

Summary
The translation of functionally active natural products into fully synthetic small molecule mimetics has remained an important process in medicinal chemistry. We recently discovered that the terpene natural product nimbolide can be utilized as a covalent recruiter of the E3 ubiquitin ligase RNF114 for use in targeted protein degradation (TPD) – a powerful therapeutic modality within modern day drug discovery. Using activity-based protein profiling-enabled covalent ligand screening approaches, we herein report the discovery of fully synthetic RNF114-based recruiter molecules that can also be exploited for PROTAC applications, and demonstrate their utility in degrading therapeutically relevant targets such as BRD4 and BCR-ABL in cells. The identification of simple and easily manipulated drug-like scaffolds that can mimic the function of a complex natural product is beneficial in further expanding the toolbox of E3 ligase recruiters, an area of great importance in drug discovery and chemical biology.
Keywords: RNF114, PROTAC, targeted protein degradation, covalent ligand, cysteine, chemoproteomics
eTOC Blurb
Using activity-based protein profiling-enabled covalent ligand screening approaches, Luo et al have discovered a fully synthetic RNF114 E3 ligase recruiter that can be used in targeted protein degradation applications.
Graphical Abstract
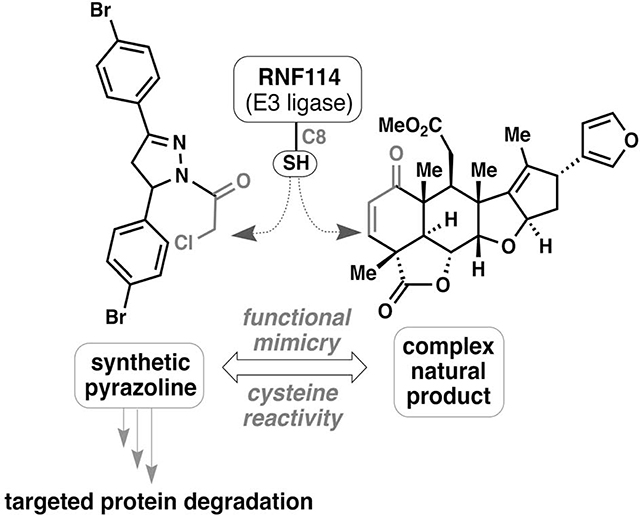
Introduction
Natural products have remained a cornerstone of drug discovery research for decades resulting in numerous FDA-approved medicines and tools for biomedical research across a wide range of therapeutic areas (Newman and Cragg, 2016). Historically, a large percentage of natural product-inspired medicines have utilized the natural product as a starting point, wherein the tools of synthetic chemistry are used to fine tune compound properties (i.e a semisynthetic approach) to achieve the desired endpoint. Alternatively, the function of natural products can also serve as motivation for medicinal chemistry, where the design of fully synthetic small molecules are less constrained by availability, synthetic manipulation limitations, and physicochemical and metabolic liabilities. Modern-day examples of this approach include the translation of the alkaloid cytisine into the smoking cessation drug varenicline, the development of the blockbuster cardiovascular drug atorvastatin from the polyketide lovastatin, and the discovery of the proteasome inhibitor carfilzomib inspired by the natural polypeptide epoxomicin (Fig. 1). Such approaches, however, are greatly facilitated by an understanding of the binding mode of the small molecule to its protein target as well as the identification of key pharmacophores within the parent natural product (Fig. 1).
Figure 1. Translation of natural product function.

Many successful drugs have recapitulated the function of natural products with fully-synthetic chemical scaffolds. Key pharmacophores are shown in red.
We recently discovered that the anti-cancer natural product nimbolide, a limonoid-type triterpene isolated from Azadirachta indica (neem), covalently reacts with an N-terminal cysteine (C8) within an intrinsically disordered region of the E3 ubiquitin ligase RNF114 in human breast cancer cells (Spradlin et al., 2019). Covalent targeting of RNF114 by nimbolide led to impaired ubiquitination of its endogenous substrate, the tumor suppressor p21, through a nimbolide-dependent competition of the RNF114-substrate binding interaction, thus providing a potential mechanism for the anti-cancer effects of this natural product. The realization that nimbolide targeted a substrate recognition domain within RNF114 suggested that nimbolide could potentially be used as a recruiter of RNF114 for targeted protein degradation (TPD) applications. Consistent with this premise, we showed that a proteolysis-targeting chimera (PROTAC) formed by linking nimbolide to a Bromodomain and extraterminal domain (BET) inhibitor JQ1 led to proteasome- and RNF114-dependent degradation of BRD4 in cells. While TPD has arisen as a powerful drug discovery paradigm for tackling the undruggable proteome by targeting intracellular proteins for proteasomal degradation rather than classic inhibition, the lack of a broad range of E3 ligase recruiters represents a known limitation in this arena (Bondeson and Crews, 2017; Chamberlain and Hamann, 2019; Lai and Crews, 2017). Indeed, while over 600 different E3 ligases have been annotated, only a small handful of these potential targets have succumbed to the PROTAC strategy. Discovering additional and more synthetically tractable E3 ligase recruiters is thus an important topic in expanding the scope of TPD and may help to address resistance mechanisms (Bond et al., 2020; Ottis et al., 2019), promote differing selectivity or kinetic profiles of degradation (Bondeson et al., 2018; Huang et al., 2018; Tong et al., 2020), and lead to cell-type or location-specific degradation.
While nimbolide has demonstrated success in TPD applications, its high molecular weight, modest chemical stability, and limited points for synthetic modification have prompted us to continually search for RNF114-based ligands for PROTAC development. Herein we realize the successful translation of the binding site of nimbolide into a simple, easily manipulated covalent small molecule. Because the N-terminal region of RNF114 that includes C8 is intrinsically disordered, structure-guided ligand discovery and optimization was not possible thus requiring unbiased approaches to ligand discovery. Activity-based protein profiling (ABPP)-enabled covalent ligand screening has been previously used to discover covalent recruiters against E3 ligases RNF4 and DCAF16 (Ward et al., 2019; Zhang et al., 2019) and has also facilitated ligand discovery against cysteines targeted by covalently-acting natural products (Grossman et al., 2017). Using this technique, we were able to discover fragments that could be used to replace nimbolide as the covalent E3 ligase recruitment module in fully functional degraders against several oncology targets.
Results
Covalent ligand screening against RNF114
To discover a fully synthetic covalent ligand that could access the same cysteine (C8) targeted by nimbolide on RNF114, we screened 318 cysteine-reactive chloroacetamide and acrylamide ligands in a gel-based competitive activity-based protein profiling (ABPP) assay, in which we competed ligands against binding of a rhodamine-functionalized cysteine-reactive iodoacetamide probe (IA-rhodamine) to pure RNF114 protein (Fig. 2a, 2b, Fig. S1, Table S1) (Bachovchin et al., 2010; Grossman et al., 2017; Ward et al., 2019). Through this screen, chloroacetamide EN219 emerged as the top hit, showing the greatest inhibition of IA-rhodamine binding to RNF114 (Fig. 2c). Dose-response studies showed that EN219 interacted with RNF114 with a 50 % inhibitory concentration (IC50) of 470 nM (Fig. 2d, 2e). EN219 also inhibited RNF114-mediated autoubiquitination and p21 ubiquitination in vitro, similarly to our previously observed findings with nimbolide (Fig. 2f). Given that EN219 possesses a stereogenic center, we also synthesized enantiomeric (R)- and (S)-EN219 derivatives (with >90% ee) (Mahé et al., 2010) and showed that these enantioenriched molecules showed similar potency for binding to RNF114 as the racemate we previously employed (Fig. S2a). We also synthesized a non-reactive acetamide analog of EN219, deschloro-EN219 (JNS 2–229), and showed that this compound no longer interacted with RNF114 (Fig. S2a). Mapping the sites of EN219 covalent modification on pure RNF114 protein by LC-MS/MS also confirmed C8 as the only site of modification (Fig. S2b). While EN219 represents an early lead compound within a drug discovery setting and recognize that through medicinal chemistry efforts, it could further be tuned, we nonetheless pursued further characterization of EN219 as a fully synthetic ligand for RNF114.
Figure 2. Covalent ligand screening against RNF114.

(a) Gel-based ABPP assay for screening covalent ligands against IA-rhodamine probe binding to pure RNF114 protein. Loss of fluorescence indicates covalent ligand binding to a cysteine on RNF114. (b) Quantified results from gel-based ABPP screen of 318 cysteine-reactive acrylamides and chloroacetamides against IA-rhodamine labeling of RNF114. DMSO vehicle or covalent ligands (50 μM) were pre-incubated with pure RNF114 protein (0.1 μg) for 30 min prior to addition of IA-rhodamine (100 nM) for 30 min at room temperature. Proteins were separated by SDS/PAGE and in-gel fluorescence was quantified. Raw gel-based ABPP data shown in Figure S1. 1. Structures of compounds screened can be found in Table S1. Data expressed as ligand/control ratio of in-gel fluorescent intensity. Shown in the inlay are the ligand/control ratios for the top 10 hits. (c) Structure of top hit EN219 with chloroacetamide cysteine-reactive warhead in red. (d) Dose-response of EN219 interaction with RNF114 by competitive gel-based ABPP. DMSO vehicle or covalent ligands were pre-incubated with pure RNF114 protein (0.1 μg) for 30 min prior to addition of IA-rhodamine (100 nM) for 30 min at room temperature. Proteins were separated by SDS/PAGE and in-gel fluorescence was quantified. Gels were also silver-stained as a loading control. (e) Percent inhibition of IA-rhodamine binding to RNF114 in (d) was quantified and 50 % inhibitory concentration (IC50) was determined to be 0.47 μM. (f) RNF114-mediated autoubiquitination and p21 ubiquitination in vitro. DMSO vehicle or EN219 (50 μM) was incubated with pure RNF114 for 30 min prior to addition of PBS or p21, ATP, and FLAG-ubiquitin (Ub) for 60 min. Proteins were separated by SDS/PAGE and blotted for FLAG. Ubiquitinated-RNF114/p21 is noted. (g) IsoTOP-ABPP analysis of EN219 in 231MFP breast cancer cells. 231MFP cells were treated in situ with DMSO vehicle or EN219 (1 μM) for 90 min. Control and treated cell lysates were labeled with IA-alkyne (100 μM) for 1 h, after which isotopically light (control) or heavy (EN219-treated) biotin-azide bearing a TEV tag was appended by CuAAC. Proteomes were mixed in a 1:1 ratio, probe-labeled proteins were enriched with avidin and digested with trypsin, and probe-modified peptides were eluted by TEV protease and analyzed by LC-MS/MS. Average light-to-heavy (control/EN219) ratios and adjusted p-values were quantified for probe-modified peptides that were present in two out of three biological replicates and plotted. Shown in red are protein and modified cysteine site for peptides that showed >2-fold control/EN219 ratio with adjusted p-value <0.05. Full data can be found in Table S2. (h) Structure of alkyne-functionalized EN219 probe. (i) EN219-alkyne pulldown of RNF114. 231MFP cells were treated with DMSO vehicle or EN219-alkyne (50 μM) for 90 min. Biotin-azide was appended to probe-labeled proteins by CuAAC and probe-labeled proteins were avidin enriched and blotted for RNF114 and negative control actin. An aliquot of input proteome from vehicle-and EN219-treated cell lysates was also subjected to blotting as an input control. Data shown in (e) are average values and individual replicate values. Data shown are from n=1 in (b) and n=3 for (d-f, i) biological replicate(s) per group. Gels shown in (d, i) are representative gels from n=3 biological replicates per group. This figure is related to Figures S1–2.
To characterize the proteome-wide interaction profile and the potential for engagement of RNF114 C8 by EN219 in cells, we next mapped the proteome-wide cysteine-reactivity of EN219 in situ in 231MFP breast cancer cells through several different approaches. First, we crudely assessed general cysteine-reactivity of EN219 compared to a previously identified promiscuous scout ligand YP 1–44 (Ward et al., 2019), and showed that EN219 did not broadly inhibit IA-rhodamine cysteine reactivity in 231MFP breast cancer cell lysate even at 250 μM, compared to YP 1–44 (Fig. S2c). We next assessed proteome-wide selectivity of EN219 by competitive isotopic tandem orthogonal proteolysis-ABPP (isoTOP-ABPP) using previously well-validated methods (Backus et al., 2016; Grossman et al., 2017; Spradlin et al., 2019; Wang et al., 2014; Weerapana et al., 2010). Cells were treated in situ with vehicle or EN219 and cells were subsequently lysed and labeled with an alkyne-functionalized iodoacetamide probe (IA-alkyne), followed by attachment of isotopically light or heavy, Tobacco Etch Virus (TEV)-cleavable biotin-azide enrichment handles through copper-catalyzed azide-alkyne cycloaddition (CuAAC). Probe-labeled proteins were enriched by avidin, digested with trypsin, and probe-modified peptides were eluted by TEV protease and analyzed by liquid chromatography-mass spectrometry (LC-MS/MS). Only 4 proteins showed >2-fold light versus heavy or control versus EN219-treated probe-modified peptide ratios with adjusted p-value <0.05 out of 686 probe-modified cysteines quantified. These proteins were RNF114 C8, TUBB1 C201, HSPD1 C442, and HIST1H3A C97, among which RNF114 was the only E3 ligase (Fig. 2g, Table S2).
To further investigate the selectivity of EN219 and to confirm EN219 engagement of RNF114 in cells, we also synthesized an alkyne-functionalized EN219 probe (EN219-alkyne) (Fig. 2h). Initially, we labeled 231MFP cells in situ with this EN219-alkyne probe and visualized labeled proteins by gel-based ABPP and investigated labeled protein bands that were competed by EN219 pre-treatment (Fig. S2d). This experiment showed about 8 potential EN219-alkyne-specific proteins that were competed by EN219 itself in cells, with several bands that were specific to EN219-alkyne (Fig. S2d). In some cases, we observed increased EN219-alkyne protein labeling of specific proteins in cells pre-treated with excess EN219, indicating increased labeling of EN219-alkyne specific off-targets when EN219-specific sites were occupied (Fig. S2d). To further elucidate these targets, we performed Tandem mass-tagging (TMT)-based quantitative proteomic profiling to identify proteins that were enriched by EN219-alkyne in situ labeling of 231MFP cells and were competed by EN219 in situ treatment (Fig. S2e, Table S3). While we did not identify RNF114 in this proteomics experiment, likely due to its relatively low abundance or other many EN219-alkyne specific targets that occluded detection of EN219-specific targets, we did identify 7 additional potential off-targets of EN219 that showed >3-fold competition with adjusted p-value <0.05 against EN219-alkyne labeling in cells (Fig. S2b; Table S3). We found 10 total targets that showed >3-fold enrichment with EN219-alkyne compared to DMSO-treated cells with adjusted p-value < 0.05 (Table S3). This number of potential EN219 off-targets would be consistent with the ~8 protein bands observed by gel-based ABPP in Fig. S2d. None of these 7–10 potential EN219 off-targets were E3 ligases. Collectively, our results suggested that EN219 was a moderately selective covalent ligand against C8 of RNF114. While we did not observe RNF114 by TMT-based proteomic experiments, RNF114 was clearly enriched from 231MFP cells treated with EN219-alkyne in situ compared to vehicle-treated controls after CuAAC-mediated appendage of biotin-azide and subsequent avidin-pulldown and RNF114 blotting (Fig. 2i).
EN219-Based RNF114 Recruiter in TPD Applications
We next tested whether EN219 could be exploited for TPD applications. We had previously demonstrated that nimbolide could be linked to the BET inhibitor ligand JQ1 to selectively degrade BRD4. Thus, we benchmarked our EN219 RNF114 recruiter by linking EN219 to JQ1 through three different linkers—ML 2–14, ML 2–31, and ML 2–32 with C4 alkyl, C7 alkyl, and polyethylene glycol (PEG4) linkers, respectively (Fig. 3a; Fig. S3a). Similar to effects seen with nimbolide-based degraders, ML 2–14 with the shortest linker showed the most robust degradation of BRD4 in 231MFP breast cancer cells compared to ML 2–31 and ML 2–32 with longer linkers, with 50 % degradation concentration (DC50) values of 36 and 14 nM for the long and short isoforms of BRD4, respectively (Fig. 3b, 3c; Fig. S3b, S3c) (Spradlin et al., 2019). EN219 treatment alone did not affect BRD4 levels, compared to ML 2–14 (Fig. S4a). This ML 2–14 mediated degradation was fully averted by pre-treating cells with the proteasome inhibitor bortezomib as well as the E1 activating enzyme inhibitor TAK-243 (Fig. 3d–3f, Fig. S4b).
Figure 3. EN219-based BRD4 degrader.

(a) Structure of ML 2–14, an EN219-based BRD4 degrader linking EN219 to BET inhibitor JQ1. (b) Degradation of BRD4 by ML 2–14. 231MFP cells were treated with DMSO vehicle or ML 2–14 for 8 h and the long and short isoforms of BRD4 and loading control actin were detected by Western blotting. (c) Percentage of BRD4 degradation quantified from (b) show 50 % degradation concentrations of 36 and 14 nM for the long and short isoforms of BRD4, respectively. (d) Proteasome-dependent degradation of BRD4 by ML 2–14. 231MFP cells were treated with DMSO vehicle or with proteasome inhibitor bortezomib (1 μM) 30 min prior to DMSO vehicle or ML 2–14 (100 nM) treatment for 8 h. BRD4 and loading control actin levels were detected by Western blotting. (e) Quantification of BRD4 levels from (d). (f) Ubiquitin activating enzyme (E1)-dependent degradation of BRD4 by ML 2–14. 231MFP cells were treated with DMSO vehicle or with E1 inhibitor TAK-243 (10 μM) 30 min prior to DMSO vehicle or ML 2–14 (100 nM) treatment for 8 h. BRD4 and loading control actin levels were detected by Western blotting. Quantification of blot can be found in Figure S4b. (g) BRD4 degradation by ML 2–14 is attenuated by nimbolide. 231MFP cells were treated with DMSO vehicle or with nimbolide (4 μM) 30 min prior to DMSO vehicle or ML 2–14 (100 nM) treatment for 8 h. (h) Quantification of BRD4 levels from (g). (i) Degradation of BRD4 in RNF114 wild-type (WT) or knockout (KO) HAP1 cells. RNF114 WT or KO HAP1 cells were treated with DMSO vehicle or ML 2–14 (1 μM) for 16 h and BRD4, RNF114, and loading control actin levels were detected by Western blotting. (j) Quantification of data from (i). (k) TMT-based quantitative proteomic data showing ML 2–14-mediated protein level changes in 231MFP cells. 231MFP cells were treated with DMSO vehicle or ML 2–14 for 8 h. Shown are average TMT ratios for all tryptic peptides identified with at least 2 unique peptides. Shown in red are proteins that showed >2-fold increased or decreased levels with ML 2–14 treatment with adjusted p-values <0.05. Data in (b-k) are from n=3 biologically independent replicates/group. Data shown in (c) are average and individual replicate values. Data shown in (e, h, j) are averages ± sem values and also individual biological replicate values. Blots shown in (b, d, f, g, i) are representative of n=3 biologically independent replicates/group. Statistical significance was calculated with unpaired two-tailed Student’s t-test and is expressed as *p<0.05 compared to vehicle-treated controls in (e, h, j) and #p<0.05 compared to ML 2–14-treated groups in (e, h) and compared to ML 2–14-treated RNF114 WT cells in (j). This figure is related to Figures S3–S4.
To further validate that ML 2–14 degradation of BRD4 was driven through RNF114, we showed that nimbolide pre-treatment completely attenuated BRD4 degradation by ML 2–14 in 231MFP breast cancer cells (Fig. 3g–3h). BRD4 degradation in HAP1 cells was also significantly attenuated in RNF114 knockout cells compared to wild-type counterparts in two independent experiments (Fig. 3i–3j, Fig. S4c–S4d). We note that these purchased RNF114 knockout cells showed residual RNF114 protein expression, likely indicating a mixed population of cells (Fig. 3i–3j). Furthermore, we showed that this loss of BRD4 protein levels was not due to transcriptional downregulation of BRD4 expression since ML 2–14 treatment in 231MFP cells did not alter BRD4 mRNA levels (Fig. S4e). TMT-based quantitative proteomic profiling of ML 2–14-mediated protein expression changes showed selective degradation of BRD3 and BRD4, but not BRD2. We also observed stabilization of two known or putative RNF114 substrates, including the tumor suppressor CDKN1A (p21) and CTGF (Fig. 3k, Table S4) (Han et al., 2013; Spradlin et al., 2019).
To further demonstrate the utility of our fully synthetic RNF114 recruiter EN219 in degrading other more challenging protein targets, we synthesized a degrader linking EN219 to the BCR-ABL inhibitor dasatinib, ML 2–23 and ML 2–22, bearing a longer PEG3 linker and a shorter C3 alkyl linker, respectively (Fig. 4a, Fig. S4f). For this particular target, ML 2–23 with the longer linker showed more robust degradation of BCR-ABL in K562 leukemia cells compared to ML 2–22, consistent with previously observed structure-activity relationships of nimbolide-based BCR-ABL degraders (Fig. 4b–4c, Fig. S4g) (Tong et al., 2020). Consistent with ML 2–23 engaging BCR-ABL in cells, we observed inhibition of CRKL phosphorylation, a downstream substrate of BCR-ABL signaling (Fig. 4b–4c). Interestingly, EN219 showed preferential degradation of BCR-ABL compared to c-ABL, compared to several previous BCR-ABL degraders utilizing cereblon or VHL recruiters that showed opposite selectivity (Fig. 4b–4c) (Burslem et al., 2019; Lai et al., 2016). This preferential degradation was also observed with the equivalent nimbolide-based degrader (Tong et al., 2020). We further showed that this loss of BCR-ABL and c-ABL was not due to transcriptional downregulation of these genes, since BCR-ABL mRNA levels remained unchanged and we observed a likely compensatory increase in c-ABL mRNA levels (Fig. S4h). Furthermore, we do not believe that the loss of BCR-ABL shown here is because of general cytotoxicity since ML 2–23 treatment at the timepoints used to assess BCR-ABL degradation only exerted relatively modest cell viability impairments in K562 cells; EN219 compromises K562 viability only at higher concentrations (Fig. S4i). While rescue experiments with proteasome inhibitors proved challenging due to the cytotoxicity of proteasome inhibitors at the long timepoints required for robust BCR-ABL degradation, we observed significant rescue of early-stage ML 2–23-mediated BCR-ABL and c-ABL degradation with pre-treatment of K562 cells with the proteasome inhibitor MG132 at a shorter time point (Fig. 4d, 4e).
Figure 4. EN219-based ABL degrader.

(a) Structure of ML 2–23, an EN219-based ABL degrader linking EN219 to ABL inhibitor dasatinib. (b) Degradation of BCR-ABL and c-ABL by ML 2–23. K562 cells were treated with DMSO vehicle or ML 2–23 for 16 h and BCR-ABL, c-ABL, p-CRKL, and loading control actin were detected by Western blotting. (c) Percentage of BCR-ABL, c-ABL, and p-CRKL expression quantified from (b). (d) Proteasome-dependent degradation of BCR-ABL and c-ABL by ML 2–23. K562 cells were treated with DMSO vehicle or with proteasome inhibitor MG132 (5 μM) 30 min prior to DMSO vehicle or ML 2–23 (1 μM) treatment for 12 h. BCR-ABL, c-ABL, and loading control actin levels were detected by Western blotting. (e) Quantification of BCR-ABL and c-ABL levels from (d). Data shown in (c, e) are averages ± sem values and also individual biological replicate values. Blots shown in (b, d) are representative of n=3 biological replicates/group. Statistical significance was calculated with unpaired two-tailed Student’s t-test and is expressed as *p<0.05 compared to vehicle-treated controls in (c, e) and #p<0.05 compared to ML 2–23-treated groups in (e). This figure is related to Figure S4.
Discussion
We previously discovered that the natural product nimbolide targets a predicted intrinsically disordered cysteine in RNF114 at a substrate recognition site (Spradlin et al., 2019). Here we report EN219 as a moderately selective covalent ligand that exploits this same binding modality. We show that EN219 can be linked to the BET inhibitor JQ1 to degrade BRD4 in a nimbolide-sensitive and proteasome- and RNF114-dependent manner. We further show that EN219 can be linked to the kinase inhibitor dasatinib to selectively degrade BCR-ABL over c-ABL in a proteasome-dependent manner in leukemia cells. These findings also further highlight that moderately selective cysteine-targeting ligands can still lead to robust protein degraders (Ward et al., 2019; Zhang et al., 2019). While EN219 represents a promising initial chemical scaffold for RNF114 recruitment, future medicinal chemistry efforts can further optimize the potency and metabolic stability of RNF114 recruiters; additionally, the facile synthesis of pyrazoline derivatives makes this a much more tractable synthetic problem when compared to the necessary chemistry to realize numerous nimbolide derivatives.
Overall, our results highlight the utility of chemoproteomic platforms for discovering chemical scaffolds that can be used as E3 ligase recruiters for TPD applications. Moreover, these studies further speak to the power of unbiased chemoproteomic approaches for identification of synthetic ligands mimicking the function of covalently acting natural products, particularly those in which structural binding information is unavailable (Nomura and Maimone, 2019).
Significance
We previously discovered that the natural product nimbolide could be used as a unique and covalent RNF114 E3 ubiquitin ligase recruiter for targeted protein degradation (TPD) applications. However, the adoption of this natural product in degraders has been hindered by the synthetic difficulty of using a complex natural product as a starting point for degrader synthesis. Here, using chemoproteomics-enabled covalent ligand screening approaches, we have discovered a more synthetically tractable covalent ligand EN219 that targets RNF114 and mimics nimbolide mode of action. We demonstrate that EN219, like nimbolide, can be incorporated into bifunctional degrader compounds to degrade neosubstrate proteins in an RNF114-dependent manner. Our study highlights the utility of covalent ligand screening in target-based screening endeavors to expand the toolbox of E3 ligase recruiters for TPD applications—an area of great important in drug discovery and chemical biology.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contract:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Daniel K. Nomura, dnomura@berkeley.edu
Materials Availability
Plasmids, compounds generated in this study will be made available upon reasonable request.
Data and Code Availability
Data generated in this study will be made available upon reasonable request. No code was developed for this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
The 231MFP cells were obtained from Prof. Benjamin Cravatt and were generated from explanted tumor xenografts of female MDA-MB-231 cells as previously described (Jessani et al., 2004). The 231MFP cells were cultured in L15 medium containing 10% FBS and maintained at 37 °C with 0% CO2. K562 chronic myeloid leukemia cell lines of female origin were purchased from ATCC. The K562 cells were cultured in Iscove’s Modified Dulbecco’s Medium containing 10% FBS and maintained at 37 °C with 5% CO2. HAP1 RNF114 wild-type and knockout cell lines of female origin were purchased from Horizon Discovery. The RNF114 knockout cell line was generated by CRISPR/Cas9 to contain a frameshift mutation in a coding exon of RNF114. HAP1 cells were grown in Iscove’s Modifed Dulbecco’s Medium in the presence of 10% FBS and penicillin/streptomycin.
METHOD DETAILS
Chemicals
Covalent ligands screened against RNF114 were purchased from Enamine LLC, including EN219. Structures of compounds screened can be found in Table S1. See Supplementary Information for synthetic methods and characterization for EN219 and degraders. Nimbolide was purchased from Cayman Chemicals (Item No. 19230).
Cell-based degrader assays
For assaying degrader activity, cells were seeded (500,000 cells for 231MFP and HAP1 cells, 1,000,000 for K562 cells) into a 6 cm tissue culture dish (Corning) in 2.0–2.5 mL of media and allowed to adhere overnight for 231MFP and HAP1 cells. The following morning, media was replaced with complete media containing the desired concentration of compound diluted from a 1,000 × stock in DMSO. For rescue studies, the cells were pre-treated with proteasome inhibitors, nimbolide, or E1 activating enzyme inhibitors 30 min prior to the addition of DMSO or degrader compounds. At the specified time point, cells were washed once with PBS on ice, before addition of 120 μL of lysis buffer (20 mM Tris-HCl at pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin) with Complete Protease Inhibitor Cocktail (Sigma) was added. The cells were incubated in lysis buffer for 5 min before scraping and transferring to microcentrifuge tubes. The lysates were then frozen at −80 °C or immediately processed for Western blotting. To prepare for Western blotting, the lysates were cleared with a 20,000g spin for 10 min and the resulting supernatant protein concentrations were quantified via BCA assay. The lysates were normalized by dilution with PBS to match the lowest concentration lysate, and the appropriate amount of 4 × Laemmli’s reducing buffer was added.
Gel-Based ABPP
Gel-Based ABPP methods were performed as previously described (Ward et al., 2019). Pure recombinant human RNF114 was purchased from Boston Biochem (K-220). RNF114 (0.25 μg) was diluted into 50 μL of PBS and 1 μL of either DMSO (vehicle) or covalently acting small molecule to achieve the desired concentration. After 30 min at room temperature, the samples were treated with 250 nM of tetramethylrhodamine-5-iodoacetamide dihydroiodide (IA-Rhodamine) (Setareh Biotech, 6222, prepared in anhydrous DMSO) for 1 h at room temperature. Incubations were quenched by diluting the incubation with 20 μL of 4 × reducing Laemmli SDS sample loading buffer (Alfa Aesar) and heated at 90 °C for 5 min. The samples were separated on precast 4–20% Criterion TGX gels (Bio-Rad Laboratories, Inc.). Fluorescent imaging was performed on a ChemiDoc MP (Bio-Rad Laboratories, Inc.). Inhibition of target labeling was assessed by densitometry using ImageJ.
EN219-alkyne probe labeling in situ and pulldown studies
Experiments were performed following an adaption of a previously described protocol (Thomas et al., 2017). The 231MFP cells were treated with either DMSO vehicle or 50 μM EN219-alkyne probe for 90 min. Cells were collected in PBS and lysed by sonication. For preparation of Western blotting samples, the lysate (1 mg of protein in 500 μl) was aliquoted per sample and then the following were added: 10 μl of 5 mM biotin picolylazide (900912 Sigma-Aldrich) and 50 μl of click reaction mix (three parts TBTA 5 mM TBTA in butanol:DMSO (4:1, v/v), one part 50 mM Cu(II)SO4 solution and one part 50 mM TCEP). Samples were incubated for 1 h at room temperature with gentle agitation. After CuAAC, proteomes were precipitated by centrifugation at 6,500 g and washed twice in ice-cold methanol (500 μl). The samples were spun in a prechilled (4 °C) centrifuge at 6,500 g for 4 min allowing for aspiration of excess methanol and subsequent reconstitution of protein pellet in 250 μl PBS containing 1.2% SDS by probe sonication. Then the proteome was denatured at 90 °C for 5 min, the insoluble components were precipitated by centrifugation at 6,500g and soluble proteome was diluted in 1.2 ml PBS (the final concentration of SDS in the sample was 0.2%) to a total volume of 1450 μl, with 50 μl reserved as input. Then 85 μl of prewashed 50% streptavidin agarose bead slurry was added to each sample and samples were incubated overnight at room temperature with gentle agitation. Supernatant was aspirated from each sample after spinning beads at 6,500 g for 2 min at room temperature. Beads were transferred to spin columns and washed three times with PBS. To elute, beads were boiled 5 min in 50 μl LDS sample buffer. Eluents were collected after centrifugation and analyzed by immunoblotting. The resulting samples were also analyzed as described below for TMT-based quantitative proteomic profiling.
LC–MS/MS analysis of pure RNF114 EN219 modification
Purified RNF114 (10 μg) in 50μl PBS was incubated 30 min at room temperature either with DMSO vehicle or EN219 (50 μM). The DMSO control was then treated with light iodoacetamide while the compound treated sample was incubated with heavy iodoacetamide for 1h each at room temperature (200 μM final concentration, Sigma-Aldrich, 721328). The samples were precipitated by addition of 12.5 μl of 100% (w/v) trichloroacetic acid and the treated and control groups were combined pairwise, before cooling to −80 °C for 1h. The combined sample was then spun for at max speed for 10 min at 4 °C, supernatant was carefully removed and the sample was washed with ice-cold 0.01 M HCl/90% acetone solution. The pellet was resuspended in 4M urea containing 0.1% Protease Max (Promega Corp. V2071) and diluted in 40 mM ammonium bicarbonate buffer. The samples were reduced with 10 mM TCEP at 60 °C for 30min. The sample was then diluted 50% with PBS before sequencing grade trypsin (1 μg per sample, Promega Corp, V5111) was added for an overnight incubation at 37 °C. The next day, the sample was centrifuged at 13,200 rpm for 30 min. The supernatant was transferred to a new tube and acidified to a final concentration of 5% formic acid and stored at −80 °C until mass spectrometry analysis.
IsoTOP-ABPP
IsoTOP-ABPP studies were done as previously reported (Spradlin et al., 2019). Cells were lysed by probe sonication in PBS and protein concentrations were measured by BCA assay35. For in situ experiments, cells were treated for 90 min with either DMSO vehicle or covalently acting small molecule (from 1,000× DMSO stock) before cell collection and lysis. Proteomes were subsequently labeled with N-5-Hexyn-1-yl-2-iodoacetamide (IA-alkyne) labeling (100 μM) for 1 h at room temperature. CuAAC was used by sequential addition of tris(2-carboxyethyl) phosphine (1 mM, Sigma), tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (34 μM, Sigma), copper(II) sμlfate (1 mM, Sigma) and biotin-linker-azide—the linker functionalized with a tobacco etch virus (TEV) protease recognition sequence as well as an isotopically light or heavy valine for treatment of control or treated proteome, respectively. After CuAAC, proteomes were precipitated by centrifugation at 6,500g, washed in ice-cold methanol, combined in a 1:1 control:treated ratio, washed again, then denatured and resolubilized by heating in 1.2% SDS–PBS to 80 °C for 5 min. Insoluble components were precipitated by centrifugation at 6,500g and soluble proteome was diluted in 5 ml 0.2% SDS–PBS. Labeled proteins were bound to avidin-agarose beads (170 μl resuspended beads per sample, Thermo Pierce) while rotating overnight at 4 °C. Bead-linked proteins were enriched by washing three times each in PBS and water, then resuspended in 6 M urea and PBS (Sigma), and reduced in TCEP (1 mM, Sigma), alkylated with iodoacetamide (18 mM, Sigma), before being washed and resuspended in 2 M urea and trypsinized overnight with 0.5 μg μl−1 sequencing grade trypsin (Promega). Tryptic peptides were eluted off. Beads were washed three times each in PBS and water, washed in TEV buffer solution (water, TEV buffer, 100 μM dithiothreitol) and resuspended in buffer with Ac-TEV protease and incubated overnight. Peptides were diluted in water and acidified with formic acid (1.2 M, Spectrum) and prepared for analysis.
Mass spectrometry analysis
Total peptides from TEV protease digestion for isoTOP-ABPP or tryptic peptides for mapping EN219 site of modification on pure RNF114 were pressure loaded onto 250 mm tubing packed with Aqua C18 reverse phase resin (Phenomenex no. 04A-4299), which was previously equilibrated on an Agilent 600 series high-performance liquid chromatograph using the gradient from 100% buffer A to 100% buffer B over 10 min, followed by a 5 min wash with 100% buffer B and a 5 min wash with 100% buffer A. The samples were then attached using a MicroTee PEEK 360 μm fitting (Thermo Fisher Scientific no. p-888) to a 13 cm laser pμlled column packed with 10 cm Aqua C18 reverse-phase resin and 3 cm of strong-cation exchange resin for isoTOP-ABPP studies. Samples were analyzed using an Q Exactive Plus mass spectrometer (Thermo Fisher Scientific) using a five-step Mμltidimensional Protein Identification Technology (MudPIT) program, using 0, 25, 50, 80 and 100% salt bumps of 500 mM aqueous ammonium acetate and using a gradient of 5–55% buffer B in buffer A (buffer A: 95:5 water:acetonitrile, 0.1% formic acid; buffer B 80:20 acetonitrile:water, 0.1% formic acid). Data were collected in datadependent acquisition mode with dynamic exclusion enabled (60 s). One full mass spectrometry (MS1) scan (400–1,800 mass-to-charge ratio (m/z)) was followed by 15 MS2 scans of the nth most abundant ions. Heated capillary temperature was set to 200 °C and the nanospray voltage was set to 2.75 kV, as previously described.
Data were extracted in the form of MS1 and MS2 files using Raw Extractor v.1.9.9.2 (Scripps Research Institute) and searched against the Uniprot human database using ProLuCID search methodology in IP2 v.3 (Integrated Proteomics Applications, Inc.) (Xu et al., 2015a). Probe-modified cysteine residues were searched with a static modification for carboxyaminomethylation (+57.02146) and up to two differential modifications for methionine oxidation and either the light or heavy TEV tags (+464.28596 or +470.29977, respectively) or the mass of the EN219 adduct. Peptides were required to be fully tryptic peptides and to contain the TEV modification. ProLUCID data was filtered through DTASelect to achieve a peptide false-positive rate below 5%. Only those probe-modified peptides that were evident across two out of three biological replicates were interpreted for their isotopic light to heavy ratios. Light versus heavy isotopic probe-modified peptide ratios are calculated by taking the mean of the ratios of each replicate paired light vs. heavy precursor abundance for all peptide spectral matches (PSM) associated with a peptide. The paired abundances were also used to calculate a paired sample t-test p-value in an effort to estimate constancy within paired abundances and significance in change between treatment and control. P-values were corrected using the Benjamini/Hochberg method.
TMT-based quantitative proteomic profiling
TMT proteomic profiling was performed as previously described (Spradlin et al., 2019).
RNF114 ubiquitination assay
Recombinant Myc-Flag-RNF114 proteins were purchased from Origene (Origene Technologies Inc., TP309752) or were purified as described previously(Spradlin et al., 2019). For in vitro auto-ubiquitination assay, 0.2 μg of RNF114 in 25 μl of TBS was pre-incubated with DMSO vehicle or the covalently acting compound for 30 min at room temperature. Subsequently, 0.1 μg of UBE1 (Boston Biochem. Inc., E-305), 0.1 μg UBE2D1 (Boston Bichem. Inc., e2–615), 5 μg of Flag-ubiquitin (Boston Bichem. Inc., u-120) in a total volume of 25 μl Tris buffer containing 2 mM ATP, 10 mM DTT and 10 mM MgCl2 were added to achieve a final volume of 50 μl. For substrate-protein ubiquitination assays, 0.1 μg of p21 (Origene) was added at this stage. The mixture was incubated at 37 °C with agitation for 1.5 h. Then, 20 μl of Laemmli’s buffer was added to quench the reaction and proteins were analyzed by Western blot assay.
Western blotting
Antibodies to RNF114 (Millipore Sigma, HPA021184), c-ABL (Santa Crus, 24–11), p-CRKL (Tyr207, Cell Signaling Technology, 3181), GAPDH (Proteintech Group Inc., 60004–1-Ig), BRD4 (Abcam plc, Ab128874), and beta-actin (Proteintech Group Inc., 6609–1-Ig) were obtained from various commercial sources and dilutions were prepared per the recommended manufacturers’ procedures. Proteins were resolved by SDS–PAGE and transferred to nitrocellulose membranes using the iBlot system (Invitrogen). Blots were blocked with 5% BSA in Tris-buffered saline containing Tween 20 (TBST) solution for 1 h at room temperature, washed in TBST and probed with primary antibody diluted in diluent, as recommended by the manufacturer, overnight at 4 °C. Following washes with TBST, the blots were incubated in the dark with secondary antibodies purchased from Ly-Cor and used at 1:10,000 dilution in 5% BSA in TBST at room temperature. Blots were visualized using an Odyssey Li-Cor scanner after additional washes. If additional primary antibody incubations were required, the membrane was stripped using ReBlot Plus Strong Antibody Stripping Solution (EMD Millipore, 2504), washed and blocked again before being re-incubated with primary antibody. Blots were quantified and normalized to loading controls using Image J.
Synthetic Methods and Characterization
(E)-tert-butyl (4-(4-(3-(4-bromophenyl)-3-oxoprop-1-en-1-yl)phenoxy)butyl)carbamate (2):
To a solution of enone 1 (226 mg, 0.75 mmol) in anhydrous DMF (8 mL) was added 4-(Boc-amino)butyl bromide (376 mg, 1.5 mmol) and K2CO3 (414 mg, 3 mmol) and the reaction mixture was stirred at 60 °C for 5 h under an atmosphere of nitrogen. Upon cooling, the inorganic salts were filtered off, the solution was diluted with EtOAc, washed with water, and the volatiles removed in vacuo. The crude material was purified by silica gel chromatography (25% EtOAc/hexanes) to yield 310 mg (87%) of 2: 1H NMR (400 MHz, CDCl3): δ 7.96 – 7.88 (m, 2H), 7.83 (d, J = 15.6 Hz, 1H), 7.72 – 7.59 (m, 4H), 7.39 (d, J = 15.6 Hz, 1H), 7.01 – 6.91 (m, 2H), 4.66 (s, 1H), 4.07 (t, J = 6.2 Hz, 2H), 3.25 (q, J = 6.8 Hz, 2H), 1.94 – 1.82 (m, 2H), 1.79 – 1.69 (m, 2H), 1.49 (s, 9H).
tert-butyl (4-(4-(3-(4-bromophenyl)-1-(2-chloroacetyl)-4,5-dihydro-1H-pyrazol-5-yl)phenoxy)butyl)carbamate (3):
To a solution of enone 2 (550 mg, 1.16 mmol) in EtOH was added hydrazine monohydrate (116 mg, 2.32 mmol) and the reaction mixture heated at 80 °C for 5 h under a nitrogen atmosphere. The reaction was cooled to room temperature, diluted with water, and extracted with DCM. The combined organic phase was dried over anhydrous magnesium sulfate and then concentrated in vacuo to ~ 5 mL. [NOTE: All of the workup procedures should be performed quickly (<1.5 h total time) and the rotovap bath kept cool as the crude product can easily undergo autooxidation]. To the concentrated DCM solution was immediately added chloroacetyl chloride (157 mg, 1.39 mmol) and triethylamine (152 mg, 1.5 mmol) at 0°C. The reaction mixture was stirred in an ice bath for 30 minutes, then warmed to room temperature and stirred overnight under N2. Upon completion of the reaction, the reaction mixture was diluted with EtOAc, washed with brine, and concentrated in vacuo. The crude was purified by silica gel column chromatography (40% EtOAc/hexanes) to yield 381 mg (58%) of 3: 1H NMR (400 MHz, CDCl3): δ 7.60 (q, J = 8.8 Hz, 4H), 7.19 – 7.10 (m, 2H), 6.87 – 6.77 (m, 2H), 5.55 (dd, J = 11.7, 4.7 Hz, 1H), 4.55 (s, 2H), 3.94 (t, J = 6.2 Hz, 2H), 3.74 (dd, J = 17.8, 11.7 Hz, 1H), 3.24 – 3.11 (m, 3H), 1.85 – 1.75 (m, 2H), 1.65 (p, J = 7.1 Hz, 2H), 1.44 (s, 9H).
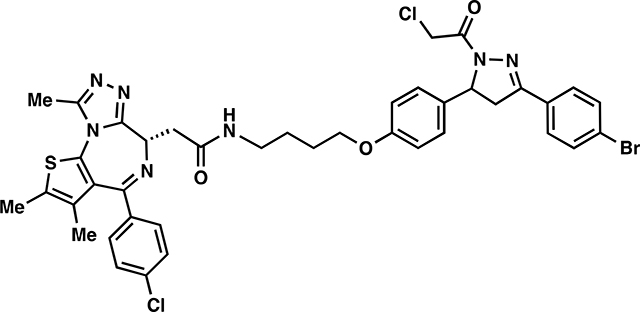
ML 2–14:
i. Chloroacetamide 3 (185 mg, 0.327 mmol) was dissolved in DCM (2.5 mL) and trifluoroacetic acid (2.5 mL) was added dropwise slowly over 20 minutes. After an additional 20 minutes of stirring, the solvent was removed in in vacuo. To remove residual TFA, the crude material was dissolved in 3mL of DCM and concentrated in vacuo and this process repeated two additional times. The deprotected amine was used directly in the next step without purification.
ii. The resulting TFA salt was dissolved in DCM (8 mL) and JQ1-acid (157 mg, 0.4 mmol), 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) (202 mg, 0.530 mmol), and N,N-Diisopropylethylamine (168 mg, 1.31 mmol) were added. The mixture was stirred overnight with monitoring by TLC (5% MeOH in DCM, 100% EtOAc). Upon completion, the reaction mixture was concentrated in vacuo and directly purified by silica gel flash column chromatography (1–5% MeOH/DCM). The eluted fractions were insufficiently pure and those containing product were combined, concentrated and purified again by flash silica chromatography (100–0% EtOAc/DCM followed by 0–5% MeOH/DCM) to afford 88.4 mg (32%) of ML 2–14: 1H NMR (400 MHz, CDCl3): δ 7.66 – 7.46 (m, 4H), 7.39 (d, J = 8.2 Hz, 2H), 7.29 (d, J = 8.3 Hz, 2H), 7.13 (d, J = 8.3 Hz, 2H), 6.86 (t, J = 5.9 Hz, 1H), 6.80 (d, J = 8.3 Hz, 2H), 5.54 (dt, J = 11.7, 4.1 Hz, 1H), 4.63 (t, J = 7.0 Hz, 1H), 4.54 (s, 2H), 3.90 (t, J = 6.1 Hz, 2H), 3.72 (dd, J = 17.8, 11.7 Hz, 1H), 3.54 (dd, J = 14.3, 7.5 Hz, 1H), 3.33 (ddt, J = 30.5, 13.3, 6.8 Hz, 3H), 3.17 (dd, J = 17.9, 4.7 Hz, 1H), 2.64 (s, 3H), 2.39 (s, 3H), 1.84 – 1.72 (m, 2H), 1.69 (d, J = 7.4 Hz, 2H), 1.65 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 170.6, 164.1, 161.4, 158.8, 155.8, 154.5, 151.4, 150.0, 136.9, 136.7, 132.9, 132.3, 132.2, 131.1, 131.0, 130.6, 130.0, 129.9, 128.9, 128.4, 127.2, 125.3, 120.7, 115.1, 67.6, 60.4, 54.7, 42.3, 42.1, 39.7, 39.4, 31.7, 26.7, 26.4, 22.8, 14.5, 14.3, 13.2, 12.0, 11.6. HRMS (ESI): calcd. C40H39BrCl2N7O3S ([M+H]+): m/z 846.1390, found: 846.1399.
(E)-tert-butyl 2-(4-(3-(4-bromophenyl)-3-oxoprop-1-en-1-yl)phenoxy)acetate (4):
To a solution of 1 (153 mg, 0.50 mmol) in anhydrous DMF (8 mL) was added tert-Butyl bromoacetate (146 mg, 0.75 mmol) and K2CO3 (138 mg, 1 mmol) and the reaction mixture was heated to 60 °C for 5 hours under an atmosphere of nitrogen. Upon cooling, the inorganic salts were filtered off, the solution was diluted with EtOAc, washed with water, and concentrated in vacuo. The crude was purified by silica gel chromatography (25% EtOAc/hexanes) to yield 150 mg (72%) of 4: 1H NMR (400 MHz, CDCl3): δ 7.97 – 7.89 (m, 2H), 7.82 (d, J = 15.6 Hz, 1H), 7.73 – 7.60 (m, 4H), 7.41 (d, J = 15.6 Hz, 1H), 7.01 – 6.93 (m, 2H), 4.61 (s, 2H), 1.54 (s, 9H).
tert-butyl 2-(4-(3-(4-bromophenyl)-1-(2-chloroacetyl)-4,5-dihydro-1H-pyrazol-5-yl)phenoxy)acetate (5):
To a solution of 4 (417 mg, 1.00 mmol) in EtOH was added hydrazine monohydrate (250 mg, 5.00 mmol) and the reaction mixture was heated at 80 °C for 5 hours under an atmosphere of nitrogen. The reaction was cooled to room temperature, diluted with water, and extracted with DCM. The combined organic phase was dried over anhydrous magnesium sulfate and then concentrated in vacuo to ~ 5 mL [NOTE: All of the workup procedures should be performed quickly (<1.5 h total time) and the rotovap bath kept cool as the crude product can easily undergo autooxidation]. To the concentrated DCM solution was immediately added chloroacetyl chloride (169 mg, 1.50 mmol) and triethylamine (303 mg, 3.00 mmol) at 0 °C under an atmosphere of nitrogen. The reaction was maintained at this temperature for 30 minutes and then warmed to room temperature and stirred overnight under N2. The reaction mixture was diluted with EtOAc, washed with brine, and concentrated in vacuo. The crude material was purified by silica gel chromatography (35 – 40 % EtOAc/hexanes) to yield 140 mg (28%) of 5: 1H NMR (400 MHz, CDCl3): δ 7.69 – 7.58 (m, 4H), 7.24 – 7.16 (m, 2H), 6.93 – 6.84 (m, 2H), 5.60 (dd, J = 11.7, 4.6 Hz, 1H), 4.58 (d, J = 1.6 Hz, 2H), 4.52 (s, 2H), 3.78 (dd, J = 17.8, 11.8 Hz, 1H), 3.23 (dd, J = 17.8, 4.7 Hz, 1H), 1.52 (s, 9H).
ML 2–22:
i. Compound 5 (15.2 mg, 0.030 mmol) was dissolved in DCM (1.5 mL) and trifluoroacetic acid (0.5 mL) added slowly over the course of 20 minutes. After stirring for an additional 20 minutes, the solvent was removed in vacuo. To remove residual TFA, the crude material was dissolved in 3mL of DCM and concentrated in vacuo and this process repeated two additional times. The crude material was used directly in the next step.
ii. The aforementioned crude carboxylic acid was dissolved in 2 mL DCM and amine 6 (15.0 mg, 0.030 mmol), 1- [Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) (17.1 mg, 0.045 mmol), and N,N-Diisopropylethylamine (193.5 mg, 1.50 mmol) were added (Tong et al., 2020). The reaction was stirred overnight monitoring by TLC (20% Methanol in DCM). Upon completion, the reaction mixture was diluted with EtOAc, washed with brine, and concentrated in vacuo. The crude material was purified by silica gel flash column chromatography (1–5% MeOH/DCM). The eluted fractions were insufficiently pure and those containing product were combined, concentrated and purified again by flash silica chromatography (100–0% EtOAc/DCM followed by 10–35% MeOH/DCM) to afford 17.1 mg (60%) of ML 2–22: 1H NMR (600 MHz, DMSO-d6) δ 11.47 (s, 1H), 9.88 (s, 1H), 8.23 (s, 1H), 8.11 (d, J = 18.2 Hz, 1H), 7.78 – 7.71 (m, 2H), 7.71 – 7.64 (m, 2H), 7.40 (dd, J = 7.8, 1.7 Hz, 1H), 7.34 – 7.23 (m, 2H), 7.18 – 7.10 (m, 2H), 6.97 – 6.88 (m, 2H), 6.06 (s, 1H), 5.54 (dd, J = 11.7, 4.7 Hz, 1H), 4.78 – 4.64 (m, 2H), 4.46 (s, 2H), 3.86 (dd, J = 18.2, 11.8 Hz, 1H), 3.50 (s, 4H), 3.31 – 3.08 (m, 4H), 2.41 (s, 6H), 2.31 (s, 2H), 2.25 (s, 3H), 1.64 (s, 2H); 13C NMR (151 MHz, DMSO) δ 167.0, 164.7, 162.8, 162.1, 161.9, 159.5, 156.6, 156.5, 154.3, 140.4, 138.4, 133.8, 133.1, 132.0, 131.4, 129.5, 129.0, 128.6, 128.4, 127.7, 126.5, 125.3, 123.7, 114.4, 82.2, 66.7, 59.2, 55.1, 54.5, 51.9, 43.1, 42.0, 41.4, 36.5, 29.5, 28.0, 25.1, 23.0, 22.0, 17.9, 13.5, 10.5; HRMS (ESI): calcd. C42H44BrCl2N10O4S ([M+H]+): m/z 933.1823, found: 933.1814.
ML 2–23:
i. Compound 5 (16.2 mg, 0.032 mmol) was dissolved in DCM (1.5 mL) and trifluoroacetic acid (0.5 mL) added slowly over the course of 20 minutes. After stirring for an additional 20 minutes, the solvent was removed in vacuo, To remove residual TFA, the crude material was dissolved in 3mL of DCM and concentrated in vacuo and this process repeated two additional times. The crude material was used directly in the next step.
ii. The aforementioned crude carboxylic acid was dissolved in DCM (2 mL) and amine 7 (19.8 mg, 0.032 mmol), 1- [Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) (18.2 mg, 0.048 mmol), and N,N-Diisopropylethylamine (206.0 mg, 1.60 mmol) were added (Tong et al., 2020). The reaction mixture was stirred overnight with monitoring by TLC (20% Methanol in DCM). Upon completion, the reaction mixture was diluted with EtOAc, washed with brine, and concentrated in vacuo. The crude material was purified by silica gel flash column chromatography (1–5% MeOH/DCM). The eluted fractions were insufficiently pure and those containing product were combined, concentrated and purified again by flash silica chromatography (100–0% EtOAc/DCM followed by 10–35% MeOH/DCM) to afford 20.1 mg (59%) of ML 2–23: 1H NMR (400 MHz, Methanol-d4) δ 8.16 (s, 1H), 7.77 – 7.71 (m, 2H), 7.61 (dd, J = 8.8, 2.3 Hz, 2H), 7.40 – 7.34 (m, 1H), 7.31 – 7.15 (m, 4H), 7.00 – 6.90 (m, 2H), 6.00 (s, 1H), 5.57 (dd, J = 11.7, 4.7 Hz, 1H), 4.73 (d, J = 13.8 Hz, 1H), 4.60 (d, J = 13.9 Hz, 1H), 4.52 (s, 2H), 4.12 (q, J = 7.1 Hz, 1H), 3.88 (dd, J = 18.2, 11.7 Hz, 1H), 3.62 (tdd, J = 13.9, 10.2, 6.7 Hz, 16H), 3.48 (t, J = 5.5 Hz, 2H), 3.19 (dd, J = 18.1, 4.8 Hz, 1H), 2.66 (dd, J = 11.8, 5.7 Hz, 6H), 2.47 (s, 3H), 2.34 (s, 3H); 13C NMR (151 MHz, DMSO) δ 167.2, 164.6, 162.8, 161.9, 159.5, 156.6, 154.4, 140.4, 137.6, 133.3, 133.1, 132.0, 131.4, 131.3, 129.1, 128.6, 128.4, 127.3, 126.5, 114.4, 69.3, 69.2, 68.4, 66.7, 59.3, 53.6, 42.0, 41.4, 41.3, 37.8, 35.3, 30.3, 29.5, 27.4, 25.1, 23.0, 22.0, 17.9, 17.6, 15.6, 13.5, 11.9, 10.5; HRMS (ESI): calcd. C47H54BrCl2N10O4S ([M+H]+): m/z 1051.2453, found: 1051.2445
EN219-alkyne:
i. Compound 5 (17.6 mg, 0.035 mmol) was dissolved in DCM (1.5 mL) and trifluoroacetic acid (0.5 mL) added slowly over the course of 20 minutes. After stirring for an additional 20 minutes, the solvent was removed in vacuo. To remove residual TFA, the crude material was dissolved in 3mL of DCM and concentrated in vacuo and this process repeated two additional times. The crude material was used directly in the next step.
ii. The aforementioned crude carboxylic acid was dissolved in DCM (2 mL) and propargylamine (2.3 mg, 0.042 mmol), 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) (19.8 mg, 0.052 mmol), and N,N-Diisopropylethylamine (223.9 mg, 1.750 mmol) were added. The reaction mixture was stirred overnight with monitoring by TLC (60% EtOAc in hexane). Upon completion, the reaction mixture was diluted with DCM, washed with brine, and concentrated in vacuo. The crude material was purified by silica gel chromatography (25 – 70 % EtOAc/hexanes) to yield 14.6 mg (88%) of EN219-alkyne: 1H NMR (400 MHz, CDCl3): δ 7.68 – 7.60 (m, 4H), 7.27 – 7.21 (m, 2H), 6.95 – 6.89 (m, 2H), 6.81 (s, 1H), 5.60 (dd, J = 11.8, 4.8 Hz, 1H), 4.64 – 4.54 (m, 2H), 4.52 (s, 2H), 4.18 (dd, J = 5.6, 2.6 Hz, 2H), 3.81 (dd, J = 17.8, 11.8 Hz, 1H), 3.22 (dd, J = 17.9, 4.8 Hz, 1H), 2.31 (t, J = 2.6 Hz, 1H); 13C NMR (101 MHz, CDCl3): δ 167.7, 163.9, 156.7, 154.3, 134.5, 132.1, 129.6, 128.1, 127.4, 125.2, 115.1, 78.9, 71.8, 67.3, 60.3, 60.0, 42.0, 41.9, 29.6, 28.7, 21.0, 14.1; HRMS (ESI): calcd. C22H19BrCl1N3O3Na ([M+Na]+): m/z 510.0191, found: 510.0186.
(E)-tert-butyl (7-(4-(3-(4-bromophenyl)-3-oxoprop-1-en-1-yl)phenoxy)heptyl)carbamate (8):
To a solution of 1 (153 mg, 0.50 mmol) in anhydrous DMF (8 mL) was added N-Boc-7-bromoheptan-1-amine (300 mg, 1.00 mmol) and K2CO3 (276 mg, 2.00 mmol). The reaction mixture was heated at 60 °C for 5 hours under an atmosphere of nitrogen. Upon cooling to room temperature, the inorganic salts were filtered off, the solution was diluted with EtOAc, washed with water, and the volatiles removed in vacuo. The crude material was purified by silica gel chromatography (25% EtOAc/hexanes) to yield 238 mg (92%) of 8: 1H NMR (400 MHz, CDCl3): δ 7.97 – 7.89 (m, 2H), 7.83 (d, J = 15.6 Hz, 1H), 7.73 – 7.59 (m, 4H), 7.39 (d, J = 15.6 Hz, 1H), 7.00 – 6.92 (m, 2H), 4.55 (s, 1H), 4.04 (t, J = 6.5 Hz, 2H), 3.16 (q, J = 6.8 Hz, 2H), 1.94 – 1.76 (m, 2H), 1.57 – 1.50 (m, 4H), 1.49 (s, 9H), 1.44 – 1.37 (m, 4H).
tert-butyl (7-(4-(3-(4-bromophenyl)-1-(2-chloroacetyl)-4,5-dihydro-1H-pyrazol-5-yl)phenoxy)heptyl)carbamate (9):
To a solution of 8 (223 mg, 0.43 mmol) in EtOH was added hydrazine monohydrate (43 mg, 0.86 mmol) and the reaction mixture heated at 80 °C for 3 h under an atmosphere of nitrogen. The reaction mixture was cooled to room temperature, diluted with water and extracted with DCM. The combined organic phase was dried over anhydrous magnesium sulfate and then concentrated in vacuo to ~ 5 mL [NOTE: All of the workup procedures should be performed quickly (<1.5 h total time) and the rotovap bath kept cool as the crude product can easily undergo autooxidation]. To the concentrated solution was quickly added chloroacetyl chloride (58 mg, 0.52 mmol) and triethylamine (57 mg, 0.56 mmol) at 0 °C. The reaction mixture was stirred in an ice bath for 30 minutes, then warmed to room temperature and stirred overnight under N2. The reaction mixture was diluted with EtOAc, washed with brine, and concentrated in vacuo. The crude was purified by silica gel chromatography (25 – 40 % EtOAc/hexanes) to yield 104 mg (40%) of 9: 1H NMR (400 MHz, CDCl3): δ 7.69 – 7.53 (m, 4H), 7.23 – 7.12 (m, 2H), 6.89 – 6.82 (m, 2H), 5.57 (dd, J = 11.7, 4.7 Hz, 1H), 4.57 (s, 2H), 3.93 (t, J = 6.5 Hz, 2H), 3.76 (dd, J = 17.8, 11.7 Hz, 1H), 3.21 (dd, J = 17.9, 4.7 Hz, 1H), 3.13 (q, J = 6.8 Hz, 2H), 1.85 – 1.70 (m, 2H), 1.55 – 1.46 (m, 4H), 1.47 (s, 9H), 1.41 – 1.32 (m, 4H).
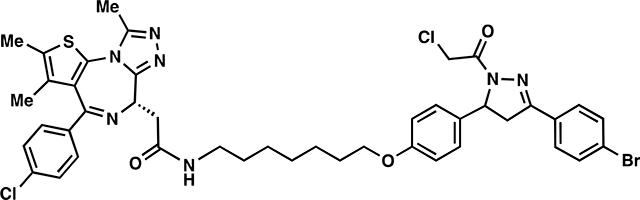
ML 2–31:
i. Compound 9 (104 mg, 0.171 mmol) was dissolved in DCM (2.5 mL) and TFA (2.5 mL) was added slowly over 20 minutes followed by stirring for an additional 20 minutes at which point the reaction mixture was concentrated in vacuo. To remove residual TFA, the crude material was dissolved in 3mL of DCM and concentrated in vacuo and this process repeated two additional times. The crude material was used without further purification for amide coupling.
ii. The aforementioned TFA salt was dissolved in DCM (8 mL) and JQ1-acid (82 mg, 0.205 mmol), 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) (97.5 mg, 0.257 mmol), and N,N-Diisopropylethylamine (552 mg, 4.28 mmol) were added. The reaction mixture was stirred overnight with monitoring by TLC (5% MeOH in DCM, 100% EtOAc). Upon completion, the reaction mixture was directly concentrated in vacuo and the crude material purified by silica gel flash chromatography (1–5% MeOH/DCM). The eluted fractions were insufficiently pure and those containing product were combined, concentrated and purified again by flash silica chromatography (100–0% EtOAc/DCM followed by 0–5% MeOH/DCM) to afford 66 mg (43%) of ML 2–31: 1H NMR (400 MHz, CDCl3): δ 7.68 – 7.57 (m, 4H), 7.48 – 7.39 (m, 2H), 7.39 – 7.32 (m, 2H), 7.22 – 7.11 (m, 2H), 6.90 – 6.81 (m, 2H), 6.67 (t, J = 5.8 Hz, 1H), 5.58 (ddd, J = 11.8, 4.7, 2.1 Hz, 1H), 4.69 – 4.61 (m, 1H), 4.59 (d, J = 2.5 Hz, 2H), 3.93 (t, J = 6.4 Hz, 2H), 3.84 – 3.67 (m, 2H), 3.56 (dd, J = 14.3, 7.4 Hz, 1H), 3.42 – 3.30 (m, 2H), 3.28 – 3.16 (m, 3H), 2.70 (s, 3H), 2.47 – 2.35 (m, 3H), 1.82 – 1.71 (m, 2H), 1.71 – 1.67 (m, 3H), 1.56 (p, J = 7.0 Hz, 2H), 1.45 – 1.43 (m, 4H); 13C NMR (151 MHz, CDCl3) δ 170.4, 163.9, 158.9, 155.7, 154.6, 149.9, 136.8, 136.6, 132.6, 132.1, 132.1, 130.9, 130.9, 130.5, 129.8, 128.7, 128.2, 127.0, 125.2, 115.1, 115.1, 114.9, 67.9, 60.2, 54.9, 54.5, 43.0, 42.2, 42.1, 39.6, 39.4, 29.7, 29.4, 29.1, 29.0, 26.8, 25.9, 18.5, 17.2, 14.4, 13.1, 12.4, 11.8; HRMS (ESI): calcd. C43H45BrCl2N7O3S ([M+H]+): m/z 888.1865, found: 888.1858.
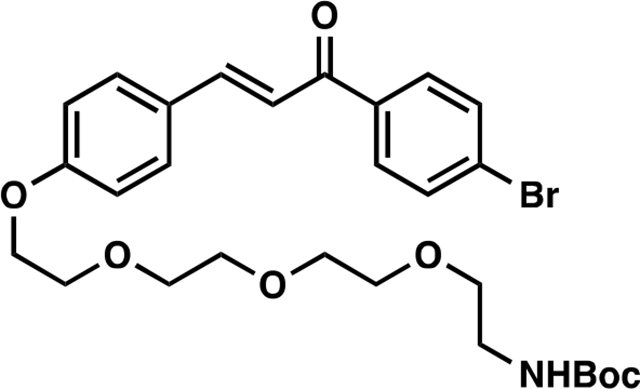
(E)-tert-butyl (2-(2-(2-(2-(4-(3-(4-bromophenyl)-3-oxoprop-1-en-1-yl)phenoxy)ethoxy)ethoxy)ethoxy)ethyl) carbamate (10):
To a solution of 1 (153 mg, 0.50 mmol) in anhydrous DMF (8 mL) was added tert-Butyl (2-(2-(2-(2-bromoethoxy)ethoxy)ethoxy)ethyl)carbamate (344 mg, 0.97 mmol) and K2CO3 (276 mg, 2.00 mmol), and the reaction mixture was stirred at 60 °C for 5 hours under an atmosphere of nitrogen. Upon cooling, the inorganic salts were filtered off, the solution was diluted with EtOAc, washed with water, and the volatiles removed in vacuo. The crude was purified by silica gel chromatography (50% EtOAc/hexanes) to yield 251 mg (88%) of 10: 1H NMR (400 MHz, CDCl3): δ 7.97 – 7.89 (m, 2H), 7.89 – 7.78 (m, 1H), 7.72 – 7.55 (m, 4H), 7.40 (d, J = 15.6 Hz, 1H), 7.04 – 6.95 (m, 2H), 5.07 (s, 1H), 4.23 (dd, J = 5.7, 4.0 Hz, 2H), 3.97 – 3.84 (m, 2H), 3.83 – 3.77 (m, 2H), 3.76 – 3.72 (m, 2H), 3.71 – 3.64 (m, 4H), 3.58 (t, J = 5.1 Hz, 2H), 3.35 (d, J = 6.3 Hz, 2H), 1.48 (s, 9H).
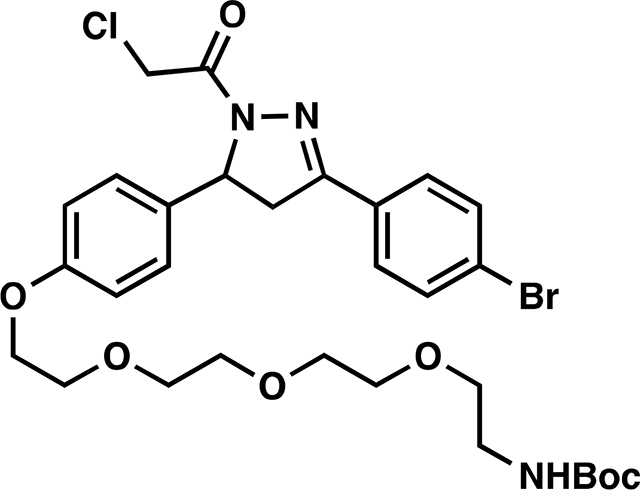
tert-butyl (2-(2-(2-(2-(4-(3-(4-bromophenyl)-1-(2-chloroacetyl)-4,5-dihydro-1H-pyrazol-5 yl)phenoxy)ethoxy)ethoxy)ethoxy)ethyl)carbamate (11):
To a solution of 10 (244 mg, 0.42 mmol) in EtOH was added hydrazine monohydrate (42 mg, 0.84 mmol) and the reaction solution was heated at 80 °C for 5 h under an atmosphere of nitrogen. Upon cooling to room temperature, the solution was diluted with water, extracted with DCM, and dried over anhydrous magnesium sulfate. The combined organic phase was concentrated in vacuo to ~ 5 mL [NOTE: All of the workup procedures should be performed quickly (<1.5 h total time) and the rotovap bath kept cool as the crude product can easily undergo autooxidation]. To the concentrated solution was quickly added chloroacetyl chloride (58 mg, 0.52 mmol) and triethylamine (57 mg, 0.56 mmol) at 0°C. The reaction mixture was stirred for 30 minutes in an ice bath and then warmed to room temperature and stirred overnight under N2. Upon completion of the reaction, the mixture was diluted with EtOAc, washed with brine, and concentrated in vacuo. The crude material was purified by silica gel chromatography (30 – 65 % EtOAc/hexanes) to yield 82 mg (30%) of 11: 1H NMR (400 MHz, CDCl3): δ 7.67 – 7.58 (m, 4H), 7.21 – 7.14 (m, 2H), 6.92 – 6.85 (m, 2H), 5.57 (dd, J = 11.7, 4.7 Hz, 1H), 5.11 (s, 1H), 4.57 (d, J = 1.1 Hz, 2H), 4.12 (dd, J = 5.6, 4.2 Hz, 2H), 3.88 – 3.83 (m, 2H), 3.75 – 3.73 (m, 2H), 3.72 – 3.69 (m, 2H), 3.67 – 3.62 (m, 4H), 3.55 (t, J = 5.1 Hz, 3H), 3.32 (q, J = 4.3, 3.4 Hz, 2H), 3.21 (dd, J = 17.8, 4.7 Hz, 1H), 1.46 (s, 9H).
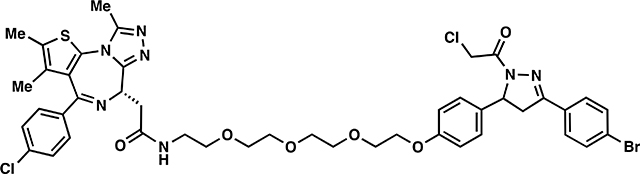
ML 2–32:
i. Compound 11 (81.7 mg, 0.120 mmol) was dissolved in DCM (2.5 mL) and trifluoroacetic acid (2.5 mL) was added slowly over 20 minutes followed by stirring for an additional 20 minutes at which point the reaction mixture was concentrated in vacuo. To remove residual TFA, the crude material was dissolved in 3mL of DCM and concentrated in vacuo and this process repeated two additional times. The crude material was used directly in the next step.
ii. The aforementioned crude material was dissolved in DCM (8 mL) and JQ1-acid (57.6 mg, 0.150 mmol), 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) (68.4 mg, 0.180 mmol), and N,N-Diisopropylethylamine (774 mg, 6.00 mmol) were added. The reaction was stirred overnight with monitoring by TLC (10% MeOH in DCM, 100% EtOAc). Upon completion, the reaction mixture was concentrated in vacuo and purified by silica gel flash chromatography (1–10% MeOH/DCM). The eluted fractions were insufficiently pure and those containing product were combined, concentrated and purified again by flash silica chromatography (100–0% EtOAc/DCM followed by 0–10% MeOH/DCM) to afford 46 mg (40%) of ML 2–32: 1H NMR (400 MHz, CDCl3): δ 7.66 – 7.57 (m, 4H), 7.45 – 7.41 (m, 2H), 7.34 (d, J = 8.5 Hz, 2H), 7.16 (dq, J = 7.9, 3.1 Hz, 2H), 6.98 (d, J = 5.1 Hz, 1H), 6.92 – 6.83 (m, 2H), 5.57 (ddd, J = 11.8, 7.3, 4.7 Hz, 1H), 4.68 (t, J = 6.9 Hz, 1H), 4.57 (s, 2H), 4.12 (dd, J = 5.7, 4.0 Hz, 2H), 3.90 – 3.84 (m, 2H), 3.78 – 3.66 (m, 10H), 3.65 – 3.58 (m, 2H), 3.54 – 3.50 (m, 2H), 3.44 – 3.37 (m, 1H), 3.20 (dd, J = 17.8, 4.7 Hz, 1H), 2.68 (d, J = 1.5 Hz, 3H), 2.42 (s, 3H), 1.69 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 170.4, 163.8, 163.7, 158.4, 155.6, 154.3, 149.7, 145.2, 136.6, 136.6, 132.9, 132.1, 131.9, 131.7, 130.8, 130.6, 130.3, 130.2, 129.9, 129.8, 128.6, 128.1, 126.9, 125.0, 114.9, 70.7, 70.5, 70.5, 70.3, 69.7, 69.6, 67.4, 60.1, 54.3, 54.3, 42.1, 41.9, 39.3, 39.0, 29.6, 14.3, 13.0, 11.7; HRMS (ESI): calcd. C44H47BrCl2N7O6S ([M+H]+): m/z 950.1863, found: 950.1852.
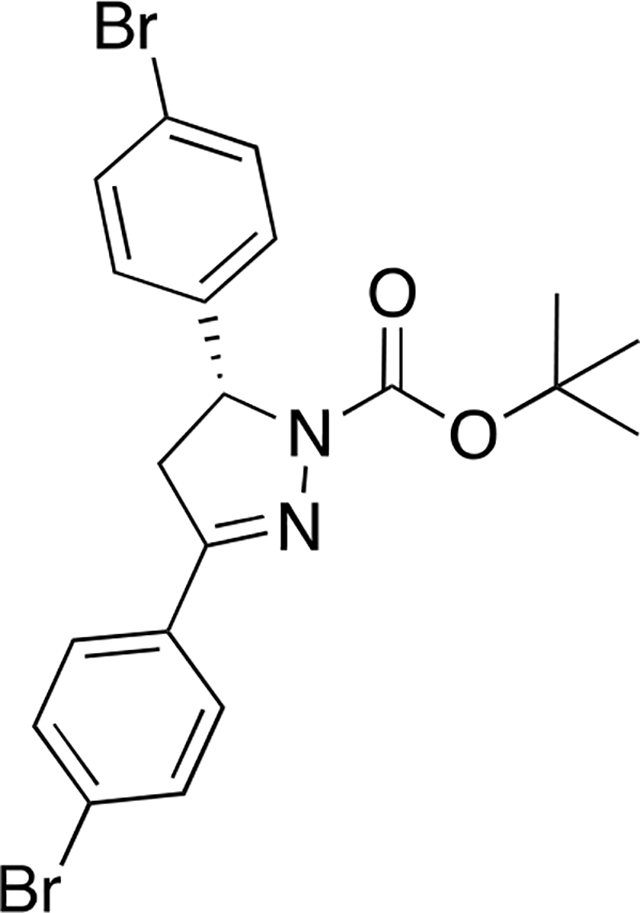
tert-butyl (R)-3,5-bis(4-bromophenyl)-4,5-dihydro-1H-pyrazole-1-carboxylate ((R)-12):
(E)-1,3-bis(4-bromophenyl)prop-2-en-1-one (109.8 mg, 0.3 mmol, 1.0 eq.), tert-butyl carbazate (43.6 mg, 0.33 mmol, 1.1 eq.), potassium phosphate (82.8 mg, 0.39 mmol, 1.3 eq.), and catalyst A (13.3 mg, 0.03 mmol, 10 mol %) were combined in THF (0.6 mL, 0.5 M) under nitrogen and stirred at 0 °C for 18h (Mahé et al., 2010). The mixture was diluted with EtOAc, filtered to remove salts, concentrated in vacuo and purified by silica gel chromatography (0–15% EtOH/Hex) to provide 94 mg (65%) of (R)-12: 1H NMR (400 MHz, Chloroform-d): δ 7.61 (d, J = 8.6 Hz, 2H), 7.55 – 7.43 (m, 4H), 7.14 – 7.08 (m, 2H), 5.31 (dd, J = 11.9, 5.1 Hz, 1H), 3.73 (dd, J = 17.5, 12.1 Hz, 1H), 3.08 (dd, J = 17.5, 5.5 Hz, 1H), 1.35 (s, 9H).
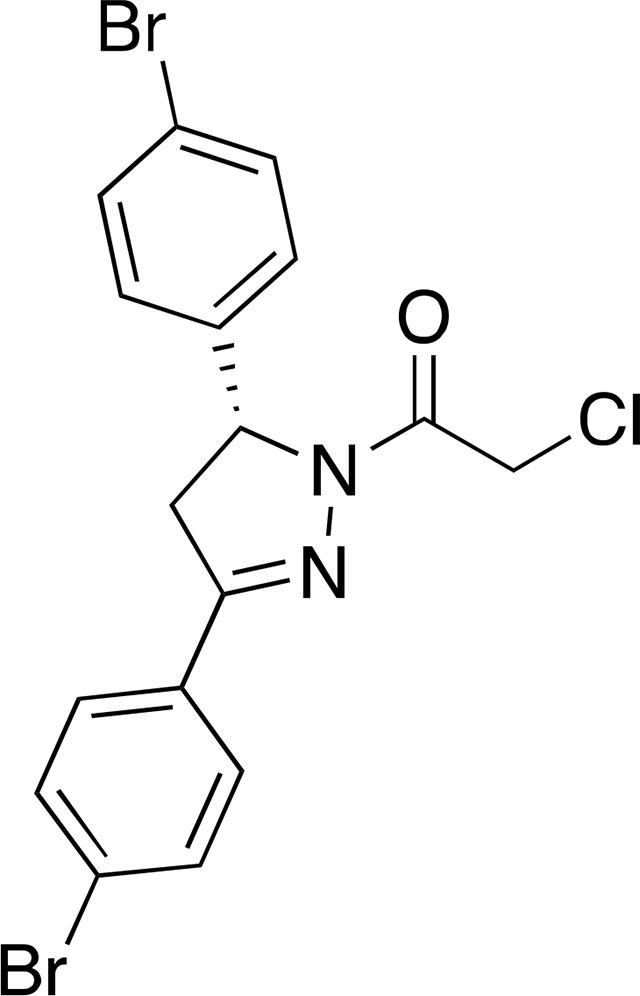
(R)-1-(3,5-bis(4-bromophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-2-chloroethan-1-one ((R)-EN219):
(R)-12 (45 mg, 0.094 mmol, 1.0 eq) was dissolved in DCM (0.47 mL, 0.2 M), cooled to 0 °C, and 4.0 M HCl in dioxane (0.234 mL, 0.94 mmol, 10.0 eq.) was added under nitrogen. The mixture was stirred at room temperature until the starting material was consumed (1.5 h), then additional DCM was added (0.47 mL, reducing to 0.1 M) and the mixture cooled to 0 °C. Chloroacetyl chloride (22 μL, 0.28 mmol, 3.0 eq.) was added followed by TEA (0.17 mL, 1.22 mmol, 13.0 eq.), and the reaction allowed to warm to room temperature and stirred for 1h. The reaction mixture was concentrated in vacuo and the crude material purified by silica gel chromatography (15% EtOAc/Hex) to yield 36 mg (85%) of (R)-EN219 as a waxy solid. The enantiomeric excess of (R)-EN219 was determined by chiral HPLC (CHIRALCEL® OD-H column, i-PrOH/hexane (10:90), flow rate 1 mL/min) and found to be 91% ee. 1H NMR (400 MHz, Chloroform-d): δ 7.63 – 7.55 (m, 4H), 7.49 – 7.43 (m, 2H), 7.11 (d, J = 8.5 Hz, 2H), 5.55 (dd, J = 11.8, 4.9 Hz, 1H), 4.60 – 4.48 (m, 2H), 3.77 (dd, J = 17.9, 11.8 Hz, 1H), 3.16 (dd, J = 17.9, 4.9 Hz, 1H); 13C NMR (101 MHz, CDCl3): δ 164.2, 154.4, 139.8, 132.4, 132.3, 129.7, 128.4, 127.7, 125.6, 122.2, 60.3, 42.1, 42.1; HRMS (ESI): calcd. C17H14ON279Br235Cl ([M+H]+): m/z 454.9156, found: 454.9160.
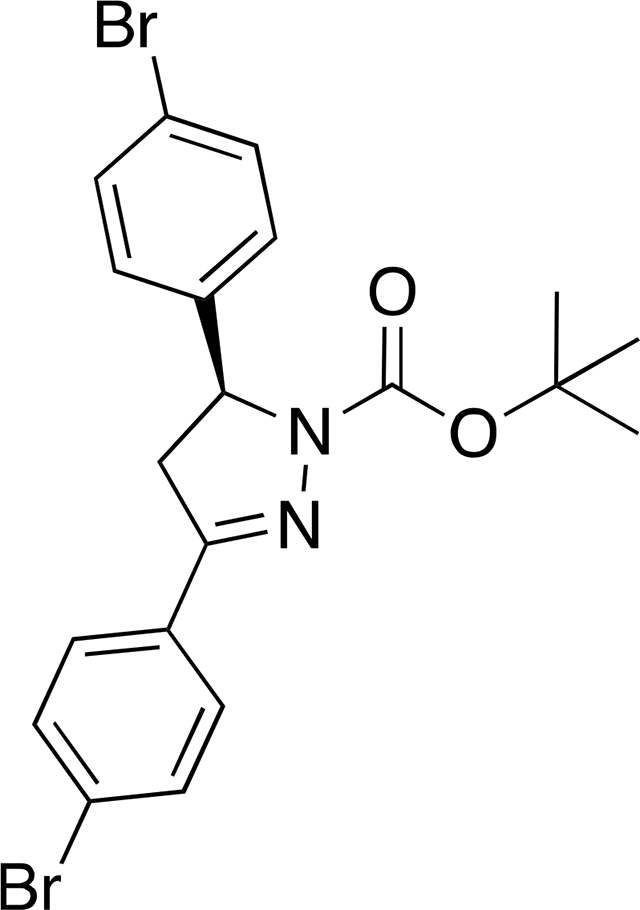
tert-butyl (S)-3,5-bis(4-bromophenyl)-4,5-dihydro-1H-pyrazole-1-carboxylate ((S)-12):
(E)-1,3-bis(4-bromophenyl)prop-2-en-1-one (116 mg, 0.32 mmol, 1.0 eq.), tert-butyl carbazate (46 mg, 0.35 mmol, 1.1 eq.), potassium phosphate (88 mg, 0.41 mmol, 1.3 eq.), and catalyst B (56 mg, 0.13 mmol, 0.4 eq) were combined in THF (2mL) under nitrogen and stirred at 0 °C for 18h (Mahé et al., 2010). The mixture was diluted with EtOAc, filtered to remove salts, concentrated and purified by silica gel chromatography (0–35% EtOAc/Hex) to provide 71 mg of (S)-12 intermediate as a yellow oil which was immediately carried forward into (S)-EN219.
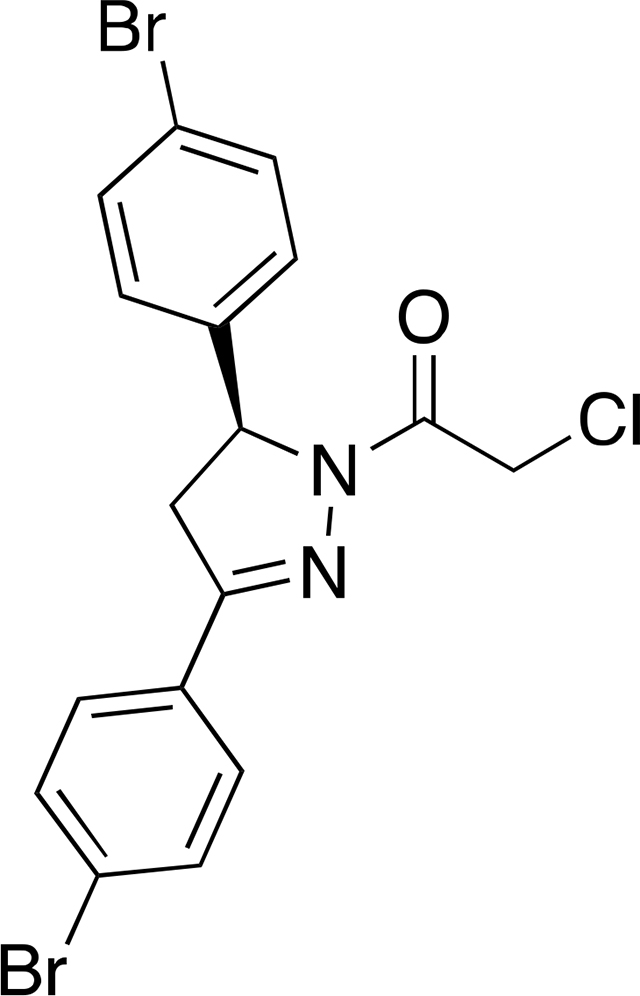
(S)-1-(3,5-bis(4-bromophenyl)-4,5-dihydro-1H-pyrazol-1-yl)-2-chloroethan-1-one ((S)-EN219):
(S)-12 (71 mg, 0.148mmol, 1.0 eq) was dissolved in DCM (4mL), cooled to 0 °C, and 4.0 M HCl in dioxane (0.370 mL, 1.48mmol mmol, 10.0 eq.) was added under nitrogen. The mixture was stirred at room temperature until the starting material was consumed (2h), then the mixture was cooled again to 0 °C. Chloroacetyl chloride (36 μL, 0.44 mmol, 3.0 eq.) was added followed by TEA (0.268 mL, 1.92 mmol, 13.0 eq.), and the reaction allowed to warm to room temperature and stirred for 30 minutes. The reaction mixture was extracted in DCM and washed with water 3x. The reaction mixture was then concentrated in vacuo and the crude material purified by silica gel chromatography (15% EtOAc/Hex) to yield 35 mg (52%) of (S)-EN219 as a light yellow solid. The enantiomeric excess of (S)-EN219 was determined by chiral HPLC (CHIRALCEL® OD-H column, i-PrOH/hexane (10:90), flow rate 1 mL/min) and found to be 91% ee. 1H NMR (600 MHz, CDCl3) δ 7.63 – 7.56 (m, 4H), 7.48 – 7.44 (m, 2H), 7.14 – 7.09 (m, 2H), 5.55 (dd, J = 11.8, 4.9 Hz, 1H), 4.56 (d, J = 13.5 Hz, 1H), 4.51 (d, J = 13.4 Hz, 1H), 3.77 (dd, J = 17.8, 11.8 Hz, 1H), 3.16 (dd, J = 17.8, 4.9 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 164.2, 154.4, 139.8, 132.4, 132.3, 129.7, 128.4, 127.7, 125.6, 122.2, 60.3, 42.1, 42.1; HRMS (ESI): calcd. C17H13ON279Br235ClNa ([M+Na]+): m/z 476.8975, found: 476.8976
Deschloro-EN219:
Hydrazine monohydrate (50% conc., 80.16 μL, 0.8 mmol, 2.0 eq.) was added to a suspension of (E)-1,3-bis(4-bromophenyl)prop-2-en-1-one (146.4 mg, 0.4 mmol, 1.0 eq.) in EtOH (1.33 mL, 0.3 M). The resulting reaction mixture was stirred at reflux temperature for 4 h before it was concentrated under reduced pressure. The crude pyrazoline was then dissolved in DCM (2mL, 0.2 M) and cooled to 0 °C. Triethylamine (167 μL, 1.2 mmol, 3.0 eq.) was added dropwise, followed by chloroacetyl chloride (42.6 μL, 0.6 mmol, 1.5 eq.). The resulting reaction mixture was stirred at ambient temperature for 1 hour before it was diluted with DCM. The organic phase was sequentially washed with satd. aq. NaHCO3 solution, brine, dried over Na2SO4, and concentrated in vacuo. Purification by column chromatography (15% EtOAc/Hex) to afforded 157 mg (96%) of deschloro-EN219: 1H NMR (400 MHz, CDCl3): δ 7.61 (q, J = 8.6 Hz, 4H), 7.49 (d, J = 8.4 Hz, 2H), 7.24 – 7.10 (m, 2H), 5.58 (dd, J = 12.3, 4.9 Hz, 1H), 3.77 (dd, J = 17.6, 12.0 Hz, 1H), 3.24 – 3.06 (m, 1H), 2.44 (s, 3H).; 13C NMR (101 MHz, CDCl3): δ 168.9, 152.7, 140.7, 132.10, 132.1, 130.2, 128.0, 127.4, 124.8, 121.7, 59.6, 42.0, 21.9; HRMS (ESI): calcd. C17H15ON279Br235 ([M+H]+): m/z 420.9546, found: 420.9550.
QUANTIFICATION AND STATISTICAL ANALYSIS
For quantification of Western blots, bands were quantified using Image J and normalized to protein loading controls. Statistical analysis was performed using a Student’s unpaired two-tailed t-test.
For isoTOP-ABPP data analysis, data were extracted in the form of MS1 and MS2 files using Raw Extractor v.1.9.9.2 (Scripps Research Institute) and searched against the Uniprot human database using ProLuCID search methodology in IP2 v.3 (Integrated Proteomics Applications, Inc.)(Xu et al., 2015b). Cysteine residues were searched with a static modification for carboxyaminomethylation (+57.02146) and up to two differential modifications for methionine oxidation and either the light or heavy TEV tags (+464.28596 or +470.29977, respectively). Peptides were required to be fully tryptic peptides and to contain the TEV modification. ProLUCID data were filtered through DTASelect to achieve a peptide false-positive rate below 5%. Only those probe-modified peptides that were evident across two out of three biological replicates were interpreted for their isotopic light to heavy ratios. For those probe-modified peptides that showed ratios greater than two, we only interpreted those targets that were present across all three biological replicates, were statistically significant and showed good quality MS1 peak shapes across all biological replicates. Light versus heavy isotopic probe-modified peptide ratios are calculated by taking the mean of the ratios of each replicate paired light versus heavy precursor abundance for all peptide-spectral matches associated with a peptide. The paired abundances were also used to calculate a paired sample t-test P value in an effort to estimate constancy in paired abundances and significance in change between treatment and control. P values were corrected using the Benjamini–Hochberg method.
Supplementary Material
Table S1. Related to Figure 2. Structures of covalent ligands screened against RNF114
Table S2. Related to Figure 2. IsoTOP-ABPP analysis of EN219 in 231MFP breast cancer cells. IsoTOP-ABPP analysis of EN219 in 231MFP breast cancer cells. 231MFP cells were treated in situ with DMSO vehicle or EN219 (1 μM) for 90 min. Control and treated cell lysates were labeled with IA-alkyne (100 μM) for 1 h, after which isotopically light (control) or heavy (EN219-treated) biotin-azide bearing a TEV tag was appended by CuAAC. Proteomes were mixed in a 1:1 ratio, probe-labeled proteins were enriched with avidin and digested with trypsin, and probe-modified peptides were eluted by TEV protease and analyzed by LC-MS/MS.
Table S3. Related to Figure 2. TMT-based proteomic analysis of EN219-alkyne pulldown competed by EN219. 231MFP cells were treated with DMSO vehicle or EN219 (20 μM) 30 min prior to treating cells with DMSO or EN219-alkyne probe (2 μM) for 90 min. Resulting cell lysates were subjected to CuAAC with biotin-azide to append a biotin enrichment handle onto EN219-alkyne labeled proteins ex situ. EN219-alkyne labeled proteins were subsequently avidin-enriched, digested with trypsin, and resulting tryptic peptides from each treatment group were labeled with TMT reagents and combined and fractionated for LC-MS/MS analysis.
Table S4. Related to Figure 3. TMT-based proteomic analysis of ML 2–14-mediated protein level changes in 231MFP cells. TMT-based quantitative proteomic data showing ML 2–14-mediated protein level changes in 231MFP cells. 231MFP cells were treated with DMSO vehicle or ML 2–14 for 8 h.
Scheme 1.

Synthetic route to degrader ML 2–14.
Scheme 2.

Synthetic route to degraders ML 2–22 and ML 2–23.
Scheme 3.

Synthetic route to degrader ML 2–31.
Scheme 4.

Synthetic route to degrader ML 2–32.
Scheme 5.

Synthetic route to enantioenriched EN219.
Scheme 6.

Synthesis of deschloro-EN219.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rb Anti-RNF114 | Sigma-Aldrich | HPA021184 |
| Mouse monoclonal c-Abl Antibody | Santa Cruz | sc-23 |
| Rb Phospho-CrkL (Tyr207) Antibody | Cell Signaling Technology | Cat#13181S |
| Mouse GAPDH Monoclonal Antibody | Proteintech Group Inc | 60004-1-Ig |
| Mouse Beta Actin Monoclonal antibody | Proteintech Group Inc | 66009-1-Ig |
| Rb Anti-Brd4 antibody | Abcam plc | Ab128874 |
| Bacterial and Virus Strains | ||
| N/A | ||
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Myc-Flag-RNF114 human recombinant protein | Origene | TP309752 |
| Recombinant protein of human cyclin-dependent kinase inhibitor 1A | Origene | TP301765 |
| Ubiquitin Activating Enzyme (UBE1) | BostonBiochem | E-305 |
| Ubiquitin-conjugating Enzyme E2D 1 (UBE2D1) | BostonBiochem | E2-615 |
| FLAG (DYKDDDDK)-Ubiquitin | BostonBiochem | U-120 |
| Dimethyl Sulfoxide | Fisher Chemical | D128-500 (CAS 67-68-5) |
| Dulbecco’s Phosphate Buffered Saline (DPBS) | Sigma-Aldrich | D8537-500mL |
| L-15 Medium | Sigma-Aldrich | L1518-500mL |
| Iscove’s Modified Dulbecco’s Medium | Sigma-Aldrich | 51471C-1000mL |
| Opti-MEM Reduced Serum Media | ThermoFisher Scientific | Cat#31985062 |
| Iodoacetamide 98% | ACROS Organics | Cat# AC122270050 |
| Iodoacetamide-13C2, 2-d2 | Sigma-Aldrich | Cat#721328 |
| Urea | Fisher Chemical | U15-500 (CAS 57-13-6) |
| ProteaseMAX Surfactant, lyophilized | Promega | REF#V2072 |
| Ammonium Bicarbonate | Fisher Chemical | A643-500 (CAS 1066-33-7) |
| TCEP 99% | Strem Chemicals | Cat#15-7400 (CAS 51805-45-9) |
| Formic Acid, 99.0+%, Optima LC/MS Grade | Fisher Chemical | A117-50 (CAS 64-18-6) |
| Sequencing Grade Modified Trypsin, Porcine | Promega | V511A |
| Covalent Ligand Library | Enamine | N/A |
| Tetramethylrhodamine-5-Iodoacetamide Dihydroiodide (IA-Rhodamine) | Setareh Biotech | 6222 (CAS 114458-99-0) |
| Laemmli SDS sample buffer, reducing (4x) | Alfa Aesar | J60015 |
| Tris(benzyltriazole methylamine) (TBTA) | Sigma-Aldrich | 678937 (CAS 510758-28-8) |
| Biotin-TEV-azide | Weerapana et. al., 2010 | N/A |
| Copper (II) Sulfate | Sigma-Aldrich | 451657 |
| AcTEV Protease | Invitrogen | 12575-015 |
| N-Hex-5-ynyl-2-iodo-acetamide (IAyne) | Chess Gmbh | 3187 |
| Spectra Multicolor Broad Range Protein Ladder | Thermo Scientific | Prod#26634 |
| Pierce Protease inhibitor Mini Tablets, EDTA-free | Thermo Scientific | Prod#A32955 |
| High Capacity Streptavidin Agarose Resin | Thermo Scientific | Ref#20357 |
| Streptavidin Agarose Resin | Thermo Scientific | Ref#20349 |
| 4-hydroxybenzaldehyde | Alfa Aesar | A13580-22 (CAS 123-08-0) |
| 3,4-Dihydro-2H-pyran | Sigma-Aldrich | D106208-100mL (CAS 110-87-2) |
| 4’-Bromoacetophenone | Sigma-Aldrich | B56404-25G (CAS 99-90-1) |
| 4-(Boc-amino) butyl bromide | Sigma-Aldrich | 90303-500mg-F (CAS 164365-88-2) |
| Hydrazine monohydrate | Sigma-Aldrich | 207942-5g (CAS 7803-57-8) |
| Chloroacetyl chloride | Sigma-Aldrich | 22880-100mL (CAS 79-04-9) |
| DIPEA | Sigma-Aldrich | D125806-100mL (CAS 7087-68-5) |
| JQ1 | eNovation Chemicals | D372464 (CAS 1268524-70-4) |
| HATU | Sigma-Aldrich | 445460-5g (CAS 148893-10-1) |
| Propargylamine | Sigma-Aldrich | P50900-1g (CAS 2450-71-7) |
| N-Boc-7-bromoheptan-1-amine | AChemBlock | P41297 (CAS 142356-34-1) |
| tert-Butyl (2-(2-(2-(2-bromoethoxy)ethoxy)ethoxy)ethyl)carbamate | Synthonix | B37532 (CAS 1076199-21-7) |
| Pyridinium p-toluenesulfonate | Sigma-Aldrich | 232238-5G (CAS 24057-28-1) |
| (E)-1,3-bis(4-bromophenyl)prop-2-en-1-one | Princeton Bio | PRIH93ED8598 (CAS 5471-96-5) |
| Nimbolide | Cayman Chemicals | 19230 (CAS 25990-37-8) |
| Critical Commercial Assays | ||
| TMT10plex Isobaric Label Reagent Set | ThermoFisher | A37725 |
| WST8 reagent | APExBio | CCK-8 |
| Monarch Total RNA Miniprep Kit | New England BioLabs | T2010S |
| ProtoScript II reverse transcriptase Kit | New England BioLabs | M0368L |
| DyNAmo HS SYBR Green Kit | Fisher Scientific | F-410L |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| 231MFP | (Jessani et al., 2004) | N/A |
| K562 | ATCC | CCL-243 |
| HAP1 | Horizon Discovery | HZGHC55905 |
| HAP1 (RNF114 Knockout) | Horizon Discovery | HZGHC55905 |
| Experimental Models: Organisms/Strains | ||
| N/A | ||
| Oligonucleotides | ||
| N/A | ||
| Recombinant DNA | ||
| N/A | ||
| Software and Algorithms | ||
| ImageJ | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| IP2 proteomics pipeline 5.0.1 | Integrated Proteomics Applications | N/A |
| Other | ||
Highlights.
We have identified a fully synthetic covalent ligand EN219 that targets RNF114
We show that EN219 mimics the mode of action exploited by natural product nimbolide
We show that EN219 can be used as an RNF114 recruiter in degrader applications
Acknowledgement
We thank the members of the Nomura Research Group and Novartis Institutes for BioMedical Research for critical reading of the manuscript. This work was supported by Novartis Institutes for BioMedical Research and the Novartis-Berkeley Center for Proteomics and Chemistry Technologies (NB-CPACT) for all listed authors. This work was also supported by the Nomura Research Group and the Mark Foundation for Cancer Research ASPIRE Award for DKN, ML, JNS, LB. This work was also supported by grants from the National Institutes of Health (R01CA240981 for DKN, TJM, ML, JNS, LB and F31CA239327 and F99CA253717 for JNS).
Declaration of Interests
JAT, JMK, MS, SMB are employees of Novartis Institutes for BioMedical Research. This study was funded by the Novartis Institutes for BioMedical Research and the Novartis-Berkeley Center for Proteomics and Chemistry Technologies. DKN is a co-founder, shareholder, and adviser for Frontier Medicines.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bachovchin DA, Ji T, Li W, Simon GM, Blankman JL, Adibekian A, Hoover H, Niessen S, and Cravatt BF (2010). Superfamily-wide portrait of serine hydrolase inhibition achieved by library-versus-library screening. Proc. Natl. Acad. Sci. U.S.A 107, 20941–20946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backus KM, Correia BE, Lum KM, Forli S, Horning BD, González-Páez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, et al. (2016). Proteome-wide covalent ligand discovery in native biological systems. Nature 534, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond MJ, Chu L, Nalawansha DA, Li K, and Crews CM (2020). Targeted Degradation of Oncogenic KRASG12C by VHL-Recruiting PROTACs. ACS Cent Sci 6, 1367–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson DP, and Crews CM (2017). Targeted Protein Degradation by Small Molecules. Annual Review of Pharmacology and Toxicology 57, 107–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson DP, Smith BE, Burslem GM, Buhimschi AD, Hines J, Jaime-Figueroa S, Wang J, Hamman BD, Ishchenko A, and Crews CM (2018). Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chemical Biology 25, 78–87.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burslem GM, Schultz AR, Bondeson DP, Eide CA, Savage Stevens SL, Druker BJ, and Crews CM (2019). Targeting BCR-ABL1 in Chronic Myeloid Leukemia by PROTAC-Mediated Targeted Protein Degradation. Cancer Res. 79, 4744–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain PP, and Hamann LG (2019). Development of targeted protein degradation therapeutics. Nat. Chem. Biol 15, 937–944. [DOI] [PubMed] [Google Scholar]
- Grossman EA, Ward CC, Spradlin JN, Bateman LA, Huffman TR, Miyamoto DK, Kleinman JI, and Nomura DK (2017). Covalent Ligand Discovery against Druggable Hotspots Targeted by Anti-cancer Natural Products. Cell Chem Biol 24, 1368–1376.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Kim Y-L, Lee K-W, Her N-G, Ha T-K, Yoon S, Jeong S-I, Lee J-H, Kang M-J, Lee M-G, et al. (2013). ZNF313 is a novel cell cycle activator with an E3 ligase activity inhibiting cellular senescence by destabilizing p21(WAF1.). Cell Death Differ. 20, 1055–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H-T, Dobrovolsky D, Paulk J, Yang G, Weisberg EL, Doctor ZM, Buckley DL, Cho J-H, Ko E, Jang J, et al. (2018). A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-kinase Degrader. Cell Chemical Biology 25, 88–99.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessani N, Humphrey M, McDonald WH, Niessen S, Masuda K, Gangadharan B, Yates JR, Mueller BM, and Cravatt BF (2004). Carcinoma and stromal enzyme activity profiles associated with breast tumor growth in vivo. Proc Natl Acad Sci U S A 101, 13756–13761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai AC, and Crews CM (2017). Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov 16, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai AC, Toure M, Hellerschmied D, Salami J, Jaime-Figueroa S, Ko E, Hines J, and Crews CM (2016). Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem. Int. Ed. Engl 55, 807–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahé O, Dez I, Levacher V, and Brière J-F (2010). Enantioselective Phase-Transfer Catalysis: Synthesis of Pyrazolines. Angewandte Chemie International Edition 49, 7072–7075. [DOI] [PubMed] [Google Scholar]
- Newman DJ, and Cragg GM (2016). Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod 79, 629–661. [DOI] [PubMed] [Google Scholar]
- Nomura DK, and Maimone TJ (2019). Target Identification of Bioactive Covalently Acting Natural Products. Curr. Top. Microbiol. Immunol 420, 351–374. [DOI] [PubMed] [Google Scholar]
- Ottis P, Palladino C, Thienger P, Britschgi A, Heichinger C, Berrera M, Julien-Laferriere A, Roudnicky F, Kam-Thong T, Bischoff JR, et al. (2019). Cellular Resistance Mechanisms to Targeted Protein Degradation Converge Toward Impairment of the Engaged Ubiquitin Transfer Pathway. ACS Chem. Biol 14, 2215–2223. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012). NIH Image to Image J: 25 years of image analysis. Nature Methods. 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradlin JN, Hu X, Ward CC, Brittain SM, Jones MD, Ou L, To M, Proudfoot A, Ornelas E, Woldegiorgis M, et al. (2019). Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat. Chem. Biol 15, 747–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JR, Brittain SM, Lipps J, Llamas L, Jain RK, and Schirle M (2017). A Photoaffinity Labeling-Based Chemoproteomics Strategy for Unbiased Target Deconvolution of Small Molecule Drug Candidates . Methods Mol. Biol 1647, 1–18. [DOI] [PubMed] [Google Scholar]
- Tong B, Spradlin JN, Novaes LFT, Zhang E, Hu X, Moeller M, Brittain SM, McGregor LM, McKenna JM, Tallarico JA, et al. (2020). A Nimbolide-Based Kinase Degrader Preferentially Degrades Oncogenic BCR-ABL. ACS Chem Biol 15, 1788–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Weerapana E, Blewett MM, and Cravatt BF (2014). A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat. Methods 11, 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CC, Kleinman JI, Brittain SM, Lee PS, Chung CYS, Kim K, Petri Y, Thomas JR, Tallarico JA, McKenna JM, et al. (2019). Covalent Ligand Screening Uncovers a RNF4 E3 Ligase Recruiter for Targeted Protein Degradation Applications. ACS Chem. Biol 14, 2430–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MBD, Bachovchin DA, Mowen K, Baker D, and Cravatt BF (2010). Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Park SK, Venable JD, Wohlschlegel JA, Diedrich JK, Cociorva D, Lu B, Liao L, Hewel J, Han X, et al. (2015a). ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J Proteomics 129, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Park SK, Venable JD, Wohlschlegel JA, Diedrich JK, Cociorva D, Lu B, Liao L, Hewel J, Han X, et al. (2015b). ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J Proteomics 129, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Crowley VM, Wucherpfennig TG, Dix MM, and Cravatt BF (2019). Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat. Chem. Biol 15, 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Related to Figure 2. Structures of covalent ligands screened against RNF114
Table S2. Related to Figure 2. IsoTOP-ABPP analysis of EN219 in 231MFP breast cancer cells. IsoTOP-ABPP analysis of EN219 in 231MFP breast cancer cells. 231MFP cells were treated in situ with DMSO vehicle or EN219 (1 μM) for 90 min. Control and treated cell lysates were labeled with IA-alkyne (100 μM) for 1 h, after which isotopically light (control) or heavy (EN219-treated) biotin-azide bearing a TEV tag was appended by CuAAC. Proteomes were mixed in a 1:1 ratio, probe-labeled proteins were enriched with avidin and digested with trypsin, and probe-modified peptides were eluted by TEV protease and analyzed by LC-MS/MS.
Table S3. Related to Figure 2. TMT-based proteomic analysis of EN219-alkyne pulldown competed by EN219. 231MFP cells were treated with DMSO vehicle or EN219 (20 μM) 30 min prior to treating cells with DMSO or EN219-alkyne probe (2 μM) for 90 min. Resulting cell lysates were subjected to CuAAC with biotin-azide to append a biotin enrichment handle onto EN219-alkyne labeled proteins ex situ. EN219-alkyne labeled proteins were subsequently avidin-enriched, digested with trypsin, and resulting tryptic peptides from each treatment group were labeled with TMT reagents and combined and fractionated for LC-MS/MS analysis.
Table S4. Related to Figure 3. TMT-based proteomic analysis of ML 2–14-mediated protein level changes in 231MFP cells. TMT-based quantitative proteomic data showing ML 2–14-mediated protein level changes in 231MFP cells. 231MFP cells were treated with DMSO vehicle or ML 2–14 for 8 h.
Data Availability Statement
Data generated in this study will be made available upon reasonable request. No code was developed for this study.