

This newly developed biological probes/proteomics method yields functional antibodies targeting previously undruggable targets.
Abstract
Several important drug targets, e.g., ion channels and G protein–coupled receptors, are extremely difficult to approach with current antibody technologies. To address these targets classes, we explored kinetically controlled proteases as structural dynamics–sensitive druggability probes in native-state and disease-relevant proteins. By using low–Reynolds number flows, such that a single or a few protease incisions are made, we could identify antibody binding sites (epitopes) that were translated into short-sequence antigens for antibody production. We obtained molecular-level information of the epitope-paratope region and could produce high-affinity antibodies with programmed pharmacological function against difficult-to-drug targets. We demonstrate the first stimulus-selective monoclonal antibodies targeting the transient receptor potential vanilloid 1 (TRPV1) channel, a clinically validated pain target widely considered undruggable with antibodies, and apoptosis-inducing antibodies selectively mediating cytotoxicity in KRAS-mutated cells. It is our hope that this platform will widen the scope of antibody therapeutics for the benefit of patients.
INTRODUCTION
Although there has been tremendous development of antibody technologies and antibody-based therapeutics over the past 20 years, it is unexpected that only 60 unique antibody therapeutics (based on target) are available to patients (1). Both G protein–coupled receptors (GPCRs) and ion channels are particularly underexploited for antibody-based therapeutics. For GPCRs, there are only two marketed monoclonal antibody therapies (1) while several are in development (2). For ion channels, there are no market-approved monoclonal antibodies (1–3). A small number of monoclonal antibodies that target ion channels with large extracellular (EC) regions (e.g., P2X channels) have been developed; however, ion channels with small EC regions, including transient-receptor potential (TRP) channels, are extremely challenging to target with antibodies (2, 4).
Current technologies for antibody development largely rely on producing antigens, either from whole or parts of proteins. When these are structurally stable in solution and maintain their native disease-relevant state and meaningful epitope structure throughout the discovery cycle, they can successfully be used with the hybridoma technology (5), phage display, and other screening-based technologies (6). These approaches have been highly successful, in particular, for soluble proteins. Another strategy for antibody development is based on predicting suitable epitope regions based on, e.g., crystal structures or a bioinformatic (in silico) evaluation of the protein sequence.
Independent of approach, poor results have been obtained for antibody development against ion channels and other multipass transmembrane proteins, typically having poorly exposed surface area. In contrast, several single-pass membrane proteins with large exposed surface area such as SLAMF7, CTLA-4, HER2, CD30, PD-L1, and PD-1 (1) have been successfully developed. The fundamental problem with multipass membrane proteins has been that the native state is not preserved unless they are embedded or anchored in a lipid membrane and, furthermore, that they have complex structural dynamics that are difficult to capture in vitro or in silico. Multipass transmembrane proteins, such as ion channels, are flexible, exhibiting transitory structural variations that are kT-driven, but also state dependent (open-closed channels, desensitization, and association/binding) (7–10). Conformational transformations may be elicited principally by (i) changes in the membrane potential of a cell (11), (ii) changes of the physical (e.g., temperature) EC environment (12), or (iii) ligand binding (13). Thus, a native protein, such as an ion channel, has a broad spectrum of different structural conformations along with different exposures of regions that may constitute opportunistic targets for ligands or drugs, including antibodies. As visualization, a simulation showing thermally induced fluctuations and motions of a TRP channel at 37°C can be found in movie S1.
Here, we present a new platform technology for antibody development against currently undruggable ion channels and other membrane proteins. This approach can produce antigens for potential epitopes identified on native-state, disease-relevant proteins in motion. The methodology encompasses a series of distinct steps, which are described in detail in the following section. In the subsequent results section, we present example data resulting from different epitope-mapping protocols as well as antibodies developed toward two targets TRPV1 and KRAS, previously undruggable with antibodies.
A protein exposes different accessible regions on its surface dynamically, and the pivotal step of the presented technology is identification of potential antibody binding sites (epitopes) on such dynamic protein structures. For this purpose, first, we use “antibody-like” (see below) proteases as freely diffusing molecular probes, adapting to the real-time dynamics and structural motion of a protein. Second, as further described below, by controlling protease activity (kinetics), only epitopes on native and disease-relevant target structure are identified. A protease specifically cleaves a peptide bond in a native, tertiary structure if said peptide bond is surface exposed (14). The binding interface of several proteases is of similar size as the binding interface of an antibody variable region (Fig. 1A) [i.e., specifically immunoglobulin G (IgG) (15–19)], and the area of the exposed target surface required for protease docking is similar to that required for antibody binding (Fig. 1B). Therefore, these proteases can, in theory, be used as druggability probes for identification of antibody binding sites. The protease, when binding to a target, provides molecular-level information on the location of its catalytic triad, within the docking area of the target protein, and yields a protease-identified cut site (PIC). The produced PICs are translated into a central point or coordinate in the epitope. The key to success lies in identifying PICs only on relevant protein structure, obtained using limited digestions to avoid target denaturation (20). Suitably, proteolytic activity (kinetics) is controlled using microfluidic flow cells (Fig. 2A) operating at low–Reynolds number flow. Membrane vesicles or cells presenting the target are immobilized on the inner surface of a microfluidic device [an example of a suitable flow cell has previously been presented in detail (21)]. The target is exposed to a protease or sequences of proteases (at a given concentration for a controlled period of time) as step functions with additional treatments, wash steps, and sample collections in-between. Cleaved-off peptides are analyzed using tandem mass spectrometry (MS/MS) (Fig. 2A). Flow cells with various surface-to-volume ratios are available to accommodate detection of peptides from small sample volumes (22, 23).
Fig. 1. Schematic drawing comparing key structural features of importance in the protease-target and antibody-target binding interfaces.
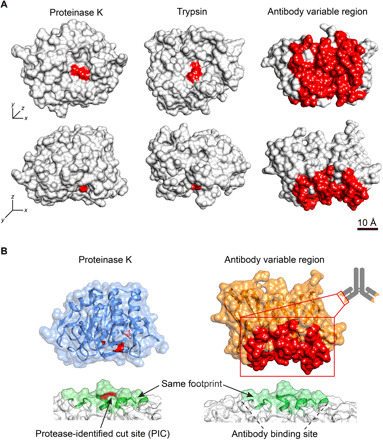
(A) Area comparison of protease and antibody binding interfaces, exemplified by the proteases proteinase K [Protein Data Bank (PDB), 2id8] and trypsin (PDB, 1h4w), respectively. The images display the bottom and side views of the respective proteases and the variable region in one antibody Fab fragment (the binding region of an antibody) (PDB, 6bae). The catalytic triad in the proteases, and the complementarity determining regions (CDR) of the antibody variable region have been colored red. (B) Binding of a protease to a protein results in a cut in the peptide sequence. Under controlled kinetics conditions in a native protein, this cut is denoted a protease-identified cut site (PIC) and can be used to identify molecular-level information of where protease binding to the target protein has occurred. Because of size similarity of the area of the protease-binding surface and the CDR of the variable region, a PIC is predictive of a region that can be bound by an antibody.
Fig. 2. Schematic drawing showing the workflow in the platform from identification of antibody binding sites to the final antibody product.
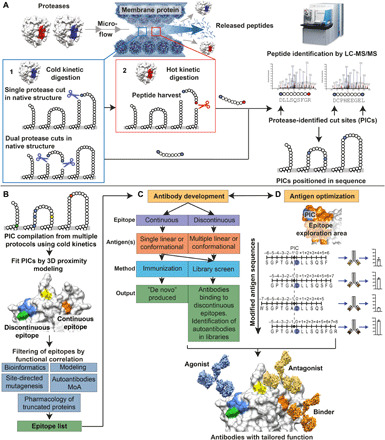
(A) The protein target is immobilized within a microfluidic flow cell, either in whole cells or in membrane-derived vesicles, enabling exposure of the protein to sequential protease digestion under controlled kinetic conditions, resulting in the release of peptides that is identified with MS/MS. A combination of distinct kinetically controlled proteolysis steps are used. (1) The protein is exposed to a protease under cold kinetic conditions that creates one or a few cuts. Release of peptides for analysis occurs if cuts are close in sequence. To also detect single cuts, (2) a protease with complementary specificity is used under hot kinetic conditions for peptide harvest. Released peptides are identified using MS/MS where cold kinetic cut sites made on the native protein are considered for further analysis. Identified cold kinetic cut sites are denoted PICs. LC-MS/MS, liquid chromatography–MS/MS. (B) Identified PICs are mapped onto a 3D structure of the target to visualize the distribution of accessible areas for antibody binding, identifying discontinuous and continuous epitopes, respectively. Epitope sequence is correlated against functional information to filter and sort epitopes that would yield binders, agonists, or antagonists. (C) Synthetic antigens mimicking the identified epitopes on the protein target are used to produce optimized antibodies using either immunization or antibody library screening (e.g., phage display). (D) Schematic of hASO where antigens and antibodies are optimized toward an epitope region through an iterative process. Small changes in antigen position/sequence/structure are introduced in each step, and the characteristics of each resulting antibody are evaluated until the desired properties (affinity, selectivity, and function) have been achieved.
Digestion protocols are tailored on the basis of target sequence. Proteases with different specificity are used in parallel to maximize amino acid sequence coverage, or they can be used sequentially to increase detection. A first step involves subjecting a target to a protease under “cold” kinetic conditions, i.e., controlled reaction rate conditions to make only one or few precision cuts in the target sequence that are predictive of surface exposure and potential antibody binding sites (Fig. 2A). To capture both single and dual cuts, a secondary protease is used under “hot” kinetic conditions to harvest peptides for identification (Fig. 2A). Primary PICs and secondary protease cut sites are easily differentiated by using first and second proteases with different sequence specificity.
Modeling and structure analysis are performed to fit PICs into a three-dimensional (3D) model, to visualize the structure of the exposed surfaces (Fig. 2B). Two types of epitopes are commonly identified. PICs, distant in the amino acid sequence, but within the same surface region of the 3D-folded protein, define a “discontinuous epitope.” PICs closely located in the amino acid sequence and within the same surface region of the folded protein define a “continuous epitope.” If an antagonistic or agonistic antibody is desired, then the epitope sequences are correlated against bioinformatic data on target function to identify epitopes overlapping or coinciding with functionally important regions (Fig. 2B).
Antibody production directed to epitopes identified through PICs and functional correlation is enabled using short synthetic peptide antigens. The antigens are designed on the basis of knowledge of the extended surface area surrounding the PICs that corresponds approximately to the surface area of an antibody-protein binding interface. Short synthetic antigens have few structural degrees of freedom and therefore result in less diverse antibodies using immunization (Fig. 2C) (24, 25). These antigens can be forced into conformations matching the target structure using peptide chemistry (e.g, cyclic, stapled, or nicked peptides). For a discontinuous epitope, multiple antigens are developed where each part, corresponding to the separate sequence segments in the discontinuous epitope, is translated into a peptide antigen. Both immunization and antibody library screening (e.g., phage display) can be used for production against a continuous epitope (Fig. 2C). With immunization, the antibody is in vivo affinity matured against the antigen and with library screening, by choosing suitable libraries; e.g., from patients with autoimmune disease, there is a possibility to select autoantibodies. For a discontinuous epitope, screening antibody libraries against multiple antigens developed for the epitope may select antibodies that bind all parts of the epitope. Subsequently, optimization of epitope-binding antibody candidates is done through a process named human antigen superoptimization (hASO) as shown in Fig. 2D. This is a systematic interrogation of the epitope area with many different antibodies, generated from a plurality of altered antigens. More specifically, antigen libraries are created, where small sequence alterations, i.e., elongations, truncations, and amino acid exchanges, are introduced into the antigens, to find a high-affinity binding antibody. This interrogation requires detailed knowledge of the epitope area, which is inherently provided by the presented technology. Ultimately, this results in an hASO-optimized antigen used to produce the final antibody with an optimal function and affinity profile. The pharmacological profile, and affinity, can be further optimized using traditional antibody engineering strategies, including in vitro affinity maturation. The end product is an antibody, tailored to elicit the optimal binding interaction toward the epitope region through detailed knowledge of both the epitope and paratope sequence, to yield the required disease-modifying functionality.
RESULTS
An example of how the level of protein digestion can be controlled by using different protease exposures to a human embryonic kidney (HEK) cell line can be seen in Fig. 3A. Here, the proteases trypsin, chymotrypsin, Asp-N, and proteinase K were used at different concentrations and incubation times. The average number of EC cut sites per protein is shown for each protease and protocol, and the protocols have been adjusted such that the lowest protease exposure yields approximately two to three cut sites per protein. In cases where a cold kinetic digestion does not generate a released fragment from the target protein for analysis, a second hot kinetic digestion is used to harvest peptides. By using proteases with different specificity, the first cut site can be identified as it has a unique molecular signature through its terminal amino acids. If trypsin (cold kinetic digestion) and Asp-N (hot kinetic digestion) are used in sequence, then we would obtain peptides where the N-terminal amino acid follows Lys/Arg and where the C-terminal amino acid is followed by Asp. Alternatively, Asp is at the N terminus, and Lys/Arg is at the C terminus. In both cases, the Lys/Arg cut resides in a region of the protein accessed by the cold protease while the protein was in a native state. As an example, we evaluated the impact of using different sequential digestion protocols with a cold kinetic digestion followed by a hot kinetic digestion, on the whole proteome level for HEK cells as can be seen in Fig. 3B. The number of EC PICs that could be detected, digested by the first protease with cold kinetic conditions, increased between 79 and 136% after addition of the second protease under hot kinetic conditions.
Fig. 3. Comparison of the number of PICs in the HEK cell membrane proteome using different digestion protocols.
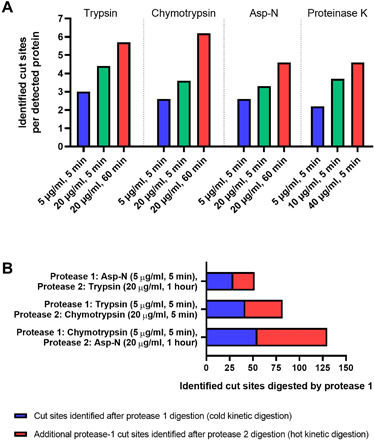
(A) Identified cut sites per detected protein with varying protease exposure. Membrane-derived vesicles from HEK cells were subjected to varying protease exposure (concentration and time) using either trypsin, chymotrypsin, Asp-N, and proteinase K. The number of identified extracellular cut sites per detected membrane protein is shown. The number of identified cut sites increases with increasing protease exposure. (B) Comparison of the number of identified cut sites after using a cold kinetic digestion (protease 1) alone or in combination with a sequential hot kinetic digestion (protease 2).
Modality-selective anti-TRPV1 antibody
TRPV1 is a clinically validated target both for acute and chronic pain and has been studied for almost two decades (26). TRPV1 is a temperature-sensitive ion channel that can be activated by several cues as described in Supplementary Text. Despite a long history as a promising drug target, small-molecule approaches, where attempts have been made to block all activity of the receptor, including heat activation, have failed because of adverse side effects such as hyperthermia or loss of heat sensation (27). Our aim was to develop modality-selective, antagonistic antibodies targeting TRPV1 to inhibit only capsaicin activation and not heat activation of the channel. It is generally believed that this modality-selective antagonism will avoid the heat-related side effects observed in clinical trials with historic small-molecule antagonists that block all TRPV1 modalities.
To probe the surface of the TRPV1 protein for potential antibody binding sites, proteolysis under cold kinetic conditions was performed on membrane vesicles derived from human TRPV1 (hTRPV1)–expressing Chinese hamster ovary (CHO) cells immobilized inside a flow cell (21, 28). It has previously been shown that TRPV1 retains capsaicin-induced activity in these membrane-derived vesicles (28). Using a digestion protocol with proteinase K (5 μg/ml) for 5 min, a total of 19 PICs were identified. Two of these were of particular interest for development of therapeutic antibodies potentially having a stimulus-selective pharmacological profile different from classical small-molecule antagonists. These were both located in the prepore EC loop preceding the pore region, as can be seen in Fig. 4A, and confirm that the prepore loop is accessible to antibody binding. A homology model of TRPV1 was created to position the PICs in 3D, where the prepore loop was determined to be highly flexible, and a potential epitope region was defined (marked in green in Fig. 4B). Cryo–electron microscopy studies have shown that activation of TRPV1 with the ligand resiniferatoxin (RTX) results in a shift in position of the prepore loop and the pore helix, leading to widening of the selectivity filter (29). Capsaicin, RTX, and other vanilloid ligands activate TRPV1 by binding into a vanilloid pocket in the transmembrane region far away from the prepore region (30). Several of these small-molecule antagonists have been shown to block all stimulus modalities, including heat activation, in TRPV1 (31). We hypothesize that binding of an antibody to the prepore loop can lead to an allosteric modulation of TRPV1 activity that is distinct from the competitive antagonism elicited by small molecules binding in the vanilloid pocket. To investigate this possibility, we created and used three different antigens, consisting of either linear or cyclic peptides, for monoclonal antibody development through immunization, and subsequently, clones were selected for further evaluation based on binding to TRPV1 using flow cytometry as can be seen in fig. S1. All three antigens resulted in monoclonal antibodies capable of binding to TRPV1 but differing in capability of inhibiting capsaicin-induced currents. To date, the clone eliciting the highest inhibition of capsaicin currents was developed using a cyclic antigen (table S2), matching the marked epitope region in Fig. 4, A and B, and the antibody was named OB1. OB1 bound to TRPV1 as observed with immunocytochemistry (Fig. 5B), and a binding median effective concentration (EC50) against the TRPV1 protein was determined to be 6 nM, using flow cytometry (Fig. 5A). OB1 inhibited both capsaicin-induced currents (Fig. 5, C and D) and currents induced by the endogenous ligand N-arachidonyl dopamine (NADA) as shown in Fig. 5E in a dose-dependent manner. With electrophysiology, it was further confirmed that 113 nM OB1 elicited 71% inhibition of 300 nM capsaicin-induced currents (Fig. 5C), 225 nM OB1 elicited 59% inhibition of 100 nM capsaicin-induced currents (Fig. 5D), and 68% inhibition of 1 μM NADA-induced currents (Fig. 5E). Using single-cell calcium imaging in combination with pulsed infrared laser heating, a 3%, nonsignificant, potentiation of heat-activated currents (Fig. 5F) was observed when comparing 225 nM OB1-treated cells to vehicle-treated cells. This experiment suggests that OB1 treatment had no effect on heat-activated currents. Together, OB1 displays a stimulus-selective multimodal pharmacological profile, inhibiting NADA and capcaisin currrents while at the same time not interfering with the ion channel’s heat-sensing capacity. OB1 is the result of interrogation of the discovered epitope region using three antigens with slightly different sequence and structure. Although still early in the development process, OB1 is a promising candidate for further preclinical development because of its high affinity and its differential pharmacological effect on TRPV1 function, comparing ligand inhibition with heat inhibition. New generations of monoclonal antibodies optimized against this epitope region hold great promise for a safe and efficacious pain therapy.
Fig. 4. Location of PICs on TRPV1 with corresponding epitope region used for development of the modality-selective anti-TRPV1 antibody OB1.
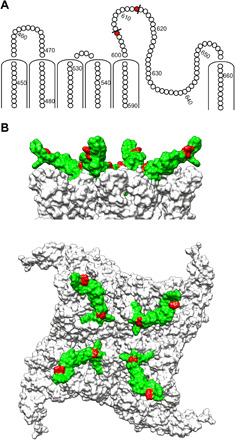
(A) Snake plot of hTRPV1 showing PICs (red) in the extracellular region, identified after a cold kinetic digestion using proteinase K. (B) Top and side view of a homology model of hTRPV1, showing the identified PICs marked in red and the epitope region translated into a synthetic antigen for development of OB1 marked in green.
Fig. 5. Pharmacological evaluation of the modality-selective anti-TRPV1 antibody OB1.
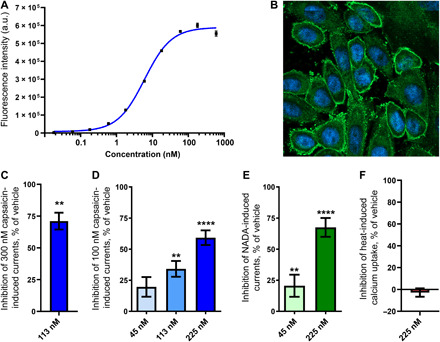
(A) Binding of OB1 to CHO cells overexpressing hTRPV1 measured using flow cytometry (n = 2). a.u., arbitrary unit. (B) Immunocytochemistry image obtained using confocal fluorescence microscopy showing binding of OB1 (green) to overexpressing hTRPV1 CHO cells. Nuclei have been colored blue with Hoechst 33342 (C) Patch-clamp recordings of 300 nM capsaicin-induced TRPV1 currents showing percentage of inhibition after treatment with 113 nM (n = 3) OB1 compared to vehicle (n = 3). Statistical significance was determined using Student’s t test. (D) Patch-clamp recordings of 100 nM capsaicin-induced TRPV1 currents showing percentage of inhibition after treatment with either 225 nM (n = 5), 113 nM (n = 4), or 45 nM (n = 5) OB1 compared to vehicle (n = 13). Statistical significance was determined using a one-way analysis of variance (ANOVA) in combination with Dunnett’s multiple comparison test. (E) Patch-clamp recordings of 1 μM NADA-induced TRPV1 currents showing percentage of inhibition after treatment with either 225 nM (n = 4) or 45 nM (n = 5) OB1 compared to vehicle (n = 13). Statistical significance was determined using a one-way ANOVA in combination with Dunnett’s multiple comparison test. (F) Fluorescence intensity recordings of heat-induced TRPV1-mediated calcium uptake showing percentage of inhibition after treatment with 225 nM (n = 6) OB1 compared to vehicle (n = 13). Statistical significance is indicated as follows: **P < 0.01 and ****P < 0.0001. Data are presented as means ± SEM.
anti-G12/13D-KRAS antibodies
KRAS is a small guanosine triphosphatase that acts as an on/off switch and is a key component in regulation of cell differentiation, proliferation, and survival (32). It is the most frequently mutated oncogene where mutated forms have been found in 22% of all cancers (33). KRAS mutations at residues G12 and G13 are dominant oncogenic mutations in human cancer and substitutions at these residues result in constitutively active guanosine 5′-triphosphate (GTP)–bound KRAS, due to reduced intrinsic GTP hydrolysis (32, 34). The most frequent KRAS substitutions to occur in cancer, in descending order, are G12D, G12V, G13D, and G12C, together comprising ~85% of all KRAS-mutated cancers (35). Although KRAS is considered a holy grail in cancer drug development, drugs that target KRAS have not been successful until the recent small-molecule efforts by Amgen (AMG510) (36) and Mirati (MRTX849) (37). AMG510 and MRTX849 covalently modify the cysteine residue in the G12C mutation and have shown promise in early clinical trials. However, AMG510 and MRTX849 can only address G12C mutant cancer, which is limited to about 12% of all KRAS mutated cancers (35). There is still a large unmet clinical need for therapies targeting additional KRAS mutations. Our aim was to explore the possibility to develop specific antibodies toward G12D- and G13D-mutated KRAS.
To probe the surface of membrane-anchored KRAS for potential antibody binding sites, we performed proteolysis under cold kinetic conditions on membrane vesicles, derived from HEK cells with an endogenous expression of KRAS. Three different protocols, as described in table S1, generated PICs, in or in close vicinity to the guanosine diphosphate (GDP)/GTP pocket as shown in Fig. 6 (A and B). The identified PICs are distributed from amino acids 10 to 21 and form a continuous epitope. Part of the amino acids 10 to 21 regions seem to be hidden by the switch I (amino acids 30 to 38) region (see fig. S2, A and B). This is a dynamic part of the protein that undergoes conformational changes between the GDP- and GTP-bound forms of KRAS (32), and there is likelihood that the identified epitope region is exposed transiently to protease and antibody binding. Because of the overlap of the epitope with the GDP/GTP pocket, this region was predicted to generate antagonistic antibodies. A linear peptide corresponding to amino acids 10 to 21 was synthesized and used for oligoclonal antibody production in rabbits. The antibodies were evaluated for their ability to inhibit KRAS-driven GTP hydrolysis in an enzymatic assay and antibody (0.25 mg/ml) inhibited KRAS (1 mg/ml) to 41% (fig. S3). Having confirmed that antibody binding to the epitope region is inhibitory, and that the epitope overlaps with the mutated residues at positions 12 and 13, we explored the possibility to develop mutant-specific antibodies. On the basis of the structural similarity of the epitope region between wild-type and G12D- and G13D-mutated KRAS (see fig. S4), antibodies named aG12/13D antibody 1 and 2 were developed using antigens with G12D and G13D substitutions. After production, enzyme-linked immunosorbent assay (ELISA) was used to evaluate the affinity of each antibody for the respective mutation using G12D and G13D peptides, and it was determined that both antibodies, produced using either a G12D or a G13D peptide as antigen, were capable of binding both peptides where aG12/13D Ab 1 shows an EC50 of 0.6 nM toward the G13D peptide and 0.7 nM toward the G12D peptide and aG12/13D Ab 2 shows an EC50 of 4.4 nM toward the G13D peptide and 2.9 nM toward the G12D peptide (see Fig. 7, A and B). The in vitro anticancer efficacy of aG12/13D antibodies was determined by evaluating their capability to cause apoptosis in four different KRAS-mutated cell lines, i.e., G12D-mutated SK-LU-1 lung adenocarcinoma cells, G13D-mutated HCT116 colon carcinoma cells, G12V-mutated SW 620 colorectal adenocarcinoma cells, G12C-mutated MIA-PA-CA pancreatic carcinoma cells, as well as wild-type KRAS cell lines, i.e., A431 epidermoid carcinoma cells, and NCI-H1975 non–small cell lung cancer cells. A noncancerous cell line CRL-1831 colon epithelial cells was also included as a control cell line in the apoptosis experiments. Although KRAS is an intracellular (IC) target, we exploited the well-known fact that many cancer cell lines, including KRAS-mutated cells, have up-regulated macropinocytosis. This uptake system has a well-documented capacity to internalize macromolecules, including proteins (38–45). We used flow cytometry and confocal microscopy experiments to evaluate internalization of Alexa Fluor 488–labeled aG12/13D antibodies in the two cell lines bearing the G12/13D mutation as well as in the two wild-type KRAS cell lines A431 and NCI-H1975 (Fig. 8). The confocal microscopy images presented in Fig. 8 (A and C) show uptake of the two aG12/13D antibodies in all the different cell lines examined. Flow cytometry experiments further confirmed these observations, and a statistically significant antibody uptake was observed in all KRAS-mutated and all wild-type KRAS cell lines, respectively, as shown in Fig. 8, B and D. In addition, antibody internalization was further confirmed in G12D- and G13D-KRAS–mutated cell lines using an Alexa Fluor 488–labeled polyclonal IgG, using flow cytometry and confocal microscopy experiments (fig. S5, A to D). In the apoptosis experiments, aG12/13D antibodies were active only in G12D- and G13D-mutated cell lines after 24 hours of treatment (Fig. 9). The antibodies did not cause apoptosis in any of the other cell lines tested, which included G12C and G12V KRAS-mutated cells, wild-type KRAS cancer cells, and noncancerous colon epithelial cells (Fig. 9). After 48 hours of treatment, we observed that the aG12/13D antibodies elicited an increased level of apoptosis in G12D- and G13D-mutated cell lines, compared to the 24-hour treatment. We also observed a small, but statistically significant, increase of apoptosis in G12V-mutated cells after 48 hours (fig. S6). None of the other cell lines showed any increase in apoptosis after 48 hours of antibody exposure (fig. S6). Together, the data show that the apoptosis-inducing effect in G12D- and G13D-mutated cells compared to wild-type KRAS cells is not a result of a quantitatively differential antibody uptake in favor of KRAS-mutated cells but rather a result from a selective inhibition of G12D- and G13D-mutated KRAS cells by the aG12/13D antibodies.
Fig. 6. Snake plot and crystal structure (surface representation) of KRAS highlighting the location of PICs in relation to the selected epitope region used for development of anti-KRAS antibodies.
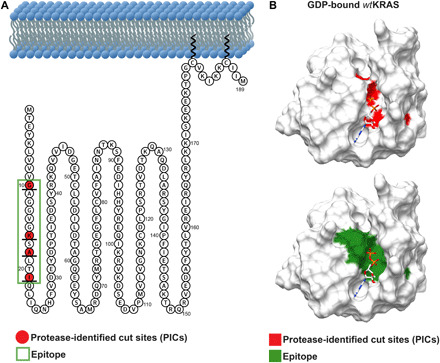
(A) Snake plot of hKRAS showing PICs (red) identified after a cold kinetic digestion and the selected epitope region marked within a green box. (B) Crystal structure of hKRAS (PDB, 4lpk) showing the identified PICs marked in red and the corresponding epitope sequence translated into a synthetic antigen for development of anti-KRAS antibodies marked in green.
Fig. 7. Determination of aG12/13D antibody 1 and 2 binding affinity by ELISA.
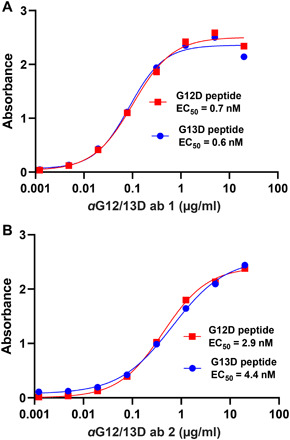
(A) ELISA measurement of aG12/13D Ab 1 against G12D and G13D peptides (n = 1). (B) ELISA measurement of aG12/13D Ab 2 against G12D and G13D peptides (n = 1).
Fig. 8. Confocal fluorescence microscopy and flow cytometry demonstrate uptake of anti-KRAS antibodies in mutated and wild-type KRAS cells.
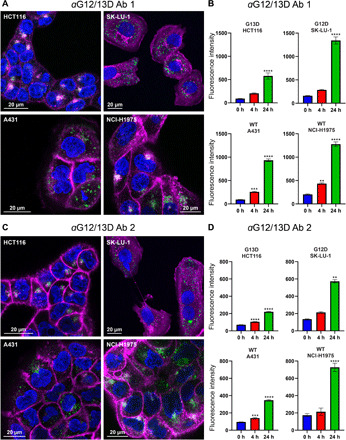
(A) Confocal microscopy images showing antibody uptake after 24 hours of treatment with 220 nM Alexa Fluor 488–conjugated aG12/13D Ab 1 in a G13D-mutated KRAS cell line HCT116, a G12D-mutated KRAS cell line SK-LU-1, and two wild-type KRAS cell lines A431 and NCI-H1975. Antibody labeling appears as green or white fluorescence. Membranes have been colored purple using CellMask plasma membrane stain and nuclei blue using Hoechst 33342. Confocal settings were optimized for each cell line, for viewing purposes. (B) Flow cytometry data showing antibody internalization at different time points after treatment with 220 nM Alexa Fluor 488–conjugated aG12/13D Ab 1 in a G13D-mutated KRAS cell line HCT116, a G12D-mutated KRAS cell line SK-LU-1, and two wild-type KRAS cell lines A431 and NCI-H1975 (n = 4). WT, wild type. (C) Confocal microscopy image showing antibody uptake after 24 hours of treatment with 220 nM Alexa Fluor 488–conjugated aG12/13D Ab 2 in a G13D-mutated KRAS cell line HCT116, a G12D-mutated KRAS cell line SK-LU-1, and two wild-type KRAS cell lines, A431 and NCI-H1975. Fluorescence from antibodies appears green or white. Membranes have been colored purple using CellMask plasma membrane stain and nuclei blue using Hoechst. Confocal settings were optimized for each cell line, for viewing purposes. (D) Flow cytometry data showing antibody internalization at different time points after treatment with 220 nM Alexa Fluor 488–conjugated aG12/13D Ab 2 in a G13D-mutated KRAS cell line HCT116, a G12D-mutated KRAS cell line SK-LU-1, and two wild-type KRAS cell lines A431 and NCI-H1975 (n = 4). Statistical significance was determined using a one-way ANOVA in combination with Dunnett’s multiple comparisons test. Statistical significance is indicated as follows: **P < 0.01, ***P < 0.001, and ****P < 0.0001. Data are presented as means ± SEM.
Fig. 9. Evaluation of the apoptosis-inducing capacity of mutation-selective anti-KRAS antibodies in a panel of KRAS-mutated, wtKRAS, and noncancerous cell lines.
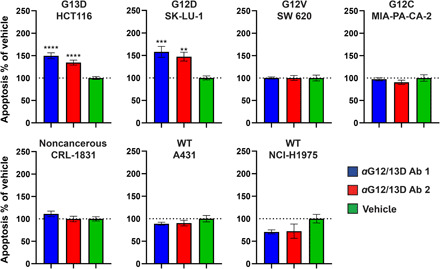
The different cell lines investigated include G13D-mutated HCT116 (colon carcinoma) cells, G12D-mutated SK-LU-1 (lung adenocarcinoma) cells, G12V-mutated SW 620 (colorectal adenocarcinoma) cells, G12C-mutated MIA-PA-CA-2 (pancreatic carcinoma) cells, cancerous wtKRAS A431 (epidermoid carcinoma) cells, cancerous wtKRAS NCI-H1975 (non–small cell lung cancer) cells, and noncancerous wtKRAS CRL-1831 (colon epithelia) cells. Cells were treated with 220 nM antibody or vehicle, and apoptosis was quantified in real time using the RealTime-Glo Annexin V Apoptosis and Necrosis Assay kit. The graphs represent the level of apoptosis normalized to vehicle, at 24 hours after addition of antibodies or vehicle to the cell cultures. For HCT116 cells, n = 10 (aG12/13D Ab 1) and 12 (vehicle, aG12/13D Ab 2). For SK-LU-1 cells, n = 8. For SW 620 cells, n = 8. For MIA-PA-CA-2 cells, n = 4. For A431 cells, n = 8. For NCI-H1975 cells, n = 4. For CRL-1831 cells, n = 6 (vehicle) and 8 (aG12/13D Ab 1 and 2). Statistical significance was determined using one-way ANOVA in combination with Dunnett’s multiple comparisons test except for SK-LU-1 cells where statistical significance was determined using a Kruskal-Wallis test in combination with Dunn’s multiple comparisons test. **P < 0.01, ***P < 0.001, and ****P < 0.0001. Data are presented as means ± SEM.
The selective effects of the antibodies on G12D and G13D mutants but not on G12C and, to a much lesser extent, on G12V mutants can possibly be explained by that the negatively charged aspartic acid residue at the G12 and G13 position in the epitope region plays an important role in the binding of the antibodies. However, the allele location at either position 12 or 13, due to the small difference in distance, seems to be of minor importance for the binding interaction. To the best of our knowledge, this is the first description of inhibitory and mutant-specific antibodies targeting KRAS. The key to this achievement was the detailed understanding of surface exposure of the epitope region and the subsequent use of short antigens for antibody production. This enabled us to direct antibody development to an accessible, functionally important region of G12D- and G13D-mutated KRAS using optimized antigens to accommodate the mutated residues.
DISCUSSION
We have here presented a novel platform technology that uses proteases under cold kinetic conditions to identify surface-exposed regions accessible to antibody binding, on native-state proteins. We have presented exemplary antibodies against two different targets that previously have been considered to be undruggable by antibodies. First, OB1 is a modality-selective, antagonistic monoclonal antibody targeting the ion channel TRPV1 without blocking the ion channel’s heat sensor. Second, we developed aG12D and aG13D antibodies, two anti-KRAS antibodies that are capable of inhibiting G12D- and G13D-mutated KRAS with selectivity over other KRAS mutations (G12V and G12C) as well as over wild-type KRAS.
Some limitations exist and further improvements can be made to the presented antibody discovery platform. First, the positions of PICs do not translate to a perfect understanding of the full binding interface between protease and target. To circumvent this limitation, several antigens around the generated PIC are developed (a schematic representation can be found in Fig. 2D). Another interesting possibility would be to use hydrogen-deuterium exchange MS, commonly used to study protein-protein interfaces, to further define the accessible area around a PIC (46). Second, short synthetic peptides can have limited structure similarity with the native target. A potential approach that would use larger antigens but still allow for precision targeting would be to first immunize animals with the full-length protein positioned in a lipid system (e.g, nanodisc) and incorporate smaller peptide antigens during screening (47–49).
To conclude, with further optimization and preclinical development, the presented anti-TRPV1 and anti-KRAS antibodies have the potential to address large unmet medical needs within the therapeutic areas of pain and oncology. Both proteins addressed herein demonstrate that the technology can discover potential new antibody therapeutics where conventional technologies have failed. Furthermore, we believe that the platform technology can be improved upon and scaled up, addressing a much larger target space. A logical approach going forward would be to try to develop antibodies against clinically and genetically validated targets that have failed because of toxicity and tolerability issues with small-molecule approaches.
MATERIALS AND METHODS
Cell culture
Adherent CHO cells with a tetracycline-regulated expression (T-REx) system of hTRPV1 were cultivated in medium [Dulbecco’s modified Eagle’s medium (DMEM)/F12 GlutaMAX] supplemented with 10% fetal bovine serum. Zeocin (350 μg/ml) and blasticidin (5 μg/ml) were used as selection antibiotics. Eighteen to 24 hours before use, the cells were incubated in medium supplemented with 10% fetal bovine serum and doxycycline (1 μg/ml) to induce TRPV1 expression. HEK293 cells were grown in Ham’s F12 medium, at 5% CO2 and 37°C, supplemented with 10% fetal calf serum and nonessential amino acids. A431 cells were cultivated using DMEM supplemented with 10% fetal bovine serum and 1% penicillin + streptomycin. HCT116 cells were cultivated using McCoy’s 5a Medium Modified supplemented with 10% fetal bovine serum and 1% penicillin + streptomycin. MIA-PA-CA-2 cells were cultivated using DMEM supplemented with 10% fetal bovine serum and 1% penicillin + streptomycin. SK-LU-1 cells were cultivated using Eagle’s minimum essential medium supplemented with 10% fetal bovine serum and 1% penicillin + streptomycin. CRL-1831 cells were cultivated using DMEM/F12 medium supplemented with 10% fetal bovine serum, 1% penicillin + streptomycin, cholera toxin (10 ng/ml), insulin (0.005 mg/ml), transferrin (0.005 mg/ml), hydrocortisone (100 ng/ml), and human recombinant EGF (20 ng/ml). The following sources were used: HCT116 (human colon carcinoma), European collection of authenticated cell cultures (ECACC) General Collection no. 91091005; SK-LU-1 (human lung adenocarcinoma), ECACC General Collection no. 93120835; SW 620 (human colon adenocarcinoma), ECACC General Collection no. 87051203; MIA-PA-CA-2 (human pancreatic carcinoma), ECACC General Collection no. 85062806; A431 (human epidermoid carcinoma), ECACC General Collection no. 85090402; NCI-H1975 (human non–small cell lung adenocarcinoma), American Type Culture Collection (ATCC) CRL5908; CRL-1831 (human normal colon epithelial), ATCC CRL-1831; and CHO cells with a T-REx system of hTRPV1 obtained from Chalmers University of Technology.
Proteoliposome preparation
Membrane derived vesicles (proteoliposomes) were prepared as previously described elsewhere (21) in buffer containing 300 mM NaCl, 10 mM tris (pH 8.0). Complete digestion protocols can be found in the Supplementary Materials.
Liquid chromatography–tandem mass spectrometry
For CHO TRPV1 studies, peptide samples were analyzed at the Proteomics Core Facility at Gothenburg University, Göteborg, Sweden. Peptides were dried and reconstituted in 15 μl of 0.1% formic acid in 3% gradient grade acetonitrile (ACN) (Merck KGaA, Darmstadt, Germany). The ACN gradient (0.2% formic acid) used was 7 to 27% (25 min), 25 to 40% (5 min), 40 to 80% (5 min), and 80% (10 min). Mass analyses were performed using an interfaced Q Exactive hybrid mass spectrometer (Thermo Fisher Scientific). Ions were created and sprayed into the mass spectrometer at a voltage of 1.8 kV and capillary temperature of 320°C in data-dependent positive-ion mode. Full scan (MS1) spectra were acquired in the Orbitrap over the mass/charge ratio (m/z) range 400 to 1600, charge range 2 to 6 at a resolution of 70,000 until an automatic gain control (ACG) target value of 1 × 106 at a maximum of 250 ms. MS/MS spectra were acquired using higher-energy collision dissociation at 30% from m/z 110 for the 10 most abundant parent ions at a resolution of 35,000 using a precursor isolation window of 2 Da until an AGC target value of 1 × 105 during an injection time of 110 ms. Dynamic exclusion during 30 s after selection for MS/MS was enabled to allow for detection of as many precursors as possible. All tandem mass spectra were searched by MASCOT (version 2.3, Matrix Science, London, UK). Thermo Proteome Discoverer v. 1.3 (Thermo Fisher Scientific) was used to validate MS/MS-based peptide and protein identifications. A false discovery rate of 1% at the peptide level was applied, with the false discovery rate determined by searching using a reversed database as decoy. The SwissProt databases used for searching TRPV1-related MS data were those released between 2015_04 (20,205 human sequences) and 2016_02 (20,199 human sequences).
For HEK293 studies, samples were analyzed on high-accuracy Orbitrap instruments. Orbitrap Fusion Tribrid, Q Exactive or Orbitrap Elite mass spectrometers (Thermo Fisher Scientific) were interfaced with Easy nanoLC 1200 liquid chromatography systems. Peptides were separated using an analytical column [300 mm by 0.075 mm inner diameter (ID)] packed with 3-μm Reprosil-Pur C18-AQ particles (Dr. Maisch, Germany) using the gradient from 5 to 7% to 30 to 32% B over 35, 50, or 75 min, followed by an increase to 100% B for 5 min at a flow of 300 nl/min. Solvent A was 0.2% formic acid in water, and solvent B was 0.2% formic acid in 80% acetonitrile. MS/MS analysis was performed in a data-dependent mode where the most intense precursor ions at charge states 2 to 7 were selected for fragmentation. Dynamic exclusion was set to 30 s. Data analysis was performed using Proteome Discoverer version 1.4 (Thermo Fisher Scientific) against SwissProt (releases 2017_01-2017_06; approximately 20,100 human entries). Mascot 2.3.2.0 (Matrix Science) was used as a search engine with precursor mass tolerance of 5 parts per million and fragment mass tolerance of 0.6 Da. Peptides were accepted with one to three missed cleavage and variable modifications of methionine oxidation (M), cysteine alkylation (C), and Peptide N Glycosidase F (PNGase F) (Asn->Asp (N)). The detected peptide threshold was set to 1% false discovery rate by searching against a reversed database.
Antibody development
Synthetic peptides, described in table S2, were synthesized and purified. The peptides were linked to keyhole limpet hemocyanin (KLH) using either a cysteine residue in the peptide or the N-terminal propargyl group. Oligoclonal KRAS antibodies were produced by immunization of specific pathogen–free rabbits with the KLH-linked peptides, followed by antigen-affinity purification of antiserum from the rabbits. Monoclonal TRPV1 antibodies were produced through immunizing mice (BALB/c and/or C57BL/6) with the KLH-linked peptides and hybridoma technology. For selection of hybridoma clones, a combination of ELISA analysis of binding to the antigen peptide and flow cytometry analysis of binding to native TRPV1 protein were used. Monoclonal antibodies were affinity purified against protein A from hybridoma supernatant. Generation of synthetic peptides, oligoclonal antibodies, and monoclonal antibodies were performed by Innovagen AB (Lund, Sweden).
Flow cytometry of antibodies binding to TRPV1
Antibody binding to CHO cells expressing TRPV1 was measured using flow cytometry (CytoFlex, Beckman Coulter). Cells were incubated with different concentrations of purified antibody solution in a total reaction volume of 50 μl for 30 min at 4°C, followed by washing in phosphate-buffered saline (PBS) with 1% bovine serum albumin (BSA) and subsequent incubation for 30 min at 4°C with 50 μl of 10 μg/ml anti-mouse IgG-Phycoerythrin (PE) secondary antibody (Invitrogen #P852). Around 100,000 cells were seeded in each well, and 10,000 to 15,000 cells were measured for each treatment. For comparison of binding to CHO cells that express TRPV1 and CHO cells that do not express TRPV1, a purified antibody concentration of 10 μg/ml was used. The negative control was secondary antibody only. For evaluation of binding EC50, OB1 concentrations of 100, 30, 10, 3, 1, 0.3, 0.1, 0.03, 0.01, and 0.003 μg/ml were used. Binding was assessed by measurement of fluorescence signal from cells and reported as a background-subtracted mean fluorescence intensity. Analysis was performed in FlowJo Software (Becton, Dickinson and Company). Four biological replicates confirming OB1 binding to TRPV1 were performed. The fluorescence-activated cell sorting titration curve was performed once.
ELISA measurements of antibodies binding to KRAS antigens
MaxiSorp, Nunc-Immuno Plates (Thermo Fisher Scientific) were coated with either G12D or G13D peptides (1 μg/ml) in PBS and incubated 1 hour at 37°C. Peptide sequences are found in table S2. Plates were washed with PBS containing 0.05% Tween 20 and incubated 1 hour at 37°C in the presence of blocking buffer [PBS, 0.5% BSA, 0.05% Tween 20 (pH 7.4)]. A serial dilution of aG12/13D Ab 1 and 2 was added in blocking buffer and incubated for 1 hour at room temperature (RT). Plates were washed with PBS containing 0.05% Tween 20, and a secondary antibody goat anti–rabbit-AP (Dako, pn D0487) was added and incubated for 1 hour at RT. Plates were washed with PBS containing 0.05% Tween 20 followed by addition of pNPP (1 mg/ml) in para-Nitrophenylphosphate (pNPP) buffer. Absorbance was measured at 405 nm.
Electrophysiological patch-clamp recordings of TRPV1 response
Whole-cell recordings were performed using a microfluidic device for patch-clamp recordings (Dynaflow, Fluicell AB, Göteborg, Sweden) together with an Axopatch 200B (Molecular Devices, USA) or HEKA EPC10 (HEKA, Germany) patch-clamp amplifiers. Pipettes (borosilicate glass capillaries, ID of 0.86 mm; GC150F-7.5, Harvard Apparatus Ltd.) and bath solutions contained IC and EC buffer, respectively. The IC buffer contained 120 mM KCl, 2 mM MgCl2, 10 mM Hepes, and 10 mM EGTA (pH 7.2). The EC buffer contained 140 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2 10 mM Hepes, and 10 mM d-glucose (pH 7.4).
The cells were clamped at −60 mV, and the current signals were recorded with a sampling frequency of 10 kHz and low-pass–filtered at 2 kHz. The patch-clamp recordings were acquired using digital/analog sampling (Axon Digidata 1550) and acquisition software (Clampex version 10.7, Molecular Devices) or, alternatively, the HEKA amplifier’s built-in digital/analog sampling and acquisition software (Patchmaster version 2.90.5, HEKA).
In experiments, current amplitudes were measured by exposing cells to capsaicin or NADA, with antibody or vehicle. The cells were exposed to the following protocols: (i) two cycles of agonist (100 nM capsaicin, 300 nM capsaicin, or 1 μM NADA) in EC buffer for 10 to 20 s (control pulses), (ii) EC buffer for 60 s, (iii) antibody or vehicle in EC buffer for 60 s (preincubation), (iv) agonist together with antibody or vehicle in EC buffer for 10 to 20 s (stimulation with antibody or vehicle present), (v) EC buffer for 120 s (washing), and (vi) agonist in EC buffer for 10 to 20 s. The time between each stimulation with agonist was 2 min. The antibody stock buffer was used as a vehicle. Measurements where the seal resistance shifted largely during treatment were excluded from analysis. Capsaicin inhibition was confirmed at two different sites. Each treatment group for the NADA experiments contain technical replicates.
Immunocytochemistry of anti-TRPV1 antibody
CHO-TRPV1 cells were incubated with 225 nM OB1 antibody in DMEM/F12 for 1 hour at RT. Cells were washed with PBS and incubated with Alexa Fluor 488 goat anti-mouse secondary antibody (0.002 mg/ml) (Invitrogen reference A32723) plus Hoechst 33342 (0.005 mg/ml) (Invitrogen reference H3570) in PBS and incubated for 1 hour at RT. Cells were subsequently washed with PBS and imaged using a Zeiss LSM 980 with Airyscan 2 at ×63 magnification with oil immersion.
Calcium imaging of TRPV1 heat response
To measure the antibodies effect on hTRPV1 heat response (42°C), an optical heating system was used to deliver heat pulses to cells. A microfluidic device, the Biopen Prime (Fluicell AB), was used to deliver the antibodies to cells.
Optical heating system
A laser heating system was used to locally increase the temperature to 42°C around selected cells. The laser heating system was built in-house by Fluicell AB. This optical local heating system is based on a CW 4W 1470-nm semiconductor diode laser (4PN-106, Seminex Corporation, USA) driven by a 20-A benchtop power source (ARO-4320, Arroyo Instruments). This delivers a localized beam to the sample (a group of cells in our case) through a 105-μm-core, 0.22-NA (numerical aperture), broadband optical fiber (M63L01, Thorlabs). The optical fiber is coupled to a 5-mm fiber optic cannula (CFMLC21L05, 105 μm, 0.22 NA, Thorlabs) so that it can be precisely positioned at any desired location in a petri dish.
A narrow beam of 1470-nm radiation, exiting the tip of the optical fiber, induces local heating of the water within its path. The extent of heating is determined by the beam intensity that is modulated by the current setting of the laser and the distance between the tip of the fiber and the sample. In this study, the current and distance are optimized to achieve a sample temperature of 42°C. The relationship between distance, applied current, and temperature was calibrated using a previously described technique (50).
Antibody delivery
A Biopen Prime (Fluicell AB) was used to deliver antibodies to cells. The Biopen is a free-standing microfluidic device that can be readily positioned using micromanipulators such that the tip can be aligned adjacent to a selected group of cells in a petri dish, to locally deliver a compound without contamination of the surrounding environment. The switching between solutions is controlled by dedicated software.
Imaging
Cells were imaged using a Bio-Rad MRC 1024 confocal unit fitted with a dual Calypso laser (Cobolt, Solna, Sweden) attached to a Nikon Diaphot 200 inverted microscope and a Nikon Plan Apo 20× dry objective (NA, 0.75; Nikon, Tokyo, Japan). Excitation wavelengths used were 491 nm (Fluo-3), and the emitted light was collected through a 522-nm filter. Images were acquired for the full view of the 20× objective. The frame rate was one image per 7 s and the pixel resolution 1024 × 1024.
Thirty minutes before imaging, the cell medium was changed to medium containing 36 μM Fluo-3-AM (F1242, Thermo Fisher Scientific), and the samples were incubated for 30 min at RT and then washed. The Biopen was positioned above a group of cells using a micromanipulator. The heat probe was positioned 10 μm above the dish bottom and at an approximately 100-μm distance from the Biopen outlet. To define which cells are exposed to solution delivery from the Biopen, an initial pulse of sulforhodamine B was delivered. After the sulforhodamine B fluorescence declined, the cells were optically heated for 7 s (42°C), and the fluorescence response from Fluo-3 was recorded. Subsequently, antibody solution was delivered for 90 s, and a second heat pulse was applied during the last 7 s of application. All molecules, including Fluo-3 AM, were dissolved in 140 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2 10 mM Hepes, and 10 mM d-glucose (pH 7.4).
Inhibition of KRAS mediated GTP hydrolysis
KRAS activity was assessed using a fluorescent phosphate-sensing probe (Thermo Fisher Scientific catalog no. PV4406) allowing real-time measurement of released phosphate from GTP hydrolysis using a microplate reader (Clariostar, BMG Labtech). Recombinant KRAS was produced by the Protein Science Facility at Karolinska Institutet. KRAS (1 mg/ml), 1 μM phosphate sensor, 6 μM GTP, antibody (0.25 mg/ml) (n = 16), or vehicle control (n = 47) were prepared in buffer (50 mM tris, 100 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 0.01% Triton X-100, and 1 mM dithiothreitol) and added to a 384-well plate, and fluorescence was measured during 4 hours.
Live cell imaging of aG12/13D antibody uptake
aG12/13D antibody 1 and 2 were labeled with Alexa Fluor 488 using a Alexa Fluor 488 protein labeling kit (catalog no. A10235, Thermo Fisher Scientific). HCT116, SK-LU-1, A431, and NCI-H1975 cells were cultured in glass bottom 96-well microplates (Corning Life Sciences). Cells were treated with 220 nM labeled aG12/13D antibody 1 or 2 for 24 hours, washed and counterstained using CellMask plasma membrane stain (Thermo Fisher Scientific) and Hoechst (Life Technologies). The fluorescence was imaged using a LSM 880 confocal microscope (Zeiss) and settings were optimized for each cell line and antibody. Images were processed in Zeiss ZEN 2.6 (blue edition).
Flow cytometry measurements of aG12/13D antibody uptake
aG12/13D antibody 1 and 2 were labeled with Alexa Fluor 488 using a Alexa Fluor 488 protein labeling kit (catalog no. A10235, Thermo Fisher Scientific). HCT116, SK-LU-1, A431 and NCI-H1975 cells were cultured in 12-well microplates (Invitrogen). Cells were treated with 220 nM labeled aG12/13D antibody 1 or 2 for 4 or 24 hours before measurements. Unstained cells were measured as a t(0) time point. Before measurements, cells were washed and harvested. Measurements were performed on a BD LSR II flow cytometer (BD Biosciences Inc.). Analysis was performed in FlowJo v.10 (BD Biosciences). For each cell line and time point, n = 4. n equals one measurement containing on average 4000 cells for HCT116, 8000 cells for SK-LU-1, 7000 cells for A431, and 4000 cells for NCI-H1975.
Antibody-induced apoptosis of anti-KRAS antibodies
The RealTime-Glo Annexin V Apoptosis and Necrosis Assay kit (Promega, catalog no. JA1011) was used to measure apoptosis in real time. Cultured cells were harvested using trypsin and transferred to L-15 assay medium, consisting of Leibovitz’s L-15 Medium (Thermo Fisher Scientific catalog no. 21083027), 10% fetal bovine serum, and 1% penicillin + streptomycin. Cells were counted and diluted to 200,000 cells/ml with L-15 assay medium. Cells were added in 96-well plates (10,000 cells per well, Costar, white with clear opaque bottom, catalog no. 3903) and incubated overnight.
Antibodies were diluted in L-15 assay medium and the detection reagent and added to the wells in a final concentration of 220 nM. The plates were the sealed with an optic film and placed in a CLARIOstar (BMG LABTECH) plate reader. Measurement of apoptosis was performed by reading of the luminescence signal.
Two to three biological replicates were performed for the following cell lines: HCT116, SK-LU-1, SW 620, CRL-1831, and A431. The NCI-H1975 and MIA-PA-CA-2 cell lines were tested once.
Statistical analysis
Electrophysiological patch-clamp recordings of TRPV1 response
For all electrophysiological measurements, the recorded amplitude of the peak during stimulation with antibody + agonist or vehicle + agonist was divided by the amplitude of the preceding peak during stimulation with agonist only, i.e., the response was expressed as a percentage of the preceding control stimulation. To take account for any effects from receptor desensitization, antibody + agonist experiments was normalized with corresponding value from vehicle + agonist experiments, i.e., remaining TRPV1 activity was expressed as “% of vehicle.” Percentage of inhibition is presented and calculated from 100% of vehicle. Normality was assessed using the Shapiro-Wilks test. n represents one cell. Statistical significance for inhibition of 300 nM capsaicin induced currents was determined using Student’s t test (t = 6.392, df = 4, and P = 0.0031). Statistical significance for inhibition of 100 nM capsaicin induced currents was determined using a one-way analysis of variance (ANOVA) in combination with Dunnett’s multiple comparison test F(3,23) = 15.06; P < 0.0001. Statistical significance for inhibition of NADA-induced currents was determined using a one-way ANOVA in combination with Dunnett’s multiple comparison test F(2,19) = 28.21; P < 0.0001.
Calcium imaging of TRPV1 heat response
Data analyses were performed in ImageJ and GraphPad Prism. The sulforhodamine B pulse visualizes which cells are reached by Biopen solution delivery and thereby defines which cell will be included in the measurement. The fluorescence intensity of these cells was measured and averaged for each time point, to obtain an average curve for the cells stimulated in one experiment. Each n corresponds to an average of the cells stimulated in one experiment and are technical replicates. The height of peak 1 was measured to determine heat response without antibody present, and the height of peak 2 was measured to determine heat response with antibody present. The ratio of the peaks for OB1 was compared to vehicle. The level of heat response after antibody treatment was normalized to vehicle. Normality was assessed using the Shapiro-Wilks test. Statistical significance for inhibition of heat-induced currents was determined to be nonsignificant using Student’s t test (t = 0.3507, df = 17, and P = 0.73).
Inhibition of KRAS-mediated GTP hydrolysis
Fluorescence values after 4 hours were compared between treatments. n represents one well. The level of KRAS response after antibody treatment was normalized to vehicle. Normality was assessed using the Shapiro-Wilks test. Statistical analysis was performed using a two-tailed Mann Whitney test (due to non-normality) (Mann-Whitney U = 72, P > 0.0001).
Flow cytometry measurements of aG12/13D antibody uptake
Mean fluorescence for each time point after antibody addition was compared to t(0). Normality was assessed using the Shapiro-Wilks test. Statistical significance was determined using a one-way ANOVA in combination with Dunnett’s multiple comparison test, except for aG12/13D Ab 2 in SK-LU-1 cells where statistical significance was determined using a Kruskal-Wallis test in combination with Dunn’s multiple comparisons test (due to non-normality). For aG12/13D Ab 1: HCT116 cells F(2, 9) = 43.7, P < 0.0001, SK-LU-1 cells F(2,9) = 213.5, P < 0.0001, A431 F(2,9) = 624.1, P < 0.0001 cells, and NCI-H1975 cells F(2,9) = 319.1, P < 0.0001. For aG12/13D Ab 2: HCT116 cells F(2, 9) = 624.3, P < 0.0001, SK-LU-1 cells Kruskal-Wallis statistic = 9.881, P = 0.0002, A431 F(2,9) = 589.7, P < 0.0001 cells, and NCI-H1975 cells F(2,9) = 63.78, P < 0.0001.
Antibody-induced apoptosis of anti-KRAS antibodies
The level of apoptosis in antibody-treated wells at 24 and 48 hours was normalized to vehicle. Wells containing visible air bubbles were excluded. Data are presented as means ± SEM. Normality was assessed using the Shapiro-Wilks test; n represents one well. Statistical significance was determined using one-way ANOVA in combination with Dunnett’s multiple comparisons test except for SK-LU-1 cells at 24 hours of treatment, CRL-183 cells at 48 hours of treatment, and MIA-PA-CA-2 cells at 48 hours of treatment, where statistical significance was determined using a Kruskal-Wallis test in combination with Dunn’s multiple comparisons test (due to non-normality). Statistics at 24 hours: SK-LU-1 cells (Kruskal-Wallis statistic = 15.61; P = 0.0004). For A431 cells F(2,21) = 1.134, P = 0.3408. For SW 620 cells F(2,21) = 0.00025; P = 0.9998. For HCT116 cells F(2,31) = 24.46, P < 0.0001. For CRL-183 cells F(2,19) = 1.271, P < 0.3033. For H1975 cells F(2,9) = 2.248, P = 0.1613. For MIA-PA-CA-2 cells F(2,9) = 0.8415, P = 0.4623. Statistics at 48 hours: SK-LU-1 cells F(2,21) = 45.09, P < 0.0001, P = 0.0004). For A431 cells F(2,21) = 0.3220, P = 0.7282. For SW 620 cells F(2,21) = 7.252, P = 0.0040. For HCT116 cells F(2,31) = 26.61, P < 0.0001. For CRL-183 cells (Kruskal-Wallis statistic = 0.2194; P = 0.9041). For MIA-PA-CA-2 cells (Kruskal-Wallis statistic = 3.038; P = 0.2345).
Acknowledgments
We thank the staff at Nanoxis Consulting AB, NMI TT Pharmaservices, Innovagen AB, and the Proteomic Core Facility at University of Gothenburg for assistance and discussions. Funding: This work was supported by Vinnova (grants 2016-04093 and 2017-02993) and the Karolinska Institute. Author contributions: C.L.T. and O.O. developed the concept and planned the study. C.L.T., M.D., and J.H. planned and analyzed the digestion experiments. A.K. and R.K. performed the digestion experiments. M.D., J.H., A.K., and R.K. planned the LS-MS/MS analysis. C.L.T. and M.D. designed and produced the antibodies. C.L.T., M.M.M., J.H., O.J., K.J., and A.K. planned and conducted the electrophysiology experiments and performed data analysis. O.J., J.B., and G.D.M.J. planned and performed the imaging experiments, and C.L.T., O.J., J.B., and J.H. performed data analysis. O.J., S.P.S., N.I., and A.C. planned, performed, and analyzed the apoptosis experiments. P.S. developed the TRPV1 homology model. A.A. and A.R. performed structure analysis of proteases and KRAS. A.R. performed computer simulations on TRP channels. A.K. and J.H. planned the flow cytometry affinity measurements. O.J., S.P.S., C.L.T., and M.-N.C. planned and performed the flow cytometry uptake experiments. C.L.T., S.P.S., and O.J. planned and analyzed the ELISA experiments. G.W., D.P., and E.J. took part in data analysis. K.J. and O.O. supported the study and participated in writing the manuscript. Competing interests: C.L.T., M.D., J.H., S.P.S., G.W., A.R., A.A., D.P., E.J., and O.O. declare financial competing interests. They are employed by Oblique Therapeutics. In addition, C.L.T., M.D., J.H., and O.O. hold shares in the company. R.K., A.K., and P.S. have received consultation fees from Oblique Therapeutics. O.O. and C.L.T. are inventors on a patent application related to this work filed by Oblique Therapeutics (no. PCT/EP2016/057164, filed 31 March 2016, and published 6 October 2016). O.O., C.L.T., and M.D. are inventors on a patent application related to this work filed by Oblique Therapeutics (no. PCT/EP2017/072001, filed 1 September 2017, and published 8 March 2018). O.O., C.L.T., M.D., and J.H. are inventors on a patent application related to this work filed by Oblique Therapeutics (no. PCT/EP2017/075532, filed 6 October 2017, and published 12 April 2018). C.L.T. and M.D. are inventors on a patent application related to this work filed by Oblique Therapeutics (no. PCT/EP2020/085629, filed 10 December 2020). C.L.T. and M.D. are inventors on a patent application related to this work filed by Oblique Therapeutics (no. PCT/EP2021/054614, filed 24 February 2021). The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Antibodies can be produced upon request under a material transfer agreement.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/16/eabe6397/DC1
REFERENCES AND NOTES
- 1.The Antibody Society (2020); www.antibodysociety.org.
- 2.Hutchings C. J., Colussi P., Clark T. G., Ion channels as therapeutic antibody targets. MAbs 11, 265–296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wulff H., Christophersen P., Colussi P., Chandy K. G., Yarov-Yarovoy V., Antibodies and venom peptides: New modalities for ion channels. Nat. Rev. Drug Discov. 18, 339–357 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haustrate A., Hantute-Ghesquier A., Prevarskaya N., Lehen’kyi V., Monoclonal antibodies targeting ion channels and their therapeutic potential. Front. Pharmacol. 10, 606 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Köhler G., Milstein C., Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497 (1975). [DOI] [PubMed] [Google Scholar]
- 6.Lad L., Clancy S., Kovalenko M., Liu C., Hui T., Smith V., Pagratis N., High-throughput kinetic screening of hybridomas to identify high-affinity antibodies using bio-layer interferometry. J. Biomol. Screen. 20, 498–507 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Frauenfelder H., Sligar S. G., Wolynes P. G., The energy landscapes and motions of proteins. Science 254, 1598–1603 (1991). [DOI] [PubMed] [Google Scholar]
- 8.Hegler J. A., Weinkam P., Wolynes P. G., The spectrum of biomolecular states and motions. HFSP J. 2, 307–313 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Latorraca N. R., Venkatakrishnan A. J., Dror R. O., GPCR dynamics: Structures in motion. Chem. Rev. 117, 139–155 (2017). [DOI] [PubMed] [Google Scholar]
- 10.L. S. Liebovitch, P. Krekora, The Physical Basis of Ion Channel Kinetics: The Importance of Dynamics, in Membrane Transport and Renal Physiology, H. E. Layton, Ed. (Springer-Verlag, New York, 2002). [Google Scholar]
- 11.D. Lipscombe, C. P. Toro, From Molecules to Networks: An Introduction to Cellular and Molecular Neuroscience: Third Edition (Academic Press, 2014). [Google Scholar]
- 12.Chowdhury S., Jarecki B. W., Chanda B., A molecular framework for temperature-dependent gating of ion channels. Cell 158, 1148–1158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.K. R. Tovar, G. L. Westbrook, Cell Physiology Source Book (2012).
- 14.Kazanov M. D., Igarashi Y., Eroshkin A. M., Cieplak P., Ratnikov B., Zhang Y., Li Z., Godzik A., Osterman A. L., Smith J. W., Structural determinants of limited proteolysis. J. Proteome Res. 10, 3642–3651 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benjamin D. C., Berzofsky J. A., East L. J., Gurd F. R. N., Hannum C., Leach S. J., Margoliash E., Michael J. G., Miller A., Prager E. M., Reichlin M., Sercarz E. E., Smith-Gill S. J., Todd P. E., Wilson A. C., The antigenic structure of proteins: A reappraisal. Annu. Rev. Immunol. 2, 67–101 (1984). [DOI] [PubMed] [Google Scholar]
- 16.Novotný J., Handschumacher M., Haber E., Bruccoleri R. E., Carlson W. B., Fanning D. W., Smith J. A., Rose G. D., Antigenic determinants in proteins coincide with surface regions accessible to large probes (antibody domains). Proc. Natl. Acad. Sci. U.S.A. 83, 226–230 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thornton J. M., Edwards M. S., Taylor W. R., Barlow D. J., Location of “continuous” antigenic determinants in the protruding regions of proteins. EMBO J. 5, 409–413 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jameson B. A., Wolf H., The antigenic index: A novel algorithm for predicting antigenic determinants. Comput. Appl. Biosci. 4, 181–186 (1988). [DOI] [PubMed] [Google Scholar]
- 19.Parker J. M., Guo D., Hodges R. S., New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: Correlation of predicted surface residues with antigenicity and x-ray-derived accessible sites. Biochemistry 25, 5425–5432 (1986). [DOI] [PubMed] [Google Scholar]
- 20.Fontana A., de Laureto P. P., Spolaore B., Frare E., Picotti P., Zambonin M., Probing protein structure by limited proteolysis. Acta Biochim. Pol. 51, 299–321 (2004). [PubMed] [Google Scholar]
- 21.Jansson E. T., Trkulja C. L., Olofsson J., Millingen M., Wikström J., Jesorka A., Karlsson A., Karlsson R., Davidson M., Orwar O., Microfluidic flow cell for sequential digestion of immobilized proteoliposomes. Anal. Chem. 84, 5582–5588 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Rajer M., Kmet M., Quantitative analysis of fine needle aspiration biopsy samples. Radiol. Oncol. 39, 269–272 (2005). [Google Scholar]
- 23.Cajulis R. S., Sneige N., Objective comparison of cellular yield in fine-needle biopsy of lymph nodes with and without aspiration. Diagn. Cytopathol. 9, 43–45 (1993). [DOI] [PubMed] [Google Scholar]
- 24.Smith K., Shah H., Muther J. J., Duke A. L., Haley K., James J. A., Antigen nature and complexity influence human antibody light chain usage and specificity. Vaccine 34, 2813–2820 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edwards B. M., Barash S. C., Main S. H., Choi G. H., Minter R., Ullrich S., Williams E., du Fou L., Wilton J., Albert V. R., Ruben S. M., Vaughan T. J., The remarkable flexibility of the human antibody repertoire; isolation of over one thousand different antibodies to a single protein, BLyS. J. Mol. Biol. 334, 103–118 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Mickle A. D., Shepherd A. J., Mohapatra D. P., Nociceptive TRP channels: Sensory detectors and transducers in multiple pain pathologies. Pharmaceuticals 9, 72 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gavva N. R., Treanor J. J. S., Garami A., Fang L., Surapaneni S., Akrami A., Alvarez F., Bak A., Darling M., Gore A., Jang G. R., Kesslak J. P., Ni L., Norman M. H., Palluconi G., Rose M. J., Salfi M., Tan E., Romanovsky A. A., Banfield C., Davar G., Pharmacological blockade of the vanilloid receptor TRPV1 elicits marked hyperthermia in humans. Pain 136, 202–210 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Trkulja C. L., Jansson E. T., Jardemark K., Orwar O., Probing structure and function of ion channels using limited proteolysis and microfluidics. J. Am. Chem. Soc. 136, 14875–14882 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Cao E., Liao M., Cheng Y., Julius D., TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504, 113–118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elokely K., Velisetty P., Delemotte L., Palovcak E., Klein M. L., Rohacs T., Carnevale V., Understanding TRPV1 activation by ligands: Insights from the binding modes of capsaicin and resiniferatoxin. Proc. Natl. Acad. Sci. U.S.A. 113, E137–E145 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.M. Cui, V. Gosu, S. Basith, S. Hong, S. Choi, Advances in Protein Chemistry and Structural Biology (2016), vol. 104. [DOI] [PubMed]
- 32.Simanshu D. K., V. Nissley D., McCormick F., RAS Proteins and their regulators in human disease. Cell 170, 17–33 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prior I. A., Lewis P. D., Mattos C., A comprehensive survey of ras mutations in cancer. Cancer Res. 72, 2457–2467 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.White Y., Bagchi A., van Ziffle J., Inguva A., Bollag G., Zhang C., Carias H., Dickens D., Loh M., Shannon K., Firestone A. J., KRAS insertion mutations are oncogenic and exhibit distinct functional properties. Nat. Commun. 7, 10647 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tate J. G., Bamford S., Jubb H. C., Sondka Z., Beare D. M., Bindal N., Boutselakis H., Cole C. G., Creatore C., Dawson E., Fish P., Harsha B., Hathaway C., Jupe S. C., Kok C. Y., Noble K., Ponting L., Ramshaw C. C., Rye C. E., Speedy H. E., Stefancsik R., Thompson S. L., Wang S., Ward S., Campbell P. J., Forbes S. A., COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Canon J., Rex K., Saiki A. Y., Mohr C., Cooke K., Bagal D., Gaida K., Holt T., Knutson C. G., Koppada N., Lanman B. A., Werner J., Rapaport A. S., San Miguel T., Ortiz R., Osgood T., Sun J. R., Zhu X., McCarter J. D., Volak L. P., Houk B. E., Fakih M. G., O’Neil B. H., Price T. J., Falchook G. S., Desai J., Kuo J., Govindan R., Hong D. S., Ouyang W., Henary H., Arvedson T., Cee V. J., Lipford J. R., The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Fell J. B., Fischer J. P., Baer B. R., Blake J. F., Bouhana K., Briere D. M., Brown K. D., Burgess L. E., Burns A. C., Burkard M. R., Chiang H., Chicarelli M. J., Cook A. W., Gaudino J. J., Hallin J., Hanson L., Hartley D. P., Hicken E. J., Hingorani G. P., Hinklin R. J., Mejia M. J., Olson P., Otten J. N., Rhodes S. P., Rodriguez M. E., Savechenkov P., Smith D. J., Sudhakar N., Sullivan F. X., Tang T. P., Vigers G. P., Wollenberg L., Christensen J. G., Marx M. A., Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C inhibitor for the treatment of cancer. J. Med. Chem. 63, 6679–6693 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y., Commisso C., Macropinocytosis in cancer: A complex signaling network. Trends Cancer. 5, 332–334 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamphorst J. J., Nofal M., Commisso C., Hackett S. R., Lu W., Grabocka E., Vander Heiden M. G., Miller G., Drebin J. A., Bar-Sagi D., Thompson C. B., Rabinowitz J. D., Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 75, 544–553 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bar-Sagi D., Feramisco J. R., Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science 233, 1061–1068 (1986). [DOI] [PubMed] [Google Scholar]
- 41.Commisso C., Davidson S. M., Soydaner-Azeloglu R. G., Parker S. J., Kamphorst J. J., Hackett S., Grabocka E., Nofal M., Drebin J. A., Thompson C. B., Rabinowitz J. D., Metallo C. M., Vander Heiden M. G., Bar-Sagi D., Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Recouvreux M. V., Commisso C., Macropinocytosis: A metabolic adaptation to nutrient stress in cancer. Front. Endocrinol. 8, 261 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakase I., Kobayashi N. B., Takatani-Nakase T., Yoshida T., Active macropinocytosis induction by stimulation of epidermal growth factor receptor and oncogenic Ras expression potentiates cellular uptake efficacy of exosomes. Sci. Rep. 5, 10300 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grimmer S., van Deurs B., Sandvig K., Membrane ruffling and macropinocytosis in A431 cells require cholesterol. J. Cell Sci. 115, 2953–2962 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Hodakoski C., Hopkins B. D., Zhang G., Su T., Cheng Z., Morris R., Rhee K. Y., Goncalves M. D., Cantley L. C., Rac-mediated macropinocytosis of extracellular protein promotes glucose independence in non-small cell lung cancer. Cancer 11, 37 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Redhair M., Clouser A. F., Atkins W. M., Hydrogen-deuterium exchange mass spectrometry of membrane proteins in lipid nanodiscs. Chem. Phys. Lipids 220, 14–22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhattacharya P., Grimme S., Ganesh B., Gopisetty A., Sheng J. R., Martinez O., Jayarama S., Artinger M., Meriggioli M., Prabhakar B. S., Nanodisc-incorporated hemagglutinin provides protective immunity against influenza virus infection. J. Virol. 84, 361–371 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leitz A. J., Bayburt T. H., Barnakov A. N., Springer B. A., Sligar S. G., Functional reconstitution of β2-adrenergic receptors utilizing self-assembling Nanodisc technology. Biotechniques 40, 601–612 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Hansen R. W., Wang X., Golab A., Bornert O., Oswald C., Wagner R., Martinez K. L., Functional stability of the human kappa opioid receptor reconstituted in Nanodiscs revealed by a time-resolved scintillation proximity assay. PLOS ONE 11, e0150658 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Węgrzyn I., Ainla A., Jeffries G. D. M., Jesorka A., An optofluidic temperature probe. Sensors 13, 4289–4302 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Caterina M. J., Schumacher M. A., Tominaga M., Rosen T. A., Levine J. D., Julius D., The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 389, 816–824 (1997). [DOI] [PubMed] [Google Scholar]
- 52.Pingle S. C., Matta J. A., Ahern G. P., Capsaicin receptor: TRPV1 a promiscuous TRP channel. Handb. Exp. Pharmacol. 179, 155–171 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Tominaga M., Caterina M. J., Malmberg A. B., Rosen T. A., Gilbert H., Skinner K., Raumann B. E., Basbaum A. I., Julius D., The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21, 531–543 (1998). [DOI] [PubMed] [Google Scholar]
- 54.Jordt S.-E., Tominaga M., Julius D., Acid potentiation of the capsaicin receptor determined by a key extracellular site. Proc. Natl. Acad. Sci. U.S.A. 97, 8134–8139 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu B., Hui K., Qin F., Thermodynamics of heat activation of single capsaicin ion channels VR1. Biophys. J. 85, 2988–3006 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Szallasi A., Blumberg P. M., Resiniferatoxin, a phorbol-related diterpene, acts as an ultrapotent analog of capsaicin, the irritant constituent in red pepper. Neuroscience 30, 515–520 (1989). [DOI] [PubMed] [Google Scholar]
- 57.Hwang S. W., Cho H., Kwak J., Lee S. Y., Kang C. J., Jung J., Cho S., Min K. H., Suh Y. G., Kim D., Oh U., Direct activation of capsaicin receptors by products of lipoxygenases: Endogenous capsaicin-like substances. Proc. Natl. Acad. Sci. U.S.A. 97, 6155–6160 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang S. M., Bisogno T., Trevisani M., Al-Hayani A., de Petrocellis L., Fezza F., Tognetto M., Petros T. J., Krey J. F., Chu C. J., Miller J. D., Davies S. N., Geppetti P., Walker J. M., di Marzo V., An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc. Natl. Acad. Sci. U.S.A. 99, 8400–8405 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ma W., Quirion R., Inflammatory mediators modulating the transient receptor potential vanilloid 1 receptor: Therapeutic targets to treat inflammatory and neuropathic pain. Expert Opin. Ther. Targets 11, 307–320 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Tian Q., Hu J., Xie C., Mei K., Pham C., Mo X., Hepp R., Soares S., Nothias F., Wang Y., Liu Q., Cai F., Zhong B., Li D., Yao J., Recovery from tachyphylaxis of TRPV1 coincides with recycling to the surface membrane. Proc. Natl. Acad. Sci. U.S.A. 116, 5170–5175 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liao M., Cao E., Julius D., Cheng Y., Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504, 107–112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gao Y., Cao E., Julius D., Cheng Y., TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 534, 347–351 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fernández-Ballester G., Ferrer-Montiel A., Molecular modeling of the full-length human TRPV1 channel in closed and desensitized states. J. Membr. Biol. 223, 161–172 (2008). [DOI] [PubMed] [Google Scholar]
- 64.Molecular Operating Environment (MOE), version 2015.10 (2016).
- 65.Lau S.-Y., Procko E., Gaudet R., Distinct properties of Ca2+-calmodulin binding to N- and C-terminal regulatory regions of the TRPV1 channel. J. Gen. Physiol. 140, 541–555 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lishko P. V., Procko E., Jin X., Phelps C. B., Gaudet R., The ankyrin repeats of TRPV1 bind multiple ligands and modulate channel sensitivity. Neuron 54, 905–918 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/16/eabe6397/DC1