

Summary
PARP1 is a Poly-ADP-ribose Polymerase (PARP) enzyme that plays a critical role in regulating DNA damage response. The main enzymatic function of PARP1 is to catalyze a protein posttranslational modification known as Poly-ADP-Ribosylation (PARylation). Human cancers with homologous recombination deficiency are highly sensitive to PARP1 inhibitors. PARP1 is aberrantly activated in many non-oncological diseases, leading to the excessive NAD+ depletion and PAR formation, thus causing cell death and tissue damage. PARP1 deletion offers a profound protective effect in the relevant animal models. However, many of the current PARP1 inhibitors also induce PARP1 trapping, which drives subsequent DNA damage, innate immune response and cytotoxicity. This minireview provides an overview of the basic biology of PARP1 trapping, and its implications in disease. Further, we also discuss the recent development of PARP1 PROTAC compounds, and their utility as “non-trapping” PARP1 degraders for the potential amelioration of non-oncological diseases driven by aberrant PARP1 activation.
Background of PARP1 and Protein Poly-ADP-Ribosylation
PARP1 is a Poly(ADP-ribose) polymerase (PARP) enzyme that is critically involved in regulating cell stress responses (Gibson and Kraus, 2012). The enzymatic function of PARP1 is to consume a cofactor, β-nicotinamide adenine dinucleotide (β-NAD+), to catalyze a reversible protein post-translational modification (PTM) known as Poly(ADP-ribosyl)ation (PARylation). PAR is a bulky and flexible polymer, whose lengths can extend to hundreds of repeating units of ADP-ribose (Curtin and Szabo, 2020; Schreiber et al., 2006). PARP1 and its mediated PARylation is critically involved in DNA damage response (DDR). PARP1 serves as an early sensor for DNA strand breaks (Liu et al., 2017). Upon the detection of DNA nicks, PARP1 becomes rapidly activated, and it consumes NAD+ to attach large amounts of PAR chains to acceptor proteins. Based on these observations, PARylation has been proposed to serve as the first wave of DDR signaling. A number of DDR proteins contain PAR-binding domains (e.g., XRCC1) and are then recruited to the DNA lesions by the protein-linked PAR chains to initiate the DNA damage repair process.
The analysis of protein PARylation has been particularly challenging, owing to the labile, heterogenous and low-abundance nature of this modification (Martello et al., 2016; Zhang et al., 2013). The characterization of the PARylated proteome has been greatly facilitated by recent development of mass spectrometry-based proteomic technologies (Bonfiglio et al., 2017; Daniels et al., 2015; Zhen and Yu, 2018). These studies have provided an unbiased overview of the landscape of protein PARylation, and show that PARylated proteins are involved in not only DDR, but also a wide array of other nuclear functions, including transcriptional control, epigenetic regulation and mRNA metabolism (Zhang et al., 2013).
PARP1 Inhibitors for the Treatment of Human Malignancies
The critical roles of PARP1 and PARylation in mediating DDR provide the rationale for developing PARP1 inhibitors (PARPi) to treat human malignancy. It was shown that cancers with BRCA1/2 mutations and other homologous recombination deficiencies (HRD) are particularly sensitive to PARPi, as a result of the synthetic lethality mechanism (Lord and Ashworth, 2017). In particular, about half of the ovarian cancers are homologous recombination deficient (owing to the mutation of BRCA1/2 and other homologous recombination genes) (Ledermann et al., 2016). Four PARPi, including Olaparib, Rucaparib, Niraparib, and Talazoparib (Table 1) have been approved by the FDA to treat certain types of breast and/or ovarian cancers (in particular, those with BRCA1/2 mutations) (de Bono et al., 2017; Kaufman et al., 2015; Sandhu et al., 2013; Swisher et al., 2017). More recent clinical studies have shown that PARPi are also highly effective against a number of other BRCA1/2-mutated cancers, regardless of their anatomical origin (e.g., prostate and pancreatic) (Shroff et al., 2018; Sigorski et al., 2020).
Table 1.
FDA-approved PARP inhibitors (PARPi) in human malignancy
| PARPi | Company | Approvals in cancer | First approval date | Single-agent dose (mg) | Cytotoxicity (IC50, nM) | PARP1 trapping potency (relative to Olaparib) |
|---|---|---|---|---|---|---|
| Talazoparib | Pfizer | Ovarian and Breast | October 2018 | 1 | 0.6 – 1.1 | High (100) |
| Niraparib | Tesaro | Ovarian | March 2017 | 300 | 2 – 35 | Middle (2) |
| Rucaparib | Clovis Oncology | Ovarian | December 2016 | 600 | 0.8 – 3.2 | Middle (1) |
| Olaparib | AstraZeneca | Ovarian, Breast, and Pancreatic | December 2014 | 300 | 1 – 19 | Middle (1) |
IC50 values are obtained from the literature and the ChEMBL database (www.ebi.ac.uk/chembl/)
Data for PARP1 trapping potency are obtained from (Hopkins et al., 2015)
All FDA-approved PARPi are NAD+-competitive, and it was initially thought that PARPi kill tumor cells by inhibiting the catalytic activity of PARP1 and blocking PARylation-dependent DDR signaling. However, later studies showed that even though these PARPi have similar PARP1-inhibitory activities in vitro, they demonstrate very different cytotoxicity against cancer cells. This effect was later mainly ascribed to the unequal PARP1 trapping abilities of these compounds (Hopkins et al., 2019; Lord and Ashworth, 2017; Murai et al., 2012; Murai and Pommier, 2019; Thomas et al., 2018) (Figure 1) (see more discussion below about PARP1 trapping). In addition to the trapping model, it is important to note that PAPR1 null cells are more sensitive to IR, Top I poisons and DNA methylating agents, in the same way that PARP1-inhibited cells are. Furthermore, inhibition of base excision repair (BER) by XRCC1 depletion is also synthetically lethal with HRD (Fan et al., 2007). Finally, because all FDA-approved PARPi are NAD+-competitive, these compounds are promiscuous that inhibit and trap other PARP family members (e.g., PARP2) (Murai et al., 2012). These results suggest that multiple factors could be involved in regulating the cytotoxicity of PARPi.
Figure 1.
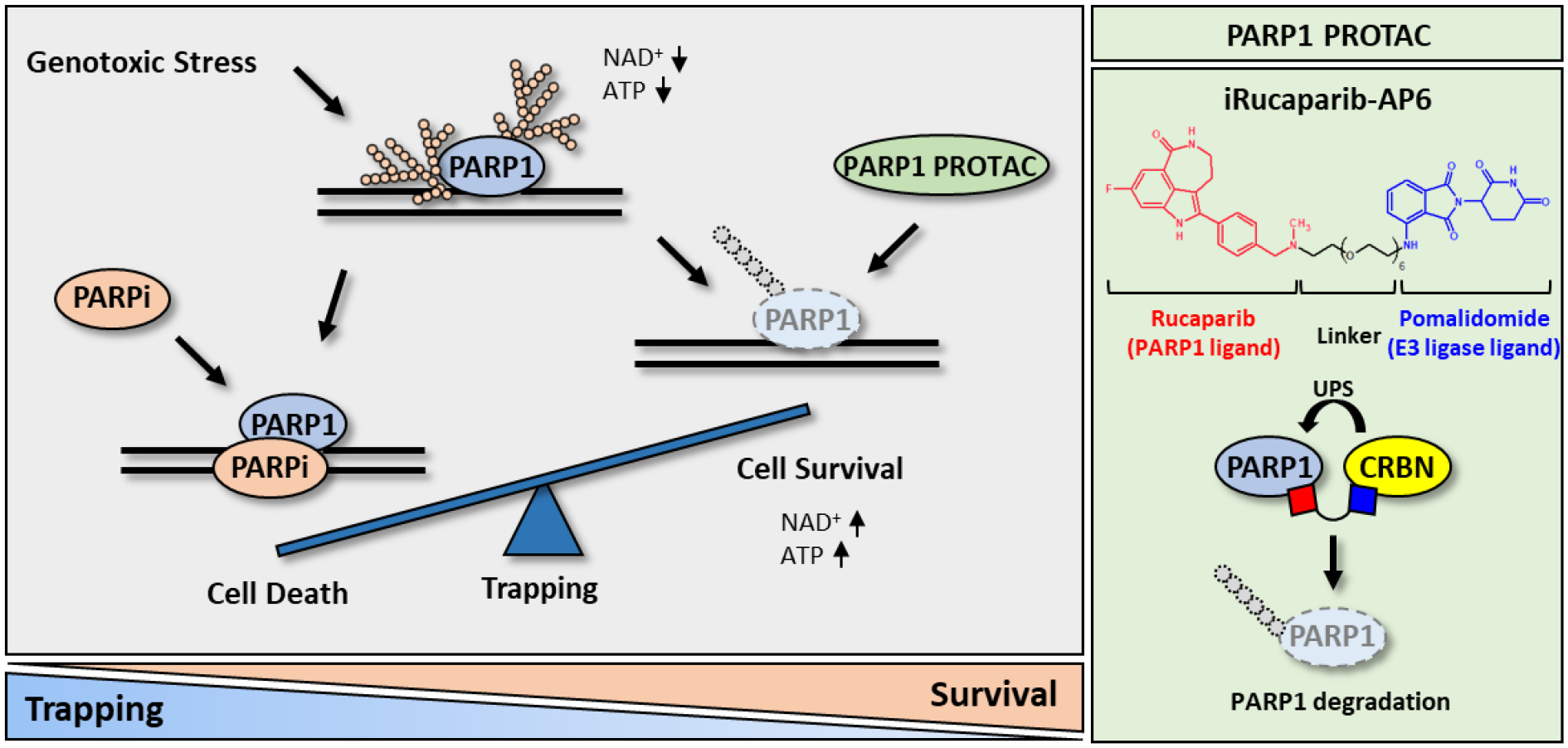
PROTAC compounds block the enzymatic activity of PARP1 without eliciting PARP1 trapping. (Left) A simplified model of PARP1 trapping, and its role in various diseases. PARP1 is hyper-activated in many non-oncological diseases. Although many of the current PARPi inhibit PARP1, they also trap PARP1, leading to DNA damage, innate immune response, and cytotoxicity. In contrast, PARP1 degraders block both the enzymatic and scaffolding effects of PARP1. (Right) The structure and mechanism of action of a recently described PARP1 PROTAC compound (i.e., iRucaparib-AP6).
The molecular underpinnings of PARP1 trapping are poorly understood. PARP1 represents one of the most abundantly PARylated proteins (Zhang et al., 2013). PARylation of PARP1 results in a dramatic change of the electrostatic and topological property of this protein, leading to its subsequent release from the DNA lesions (Satoh and Lindahl, 1992). Upon the treatment of PARPi, PARP1 is unable to PARylate itself, which could result in the formation of a trapped PARP1-DNA-PARPi complex (Lord and Ashworth, 2017; Murai et al., 2012). These studies indicate that PAR polymers are not only necessary for the recruitment of XRCC1, but also for the electrostatic repulsion of PARP1 from the damaged DNA. Besides the PAR-mediated steric hinderance model, PARPi could induce PARP1 trapping as a result of their allosteric binding effect. Indeed, the clinically approved PARPi differ in their allosteric binding to PARP1, leading to the unequal PARP1 trapping activities (Zandarashvili et al., 2020). In particular, it has been shown that compared to the other PARPi, Talazoparib is about 100-fold more potent at PARP1 trapping (Hopkins et al., 2019; Lord and Ashworth, 2017; Murai et al., 2012; Murai and Pommier, 2019). Accordingly, Talazoparib also demonstrates the highest cytotoxicity among the FDA-approved PARPi. It is important to note that from a structure point of view, Talazoparib resembles a prototype PARPi, 4-amino-1,8-naphthalimide, which also functions as a DNA intercalating compound that inhibits topoisomerases (Banasik et al., 1992; Tomczyk and Walczak, 2018). Whether Talazoparib kill cancer cells via both PARP1 inhibition/trapping and DNA intercalation is poorly understood. Finally, a recent study showed that even in the presence of PARPi, different PARP1 molecules are recruited to, and exchange rapidly at DNA lesions, owing to delayed DNA repair. These results raise the intriguing hypothesis that PARPi-mediated formation of persistent PARP1 foci may not be caused by the physical stalling of PARP1 (Shao et al., 2020).
It is being increasingly appreciated that PARPi also play an important role in modulating immune signaling in cancers. PARPi treatment is known to induce the formation of cytosolic double-strand DNA (dsDNA) and micronuclei. These species are detected by the cytosolic DNA sensor, cGAS (cyclic GMP-AMP synthetase), which then leads to the activation of cGAS-STING signaling and downstream innate immune response. PARPi induce the activation of cGAS-STING signaling in both BRCA-mutated and -wild type cells, suggesting that the immunomodulatory roles of PARPi might not be affected by the HRD status of the cell (Chabanon et al., 2019; Ding et al., 2018; Kim et al., 2020; Shen et al., 2019). The coordinated activation of both local and systemic immune response has emerged as an important contributor to the antitumor effects of PARPi. It is important to note that the presence of the PARP1 protein with uncompromised DNA-binding activities is required for PARPi-induced innate immune response. These results indicate that similar to the cytotoxicity of PARPi, the immunomodulatory roles of PARPi are primarily mediated by the PARP1 trapping, rather than PARP1 inhibition activities of these compounds (Kim et al., 2020).
PARP1 and PARylation-dependent Signaling in Regulating Cell Death
Although PARPi have shown impressive clinical efficacy against cancer, aberrant activation of PARP1 and PARylation-mediated signaling is tightly linked to the pathogenesis of many non-oncological human diseases (Dawson and Dawson, 2017; Fatokun et al., 2014; Koh et al., 2005; Martire et al., 2015). Indeed, pathological hyperactivation of PARP1 has been demonstrated in various ischemia-reperfusion (I/R) injuries of the brain, heart, skeletal muscle, retina, gut, and kidney (Crawford et al., 2010; Kovacs et al., 2019; Love et al., 1999; Pacher and Szabó, 2007; Yoon and Kim, 2015). During I/R injuries, genotoxic stress resulting from reactive oxygen/nitrogen species is known to induce DNA damage, PARP1 activation and the subsequent tissue damage (Virag and Szabo, 2002).
PARP1 has also been shown to be implicated in various forms of neurological disorders (Andrabi et al., 2006; Xu et al., 2016). In particular, pathologic protein aggregates, including alpha-synuclein and amyloid peptides can activate PARP1, and the resulting PAR polymers, in turn, accelerates the formation of protein aggregates, forming a feed-forward loop that causes neuronal cell death (Kam et al., 2018). Aberrant PAR accumulation has also been found in the brains and cerebrospinal fluid (CSF) of Alzheimer’s disease (AD) and Parkinson’s disease (PD) patients, with a positive correlation between higher PAR levels and disease duration/progression (Burguillos et al., 2011; Kam et al., 2018). A common molecular underpinning that drives the aberrant PARP1 activation in I/R injury and neurodegenerative diseases is the DNA damage induced by the relevant genotoxic agents. In the context of neurodegenerative diseases, protein aggregates (e.g., alpha-synuclein) activate nitric oxide synthase (NOS), leading to the generation of reactive nitrogen species (e.g., nitric oxide and peroxynitrite) and subsequently, DNA damage and PARP1 activation.
Several mechanisms have been proposed to address how the aberrant activation of PARP1 causes cell death. First, PAR has been shown to facilitate the release of AIF (apoptosis-inducing factor) from mitochondria where it interacts with a nuclease called MIF (migration inhibitory factor). This protein complex translocates to the nucleus where its binds to DNA, leading to DNA fragmentation and a form of programmed cell death known as parthanatos (Wang et al., 2016). Second, PARP1 overactivation can also kill cells by altering their energy metabolism (Andrabi et al., 2014; Canto et al., 2015). Specifically, PARP1 is one of the most abundant nuclear enzymes, and it becomes highly active upon sensing genotoxic stress. It has been shown that cellular NAD+ levels could decrease by more than 80% within 15 min of PARP1 hyperactivation (e.g., during genotoxic stress). ATP is then channeled through the salvage pathway to replenish the cellular NAD+ pool. This results in the eventual cellular energy crisis and cell death (e.g., necrosis). Finally, recent studies suggest that protein PARylation might be linked to a class of subcellular organelles called “biomolecular condensates” (Altmeyer et al., 2015; Duan et al., 2019). These biomolecular condensates are highly dynamic structures that are not enclosed by any physical barriers. Instead, they originate from biological polymers that undergo phase transition, as a result of clustering-induced self-assembly. Biomolecular condensates contain many RNA-binding proteins (e.g., FUS, TAF15 and hnRNPs) that are known to be PARylated (Zhang et al., 2013). Importantly, mutations of these RNA-binding proteins are frequently found in neurodegenerative diseases (Duan et al., 2019). It is perhaps not surprising that PAR chains could function as a nucleating factor that promotes the aggregation of these proteins, and in doing so, seeds the formation of biomolecular condensates (Leung, 2020). The resulting insoluble fibrils, in turn, could cause further DNA damage and PARP1 activation, thereby creating a feed-forward loop to accelerate protein aggregation and neuronal cell death (Kam et al., 2018).
“Non-Trapping” PARP1 Degraders for the Treatment of Non-Oncological Human Disease
Genetic deletion of PARP1 offers profound tissue protection in animal models of I/R injury (e.g., stroke, cardiac infarction and ischemic renal injury) (Eliasson et al., 1997; Yang et al., 2000; Zheng et al., 2005) and neurological disorders (e.g., Parkinson’s disease, Amyotrophic lateral sclerosis, Cerebellar ataxia and Neuronal injury) (Brochier et al., 2015; Cardinale et al., 2015; Hoch et al., 2017; Kam et al., 2018; Mandir et al., 1999; Yokoyama et al., 2010). These studies point to PARP1 as a viable therapeutic target for the amelioration of these non-oncological diseases. It was shown that the treatment with a novel PARP1 inhibitor (MP-124) significantly reduced the infarct volume and the associated neurological deficits in a non-human primate model of ischemic stroke (Matsuura et al., 2011). In addition, it has been shown that veliparib (a PARPi with moderate PARP1 trapping activities) blocks PAR formation, and in doing so, prevents alpha-synuclein preformed fibrils (PFFs)-induced dopamine (DA) neuron death (Kam and Mao, 2018). The potential functions of PARPi as cytoprotective agents (e.g., for stroke) are also being studied in a number of clinical trials (Curtin and Szabo, 2020).
Despite this exciting progress, it is important to recognize that many of the current PARPi possess both PARP1 inhibition and PARP1 trapping activities (Figure 1). While they block the enzymatic activity of PARP1 and thus reduce NAD+ consumption and PAR formation, PARP1 trapping could have a series of deleterious effects, including the induction of DNA damage, innate immune response, and eventually, cell death. Towards this, therapeutic strategies that mimic PARP1 genetic depletion (i.e., inhibit PARP1 without causing PARP1 trapping) will likely have substantial potential in these non-malignant human diseases. It is also important to note that PARP1 knockout mice are viable and they develop normally (Wang et al., 1995). These results suggest that PARP1 is dispensable, and the depletion of the PARP1 protein, at least temporarily, should have minimal side effects. Furthermore, the risk of impaired DDR as a result of PARP1 inhibition is also mitigated as the relevant patients usually do not have HRD.
A gene can be inactivated at the DNA, RNA or protein level, potentially by using CRISPR-Cas9, RNAi (RNA interference) or PROTAC (Proteolysis targeting chimera), respectively. Among them, PROTAC has recently emerged as a valuable tool for the chemical knockdown of a protein of interest (POI) (Sakamoto et al., 2001). PROTAC compounds are heterobifunctional small molecules that consist of a POI binder, an E3 ubiquitin ligase recruiting ligand, and an interval linker (Figure 1, Right). These compounds bring a POI and a E3 into proximity, leading to the ubiquitination and subsequent degradation of the target protein via the ubiquitin-proteasome system (UPS) (Toure and Crews, 2016). Compared to RNAi or the CRISPR-Cas9 technology, a unique feature of PRTOAC therefore is that it induces the reversible degradation of POI at the post-translational level.
PARP1 PROTAC compounds degrade PARP1 (Cao et al., 2020; Wang et al., 2019; Zhang et al., 2020; Zhao et al., 2019), and in doing so, could be used to block both catalytic activity and scaffolding functions (i.e., non-enzyme related) of PARP1 (Figure 1, Right). The strategy is suited for the prevention of PARP1 trapping, and its associated effects on DNA damage, innate immune signaling, and cell death. Wang et al. reported the development of PARP1 PROTAC compounds that successfully induce the degradation of this protein (Wang et al., 2019) (Figure 2). They performed a systematic evaluation of the warhead (i.e., using different clinically relevant PARP1 inhibitors), the linker (i.e., lengths, conjugation sites and chemistries) and the E3 binder (i.e., ligands for cereblon (CRBN) and von Hippel-Lindau (VHL). It was found that all of these attributes contribute critically to the efficacy of a PARP1 PROTAC compound. These studies eventually led to the identification of iRucaparib-AP6 as a highly potent and selective PARP1 degrader (DC50 = 82 nM). iRucaparib-AP6 is cell membrane-permeable, which potently blocks the enzymatic activity of PARP1 both in vitro and in intact cells. In doing so, it prevents PAR formation and NAD+/ATP depletion induced by various genotoxic agents. Unlike a regular PARPi, iRucaparib-AP6 does not cause DDR, cell death or the activation of the innate immune signaling (Kim et al., 2020; Wang et al., 2019) (Figure 2). It was shown that iRucaparib-AP6 is able to protect primary cardiomyocytes and C2C12 muscle cells against various genotoxic agents (Wang et al., 2019). Finally, to mitigate oxidative damage during infarction and ischemic injury, antioxidants (Amato et al., 2019; Rodrigo et al., 2013) could be used in combination with PARP1 PROTAC compounds to both offer protection (i.e., prevent DDR-induced genotoxic stress) and block adverse effects associated with PARP1 trapping.
Figure 2.
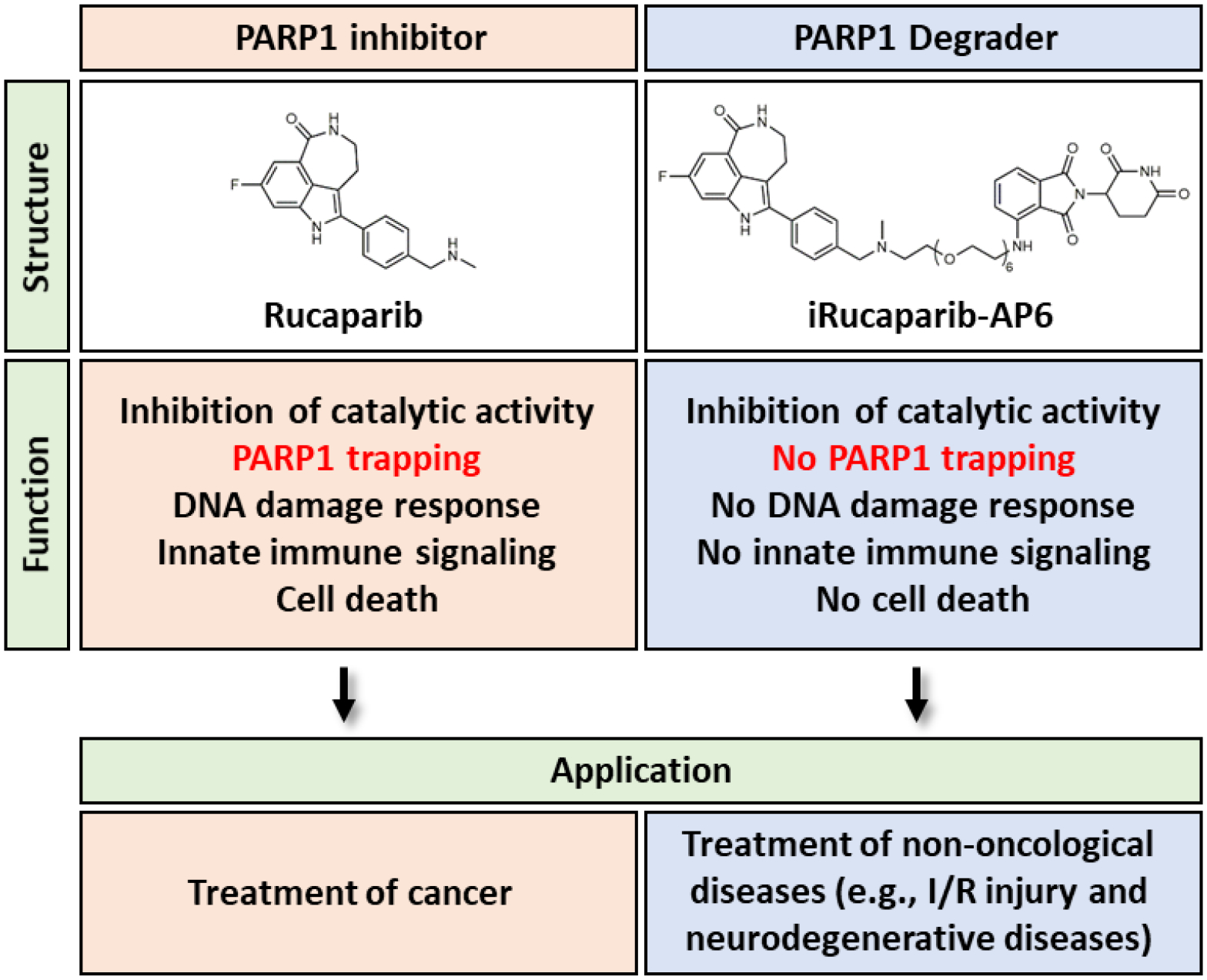
Comparison between a PARP1 inhibitor and a PROTAC-based PARP1 degrader.
PROTACs are usually “beyond rule of five (bRo5)” molecules due to their unique molecular architecture (Churcher, 2018; Edmondson et al., 2019). Whereas the general DMPK properties of PROTACs are still not fully understood (Watt et al., 2019), recent studies suggest that, against conventional wisdom, good bioavailability can be achieved with this class of molecules (Cantrill et al., 2020; Pike et al., 2020). For example, ARV-110 is an oral androgen receptor PROTAC that showed efficacy in a phase 1 clinical trial (NCT03888612). The mechanistic difference in chemical inhibitors and degraders also allows PROTACs to extend the functional duration beyond that of normal small-molecule drugs. Exceptionally long-lasting effects can be observed when the resynthesis of the target protein is slow (Mares et al., 2020). However, the pharmacokinetics (PK) and pharmacodynamics (PD) relationship of PROTAC molecules is complicated and requires special considerations. In particular, the PROTAC molecules would initially act as inhibitors when dosed too high, and only start to function as degraders after the drug concentration falls into the effective range. This “hook effect” may lead to potential toxicity issues during this “incubation period” (Pettersson and Crews, 2019). Additionally, metabolic cleavage of the linker will also generate competing inhibitors to attenuate degradation and provide cytotoxicity. Finally, the degradation of a target protein in vivo by PROTAC compounds is often affected by the efficiency of the proteasomal machinery (e.g., tissue-specific expression of the relevant E3 ligase), and therefore could be context-dependent. Future development of a PROTAC-based PARP1 degrader should focus on understanding the in vivo efficacy and PK/PD relationship and optimizing the linker.
Concluding Remark
Although the role of PARP1 as a therapeutic target for HRD-deficient cancers has been well established, hyperactive PARP1 signaling is also critically involved in the pathogenesis of many non-oncological human diseases. Genetic deletion of PARP1 offers dramatic tissue protection by reducing NAD+ consumption and PAR formation, and thus ameliorating cell death (e.g., necrosis and parthanatos). Many of the current PARPi possess both PARP1 inhibition and PARP1 trapping activities. While PARP1 trapping is desirable for the treatment of cancers, it could counteract the tissue protective effect resulting from the catalytic inhibition of PARP1. In this regard, PARP1 PROTAC compounds represent PARP1 degraders that block the catalytic activity of PARP1 without eliciting PARP1 trapping. Compared to regular small molecule compounds, PROTACs have larger molecular weights (i.e., incorporation of the linker and the E3 binder), but act in a catalytic mode (i.e., the PROTAC molecule is recycled to repeat the degradation events). This results in their non-traditional adsorption/distribution, as well as PK/PD properties. However, a number of PROTAC agents have recently entered clinical trials, with promising profiles of drug-like properties, safety and efficacy (Mullard, 2019). PARP1 PROTACs thus offer a highly promising strategy for the potential amelioration of the various non-oncological, pathological conditions caused by PARP1 hyperactivation. Further development is needed to establish the full therapeutic potential of these compounds.
Acknowledgements
We thank members of the Yu and Chen labs for helpful discussions. This work was supported, in part, by grants from the NIH (R01GM122932 and R35GM134883 to Y.Y., and R01CA226419 to C.C.) and Welch foundation (I-1800 to YY).
Footnotes
Declaration of Interests
A provisional patent application on the PARP degraders and technologies described herein was previously filed (Y.Y. and C.C.).
References
- Altmeyer M, Neelsen KJ, Teloni F, Pozdnyakova I, Pellegrino S, Grofte M, Rask MD, Streicher W, Jungmichel S, Nielsen ML, et al. (2015). Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat Commun 6, 8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato A, Terzo S, and Mulè F (2019). Natural Compounds as Beneficial Antioxidant Agents in Neurodegenerative Disorders: A Focus on Alzheimer’s Disease. Antioxidants (Basel, Switzerland) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, et al. (2006). Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A 103, 18308–18313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi SA, Umanah GK, Chang C, Stevens DA, Karuppagounder SS, Gagne JP, Poirier GG, Dawson VL, and Dawson TM (2014). Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Natl Acad Sci U S A 111, 10209–10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banasik M, Komura H, Shimoyama M, and Ueda K (1992). Specific inhibitors of poly(ADP-ribose) synthetase and mono(ADP-ribosyl)transferase. The Journal of biological chemistry 267, 1569–1575. [PubMed] [Google Scholar]
- Bonfiglio JJ, Colby T, and Matic I (2017). Mass spectrometry for serine ADP-ribosylation? Think o-glycosylation! Nucleic Acids Res 45, 6259–6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochier C, Jones JI, Willis DE, and Langley B (2015). Poly(ADP-ribose) polymerase 1 is a novel target to promote axonal regeneration. Proc Natl Acad Sci U S A 112, 15220–15225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burguillos MA, Hajji N, Englund E, Persson A, Cenci AM, Machado A, Cano J, Joseph B, and Venero JL (2011). Apoptosis-inducing factor mediates dopaminergic cell death in response to LPS-induced inflammatory stimulus: evidence in Parkinson’s disease patients. Neurobiol Dis 41, 177–188. [DOI] [PubMed] [Google Scholar]
- Canto C, Menzies KJ, and Auwerx J (2015). NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab 22, 31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantrill C, Chaturvedi P, Rynn C, Petrig Schaffland J, Walter I, and Wittwer MB (2020). Fundamental aspects of DMPK optimization of targeted protein degraders. Drug Discov Today 25, 969–982. [DOI] [PubMed] [Google Scholar]
- Cao C, Yang J, Chen Y, Zhou P, Wang Y, Du W, Zhao L, and Chen Y (2020). Discovery of SK-575 as a Highly Potent and Efficacious Proteolysis-Targeting Chimera Degrader of PARP1 for Treating Cancers. 63, 11012–11033. [DOI] [PubMed] [Google Scholar]
- Cardinale A, Paldino E, Giampa C, Bernardi G, and Fusco FR (2015). PARP-1 Inhibition Is Neuroprotective in the R6/2 Mouse Model of Huntington’s Disease. PLoS One 10, e0134482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabanon RM, Muirhead G, Krastev DB, Adam J, Morel D, Garrido M, Lamb A, Henon C, Dorvault N, Rouanne M, et al. (2019). PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J Clin Invest 129, 1211–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churcher I (2018). Protac-Induced Protein Degradation in Drug Discovery: Breaking the Rules or Just Making New Ones? Journal of medicinal chemistry 61, 444–452. [DOI] [PubMed] [Google Scholar]
- Crawford RS, Albadawi H, Atkins MD, Jones JE, Yoo HJ, Conrad MF, Austen WG Jr., and Watkins MT (2010). Postischemic poly (ADP-ribose) polymerase (PARP) inhibition reduces ischemia reperfusion injury in a hind-limb ischemia model. Surgery 148, 110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin NJ, and Szabo C (2020). Poly(ADP-ribose) polymerase inhibition: past, present and future. Nat Rev Drug Discov 19, 711–736. [DOI] [PubMed] [Google Scholar]
- Daniels CM, Ong SE, and Leung AK (2015). The Promise of Proteomics for the Study of ADP-Ribosylation. Mol Cell 58, 911–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, and Dawson VL (2017). Mitochondrial Mechanisms of Neuronal Cell Death: Potential Therapeutics. Annu Rev Pharmacol Toxicol 57, 437–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bono J, Ramanathan RK, Mina L, Chugh R, Glaspy J, Rafii S, Kaye S, Sachdev J, Heymach J, Smith DC, et al. (2017). Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov 7, 620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Kim HJ, Wang Q, Kearns M, Jiang T, Ohlson CE, Li BB, Xie S, Liu JF, Stover EH, et al. (2018). PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1- Deficient Ovarian Cancer. Cell Rep 25, 2972–2980 e2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y, Du A, Gu J, Duan G, Wang C, Gui X, Ma Z, Qian B, Deng X, Zhang K, et al. (2019). PARylation regulates stress granule dynamics, phase separation, and neurotoxicity of disease-related RNA-binding proteins. Cell Res 29, 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmondson SD, Yang B, and Fallan C (2019). Proteolysis targeting chimeras (PROTACs) in ‘beyond rule-of-five’ chemical space: Recent progress and future challenges. Bioorg Med Chem Lett 29, 1555–1564. [DOI] [PubMed] [Google Scholar]
- Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, et al. (1997). Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med 3, 1089–1095. [DOI] [PubMed] [Google Scholar]
- Fan J, Wilson PF, Wong HK, Urbin SS, Thompson LH, and Wilson DM 3rd (2007). XRCC1 down-regulation in human cells leads to DNA-damaging agent hypersensitivity, elevated sister chromatid exchange, and reduced survival of BRCA2 mutant cells. Environmental and molecular mutagenesis 48, 491–500. [DOI] [PubMed] [Google Scholar]
- Fatokun AA, Dawson VL, and Dawson TM (2014). Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br J Pharmacol 171, 2000–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, and Kraus WL (2012). New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol 13, 411–424. [DOI] [PubMed] [Google Scholar]
- Hoch NC, Hanzlikova H, Rulten SL, Tetreault M, Komulainen E, Ju L, Hornyak P, Zeng Z, Gittens W, Rey SA, et al. (2017). XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature 541, 87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins TA, Ainsworth WB, Ellis PA, Donawho CK, DiGiammarino EL, Panchal SC, Abraham VC, Algire MA, Shi Y, Olson AM, et al. (2019). PARP1 Trapping by PARP Inhibitors Drives Cytotoxicity in Both Cancer Cells and Healthy Bone Marrow. Mol Cancer Res 17, 409–419. [DOI] [PubMed] [Google Scholar]
- Kam TI, and Mao X (2018). Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson’s disease. 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam TI, Mao X, Park H, Chou SC, Karuppagounder SS, Umanah GE, Yun SP, Brahmachari S, Panicker N, Chen R, et al. (2018). Poly(ADP-ribose) drives pathologic alpha-synuclein neurodegeneration in Parkinson’s disease. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, Mitchell G, Fried G, Stemmer SM, Hubert A, et al. (2015). Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 33, 244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Wang XD, and Yu Y (2020). PARP1 inhibitors trigger innate immunity via PARP1 trapping-induced DNA damage response. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh DW, Dawson TM, and Dawson VL (2005). Poly(ADP-ribosyl)ation regulation of life and death in the nervous system. Cell Mol Life Sci 62, 760–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs K, Vaczy A, Fekete K, Kovari P, Atlasz T, Reglodi D, Gabriel R, Gallyas F, and Sumegi B (2019). PARP Inhibitor Protects Against Chronic Hypoxia/Reoxygenation-Induced Retinal Injury by Regulation of MAPKs, HIF1α, Nrf2, and NFκB. Invest Ophthalmol Vis Sci 60, 1478–1490. [DOI] [PubMed] [Google Scholar]
- Ledermann JA, Drew Y, and Kristeleit RS (2016). Homologous recombination deficiency and ovarian cancer. European journal of cancer (Oxford, England : 1990) 60, 49–58. [DOI] [PubMed] [Google Scholar]
- Leung AKL (2020). Poly(ADP-ribose): A Dynamic Trigger for Biomolecular Condensate Formation. Trends Cell Biol 30, 370–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Vyas A, Kassab MA, Singh AK, and Yu X (2017). The role of poly ADP-ribosylation in the first wave of DNA damage response. Nucleic Acids Res 45, 8129–8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, and Ashworth A (2017). PARP inhibitors: Synthetic lethality in the clinic. Science 355, 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love S, Barber R, and Wilcock GK (1999). Neuronal accumulation of poly(ADP-ribose) after brain ischaemia. Neuropathol Appl Neurobiol 25, 98–103. [DOI] [PubMed] [Google Scholar]
- Mandir AS, Przedborski S, Jackson-Lewis V, Wang ZQ, Simbulan-Rosenthal CM, Smulson ME, Hoffman BE, Guastella DB, Dawson VL, and Dawson TM (1999). Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc Natl Acad Sci U S A 96, 5774–5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mares A, Miah AH, Smith IED, Rackham M, and Thawani AR (2020). Extended pharmacodynamic responses observed upon PROTAC-mediated degradation of RIPK2. 3, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martello R, Leutert M, Jungmichel S, Bilan V, Larsen SC, Young C, Hottiger MO, and Nielsen ML (2016). Proteome-wide identification of the endogenous ADP-ribosylome of mammalian cells and tissue. Nat Commun 7, 12917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martire S, Mosca L, and d’Erme M (2015). PARP-1 involvement in neurodegeneration: A focus on Alzheimer’s and Parkinson’s diseases. Mech Ageing Dev 146–148, 53–64. [DOI] [PubMed] [Google Scholar]
- Matsuura S, Egi Y, Yuki S, Horikawa T, Satoh H, and Akira T (2011). MP-124, a novel poly(ADP-ribose) polymerase-1 (PARP-1) inhibitor, ameliorates ischemic brain damage in a non-human primate model. Brain Res 1410, 122–131. [DOI] [PubMed] [Google Scholar]
- Mullard A (2019). Arvinas’s PROTACs pass first safety and PK analysis. Nat Rev Drug Discov 18, 895. [DOI] [PubMed] [Google Scholar]
- Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, and Pommier Y (2012). Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer research 72, 5588–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, and Pommier Y (2019). PARP Trapping Beyond Homologous Recombination and Platinum Sensitivity in Cancers. Annual Review of Cancer Biology 3, 131–150. [Google Scholar]
- Pacher P, and Szabó C (2007). Role of poly(ADP-ribose) polymerase 1 (PARP-1) in cardiovascular diseases: the therapeutic potential of PARP inhibitors. Cardiovasc Drug Rev 25, 235–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson M, and Crews CM (2019). PROteolysis TArgeting Chimeras (PROTACs) - Past, present and future. Drug Discov Today Technol 31, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike A, Williamson B, Harlfinger S, Martin S, and McGinnity DF (2020). Optimising proteolysis-targeting chimeras (PROTACs) for oral drug delivery: a drug metabolism and pharmacokinetics perspective. Drug Discov Today. [DOI] [PubMed] [Google Scholar]
- Rodrigo R, Libuy M, Feliú F, and Hasson D (2013). Molecular basis of cardioprotective effect of antioxidant vitamins in myocardial infarction. BioMed research international 2013, 437613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, and Deshaies RJ (2001). Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A 98, 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, Hylands L, Riisnaes R, Forster M, Omlin A, et al. (2013). The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol 14, 882–892. [DOI] [PubMed] [Google Scholar]
- Satoh MS, and Lindahl T (1992). Role of poly(ADP-ribose) formation in DNA repair. Nature 356, 356–358. [DOI] [PubMed] [Google Scholar]
- Schreiber V, Dantzer F, Ame JC, and de Murcia G (2006). Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 7, 517–528. [DOI] [PubMed] [Google Scholar]
- Shao Z, Lee BJ, Rouleau-Turcotte E, Langelier MF, Lin X, Estes VM, Pascal JM, and Zha S (2020). Clinical PARP inhibitors do not abrogate PARP1 exchange at DNA damage sites in vivo. Nucleic Acids Res 48, 9694–9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, Yap TA, Mills GB, and Peng G (2019). PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res 79, 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff RT, Hendifar A, McWilliams RR, Geva R, Epelbaum R, Rolfe L, Goble S, Lin KK, Biankin AV, Giordano H, et al. (2018). Rucaparib Monotherapy in Patients With Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis Oncol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigorski D, Iżycka-Świeszewska E, and Bodnar L (2020). Poly(ADP-Ribose) Polymerase Inhibitors in Prostate Cancer: Molecular Mechanisms, and Preclinical and Clinical Data. Target Oncol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, Konecny GE, Coleman RL, Tinker AV, O’Malley DM, et al. (2017). Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol 18, 75–87. [DOI] [PubMed] [Google Scholar]
- Thomas A, Murai J, and Pommier Y (2018). The evolving landscape of predictive biomarkers of response to PARP inhibitors. J Clin Invest 128, 1727–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomczyk MD, and Walczak KZ (2018). l,8-Naphthalimide based DNA intercalators and anticancer agents. A systematic review from 2007 to 2017. European journal of medicinal chemistry 159, 393–422. [DOI] [PubMed] [Google Scholar]
- Toure M, and Crews CM (2016). Small-Molecule PROTACS: New Approaches to Protein Degradation. Angew Chem Int Ed Engl 55, 1966–1973. [DOI] [PubMed] [Google Scholar]
- Virag L, and Szabo C (2002). The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev 54, 375–429. [DOI] [PubMed] [Google Scholar]
- Wang S, Han L, Han J, Li P, Ding Q, Zhang QJ, Liu ZP, Chen C, and Yu Y (2019). Uncoupling of PARP1 trapping and inhibition using selective PARP1 degradation. Nature chemical biology 15, 1223–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, An R, Umanah GK, Park H, Nambiar K, Eacker SM, Kim B, Bao L, Harraz MM, Chang C, et al. (2016). A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZQ, Auer B, Stingl L, Berghammer H, Haidacher D, Schweiger M, and Wagner EF (1995). Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev 9, 509–520. [DOI] [PubMed] [Google Scholar]
- Watt GF, Scott-Stevens P, and Gaohua L (2019). Targeted protein degradation in vivo with Proteolysis Targeting Chimeras: Current status and future considerations. Drug Discov Today Technol 31, 69–80. [DOI] [PubMed] [Google Scholar]
- Xu JC, Fan J, Wang X, Eacker SM, Kam TI, Chen L, Yin X, Zhu J, Chi Z, Jiang H, et al. (2016). Cultured networks of excitatory projection neurons and inhibitory interneurons for studying human cortical neurotoxicity. Sci Transl Med 8, 333ra348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Zingarelli B, and Szabó C (2000). Effect of genetic disruption of poly (ADP-ribose) synthetase on delayed production of inflammatory mediators and delayed necrosis during myocardial ischemia-reperfusion injury. Shock 13, 60–66. [DOI] [PubMed] [Google Scholar]
- Yokoyama H, Kuroiwa H, Tsukada T, Uchida H, Kato H, and Araki T (2010). Poly(ADP-ribose)polymerase inhibitor can attenuate the neuronal death after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity in mice. J Neurosci Res 88, 1522–1536. [DOI] [PubMed] [Google Scholar]
- Yoon SP, and Kim J (2015). Poly(ADP-ribose) polymerase 1 activation links ischemic acute kidney injury to interstitial fibrosis. J Physiol Sci 65, 105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandarashvili L, Langelier MF, Velagapudi UK, Hancock MA, Steffen JD, Billur R, Hannan ZM, Wicks AJ, Krastev DB, Pettitt SJ, et al. (2020). Structural basis for allosteric PARP-1 retention on DNA breaks. Science 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang J, Ding M, and Yu Y (2013). Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nat Methods 10, 981–984. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Chang X, Zhang C, Zeng S, Liang M, Ma Z, Wang Z, Huang W, and Shen Z (2020). Identification of probe-quality degraders for Poly(ADP-ribose) polymerase-1 (PARP-1). Journal of enzyme inhibition and medicinal chemistry 35, 1606–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Lan T, Su S, and Rao Y (2019). Induction of apoptosis in MDA-MB-231 breast cancer cells by a PARP1-targeting PROTAC small molecule. Chemical communications (Cambridge, England) 55, 369–372. [DOI] [PubMed] [Google Scholar]
- Zhen Y, and Yu Y (2018). Proteomic Analysis of the Downstream Signaling Network of PARP1. Biochemistry (Mosc) 57, 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Devalaraja-Narashimha K, Singaravelu K, and Padanilam BJ (2005). Poly(ADP-ribose) polymerase-1 gene ablation protects mice from ischemic renal injury. Am J Physiol Renal Physiol 288, F387–398. [DOI] [PubMed] [Google Scholar]