

Netrin-1 reduction activates AEP that cleaves UNC5C receptor, contributing to AD pathogenesis.
Abstract
Netrin-1, a family member of laminin-related secreted proteins, mediates axon guidance and cell migration during neural development. T835M mutation in netrin receptor UNC5C predisposes to the late-onset Alzheimer’s disease (AD) and increases neuronal cell death. However, it remains unclear how this receptor is molecularly regulated in AD. Here, we show that δ-secretase selectively cleaves UNC5C and escalates its proapoptotic activity, facilitating neurodegeneration in AD. Netrin deficiency activates δ-secretase that specifically cuts UNC5C at N467 and N547 residues and enhances subsequent caspase-3 activation, additively augmenting neuronal cell death. Blockade of δ-secretase cleavage of UNC5C diminishes T835M mutant’s proapoptotic activity. Viral expression of δ-secretase–truncated UNC5C fragments into APP/PS1 mice strongly accelerates AD pathologies, impairing learning and memory. Conversely, deletion of UNC5C from netrin-1–depleted mice attenuates AD pathologies and rescues cognitive disorders. Hence, δ-secretase truncates UNC5C and elevates its neurotoxicity, contributing to AD pathogenesis.
INTRODUCTION
Alzheimer’s disease (AD) is the most common dementia and an age-dependent neurodegenerative disorder. The characteristic pathological hallmarks include extracellular senile plaques, which are predominantly composed of amyloid-β (Aβ) peptide deposits, and intraneuronal fibrillary tangles (NFT), which are mainly composed of hyperphosphorylated and truncated microtubule-associated protein Tau, accompanied by extensive neuronal loss and chronic neuroinflammation (1). Aβ results from the sequential proteolytic cleavage of transmembrane APP (amyloid precursor protein) by β-secretase (BACE1) and γ-secretases.
Netrin and its receptors Unc5 and DCC (deleted in colorectal carcinoma) regulate axon guidance and cell migration (2). In both invertebrates and vertebrates, UNC5 receptors facilitate chemorepulsion away from a netrin source, whereas DCC promotes chemoattraction to a netrin source (3). Netrin-1, a secreted laminin family trophic factor, has anti-inflammatory and antiapoptotic effects and has a key role in neurogenesis and morphogenesis of neural structures (4). Accumulating evidence supports that netrin-1 is involved in AD pathology. For instance, netrin-1 improves the Aβ-mediated suppression of memory and synaptic plasticity (5, 6). Netrin-1 is reduced in Aβ42-induced AD model rats (7). Noticeably, netrin-1 binds APP and modulates its signaling, triggering amyloid precursor protein intracellular domain (AICD)-dependent gene transcription, and this interaction represses Aβ peptide production in the brain slices from AD model mice. Furthermore, netrin-1 brain administration ameliorates AD pathologies (8). APP directly interacts with DCC receptor in the presence of netrin-1 and enhances netrin-1–mediated DCC intracellular signaling, such as MAPK (mitogen-activated protein kinase) activation. Inactivation of APP in mice is associated with reduced commissural axon outgrowth. Thus, APP functionally acts as a coreceptor for DCC to mediate axon guidance (9). Recently, several rare genetic variants have been identified in APP, TREM2 (triggering receptor expressed on myeloid cells 2), and UNC5C (a human homolog of Unc5h3) that affect risk for AD (10). A rare coding mutation, T835M, in UNC5C that is segregated with disease in an autosomal dominant pattern in two families enriched for late-onset AD (LOAD) has been reported. T835M alters a conserved residue in the hinge region of UNC5C, and this mutation leads to increased cell death in several cell types, including neurons (11). T835M-UNC5C–induced neuronal cell death is mediated by an intracellular death-signaling cascade, consisting of death-associated protein kinase 1 (DAPK1)/protein kinase D/apoptosis signal-regulating kinase 1 (ASK1)/c-Jun N-terminal kinase (JNK)/reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase/caspases, which merges at ASK1 with a death-signaling cascade, mediated by APP. Notably, netrin-1 binds to APP and partially inhibits the death-signaling cascade, induced by APP (12). Because of the enriched hippocampal expression of UNC5C in the adult nervous system, it has been proposed that T835M UNC5C contributes to the risk of AD by increasing susceptibility to neuronal cell death, particularly in vulnerable regions of the AD brain (11). The brain structure is associated with the polymorphisms of UNC5C in AD. UNC5C genetic variations affect the neuroimagings in AD (13) and are associated with cerebral amyloid angiopathy, indicating that this receptor is indeed implicated in AD pathology (14).
δ-Secretase is an acidosis-activated asparagine endopeptidase (AEP). It is located in the endo-lysosomes, and its activation is mediated by pH-dependent autocleavage (15, 16). We found that AEP cleaves SET, a deoxyribonuclease inhibitor, elevates DNA nicking, and promotes neuronal cell death (17). Moreover, SET acts as a PP2B inhibitor, and AEP cleavage of SET elevates Tau hyperphosphorylation in AD (18). Recently, we have shown that AEP is escalated and activated in an age-dependent manner. It acts as a δ-secretase that simultaneously cleaves both APP and Tau, facilitating senile plaques and NFT pathologies via enhancing Aβ production and Tau N368 aggregation. Depletion of δ-secretase diminishes AD pathologies in AD mouse models (19, 20). Notably, Tau N368 concentrations in cerebrospinal fluid (CSF) samples from patients with AD longitudinally correlate with Tau PET (positron emission tomography) signals in AD brains, in alignment with the cognitive defects (21). In the current study, we report that netrin-1 reduction in AD brains triggers δ-secretase activation, which subsequently cleaves UNC5C receptor on N467 and N547 residues, augmenting caspase-3 activities and neuronal apoptosis. Overexpression of δ-secretase–truncated UNC5C fragments in the hippocampus of APP/PS1 mice accelerates AD pathologies and cognitive defects, whereas knocking down UNC5C or blunting UNC5C cleavage by δ-secretase mitigates netrin deficiency–elicited AD pathologies and learning and memory impairment. Therefore, δ-secretase selectively cuts UNC5C receptor and mediates its pathological effects in AD pathogenesis.
RESULTS
δ-Secretase selectively cuts UNC5C at N467 and N547 residues
UNC5C T835M mutant predisposes to LOAD and increases neuronal cell death (11, 12). Because δ-secretase is activated in AD and proteolytically cleaves numerous effectors, we tested whether δ-secretase also cleaves UNC5C receptor. In vitro cleavage assays revealed that hemagglutinin (HA)-UNC5C was selectively cleaved in the brain lysates with pH 6.0, not 7.4. Knockout (KO) of δ-secretase abolished UNC5C fragmentation, indicating that acidosis-activated δ-secretase mediates UNC5C fragmentation (Fig. 1, A and B). Inhibition of δ-secretase with peptide inhibitor AENK but not control peptide AEQK in pH 6.0 brain lysates antagonized δ-secretase truncation of UNC5C (Fig. 1, C and D). To further validate that δ-secretase protease activity is implicated in fragmenting UNC5C, we transfected human embryonic kidney (HEK) 293 cells with glutathione S-transferase (GST)–tagged δ-secretase wild-type (WT) or enzymatic-dead C189S mutant. Overexpression of protease inactive C189S mutant robustly dampened UNC5C cleavage as compared with WT (Fig. 1E). To assess whether δ-secretase directly cuts UNC5C, we purified both recombinant proteins and performed in vitro cleavage assay and found that active δ-secretase directly cleaved UNC5C proteins into numerous fragments, fitting with the findings in cellular or brain tissue lysates (Fig. 1F). Proteomic analysis revealed that N467 and N547 were two noticeable cutting sites on UNC5C receptor (Fig. 1G). Mutation of either N467 or N547 residue into A selectively abolished one of the UNC5C fragments below 100 kDa, and mutation of both of amino acids suppressed these two band truncations (Fig. 1H). Because UNC5C receptor has numerous δ-secretase cleavage sites, it is worth noting that the cutting sites for the prominent 75- and 80-kDa bands were not successfully mapped out. Accordingly, we generated rabbit polyclonal anti–UNC5C N467 and anti–UNC5C N547 antibodies, which recognized the specific truncates with correct molecular weights (fig. S1A). Mutation of N into A abrogated antibodies’ signals against UNC5C cleavage at N467 or N547 or both locations (fig. S1B). N467 antibody is more specific to recognize the fragment versus the full-length UNC5C. Moreover, preincubation of the anti-serum with UNC5C 459–467 peptide, the antigen for generating anti–UNC5C N467, but not the scrambled control peptide, completely abrogated the immunohistochemistry (IHC) signals on AD brain sections (fig. S1C), underscoring the antibody against UNC5C N467 is specific. In addition, we also validated the specificity of anti–UNC5C N467 and C468 antibodies in 3xTg-AEP WT and 3xTg-AEP KO mice brains (fig. S1D). However, C468 antibody recognized the full length of UNC5C and cleaved fragments at the same time. Therefore, it was not selected for further validation. Furthermore, UNC5C receptor but not UNC5B or D was selectively shredded by active δ-secretase (fig. S1E). Hence, UNC5C receptor is cleaved by δ-secretase at N467 and N547 residues, and anti–UNC5C N467 specifically recognizes the truncated fragment.
Fig. 1. δ-Secretase cleaves UNC5C at N467 and N547 residues in vitro.
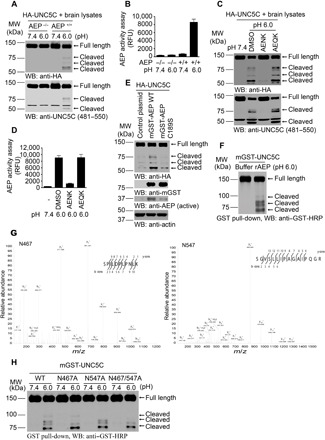
(A and B) HA-UNC5C was incubated with brain lysates derived from WT (AEP+/+) or AEP KO (AEP−/−) mice at pH 7.4 or 6.0, respectively. Western blot (WB) showed that UNC5C was cleaved at pH 6.0 by AEP+/+ brain lysates when AEP was activated. (B; means ± SEM; n = 3). (C and D) The proteolysis of UNC5C is blocked by Fmoc-Ala-Glu-Asn-Lys-NH2 (AENK) peptide, but not by Fmoc-Ala-Glu-Gln-Lys-NH2 (AEQK). δ-Secretase activity was inhibited by AENK (D; means ± SEM; n = 3). (E) Mutant of δ-secretase C189 diminishes UNC5C cleavage. HEK293 cells cotransfected with HA-UNC5C and mGST-AEP WT, mGST-AEP C189S, or control plasmid were incubated in buffer (pH 6.0) at 37°C for 45 min. (F) WB showed the processing of purified GST-UNC5C by recombinant δ-secretase (rAEP). (G) Mass spectrometry analysis of recombinant UNC5C fragmented by δ-secretase. The detected peptide sequences indicated that N467 and N547 were the two main cleavage sites. The detected peptides of UNC5C are 468–478 and 548–563, respectively. The sequences are shown in the upper right of each image. (H) Cleavage of mutant UNC5C by rAEP. UNC5C cleavage was analyzed by WB after GST–UNC5C WT, N467A, N547A, or N467A/N547A mutant was incubated with rAEP.
Netrin-1 is reduced in AD brains, associated with UNC5C cleavage by active δ-secretase
To examine whether netrin-1 and δ-secretase are altered in human AD brains, we conducted immunofluorescence (IF) costaining and found that δ-secretase was evidently elevated in AD brains compared with healthy control (HC) brains, and netrin-1 was notably reduced in AD brains versus control, inversely correlated with prominent δ-secretase escalation (Fig. 2A). Quantification of δ-secretase enzymatic activities validated this finding (Fig. 2B). To examine whether UNC5C is also proteolytically cleaved in human AD brains, we performed immunoblotting with brain lysates and found that netrin-1 was evidently reduced in human AD brains but not HCs; subsequently, caspase-3 and δ-secretase were activated. As a result, UNC5C was strongly cleaved in AD brains, which was revealed by both anti–UNC5C N467 and its C468 antibodies. Notably, C468 antibody recognized both full length (FL) and truncated UNC5C receptor fragments. As a positive control, Tau N368 truncate was pronouncedly escalated in AD brains by active δ-secretase (Fig. 2C). Correlated with augmented UNC5C N467 fragments in both the cortex and hippocampus, quantification of IHC analysis showed that UNC5C N467 was strongly elevated in AD but not HC brains (Fig. 2D). IF staining indicated extensive senile plaques in AD brains. Noticeably, truncated UNC5C N467 localized in ThS (thioflavin S)–positive aggregates (Fig. 2E). To further investigate UNC5C receptor cleavage in human AD brains, we extended our study into human AD and HC induced pluripotent stem cell (iPSC)–differentiated neurons. Immunoblotting demonstrated that netrin-1 was reduced in AD samples, coupled with δ-secretase and caspase-3 activation. Consequently, UNC5C was potently cleaved in AD neurons as compared with HC neurons (Fig. 2F).
Fig. 2. Netrin-1 is reduced in AD brains, associated with UNC5C cleavage by active δ-secretase.
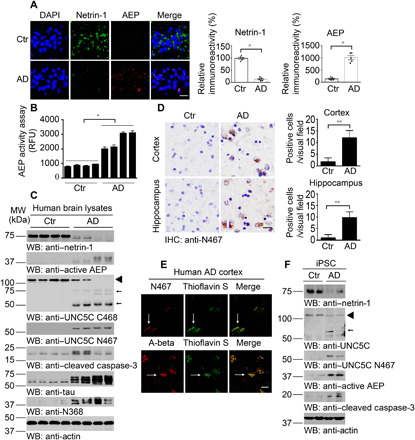
(A) IF staining of netrin-1 (green) and δ-secretase (red) in the hippocampus of AD human brain slides. Quantification of relative immunoreactivity is shown (means ± SEM; n = 5). Scale bar, 20 μm. (B) δ-Secretase activity assay of AD and age-matched normal human brain lysates (means ± SEM; n = 4). RFU, relative fluorescence units. *P < 0.05 by two-tailed t test. (C) WB detection of netrin-1 deprivation, caspase-3 and δ-secretase activation, UNC5C fragments in human brain samples of AD and age-matched controls. ◄, full length; ←, cleaved. (D) IHC staining showed UNC5C N467 fragment in human AD brains. Means ± SEM; n = 3, **P < 0.01 by two-tailed t test. Scale bar, 20 μm. (E) IF staining showed the colocalization of N467 (red) and ThS (green) or A-beta (red) and ThS in the cortex of AD human brains. UNC5C N467 is distributed in amyloid plaques. Scale bar, 20 μm. (F) WB detection of netrin-1 deprivation, caspase-3 and δ-secretase activation, and UNC5C fragments in iPSC from patients with AD but not healthy controls. ◄, full length; ←, cleaved.
To further explore whether netrin-1 and UNC5C proteolytic fragmentation occurs in the AD mouse model, we monitored different ages of WT and 3xTg mice. Immunoblotting showed that netrin-1 was progressively diminished with ages. By contrast, δ-secretase was gradually activated, resulting in extensive UNC5C receptor truncation and N467 fragment escalation. Caspase-3 was also increasingly activated during aging (fig. S2A). IF costaining also validated the opposite relationship between netrin-1 reduction and δ-secretase augmentation in the different ages of brain sections of 3xTg (fig. S2B). Therefore, netrin-1 is selectively deficient in human AD brains, leading to δ-secretase activation and selective UNC5C receptor proteolytic fragmentation.
UNC5C cleavage by δ-secretase mediates its cytotoxicity
Since UNC5C is the substrate for both caspase-3 and δ-secretase, we wondered whether these two proteases affect each other’s proteolytic actions. Caspase-3 cleaves UNC5C receptor at D415. Mutation of D415 into A did not affect δ-secretase fragmentation of UNC5C (Fig. 3, A and B). On the other hand, blockade of δ-secretase cleavage of UNC5C showed that N467/547A mutant exhibited comparable cleavage effect as WT by active caspase-3, although the D415A mutant only blunted one of two truncates from active caspase-3, suggesting that caspase-3 cleaves UNC5C receptor at two locations in addition to the D415 site (Fig. 3C). Hence, blockage of either caspase-3 or δ-secretase cleavage sites on UNC5C does not influence the other protease fragmentation of UNC5C. Because caspase-3 cutting site at the D415 site is upstream of the N467 and N547 residues, to assess whether caspase-3 cleavage on UNC5C facilitates δ-secretase subsequent fragmentation, we conducted an in vitro assay that showed UNC5C amino acid 416–913 fragment yielded more fragments from active δ-secretase than FL (Fig. 3D), indicating that caspase-3 cleavage of UNC5C accelerates the resultant further fragment truncation by δ-secretase.
Fig. 3. δ-Secretase cleavage of UNC5C mediates T835 mutant’s cytotoxicity.
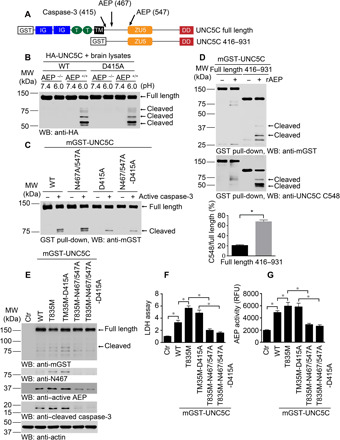
(A) Schematic diagram shows the cutting sites on UNC5C by caspase-3 and δ-secretase. IG, immunoglobulin-like domain; T, type I thrombospondin motifs; TM, transmembrane domain; ZU5, ZU5 domain; DD, death domain. (B) HA-UNC5C (WT and D415A) were incubated with brain lysates derived from WT (AEP+/+) or AEP KO (AEP−/−) mice at pH 7.4 or 6.0, respectively. WB showed that D415A mutation did not affect UNC5C cleaved by δ-secretase. (C) Purified mGST-UNC5C (WT, N467/547A, D415A, and N467/547A-D415A) were incubated with active caspase-3 in caspase assay buffer. WB showed that N467/547A mutation did not affect UNC5C cleaved by caspase-3. (D) Truncated UNC5C by caspase-3 (UNC5C 416–931) was prone to be cleaved by δ-secretase compared with full length, promoting more C-terminal fragments (death domain) production (means ± SEM; n = 3). **P < 0.01 by Student’s t test. (E) WB showed that UNC5C with T835M mutant was truncated into fragments when expressed in SH-SY5Y cells, inducing cell death. δ-Secretase cleavage of T835M was indispensable for UNC5C to mediate cell death. (F) Lactate dehydrogenase (LDH) assay showed the cytotoxicity of above mutated UNC5C (means ± SEM; n = 3). (G) δ-Secretase activity assay (means ± SEM; n = 3). *P < 0.05 by one-way analysis of variance (ANOVA) followed by Tukey’s multiple-comparison test for (F) and (G).
T835M mutation potentiates UNC5C-provoked cell death (12). To explore whether UNC5C cleavage by δ-secretase mediates its cytotoxicity effect, we prepared numerous mutants in T835M UNC5C and evaluated their activities in cell death. Compared with vector control, UNC5C WT overexpression promoted both δ-secretase and caspase-3 activation and notably augmented cell death. As expected, T835M mutant further escalated its cytotoxicity, which was associated with higher δ-secretase and caspase-3 activities. Notably, blunting caspase-3 cleavage in T835M/D415A mutant did not diminish either protease activity or cell death. By contrast, blockade of δ-secretase cleavage of UNC5C with T835M/N467/547A notably repressed both protease activation and cell death. Similar effects were observed with both proteases-resistant T835M/N467/547/A-D415A mutant (Fig. 3, E to G). Hence, these findings indicate that δ-secretase fragmentation is indispensable for UNC5C cytotoxicity. It was worth noting that UNC5C N467 fragmentation was progressively elevated from UNC5C WT to T835M and T835M-D415A, correlating with active δ-secretase auto-cleavage activation pattern.
To examine the biological functions of δ-secretase–truncated UNC5C fragments, we infected primary neurons with viruses expressing various green fluorescent protein (GFP)–tagged UNC5C truncates. IF costaining revealed the apoptosis in MAP2-positive neurons was escalated by UNC5C WT, which was notably reduced in δ-secretase–resistant UNC5C N467/547A cells. However, lactate dehydrogenase (LDH) assay and TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling) staining demonstrated that both the N-terminal fragments 1–467 and 1–547 and their correspondent C-terminal 468–931 and 548–931 truncates exhibited stronger neuronal death–promoting effects than FL UNC5C receptor. Concomitantly, these fragments activated caspase-3 more pronouncedly than UNC5C WT or N467/547A mutant (fig. S3, A to E). δ-Secretase enzymatic activities tightly echoed neuronal cell death effects (fig. S3F). Thus, δ-secretase cleavage of UNC5C is required for UNC5C-induced caspase-3 activation and cytotoxicity. However, caspase-3–mediated D415 cleavage of UNC5C is unessential for δ-secretase activation or cytotoxicity of UNC5C T835M.
Netrin-1 deprivation elicits δ-secretase and caspase-3 activation and neuronal cell death via both UNC5C and DCC
To test the roles of caspases and δ-secretase in netrin-1 deprivation–induced neuronal cell death, we pretreated neurons with pan-caspase inhibitor FMK001 or δ-secretase–specific inhibitor CPA (AEP inhibitor Compound 11A) , followed by treatment with DCC-4Fbn, which is a recombinant protein from DCC extracellular domain binding netrin-1 (22). On the other hand, we also used netrin-1 monoclonal antibody 2F5 to neutralize secreted netrin-1 in the media. Antagonizing secreted netrin-1 in the neuronal medium with 2F5 or DCC-4Fbn strongly reduced netrin-1 in primary neurons, resulting in δ-secretase activation that cleaved UNC5C receptor at N467. By contrast, inhibition of caspases or δ-secretase potently repressed caspase-3 and δ-secretase activation, abolishing UNC5C N467 fragmentation (Fig. 4A). Bright-field images of neurons taken with a light microscope provided a more intuitive impression induced by different treatments. Netrin-1 deletion induced neuronal dendrite reduction and cell loss, which could be neutralized by inhibiting AEP (Fig. 4B). Quantification demonstrated that both 2F5 and DCC-4Fbn significantly induced neuronal cell death. Noticeably, DCC-4Fbn–triggered cell death was strongly suppressed by either pan-caspase or δ-secretase inhibitor. δ-Secretase enzymatic activities were in alignment with cell death effects (Fig. 4, C and D). These findings suggest that caspase-3 and δ-secretase might directly regulate each other’s protease activity. In vitro cleavage assay with purified GST–caspase-3 recombinant proteins as substrate showed that active δ-secretase directly cut GST–caspase-3, enhancing active caspase-3 levels (fig. S4A). To further interrogate the relationship between caspase-3 and δ-secretase in truncating UNC5C, we pretreated primary neuronal cultures with specific caspase-3 or pan-caspase inhibitor, respectively, followed by netrin-1 withdrawal. Caspase-3 activation was completely blunted by both inhibitors, so was UNC5B cleavage, which is cut by caspase-3 at D412 (23). In contrast, δ-secretase was still proteolytically activated by netrin-1 neutralization even in the presence of caspase-3 inhibitor, whereas its activation was substantially antagonized by pan-caspase inhibitor. Noticeably, UNC5C fragmentation patterns tightly coupled with δ-secretase activities. As positive control, APP N586 and C586 truncation oscillated with upstream active δ-secretase (fig. S4B). Therefore, δ-secretase proteolytic activation is predominantly mediated by other caspases, but not caspase-3. Netrin-1 deficiency elicits δ-secretase activation, leading to UNC5C fragmentation and neuronal cell death. In addition, chronic oral treatment of 3xTg mice with δ-secretase inhibitor CPA notably inhibited AEP activation and UNC5C fragmentation in vivo (fig. S4C).
Fig. 4. Netrin-1 deprivation induces UNC5C cleaved by δ-secretase, triggering primary neuronal apoptosis.
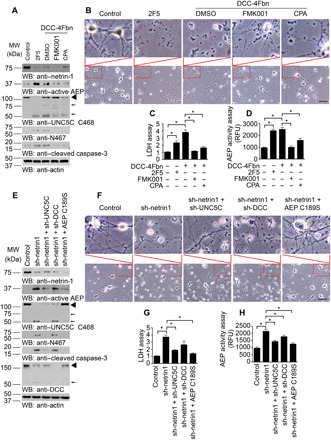
(A) Primary neurons were pretreated with pan-caspase inhibitor (FMK001, 100 nM) or δ-secretase specific inhibitor (Compound 11A, CPA; 100 nM) 30 min before 2F5 (10 μg/ml), DCC-4Fbn (10 μg/ml), or control vehicle treatments. Twenty-four hours later, neurons were collected for detecting netrin-1, AEP, UNC5C, and caspase-3 expression by WB. 2F5, netrin-1 monoclonal antibody to neutralize netrin-1; DCC-4Fbn, a recombinant protein from DCC extracellular domain; Actin, loading control; MW, molecular weight. ◄, full length; ←, cleaved. (B) Morphological characteristics of neurons under an optical microscope. The upper image is a larger version of the lower image in the indicator box. Scale bar, 100 μm. (C) LDH assay with neuronal mediums. (D) δ-Secretase activity in the cell lysates. (E) Primary neurons were infected with different viruses at day 5. Control: AAV-GFP; sh-Ntn1: Ntn1 AAV siRNA Pooled Virus; sh-UNC5C: Unc5c AAV siRNA Pooled Virus; sh-DCC: DCC AAV siRNA Pooled Virus; δ-secretase C189S: AAV-δ-secretase C189S. Nine days later, neurons were collected for detecting netrin-1, AEP, UNC5C, DCC, and caspase-3 expression by WB. ◄, full length; ←, cleaved. (F) Morphological characteristics of neurons under an optical microscope. The upper image is a larger version of the lower image in the indicator box. Scale bar, 100 μm. (G) LDH assay with neuronal mediums. (H) δ-Secretase activity in the cell lysates. For (C), (D), (G), and (H), data are means ± SEM, representatives of three independent experiments; *P < 0.05 by one-way ANOVA followed by Tukey’s multiple-comparison test.
To explore the role of netrin receptors in netrin-1 deletion–triggered δ-secretase activation, we treated primary neurons with viruses expressing the specific short hairpin RNAs (shRNAs). Specifically, we infected neurons with virus expressing netrin-1 shRNA in the presence of DCC or UNC5C shRNAs or δ-secretase C189S mutant. Immunoblotting indicated that knockdown of netrin-1 strongly triggered caspase-3 activation in neurons. Notably, deletion of UNC5C but not DCC substantially blocked caspase-3 activation. Moreover, inhibition of δ-secretase with C189S mutant completely suppressed caspase-3 activation, fitting with observations that active δ-secretase cuts caspase-3 and regulates its activation (Fig. 4 and fig. S4A). Accordingly, deletion of UNC5C or blockade of δ-secretase utterly diminished UNC5C N467 cleavage induced by netrin-1 depletion (Fig. 4E). Neuronal dendrite reduction and cell loss induced by netrin-1 deletion could be neutralized by inhibiting AEP or knocking down netrin-1 receptors, especially UNC5C (Fig. 4F). Quantification showed that netrin-1 deprivation–induced cell death was significantly blunted in UNC5C knockdown or δ-secretase–inhibited cells, and their cell death inhibitory effects were stronger than DCC deletion. δ-Secretase enzymatic activities correlated with cell death effects (Fig. 4, F to H). Therefore, netrin-1 deprivation elicits δ-secretase activation, leading to prominent neuronal cell death. Blockade of δ-secretase cleavage of UNC5C T835M strongly attenuates its proapoptotic effect.
Blockade of UNC5C cleavage by δ-secretase reduces netrin deficiency–elicited amyloid pathologies and cognitive deficits
To investigate the biological roles of netrin-1 deficiency–induced δ-secretase cleavage of UNC5C receptor, we used netrin-1 f/f mice (3 months old) and injected Adeno-associated virus (AAV)-Cre virus into the hippocampus of these mice in the presence of virus expressing shRNA against UNC5C, DCC, or inactive δ-secretase C189S. Moreover, we also included two more groups with virus expressing UNC5C WT or δ-secretase–resistant N467/547A mutant. Three months later, we performed immunoblotting with the brain lysates and found that netrin-1 was robustly deleted compared with control. Consistent with in vitro neuronal findings, netrin-1 deletion triggered δ-secretase activation, which was blunted when UNC5C but not DCC was knocked down. As expected, C189S notably antagonized netrin-1 deficiency–elicited δ-secretase activation. Nevertheless, overexpression of UNC5C WT but not δ-secretase–resistant mutant further escalated netrin-1 reduction–incurred δ-secretase activities. Consequently, UNC5C receptor was evidently cleaved by δ-secretase at N467, when netrin-1 was wiped out, and deletion of UNC5C but not DCC receptor diminished UNC5C fragmentation. Furthermore, overexpression of inactive δ-secretase C189S mutant greatly repressed endogenous UNC5C receptor cleavage; in contrast, UNC5C N467 was prominently cleaved in UNC5C WT but not N467/547A mutant. As a positive control for δ-secretase activation, Tau N368 oscillated with the same pattern. It is worth noting that caspase-3 was clearly activated when netrin-1 was deleted; however, knockdown of UNC5C or DCC or overexpression of inactive C189S strongly suppressed active caspase-3. Netrin-1 deficiency–elicited active caspase-3 was further elevated in the brain overexpressing UNC5C WT and δ-secretase–resistant mutant, with the former stronger than the latter (Fig. 5, A and B). δ-Secretase cleaves APP at the N585 residue, promoting Aβ production (20). IF costaining on the brain section demonstrated that mouse Aβ signals in the hippocampus tightly coupled with UNC5C N467 fluorescence activities (Fig. 5C). Active δ-secretase fluorescent signals correlated with UNC5C N467 levels (fig. S5A). IHC staining of UNC5C N467 validated immunoblotting observations (fig. S5B). Together, these findings support that netrin deficiency–mediated δ-secretase activities regulate both UNC5C fragmentation and Aβ production. Quantification of mouse Aβ40 and Aβ42 showed that their concentrations firmly coupled with δ-secretase protease activities (Fig. 5, D and E). Silver staining indicated that extensive aggregates were accumulated in the hippocampal neurons when netrin-1 was deleted. The aggregate abundance correlated with δ-secretase activities (fig. S5C). Golgi staining showed that dendritic spines were inversely coupled with Aβ concentrations (fig. S5D). We made similar observations with the hippocampal synapses, which were analyzed by electronic microscopy (EM) (Fig. 5, F and G).
Fig. 5. Blockade of UNC5C cleavage by δ-secretase reduces netrin deficiency–elicited amyloid pathologies and cognitive deficits.
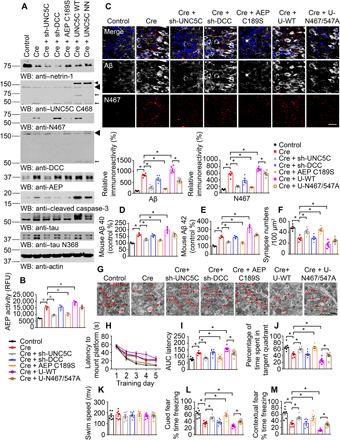
Netrin-1 deprivation in the hippocampus of netrin f/f mice induced amyloid pathologies, which could be improved by decreasing the expression of UNC5C and inactivating AEP. (A) WB showed the processing of netrin-1 deprivation by injecting AAV-Cre virus into the hippocampus and UNC5C fragmentation induced by subsequent δ-secretase activation in netrin f/f mice. The hippocampal lysates were detected with various indicated antibodies. Actin, loading control. ◄, full length (transfected and endogenous UNC5C); ←, cleaved. Control: AAV-GFP; Cre: AAV-Cre; sh-UNC5C: Unc5c AAV siRNA Pooled Virus; sh-DCC: DCC AAV siRNA Pooled Virus; AEP C189S: AAV-AEP C189S; UNC5C WT: lentivirus (LV)–UNC5C; UNC5C NN: LV–UNC5C N467/547A. These viruses were injected into both sides of the hippocampus. (B) δ-Secretase activity in the hippocampus of netrin f/f mice. Data are means ± SEM; n = 3 mice per group. (C) IF signals of anti-Aβ (white) and anti-N467 (red) were detected in the hippocampus infected with various indicated viruses. The nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Quantification of relative immunoreactivity was shown (means ± SEM; n = 5). Scale bar, 20 μm. (D and E) Enzyme-linked immunosorbent assay (ELISA) quantification of Aβ in the brain lysates of above mice. Data were means ± SEM; n = 3. (F and G) The synaptic density in the hippocampus of above mice determined by electron microscopy (means ± SEM; n = 6). Scale bar, 1 μm. (H and I) MWM analysis as time to platform (latency, seconds) and integrated latency (AUC) for above mice (means ± SEM; n = 7). (J and K) Probe trail and swim speed of MWM test (means ± SEM; n = 7). (L and M) Fear conditioning test including cued fear conditioning test and contextual fear conditioning test (means ± SEM; n = 7). For (B) to (F) and (H) to (M), *P < 0.05 by one-way ANOVA followed by Tukey’s multiple-comparison test.
Next, we monitored the learning and memory behaviors with both the Morris water maze (MWM) and contextual fear conditioning assays. MWM showed that deletion of netrin-1 significantly decreased the cognitive functions, which was restored by deletion of UNC5C but not DCC. Furthermore, δ-secretase C189S also rescued netrin-1 reduction–elicited cognitive dysfunctions. However, overexpression of UNC5C WT but not δ-secretase–resistant mutant worsened the latency as compared with Cre alone, indicating the further learning function impairment (Fig. 5, H and I). The probe assay with times in the target quadrant revealed similar patterns, and overexpression of the N467/547A mutant showed significantly improved memory as compared with UNC5C WT, which exacerbated the memory deficits as compared with Cre alone (Fig. 5J). All of the mice exhibited comparable swim speeds, indicating that the locomotive function remained intact among the groups (Fig. 5K). We made similar observations in cued fear conditioning and contextual fear conditioning tests (Fig. 5, L and M). Hence, netrin-1 deficiency escalates δ-secretase cleavage of UNC5C, promoting AD pathologies and cognitive disorders, and blockade of UNC5C cleavage by δ-secretase alleviates the deficits.
To further assess netrin-1 reduction and its receptors’ pathological roles in AD pathologies, we used APP/PS1 mice. We injected AAV virus expressing shRNA to delete netrin-1 in the presence of shRNAs against UNC5C or DCC, respectively. Again, netrin-1 reduction triggered δ-secretase activation and UNC5C N467 cleavage, which was abolished when UNC5C was eliminated. Notably, caspase-3 was prominently activated in all of the brains when netrin-1 was deleted regardless of UNC5C or DCC knockdown. As a control, Tau N368 fragmentation fitted with active δ-secretase levels and activities. (fig. S6, A and B). IF costaining supported that Aβ aggregates were highly increased in the brains when netrin-1 or netrin/DCC was deleted, whereas senile plaques were strongly reduced when UNC5C receptor was also eliminated in netrin-1–deficient brains (fig. S6C). Quantification showed that both mouse and human Aβ40 and Aβ42 concentrations were closely correlated with δ-secretase activities (fig. S6, D to G). The hippocampal synapses analyzed by EM displayed a similar pattern (fig. S6, H and I). Cognitive behavioral tests with MWM and fear conditioning assays demonstrated that netrin-1 deficiency–elicited cognitive disorders were significantly attenuated by UNC5C deletion but not DCC depletion (fig. S6, J to O). Thus, netrin-1 reduction in APP/PS1 mice induces δ-secretase activation and UNC5C N467 cleavage, leading to AD pathologies and cognitive deficits.
δ-Secretase–truncated UNC5C fragments promote neuronal degeneration and cognitive disorders in APP/PS1 mice
To assess δ-secretase–cleaved UNC5C fragments’ pathological roles in AD onset, we injected various virus expressing GFP-tagged UNC5C truncates into the hippocampus of APP/PS1 mice. As expected, δ-secretase and caspase-3 were strongly activated, associated with Tau N368 cleavage, fitting with δ-secretase protease activities. In alignment with in vitro neuronal findings, UNC5C truncates exhibited stronger effect than FL UNC5C in δ-secretase and caspase-3 activation (Fig. 6, A and B). IF costaining with anti-Aβ/ThS demonstrated that δ-secretase–truncated UNC5C elicited significantly more abundant amyloid plaques than UNC5C FL, which was substantially more copious than control (Fig. 6, C to E). Quantification demonstrated that both human and mouse Aβ40 and Aβ42 concentrations were greatly increased by UNC5C fragments, correlated with δ-secretase activation patterns (Fig. 6, F to J). IHC staining revealed that Aβ were prominently aggregated after overexpressing UNC5C FL or its δ-secretase–cleaved fragments with the greatest effects in C-terminal 468–931 and 548–931 truncates (fig. S7A). Neuronal apoptosis with TUNEL staining showed a similar effect, consistent with active caspase-3 levels in the immunoblotting (fig. S7B). Silver staining also disclosed demonstrable Aβ aggregates in the hippocampus expressing UNC5C receptor or its δ-secretase–cleaved fragments, with the most abundant levels in the C-terminal truncated samples (fig. S7C). Quantification of dendritic spines analyzed by Golgi staining (fig. S7D) and synapses studied by EM (Fig. 6, J and K) inversely correlated with δ-secretase protease activities. Cognitive behavioral tests showed that learning and memory were significantly impaired by overexpression of UNC5C receptor or its δ-secretase–cleaved truncates, with the C-terminal fragments 468–931 and 548–931 exhibiting the worst activities (Fig. 6, L to Q). Hence, δ-secretase–truncated UNC5C fragments trigger δ-secretase and caspase-3 activation and neuronal apoptosis, facilitating AD pathologies and cognitive disorders. Notably, the death domain–containing C-terminal fragments have the strongest effect.
Fig. 6. Fragments of UNC5C cleaved by δ-secretase induce neuronal cell death in the hippocampus of APP/PS1 mice.
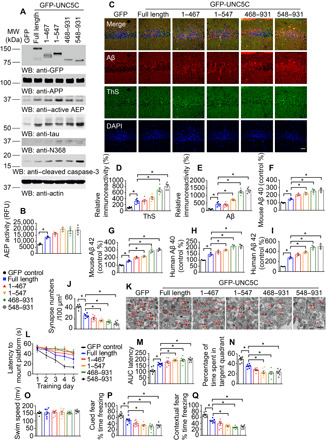
(A) WB showed the expression of UNC5C full-length, UNC5C 1–467, 1–547, 468–931, and 548–931 fragments in the hippocampus. The hippocampal lysates were detected with various indicated antibodies. Actin, loading control. Control: LV-GFP; full length: LV–GFP-UNC5C full length; 1–467: LV–GFP-UNC5C 1–467; 1–547: LV–GFP-UNC5C 1–547; 468–931: LV–GFP-UNC5C 468–931; 468–931: LV–GFP-UNC5C 468–931. (B) δ-Secretase activity in the hippocampus of above mice. Data were means ± SEM; n = 3 mice per group. (C) IF signals of ThS (green) and anti-Aβ (red) were detected in the hippocampus injected with various viruses. The nuclei were stained with DAPI. Scale bar, 30 μm. (D and E) Quantification of relative immunoreactivity was shown. (F to I) ELISA quantification of Aβ in the brain lysates of above mice. Data were means ± SEM; n = 3. (J and K) The synaptic density in the hippocampus of above mice determined by electron microscopy (means ± SEM; n = 6). Scale bar, 1 μm. (L and M) MWM analysis as time to platform (latency, s) and integrated latency (AUC) for above mice (means ± SEM; n = 7). (N and O) Probe trail and swim speed of MWM test (means ± SEM; n = 7). (P and Q) Fear conditioning test including cued fear conditioning test and contextual fear conditioning test (means ± SEM; n = 7). *P < 0.05 by one-way ANOVA followed by Tukey’s multiple-comparison test.
DISCUSSION
The DCC and UNC5H (human homologs also called UNC5A-C) netrin-1 receptors belong to the functional dependence receptors family, which share the ability to induce apoptosis in the absence of their ligands and promote cell survival upon binding to the ligands (23, 24). UNC5s or DCC, when expressed in the absence of netrin-1, induces apoptosis, whereas the presence of netrin-1 is sufficient for blocking this proapoptotic activity (25, 26). The cleavage sites of UNC5s and DCC receptors are recognized in vitro by caspase-3 (at position D412 for UNC5H2, D415 for UNC5H3, and D1290 for DCC). Mutations of the cleavage sites prevent the proapoptotic activity of these receptors, suggesting that cleavage is a prerequisite for some of cell death induction by releasing/exposing a proapoptotic domain named addiction dependence domain (ADD) lying in the intracellular domain (ICD) of DCC or UNC5H (25, 26). In the current research, we demonstrate that UNC5C receptor acts as a specific substrate for δ-secretase and facilitates neurodegeneration upon proteolytic fragmentation. δ-Secretase cleaves UNC5C in the ICD after caspase-3 cleavage site D415, yielding several truncates (Fig. 3). In human AD brains, it is robustly shredded by active δ-secretase, correlating with evident netrin-1 reduction in the patients as compared with HCs (Fig. 2). Although the ICD domain of the UNC5C receptor can be cut by both caspase-3 and δ-secretase, it appears that mutation of D415 into A does not interfere with δ-secretase cleavage of the domain. On the other hand, blockade of δ-secretase fragmentation on ICD with N467/547A mutation does not affect active caspase-3 truncation of this region either (Fig. 3C). Nevertheless, truncation at the D415 site by caspase-3 provides a better substrate to allow δ-secretase to shred the resultant 416–931 fragment more efficiently (Fig. 3D), indicating that caspase-3 cleavage of UNC5C facilitates its subsequent truncation by active δ-secretase. In vitro cleavage and cellular assays support that δ-secretase somehow mediates caspase-3 activation. For instance, purified active δ-secretase directly cuts GST–caspase-3 recombinant proteins (fig. S4). Inhibition of δ-secretase with its specific inhibitor CPA blocks netrin-1 deprivation–induced caspase-3 activation. Moreover, enzymatic-dead δ-secretase C189S antagonizes netrin-1 deletion–elicited caspase-3 activation (Fig. 4).
δ-Secretase is also called legumain, which resides in the endo-lysosomes (27). Its activation is autocatalytic, requires sequential removal of C- and N-terminal propeptides at different pH thresholds, and is bimolecular (15). It has been proposed that inactive 56-kDa zymogen is progressively converted to active 46-kDa δ-secretase by sequential autocatalytic cleavage at N323 followed by D25 cleavage by caspases. Subsequent C-terminal trimming by other lysosomal proteases yields the final mature 36-kDa form. Autoactivation is highly pH dependent and may, in part, be controlled by endosomal acidification or progress through the endo-lysosome system (15). X-ray crystallography structural comparisons between δ-secretase and caspases reveal similarities in the composition of key residues, caspase-like catalytic domain, and the catalytic mechanism (16). Intriguingly, the main physiological role of plant legumain is to mediate apoptosis, exerting caspase-like activities/specificities (28, 29). Although δ-secretase mainly resides in the endo-lysosomes and is activated under acidic pH (30), we have reported that δ-secretase is phosphorylated by SRPK2 on S226, triggering its cytoplasmic translocation and protease activation in AD brains. On the other hand, brain-derived neurotrophic factor (BDNF)–activated Akt phosphorylates δ-secretase on T322 and suppresses its protease activity (31, 32). Deficiency of BDNF in AD brains elicits its activation and Tau N368 fragmentation that binds TrkB receptors and inhibits the neurotrophic signalings (33, 34). In this report, we show that δ-secretase cleaves transmembrane UNC5C receptor on multiple intracellular sites. Conceivably, the cytoplasm translocated, and the S226-phosphorylated δ-secretase cleaves the UNC5C receptor at the plasma membrane. It remains unknown the pathological UNC5C proteolytic cleavage sequence in the brain by active caspases and δ-secretase. Because caspase-3–truncated ICD 416–931 is more readily cut by δ-secretase compared with FL UNC5C, imaginably, active caspase-3 may cut at D415 first, and the resultant 416–931 is subsequently shredded by active δ-secretase more efficiently at N467 and N547 residues, respectively, yielding cytotoxic C-terminal 468–931 and 548–931 fragments. Consequently, these fragments promote further activation of δ-secretase and caspase-3, resulting in escalated neuronal apoptosis in vitro and in mouse brains (Figs. 4 to 6 and figs. S6 and S7). In alignment with the augmented neuronal degeneration, in netrin f/f mice and APP/PS1 mice, deletion of endogenous netrin-1 in the hippocampus greatly activates δ-secretase that cleaves UNC5C at N467 and promotes Aβ production. Concomitantly, the dendritic spines and synapses are highly reduced, and these activities are solely dependent on UNC5C but not DCC receptor, as deletion of UNC5C but not DCC rescues the effects (Fig. 5 and fig. S6). Notably, inactivation of δ-secretase via C189S also abolishes netrin deficiency–induced AD pathologies and cognitive deficit, supporting that netrin reduction–triggered δ-secretase plays a critical role in these processes. Nonetheless, overexpression of δ-secretase–resistant UNC5C N467/547A mutant only partially restores AD pathologies and learning and memory dysfunctions, although it displays improved activities versus UNC5C WT (Fig. 5 and fig. S5). Presumably, several reasons may explain why δ-secretase–uncleavable UNC5C mutant does not fully inhibit netrin-1 deficiency–elicited AD pathologies and cognitive defects. First, there are multiple δ-secretase cutting sites on the ICD of UNC5C, only two were mapped out, and N467/547A mutation cannot completely suppress UNC5C cleavage by active δ-secretase. Second, UNC5C ICD can be cut by caspase-3 at two sites, and blockade of δ-secretase cleavage on UNC5C cannot prevent caspase-3 fragmentation of the receptor. Third, although the UNC5C receptor is highly expressed in the hippocampus (35) and it is selectively cut by δ-secretase but not other netrin receptors UNC5B or UNC5D (fig. S1), netrin-1 reduction–activated δ-secretase may cleave numerous other substrates including APP, Tau, and SET, contributing to AD pathologies. The pathologic significance of UNC5C cleavage by active δ-secretase is manifested in Fig. 6, where we show that these fragments strongly promote neuronal apoptosis and escalates δ-secretase activities, leading to AD pathology escalation and cognitive disorders in APP/PS1.
UNC5C plays a crucial role in mediating axon repulsion of neuronal growth cones and cell migration in the developing nervous system (23). Dysfunctional UNC5C causes neurons to misroute and fail to receive survival signals, ultimately triggering cell death (26). With recent advances in whole-exome sequencing and whole-genome sequencing technology, among numerous netrin receptors, only UNC5C is associated with AD. Six single nucleotide polymorphisms (SNPs) in UNC5C have been reported to increase the risk of LOAD (36). Wetzel-Smith et al. (11) revealed that a UNC5C variant (T835M) notably increases the levels of extracellular tau proteins and decreases the cell survival in in vitro cell models. In alignment with the observations, we also find that overexpression GFP-UNC5C FL or its fragments primarily elevates Tau levels in the brain (Fig. 6A), associated with δ-secretase–mediated Tau N368 fragmentation. We show that blockade of δ-secretase in UNC5C T835M substantially represses its pro–cell death activities (Fig. 3).
The altered UNC5C expression is associated with many types of cancers including colorectal, breast, stomach, lung, ovary, uterus, or kidney cancers (37). UNC5C expression is down-regulated in a large fraction of human colorectal cancers, mainly through promoter methylation. Moreover, in mice, inactivation of UNC5C is associated with increased intestinal tumor progression and a decrease in tumor cell apoptosis. The loss of UNC5C expression observed in human colorectal cancer is a selective advantage for tumor progression, in agreement with the dependence receptor hypothesis. Thus, UNC5C receptor is a tumor suppressor that regulates sporadic colorectal cancer (38). Previously, we have reported that netrin-1 stimulates the association between UNC5H2 with phosphoinositide-3 kinase enhancer L (PIKE-L) and promotes neuronal survival via activating PI3K (phosphatidylinositol 3-kinase)/Akt signaling (39). In agreement with this notion, netrin-1 is reduced in AD brains (Fig. 2), UNC5C receptor is proteolytically truncated by active δ-secretase and caspases, triggering neuronal cell death. However, compared with UNC5C WT, overexpression of δ-secretase uncleavable UNC5C N467/547A mutant induces less neuronal apoptosis in vitro and in vivo (Fig. 5 and fig. S3), indicating that fragmented UNC5C contributes to neuronal degeneration in the hippocampus, fitting with its abundance in these brain regions (11). Consistent with this concept, it has been reported that netrin-1 interrupts Aβ amplification and increases sAPPα, improving cognition in AD mouse model (40). Nevertheless, recent proteomic analysis showed that netrin-1 is elevated in AD brains (41), which is contradictory to what we have found in the AD brains (Fig. 2 and fig. S2). The reason accounting for this discrepancy remains unclear. Presumably, netrin-1 is aberrantly enriched in some brain regions, which was analyzed in the previous liquid chromatography–mass spectrometry (LC-MS) analysis. Clearly, further investigation into these issues is warranted by our findings and the previous studies. The presence of UNC5C cleaved fragments in the amyloid plaque suggests that they may be secreted and deposited into the extracellular proteinaceous inclusions. Our findings that AEP cleaves APP, Tau, and UNC5C, etc., and regulates the pathogenesis in AD suggest that AEP inhibitors may be beneficial for treating this devastating disease. Together, these reports strongly support that netrin signaling implicates in AD pathogenesis. Identification of UNC5C receptor selective fragmentation by active δ-secretase, triggered by netrin-1 deficiency, provides an innovative insight into the pathological roles of the netrin/UNC5C pathway contributing to AD.
MATERIALS AND METHODS
Mice, rats, and cell lines
WT C57BL/6J mice, 3xTg mice, Netrin-1flox/flox mice, and APP/PS1 mice were ordered from the Jackson laboratory (stock nos. 000664, 004807, 028038, and 004462, respectively). AEP KO mice were generated as previously reported (19). 3xTg mice were crossed with AEP−/− mice to generate 3xTg/AEP−/− mice. All mice were housed in standard conditions at 22°C and 12-hour light-dark cycle with free access to water and food. Animal handling and care was performed following the Emory Medical School guidelines and National Institutes of Health animal care guidelines. The sample size was set on the basis of the result from Power and Precision software (BioStat). Mice were assigned to different groups according to a random number table. Investigators were blinded to the group information during the animal experiments. The rats for primary cortical neurons were ordered from the Jackson laboratory. The protocols for these animal experiments were reviewed and approved by the Emory Institutional Animal Care and Use Committee. HEK293 cells [American Type Culture Collection (ATCC), CRL-1573] were cultured in high-glucose Dulbecco’s minimum Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) and penicillin (100 U/ml)–streptomycin (100 μg/ml; all from HyClone). SH-SY5Y (ATCC, CRL-2266) cells were cultured in advanced DMEM/F12 (Gibco) supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml). Cells were incubated in a humidified atmosphere of 5% CO2 at 37°C.
Human tissue samples
Frozen brain samples and tissue sections were from postmortem brains of five AD cases (age, 74.3 ± 13.3 years, means ± SD) and five nondemented controls (age, 74.6 ± 12.7 years) from Emory Alzheimer’s Disease Research Center. Informed consents were obtained from all subjects. The study was reviewed and approved by the Emory University CND Tissue Committee. The diagnosis of AD is based on criteria of the Consortium to Establish a Registry for AD and the National Institute on Aging. Diagnoses were verified by the presence of neurofibrillary tangles and amyloid plaques in formalin-fixed tissue. The postmortem interval in the AD group was similar to that in the control group.
Differentiation of human iPSC-derived neural stem cells into neurons
Human iPSC-derived neural stem cells (NSCs) were purchased from Axol Bioscience (Cambridge, UK). Information about the donors is readily available online (www.axolbio.com/). We used iPSC-derived NSCs obtained from two donors: ax0111 from AD patient with ApoE4/4 genotype, and ax0112 from AD patient with ApoE3/3 genotype. Neuron differentiation from NSCs was accomplished by culturing on PLO/laminin-coated plates in neuronal differentiation medium, which was composed of DMEM/F12 and neurobasal medium (1:1) supplemented with N2, B27, BDNF (20 ng/ml), GDNF (glial cell line–derived neurotrophic factor) (20 ng/ml), NT3 (10 ng/ml), insulin-like growth factor (10 ng/ml), ascorbic acid (200 μM) (all from STEMCELL Technologies), and dbcAMP (100 nM) (Sigma-Aldrich).
Antibodies and reagents
Antibodies to the following targets were used: anti–HA–horseradish peroxidase (HRP) (Santa Cruz Biotechnology, sc-7392), anti–GST-HRP (Sigma-Aldrich, A7340), anti-GFP (Santa Cruz Biotechnology, sc-9996), anti–UNC5C 481–550 (Santa Cruz Biotechnology, sc-135077), anti–β-actin (Abcam, ab8227), anti-AEP antibody (Cell Signaling Technology, no. 93627), anti-AEP antibody clone 6E3 (from C. Watts, University of Dundee), anti–cleaved caspase-3 (Cell Signaling Technology, no. 9661), anti–netrin-1 (Abcam, ab126729), anti–UNC5C N467 (Covance), anti–UNC5C N547 (Covance), anti–UNC5C C468 (Covance), anti–UNC5C C548 (Covance), anti-Tau (Thermo Fisher Scientific, MN1000), anti–Tau N368 (Ye Lab), anti-DCC (Santa Cruz Biotechnology, sc-6535), anti-flag (Sigma-Aldrich, F3165), anti-MAP2 (Sigma-Aldrich, M9942), anti-Aβ (BioLegend, 4G8), anti–Protein A/G PLUS Agarose (Santa Cruz Biotechnology, sc-200), Glutathione Sepharose 4B (GE Healthcare, 17075601), and anti-UNC5D (R&D Systems, AF1429).
Reagents: AEP substrate Z-Ala-Ala-Asn-AMC (Bachem, 4033201), UNC5C 459-467 peptide antigen (Covance), 4′,6-diamidino-2-phenylindole (Sigma-Aldrich, D9542), silver nitrate (Sigma-Aldrich, catalog 209139), Mouse and Rabbit Specific HRP/DAB IHC Detection Kit (Abcam, ab236466), CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega, G1780), 2F5 (AdipoGen, AG-27B-0018PF-C100), DCC-4Fbn (Ye Lab), Recombinant Mouse Netrin-1 Protein (R&D Systems, 1109-N1/CF), Phusion High-Fidelity PCR Kit (NEB, E0553L), Recombinant Human Caspase-3 Protein (Novus, H00000836-P02), AENK (Millipore, 5.33796.0001), Pan Caspase Inhibitor Z-VAD-FMK (R&D Systems, FMK001), Amyloid beta 40 Mouse ELISA Kit (Thermo Fisher Scientific, KMB3481), Amyloid beta 42 Mouse ELISA Kit (Thermo Fisher Scientific, KMB3441), Amyloid beta 40 Human ELISA Kit (Thermo Fisher Scientific, KHB3481), Amyloid beta 42 Human ELISA Kit (Thermo Fisher Scientific, KHB3441), In Situ Cell Death Detection Kit, TMR red (Roche, 12156792910), QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent, 210519), and Recombinant Legumain Protein (Sino Biological, 50051-M07H). The recombinant AEP was first activated by incubation in activation buffer (0.1 M NaOAc, 0.1 M NaCl, pH 4.5) at 37°C for 4 hours, AEP inhibitor Compound 11A (CPA, Ye Lab), Lipofectamine 3000 (Invitrogen, L3000008), and Affi-Gel 10 (Bio-Rad, 153-6046). All other chemicals not included above were purchased from Sigma-Aldrich.
Plasmids and viral vectors
HA-UNC5C plasmid was a gift from P. Mehlen’s laboratory (42). Human UNC5C (NP_003719) VersaClone cDNA was purchased from R&D Systems. In addition, mGST-UNC5C plasmids were recombined using Phusion High-Fidelity PCR Kit. All of the mutations for UNC5C were introduced using site-directed mutagenesis kit (Agilent Technologies). The toxicity of human UNC5C with T835M mutation was confirmed in SH-SY5Y cells using Lipofectamine 3000 (Invitrogen) for the transfection. The primers sequences are presented in table S1. Lentivirus package for the full-length, truncated, and site-mutated UNC5C and AAV package for AEP C189S were prepared by F.P.M.’s laboratory. All DNA sequencing was conformed in the Eurofins Genomics LLC. Flag-UNC5B-D plasmids were ordered from Addgene, and they were gifts from W. Wojtowicz (Addgene plasmid nos. 72195, 72196, and 72197). AAV-GFP, AAV-Cre, UNC5C AAV siRNA Pooled Virus, DCC AAV siRNA Pooled Virus, and Ntn1 AAV siRNA Pooled Virus were purchased from the abm company.
In vitro cleavage assay
To assess the cleavage of UNC5C by AEP in vitro, HEK293 cells were transfected with HA-UNC5C plasmid by the calcium phosphate precipitation method. Forty-eight hours after transfection, the cells were collected, washed in phosphate-buffered saline (PBS), lysed in buffer [50 mM sodium citrate, 5 mM dithiothreitol (DTT), 0.1% CHAPS, pH 5.5, 0.5% Triton X-100] for 5 min, and centrifuged for 15 min at 15,000g at 4°C. The supernatant was incubated with mouse brain lysates under pH 7.4 or 6.0 at 37°C for 30 min. To test the effect of AEP inhibitor on the cleavage of UNC5C by AEP, AENK peptide inhibitor and inactive control AEQK were used. To further verify UNC5C cleaved by AEP, HEK293 cells cotransfected with HA-UNC5C and mGST-AEP WT, mGST-AEP C189S, or control plasmid. Forty-eight hours after transfection, the cell lysates were incubated in reaction buffer (pH 6.0) at 37°C for 45 min. To measure the cleavage of purified UNC5C fragments by AEP, GST-tagged UNC5C was purified with glutathione beads. The purified UNC5C protein was incubated with recombinant AEP protein (5 μg/ml) in AEP assay buffer (50 mM sodium citrate, 5 mM DTT, 0.1% CHAPS, and 0.5% Triton X-100, pH 6.0) at 37°C for 60 min.
To clarify the cleavage of other netrin-1 receptors by AEP in vitro, flag-UNC5B, flag-UNC5C, and flag-UNC5D plasmids were used in the experiments. The procedure of cleavage assay is same with above description.
To verify whether caspase-3 is a substrate of AEP, mGST–caspase-3 protein was incubated with recombinant AEP protein (5 μg/ml) in AEP assay buffer (50 mM sodium citrate, 5 mM DTT, 0.1% CHAPS, and 0.5% Triton X-100, pH 6.0) at 37°C for 45 min.
To investigate the relationship of AEP and caspase-3 in cleaving UNC5C, we developed D415A and N467/547A mutations. Purified mGST–UNC5C WT, N467/547A, D415A, and N467/547A-D415A proteins were incubated with active caspase-3 (5 μg/ml) in caspase buffer (100 mM NaCl, 50 mM Hepes, 10 mM DTT, 1 mM EDTA, 10% glycerol, 0.1% CHAPS, pH 7.4) at 37°C for 50 min. All the samples were boiled in 1× SDS loading buffer and then analyzed by immunoblotting.
AEP/δ-secretase activity assay
Cell lysates or brain tissue homogenates were incubated in 200 μl of assay buffer (20 mM citric acid, 60 mM Na2HPO4, 1 mM EDTA, 0.1% CHAPS, and 1 mM DTT, pH 6.0) containing 20 μM AEP substrate Z-Ala-Ala-Asn-AMC. AMC released from substrate cleavage was quantified by measuring at 460 nm in a fluorescence plate reader at 37°C for 1 hour in kinetic mode. The activity of AEP was expressed as the reading at 1 hour minus the baseline reading.
Mass spectrometry analysis
Protein samples were in-gel digested with trypsin. Peptide samples were resuspended in loading buffer (0.03% trifluoroacetic acid, 0.1% formic acid, and 1% acetonitrile) and loaded onto a 20-cm nano–high-performance liquid chromatography column (internal diameter, 75 μm) packed with Reprosil-Pur 120 C18-AQ 1.9-μm beads (Dr. Maisch) and eluted over a 2-hour 1 to 50% buffer B reverse-phase gradient (buffer A, 1% acetonitrile and 0.1% formic acid in water; buffer B, 0.1% formic acid in acetonitrile) generated by a Dionex RSLCnano UPLC system (Thermo Fisher Scientific). Peptides were ionized with 2.0-kV electrospray ionization voltage from a nano-ESI source on an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific). Data-dependent acquisition of MS spectra at 120,000 resolution (full width at half maximum) and tandem mass spectrometry (MS/MS) spectra were obtained in the Orbitrap after electron-transfer dissociation with supplemental activation with high energy (EThcD) for peptide masses corresponding to the peptide SGVSLLIPAGAIPQGR (548–563) and SPILDPLPNLK (468–478). To identify AEP-cleavage sites in human UNC5C, Proteome Discoverer 2.0 (PD) was used to search and match MS/MS spectra to a complete human proteome database (NCBI reference sequence revision 62, with 68,746 entries) with a ±10-ppm mass accuracy threshold and allowable cleavages at glutamates and asparagines. A percolator was used to filter the peptide spectral matches to a false discovery rate of <1%. All MS/MS spectra for putative AEP-generated UNC5C cleavage sites were manually inspected (43).
Generation of AEP-derived UNC5C fragment antibodies
The anti–UNC5C N467, anti–UNC5C N547, anti–UNC5C C468, and anti–UNC5C C548 antibodies were developed by immunizing rabbits with the following peptides: Ac-CVSDKIPMTN-OH (anti–UNC5C N467), Ac-CLGGHLIIPN-OH (anti–UNC5C N547), H2N-SPILDPLPNC-amide (anti–UNC5C C468), and H2N-SGVSLLIPAC-amide (anti–UNC5C C548), respectively. The antiserum was pooled, and the titers against the immunizing peptides were determined by enzyme-linked immunosorbent assay (ELISA). The maximal dilution showing a positive response using chromogenic substrate for HRP was >1:750,000. The immunoactivity of the antiserum was further verified by IHC and Western blot, and then the antiserum was purified with Affi-Gel 10.
Stereotaxic injection of the viral vectors
Three-month-old netrin-1flox/flox mice and APP/PS1 mice were anesthetized with isoflurane (Piramal Healthcare). Meloxicam (2 mg/kg) was injected subcutaneously for analgesics (Loxicom, Norbrook). Bilateral intracerebral injection of 2 μl of viral vectors was performed stereotactically at coordinates (relative to bregma; posterior, 1.94 mm; lateral, 1.4 mm; and ventral, 2.2 mm) using 10-μl glass syringes with a fixed needle at a rate of 0.5 μl/min. The needle stayed in place for 5 min before it was removed slowly (not less than 2 min). The mice were placed on a heating pad until they began to recover from the surgery.
Western blot analysis
The human tissue or mouse brain tissue samples were lysed in lysis buffer (40 mM NaCl, 1 mM EDTA, 50 mM tris, pH 7.4, 0.5% Triton X-100, 1.5 mM Na3VO4, 50 mM NaF, 10 mM sodium pyrophosphate, and 10 mM sodium β-glycerophosphate, supplemented with protease inhibitors cocktail) on ice, and centrifuged at 16,000g at 4°C for 20 min. The supernatant was boiled in 1× SDS loading buffer. After SDS–polyacrylamide gel electrophoresis, the samples were transferred to nitrocellulose membrane. Western blot analysis was carried out with various antibodies. Images have been cropped for presentation.
Immunostaining
Paraffin-embedded human brain sections and free-floating mouse brain sections were detected by immunostaining with a variety of antibodies. Paraffin-embedded human brain sections were dewaxed with xylene, treated with gradient alcohol, and washed with PBS. Then, these sections were treated with 3% H2O2 at room temperature for 10 min, and washed three times with PBS. After antigen retrieval in boiling sodium citrate buffer, the sections were blocked in 1% bovine serum albumin (BSA) containing 0.3% Triton X-100 for 60 min and incubated with primary antibodies at 4°C overnight. The signal for immunochemistry was developed using the Histostain-SP kit. The signal for IF was developed using secondary antibody, Alexa Fluor 594, Alexa Fluor 488, or Cyanine5. TUNEL assay was performed according to the instructions. For ThS staining, the sections were incubated for 8 min with 0.0125% ThS in 50% ethanol, and washed with 50% ethanol and distilled water. The sections were covered with a glass cover along with using mounting solution and examined under an optical microscope or fluorescence microscope. The researchers who performed immunostaining and analysis were blinded to the group allocations.
Electron microscopy
Synaptic density in the hippocampus of mice was determined by electron microscopy. After deep anesthetization with CO2, mice were perfused transcardially with 3% paraformaldehyde and 2% glutaraldehyde in PBS. Hippocampal slices were postfixed in cold 1% OsO4 for 1 hour. Samples were prepared and examined using standard procedures. Ultrathin sections (90 nm) were stained with uranyl acetate and lead acetate and viewed at 100 kV in a JEOL 200CX electron microscope. Synapses were identified by the presence of synaptic vesicles and postsynaptic densities.
Golgi staining
Mouse brains were fixed in 10% formalin for 24 hours and then immersed in 3% potassium bichromate for 7 days in the dark. The solution was changed every day. Then, the brains were transferred into 2% silver nitrate solution and incubated in the dark for 24 hours. Vibratome sections were cut at 50 μm, air dried for 10 min, dehydrated through 95 and 100% ethanol, cleared in xylene, and mounted with coverslips.
ELISA quantification of Aβ
To detect the concentration of Aβ in the mice brain, the brain tissues were homogenized in 8× mass of 5 M guanidine HCl/50 mM tris-HCl (pH 8.0) and then incubated for 3 hours at room temperature. The lysates were diluted with cold reaction buffer (PBS with 0.03% Tween 20, 5% BSA, and supplemented with protease inhibitor cocktail) and then centrifuged at 16,000g at 4°C for 20 min. The Aβ in the brain samples were analyzed using Amyloid beta 40 Mouse ELISA Kit (Thermo Fisher Scientific, KMB3481), Amyloid beta 42 Mouse ELISA Kit (Thermo Fisher Scientific, KMB3441), Amyloid beta 40 Human ELISA Kit (Thermo Fisher Scientific, KHB3481), and Amyloid beta 42 Human ELISA Kit (Thermo Fisher Scientific, KHB3441) according to the instructions. The concentrations of Aβ were determined by comparison with the standard curve.
Morris water maze
Three months after the viral vector injection, netrin-1flox/flox mice and APP/PS1 mice were trained in a round and water-filled tub (52-inch diameter) in an environment rich with extra maze cues. An invisible escape platform was located 1 cm below the water surface in a fixed spatial location independent of a subject’s beginning position on a particular trial. In this manner, mice needed to use other maze cues to determine the location of platform. At the beginning of each trial, the subject was placed in the water maze with its paws touching the wall from one of four different starting positions (N, S, E, and W). Each one was given four trials per day lasting for five consecutive days with a 15-min intertrial interval. The maximum trial time was 60 s, and the subject was manually guided to the platform if it did not arrive by the allotted time. Once reaching the invisible escape platform, the subject was left on it for an additional 5 s to allow survey for the cues in the environment to guide future navigation to the platform. After each trial, the mouse was dried and kept in a cage filled with paper towels to allow the subject to dry off. The temperature of the water was maintained between 22° and 25°C. After 5 days of task acquisition, the platform was removed, and a probe trial over 60 s was performed testing the percentage of time spent in the quadrant, which previously contained the escape platform. All trials were analyzed for latency and swim speed.
Fear conditioning test
On day 1, the subject was placed in the fear conditioning apparatus (7-inch D × 7-inch W × 12-inch H, Coulbourn) composed of plexiglass and a metal shock grid floor and allowed to explore the enclosure for 3 min. After this habituation period, three conditioned stimulus (CS)–unconditioned stimulus (US) cycles were presented with a 1-min intertrial interval. The CS was composed of a 20-s 85-db tone, and US was composed of 2 s of a 0.5-mA footshock, which was coterminate with each CS presentation. One minute following the last CS-US presentation, mice were put back to their home cage. On day 2, the subjects were performed with a context test, during which the mouse was placed in the same chamber used on day 1, and the amount of freezing was recorded using a camera with the software provided by Coulbourn. No shock was given during the context test. On day 3, a tone test was performed, during which mice were exposed to the CS in a novel compartment. At the beginning, the subject was allowed to explore the new context for 2 min. Then, the 85-db tone was presented for 6 min, and the amount of freezing was recorded in the same way (19).
Primary neuron cultures
Primary cortical neurons of rats were cultured as previously described (19). To measure the effect of netrin-1 deprivation on primary neurons and the role of AEP during this process, neurons cultured 7 days in vitro (DIV 7) were pretreated with pan-caspase inhibitor (FMK001, 100 nM) or AEP (δ-secretase)–specific inhibitor (Compound 11A, CPA; 100 nM) 30 min before 2F5 (10 μg/ml), DCC-4Fbn (10 μg/ml), or control vehicle treatment. Twenty-four hours later, neurons were collected for detecting netrin-1, AEP, UNC5C, and caspase-3 expression and AEP activity. The medium was collected for LDH assay according to the instruction. To further verify the role of netrin-1 receptors and AEP during this process, primary neurons cultured 13 days in vitro (DIV 13) were infected with different viruses at day 5 (AAV-GFP, Ntn1 AAV siRNA Pooled Virus, Unc5c AAV siRNA Pooled Virus, DCC AAV siRNA Pooled Virus, and AAV-AEP C189S); 9 days later, neurons and medium were collected for further detection.
Statistical analysis
Statistical analysis was performed using either Student’s t test (two-group comparison) or one-way analysis of variance followed by Tukey’s multiple-comparison test (more than two groups), and differences with P value less than 0.05 were considered significant.
Acknowledgments
Funding: This work is supported by a grant from the National Institutes of Health (RF1, AG051538) to K.Y. and the State Key Program of the National Natural Science Foundation of China (no. 81671051) to Z.Z. This study was supported in part by the Rodent Behavioral Core (RBC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities. We thank ADRC at Emory University for human patients with AD and HC samples (P30 AG066511). Additional support was provided by the Viral Vector Core of the Emory Neuroscience NINDS Core Facilities (P30NS055077). Further support was provided by the Georgia Clinical and Translational Science Alliance of the National Institutes of Health under Award Number UL1TR002378. Author contributions: K.Y. conceived the project, designed the experiments, analyzed the data, and wrote the manuscript. G.C., Z.W., and E.H.A. designed and performed most of the experiments and analyzed the data. S.S.K. conducted stereotaxic injection of the viral vectors. Y.X. prepared iPSC-derived neurons. X.L. prepared primary neurons and assisted with in vivo and in vitro experiments. I.M.S. and F.P.M. provided the AAV-AEP C189S viral vectors. Z.Z. assisted with data analysis and interpretation and critically read the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/16/eabe4499/DC1
REFERENCES AND NOTES
- 1.Holtzman D. M., Morris J. C., Goate A. M., Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 3, 77sr71 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore S. W., Tessier-Lavigne M., Kennedy T. E., Netrins and their receptors. Adv. Exp. Med. Biol. 621, 17–31 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Hong K., Hinck L., Nishiyama M., Poo M. M., Tessier-Lavigne M., Stein E., A ligand-gated association between cytoplasmic domains of UNC5 and DCC family receptors converts netrin-induced growth cone attraction to repulsion. Cell 97, 927–941 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Bradford D., Cole S. J., Cooper H. M., Netrin-1: Diversity in development. Int. J. Biochem. Cell Biol. 41, 487–493 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Shabani M., Haghani M., Tazangi P. E., Bayat M., Shid Moosavi S. M., Ranjbar H., Netrin-1 improves the amyloid-β-mediated suppression of memory and synaptic plasticity. Brain Res. Bull. 131, 107–116 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Zamani E., Parviz M., Roghani M., Mohseni-Moghaddam P., Key mechanisms underlying netrin-1 prevention of impaired spatial and object memory in Aβ1-42 CA1-injected rats. Clin. Exp. Pharmacol. Physiol. 46, 86–93 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Sun L., Ju T., Wang T., Zhang L., Ding F., Zhang Y., An R., Sun Y., Li Y., Lu Y., Zhang X., Chi L., Decreased netrin-1 and correlated Th17/Tregs balance disorder in Aβ1–42 induced Alzheimer’s disease model rats. Front. Aging Neurosci. 11, 124 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lourenço F. C., Galvan V., Fombonne J., Corset V., Llambi F., Müller U., Bredesen D. E., Mehlen P., Netrin-1 interacts with amyloid precursor protein and regulates amyloid-β production. Cell Death Differ. 16, 655–663 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rama N., Goldschneider D., Corset V., Lambert J., Pays L., Mehlen P., Amyloid precursor protein regulates netrin-1-mediated commissural axon outgrowth. J. Biol. Chem. 287, 30014–30023 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ridge P. G., Hoyt K. B., Boehme K., Mukherjee S., Crane P. K., Haines J. L., Mayeux R., Farrer L. A., Pericak-Vance M. A., Schellenberg G. D., Kauwe J. S. K.; Alzheimer’s Disease Genetics Consortium (ADGC) , Assessment of the genetic variance of late-onset Alzheimer’s disease. Neurobiol. Aging 41, 200.e13–200.e20 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wetzel-Smith M. K., Hunkapiller J., Bhangale T. R., Srinivasan K., Maloney J. A., Atwal J. K., Sa S. M., Yaylaoglu M. B., Foreman O., Ortmann W., Rathore N., Hansen D. V., Tessier-Lavigne M.; Alzheimer’s Disease Genetics Consortium, Mayeux R., Pericak-Vance M., Haines J., Farrer L. A., Schellenberg G. D., Goate A., Behrens T. W., Cruchaga C., Watts R. J., Graham R. R., A rare mutation in UNC5C predisposes to late-onset Alzheimer’s disease and increases neuronal cell death. Nat. Med. 20, 1452–1457 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hashimoto Y., Toyama Y., Kusakari S., Nawa M., Matsuoka M., An Alzheimer disease-linked rare mutation potentiates netrin receptor uncoordinated-5C-induced signaling that merges with amyloid β precursor protein signaling. J. Biol. Chem. 291, 12282–12293 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun J.-H., Wang H.-F., Zhu X.-C., Yu W.-J., Tan C.-C., Jiang T., Tan M.-S., Tan L., Yu J.-T.; Alzheimer’s Disease Neuroimaging Initiative , The impact of UNC5C genetic variations on neuroimaging in Alzheimer’s disease. Mol. Neurobiol. 53, 6759–6767 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Yang H.-S., White C. C., Chibnik L. B., Klein H.-U., Schneider J. A., Bennett D. A., De Jager P. L., UNC5C variants are associated with cerebral amyloid angiopathy. Neurol. Genet. 3, e176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li D. N., Matthews S. P., Antoniou A. N., Mazzeo D., Watts C., Multistep autoactivation of asparaginyl endopeptidase in vitro and in vivo. J. Biol. Chem. 278, 38980–38990 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Zhao L., Hua T., Crowley C., Ru H., Ni X., Shaw N., Jiao L., Ding W., Qu L., Hung L.-W., Huang W., Liu L., Ye K., Ouyang S., Cheng G., Liu Z.-J., Structural analysis of asparaginyl endopeptidase reveals the activation mechanism and a reversible intermediate maturation stage. Cell Res. 24, 344–358 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Z., Jang S.-W., Liu X., Cheng D., Peng J., Yepes M., Li X.-j., Matthews S., Watts C., Asano M., Hara-Nishimura I., Luo H. R., Ye K., Neuroprotective actions of PIKE-L by inhibition of SET proteolytic degradation by asparagine endopeptidase. Mol. Cell 29, 665–678 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Basurto-Islas G., Gu J.-H., Tung Y. C., Liu F., Iqbal K., Mechanism of tau hyperphosphorylation involving lysosomal enzyme asparagine endopeptidase in a mouse model of brain ischemia. J. Alzheimers Dis. 63, 821–833 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Zhang Z., Song M., Liu X., Kang S. S., Kwon I.-S., Duong D. M., Seyfried N. T., Hu W. T., Liu Z., Wang J.-Z., Cheng L., Sun Y. E., Yu S. P., Levey A. I., Ye K., Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat. Med. 20, 1254–1262 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z., Song M., Liu X., Kang S. S., Duong D. M., Seyfried N. T., Cao X., Cheng L., Sun Y. E., Yu S. P., Jia J., Levey A. I., Ye K., Delta-secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer’s disease. Nat. Commun. 6, 8762 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leuzy A., Cicognola C., Chiotis K., Saint-Aubert L., Lemoine L., Andreasen N., Zetterberg H., Ye K., Blennow K., Höglund K., Nordberg A., Longitudinal tau and metabolic PET imaging in relation to novel CSF tau measures in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 46, 1152–1163 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paradisi A., Maisse C., Coissieux M.-M., Gadot N., Lépinasse F., Delloye-Bourgeois C., Delcros J.-G., Svrcek M., Neufert C., Fléjou J.-F., Scoazec J.-Y., Mehlen P., Netrin-1 up-regulation in inflammatory bowel diseases is required for colorectal cancer progression. Proc. Natl. Acad. Sci. U.S.A. 106, 17146–17151 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mehlen P., Guenebeaud C., Netrin-1 and its dependence receptors as original targets for cancer therapy. Curr. Opin. Oncol. 22, 46–54 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Mehlen P., Furne C., Netrin-1: When a neuronal guidance cue turns out to be a regulator of tumorigenesis. Cell. Mol. Life Sci. 62, 2599–2616 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mehlen P., Rabizadeh S., Snipas S. J., Assa-Munt N., Salvesen G. S., Bredesen D. E., The DCC gene product induces apoptosis by a mechanism requiring receptor proteolysis. Nature 395, 801–804 (1998). [DOI] [PubMed] [Google Scholar]
- 26.Llambi F., Causeret F., Bloch-Gallego E., Mehlen P., Netrin-1 acts as a survival factor via its receptors UNC5H and DCC. EMBO J. 20, 2715–2722 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dall E., Brandstetter H., Structure and function of legumain in health and disease. Biochimie 122, 126–150 (2016). [DOI] [PubMed] [Google Scholar]
- 28.del Pozo O., Lam E., Caspases and programmed cell death in the hypersensitive response of plants to pathogens. Curr. Biol. 8, R896–R897 (1998). [DOI] [PubMed] [Google Scholar]
- 29.De Jong A. J., Hoeberichts F. A., Yakimova E. T., Maximova E., Woltering E. J., Chemical-induced apoptotic cell death in tomato cells: Involvement of caspase-like proteases. Planta 211, 656–662 (2000). [DOI] [PubMed] [Google Scholar]
- 30.Chen J. M., Dando P. M., Rawlings N. D., Brown M. A., Young N. E., Stevens R. A., Hewitt E., Watts C., Barrett A. J., Cloning, isolation, and characterization of mammalian legumain, an asparaginyl endopeptidase. J. Biol. Chem. 272, 8090–8098 (1997). [DOI] [PubMed] [Google Scholar]
- 31.Wang Z.-H., Liu P., Liu X., Manfredsson F. P., Sandoval I. M., Yu S. P., Wang J.-Z., Ye K., Delta-secretase phosphorylation by srpk2 enhances its enzymatic activity, provoking pathogenesis in Alzheimer’s disease. Mol. Cell 67, 812–825.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Z.-H., Wu W., Kang S. S., Liu X., Wu Z., Peng J., Yu S. P., Manfredsson F. P., Sandoval I. M., Liu X., Wang J.-Z., Ye K., BDNF inhibits neurodegenerative disease-associated asparaginyl endopeptidase activity via phosphorylation by AKT. JCI Insight 3, e99007 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Z.-H., Xiang J., Liu X., Yu S. P., Manfredsson F. P., Sandoval I. M., Wu S., Wang J.-Z., Ye K., Deficiency in BDNF/TrkB neurotrophic activity stimulates δ-secretase by upregulating C/EBPβ in Alzheimer’s disease. Cell Rep. 28, 655–669.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiang J., Wang Z.-H., Ahn E. H., Liu X., Yu S.-P., Manfredsson F. P., Sandoval I. M., Ju G., Wu S., Ye K., Delta-secretase-cleaved Tau antagonizes TrkB neurotrophic signalings, mediating Alzheimer’s disease pathologies. Proc. Natl. Acad. Sci. U.S.A. 116, 9094–9102 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim D., Ackerman S. L., The UNC5C netrin receptor regulates dorsal guidance of mouse hindbrain axons. J. Neurosci. 31, 2167–2179 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Q., Wang B.-L., Sun F.-R., Li J.-Q., Cao X.-P., Tan L., The role of UNC5C in Alzheimer’s disease. Ann. Transl. Med. 6, 178 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thiébault K., Mazelin L., Pays L., Llambi F., Joly M.-O., Scoazec J.-Y., Saurin J.-C., Romeo G., Mehlen P., The netrin-1 receptors UNC5H are putative tumor suppressors controlling cell death commitment. Proc. Natl. Acad. Sci. U.S.A. 100, 4173–4178 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bernet A., Mazelin L., Coissieux M.-M., Gadot N., Ackerman S. L., Scoazec J.-Y., Mehlen P., Inactivation of the UNC5C Netrin-1 receptor is associated with tumor progression in colorectal malignancies. Gastroenterology 133, 1840–1848 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang X., Jang S.-W., Okada M., Chan C.-B., Feng Y., Liu Y., Luo S.-W., Hong Y., Rama N., Xiong W.-C., Mehlen P., Ye K., Netrin-1 mediates neuronal survival through PIKE-L interaction with the dependence receptor UNC5B. Nat. Cell Biol. 10, 698–706 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spilman P. R., Corset V., Gorostiza O., Poksay K. S., Galvan V., Zhang J., Rao R., Peters-Libeu C., Vincelette J., Geehan A. M., Dvorak-Ewell M., Beyer J., Campagna J., Bankiewicz K., Mehlen P., John V., Bredesen D. E., Netrin-1 interrupts amyloid-β amplification, increases sAβPPα in vitro and in vivo, and improves cognition in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 52, 223–242 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Bai B., Wang X., Li Y., Chen P.-C., Yu K., Dey K. K., Yarbro J. M., Han X., Lutz B. M., Rao S., Jiao Y., Sifford J. M., Han J., Wang M., Tan H., Shaw T. I., Cho J.-H., Zhou S., Wang H., Niu M., Mancieri A., Messler K. A., Sun X., Wu Z., Pagala V., High A. A., Bi W., Zhang H., Chi H., Haroutunian V., Zhang B., Beach T. G., Yu G., Peng J., Deep multilayer brain proteomics identifies molecular networks in Alzheimer’s disease progression. Neuron 105, 975–991.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Delloye-Bourgeois C., Fitamant J., Paradisi A., Cappellen D., Douc-Rasy S., Raquin M.-A., Stupack D., Nakagawara A., Rousseau R., Combaret V., Puisieux A., Valteau-Couanet D., Bénard J., Bernet A., Mehlen P., Netrin-1 acts as a survival factor for aggressive neuroblastoma. J. Exp. Med. 206, 833–847 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Z., Kang S. S., Liu X., Ahn E. H., Zhang Z., He L., Iuvone P. M., Duong D. M., Seyfried N. T., Benskey M. J., Manfredsson F. P., Jin L., Sun Y. E., Wang J.-Z., Ye K., Asparagine endopeptidase cleaves α-synuclein and mediates pathologic activities in Parkinson’s disease. Nat. Struct. Mol. Biol. 24, 632–642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/16/eabe4499/DC1