

Abstract
Striated muscle responds to mechanical overload by rapidly up-regulating the expression of the cardiac ankyrin repeat protein, CARP, which then targets the sarcomere by binding to titin N2A in the I-band region. To date, the role of this interaction in the stress response of muscle remains poorly understood. Here, we characterise the molecular structure of the CARP-receptor site in titin (UN2A) and its binding of CARP. We find that titin UN2A contains a central three-helix bundle fold (ca 45 residues in length) that is joined to N- and C-terminal flanking immunoglobulin domains by long, flexible linkers with partial helical content. CARP binds titin by engaging an α-hairpin in the three-helix fold of UN2A, the C-terminal linker sequence, and the BC loop in Ig81, which jointly form a broad binding interface. Mutagenesis showed that the CARP/N2A association withstands sequence variations in titin N2A and we use this information to evaluate 85 human single nucleotide variants. In addition, actin co-sedimentation, co-transfection in C2C12 cells, proteomics on heart lysates, and the mechanical response of CARP-soaked myofibrils imply that CARP induces the cross-linking of titin and actin myofilaments, thereby increasing myofibril stiffness. We conclude that CARP acts as a regulator of force output in the sarcomere that preserves muscle mechanical performance upon overload stress.
Keywords: nuclear magnetic resonance, hydrogen-deuterium exchange mass spectrometry, muscle stress response, actin cytoskeleton, sarcomere mechanics
Introduction
The sarcomere is the contractile unit of skeletal and cardiac striated muscle. Its homeostatic balance conditions the mechanical performance of muscle in response to a variety of stressors derived from mechanical load, metabolic or immunogenic processes, ageing and chronic disease, among others. The heart responds to such stressors by reactivating a foetal gene program that evokes sarcomere remodelling, metabolic reprogramming, and reactivation of signalling pathways otherwise suppressed in the postnatal heart.1 The Cardiac Ankyrin Repeat Protein, CARP/Ankrd1, is the best studied representative of the MARP protein family and a central member of the foetal heart program.2 Its levels are rapidly up-regulated in the postnatal heart in stress states, including the clinical heart failure that ensues upon dilated or arrhythmogenic right ventricular cardiomyopathy.3,4 CARP is also central to skeletal muscle, where its expression increases rapidly, from near zero levels, under stress conditions such as eccentric contractions or unilateral diaphragm denervation induced passive stretch.5,6 Taken together, clinical and experimental correlative studies show that the increase of CARP levels is a universal stress response of striated muscle. Yet, the physiological outcome of CARP up-regulation remains to date poorly understood.
An additional response of muscle to stress is the remodelling of the sarcomeric titin filament. Titin is a gigantic multi-domain repeat protein that spans half the sarcomere, from the Z-disc to the M-line. It provides elasticity to the sarcomere through the molecular springs of its I-band region and it exists in many differentially-spliced isoforms, which are characterised by distinct levels of compliance.7,8 The ratio of titin isoforms in the sarcomere determines myofibrillar stiffness.9 This ratio is dynamically adjusted during muscle development, in disease and in response to stress and mechanical demand. Titin remodelling is, thereby, a critical mechanism for the modulation of muscle performance.7,8 The N2A element of titin, located in its spring region, is the primary binding site of CARP in the sarcomeric I-band and is responsible for the recruitment of CARP to the sarcomere upon overexpression of the latter.10–12 Titin-N2A is composed of four immunoglobulin (Ig) domains (Ig80, Ig81, Ig82 and Ig83) and a unique helical sequence of ~100 residue length (UN2A) located between domains Ig80 and Ig81 (Figure 1).12,13 The main CARP binding site in titin resides in the unique UN2A sequence,10,11 but Ig81 contributes a second binding site that confers high affinity to the interaction.12 CARP binds the dual domain UN2A-Ig81 through its C-terminal (~210 residues) ankyrin repeat domain. The ankyrin repeat motif consists of ~33 residues that fold into two short α-helices linked by a tight turn, forming an α-hairpin structure. Ankyrin repeats pack consecutively into an array, resulting in an elongated 3D-fold with a slightly concave shape: the ankyrin repeat fold.14 The ankyrin repeat domain from CARP is predicted to contain 6 ankyrin repeats.12 As CARP is known to self-dimerize, its interaction with titin N2A has been speculated to lead to the cross-linking of titin molecules in the sarcomere, likely contributing to provide stretch resilience to the myofibril.11 Interestingly, in the failing human heart, the remodelling of titin ratios causes a net sarcomeric enrichment of the N2A-containing isoform N2BA. Thus, CARP up-regulation and increased sarcomeric ratios of N2A-containing titin might converge into a same pathophysiological pathway in the heart. In healthy postnatal skeletal muscle, N2A-containing titin is typically dominant.8 To date, the potential modulation of titin-based stiffness by CARP in either muscle type remains largely unexplored.
Figure 1: Titin N2A.
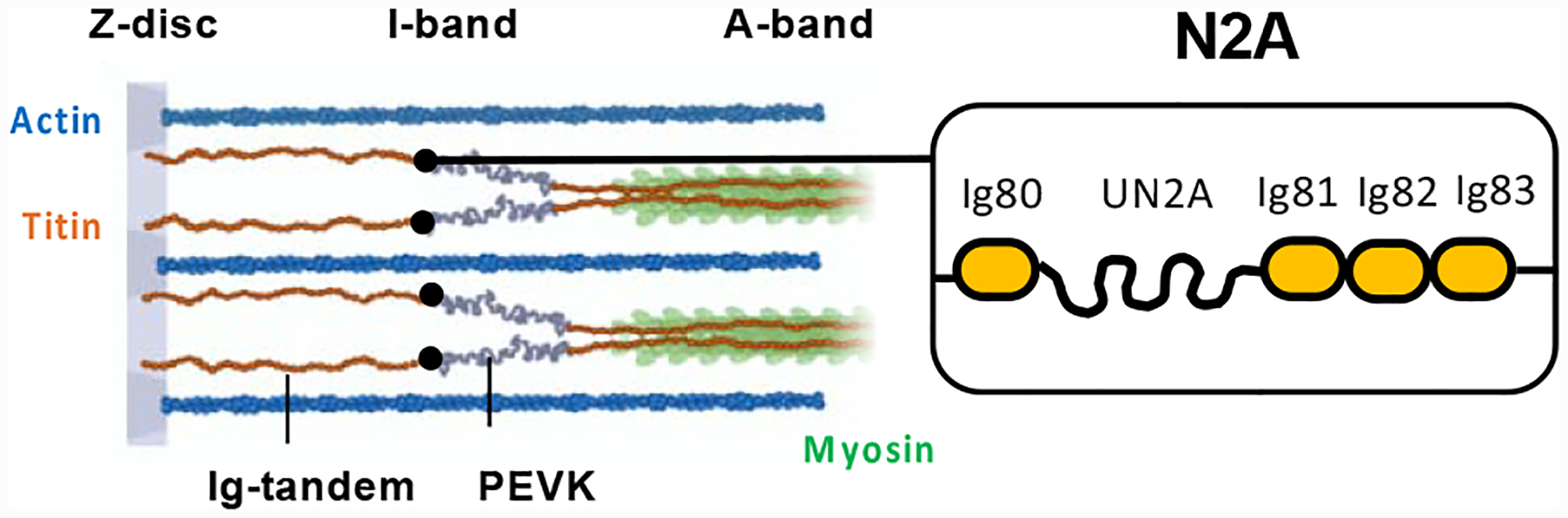
Sarcomeric location and domain composition of titin N2A (Ig, immunoglobulin domain; UN2A, unique sequence).
Despite the suspected importance of the CARP/N2A interaction in muscle physiology, little knowledge currently exists of its molecular principles and functional role. In this work, we sought to investigate the CARP/N2A association at the molecular level in vitro as well as to explore its potential functional outcome ex vivo. Our data indicate that CARP induces the cross-linking of titin and actin myofilaments in the sarcomere in situ, thereby conferring mechanical stiffness and stability to the sarcomere under stress conditions. This proposed mechanism provides a rational for the protective role of CARP in muscle.
Results
UN2A has a small structured central segment flanked by long flexible linker sequences
To investigate the CARP/N2A interaction at the molecular level, first we analysed the CARP-receptor site in titin, UN2A-Ig81. While the 3D-structure of Ig81 is available,12 the primary binding site - UN2A - remains uncharacterised. Here, we deciphered the structure of UN2A by identifying its secondary structure content on a per residue basis using NMR spectroscopy, confirming sequence disorder using hydrogen/deuterium exchange mass spectrometry (HDX-MS) and diffusion coefficient calculation by NMR and, finally, calculating its 3D-structure using NMR-guided ab initio modelling.
To this effect, two-dimensional heteronuclear 1H-15N HSQC spectra and three-dimensional HNCO, HNCA (Cɑ), HNCACB and HN(CO)CACB spectra were acquired. These spectra provided in total five chemical shifts (H, N, C=O, Cα and Cβ) per residue. The 1H-15N HSQC spectrum of UN2A showed 90 resolved cross-peaks, 75 of which were assigned to individual residues (Figure 2A,B; Figure 3). Considering additional spectra, chemical shift information was obtained for 91 residues in total. Most of the assigned residues were located in the central region of UN2A (residues 34–73; to ease structural annotation, residue D9472 in UniprotKB Q8WZ42 is residue D1 throughout this work), while many residues in the N- and C-terminal sequence portions remained partially unassigned due to signal overlap. The latter is due to chemical shift similarity that reflects a similar chemical environment for the residues affected which, in turn, is indicative of structural disorder in these regions. The chemical shifts obtained were then used for the calculation of secondary structure at the residue level using TALOS-N15, which estimates the secondary structure content of a target protein based on the similarity of its experimental chemical shifts and sequence to those of a database of 3D-structures. In addition, TALOS-N calculates RCI-S2 values per residue based on experimental backbone chemical shifts.16 RCI-S2 values (ranging from zero to one, where one signifies maximally rigid) reflect residue rigidity in the picosecond-to-nanosecond time scale. In this work, TALOS-N was used to calculate the secondary structure conformation and RCI-S2 values for 91 residues in the UN2A sequence. In this way, 46 residues were estimated to adopt α-helical conformation, 43 residues were predicted to be in random coil formation and 2 residues in β-sheet; 45 residues showed S2<0.7, indicating inherently high flexibility (Figure 3). Notably, the 46 α-helical residues mainly clustered in the central region of UN2A (residues 34–73). RCI-S2 values for these residues were high (>0.7), indicating a well-structured segment. In contrast, flexible residues (S2<0.7) largely mapped to the N- and C-terminal sequences of UN2A (residues 1–33 and 74–110), which also had low secondary structure content, suggesting that these terminal regions are poorly ordered.
Figure 2: NMR characterization of titin N2A.
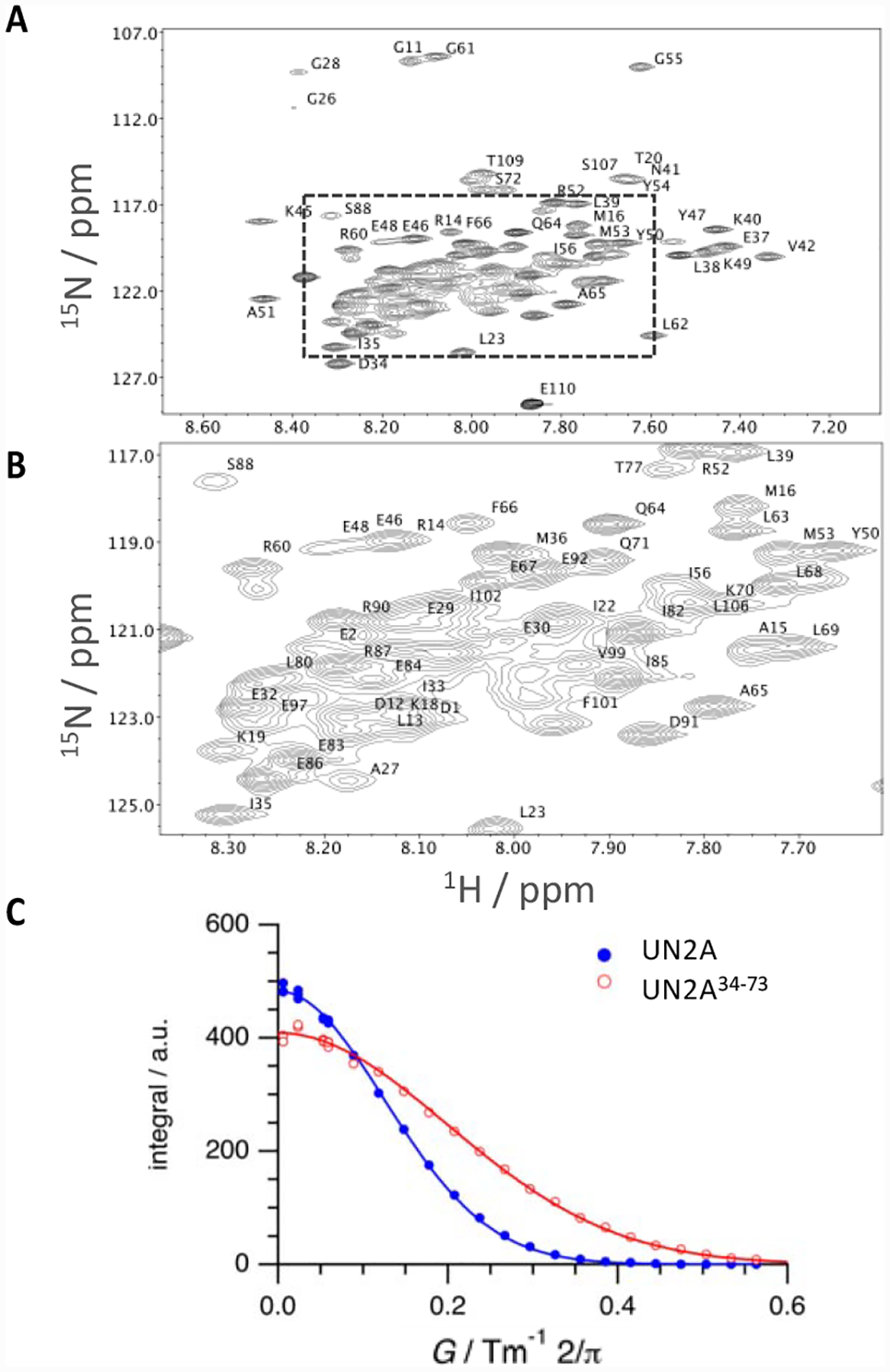
A., B. 1H-15N TROSY-HSQC NMR spectrum of UN2A. Assigned peaks are labelled. Entire spectral range (A) and (B) zoom into the region selected in A (box) are shown; C. Diffusion profiles acquired for UN2A (closed circles, blue) and its central segment, UN2A34−73 (open circles, red) and data fitting by applying Stejskal-Tanner equation (straight lines).
Figure 3: Structural analysis of titin UN2A-Ig81.
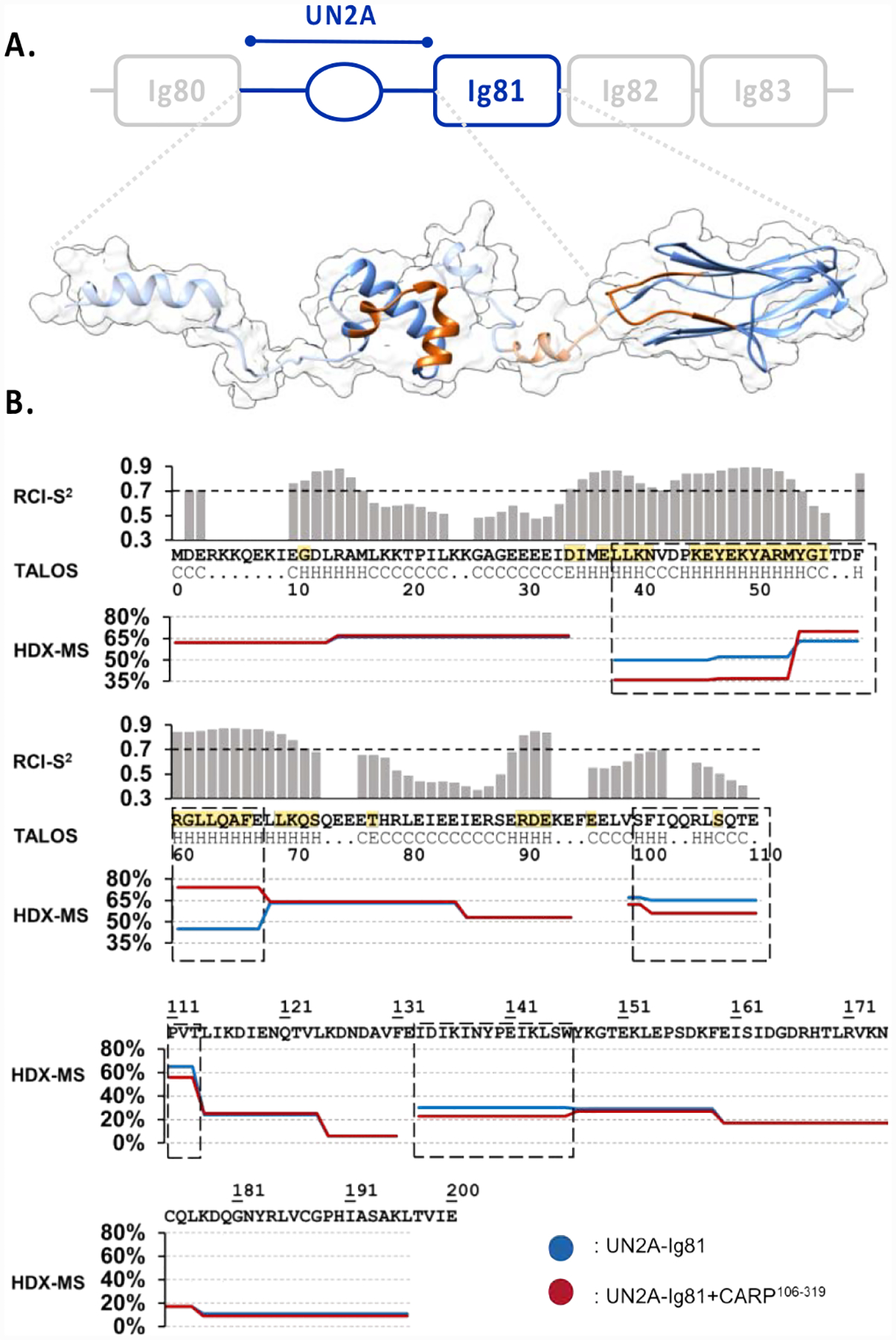
A. Proposed 3D-model of UN2A-Ig81. The experimentally determined, structured domains (UN2A34−73 and Ig81) are shown as solid ribbons, where UN2A34−73 corresponds to the top NMR-guided CS-ROSETTA model and Ig81 is PDB entry 5JOE. Predicted segments are displayed as a semi-transparent ribbon based on TALOS-N output. The model shown is a representative conformation among those coexisting in solution as a result of the flexible semi-helical linker segments. No rotational order on the orientation of domains is implied. The CARP-binding interface identified using HDX-MS is shown in orange; B. NMR-based TALOS-N analysis of UN2A (upper panel). The secondary structure of each residue is below the UN2A sequence (central panel): H indicates α-helix, E is β-strand and C is coil. RCI-S2 values are plotted in histogram format, where a value <0.7 is considered to indicate inherent residue flexibility. Assigned secondary structure and RCI-S2 values indicate the existence of an ordered, helical central fold in the UN2A sequence flanked by flexible N- and C- terminal tails. Residues identified by NMR to be influenced by CARP binding are in yellow. HDX-MS DRFU values for UN2A-Ig81 (lower panel) after 0.5min incubation time for both free (blue) and CARP-bound (red) samples. Sequences boxed (dash line) show significant differences in DRFU values, which is indicative of their participation in CARP binding.
To further characterize order/disorder in titin UN2A, we studied the dual UN2A-Ig81 construct by HDX-MS. In this method, the protein medium is supplemented with D2O and the exchange of hydrogen atoms comprising the protein with the deuterium of the medium is measured as a function of incubation time. Once the reaction has been quenched to pause the exchange, the sample is proteolyzed and the resulting peptides are analysed by MS. By mapping the relative fractional deuterium uptake (RFDU) of the peptides onto the protein sequence, residues in a protein core and in protein-protein binding interfaces can be identified as these display reduced RFDU values.17 In UN2A-Ig81, the two domain components have a comparable molecular mass (UN2A: 13.3 kDa; Ig81: 10.6 kDa), are joined by a flexible linker and do not interact with each other significantly as shown by size exclusion chromatography (Figure S1). Being part of a same construct and, thereby, assayed under the same experimental conditions of pH and temperature, the RFDU values of the individual UN2A and Ig81 domains are directly comparable. The crystal structure of Ig8112 confirmed that this domain folds into a compact β-sandwich, as characteristic of the immunoglobulin fold. Therefore, H/D exchange values for Ig81 can serve here as a direct reference for a small globular fold. The HDX-MS data (Figure 3B) recorded from UN2A-Ig81 had an excellent sequence coverage (ca 93%) and, thus, reported comprehensively on both UN2A and Ig81 domains. RFDU values for the well-folded Ig81 domain were 10–25% overall, with the long BC-loop having the highest RFDU at 30%. In contrast, UN2A had notably higher values: residues 38–67 in the central helical segment had an average RFDU of ~45% and peptides at both termini (residues 1–34 and 68–84) showed an elevated value of 64%. These data revealed that, compared to Ig81, UN2A is overall loosely packed and confirmed that the best-ordered region of UN2A is its central segment.
To confirm that the central three-helix bundle fold in UN2A is stable in solution, we investigated the overall size of full-length UN2A and UN2A34−73 by determining their respective hydrodynamic radii, RH, from diffusion coefficients (D) measured by NMR spectroscopy (Figure 2C). The RH value describes the size of a protein by considering it as a hydrated sphere of radius RH. Experimental RH values can then be compared with those predicted for the polypeptide chain in a fully folded state or as entirely unstructured ensemble (see Methods), which permits concluding on the compact nature of the fold in solution.18 In this work, the diffusion coefficient measured for UN2A was D = (9.5±0.1)×10−11 m2s−1, corresponding to a RH = 25.5 Å, while the values for UN2A34−73 were D = (1.56±0.03) ×10−10 m2 s−1 and RH = 15.5 Å. Predicted RH values for folded and unfolded states of UN2A were RH = 18.8 Å (folded) and RH = 32.2 Å (unstructured) and for UN2A34−73, RH = 14.6 Å (folded) and RH = 19.5 Å (unstructured). The comparison of experimental and theoretical RH values indicated that UN2A comprises both compactly folded and unstructured segments, supporting the conclusions drawn from RCI-S2 and RDFU values. In contrast, values for UN2A34−73 supported the view that this central segment forms a compact fold in solution.
The structured central segment of UN2A folds into a three-helix bundle
As the low chemical shift dispersion of UN2A prevented the acquisition of through-space NOE restraints, its structure calculation was performed by NMR-guided ab initio modelling in CS-ROSETTA.19 This software predicts the structure of a target protein guided by experimental backbone chemical shifts acquired by NMR spectroscopy. It is documented that the presence of disordered segments in a sequence deteriorates fragment picking and negatively affect the overall modelling performance of ROSETTA.20 Thus, to palliate the negative effects of the flexible regions identified, four increasingly complete sequence segments of UN2A were modelled in this study: UN2A34−73 (representing the most structured central segment), UN2A11−73 and UN2A34−110 (corresponding to the central fold extended N- or C-terminally by 23 or 37 residues, respectively) and full-length UN2A1–110.
CS-ROSETTA modelling of the central segment, UN2A34−73, successfully yielded a converged model with the top ten models displaying an RMSDCα = 2.3 Å (RMSDCα: root-mean-square deviation for Cα atoms), which is close to the standard convergence threshold of 2.0 Å (Figure S2). In addition, the 32500 decoys calculated by CS-ROSETTA were grouped in CALIBUR,21 which clusters individual conformers according to their RMSDCα to find subpopulations representing the most frequently modelled conformations. The largest model cluster (containing the most frequently calculated conformation) selected with CALIBUR contained 10216 decoys, with a RMSDCα value of 0.63 Å for the ten best models. For comparison, we attempted the modelling of UN2A34−73 using CS-ROSETTA in the absence of experimental chemical shift information. The calculated models in this case did not display convergence (RMSDCα = 5.44 Å for ten best models). Thus, our modelling results confirmed the known fact that experimental chemical shifts restrict the possible conformational space of the backbone and, therefore, help the selection of high-quality fragments in ROSETTA, improving modelling performance.22 The calculated consensus model for UN2A34−73 showed that it folds into a three-helix bundle, where the two N-terminal α-helices are approximately parallel to each other forming an α-hairpin and a third, C-terminal helix packs diagonally against the α-hairpin (Figure 3A; S2). This fold closely resembles that of the m-domain from cardiac MyBP-C3 (PDB: 2LHU),23 with which UN2A34−73 shares high sequence conservation (40% identity, 71% similarity).12 As models for MyBP-C3 and UN2A34−73 in this work have been calculated independently (see Methods), the fold agreement supports the validity of the NMR-guided modelling of UN2A34–73.
Attempts to calculate structural models in CS-ROSETTA for UN2A11−73, UN2A34−110 and full-length UN2A were unsuccessful. This outcome was not unexpected as NMR data were largely absent for N- and C-terminal sequences and, there where NMR data were present, high RCI-S2 values indicated structural disorder, which adversely affects modelling.20
In brief, we concluded that titin-UN2A forms a small tri-helical fold of ca 45 amino acid length, with the flanking sequences being long flexible linkers to the neighbouring Ig80 and Ig81 domains. These data and the crystal structure of Ig8112 allow us to propose now a structural model for the UN2A-Ig81 CARP-binding site in titin (Figure 3A).
The CARP binding interface on titin-N2A spans UN2A, linker and I81 components
To identify the CARP interface on titin at the residue level, we studied CARP106−319/UN2A-Ig81 binding using HDX-MS to analyse the differences in the RFDU values of free and complexed samples. This work used the C-terminal ankyrin-repeat fraction of CARP, CARP106−319, as this is known to mediate the interaction with titin.11,12 The result showed that the CARP binding site in titin comprises the central helical segment of UN2A, its C-terminal linker to Ig81, and the BC-loop in Ig81 (residues 133–146) (Figure 3; S3). Of particular note, the BC-loop in Ig81 is a uniquely extended feature not present in other Ig domains from titin.12 Titin Ig domains are classified into two well-characterised subtypes, termed N-conserved and N-variable, based on the structural features of the N-terminal pole of their β-sandwich fold.24 The long BC-loop in the N-terminal pole of Ig81 does not match the features of either subtype and it is unique among titin domains. Our finding contributes to support the view that individualized loop features in otherwise repetitive titin Ig domains have evolved to support the recruitment of sarcomeric partners to specific loci of the titin protein.25 A result of further interest was that, in UN2A34−73, a sequence segment had decreased RFDU values (38-LLKNVDPKEYEKYARM-53), while another showed increased values (54-YGITDFRGLLQAFE-67). The decreased H/D exchange results from a protection of the sequence by CARP binding, while the increased exchange suggests that that segment becomes displaced and more exposed to solvent upon CARP binding. This indicated that CARP binding induces a conformational change in UN2A.
A validation of the CARP-interface on titin-UN2A was sought by NMR and analytical size exclusion chromatography. In NMR, 15N-labeled UN2A was stepwise titrated with increasing concentrations of non-labelled CARP106−319 and the binding monitored in 1H-15N HSQC NMR spectra. The change in cross signals (Figure S4) allowed to identify residues with both a reduced signal intensity and a change in their chemical shifts. The result revealed that most binding-sensitive residues mapped to the helical UN2A34−73 segment, with a few contacts also present in the linker sequence C-terminal to UN2A (Figure 3A). The contribution of the N- and C-terminal flexible UN2A linker sequences to CARP binding was further tested using size exclusion chromatography that monitored binding of CARP106−319 to UN2A-Ig81, UN2A and the truncated variants UN2A34−110 and UN2A34−73 (Figure 4A–D). The role of Ig81 in CARP binding has been previously reported.12 The chromatograms revealed that full-length UN2A and UN2A34−110 bind to CARP106−319 comparably, but that UN2A34−73 forms a weaker complex that co-segregates in the column only partially. This result confirmed the involvement of the UN2A C-terminal linker sequence in CARP binding, while showing that the N-terminal linker is dispensable for the interaction. In conclusion, data from HDX-MS, NMR and chromatography showed that the binding interface of CARP in titin-N2A comprises the central structured segment of UN2A, the linker to Ig81, and the BC loop in Ig81.
Figure 4: Exploration of the titin/CARP interface through mutagenesis.
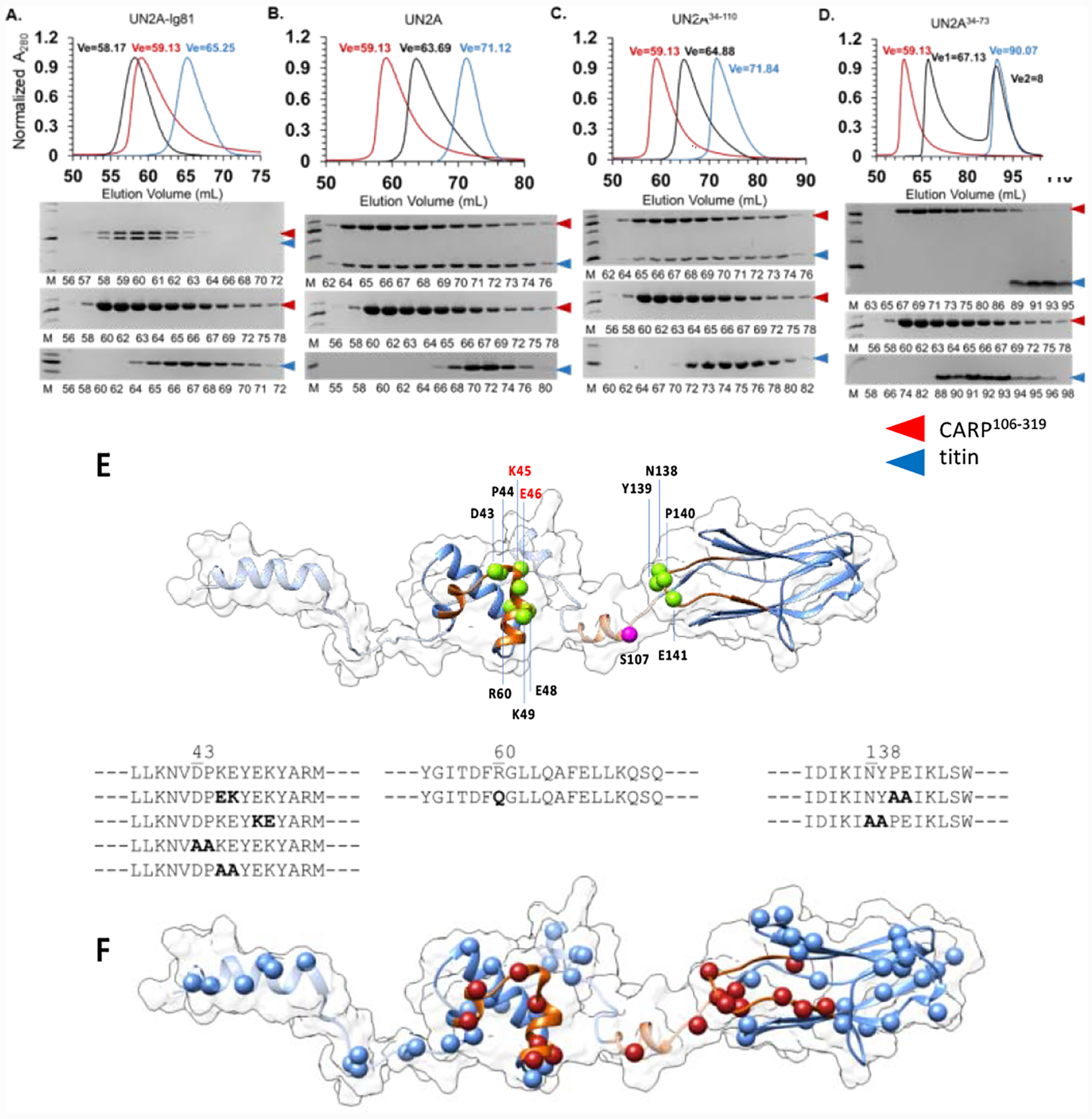
The binding of CARP106−319 to (A) UN2A-Ig81, (B) UN2A, (C) UN2A34−110, (D) UN2A34−73 was monitored by size exclusion chromatography. Complexes were formed by mixing individual proteins in a 1:1 molar ratio and subsequently applied to a Superdex 75 16/600 column (GE Healthcare) equilibrated in 25mM HEPES pH 7.5, 100mM NaCl, 1mM TCEP. Chromatograms were normalized to ease comparison. Chromatograms revealed that UN2A-Ig81, full-length UN2A and UN2A34−110 fully co-segregated with CARP106−319, indicating that the N-terminal flexible linker sequence of UN2A is not required for CARP binding. However, the diminished co-segregation of the UN2A34−73 variant with CARP106−319 confirmed that the C-terminal linker sequence contributes to the interaction; E. Sequence alterations introduced in titin by site-directed mutagenesis in this study. For reference, the phosphorylation site by protein kinase A (S107) previously identified34,51 is shown in magenta; F. Human single nucleotide variants from the gnomAD database mapped onto the 3D-model of titin UN2A-Ig81. Residues in the CARP-binding interface are shown in red. (Single nucleotide variants are listed in Table S1).
Previously, we reported the characterization of UN2A-I81 using SAXS.12 The radius of gyration (Rg), the molecular length (Dmax), the pair distance distribution function P(r) and the calculated SAXS-based molecular envelope agreed in revealing that UN2A-I81 adopts an extended molecular conformation in solution, where domains do not stack onto each other (as in a clamshell-like state). In the extended conformation of UN2A-I81, the CARP binding elements must be roughly co-linear, forming a broad surface. An initial SAXS-based low-resolution molecular envelope on the CARP:UN2A-I81 complex12 hinted at a potential binding of CARP at the junction of the UN2A and Ig81 domains. The identification of molecular binding interfaces in the current work is in full agreement with those earlier insights and permit now the confident interpretation of the low-resolution molecular shapes.
The binding interface of titin on CARP106−319 was also identified using HDX-MS. Interestingly, no difference on H/D exchange was observed between free and complexed CARP (Figure S5). This indicated that the same interface was involved in dimer formation and titin binding. A previous quantitative determination of the oligomeric state of CARP106−319, N2A and the CARP106−319/N2Acomplex, which used size exclusion chromatography with multiangle laser light scattering (SEC-MALLS) to establish their molecular mass12, revealed that CARP106−319 in isolation forms dimers but that these dissociate upon complexation with N2A, with the CARP106−319/N2A complex having a 1:1 stoichiometry. Taken together, the data show that titin binding not only breaks the CARP dimer as shown by SEC-MALLS, but that it also engages the same CARP surface as revealed now by HDX-MS. Thus, we deduced that the dimerization interface and the titin-N2A binding interface on CARP coincide so that N2A outcompetes CARP subunits by docking to their dimerization interface.
Susceptibility of the CARP/N2A interface to sequence alteration
To identify critical contact residues in the CARP/N2A interface, we sought to ablate the interaction by site-directed mutagenesis. Ten UN2A or UN2A-Ig81 variants containing single, double or quadruple residue exchanges in contact sequences were generated and their ability to bind CARP studied. In particular, UN2A variants sampled a DPKEYEK motif in the α-hairpin that is highly conserved in vertebrate titin (BLASTP of >1200 titin sequences), and the BC loop in Ig81. Namely, we studied variants UN2AK45E/E46K, UN2AE48K/K49E, UN2AD43A/P44A, UN2AK45A/E46A and UN2AR60Q (the latter mutation is located in the segment displaced upon CARP binding and has been linked to hypertrophic cardiomyopathy26); as well as UN2A-Ig81 variants that had a mutated helical segment and/or a mutated Ig81-BC loop: UN2A-Ig81K45E/E46K, UN2A-Ig81K45E/E46K/P140A/E141A, UN2A-Ig81P140A/E141A, UN2A-Ig81N138A/Y139A and UN2A-Ig81R60Q (overview of mutations in Figure 4E). As CARP106−319 changes its oligomeric state upon N2A binding from dimer (in isolation) to single subunit (in complex with N2A),12 interaction measurements using quantitative methods (as biolayer interferometry) proved unsuccessful. Thus, we used analytical size exclusion chromatography in this work. Notably, all variants – at exception of UN2AK45E/E46K – cosegregated with CARP106−319 in gel filtration as the wild-type sample. UN2AK45E/E46K (carrying a double charge reversal) showed a modest reduction in binding, verifying the involvement of these residues in CARP binding. However, UN2A-Ig81K45E/E46K co-segregated with CARP106−319 in gel filtration as the wild-type, showing that the presence of Ig81 was compensatory and rescued the interaction. It could be concluded that no set of mutations abolished CARP binding or weakened the interaction significantly.
The unexpected robustness of the CARP/N2A association led us to investigate the disruptive potential of 85 human single nucleotide variants in UN2A-Ig81 identified in the nonadditive databases gnomAD v2.1.1 and v3 (Figure 4F; Table S1). Of these, 61 variants occurred in folded segments of titin (UN2A34−73 or Ig81) and 24 variants were on the CARP interface. An analysis of all single nucleotide variants based on the structural knowledge of UN2A-Ig81 and its CARP interface that used FoldX27 and Missense3D28 indicated that none of the 85 variants has the potential to abolish the CARP/N2A interaction and that any disruptions to the complex might not be critical (Table S1). Two variants (rs1046404085, W9617C; rs1183241604, G9659D) appear to have the potential to destabilise Ig81 and thereby modestly weaken (but not abolish) CARP binding.
CARP induces the binding of titin N2A to F-actin
CARP is speculated to provide stability to the sarcomere by cross-linking titin filaments in the sarcomeric I-band.11 Here, we asked whether such cross-linking could also involve the main I-band component, filamentous actin (F-actin). Specifically, we used co-sedimentation assays to test the binding of UN2A and UN2A-Ig81 to F-actin in the presence or absence of CARP and/or calcium. In such assay (that uses actin extracted from rabbit muscle), the soluble titin samples remain in the supernatant fraction upon high speed centrifugation (100 000 g), while the large filamentous actin sediments and is found in the pelleted fraction. Upon association with F-actin, proteins that would normally remain in the soluble fraction upon centrifugation, will co-sediment and appear in the pellet. We found that UN2A and UN2A-Ig81 did not co-sediment with actin, but that they would do so in the presence of CARP (Figure 5A). A dependence of the results on calcium was not evident (Figure S6). These data pointed to the possibility of CARP acting as a cross-linker of titin and actin filaments in the sarcomere.
Figure 5: Titin-N2A binds F-actin in a CARP-dependent fashion.
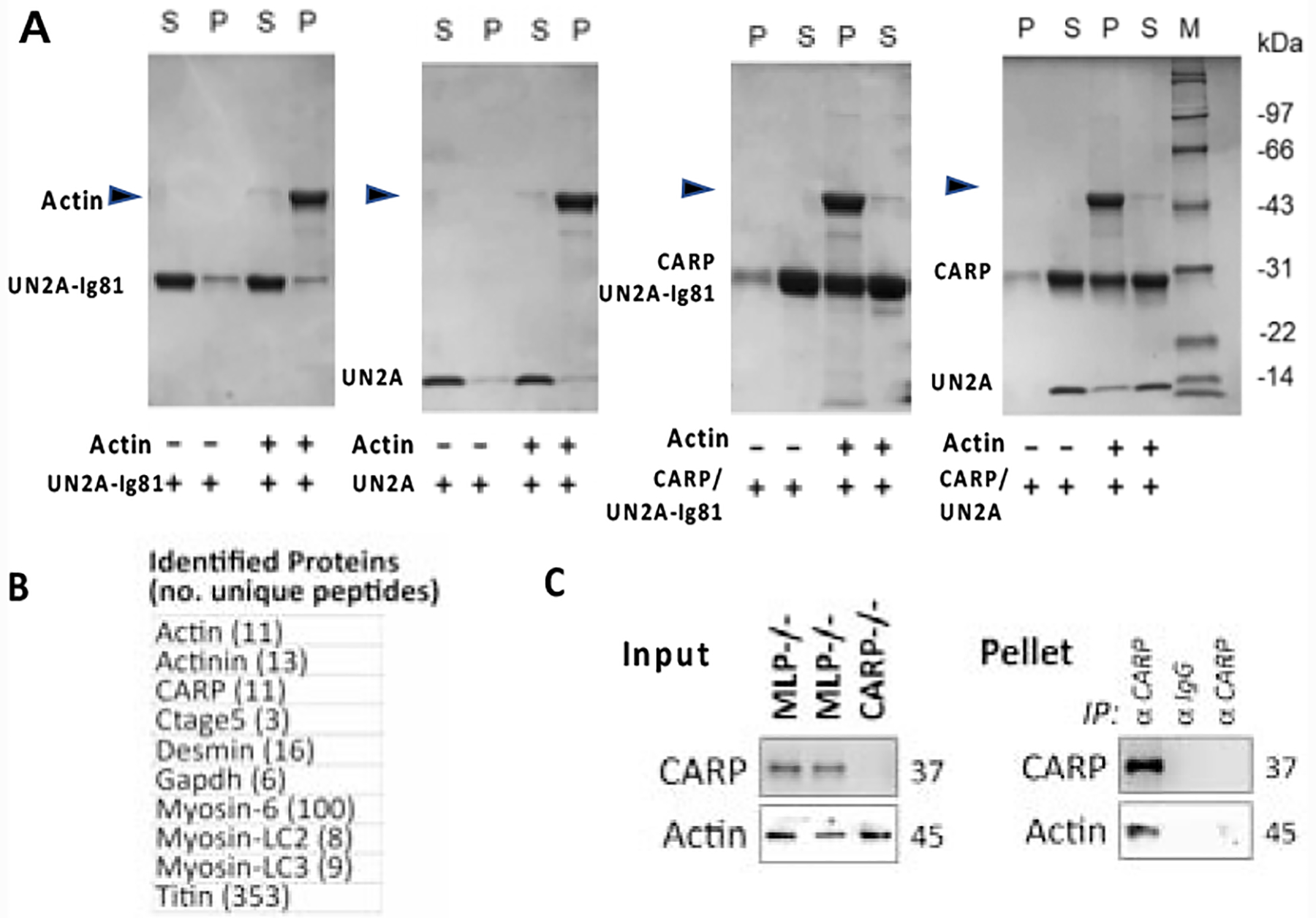
A. Actin co-sedimentation assays of UN2A-Ig81, UN2A, CARP106−319/UN2A-Ig81 and CARP106−319/UN2A. In the presence of CARP106−319, titin segments co-sediment with actin. (SDS-PAGEs and calcium dependence analysis are shown in Figure S7); B. and C. CARP immunoprecipitation from heart lysates. (B) Proteins unique to MLP−/− KO hearts identified using LC-MS/MS analysis are listed (proteins identified in both samples were excluded). Identity of proteins and number of unique peptides is stated. (C) immunoblot analysis of whole heart lysates (input) and immunoprecipitated proteins (IP pellet). Actin co-immunoprecipitate with CARP only in whole cardiac lysates from MLP−/− KO hearts, but not in lysates of CARP−/− KO hearts, or when rabbit IgG isotype controls were used instead of anti-CARP antibodies.
Next, we validated the CARP/titin/actin association in transfected C2C12 myoblasts (Figure 6). We used full-length Cherry-CARP or Cherry-CARP106−319 and titin GFP-Ig80-UN2A-Ig81. When transfected as single proteins, Cherry-CARP formed distinct puncta, while GFP-Ig80-UN2A-Ig81 displayed a diffuse localisation; none of the isolated samples associated with F-actin. Upon co-transfection, a subpopulation of Cherry-CARP and GFP-Ig80-UN2A-Ig81 was recruited to F-actin around the cell nucleus, resulting in a changed actin filament architecture (Figure 6A). Co-transfection of GFP-Ig80-UN2A-Ig81 and Cherry-CARP106−319 showed that both proteins strongly co-localised with actin filaments throughout the cell (Figure 6B). This result supports the conclusion that CARP/titin-N2A associate with F-actin.
Figure 6: Association of titin/CARP with F-actin in cells.
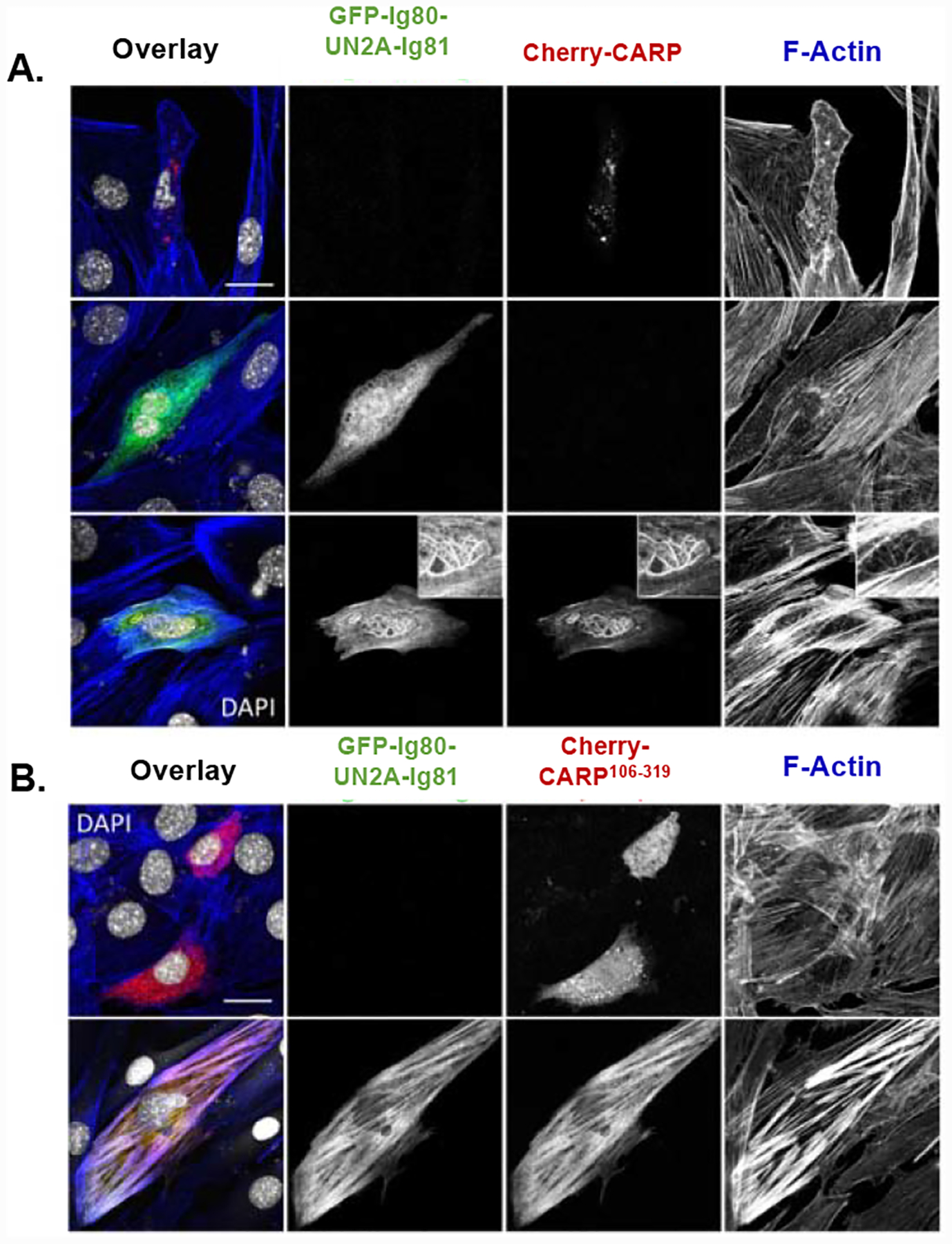
Transfection of CARP and titin-N2A constructs into C2C12 myoblasts. Cells were cotransfected with (A) Cherry-CARP and/or GFP-Ig80-UN2A-Ig81 as well as (B) Cherry-CARP106−319 and GFP-Ig80-UN2A-Ig81. Endogenous F-actin was stained with Alexa-647-Phalloidin, cell nuclei with DAPI. (Scale bar = 20μm).
To test the physiological validity of this deduction, particularly in the heart where CARP is most commonly studied, we performed immunoprecipitation of endogenous CARP in whole heart lysates from transgenic mice, followed by identification of precipitated proteins using proteomic mass spectrometry analysis. We used both MLP−/− 29 and CARP−/− 30 transgenic knockout mice; the former where chosen as they display high levels of CARP protein expression, while the latter are a null mutant that served as a control for unspecific protein binding to the anti-CARP antibody. The results (Figure 5B; S7) confirmed the association of CARP with both titin and actin, with other sarcomeric proteins also being identified (including myosin and α-actinin that are also actin-associated proteins). We verified the CARP/actin interaction in heart lysates by testing for the presence of cardiac actin in CARP immunoprecipitates from MLP−/− KO mice using immunoblotting. CARP/actin binding was found in MLP−/− heart lysates when precipitated with CARP, but not with unspecific rabbit IgG, or in CARP−/− lysates (Figure 5C).
CARP increases myofibrillar stiffness
Finally, we considered that a CARP/titin/F-actin interaction might plausibly cause a modulation of sarcomere mechanics. To gain an insight into this question, we asked whether the exposure of myofibrils to CARP106−319 solutions might alter their passive tension. In this study, we used skeletal muscle fibres from mice as these are rich in N2A-containing titin, while cardiac tissue of small vertebrates is largely depleted of this titin isoform with isoform N2B being dominant instead.7,8 Thus, we studied permeabilised skinned psoas muscle fibres before and after incubation in a relaxing solution containing CARP. Muscle fibres were subjected to an 8-step, passive stretch-hold ramp protocol from 2.2 to 3.0μm sarcomere length (SL), with steady-state force measured at each step. A similar sized sham protein, the non-muscle mCitrine fluorescent protein, was used as a control. Remarkably, the results (Figure 7A) showed that CARP incubation increases passive tension 20–40% at all SLs, suggesting that CARP induces a notable increase in fibre stiffness through its interaction with sarcomeric components. This finding points to CARP having a mechanical role in the sarcomere by acting as a modulator of the stretch response. We attempted to confirm that the observed effect was due to a direct effect of CARP on F-actin by pursuing the extraction of F-actin with gelsolin. However, gelsolin extraction is known to be effective only in cardiac muscle31 and invertebrate skeletal muscle,32 but does not sever F-actin in the skeletal muscle fibres. In vertebrate skeletal muscle, actin is protected by nebulin and its resistance to gelsolin extraction is well documented.33 Thus, F-actin extraction could not be employed as validation method in this study. Nevertheless, our data prompt now a further investigation in vivo and in fibres of CARP’s role as a rapid stress-induced modulator of sarcomere mechanics.
Figure 7: Myofiber stiffening by CARP.
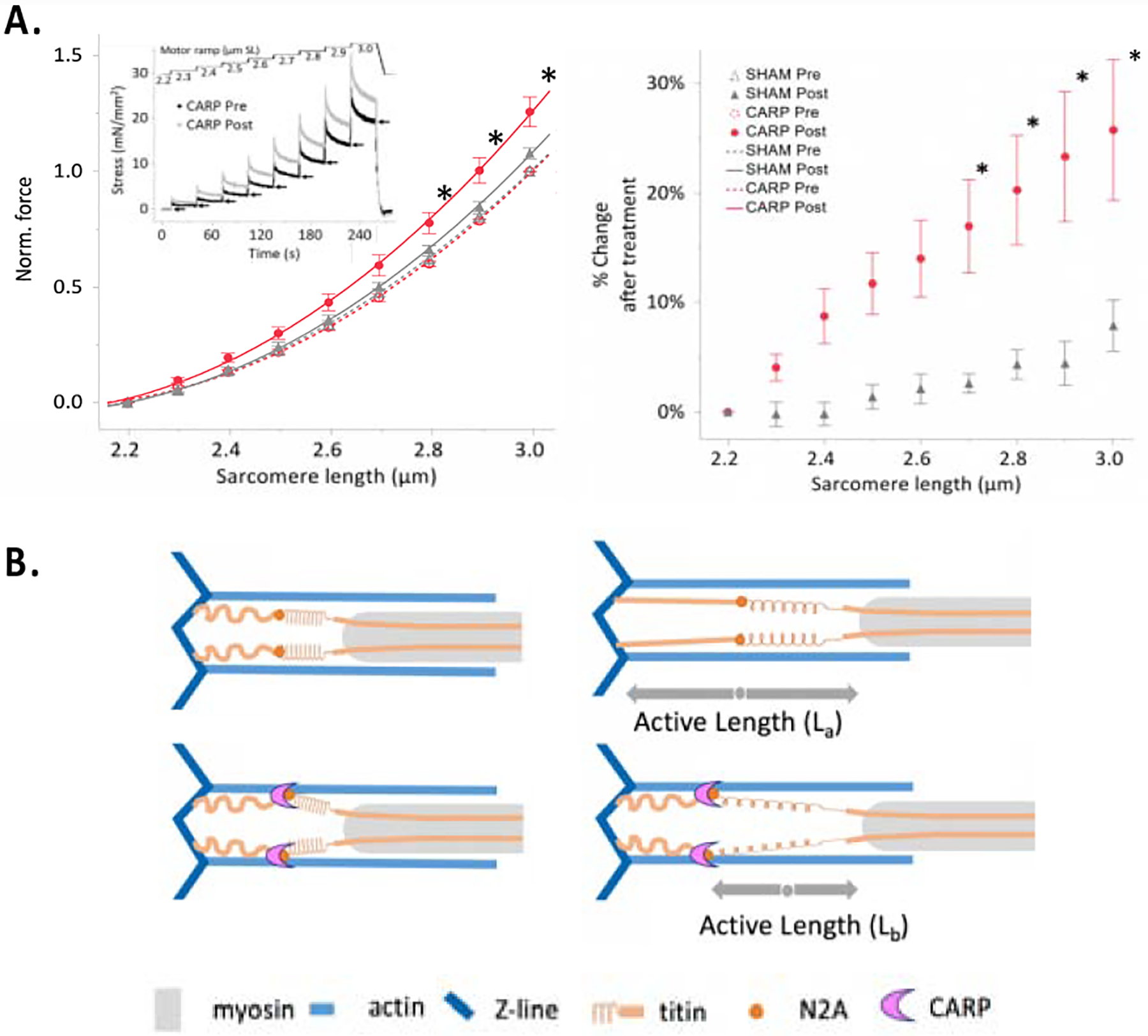
A. Passive force of permeabilised fiber bundles. (Left) The passive force-SL curves from fibers pre- and post-treatment with CARP (n = 10) or sham (n = 9) proteins. Data are fitted with second order polynomials (R2 > 0.92) and surrounded by the 95% confidence interval of the fit. Inset, mechanics protocol. Arrows indicate quasi-steady-state forces that were used to calculate data points in main figure. (Right) The percent change in force after treatment of CARP or the sham protein mCitrine, relative to before treatment. *P < 0.01, CARP treated vs all else (mean ± s.e.m); B. Proposed mechanical role of a potential CARP-based clamp. The CARP-mediated cross-linking of titin and actin at the N2A locus might effectively incapacitate the extension of titin’s proximal spring, enhancing the contribution of the stiffer PEVK region. Thereby, CARP could cause titin isoforms to perform as shorter, stiffer variants.
Discussion
The titin-N2A element in the elastic sarcomeric I-band is located at the junction of two main spring regions of titin: the proximal poly-Ig tandem and the PEVK-rich region. Although initially viewed as a mechanical element, a recent in vitro study that used atomic force microscopy has shown that the compliance of UN2A has a negligible effect on the passive force that titin generates – both as single titin chain or in its CARP complexed form.34 This indicates that UN2A is not a mechanical component of titin per se. This view is further strengthened by the structural analysis of UN2A in this study, that reveals it to consist of a small (ca 45 amino acid length) helical fold flanked by long, flexible, helix-containing sequences that connect it to neighbouring Ig domains (Figure 3A). UN2A must now be deemed improbable to have a primary mechanical function per se in muscle. Instead, our findings point to a contribution of titin N2A to muscle mechanics exerted via its binding of CARP, that appears to induce cross-linking of the titin/actin filaments. Our data reveal that CARP induces the formation of CARP/titin-N2A/actin complexes in vitro, in cells and in heart lysates and that CARP increases the passive tension of myofibrils when added exogenously. We reason speculatively that a CARP-clamp at the N2A locus would effectively shorten the length of titin’s elastic fraction by incapacitating the extension of its proximal I-band poly-Ig tandems (Figure 7B). Thereby, CARP binding would swiftly confine the mechanical compliance of titin into its stiffer PEVK-rich subregion, C-terminal to the N2A element. Thus, CARP might increase sarcomere stiffness by effectively transforming pliant titin isoforms into effectually shorter and stiffer variants. Furthermore, by acting as a modulator of stretch, CARP might influence a variety of mechano-activated pathways, contributing to overall protective outcomes in muscle as e.g preventing the apoptosis of cardiomyocytes triggered by stretch overload.35 In this regard, it must also be noted that titin N2A is a signalling hub that recruits various proteins such as the methyltransferase Smyd2, chaperone HSP90, P94/calpain3 and myopalladin.36 It is plausible that CARP links the functions of these proteins at the N2A locus to the mechanical state of the sarcomere. In addition, the rapid CARP-controlled response mechanism could reinforce upon stress a recently speculated basal mechanism, where domain Ig83 in N2A allegedly reduces the compliance of titin via a Ca2+-dependent interaction with actin.37,38 Jointly, the CARP- and Ca2+-dependent mechanisms reveal the titin N2A locus as a central regulator of sarcomere mechanics. Exploring the interplay of N2A-based mechanisms, their distinct regulation and physiological time courses will bring essential knowledge of the (patho)physiology of mechanical adaptation in muscle.
Finally, our model provides a rationale to the correlative enrichment in titin N2BA isoforms and CARP in failing human hearts, which are under biomechanical stress from increased ventricular volumes and pressure. Increased ratios of N2BA titin reduce passive tension.7,8 However, CARP up-regulation could counteract this pathological increase in compliance, acting to stabilise the sarcomere and preserve beating force in hearts. Skeletal muscle from mice lacking all three muscle ankyrin repeat proteins (CARP, MARP, DARP) shows a decrease in fibre stiffness, supporting this deduction.39 N2A-containing titin is dominant in postnatal skeletal muscle, and so the stiffness of the latter can be strongly influenced by CARP up-regulation upon stress. Stress states in skeletal muscle share many similarities with cardiomyocytes under dilated cardiomyopathy and, thus, point to the N2A/CARP node as a fundamental and global mechanism of striated muscle stress response.
Methods
Expression clones
The expression vectors of human CARP106−319 (residues 106–319; UniprotKB Q15327), UN2A (residues 9472–9581) and UN2A-Ig81 (residues 9472–9671) from human titin (UniprotKB Q8WZ42) have been previously reported.12 (To ease the structural annotation of UN2A, residue D9472 from titin is taken as residue D1 throughout this work). The truncated variant UN2A34−110 (residues 9505–9581) was produced by polymerase chain reaction (PCR), where the coding insert was introduced into the vector pET-trx1a (EMBL) using NcoI and KpnI restriction sites. The pET-trx1a vector adds a His6-Tag, a thioredoxin domain and a tobacco etch virus protease recognition sequence N-terminal to the target gene. UN2A34−110 was then used as starting point to generate UN2A34−73 (residues 9505–9544), where the codon for residue E74 was mutated into a stop codon (TAA) using PCR according to the New England Biolabs Q5 site directed mutagenesis protocol. Single amino acid mutations and codon terminations in variants UN2AK45E/E46K, UN2AE48K/K49E, UN2AD43A/P44A, UN2AK45A/E46A, UN2AR60Q, UN2A-Ig81K45E/E46K, UN2A-Ig81K45E/E46K/P140A/E141A, UN2A-Ig81P140A/E141A, UN2A-Ig81N138A/Y139A and UN2A-Ig81R60Q were introduced following the same protocol.
For confocal visualisation during myoblast studies, full-length CARP, CARP106−319 or titin Ig80-UN2A-Ig81 (residues 9353–9671) were amplified by PCR and subcloned into the mammalian expression vectors pEGFP-C1 (Clontech) or pmCherry-C1, as described previously.11 The pmCherry-C1 vector leading to the mCherry-tagged constructs was generated by in-frame replacement of GFP in pEGCP-C1 by mCherry.
All constructs were confirmed by sequencing.
Recombinant protein production
Protein samples were produced in E. coli Rosetta (DE3) cells (Merck Millipore) as previously described.12 In brief, cells were grown at 37°C in Luria-Bertani medium supplemented with 25μg/ml kanamycin and 34μg/ml chloramphenicol up to OD600=0.6. Protein expression was induced with 0.5mM isopropyl β-D-1-thiogalactopyranoside and cells further cultured overnight at 18°C. Cells were harvested by centrifugation and lysed by sonication in 25mM HEPES pH 7.5, 300mM NaCl, 1mM dithiothreitol, supplemented with an EDTA-free protease inhibitor cocktail (Roche Applied Science). Lysates were clarified by centrifugation and proteins isolated from the supernatant by Ni2+-affinity chromatography on a Histrap™ HP 5mL column (GE healthcare) in lysis buffer. Tag removal was by cleavage with TEV protease. Cleaved samples were further purified by reverse metal affinity chromatography and size exclusion chromatography, the latter used a HiLoad Superdex 75 16/60 column (GE healthcare) equilibrated in 25mM HEPES pH7.5, 100mM NaCl, 1mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP).
For NMR analysis, 13C and 15N isotopically labelled UN2A samples were produced in E. coli Rosetta (DE3) cells (Merck Millipore) grown in M9 minimal media containing 14.6g Na2HPO4, 5.4g KH2PO4, 1g 15NH4Cl, 2mM MgSO4, 15μM CaCl2, 1.5μM MnCl2, 0.5μM FeSO4 and 2g Glucose-13C. For 2H, 13C and 15N isotopic labeling of UN2A, the culture media was further supplemented with 100% D2O. Doubly (13C and 15N) and triply (2H, 13C and 15N) labeled UN2A samples were purified as described above and concentrated up to 1.2mM in 30mM phosphate (Na2HPO4/NaH2PO4) pH 7.5, 100mM NaCl and 5% [v/v] D2O.
For the production of CARP106−319 in complex with N2A construct variants, individual samples were produced independently in a purified state and subsequently mixed at 1:1 molar ratio. Complexed samples were further purified by size exclusion chromatography using a HiLoad Superdex 75 16/600 column (GE Healthcare) in 25mM HEPES pH7.5, 100mM NaCl, 1mM TCEP.
All samples were stored at 4 °C until further use.
NMR spectroscopy
Triply (2H, 13C and 15N) labelled UN2A samples were used for acquiring TROSY edited versions of two-dimensional 1H-15N heteronuclear single quantum correlation (HSQC) spectra and three-dimensional 2H decoupled HNCO, HNCA, HNCACB and HHN(CO)CACB spectra to accomplish backbone assignment. All NMR spectra were collected at T=298K on a Bruker Avance III 600MHz spectrometer equipped with a TCI cryo-probe. One-dimensional NMR spectra were processed and analysed with TopSpin software40 whereas two- and three-dimensional data were processed using nmrPipe41 and assigned with nmrViewJ42. The estimated Ramachandran assignment of dihedral angles (ϕ, ψ) was predicted for each residue in the UN2A sequence from backbone resonances based on chemical shifts using the TALOS-N software.15 In addition, TALOS-N was used to estimate random coil index (RCI)-S2 values per residue based on experimental backbone chemical shifts.16
The interaction between non-isotopically labelled CARP106−319 and 15N-UN2A was monitored by the acquisition of a series of two-dimensional 1H-15N HSQC spectra (TROSY edited version). Molar ratios of CARP106−319 and UN2A samples were measured by NMR by keeping the total amount of UN2A constant and varying the amount of CARP106−319 added in the molar ratios, n, of n = 0, n = 0.13, n = 0.26, n = 0.43, n = 0.66, n = 1, n = 1.5 and n = 2. The titration data were acquired at T=298K and data processing was done as described above.
Chemical shift-based model calculation in CS-ROSETTA
The calculation of 3D-models was performed on the scientific computer cluster of the University of Konstanz (SCCKN; https://www.scc.uni-konstanz.de) using CS-ROSETTA v3.6 and ROSETTA v3.8.19 For this, chemical shifts derived from NMR data were input into CS-ROSETTA in TALOS-N format as corresponding to UN2A sequences with different levels of truncation: UN2A, UN2A11−74, UN2A34−110 and UN2A34–73. For residues unassigned in NMR spectra and, therefore, lacking chemical shifts, fragment picking was solely based on sequence similarity. The fragments were then used for ROSETTA Monte Carlo assembly and a total of 32500 model decoys generated per UN2A sequence. The energy of each decoy was then re-scored to include an experimental potential accounting for chemical shift data.19 The ten models with the lowest total energy were selected. The modelling was considered to have converged if the Cα-atom root-mean-square deviation (RMSDCα) of the ten top conformers against the best model was lower than 2.0Å. Disordered parts of the protein with S2 <0.7 remained untrimmed and were part of the calculated models. Importantly, to avoid modelling bias, the ROSETTA fragment library used in this study did not include MyBP-C3 (PDB: 2LHU), with which UN2A shares sequence similarity.12
To further evaluate modelling results, decoys calculated with CS-ROSETTA were subjected to cluster analysis using CALIBUR that reported the three largest conformational subpopulations.21 The ten lowest-energy decoys of the first and largest cluster were used to calculate an RMSDCα value. The stereochemistry of selected models was assessed using MOLPROBITY.43
Hydrogen/deuterium exchange mass spectrometry (HDX-MS)
HDX-MS was performed on CARP106−319, UN2A-Ig81 and the CARP106−319/UN2A-Ig81 complex using a Waters Synapt G2Si spectrometer equipped with a nanoACQUITY UPLC system with HDX technology and a LEAP autosampler. Individual proteins were purified by size exclusion chromatography in 25mM phosphate pH 7.5, 100mM NaCl and 1mM TCEP, immediately prior to analysis. The final concentration was 5μM for each sample. For each deuteration time, 4μL sample was equilibrated at 25°C for 5min and then mixed with 56μL D2O buffer containing 25mM phosphate pH 7.5, 100mM NaCl, 1mM TCEP for 0, 0.5, 1, 2, or 5 min. The exchange was quenched with an equal volume of quench solution (3M guanidine, 0.1% formic acid pH 2.66).
The quenched sample (50μL) was injected into the sample loop, followed by digestion on an in-line pepsin column (Pierce Inc.) at 15°C. The resulting peptides were captured on a BEH C18 Vanguard pre-column, separated by analytical chromatography (Acquity UPLC BEH C18, 1.7μM, 1.0 × 50mm, Waters Corporation) using a 7–85% acetonitrile gradient in 0.1% formic acid over 7.5min, and electrosprayed into the Waters SYNAPT G2Si quadrupole time-of-flight mass spectrometer. The mass spectrometer was set to collect data in the Mobility, ESI+ mode; mass acquisition range of 200–2000 (m/z); scan time 0.4s. Continuous lock mass correction was accomplished with infusion of leu-enkephalin (m/z = 556.277) every 30s (mass accuracy of 1ppm for calibration standard). For peptide identification, the mass spectrometer was set to collect data in MSE, ESI+ mode. The peptides were identified from triplicate MSE analyses of 10μM sample, and data were analysed using PLGS 3.0 (Waters Corporation). Peptide masses were identified using a minimum number of 250 ion counts for low energy peptides and 50 ion counts for their fragment ions. The peptides identified in PLGS were then analysed in DynamX 3.0 (Waters Corporation) and the deuterium uptake was corrected for back-exchange as previously described.44 The relative deuterium uptake for each peptide was calculated by comparing the centroids of the mass envelopes of the deuterated samples vs. the undeuterated controls as reported.45 The experiments were performed in triplicate; independent replicates of the triplicate experiment were performed to verify the results.
Determination of diffusion coefficients by NMR spectroscopy
Diffusion NMR data were recorded at T=298 K by using a pulse sequence comprising a stimulated echo assisted by bipolar gradients, G,46 employing a diffusion time (Δ) of 120 ms and a gradient length (δ) of 10 ms (UN2A) as Δ=80 ms and δ=6 ms (UN2A34−73) along the z axis. Gradients were calibrated as described.47 Integrals for proton signals (I) were determined in the spectral range between 0.5 and 4.0 ppm (UN2A) as 8.3 and 6.5 ppm (UN2A34−73) and used for the determination of the diffusion coefficient, D, according to
where γ is the gyromagnetic ratio of protons. Samples used for diffusion coefficient determination were concentrated to 200 μM (UN2A) and 30 μM (UN2A34−73), respectively. The hydrodynamic radius (RH) was calculated using the Stokes-Einstein equation:
in which kB is the Boltzmann constant and η = 0.9 mPa•s at T = 298 K. For comparison, the theoretical RH value of the protein assuming a properly globular fold was calculated using:48
where N represents the number of residues comprising the protein under investigation. The RH value that considers the protein as an unstructured, unfolded chain was calculated applying:48
Analysis of single nucleotide variants
Single nucleotide variants in UN2A-Ig81 and their corresponding frequencies were obtained from gnomAD v2.1.1 and v3. Meta-SNP (http://snps.biofold.org/meta-snp) was used to predict the damage potential of the variants. Meta-SNP produces a consensus prediction based on PANTHER, PhD-SNP, SIFT and SNAP predictors. It outputs a reliability index (RI) for each variant, where predictions with RI ≥5 are on average ~87% accurate. Further, single nucleotide variants and the in vitro interface mutations generated in this study were analysed using Missense3D (http://www.sbg.bio.ic.ac.uk/~missense3d) that estimates the impact of the exchange on the protein structure.28 Those variants and in vitro mutations that localised to domain I81 (PDB: 5JOE) were also analysed using FoldX.27 FoldX quantitatively estimates the contribution of residue interactions to protein stability. The free energy of folding (ΔG) of the protein is calculated before and after mutation and the difference between the wildtype and mutant structure (ΔΔG) indicates the relative destabilisation of the change. Positive differences indicate destabilising changes and negative differences are stabilising. ΔΔG values over 2–2.5kcal/mol are generally considered as destabilising.
Actin co-sedimentation
Actin was prepared from rabbit psoas muscle.49 G-actin was centrifuged at 100,000g for 30min and the supernatant was polymerised in 50mM NaCl, 20mM MOPS (pH7.0), 5mM MgCl2. Proteolytic inhibitors were added to the polymerised actin as CARP106−319 is sensitive to proteolysis. Titin UN2A-Ig81, UN2A and CARP106−319 were centrifuged at 100,000g for 30min immediately before the assay. UN2A-Ig81 or UN2A or CARP106−319 (20μM) were added to actin (10μM) in a total volume of 100μl of buffer. Assay buffer was 50mM NaCl, 20mM MOPS (pH7.0), 5mM MgCl2 and 2mM CaCl2 or 2mM EGTA. A similar assay was performed with UN2A-Ig81 plus CARP106−319 (total 20μM) or UN2A plus CARP106−319 (total 20μM). Samples were incubated at 23°C for 1h and centrifuged at 100,000g for 30min. Supernatant and pellet fractions were analysed by SDS-PAGE with 12% acrylamide gels.
Co-immunoprecipitation of CARP interacting proteins
Identification of CARP interacting proteins by mass-spectrometry was performed by immuno-precipitating endogenous CARP from whole cardiac lysates of muscle lim protein (MLP/Csrp3) transgenic knockout mice29 or CARP knockout mice.30 Mice were maintained in a temperature and light-controlled room (22°C, 14h light/10h dark) and fed ad libitum with a standard diet. Mice were euthanised using carbon dioxide, followed by cervical dislocation and hearts harvested. Heart tissues were lysed into lysis buffer (300mM NaCl, 20mM Tris-HCl pH8, 1% Triton X-100, 0.1% SDS, 1x Protease Inhibitor Cocktail (Roche), 2mM DTT) and sonicated on ice for 2min. Soluble proteins were separated from insoluble cellular debris by centrifugation for 10min at 15,000rpm at 4°C. The supernatant was transferred into a new container and pre-cleared with Protein-G linked magnetic beads (Dynabeads, Life Technologies) for 1h at 4°C on a shaker to remove proteins that bind non-specifically to the beads. After removal of beads, 1μg of CARP antibody (see below) was added to the lysates and incubated overnight at 4°C. Following the incubation, Protein-G coupled magnetic beads were added to the lysate and incubated on a shaker for 3h at 4°C. Beads were washed 3 times with ice-cold 1 × phosphate buffered saline, supplemented with 0.2% NP-40, followed by 2 washes with ice-cold 10mM Tris-HCl pH8, and 2 washes with ice-cold distilled water. After removal of wash buffer, beads with bound complexes were analysed by mass-spectrometry for identification of proteins or SDS-PAGE followed by silver stain (Sigma Aldrich) for visualisation of proteins or immunoblot analysis for analysis of interaction partners.
For immunoprecipitation and immunoblot experiments the following primary antibodies were used: CARP (polyclonal rabbit, Ankrd1-1; Myomedix, Germany); cardiac actin (monoclonal mouse, clone Ac1–20.4.2, Progen Biotechnik); Rabbit IgG isotype control (Thermo Fisher; 10500C). Secondary antibodies used in immunoblot detection were from Cell Signaling (horseradish peroxidase linked -anti-rabbit IgG: #7074, -anti-mouse IgG: #7076 and -anti-rabbit light chain specific: # 93702).
All procedures involving animals were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of California San Diego (S13009) and were performed conforming to the NIH Guide for the Care and Use of Laboratory Animals.
Proteomics LC-MS/MS analysis
For identification of proteins in co-immunoprecipitates of CARP, 25μl 5% RapiGest (Waters Corporation, 5 Pack of 1mg Vials, 186001860) in 10 × TNE buffer (100mM Tris-HCl, 2M NaCl, 10mM EDTA, pH 7.4) was mixed with the beads-bound immunocomplexes dissolved in 225μl distilled water. The sample was boiled for 5min after which the magnetic beads were removed and 375μl water was added. TCEP was added to the final concentration of 1mM and incubated at 37°C for 30min. Iodoacetamide was then added to a final concentration of 2mM and incubated at 37°C for 30min. Before the addition of trypsin, TCEP was added to a final concentration of 2mM. Trypsin was then added to a final concentration of 1:50 trypsin:protein and incubated overnight at 37°C. To remove RapiGest, HCl was added to a final concentration of 250mM and incubated at 37°C for 1h. The sample was then centrifuged at 14,000 rpm at 4°C for 30min. The supernatant was removed from the pellet into a fresh tube. The peptides were then extracted using Aspire RP30 desalting columns (Thermo Scientific) before injection into the mass spectrometer. Trypsin-digested peptides were analysed by liquid chromatography (LC)-MS/MS with nanospray ionization. All nanospray ionization experiments were performed using a QSTAR-Elite hybrid mass spectrometer (ABSCIEX) interfaced to a nanoscale reversed-phase high-pressure liquid chromatography (Tempo) using a 10cm-180 ID glass capillary packed with 5μm C18 Zorbax™ beads (Agilent). The buffer compositions were as follows: Buffer A was composed of 98% H2O, 2% acetonitrile, 0.2% formic acid, 0.005% trifluoroacetic acid; Buffer B was composed of 100% acetonitrile, 0.2% formic acid, and 0.005% trifluoroacetic acid. Peptides were eluted from the C-18 column into the mass spectrometer using a linear gradient of 5–60% Buffer B over 60 min at 400μl/min. LC-MS/MS data were acquired in a data-dependent fashion by selecting the 6 most intense peaks with charge state of 2 to 4 that exceeds 20 counts, with exclusion of former target ions set to 120 seconds and the mass tolerance for exclusion set to 100ppm. Time-of-flight MS were acquired at m/z 400 to 1800 Da for 0.5s with 12 time bins to sum. MS/MS data were acquired from m/z 50 to 2,000Da by using “enhance all” function and 24 time bins to sum, dynamic background subtract, automatic collision energy, and automatic MS/MS accumulation with the fragment intensity multiplier set to 6 and maximum accumulation of 2s before returning to the survey scan. Peptide identifications were made using Mascot (Matrix Sciences) and paragon algorithm executed in Protein Pilot 3.0 (ABSCIEX).
Protein expression and localisation in C2C12 cells
To study the colocalisation of titin and CARP samples with actin filaments in the cell context, expression clones were transfected into C2C12 cells using Lipofectamine 2000 (Thermo Fisher). Following 3 days of expression, cells were fixed with 4% paraformaldehyde in phosphate buffered saline for 5min at room temperature. Cells were permeabilised using 1x phosphate buffered saline supplemented with 0.2% Triton X-100 for 5min and subsequently stained with 4′,6-diamidino-2-phenylindole (DAPI) and Alexa-647 Phalloidin (Thermo Fisher) dissolved in gold buffer (20mM Tris-HCl pH7.5, 155mM NaCl, 2mM ethylene glycol tetraacetic acid, 2mM MgCl2, 5% bovine serum albumin) for 1h. After washing cells for 3 times with 1x PBS, cells were mounted in fluorescent mounting medium (DAKO) and processed for imaging on an Olympus Fluoview 1000 confocal microscope or Leica SP5 confocal microscope in sequential scanning mode using a 40x or 63x oil-immersion objective and zoom rates between 1 and 4.
Force measurements on single myofibers
Male C57BL/6N mice (Charles River Laboratories, Wilmington, USA) were used at the age of 5–8 weeks. Animals were anesthetised with isoflurane 100% (v/v), rimadyl 4 mg/kg body weight and 250 I.U. heparin sodium anticoagulated. They were killed by cervical dislocation and permeabilised (“skinned”) psoas fibres were prepared from psoas muscles using standard techniques.50 On the day of experiments, muscles were removed from the storage solution and vigorously washed in relaxing solution (2.5mM ATP, 14.5mM phosphocreatine, 20mM MOPS, 5mM K2EGTA, 2.5mM magnesium acetate, 170mM K-propionate, pH 7.2) on ice. Small bundles of 3–8 fibres were separated from the muscle and attached lengthwise to a piezomotor on one end and a force transducer on another via aluminium clamps (Scientific Instruments, Heidelberg, Germany). Force data were recorded at 1000 Hz at room temperature. Each fibre bundle was suspended in a bath of relaxing solution and initially set to slack sarcomere length (SL; ~2.2μm), which was measured by laser diffraction. Length changes were accomplished by manual or computer-driven means. For passive tension measurements, fibres were stretched from slack length in equidistant 0.1μm SL steps between 2.2 and 3.0μm at a rate of 0.1μm SL per second. After each step, force relaxation was allowed for a 60s hold period. The force value at the end of the hold time was used for analysis. For each preparation, fibers were subjected to stretch-hold two times, once before and once after a 10min incubation with recombinant CARP106−319 or mCitrine protein (control) (0.1μg/mL in both cases). Force values were normalised to the maximum force reached at the end of the last stretch step in control fibres.
Statistics were calculated using a two-way full factor ANOVA conducted on force data with main fixed effects SL, condition (pre-treatment / post-treatment / pre-sham / post-sham), and a random (repeated measures) effect for individual. Significant main effects were further assessed with a Tukey Highly Significantly Different multiple comparison procedure. Data were best Box-Cox transformed to meet assumptions of normality and homogenous variance. *P < 0.01, CARP treated vs all else (mean ± s.e.m).
Muscle fibre bundle preparation from post-mortem mice followed the locally approved guidelines of the Animal Care and Use Committee of the University of Münster. All procedures conformed to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes.
Supplementary Material
Acknowledgements
We acknowledge the financial support of the Leducq Foundation (TNE-13CVD04), AFM-Téléthon (21436), EU-RISE-H2020 “Muscle relief”, NIH (HL128457, P30AR061303, S10-OD021724, S10-OD016234, NS047101), American Heart Association (17UFEL33520004), Swedish Hjärt-Lungfonden (20180199), the Wallenberg Foundation, the Swedish Research Council (2016/82), the Swedish Society for Medical Research (S150086), ERC (804418) and BBSRC (BB/M00676X/1). JB and JRF are supported by EU Marie Sklodowska-Curie Individual Fellowships (Titin Signals, 656636) and (TTNPred, 753054), respectively.
Abbreviations
- CARP
cardiac ankyrin repeat protein
- DAPI
4′,6-diamidino-2-phenylindole
- EDTA
Ethylenediaminetetraacetic acid
- GFP
green fluorescent protein
- HDX-MS
Hydrogen/Deuterium exchange mass spectrometry
- HSQC
heteronuclear single quantum correlation
- Ig
immunoglobulin
- LC-MS
Liquid chromatography–mass spectrometry
- MARP
muscle ankyrin repeat protein
- MLP
muscle Lim protein
- MS
Mass spectrometry
- MyBP-C3
Myosin Binding Protein C3
- NMR
Nuclear magnetic resonance
- NOE
nuclear Overhauser effect
- PCR
polymerase chain reaction
- RCI
random coil index
- RFDU
relative fractional deuterium uptake
- RMSD
root-mean-square deviation
- TCEP
Tris(2-carboxyethyl)phosphine hydrochloride
- UN2A
unique N2A sequence
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession numbers
NMR data for UN2A are deposited with the Biological Magnetic Resonance Data Bank (BMRB ID 50117) and mass spectrometry data with PRIDE (ID PXD017426). Protein structure coordinates of UN2A34−73 are deposited with the Protein Data Bank (ID 7NIP).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Taegtmeyer H, Sen S, Vela D, (2010). Return to the fetal gene program: a suggested metabolic link to gene expression in the heart. Ann NY Acad Sci, 1188, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuo H, Chen J, Ruiz-Lozano P, Zou Y, Nemer M, Chien KR, (1999). Control of segmental expression of the cardiac-restricted ankyrin repeat protein gene by distinct regulatory pathways in murine cardiogenesis. Development, 126, 4223–4234. [DOI] [PubMed] [Google Scholar]
- 3.Zhang N, Xie XJ, Wang JA, (2016). Multifunctional protein: cardiac ankyrin repeat protein. J Zhejiang Univ Sci B, 17, 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kojic S, Radojkovic D, Faulkner G, (2011). Muscle ankyrin repeat proteins: their role in striated muscle function in health and disease. Crit Rev Clin Lab Sci, 48, 269–294. [DOI] [PubMed] [Google Scholar]
- 5.Barash IA, Mathew L, Ryan AF, Chen J, Lieber RL, (2004). Rapid muscle-specific gene expression changes after a single bout of eccentric contractions in the mouse. Am J Physiol Cell Physiol, 286, C355–C364. [DOI] [PubMed] [Google Scholar]
- 6.van der Pijl R, Strom J, Conijn S, Lindqvist J, Labeit S, Granzier H, Ottenheijm C, (2018). Titin-based mechanosensing modulates muscle hypertrophy. J Cachexia Sarcopenia Muscle, 9, 947–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neagoe C, Opitz CA, Makarenko I, Linke WA, (2003). Gigantic variety: expression patterns of titin isoforms in striated muscles and consequences for myofibrillar passive stiffness. J Muscle Res Cell Motil, 24, 175–189. [DOI] [PubMed] [Google Scholar]
- 8.Linke WA, Hamdani N, (2014). Gigantic business: titin properties and function through thick and thin. Circ Res, 114, 1052–1068. [DOI] [PubMed] [Google Scholar]
- 9.Cazorla O, Freiburg A, Helmes M, Centner T, McNabb M, Wu Y, Trombitás K, Labeit S, Granzier H, (2000). Differential expression of cardiac titin isoforms and modulation of cellular stiffness. Circ Res, 86, 59–67. [DOI] [PubMed] [Google Scholar]
- 10.Miller MK, Bang ML, Witt CC, Labeit D, Trombitas C, Watanabe K, Granzier H, McElhinny AS, Gregorio CC, Labeit S, (2003). The muscle ankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family of titin filament-based stress response molecules. J Mol Biol, 333, 951–964. [DOI] [PubMed] [Google Scholar]
- 11.Lun AS, Chen J, Lange S, (2014). Probing muscle ankyrin-repeat protein (MARP) structure and function. Anat Rec (Hoboken), 297, 1615–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou T, Fleming JR, Franke B, Bogomolovas J, Barsukov I, Rigden DJ, Labeit S, Mayans O, (2016). CARP interacts with titin at a unique helical N2A sequence and at the domain Ig81 to form a structured complex. FEBS Lett, 590, 3098–3110. [DOI] [PubMed] [Google Scholar]
- 13.Tiffany H, Sonkar K, Gage MJ, (2017). The insertion sequence of the N2A region of titin exists in an extended structure with helical characteristics. Biochim Biophys Acta Proteins Proteom, 1865, 1–10. [DOI] [PubMed] [Google Scholar]
- 14.Sedgwick SG, Smerdon SJ, (1999) The ankyrin repeat: a diversity of interactions on a common structural framework. Trends Biochem Sci. 24, 311–316. [DOI] [PubMed] [Google Scholar]
- 15.Shen Y, Bax A, (2013). Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J Biomol NMR 56, 227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berjanskii MV, Wishart DS, (2008). Application of the random coil index to studying protein flexibility. J Biomol NMR, 40, 31–48. [DOI] [PubMed] [Google Scholar]
- 17.Kostyukevich Y, Acter T, Zherebker A, Ahmed A, Kim S, Nikolaev E, (2018). Hydrogen/deuterium exchange in mass spectrometry. Mass Spectrom Rev, 37, 811–853. [DOI] [PubMed] [Google Scholar]
- 18.Macchioni A, Ciancaleoni G, Zuccaccia C, Zuccaccia D., (2008). Determining accurate molecular sizes in solution through NMR diffusion spectroscopy. Chem Soc Rev 37(3):479–89. [DOI] [PubMed] [Google Scholar]
- 19.Shen Y, Vernon R, Baker D, Bax A, (2009). De novo protein structure generation from incomplete chemical shift assignments. J Biomol NMR, 43, 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen Y, Lange O, Delaglio F, Rossi P, Aramini JM, Liu G, et al. , (2008). Consistent blind protein structure generation from NMR chemical shift data. Proc Natl Acad Sci. USA 105, 4685–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li SC, Ng YK, (2010). Calibur: a tool for clustering large numbers of protein decoys. BMC Bioinformatics, 11, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vernon R, Shen Y, Baker D, Lange OF, (2013). Improved chemical shift based fragment selection for CS-Rosetta using Rosetta3 fragment picker. J Biomol NMR, 57, 117–127. [DOI] [PubMed] [Google Scholar]
- 23.Howarth JW, Ramisetti S, Nolan K, Sadayappan S, Rosevear PR, (2012). Structural insight into unique cardiac myosin-binding protein-C motif: a partially folded domain. J Biol Chem, 287, 8254–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marino M, Svergun D, Kreplak L, Konarev PV, Maco B, Labeit D, Mayans O, (2005). Poly-Ig tandems from I-band titin share extended domain arrangements irrespective of the distinct features of their modular constituents. J Muscle Res Cell Motil, 26, 355–365. [DOI] [PubMed] [Google Scholar]
- 25.Zacharchenko T, von Castelmur E, Rigden DJ, Mayans O, (2015). Structural advances on titin: towards an atomic understanding of multi-domain functions in myofilament mechanics and scaffolding. Biochem Soc Trans, 43, 850–855. [DOI] [PubMed] [Google Scholar]
- 26.Arimura T, Bos JM, Sato A, Kubo T, Okamoto H, Nishi H, Harada H, Koga Y, Moulik M, Doi YL, Towbin JA, Ackerman MJ, Kimura A, (2009). Cardiac ankyrin repeat protein gene (ANKRD1) mutations in hypertrophic cardiomyopathy. J Am Coll Cardiol, 54, 334–42. [DOI] [PubMed] [Google Scholar]
- 27.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L, (2005). The FoldX web server: an online force field. Nucleic Acids Res, 33, W382–W388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ittisoponpisan S, Islam SA, Khanna T, Alhuzimi E, David A, Sternberg MJE, (2019). Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated? J Mol Biol, 431, 2197–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lange S, Gehmlich K, Lun AS, Blondelle J, Hooper C, Dalton ND, Alvarez EA, Zhang X, Bang ML, Abassi YA, Dos Remedios CG, Peterson KL, Chen J, Ehler E, (2016). MLP and CARP are linked to chronic PKCα signalling in dilated cardiomyopathy. Nat Commun, 7, 12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barash IA, Bang ML, Mathew L, Greaser ML, Chen J, Lieber RL, (2007) Structural and regulatory roles of muscle ankyrin repeat protein family in skeletal muscle. Am J Physiol Cell Physiol 293, C218–C227. [DOI] [PubMed] [Google Scholar]
- 31.Linke WA, Ivemeyer M, Labeit S, Hinssen H, Rüegg JC, Gautel M, (1997). Actin-titin interaction in cardiac myofibrils: probing a physiological role. Biophys J, 73, 905–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kulke M, Neagoe C, Kolmerer B, Minajeva A, Hinssen H, Bullard B, Linke WA, (2001). Kettin, a major source of myofibrillar stiffness in Drosophila indirect flight muscle. J Cell Biol, 154, 1045–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonsior S, Hinssen H, (1995). Exogenous gelsolin binds to sarcomeric thin filaments without severing. Cell Motil Cytoskeleton, 31, 196–206. [DOI] [PubMed] [Google Scholar]
- 34.Lanzicher T, Zhou T, Saripalli C, Keschrumrus V, Smith III JE, Mayans O, Sbaizero O, Granzier H, (2020). Single-Molecule Force Spectroscopy on the N2A Element of Titin: Effects of Phosphorylation and CARP. Front Physiol, 11, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shyu KG, (2009). Cellular and molecular effects of mechanical stretch on vascular cells and cardiac myocytes. Clin Sci (Lond), 116, 377–389. [DOI] [PubMed] [Google Scholar]
- 36.Nishikawa K, Lindstedt SL, Hessel A, Mishra D, (2020). N2A Titin: Signaling Hub and Mechanical Switch in Skeletal Muscle. Int J Mol Sci. 21, 3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dutta S, Tsiros C, Sundar SL, Athar H, Moore J, Nelson B, Gage MJ, Nishikawa K, (2018). Calcium increases titin N2A binding to F-actin and regulated thin filaments. Sci Rep, 8, 14575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nishikawa K, Dutta S, DuVall M, Nelson B, Gage MJ, Monroy JA, (2019). Calcium-dependent titin-thin filament interactions in muscle: observations and theory. J Muscle Res Cell Motil, 41, 125–139. [DOI] [PubMed] [Google Scholar]
- 39.Bang ML, Gu Y, Dalton ND, Peterson KL, Chien KR, Chen J, (2014). The muscle ankyrin repeat proteins CARP, Ankrd2, and DARP are not essential for normal cardiac development and function at basal conditions and in response to pressure overload. PLoS One, 9, e93638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clos LJ II, Jofre MF, Ellinger JJ, Westler WM, Markley JL, (2013). NMRbot: Python Scripts Enable High-Throughput Data Collection on Current Bruker BioSpin NMR Spectrometers. Metabolomics, 9, 558–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A, (1995). NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- 42.Johnson BA, Blevins RA, (1994). NMR View: a computer program for visualization and analysis of NMR data. J Biomol NMR, 4, 603–614. [DOI] [PubMed] [Google Scholar]
- 43.Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC, (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Cryst, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramsey KM, Dembinski HE, Chen W, Ricci CG, Komives EA, (2017). DNA and IκBα Both Induce Long-Range Conformational Changes in NFκB. J Mol Biol, 429, 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wales TE, Fadgen KE, Gerhardt GC, Engen JR, (2008). High-speed and high-resolution UPLC separation at zero degrees. Celsius Anal Chem, 80, 6815–6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones JA, Wilkins DK, Smith LJ, Dobson CM, (1997). Characterization of protein unfolding by NMR diffusion measurements. J Biomol NMR 10, 199–203. [Google Scholar]
- 47.Berger S, Braun S, (2004). 200 and More NMR Experiments. A Practical Course. Wiley-VHC, Weinheim, Germany. [Google Scholar]
- 48.Wilkins DK, Grimshaw SB, Receveur V, Dobson CM, Jones JA, Smith LJ, (1999). Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry 38, 16424–31. [DOI] [PubMed] [Google Scholar]
- 49.Spudich JA, Watt S, (1971). The regulation of skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J Biol Chem, 246, 4866–4871. [PubMed] [Google Scholar]
- 50.Joumaa V, Herzog W, (2014). Calcium sensitivity of residual force enhancement in rabbit skinned fibers. Am J Physiol Cell Physiol, 307, C395–C401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adams M, Fleming JR, Riehle E, Zhou T, Zacharchenko T, Markovic M, Mayans O, (2019). Scalable, Non-denaturing Purification of Phosphoproteins Using Ga3+-IMAC: N2A and M1M2 Titin Components as Study case. Protein J 38, 181–189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.