+;+/−, 4 −/− dissociated cortices. B, C, n = 5 +/+;+/−, 5 −/− dissociated cortices. F, n = 4 +/+;+/− and 4 −/− coverslips from 2 embryos each. H, n = 5 +/−, 5 −/− coverslips from 3 embryos each. Scale bars: D, G, 10 µm. **p < 0.01. ***p < 0.001. A, F, H, t test.
For immunofluorescence on coverslips of dissociated cortical progenitors and MEFs (see Figs. 4, 7, 8, 10, and 12), a similar protocol was used but with primary antibodies applied for 3 h at room temperature. Antigen retrieval was not used in dissociated progenitors, except in the case of BrdU and Tbr2 immunostaining; coverslips were immersed in 0.07 m NaOH, pH 13, for 2 min before permeabilization. Coverslips were mounted on Superfrost Plus slides with Fluoro-Gel (Electron Microscopy Sciences, 17985-10).
Figure 4.

Cep55 protein localizes in late-stage midbodies of NSCs, BPs, and MEFs. A, B, Representative images of Cep55+/− control and Cep55−/− E14.5 NSCs undergoing cytokinesis that were cultured for 1 day in vitro (DIV) fixed and immunostained for endogenous Cep55 (red), Aurora kinase B (green), Nestin for NSCs (gray), and DAPI. Cep55 is not detectable in mitotic spindles or centrosomes in metaphase, anaphase, or early midbody stage NSCs but accumulates in the central bulge of late midbodies and in MBRs in control (A) but is undetectable in KO (B) NSCs. C, Quantification of endogenous Cep55 immunofluorescence signal intensity in control and −/− NSC midbodies. D, Cep55 in a late-stage midbody of a BP from control brain (left). Cep55 does not localize to midbodies of BPs from KO brains (right). E, Cep55 signal was not detected at centrosomes, marked by pericentrin, in NSCs (open arrows), but is detected in a nearby MBR (closed arrow). F, G, Cep55+/+ MEFs show a similar pattern of Cep55 localization as NSCs, with accumulation only in late-stage midbodies (F), but is undetectable in −/− MEF midbodies (G). Scale bars: A, metaphase and early midbody: 5 µm; D, 4 µm; E, 5 µm; F, 10 µm; inset, 5 µm. ****p <0.0001, t test.
Figure 7.
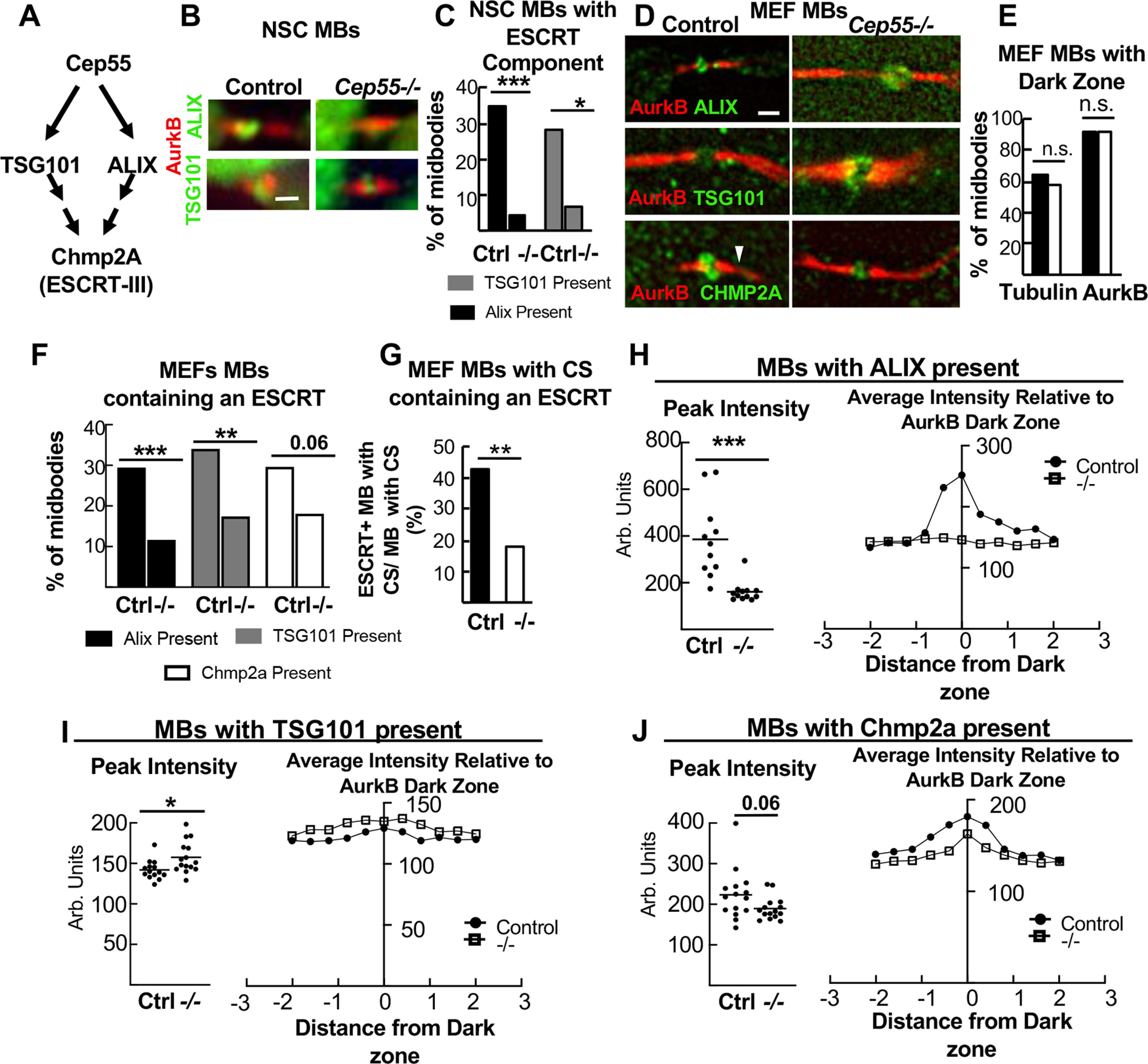
Cep55 KO NSCs and MEFs have reduced, but not eliminated, ESCRT recruitment to midbodies. A, Model from the literature for Cep55 recruiting ESCRTs in midbodies (MBs). B, Representative images of MBs of NSCs, from control or Cep55−/− E14.5 embryos, immunostained for ALIX or TSG101. C, −/− NSC MBs are much less likely to have detectable Alix or TSG101. Alix: n = 49 NSC control MBs (3 animals, 3 coverslips); n = 46 −/− MBs (2 animals, 3 coverslips); TSG101: n = 32 control (2 animals, 2 coverslips), n = 45 −/− (3 animals, 3 coverslips); all, 1 DIV. D, Representative images of MBs of 1 DIV MEFs, from control or −/− E14.5 embryos, immunostained for ALIX, TSG101, or Chmp2A. Arrowhead indicates constriction site (CS). E, Central dark zones were detected at normal percentage in −/− MEF MBs, using α-tubulin or AurkB immunostaining. n = 402 control, 355 −/− MBs (5 animals, 6 coverslips each). F, Cep55−/− MEF midbodies are less likely to have detectable Alix, TSG101, or Chmp2a than controls. Alix: n = 153 control, 129 −/− MBs (4 animals, 4 coverslips each); Tsg101: n = 141 control, 132 −/− MBs (4 animals, 4 coverslips each); Chmp2a: n = 108 control, 105 −/− MBs (3 animals, 3 coverslips each). G, The % of MBs with CSs that contain detectable ESCRT is decreased in −/− MBs. n (MBs with CSs) = 110 control, 95 −/− (5 animals, 11 coverslips each). H, I, MBs with ESCRT present were analyzed by fluorescence intensity line scans through the central dark zones. Alix peak is severely reduced in −/− MBs, but Tsg101 and Chmp2a peaks are not. Alix: n = 11 control, −/− MBs (2 animals, 2 coverslips, 1 experiment); Tsg101: n = 14 control, 15 −/−MBs (2 animals, 2 coverslips, 1 experiment); Chmp2a: n = 15 control, −/− MBs (2 animals, 2 coverslips, 1 experiment). Scale bars: B, D, 1 µm. *p < 0.05; **p < 0.01; ***p < 0.001; C-G, Fisher's exact test; H-J, t test.
Figure 10.
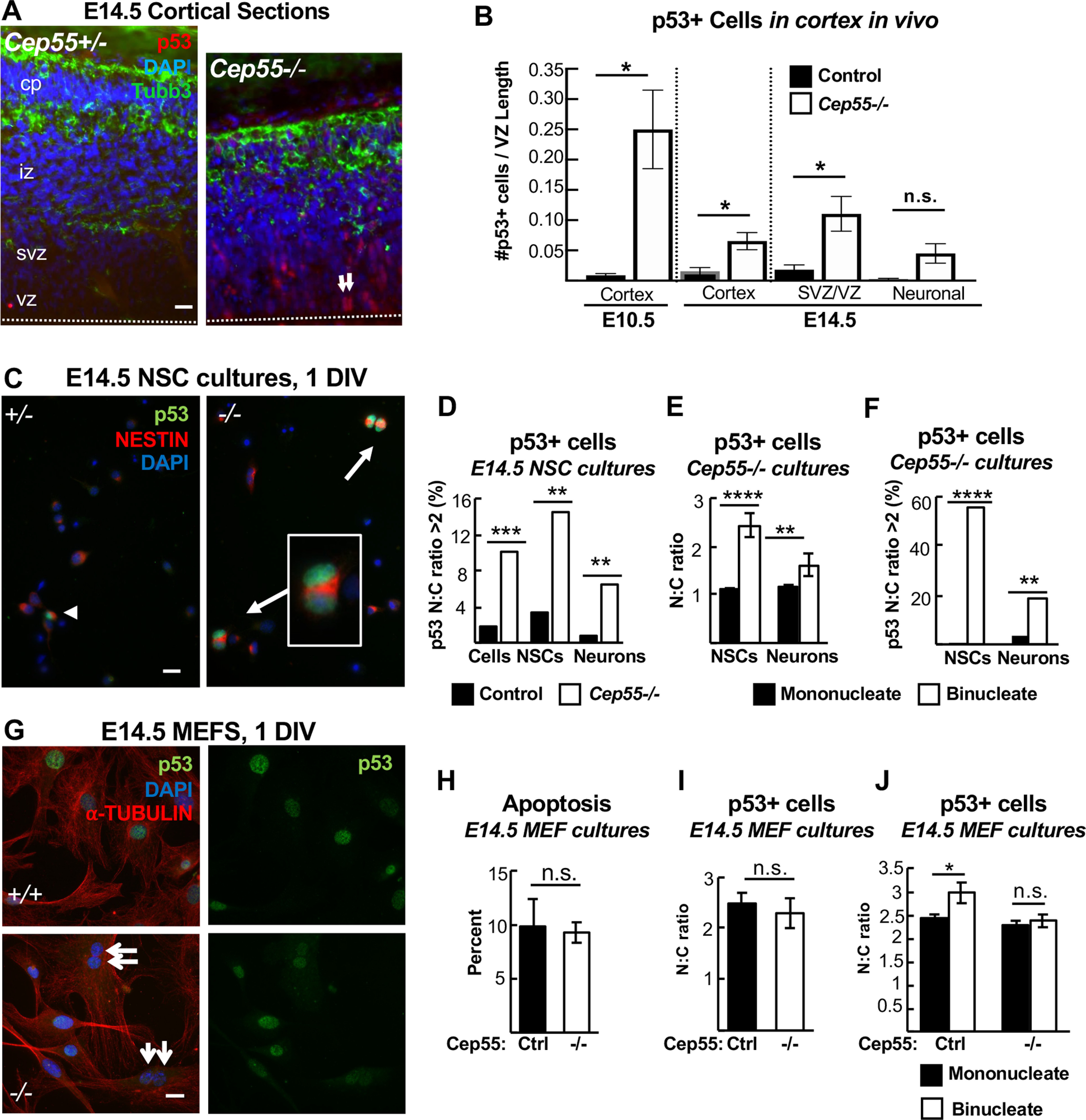
p53 nuclear accumulation is increased in Cep55 KO binucleate NSCs and neurons, but not in binucleate MEFs. A, B, Cortical sections immunostained for p53 and Tubb3 show almost no p53+ cells in controls but many in Cep55−/− cortices. Arrows indicate paired nuclei with p53 expression. B, p53+ cell counts are increased in Cep55−/− cortices at E10.5, and at E14.5, primarily in the vz/svz. C, Images of dissociated NSCs cultured 1 DIV show p53+ binucleate NSCs from −/− cortices. D, Cells with a p53 N:C ratio of >2 are greatly increased in −/− cultures, both NSCs and neurons. E, In −/− NSC cultures, the mean N:C ratio of p53 intensity is ∼1 in mononucleate NSCs or neurons, but significantly higher in binucleate progenitors and neurons. F, In −/− cultures, over half of binucleate NSCs have a p53 N:C ratio >2, compared with only 1% of mononucleate NSCs. Among binucleate neurons, ∼20% have a p53 N:C ratio of >2 versus only 2% of mononucleate neurons. H, Apoptosis (CC3+) is not increased in −/− primary MEFs cultured 1 DIV. I, The N:C ratio of p53 signal (p53, green, G) is not different in −/− MEFs compared with controls. J, Binucleate −/− MEFs (G, arrows) did not have an increased p53 N:C ratio compared with mononucleates. A, Dashed line indicates apical membrane. B, n = 3 +/+;+/− and 3 −/− mice at each age. D-F, n = 4 +/+ or +/− and 4 −/− coverslips from 2 embryos each; 124 control and 137 −/− NSCs; 143 −/− and 184 −/− neurons for N:C ratios. H, n = 3 control and 3 −/− coverslips from 3 embryos each. I, n = 195 control and 232 −/− cells from 3 control and −/− coverslips and embryos each. J, n = 153 mononucleate and 25 binucleate control cells and 158 mononucleate and 68 binucleate −/− cells from 3 coverslips and 3 embryos each. Scale bars: A, C, G, 20 µm. *p < 0.05. **p < 0.01. ***p < 0.001. ****p < 0.0001. All experiments, t test.
Paraffin-embedded brains were sectioned and stained with H&E by the UVA Research Histology Core for Figures 1 and 2, and Extended Data Figure 11-1.
Figure 2.
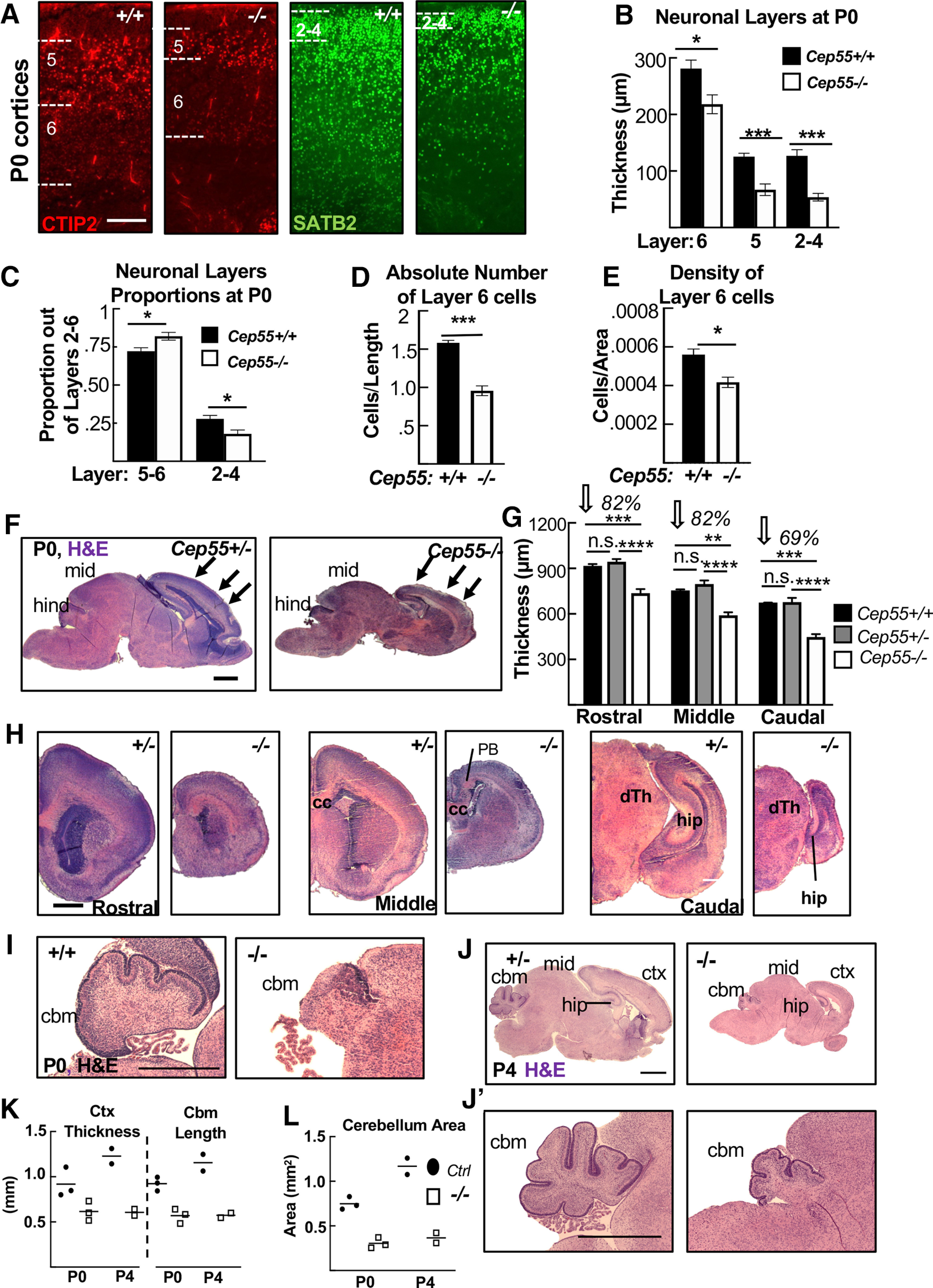
Disruption of cortical and cerebellar structure in Cep55 KO brains. A, Representative images of +/+ and −/− cortical sections labeled with antibodies to Ctip2, marking layers 6 (faint staining) and 5 (bright staining), and Satb2, marking layers 2-4. B, All neuronal layers are significantly thinner in −/− cortices at P0. C, Layers 5-6 occupy a larger proportion of cp, whereas layers 2-4 occupy a decreased proportion of the cp in −/− cortices. D, The absolute number of layer 6 cells (no. of nuclei/length of cortex) is reduced by 40% in −/− cortices. E, −/− layer 6 neurons are 25% less dense than WT cells (no. of nuclei/area). B, C, n = 5 +/+ and 4 −/− mice. D, E, n = 3 +/+ and 3 −/− mice. F, H, Representative Cep55+/− and −/− sagittal (F) and coronal (H) sections stained with H&E. F, Arrows indicate approximate locations of rostral, middle, and caudal, coronal sections in C. mid, Midbrain; hind, hindbrain; PB, Probst bundles of axons; hip, hippocampus; cc, corpus callosum; dTh, dorsal Thalamus. G, The mean cortical thickness in −/− brains is 82% of normal in rostral and middle sections, and only 69% of normal in caudal sections. G, n = 3 +/+, 5 +/−, and 5 −/− mice. I-J′, Representative control and −/− sagittal sections of P0 (I) and P4 (J,J′) brains. ctx, Cortex; cbm, cerebellum. K, L, Cortex thickness, cbm length, and cbm area are reduced in −/− mice at P0 and P4. P0, n = 2 +/+, 1 +/−, and 3 −/− pups; P4, n = 1 +/+, 1 +/−, and 2 −/− pups. Scale bars: A, 100 µm; F, H, I, 500 µm; J, J′, 1 mm. *p < 0.05. **p < 0.01. ***p < 0.001. ****p < 0.0001. F, One-way ANOVA. others: t test.
Cortical cell cultures
Cells were dissociated from E14.5 cortices following a protocol adapted from Sally Temple's laboratory (Qian et al., 1998). The Worthington Papain Dissociation Kit was used to dissociate cells (Worthington Biochemical, catalog #LK003150). Cells were cultured in DMEM with Na-pyruvate, L-glutamine, B-27, N2, N-acetyl-cysteine, and basic fibroblast growth factor (Final Culture Media). For Figure 12, 10 μm BrdU was added to the cultures at plating and then washed out after 6 h. After 24 or 48 h, cells were fixed by adding an equal volume of room temperature 8% PFA for 5 min to cell media, followed by removal of media and addition of −20°C cold methanol for 5 min.
MEF cultures
Embryos from timed pregnant females were collected at E14.5. Embryos were decapitated, internal organs were removed from body cavities, and remaining tissue was triturated with a 1 ml pipette and incubated in 1 ml of 0.25% trypsin-EDTA for 25 min at 37°C. The tissue was further triturated and tubes were spun at 1000 rpm for 5 min, and the medium was aspirated off. The pellet was resuspended in DMEM/20% FBS with penicillin/streptomycin added. The resulting cells from each embryo were plated in two 10 cm plates in DMEM/10% FBS. The next day, media was replaced. When approaching confluence, cells were trypsinized and plated on fibronectin-coated coverslips, grown overnight, and fixed with 4% PFA for 10 min or cold methanol for 10 min at 24 or 48 h.
Cortical slabs for apical membrane view
Cortical slabs were prepared as previously described (Janisch and Dwyer, 2016). The meninges and skull were removed to expose the brain in E12.5 and E14.5 embryos, followed by fixation with 2% PFA for 20 min. Next, apical slabs were made by pinching off cortices, flipping so that the apical surface was upright, and trimming to flatten the slab. Slabs were fixed for another 2 min with 2% PFA followed by blocking with 5% normal goat serum for 1 h. Primary antibodies were applied for 1 h at room temperature and then moved to 4°C overnight. The next day, after three 10 min PBS washes, secondary antibodies and DAPI were applied at a concentration of 1:200 for 30 min. After two more 10 min PBS washes, slabs were coverslipped with VectaShield fluorescent mounting medium (Vector Laboratories, H-1000) and imaged. z-stack depth was 8-20 μm, and step size was 0.5 μm.
Flow cytometry
Cells from E15.5 brains were dissociated using the Papain Dissociation Kit (Worthington Biochemical). Single-cell suspensions were obtained by filtering through a 40 μm filter (BD Falcon Cell Strainer, blue nylon mesh, catalog #352340). For propidium iodide staining, cells were resuspended in 500 μl PBS and added to 4.5 ml ice-cold 70% ethanol for at least 2 h. Samples were stored at 4°C. Fixed cells were rinsed in PBS and resuspended in 1 ml solution containing 100 μg/ml RNase A, 0.1% Triton X-100, and 50 μg/ml propidium iodide, and incubated at room temperature for 30 min in the dark. For Ki-67 and DAPI analysis, single-cell suspensions of E15.5 brains (n = 3 pairs of Cep55 mutants and littermate controls) were fixed in 1.5% PFA for 15 min on ice. Cells were then washed twice with FACS buffer (2% BSA, 1 mm EDTA, 0.01% sodium azide, PBS) and permeabilized in 0.1% Triton X-100 in PBS for 30 min on ice. Cells were washed twice in FACS buffer, and 2 × 106 cells were incubated in 100 μl FACS buffer with 1 μg/ml DAPI and 2 μl anti-Ki-67 antibody (monoclonal rat anti-Ki-67 AlexaFluor-647, clone SolA15, eBioscience). Following three washes with FACS buffer, fluorescence was measured using a FACSCanto II flow cytometer (Becton Dickinson). At least 20,000 events were collected per sample. Data were analyzed using FlowJo software (TreeStar).
Abscission live imaging and analysis
This method was previously described by McNeely and Dwyer (2020), based on first establishing SiR-tubulin for abscission duration studies in HeLa cells (Janisch et al., 2018). Briefly, E13.5 slabs were prepared as described above and placed apical surface down in a glass-bottom dish (MatTek, P35G-1.0-20-C) containing 50 nM SiR-Tubulin (Cytoskeleton, CY-SC002) in Final Culture Medium (described above). Each dish was placed in a humidifying chamber and into a 37°C incubator with 5% CO2 overnight (∼15 h). The next day, the cortices were removed from the incubator, and prepared for imaging by adding Matrigel (Corning, 356237; 1:3 dilution in Final Culture Medium) and a coverslip (Fisher 12-545-100) over the top of the cortical slab explant. Matrigel was allowed to solidify for 5 min in the incubator before final culture medium with 2% 4-(2-hydroxyl)−1-piperazineethanesulfonic acid (Invitrogen, 15630080) was added to the dish; then, imaging was performed. An Applied Precision (GE Healthcare) DeltaVision with a heating plate and 40× objective (numerical aperture 1.53) was used for time-lapse image acquisition; z stacks of images ∼10 μm deep (z steps 0.4 μm) were taken every 15 min for abscission for up to 6 h. To minimize phototoxicity, the neutral density filter was set at ≤32%, and the exposure time was kept to a minimum (<0.1 ms per slice). Deconvolved full z stacks and maximum intensity projection images were analyzed using ImageJ. For abscission duration, time 0 was midbody formation as ascertained by SiR-Tubulin appearance (compact microtubule bundles). Abscission completion was scored as the time point when there was complete removal of microtubules on a midbody flank ascertained when the SiR-Tubulin signal intensity decreased to background level. Midbody membrane scission was shown to be temporally coincident with midbody flank microtubule disassembly by several previous publications using differential interference contrast or phase imaging of cell lines in two-dimensional dissociated cultures (Steigemann et al., 2009; Elia et al., 2011; Lafaurie-Janvore et al., 2013); however, here, we cannot rule out the possibility that the midbody plasma membrane might remain connected for some period of time after the microtubules are gone.
Antibodies
Antibodies used in this analysis are as follows: mouse monoclonal anti-mouse citron kinase (CitK, 1:100; 611367, BD Biosciences), mouse polyclonal anti-human Cep55 (1:00, H00055165-B01P, Abnova), mouse monoclonal anti-mouse Cep55 (1:100 for immunofluorescence experiments, sc-377018, Santa Cruz Biotechnology), rabbit polyclonal anti-human cleaved-caspase 3 (CC3; 1:250, 9661s, Cell Signaling Technology), rat monoclonal anti-mouse Tbr2 (1:200, 14-4875, eBioscience/Thermo Fisher Scientific), rabbit polyclonal anti-mouse Pax6 (1:200, PRB-278P, BioLegend), mouse monoclonal anti-rat Aurora B kinase (1:300, 611082 BD Biosciences), rabbit monoclonal anti-human Aurora B kinase (1:100, ab2254, Abcam), rat monoclonal α-tubulin (1:300, NB600-506, Novus Biologicals), rabbit monoclonal anti-human phospho-histone H3 (PH3; 1:200, 3458, Cell Signaling Technology), chicken polyclonal anti-mouse Nestin (1:600, NES, Aves Labs), rat monoclonal anti-human Ki67 (1:100, 14-5698, eBioscience), rabbit polyclonal anti-mouse p53 (1:500, NCL-L-p53-CM5p, Leica Biosystems), rat monoclonal anti-human Ctip2 (1:400, 18465, Abcam), rabbit polyclonal anti-mouse Tbr1 (1:200, 31940, Abcam), rabbit monoclonal Satb2 (1:200 Ab92446, Abcam), rabbit polyclonal pericentrin (1:500, 92371, BioLegend), mouse monoclonal Tubb3 (Tuj1) (1:500, 801201, BioLegend), mouse monoclonal PH3 (Ser10) (1:200, 9706, Cell Signaling Technology), Phalloidin Oregon Green or 568 (1:50, 07466, Al2380, Invitrogen), chicken polyclonal anti-human α-tubulin (1:100, ab89984, Abcam), mouse monoclonal Alix (1:100, SC-53538, Santa Cruz Biotechnology), mouse monoclonal Tsg101 (1:100, SC-7964, Santa Cruz Biotechnology), rabbit polyclonal CHMP2A (1:100, 10 477-1-AP Proteintech), rat monoclonal Zo-1 (1:50, R26.4DC, DSHB), polyclonal rabbit anti Zo-1 (1:50 61-7300, rabbit, Invitrogen/Thermo Fisher Scientific), and mouse monoclonal anti-mouse BrdU, clone B44 (20 µl per 50 µl, 347580, BD Biosciences). All antibodies were validated for the application used in multiple previous publications.
Imaging and statistical analysis
Images used for data analysis in Figures 2A–E, 7B, 8D–G, 9A–D, M, P, 10, 11D, 12C–J, and Extended Data Figure 11-1F were collected on either a Carl Zeiss Axio ImagerZ1 microscope with AxioCam MRm or a Carl Zeiss AxioObserver fluorescent widefield inverted scope microscope. Images in Figures 3A–C,F–L, 4-6, 7D, 9E–L, 11F–O, and 12A were taken on an inverted DeltaVision with TrueLight deconvolution microscope with softWoRx Suite 5.5 image acquisition software (Applied Precision (GE Healthcare). A Leica Microsystems MZ16F microscope with DFC300FX camera was used for images in Figures 1, 2F–L, 3D, E, 11A, and Extended Data Figures 1-1 and 11-1A-E. Control and mutant fluorescence images were collected with the same exposure times and on the same day. All image analysis was performed in ImageJ/Fuji, and any changes to brightness and contrast were applied uniformly across images. Statistical analyses were performed using Excel (Microsoft) or GraphPad Prism software. The sample sizes were predetermined based on our laboratory's previous experience with cortical development analyses and others' published results. After obtaining pilot data, power analyses were performed, if necessary, to determine whether the number of samples obtained was high enough for the effect size seen. NSC cultures that were unhealthy were not imaged and analyzed, but no other data were excluded from the analysis. No randomization or blinding was used as no experimental manipulation was applied other than genetic KOs. Genotyping was performed after collection of embryos to determine genetic status. Statistical tests used are specified in each figure legend. For each sample set, a statistical test of normality was performed using GraphPad PRISM software. Parametric tests were used when sample sets had a normal distribution, and nonparametric tests were used when sample sets did not have a normal distribution. Variance was calculated and was typically similar between groups. All graphs with error bars are mean ± SEM.
Figure 11.
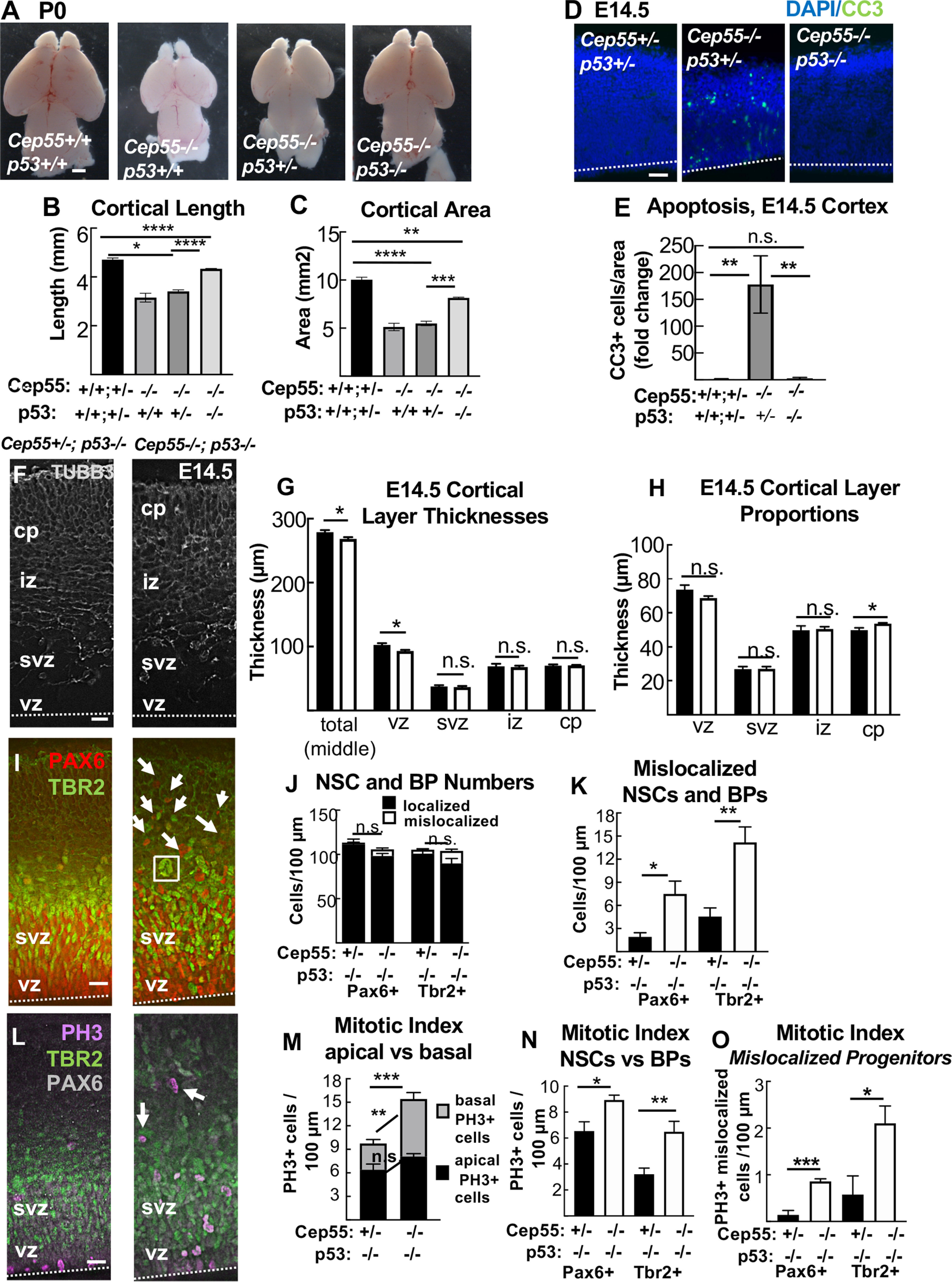
p53 KO blocks apoptosis and partially rescues brain size but exacerbates progenitor defects of Cep55 KO. A–C, Codeletion of p53 partially rescues microcephaly in Cep55−/− mice. B, C, Homozygous p53 KO shows partial rescue of Cep55−/− cortical length and area. D, E, Apoptosis is fully blocked in Cep55−/−;p53−/− dKOs. F, Representative images of E14.5 cortical sections from control (Cep55+/−;p53−/−) and dKOs immunostained for neuron marker Tubb3 (gray) show cp and axons in the iz. In dKO images, there is disorganization of the cp and iz boundary. G, Mean thicknesses of total cortex and vz are slightly decreased in dKO brains. H, The dKO cp occupies proportionally more of cortical width than in controls. I, Cortical sections stained for NSC marker Pax6 (red) and BP marker Tbr2 (green) show NSCs and BPs in the vz and svz of dKO brains are more disorganized, with many nuclei mislocalized basally above the svz (arrows), and some large nuclei (square). J, K, There is no difference in the number of total or localized NSCs (Pax6+) or BPs (Tbr2+) per cortical length in dKOs. L, PH3 (magenta) immunostaining is used to mark cells in mitosis. Mislocalized mitotic cells are noted in dKO cortices (arrows). M, dKO cortices show increased numbers of mitotic cells, with a normal number of mitotic cells at the apical membrane but an increased number basally. N, The mitotic index of both NSCs (Pax6+) and BPs (Tbr2+) is significantly increased in dKO cortices. O, There are increased numbers of mitotic NSCs mislocalized above the apical membrane, and mitotic BPs mislocalized above the svz, in dKO sections. D, F, I, L, Dashed line indicates apical membrane. B, C, n = 11 controls (Cep55+/+,+/−;p53+/+,+/−), 2 Cep55−/−;p53+/+, 5 Cep55−/−;p53+/−, 3 Cep55−/−;p53−/−. E, n = 3 controls (Cep55+/+,+/−;p53+/+,+/−), 2 Cep55−/−;p53+/+, 3 Cep55−/−;p53−/−. F-O, n = 5 control (Cep55+/+,+/−;p53−/−) and 5 Cep55−/−; p53−/− brains. Scale bars: A, 0.5 mm; D, 40 µm; F, I, L, 20 µm. *p < 0.05. **p < 0.01. ***p < 0.001. ****p < 0.0001. B, C, E, One-way ANOVA; others, t test.
Results
Cep55 KO results in microcephaly with thin neuronal and axonal layers
The mutation of Cep55 used in this analysis is a 600 base pair (bp) deletion encompassing all of exon 6 and flanking intronic sequence, generated by the CMMR (see Materials and Methods). The total murine Cep55 gene consists of 9 exons encoding a protein of 462 amino acids (AA) (Fig. 1A). Cep55 protein domains include two coiled-coil regions (CC1 and CC2) surrounding the ESCRT- and Alix-binding region, and two ubiquitin binding domains in the C-terminus (Morita et al., 2007; Lee et al., 2008; Said Halidi et al., 2019). This deletion is predicted to result in a frameshift starting at AA 227, resulting in multiple premature stop codons starting after AA 237. The Cep55 protein is undetectable by immunoblot of homozygous mutant tissue lysates (Fig. 1B).
Given that Cep55 was reported to be essential for mammalian cell division (Fabbro et al., 2005; Zhao et al., 2006), we were surprised to discover that Cep55−/− mice are born alive at expected Mendelian ratios, and appear grossly normal with a slightly small body (Fig. 1C). However, while the pups survive postnatally, they fail to thrive and usually die before weaning (Extended Data Fig. 1-1A-C). Upon inspection, Cep55−/− pups have slightly flattened heads (Fig. 1C, arrow; Extended Data Fig. 1-1D), and a 10% reduction in body length (Fig. 1D). Removing the brains from the heads revealed that Cep55−/− mice have severe microcephaly, with a 50% reduction in cortical area and 30% reduction in cortical length on postnatal day 0 (P0) (Fig. 1E,G,H). Dissection of prenatal brains showed a similar reduction in brain size compared with controls at embryonic day (E)14.5 and a smaller (8%) reduction at age E12.5 (Fig. 1F–H). Cortical size is disproportionately reduced compared with body size (Fig. 1I). However, eye size is normal in Cep55−/− pups (Extended Data Fig. 1-1D–F). Thus, Cep55 loss in mice appears to more drastically disrupt brain development than body development, and the reduction in cortical growth correlates with the onset of neurogenesis around E12.5.
In addition to reduced brain size, Cep55−/− mice have reduced cortical thickness at P0 (Fig. 1J). To begin to address the cellular cause of the thinner cortex in Cep55 KOs, we compared the thicknesses of individual cortical layers in P0 control and KO brains. Cortical stem (NSC) and progenitor cells reside in the ventricular zone (vz) and subventricular zone (svz), whereas postmitotic neurons reside in the cortical plate (cp) and project their axons in the intermediate zone (iz). The cp and iz of Cep55−/− cortices are significantly reduced in thickness, whereas the vz and svz are not (Fig. 1J,K), suggesting a deficit of neurons. To examine the neuronal layers within the cp, we labeled control and Cep55−/− cortical sections with Ctip2, for layers 5 and 6, and Satb2, for layers 2-4 (Fig. 2A). Cep55−/− cortical layers are ordered similarly to controls, but all are reduced in thickness (Fig. 2B). As a proportion of the total cp, the deep layers 5-6 of Cep55−/− brains occupy more space, whereas the upper layers 2-4 occupy less (Fig. 2C). Interestingly, both the absolute number and the density of nuclei in layer 6 are significantly reduced (Fig. 2D,E). Together, these data show that the reduced thickness of Cep55−/− cortices at birth is attributable to reduced thickness of all neuronal layers and reduced number of neurons.
To determine whether Cep55−/− mice have differential effects in different brain regions, we analyzed P0 cross sections. We noted a reduction in size of all brain regions, including the forebrain, midbrain, and hindbrain (Fig. 2F). Interestingly, while cortical thickness is reduced globally, the caudal cortex is the most severely affected (Fig. 2G,H). Cep55−/− mice also have smaller cerebellum area, with reduced foliation and little growth postnatally (Fig. 2I–L). These data show that Cep55 is required for growth of multiple parts of the brain, but also reveal regional differences in the requirement for Cep55 function.
Cep55 KO brains exhibit cortical layer disorganization and altered NSC numbers during corticogenesis
To elucidate the developmental defects leading to smaller brain size at birth, we examined earlier stages of cortical development. Because Cep55−/− brains are already dramatically reduced in size at E14.5, and this is an age when the cp is forming, we analyzed numbers and positions of NSCs, basal progenitors (BPs), and neurons at this age. By labeling E14.5 sections with neuronal tubulin (Tubb3), we noted the cp was already thinner in Cep55−/− cortices, and the iz layer of axons was not distinct (Fig. 3A). Cep55−/− cortical thickness is reduced by 12% at E14.5 (compared with 25% at P0; Fig. 1K), and this reduction is in the neuronal layers (cp and iz) (Fig. 3B,C). Analyses of H&E-stained sections confirmed a thinner cp and iz, and showed a slightly thicker marginal zone (mz) (Fig. 3D,E).
The neurons of the cp are daughters of cell divisions of NSCs and BPs. NSCs divide many times, first symmetrically to multiply the NSCs, and then asymmetrically to produce BPs and neurons. BPs divide symmetrically to produce two neurons. Neuron daughters exit the cell cycle and differentiate. To determine whether the organization and numbers of NSCs and BPs in E14.5 Cep55−/− cortices are altered, we used Pax6 and Tbr2 antibodies to mark them, respectively (Fig. 3F). We observe a decrease in the number of NSCs in Cep55−/− cortices, while the number of BPs is normal (Fig. 3G). Notably, NSC and BP positioning appears disorganized, with a small but significant number of NSC nuclei mislocalized at positions basal to the svz (Fig. 3F, arrows; Fig. 3G,H).
Cep55 is not thought to have a primary role in mitosis (Fabbro et al., 2005). To check this, we measured mitotic index in KO brains, using PH3 (Fig. 3I). There appears to be a trend for increased numbers of mitotic cells in Cep55−/− cortical sections (6.69 vs 9 cells per 100 µm; p = 0.11) (Fig. 3J); however, the increase is in basally rather than apically positioned mitotic nuclei, and is because of an increase in the mitotic index of BPs, not NSCs (Fig. 3K). This suggests that there is not a primary defect in mitosis duration in Cep55−/− NSCs.
We next investigated whether the cellular phenotypes observed in Cep55−/− cortices at E14.5 begin at younger ages. At E12.5, when there is only a thin neuronal layer (preplate), Cep55−/− cortical thickness and preplate thickness were not significantly different from controls, although the preplate was slightly thicker proportionally (Fig. 3L–O). At the same time, the number of BPs in Cep55−/− cortices is slightly decreased, suggesting that there is a deficit in their production (Fig. 3P). Overall, these data demonstrate that Cep55 is required for early stages of corticogenesis, with subtle abnormalities apparent by E12.5, resulting in more dramatic deficits in progenitors and neurons at E14.5. These could reflect direct effects of Cep55 loss, or also indirect effects of NSCs on their daughter cells.
Cep55 is expressed in proliferative cells in the brain and body, and is specifically detected in midbodies during late abscission
Having found defects in Cep55−/− neurons, NSCs, and BPs, we needed to ascertain in which of these cell types Cep55 is expressed. Previous RNA sequencing experiments found Cep55 mRNA expressed in proliferating cell types in early mouse embryos (Gao et al., 2017), and in the murine cerebral cortex at E14.5 and P0 (Loo et al., 2019). RNA ISH shows that Cep55 is expressed in proliferative zones of the cortex and eye at E14.5, but is not detectable in neuronal layers. Lower expression in other proliferating tissues is observed (Visel et al., 2004) (Extended Data Fig. 4-1A-C). We tested for endogenous Cep55 protein expression and subcellular localization by immunohistochemistry on control and KO cells, and detected Cep55 protein in dividing NSCs of control embryos, but not KOs (Fig. 4A–C). Interestingly, Cep55 is specifically detected only at late stages of abscission: in late midbodies at the central bulge of the midbody, and in the postabscission MBRs. No specific Cep55 signal was detected in metaphase, anaphase, or early midbodies. In BPs from control brains, Cep55 has the same localization pattern but is absent in BPs from KO brains (Fig. 4D). We do not detect Cep55 protein at centrosomes (Fig. 4E), concurring with others (Zhao et al., 2006; Bastos and Barr, 2010).
To investigate whether Cep55 is expressed in cells of non-neural tissues, we dissociated MEFs from E14.5 control and Cep55−/− embryo bodies. We noted the same expression pattern in MEFs as in NSCs, with detectable signal above background only in late-stage midbodies of control MEFs (Fig. 4F,G). Thus, we find that Cep55 is expressed in multiple proliferating embryonic cell types (NSCs, BPs, and MEFs) and that it accumulates specifically at the late stage of abscission in the central bulge of the midbody.
Midbody defects in fixed cortices of Cep55 KOs are consistent with delays in NSC abscission
Based on the specific localization of Cep55 to late-stage NSC midbodies, as well as the prior reports of a requirement for Cep55 in completion of abscission in cell lines, we hypothesized that microcephaly in Cep55−/− mice is because of a primary defect in abscission completion. However, the growth of the brain (albeit less than normal) and almost normal body size of Cep55 KOs suggest that abscission is completing in most cells. To begin to address whether abscission processes in Cep55−/− brains are normal, we used a fixed cortical slab preparation in which we can image and quantitatively analyze many NSCs undergoing mitosis and their midbodies at different stages of abscission (Fig. 5A,B) (Janisch et al., 2013; Janisch and Dwyer, 2016). By labeling the NSC apical cell junctions with ZO-1, we observed that Cep55−/− endfeet are more variable in size, with many abnormally large endfeet (Fig. 5C). Indeed, quantification shows a striking decrease in the density of apical endfeet (Fig. 5D). Since the apical membrane expands during mitosis, we quantified the apical mitotic index of NSCs, using PH3, but it is not altered in Cep55−/− cortices (Fig. 5C, left panels, Fig. 5E), consistent with our finding in cross sections (Fig. 3J). However, the midbody index, the percent of NSCs in abscission, is increased by ∼25% at E14.5 (Fig. 5C, right panels, Fig. 5F). These data suggest that, in Cep55 KO cortices, NSCs may take longer to complete abscission. As the abscission process proceeds, midbodies mature by becoming thinner, and then forming constriction sites on each flank, where microtubule disassembly and membrane scission occurs (Guizetti et al., 2011). We observe fewer midbodies with constriction sites in Cep55−/− brains (Fig. 5G), and more short midbodies (Fig. 5H). These data suggest that, without Cep55, midbodies are able to compact their microtubules to become thinner as they mature, but have a defect in making the constriction sites, and abscission duration is longer than normal.
Figure 5.
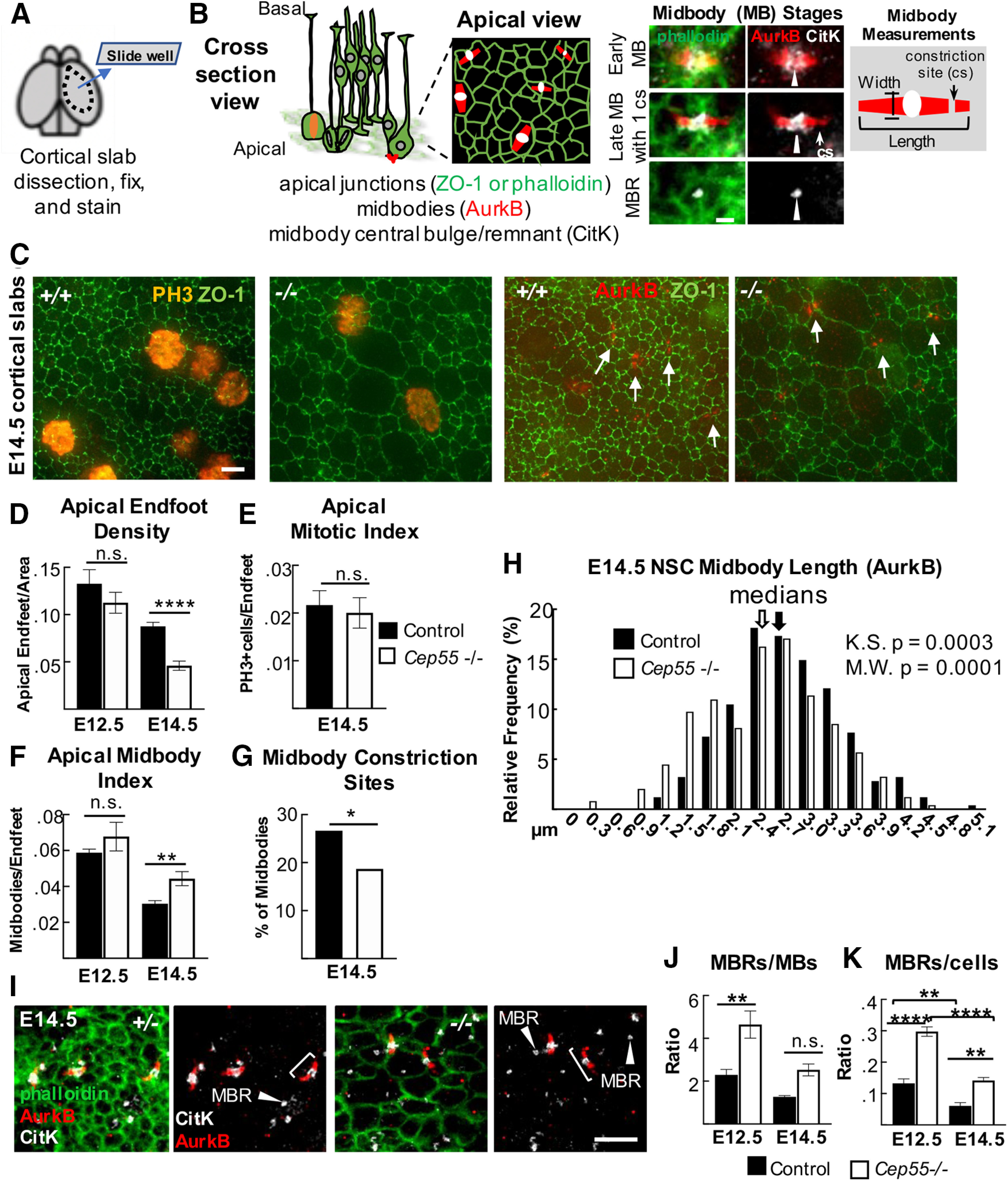
Cep55 KOs display NSC midbody defects in fixed cortical slab preparations. A, B, Schematics of cortical slab dissection, cross section versus apical membrane views of NSCs undergoing cytokinesis. Images of midbodies (MB) at different stages of the abscission process: early (wider), late-stage (thinner), and an MBR. C, Representative images of E14.5 cortical slabs immunostained for apical junctions (ZO-1), mitotic chromatin (PH3+), or MBs (AurkB +, arrows). D, Mean apical endfoot density is reduced in −/− slabs. E12.5: n = 4 +/− slabs (4 brains), 4 −/− slabs (4 brains). E14.5: n = 8 control slabs (6+/+, 2+/− brains) and 7 −/− slabs (6 brains). E, F, Mitotic index is normal, but MB index is significantly increased. E, n = 6 control slabs (4 +/+, 2 +/− brains), 6 −/− slabs (5 brains). F, E12.5: n = 4 +/− slabs (4 brains), 4 −/− slabs (4 brains). E14.5: n = 6 control slabs (4 +/+, 2 +/− brains), 5 −/− slabs (5 brains). G, A smaller percentage of −/− MBs have visible cs. H, −/− MBs tend to be shorter than controls (measured by AurkB). Medians: 2.7 µm for +/+, 2.5 µm for −/−. Bin = 0.3 µm. G, H, n = 353 control MBs (5 +/+, 2 +/− brains), 246 −/− MBs (5 brains). I, Cortical slabs immunostained for AurkB to mark pre-abscission MB flanks (brackets), and CitK to mark MB bulges and post-abscission MBRs (arrowheads). J, K, MBRs are increased in −/− brains, normalized to MB number or NSC (endfoot) number. J, K, E12.5 and E14.5: n = 4 control slabs (2 +/+, 2 +/− brains), 4 −/− slabs (4 brains). Scale bars: B, 1 µm; C, 2 µm; I, 5 µm. *p < 0.05. **p < 0.01. ****p < 0.0001. D, F, G, t test, J, K, ANOVA; E, Fisher's exact test.
Since Cep55 protein is abundant in MBRs (Fig. 4A), we investigated whether Cep55 loss alters MBR numbers at the apical membranes of NSCs, using CitK as a marker (Gruneberg et al., 2006; Ettinger et al., 2011). Remarkably, there are approximately twice as many MBRs present on the apical membranes of Cep55−/− brains as in control brains, whether normalized to midbody number or cell number (Fig. 5I–K). This could be another manifestation of delayed abscission, at a late stage when the flanks are no longer detectable with Aurora B staining. It could also represent a role of Cep55 in MBR disposal. Together, these analyses in fixed brains show midbody abnormalities that are consistent with delayed abscission of NSCs when Cep55 is absent.
Cep55 KO NSCs can complete microtubule disassembly in abscission, but it is delayed
To directly test whether abscission is delayed or fails in NSCs of Cep55−/− cortex, we performed time-lapse imaging of abscission in vivo, in live cortical slab explants, using a method we developed previously (McNeely and Dwyer, 2020). By dissecting cortical slabs from embryonic brains of a membrane-GFP mouse line, and incubating them with the cell-permeable far-red fluorescent microtubule dye SiR-tubulin, we can image individual NSCs at the apical membrane undergoing cytokinesis from furrowing to abscission. As the cleavage furrow ingresses toward the apical membrane, the midbody forms as a tight bundle of microtubules, the central bulge becomes apparent, and then microtubule disassembly occurs on each midbody flank independently (Fig. 6A). We previously showed that, in control E13.5 NSCs in vivo, abscission takes on average 50 min to complete, with a wide range of times observed, and microtubule disassembly occurs on both sides of the midbody in the majority of divisions (McNeely and Dwyer, 2020).
Figure 6.

Cep55 KO causes delayed midbody microtubule disassembly during NSC abscission. A, Schematics and time-lapse images of an E13.5 NSC in a cortical slab explant undergoing midbody (MB) abscission. Microtubules are labeled by SiR-Tubulin. Distinct steps shown are as follows: MB formation, flank 1 thinning, microtubule disassembly on flank 1 (first abscission), and microtubule disassembly on flank 2 (second abscission). After bilateral flank disassembly, the MBR is left at the apical membrane. B–D, Time from MB formation to complete first abscission and second abscission are increased in Cep55−/− NSCs, but there is no change in the percentage of bilateral abscissions detected. E, F, Cumulative frequency plots for the first abscission and second abscission show the curves are shifted to the right in −/− NSCs. G, Control NSC with a bipolar spindle (asterisks at spindle poles) proceeded to form a bipolar midbody at 60 min. Cep55−/− NSC with a tripolar spindle (asterisk at spindle poles) proceeded to form a tripolar midbody at 105 min (arrowheads point to midbody “flanks”). H, Tripolar spindles are increased in −/− cortices. B, C, E, n = 54 control cells (2 +/+, 1 +/− slabs), 46 −/− cells (4 slabs). D, F, n = 43 control cells, 39 −/− cells. G, n = 71 control cells (2 +/+, 1 +/− slabs) and 86 −/− cells (4 slabs). Scale bars: A, 1 µm; G, 10 µm. *p < 0.05. **p < 0.01. B, D, t test. E, F, Kolmogorov–Smirnov and Mann–Whitney tests. C, H, Fisher's exact test.
Using this method, we quantified several aspects of the abscission process in Cep55−/− NSCs: midbody formation, midbody structure, and time to abscission (ascertained as microtubule disassembly) on one or both flanks. Unexpectedly, we found that the vast majority (92%) of Cep55−/− NSCs are able to complete abscission. However, these KO NSCs do have a significant increase in average time to the first abscission (delay of 23 min; Fig. 6B). They are also able to complete bilateral abscissions: 80% of both control and Cep55−/− NSCs have observable second abscissions on the other midbody flank (Fig. 6C). But again, the time to the second abscission is significantly increased in KO NSCs (Fig. 6D). Cumulative frequency plots illustrate the slower abscission kinetics in the Cep55−/− NSCs (Fig. 6E,F).
We did observe a small number of Cep55 KO NSCs that displayed abnormalities at an earlier step of cytokinesis. Notably, there is a significant increase in the number of Cep55−/− NSCs that have tripolar mitotic spindles (7%, 6 of 81 KO NSCs imaged vs 0 of 71 in controls) (Fig. 6G,H). Of these 6 NSCs with tripolar spindles, 4 progressed into tripolar midbodies, which remarkably were still able to undergo microtubule disassembly, 1 regressed, and 1 drifted out of the imaging field. We observed a few Cep55−/− NSCs that had bipolar spindles that initiated a furrow but did not complete (4 of 50, compared with 0 of 54 control NSCs). In 3 of these NSCs, the furrow only partly ingressed and then regressed, so the midbody never formed, while in the other, the midbody formed, but then the membrane regressed. Together with our preceding data on Cep55 protein localization and fixed midbody abnormalities, these findings indicate that the primary function of Cep55 in NSCs is to promote timely abscission, with particular effects on the kinetics of late steps, such as constriction site formation and microtubule disassembly.
Cep55 KO NSCs and MEFs have decreased but not eliminated ESCRT recruitment to late-stage midbodies
Since Cep55−/− NSCs have delayed abscission, and Cep55 is thought to recruit other abscission proteins, we wanted to determine whether their localization was altered. The current model for the mechanism of abscission, developed from mammalian cell lines and invertebrates, proposes that, at the late stage of abscission, ESCRT components are recruited to the midbody, and then form helical filaments extending from the central bulge to constriction sites, compacting microtubules and pulling the plasma membrane close enough for membrane scission (Guizetti et al., 2011; Christ et al., 2017; Goliand et al., 2018; Stoten and Carlton, 2018). Cep55 is thought to be necessary to recruit this abscission machinery to the midbody in human cells through interactions with ESCRT-I/TSG101 and Alix. Then TSG101 and Alix both recruit ESCRT-III, which assemble into filaments (Morita et al., 2007; Carlton et al., 2008; Lee et al., 2008; Christ et al., 2016) (Fig. 7A). Notably, Cep55 is absent in invertebrate genomes, yet their midbodies still recruit ESCRTs to mediate abscission. Therefore, we asked whether abscission in Cep55−/− NSCs is accomplished with or without the ESCRT machinery. First, we tested for the localization of endogenous Tsg101 and Alix in midbodies of control and Cep55−/− NSCs. Approximately one-third of control NSC midbodies have Alix or Tsg101 present at the midbody center, reflecting that recruitment is temporally regulated and occurs only at late abscission stages. Among Cep55−/− NSC midbodies, however, only ∼5% had detectable Alix or Tsg101 (Fig. 7B,C). This suggests that, in the absence of Cep55, ESCRT component recruitment may be delayed, or total protein recruitment may be decreased.
Next, we used MEFs to further analyze midbodies and ESCRT recruitment when Cep55 is absent, since MEF cells and midbodies are much flatter than NSCs and easier to image. Control MEFs exhibit identical Cep55 localization in late-stage midbodies as NSCs do (Fig. 4F). Additionally, an antibody against the downstream ESCRT-III component, Chmp2a, works on MEFs (Fig. 7D). Cep55 was previously suggested to be required for formation of the dense midbody matrix in HeLa cells, which makes the central bulge appear as an unlabeled “dark zone” with α-tubulin or Aurora B antibody immunostaining (Zhao et al., 2006). We tested this in MEFs, but the dark zones were equally detected in Cep55−/− MEFs as controls (Fig. 7E). Next, we tested Alix and Tsg101 recruitment and found, similarly to NSCs, the percentages of Cep55−/− MEF midbodies with detectable endogenous Alix and TSG101 are significantly decreased compared with controls (Fig. 7D,F). There is also a trend for reduced midbodies with the ESCRT-III component Chmp2a detectable (Fig. 7F). Interestingly, if we just consider midbodies that have formed constriction sites, ∼40% of control MEFs have ESCRT localization, versus 15% of the Cep55−/− MEFs, suggesting that constriction sites can form before or independently of ESCRT recruitment (Fig. 7G).
Next, we asked whether the minority of Cep55−/− MEF midbodies with detectable ESCRTs had normal distribution of that ESCRT within the midbody. We evaluated ESCRT protein signal intensity using line scans drawn lengthwise along the midbody. In controls, a peak of ESCRT intensity is seen, as expected, in the central matrix/“dark zone” between the Aurora B-stained flanks (Fig. 7H–J, controls). In Cep55−/− midbodies that had detectable Alix, the average peak intensity of Alix fluorescence is significantly decreased, and the peak of intensity at the dark zone is lost (Fig. 7H). Surprisingly, for Tsg101-containing Cep55−/− midbodies, there is not a decrease but a slight increase in peak intensity of Tsg101, and the intensity pattern is similar to controls (Fig. 7I). We found a trend for decreased peak Chmp2a fluorescence compared with controls (Fig. 7J). This suggests the interesting possibility that Alix and Tsg101 recruitments are affected differently by loss of Cep55. Together, these data indicate that Cep55 is not essential for ESCRT component recruitment to the midbody in NSCs and MEFs but suggests that Cep55 may increase or accelerate their recruitment to ensure efficient abscission.
Cep55 KO mice have increased numbers of binucleate cortical cells and MEFs
Our analyses of midbodies and abscission in NSCs of the Cep55 KO so far suggest that most NSCs can complete abscission in the absence of Cep55, but that the process is slower and a subset of cells may fail. To assay cytokinesis success and failure in a larger sample of cells, we analyzed DNA content of embryonic cortical cells with flow cytometry. Dissociating cells of E15.5 cortices and labeling DNA with propidium iodide, we found 30% of Cep55−/− cortical cells have 4N DNA content compared with 2% in controls, a 15-fold increase (Fig. 8A). An increase in cells with 4N DNA content could come from either an arrest in G2/M phase, or failed cytokinesis resulting in formation of a binucleate or tetraploid progenitor or neuron. Since we did not see significantly increased numbers of PH3+ cells in Cep55−/− cortices (Fig. 3J), this suggests the presence of binucleate/tetraploid cells. To investigate whether cells with 4N DNA content were progenitors or neurons, we used Ki67 and DAPI colabeling to differentiate cycling progenitor cells (Ki67+) from noncycling neurons (Ki67–) cells. Interestingly, we observed an increased 4N DNA peak in both Ki67+ (Fig. 8B) and Ki67– Cep55−/− cells (Fig. 8C). In controls, there is no 4N DNA peak in Ki67– cells because neurons are postmitotic (Fig. 8C, left). These data suggest that, in Cep55−/− cortices, there are significant numbers of binucleate neurons and NSCs.
To further investigate whether there are binucleate NSCs and/or neuron populations in Cep55−/− cortices, we imaged dissociated, cultured cortical cells from E14.5 cortices. We used Nestin immunostaining to mark NSCs, and actin (phalloidin) and DAPI staining to differentiate cells with one or two nuclei. Indeed, we observed increased percentages of binucleate cells in Cep55−/− cultures, including ∼24% of NSCs (Nestin+) and 12% of neurons (Nestin–) (Fig. 8D–F). These data show that Cep55 KO results in significant numbers of binucleate NSCs and neurons in the brain.
Since the brain size is more severely affected than body size in Cep55 KOs (Fig. 1I), We wondered whether binucleation is a consequence of Cep55 loss in non-neural cell types too. Cep55 is localized in embryonic fibroblast midbodies in a similar distribution as seen in NSCs (Fig. 4F), and its loss causes decreased recruitment of ESCRTs (Fig. 7F). Indeed, we do observe a doubling of binucleate cells in Cep55−/− MEF cultures compared with controls (Fig. 8G,H). Thus, while the numbers of binucleate cells are not as dramatic as seen in NSC cultures, Cep55 is important for ensuring abscission success in MEFs as well.
Apoptotic cells are increased in Cep55 KO cortex but not non-neural tissues during embryogenesis
Abscission defects and binucleation have been linked to apoptosis of NSCs and neurons in other genetic mouse microcephaly mutants (Bianchi et al., 2017; Moawia et al., 2017; Little and Dwyer, 2019). The disorganization and reduced cell numbers we noted in Cep55−/− cortices suggest some cells could be dying. To assay for apoptosis, we labeled cortical sections with antibodies to cleaved caspase-3 (CC3). In control developing cortex, apoptotic cells are only rarely detected, but there is a striking increase in apoptotic cells in Cep55−/− cortices (Fig. 9A). Apoptosis is most increased in the proliferative zones, but is also increased in the cp (neuronal layer) (Fig. 9B). This suggests widespread apoptosis in multiple cortical cell types. Since brain size is already significantly decreased at E14.5, we looked at an earlier age for apoptosis. Indeed, apoptosis is dramatically increased in Cep55−/− cortical epithelium at E10.5, when the cortex is virtually all NSCs (Fig. 9C,D). Consistent with this, the percent of apoptotic nuclei drops by half from E12.5 to E14.5, as the proportion of neurons in the cortex increases (Fig. 9F). To confirm whether neurons and BPs undergo apoptosis in Cep55−/− cortices, we performed colabeling of CC3 with Tubb3, Satb2, Ctip2, and Tbr2; ∼35% of Cep55−/− apoptotic cells at E12.5 and 45% at E14.5 are neurons (Tubb3+), with ∼10% of both Satb2+ and Ctip2+ neurons undergoing apoptosis (Fig. 9E,G,H). BPs (Tbr2+) comprise 12% and 20% of apoptotic cells at E12.5 and E14.5, respectively, with 5%-10% of them undergoing apoptosis (Fig. 9I–L). Therefore, we conclude that apoptosis is highest in NSCs at early ages, but that death of BPs and neurons also occurs.
We wondered whether a lack of apoptosis in noncortical tissues could explain the less severe body size in Cep55−/− mice compared with brain size. Alternatively, apoptosis could occur in these tissues, but proliferation increases to compensate. To address this question, we labeled E10.5 whole-embryo sections with CC3. Interestingly, apoptosis is observed throughout CNS tissues (Fig. 9M, spinal cord, and hindbrain and midbrain, not shown) but is not seen in the rest of the body, including the heart precursor, the forelimb, and the retina (quantification; Fig. 9N). There are many more mitotic cells in the developing cortex than in these tissues in Cep55−/− mice (Fig. 9O); however, even when normalized to the number of mitotic cells, apoptosis is specifically increased in the Cep55−/− cortex, not in the other tissues (Fig. 9P). These data suggest the interesting possibility that a distinct apoptotic response occurs in CNS tissues after Cep55 loss, that does not occur in other proliferating tissues.
p53 nuclear expression is increased in Cep55 KO binucleate cortical cells, but not in binucleate MEFs
We previously showed that NSC apoptosis in a different abscission mutant, in the kinesin Kif20b, was mediated by the tumor suppressor p53 (Little and Dwyer, 2019). To determine whether p53 elevation occurs in Cep55−/− cortices, we labeled E14.5 cortical sections with antibodies to p53. Indeed, while virtually no cells with bright nuclear p53 accumulation are observed in control sections, greatly increased numbers of p53+ cells are seen in Cep55−/− sections (Fig. 10A,B). We noticed these cells throughout the cortex, but especially increased in proliferative zones (vz/svz). While some p53+ cells appear as single nuclei, others appear to be paired nuclei (Fig. 10A, arrows). To further delineate in which Cep55−/− cell p53 expression occurred, we used dissociated cortical cell cultures. We observed a fivefold increase in the number of cells with a nuclear:cytoplasmic (N:C) ratio of ≥2, indicative of p53 activation as it acts in the nucleus (Fig. 10C,D).
Interestingly, just as apoptosis is increased in both NSCs and neurons in Cep55−/− cortex, we found increased nuclear p53 expression in both cell types (Fig. 10D). While cytokinetic defects would occur only in dividing NSCs, we reasoned that a failed cytokinesis event could result in the formation of a binucleate daughter cell, either progenitor cell or neuron. Indeed, we had observed both binucleate NSCs and neurons in Cep55−/− cultures (Fig. 8F). To investigate whether binucleation was associated with p53 activation and apoptosis in progenitor cells and neurons, we colabeled cells with Nestin, phalloidin, and p53. Indeed, there is an increased N:C ratio of p53 expression in binucleate Cep55−/− NSCs and neurons compared with mononucleate cells (Fig. 10E). Furthermore, almost no mononucleate cells have p53 N:C ratios > 2, while >50% of binucleate progenitors and 20% of binucleate neurons do (Fig. 10F). These data suggest the existence of a p53-dependent pathway for apoptosis of binucleate cells in the cortex, that may be most sensitive in NSCs.
Our previous figures showed that apoptosis is increased in the brain but not body tissues of Cep55 KOs. Cep55−/− MEFs have defective ESCRT recruitment during abscission, and increased binucleation. Therefore, we asked whether they also had increased p53 expression or apoptosis. Surprisingly, the answer appears to be no: neither apoptosis nor p53 levels are increased in Cep55−/− MEF cultures (Fig. 10G–I). Furthermore, binucleate Cep55−/− MEFs do not have any detectable difference in p53 expression compared with mononucleate MEFs (Fig. 10J). These data suggest that the p53-dependent apoptotic response to binucleation/tetraploidy is regulated differently in various cell types, contributing to the dramatic tissue-level differences in phenotypic severity observed in germline Cep55 KOs.
p53 codeletion partially rescues Cep55 KO microcephaly
To test the hypothesis that p53 activation is the cause of apoptosis and microcephaly in Cep55−/− mice, we crossed the p53 KO to the Cep55 KO. Mouse KOs of p53 have normal brain size and structure at birth, with almost no consequences for development (Jacks et al., 1994; Insolera et al., 2014). As expected, Cep55−/− mice with WT p53 status exhibit microcephaly (Fig. 11A, second panel). We detected no difference in brain size with heterozygous deletion of p53 (Fig. 11A, third panel). However, complete deletion of p53 partially rescues brain size (Fig. 11A, right-most panel; Fig. 11B,C). Cep55−/−;p53−/− (dKO) cortical length is 21% longer than Cep55−/−, but still 10% shorter than WT controls; cortical area is 32% larger than Cep55−/−, but still 19% smaller than WT controls (Fig. 11B,C). Next, we evaluated whether deletion of p53 prevents the massive apoptosis in the Cep55 KO. Indeed, apoptosis in Cep55−/− mice is p53-dependent (Fig. 11D,E). Therefore, preventing apoptosis partially rescues brain size in Cep55−/− mice, but not to WT size. These results highlight the importance of Cep55 function for sufficient NSC proliferation, since even with apoptosis prevention, brain growth is impaired.
We earlier noted that Cep55−/− mice suffer postnatal lethality starting in the second week of life, and have reduced postnatal growth (Extended Data Fig. 1-1A,B). Therefore, we investigated whether lethality or body size is improved in dKO mice, as brain size is. Surprisingly, the postnatal survival curve of dKO mice is not improved compared with Cep55 single KOs, and body weight and size are still severely reduced (Extended Data Fig. 11-1A-C). Thus, there are disparities in both the consequences of Cep55 loss for development of different tissues, and in the degree of p53 KO rescue.
p53 codeletion partially rescues thinner neuronal layers in Cep55 KO mice but exacerbates progenitor defects
We next wanted to use the dKO to test whether there were cellular consequences for Cep55 loss during corticogenesis, apart from apoptosis. We showed above that Cep55−/− cortices have thinner neuronal layers. To determine whether this phenotype is p53-dependent, we examined cortical layering in dKO cortices. At E14.5, dKO mice had slightly reduced cortical thickness compared with p53−/− controls, but no reduction in the cp or iz (Fig. 11F,G), suggesting that the thin neuronal layers in Cep55−/− embryos were secondary to early apoptosis. Instead, the vz thickness was ∼10% reduced in dKOs, and the cp was proportionally slightly thicker (Fig. 11H). However, by P0, the cortical thickness of dKOs is reduced by 25% because of significantly thinner iz and cp (Extended Data Fig. 11-1D,E,G). Proportionally, layers 5-6 are increased and layers 2-4 are decreased in dKOs (Extended Data Fig. 11-1F,H), as was also observed in Cep55 single KOs (Fig. 2H–J). Together, these data indicate that apoptosis inhibition by p53 KO can partially rescue the thickness of neuronal layers, but not the increased proportion of deep layer neurons.
We then tested whether p53 KO would rescue the NSC and BP phenotypes observed in Cep55−/− cortices. At E14.5, Cep55 KOs have disorganization of the vz and svz, with reduced NSCs, some mislocalized NSCs, and increased mitotic index of BPs (Fig. 3). In dKOs, there is no difference in the total number of NSCs or BPs (Fig. 11I,J); however, there are increased numbers of mislocalized NSCs and BPs, even more than in Cep55 single KOs (Fig. 11I, arrows; Fig. 11K). Quantifying PH3+ cells, the mitotic index in dKOs was increased in cells located basally but not apically, similar to the single Cep55 KOs (Fig. 11L,M). However, the dKO has increased mitotic indices in both NSCs and BPs (Fig. 11N), correlating with many mislocalized NSCs and BPs (Fig. 11O). Additionally, we observed many abnormally large nuclei in dKO cortices, suggesting survival of binucleate cells (example Fig. 11I, square). These data reveal these NSC and BP phenotypes to be consequences of Cep55 loss rather than secondary to p53 activation or the apoptotic process.
p53-independent cell cycle exit of NSCs occurs in Cep55 KOs
We wanted to further explore why p53 KO does not fully rescue brain size in Cep55 KOs, and the consequences of Cep55 loss aside from apoptosis. We noted earlier that Cep55 KOs have a proportionately thicker preplate at embryonic ages, with increased proportion of deep layer neurons at birth (Figs. 2C, 3O). This suggested the hypothesis that Cep55 loss causes some daughter cells of NSC divisions to prematurely exit the cell cycle to become postmitotic neurons, thereby depleting the early progenitor pool. To test this, we used (Bromo-deoxyUridine (BrdU) labeling followed by analysis 1 d later to map the fates of daughter cells of NSC divisions in Cep55 KO brains at E12.5. We scored daughter cells as neurons (Tubb3+, exited cell cycle), BPs (Tbr2+), or presumed NSCs (BrdU+ only). Strikingly, we found that neuron daughters from those divisions were tripled in the Cep55 KOs (Fig. 12A,B).
NSCs can produce various daughter fates from both symmetric and asymmetric divisions, which cannot be analyzed individually by in vivo BrdU fate mapping. To determine whether Cep55 loss alters the specific types of NSC divisions, we analyzed individual divisions with dissociated cultures of E12.5 cells with BrdU, fixed after 1 d, and identified pairs of daughter cells produced from NSC divisions that day (Fig. 12C). We classified daughter cells as arising from asymmetric, neurogenic symmetric, or proliferative symmetric divisions (Fig. 12C,D). There was no difference in the numbers of symmetric versus asymmetric divisions in Cep55 KO cultures. However, there was a 41% increase in neurogenic symmetric divisions at the expense of proliferative symmetric divisions (Fig. 12E). Accordingly, cell cycle exit was significantly increased (Fig. 12F). There was no change in the number of BPs produced, suggesting that BPs do not account for this effect (Fig. 12G,H). To test whether the excess cell cycle exit in Cep55 KO NSCs requires p53, we performed the same assay with Cep55;p53 dKOs. Interestingly, in the dKO cultures, there are still increased neurogenic symmetric divisions at the expense of proliferative symmetric divisions, and there is still increased cell cycle exit compared with controls (Fig. 12I,J). Together, these data show that loss of Cep55 causes increased cell cycle exit of NSC daughter cells to become postmitotic neurons, through a pathway independent of p53, and suggests this may contribute to the microcephaly phenotype.
Discussion
Here we investigated the roles of the cytokinetic abscission regulator Cep55 in embryonic development, particularly in the brain. Data from cell lines had led to the model that Cep55 is essential for the recruitment of ESCRT components to the midbody, to complete abscission. This model predicted that KO of Cep55 would cause early embryonic lethality. However, we found that, while some cells fail abscission in the absence of Cep55, most succeed, and embryos develop. ESCRT recruitment is reduced but not abolished in Cep55−/− midbodies. We find that Cep55 acts to ensure the speed and success rate of abscission, and that it is essential for proper brain growth (working model, Fig. 12K). NSCs that fail abscission and become binucleate activate p53 and apoptosis, but embryonic fibroblasts do not. p53 codeletion to prevent apoptosis partially rescues Cep55−/− brain size, but not body size. In addition to apoptosis, Cep55 KO NSCs also display p53-independent increases in cell cycle exit. These tissue-specific consequences elucidate why brain development is especially vulnerable to defects in cytokinesis, and underline the need for studying abscission regulation in vivo.
Cep55 mutants serve as models for an emerging spectrum of human brain malformations. Human mutations in Cep55 also cause severe defects in CNS development and relative sparing of other tissues. Different alleles identified so far cause a range of phenotypic severity. Three different nonsense mutations, when homozygous, resulted in lethal hydranencephaly, a fluid-filled skull with almost no brain (Bondeson et al., 2017; Frosk et al., 2017; Rawlins et al., 2019). These may be null alleles if nonsense-mediated decay occurs, but absence of Cep55 protein was not confirmed. Compound heterozygotes for one nonsense allele and one missense allele have microcephaly, a small brain with normal structure. Finally, splicing mutations in Cep55 result in micro-lissencephaly, a small brain with fewer gyri. The latter two classes of patients survived (Barrie et al., 2020). Unlike humans and mice with Cep55 mutations, a zebrafish Cep55 mutant has small eyes (Jeffery et al., 2015; Yanagi et al., 2019). The developing eye has NSCs similar to those in the brain, so it is unclear why the human and mouse do not have small eyes on Cep55 loss. A possible explanation is that fish have larger eyes than forebrains, so their eye growth may require the abscission speed and accuracy that Cep55 ensures. Together, these data suggest that Cep55 evolved in vertebrates to help build bigger more complex nervous systems.
KO of Cep55 enabled us to est the model that Cep55 is required to recruit ESCRT components to the midbody, and that this recruitment is necessary for abscission completion (Stoten and Carlton, 2018). Abscission involves both membrane scission and microtubule disassembly to sever the intercellular bridge; ESCRT components are thought to couple these processes together temporally, mediating membrane scission and recruiting the microtubule-severing protein spastin (Steigemann et al., 2009; Guizetti et al., 2011). Our findings appear to challenge two aspects of this model. First, we find localized ESCRT components in some Cep55−/− midbodies, suggesting that Cep55 is not the sole recruiter of ESCRTs to the midbody. One candidate from Drosophila melanogaster, which lacks Cep55, is MKLP1 (Lie-Jensen et al., 2019). In mammalian cells, additional candidates could be ESCRT-recruiting proteins for other cellular processes (Campsteijn et al., 2016). Bypassing ESCRTs altogether may be possible, as reported in Caenorhabditis elegans (Konig et al., 2017). Second, we find that, without Cep55, microtubule disassembly during abscission does not fail but is delayed. In HeLa cell cultures, Cep55 depletion caused greatly increased midbody index, and cells remained at midbody stage for hours before either regressing or finally succeeding (Fabbro et al., 2005; Zhao et al., 2006). By contrast, in NSCs in vivo, we only observed a small increase in midbody index, and only a slight delay in microtubule disassembly in Cep55−/− brains. These findings suggest the intriguing possibility that microtubule disassembly and membrane scission are uncoupled in the Cep55−/− mouse, and that membrane scission is more severely delayed. Microtubule severing proteins might be recruited through post-translation modifications of tubulin instead of via ESCRTs (McNally and Roll-Mecak, 2018). This could explain why we observe excess MBRs at the apical membrane in KO brains: they could be tethered by a hollow membrane bridge. Alternatively, increased MBRs in Cep55−/− mice could indicate a problem in MBR disposal or engulfment. Clearly, more work from many model systems is needed to understand the exact roles of Cep55 at the molecular level, the mechanisms that may recruit ESCRTs to the midbody besides Cep55, and whether there are alternate pathways for membrane scission or microtubule disassembly.
While other work has suggested that NSCs have more sensitive apoptotic pathways compared with other cell types, to our knowledge, this is the first demonstration that a particular cell division defect (binucleation) produces different p53 responses in neural versus non-neural cells in vivo. We find increased p53 expression and apoptosis in Cep55−/− binucleate NSCs, but not binucleate MEFs, and increased apoptosis in Cep55−/− CNS tissues, but not in other embryonic body tissues. Furthermore, we show that prevention of p53-dependent apoptosis partially rescues brain size in Cep55−/− mice but does not improve body size or survival. It is likely that, in multiple Cep55−/− cell types, abscission delays and failures hinder proliferation, resulting in decreased growth of body tissues, but that in NSCs they additionally result in p53-dependent apoptosis. The explanation for this might be that some tissues have a higher tolerance for failed cytokinesis. Indeed, some mammalian tissues, such as the liver and heart, have binucleate cells normally (Guidotti et al., 2003; Paradis et al., 2014), and syncytia development by incomplete cytokinesis is part of testis development (Ren and Russell, 1991; Greenbaum et al., 2011). Our findings are consistent with recent evidence for variable sensitivity of developing tissues to p53 overexpression (Bowen et al., 2019). Since cortical NSCs undergo many rounds of cell divisions to stochastically create lineage trees of different daughter cell types (Dwyer et al., 2016; Llorca et al., 2019), even low rates of apoptosis early in development can have profound effects on brain growth. Conversely, survival of binucleate or aberrant NSCs that should have died can perhaps be even more damaging to cortical structure, as we saw in the Cep55;p53 dKO brains, which had many mislocalized mitotic NSCs and BPs.
In addition to p53-dependent apoptosis, we found cell cycle exit is another altered fate outcome of NSC divisions in Cep55−/− brains. This occurs through an unknown non-p53 pathway. Similarly, abscission defects in the Kif20b mutant also increased cell cycle exit (McNeely and Dwyer, 2020), and delay in prometaphase was reported to cause both p53-dependent apoptosis and p53-independent cell cycle exit in NSCs (Pilaz et al., 2016). The mechanism is unknown, but in cortical NSC divisions, daughter cell fate choices happen in the few hours after mitosis, concurrently with abscission. In Cep55−/− cortices, the abscission delays, MBR accumulation, altered apical endfoot size, or binucleation could all influence the choice of cell cycle exit or reentry.
During the writing of this manuscript, another research group published the phenotype of a Cep55 KO mouse (Tedeschi et al., 2020). They similarly found that Cep55−/− mice have microcephaly disproportionate to body size, reductions in NSCs and BPs at early ages, an accumulation of binucleate NSCs, and apoptosis specific to neural tissues. However, we performed additional analyses that provide more information on the role of Cep55 in corticogenesis, and cause us to differ with some of their interpretations. First, we differ in that we find ESCRTs are sometimes recruited, albeit reduced, in Cep55−/− cells. Second, we identify specific abscission defects in Cep55−/− brains, not simply binucleate cells: we find abscission delay, midbody structural defects, and MBR accumulation. Importantly, we are the first to show that Cep55 is not required for microtubule disassembly during abscission. Future work will determine whether Cep55−/− NSCs are completing abscission without ESCRT components, or whether recruitment is simply reduced or delayed. Third, we do find evidence of a role for Cep55 in abscission of non-neural cell divisions, suggesting that Cep55 and the ESCRT system is not dispensable for non-neural cells. Fourth, we identify contrasting p53 responses to cytokinetic defects in different cell types as a potential mechanism leading to disparate secondary consequences and tissue phenotypes. Our data support that cell division defects and apoptosis are indeed related, with increased p53 expression occurring in binucleate Cep55−/− cortical cells, and exacerbated mitotic indices in the Cep55;p53 dKO brains when apoptosis was prevented. Fifth, we show that p53 KO partly rescues the microcephaly but that p53-independent cell cycle exit and cortical disorganization still occur. The two KO alleles are highly similar: both involve deletion of exon 6, causing premature stop codons, and are likely null alleles. We cannot rule out some remaining N-terminal protein fragment, but it would not localize to the midbody. Future work by many groups with complimentary approaches will be needed to understand Cep55 mutant phenotypes in humans and other organisms, how Cep55 evolved in vertebrates, and the complex regulation of cytokinesis in developing tissues.
Cep55 KO mice have normal eye size and can survive postnatally, but exhibit failure to thrive and peri-weaning lethality. (A–C) Cep55−/− mice exhibit failure to thrive and pre-weaning lethality. (A) Most −/− pups die in the second or third week of life, and none survived past 25 d. n = 46 +/+ or +/− controls, 12 −/− pups. (B–C) Cep55−/− mice fail to gain weight between 6 and 10 d postnatally and are significantly smaller than controls. Day 6 n= 13 controls and 6 −/− pups; day 8 n = 26 controls and 13 −/− pups; day 10 n= 11 controls and 4 −/− pups. (D) Examples of postnatal day 1 control and −/− heads and dissected eyes. Note flatter head shape of −/− mice apparent in the outline. (E) Average eye diameter is not significantly changed in −/− pups. (F) Normalizing eye diameter to length of head showed no change between control and −/− pups. n= 6 +/+, 9 +/−, 5 −/− pups. Scale bar: C, 5 mm. **p <0.01, ****p <0.0001. B: t test, E, F: One-way ANOVA. Download Figure 1-1, TIF file (7.8MB, tif) .
Cep55 mRNA is expressed in proliferating embryonic tissues, including germinal zones of the nervous system. (A–C) Images of RNA ISH for Cep55 mRNA on sagittal sections of E14.5 mouse embryos. Cep55 is expressed in germinal zones of the nervous system including brain and eye, as well as widespread in proliferating embryonic tissues. Structures of interest are labeled in whole embryo (A–C) and in zoomed forebrain (A'-B') and eye (C') images. mid: midbrain; hind: hindbrain; ob: olfactory bulb; lat vent: lateral ventricle. (Images from GenePaint, https://gp3.mpg.de) (Visel et al., 2004). Download Figure 4-1, TIF file (7.7MB, tif) .
p53 deletion does not rescue lethality or cortical layer defects of Cep55 KO. (A–C) p53 codeletion does not improve Cep55−/− pups' postnatal survival (A) or body weight (B, C) (D) Representative images of P0 control (Cep55+/−;p53−/−) and dKO (Cep55−/−;p53−/−) coronal sections at level of hippocampus, stained with H&E. (E) Mean total cortical thickness and iz thickness is significantly reduced in dKO brains at P0. (F) Representative images of P0 control and dKO cortical sections labeled with antibodies to Ctip2, marking layers 6 (faint staining) and 5 (bright staining). (G) Layers 2–6 comprising the cp, and layer 6 is significantly thinner in dKO cortices at P0. (H) Layers 5-6 occupy a larger proportion of cp while layers 2-4 occupy a decreased proportion of the cp in dKO cortices compared with controls. A and B, n = 15 controls, 4 Cep55+/−;p53−/− and 3 Cep55−/−;p53−/−. E: n = 4 Cep55+/+;p53−/−, 3 Cep55−/−p53−/− mice. G,H: n= 3 Cep55 +/+;−/−p53−/− and Cep55−/−p53−/− mice. Scale bars: D: 100 µm, F: 40 µm. *p < 0.05, **p < 0.01. B, one-way ANOVA E,G,H: t test. Download Figure 11-1, TIF file (8.9MB, tif) .
Footnotes
This work was supported by National Institutes of Health Grants RO1NS076640, R21NS106162, and R01HD102492 to N.D.D., F30HD093290 to J.N.L., and UVA Cell and Molecular Biology Training Grant T32GM008136 to J.N.L. We thank Bettina Winckler, Xiaowei Lu, Ann Sutherland, Michael McConnell, Todd Stukenberg, and their laboratories for advice and discussion; Adriana Ehlers, Gabrielle Wolfe, and Naaz Daneshvar for help with cryosectioning and taking images; the Canadian Mouse Mutant Repository for use of the Cep55 allele sperm samples; UVA Genetically Enginereed Murine Model Core for in vitro fertilization services; and UVA Flow Cytometry Core for use of flow cytometry facilities.
The authors declare no competing financial interests.
References
- Addi C, Bai J, Echard A (2018) Actin, microtubule, septin and ESCRT filament remodeling during late steps of cytokinesis. Curr Opin Cell Biol 50:27–34. 10.1016/j.ceb.2018.01.007 [DOI] [PubMed] [Google Scholar]
- Addi C, Presle A, Fremont S, Cuvelier F, Rocancourt M, Milin F, Schmutz S, Chamot-Rooke J, Douche T, Duchateau M, Giai Gianetto Q, Salles A, Menager H, Matondo M, Zimmermann P, Gupta-Rossi N, Echard A (2020) The Flemmingsome reveals an ESCRT-to-membrane coupling via ALIX/syntenin/syndecan-4 required for completion of cytokinesis. Nat Commun 11:1941. 10.1038/s41467-020-15205-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrie ES, Overwater E, van Haelst MM, Motazacker MM, Truxal KV, Crist E, Mostafavi R, Pivnick EK, Choudhri AF, Narumanchi T, Castelluccio V, Walsh LE, Garganta C, Gastier-Foster JM (2020) Expanding the spectrum of CEP55-associated disease to viable phenotypes. Am J Med Genet A 182:1201–1208. 10.1002/ajmg.a.61512 [DOI] [PubMed] [Google Scholar]
- Bastos RN, Barr FA (2010) Plk1 negatively regulates Cep55 recruitment to the midbody to ensure orderly abscission. J Cell Biol 191:751–760. 10.1083/jcb.201008108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi FT, Tocco C, Pallavicini G, Liu Y, Vernì F, Merigliano C, Bonaccorsi S, El-Assawy N, Priano L, Gai M, Berto GE, Chiotto AM, Sgrò F, Caramello A, Tasca L, Ala U, Neri F, Oliviero S, Mauro A, Geley S, et al. (2017) Citron kinase deficiency leads to chromosomal instability and TP53-sensitive microcephaly. Cell Rep 18:1674–1686. 10.1016/j.celrep.2017.01.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson ML, Ericson K, Gudmundsson S, Ameur A, Ponten F, Wesstrom J, Frykholm C, Wilbe M (2017) A nonsense mutation in CEP55 defines a new locus for a Meckel-like syndrome, an autosomal recessive lethal fetal ciliopathy. Clin Genet 92:510–516. 10.1111/cge.13012 [DOI] [PubMed] [Google Scholar]
- Bowen ME, McClendon J, Long HK, Sorayya A, Van Nostrand JL, Wysocka J, Attardi LD (2019) The spatiotemporal pattern and intensity of p53 activation dictates phenotypic diversity in p53-driven developmental syndromes. Dev Cell 50:212–228.e216. 10.1016/j.devcel.2019.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campsteijn C, Vietri M, Stenmark H (2016) Novel ESCRT functions in cell biology: spiraling out of control? Curr Opin Cell Biol 41:1–8. 10.1016/j.ceb.2016.03.008 [DOI] [PubMed] [Google Scholar]
- Carlton JG, Agromayor M, Martin-Serrano J (2008) Differential requirements for Alix and ESCRT-III in cytokinesis and HIV-1 release. Proc Natl Acad Sci USA 105:10541–10546. 10.1073/pnas.0802008105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaigne A, Labouesse C, White IJ, Agnew M, Hannezo E, Chalut KJ, Paluch EK (2020) Abscission couples cell division to embryonic stem cell fate. Dev Cell 55:195–208.e195. 10.1016/j.devcel.2020.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ L, Wenzel EM, Liestol K, Raiborg C, Campsteijn C, Stenmark H (2016) ALIX and ESCRT-I/II function as parallel ESCRT-III recruiters in cytokinetic abscission. J Cell Biol 212:499–513. 10.1083/jcb.201507009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ L, Raiborg C, Wenzel EM, Campsteijn C, Stenmark H (2017) Cellular functions and molecular mechanisms of the ESCRT membrane-scission machinery. Trends Biochem Sci 42:42–56. 10.1016/j.tibs.2016.08.016 [DOI] [PubMed] [Google Scholar]
- Connell JW, Lindon C, Luzio JP, Reid E (2009) Spastin couples microtubule severing to membrane traffic in completion of cytokinesis and secretion. Traffic 10:42–56. 10.1111/j.1600-0854.2008.00847.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowell EF, Gaffuri AL, Gayraud-Morel B, Tajbakhsh S, Echard A (2014) Engulfment of the midbody remnant after cytokinesis in mammalian cells. J Cell Sci 127:3840–3851. 10.1242/jcs.154732 [DOI] [PubMed] [Google Scholar]
- Di Cunto F, Imarisio S, Hirsch E, Broccoli V, Bulfone A, Migheli A, Atzori C, Turco E, Triolo R, Dotto GP, Silengo L, Altruda F (2000) Defective neurogenesis in citron kinase knockout mice by altered cytokinesis and massive apoptosis. Neuron 28:115–127. 10.1016/s0896-6273(00)00090-8 [DOI] [PubMed] [Google Scholar]
- Dwyer ND, Manning DK, Moran JL, Mudbhary R, Fleming MS, Favero CB, Vock VM, O'Leary DD, Walsh CA, Beier DR (2011) A forward genetic screen with a thalamocortical axon reporter mouse yields novel neurodevelopment mutants and a distinct Emx2 mutant phenotype. Neural Dev 6:3. 10.1186/1749-8104-6-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer ND, Chen B, Chou SJ, Hippenmeyer S, Nguyen L, Ghashghaei HT (2016) Neural stem cells to cerebral cortex: emerging mechanisms regulating progenitor behavior and productivity. J Neurosci 36:11394–11401. 10.1523/JNEUROSCI.2359-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia N, Sougrat R, Spurlin TA, Hurley JH, Lippincott-Schwartz J (2011) Dynamics of endosomal sorting complex required for transport (ESCRT) machinery during cytokinesis and its role in abscission. Proc Natl Acad Sci USA 108:4846–4851. 10.1073/pnas.1102714108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettinger AW, Wilsch-Brauninger M, Marzesco AM, Bickle M, Lohmann A, Maliga Z, Karbanova J, Corbeil D, Hyman AA, Huttner WB (2011) Proliferating versus differentiating stem and cancer cells exhibit distinct midbody-release behaviour. Nat Commun 2:503. 10.1038/ncomms1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbro M, Zhou BB, Takahashi M, Sarcevic B, Lal P, Graham ME, Gabrielli BG, Robinson PJ, Nigg EA, Ono Y, Khanna KK (2005) Cdk1/Erk2- and Plk1-dependent phosphorylation of a centrosome protein, Cep55, is required for its recruitment to midbody and cytokinesis. Dev Cell 9:477–488. 10.1016/j.devcel.2005.09.003 [DOI] [PubMed] [Google Scholar]
- Frosk P, Arts HH, Philippe J, Gunn CS, Brown EL, Chodirker B, Simard L, Majewski J, Fahiminiya S, Russell C, Liu YP, Hegele R, Katsanis N, Goerz C, Del Bigio MR, Davis EE (2017) A truncating mutation in CEP55 is the likely cause of MARCH, a novel syndrome affecting neuronal mitosis. J Med Genet 54:490–501. 10.1136/jmedgenet-2016-104296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Jia G, Li A, Ma H, Huang Z, Zhu S, Hou Y, Fu X (2017) RNA-Seq transcriptome profiling of mouse oocytes after in vitro maturation and/or vitrification. Sci Rep 7:13245. 10.1038/s41598-017-13381-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goliand I, Adar-Levor S, Segal I, Nachmias D, Dadosh T, Kozlov MM, Elia N (2018) Resolving ESCRT-III spirals at the intercellular bridge of dividing cells using 3D STORM. Cell Rep 24:1756–1764. 10.1016/j.celrep.2018.07.051 [DOI] [PubMed] [Google Scholar]
- Green RA, Paluch E, Oegema K (2012) Cytokinesis in animal cells. Annu Rev Cell Dev Biol 28:29–58. 10.1146/annurev-cellbio-101011-155718 [DOI] [PubMed] [Google Scholar]
- Greenbaum MP, Iwamori T, Buchold GM, Matzuk MM (2011) Germ cell intercellular bridges. Cold Spring Harb Perspect Biol 3:a005850. 10.1101/cshperspect.a005850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruneberg U, Neef R, Li X, Chan EH, Chalamalasetty RB, Nigg EA, Barr FA (2006) KIF14 and citron kinase act together to promote efficient cytokinesis. J Cell Biol 172:363–372. 10.1083/jcb.200511061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti JE, Bregerie O, Robert A, Debey P, Brechot C, Desdouets C (2003) Liver cell polyploidization: a pivotal role for binuclear hepatocytes. J Biol Chem 278:19095–19101. 10.1074/jbc.M300982200 [DOI] [PubMed] [Google Scholar]
- Guizetti J, Schermelleh L, Mantler J, Maar S, Poser I, Leonhardt H, Muller-Reichert T, Gerlich DW (2011) Cortical constriction during abscission involves helices of ESCRT-III-dependent filaments. Science 331:1616–1620. 10.1126/science.1201847 [DOI] [PubMed] [Google Scholar]
- Hayashi S, Lewis P, Pevny L, McMahon AP (2002) Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech Dev 119 Suppl 1:S97–S101. 10.1016/S0925-4773(03)00099-6 [DOI] [PubMed] [Google Scholar]
- Hu CK, Coughlin M, Mitchison TJ (2012) Midbody assembly and its regulation during cytokinesis. Mol Biol Cell 23:1024–1034. 10.1091/mbc.E11-08-0721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insolera R, Bazzi H, Shao W, Anderson KV, Shi SH (2014) Cortical neurogenesis in the absence of centrioles. Nat Neurosci 17:1528–1535. 10.1038/nn.3831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA (1994) Tumor spectrum analysis in p53-mutant mice. Curr Biol 4:1–7. 10.1016/s0960-9822(00)00002-6 [DOI] [PubMed] [Google Scholar]
- Janisch KM, Dwyer ND (2016) Imaging and quantitative analysis of cytokinesis in developing brains of Kinesin-6 mutant mice. Methods Cell Biol 131:233–252. 10.1016/bs.mcb.2015.06.008 [DOI] [PubMed] [Google Scholar]
- Janisch KM, Vock VM, Fleming MS, Shrestha A, Grimsley-Myers CM, Rasoul BA, Neale SA, Cupp TD, Kinchen JM, Liem KF Jr, Dwyer ND (2013) The vertebrate-specific Kinesin-6, Kif20b, is required for normal cytokinesis of polarized cortical stem cells and cerebral cortex size. Development 140:4672–4682. 10.1242/dev.093286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janisch KM, McNeely KC, Dardick JM, Lim SH, Dwyer ND (2018) Kinesin-6 KIF20B is required for efficient cytokinetic furrowing and timely abscission in human cells. Mol Biol Cell 29:166–179. 10.1091/mbc.E17-08-0495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery J, Neyt C, Moore W, Paterson S, Bower NI, Chenevix-Trench G, Verkade H, Hogan BM, Khanna KK (2015) Cep55 regulates embryonic growth and development by promoting Akt stability in zebrafish. FASEB J 29:1999–2009. 10.1096/fj.14-265090 [DOI] [PubMed] [Google Scholar]
- Konig J, Frankel EB, Audhya A, Muller-Reichert T (2017) Membrane remodeling during embryonic abscission in Caenorhabditis elegans. J Cell Biol 216:1277–1286. 10.1083/jcb.201607030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafaurie-Janvore J, Maiuri P, Wang I, Pinot M, Manneville JB, Betz T, Balland M, Piel M (2013) ESCRT-III assembly and cytokinetic abscission are induced by tension release in the intercellular bridge. Science 339:1625–1629. 10.1126/science.1233866 [DOI] [PubMed] [Google Scholar]
- Lee HH, Elia N, Ghirlando R, Lippincott-Schwartz J, Hurley JH (2008) Midbody targeting of the ESCRT machinery by a noncanonical coiled coil in CEP55. Science 322:576–580. 10.1126/science.1162042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenhart KF, DiNardo S (2015) Somatic cell encystment promotes abscission in germline stem cells following a regulated block in cytokinesis. Dev Cell 34:192–205. 10.1016/j.devcel.2015.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Bielas SL, Zaki MS, Ismail S, Farfara D, Um K, Rosti RO, Scott EC, Tu S, Chi NC, Gabriel S, Erson-Omay EZ, Ercan-Sencicek AG, Yasuno K, Çağlayan AO, Kaymakçalan H, Ekici B, Bilguvar K, Gunel M, Gleeson JG (2016) Biallelic mutations in citron kinase link mitotic cytokinesis to human primary microcephaly. Am J Hum Genet 99:501–510. 10.1016/j.ajhg.2016.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie-Jensen A, Ivanauskiene K, Malerod L, Jain A, Tan KW, Laerdahl JK, Liestol K, Stenmark H, Haglund K (2019) Centralspindlin recruits ALIX to the midbody during cytokinetic abscission in Drosophila via a mechanism analogous to virus budding. Curr Biol 29:3538–3548.e3537. 10.1016/j.cub.2019.09.025 [DOI] [PubMed] [Google Scholar]
- Little JN, Dwyer ND (2019) p53 deletion rescues lethal microcephaly in a mouse model with neural stem cell abscission defects. Hum Mol Genet 28:434–447. 10.1093/hmg/ddy350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorca A, Ciceri G, Beattie R, Wong FK, Diana G, Serafeimidou-Pouliou E, Fernandez-Otero M, Streicher C, Arnold SJ, Meyer M, Hippenmeyer S, Maravall M, Marin O (2019) A stochastic framework of neurogenesis underlies the assembly of neocortical cytoarchitecture. Elife 8:e51381. 10.7554/eLife.51381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo L, Simon JM, Xing L, McCoy ES, Niehaus JK, Guo J, Anton ES, Zylka MJ (2019) Single-cell transcriptomic analysis of mouse neocortical development. Nat Commun 10:134. 10.1038/s41467-018-08079-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makrythanasis P, Maroofian R, Stray-Pedersen A, Musaev D, Zaki MS, Mahmoud IG, Selim L, Elbadawy A, Jhangiani SN, Coban Akdemir ZH, Gambin T, Sorte HS, Heiberg A, McEvoy-Venneri J, James KN, Stanley V, Belandres D, Guipponi M, Santoni FA, Ahangari N, et al. (2018) Biallelic variants in KIF14 cause intellectual disability with microcephaly. Eur J Hum Genet 26:330–339. 10.1038/s41431-017-0088-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally FJ, Roll-Mecak A (2018) Microtubule-severing enzymes: from cellular functions to molecular mechanism. J Cell Biol 217:4057–4069. 10.1083/jcb.201612104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeely KC, Dwyer ND (2020) Cytokinesis and postabscission midbody remnants are regulated during mammalian brain development. Proc Natl Acad Sci USA 117:9584–9593. 10.1073/pnas.1919658117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mierzwa B, Gerlich DW (2014) Cytokinetic abscission: molecular mechanisms and temporal control. Dev Cell 31:525–538. 10.1016/j.devcel.2014.11.006 [DOI] [PubMed] [Google Scholar]
- Moawia A, Shaheen R, Rasool S, Waseem SS, Ewida N, Budde B, Kawalia A, Motameny S, Khan K, Fatima A, Jameel M, Ullah F, Akram T, Ali Z, Abdullah U, Irshad S, Höhne W, Noegel AA, Al-Owain M, Hörtnagel K, et al. (2017) Mutations of KIF14 cause primary microcephaly by impairing cytokinesis. Ann Neurol 82:562–577. 10.1002/ana.25044 [DOI] [PubMed] [Google Scholar]
- Morita E, Sandrin V, Chung HY, Morham SG, Gygi SP, Rodesch CK, Sundquist WI (2007) Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J 26:4215–4227. 10.1038/sj.emboj.7601850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L (2007) A global double-fluorescent Cre reporter mouse. Genesis 45:593–605. 10.1002/dvg.20335 [DOI] [PubMed] [Google Scholar]
- Paradis AN, Gay MS, Zhang L (2014) Binucleation of cardiomyocytes: the transition from a proliferative to a terminally differentiated state. Drug Discov Today 19:602–609. 10.1016/j.drudis.2013.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterman E, Gibieza P, Schafer J, Skeberdis VA, Kaupinis A, Valius M, Heiligenstein X, Hurbain I, Raposo G, Prekeris R (2019) The post-abscission midbody is an intracellular signaling organelle that regulates cell proliferation. Nat Commun 10:3181. 10.1038/s41467-019-10871-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilaz LJ, McMahon JJ, Miller EE, Lennox AL, Suzuki A, Salmon E, Silver DL (2016) Prolonged mitosis of neural progenitors alters cell fate in the developing brain. Neuron 89:83–99. 10.1016/j.neuron.2015.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Goderie SK, Shen Q, Stern JH, Temple S (1998) Intrinsic programs of patterned cell lineages in isolated vertebrate CNS ventricular zone cells. Development 125:3143–3152. [DOI] [PubMed] [Google Scholar]
- Rawlins LE, Jones H, Wenger O, Aye M, Fasham J, Harlalka GV, Chioza BA, Miron A, Ellard S, Wakeling M, Crosby AH, Baple EL (2019) An Amish founder variant consolidates disruption of CEP55 as a cause of hydranencephaly and renal dysplasia. Eur J Hum Genet 27:657–662. 10.1038/s41431-018-0306-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren HP, Russell LD (1991) Clonal development of interconnected germ cells in the rat and its relationship to the segmental and subsegmental organization of spermatogenesis. Am J Anat 192:121–128. 10.1002/aja.1001920203 [DOI] [PubMed] [Google Scholar]
- Said Halidi KN, Fontan E, Boucharlat A, Davignon L, Charpentier M, Boulle M, Weil R, Israel A, Laplantine E, Agou F (2019) Two NEMO-like ubiquitin-binding domains in CEP55 differently regulate cytokinesis. iScience 20:292–309. 10.1016/j.isci.2019.08.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skop AR, Liu H, Yates J, Meyer BJ, Heald R (2004) Dissection of the mammalian midbody proteome reveals conserved cytokinesis mechanisms. Science 305:61–66. 10.1126/science.1097931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steigemann P, Wurzenberger C, Schmitz MH, Held M, Guizetti J, Maar S, Gerlich DW (2009) Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell 136:473–484. 10.1016/j.cell.2008.12.020 [DOI] [PubMed] [Google Scholar]
- Stoten CL, Carlton JG (2018) ESCRT-dependent control of membrane remodelling during cell division. Semin Cell Dev Biol 74:50–65. 10.1016/j.semcdb.2017.08.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedeschi A, Almagro J, Renshaw MJ, Messal HA, Behrens A, Petronczki M (2020) Cep55 promotes cytokinesis of neural progenitors but is dispensable for most mammalian cell divisions. Nat Commun 11:1746. 10.1038/s41467-020-15359-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Thaller C, Eichele G (2004) GenePaint.org: an atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res 32: D552–D556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagi K, Sone R, Ohga R, Kawahara A (2019) Involvement of the centrosomal protein 55 (cep55) gene in zebrafish head formation. Genes Cells 24:642–649. 10.1111/gtc.12715 [DOI] [PubMed] [Google Scholar]
- Zhao WM, Seki A, Fang G (2006) Cep55, a microtubule-bundling protein, associates with centralspindlin to control the midbody integrity and cell abscission during cytokinesis. Mol Biol Cell 17:3881–3896. 10.1091/mbc.e06-01-0015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cep55 KO mice have normal eye size and can survive postnatally, but exhibit failure to thrive and peri-weaning lethality. (A–C) Cep55−/− mice exhibit failure to thrive and pre-weaning lethality. (A) Most −/− pups die in the second or third week of life, and none survived past 25 d. n = 46 +/+ or +/− controls, 12 −/− pups. (B–C) Cep55−/− mice fail to gain weight between 6 and 10 d postnatally and are significantly smaller than controls. Day 6 n= 13 controls and 6 −/− pups; day 8 n = 26 controls and 13 −/− pups; day 10 n= 11 controls and 4 −/− pups. (D) Examples of postnatal day 1 control and −/− heads and dissected eyes. Note flatter head shape of −/− mice apparent in the outline. (E) Average eye diameter is not significantly changed in −/− pups. (F) Normalizing eye diameter to length of head showed no change between control and −/− pups. n= 6 +/+, 9 +/−, 5 −/− pups. Scale bar: C, 5 mm. **p <0.01, ****p <0.0001. B: t test, E, F: One-way ANOVA. Download Figure 1-1, TIF file (7.8MB, tif) .
Cep55 mRNA is expressed in proliferating embryonic tissues, including germinal zones of the nervous system. (A–C) Images of RNA ISH for Cep55 mRNA on sagittal sections of E14.5 mouse embryos. Cep55 is expressed in germinal zones of the nervous system including brain and eye, as well as widespread in proliferating embryonic tissues. Structures of interest are labeled in whole embryo (A–C) and in zoomed forebrain (A'-B') and eye (C') images. mid: midbrain; hind: hindbrain; ob: olfactory bulb; lat vent: lateral ventricle. (Images from GenePaint, https://gp3.mpg.de) (Visel et al., 2004). Download Figure 4-1, TIF file (7.7MB, tif) .
p53 deletion does not rescue lethality or cortical layer defects of Cep55 KO. (A–C) p53 codeletion does not improve Cep55−/− pups' postnatal survival (A) or body weight (B, C) (D) Representative images of P0 control (Cep55+/−;p53−/−) and dKO (Cep55−/−;p53−/−) coronal sections at level of hippocampus, stained with H&E. (E) Mean total cortical thickness and iz thickness is significantly reduced in dKO brains at P0. (F) Representative images of P0 control and dKO cortical sections labeled with antibodies to Ctip2, marking layers 6 (faint staining) and 5 (bright staining). (G) Layers 2–6 comprising the cp, and layer 6 is significantly thinner in dKO cortices at P0. (H) Layers 5-6 occupy a larger proportion of cp while layers 2-4 occupy a decreased proportion of the cp in dKO cortices compared with controls. A and B, n = 15 controls, 4 Cep55+/−;p53−/− and 3 Cep55−/−;p53−/−. E: n = 4 Cep55+/+;p53−/−, 3 Cep55−/−p53−/− mice. G,H: n= 3 Cep55 +/+;−/−p53−/− and Cep55−/−p53−/− mice. Scale bars: D: 100 µm, F: 40 µm. *p < 0.05, **p < 0.01. B, one-way ANOVA E,G,H: t test. Download Figure 11-1, TIF file (8.9MB, tif) .