

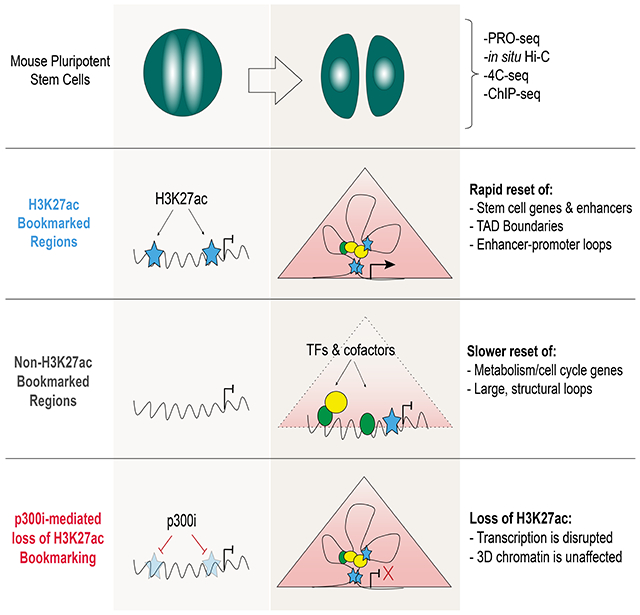
SUMMARY
During self-renewal, cell type-defining features are drastically perturbed in mitosis and must be faithfully reestablished upon G1 entry; a process that remains largely elusive. Here, we characterized at a genome-wide scale the dynamic transcriptional and architectural resetting of mouse pluripotent stem cells (PSCs) upon mitotic exit. We captured distinct waves of transcriptional reactivation with rapid induction of stem cell genes and transient activation of lineage-specific genes. Topological reorganization at different hierarchical levels also occurred in an asynchronous manner and showed partial coordination with transcriptional resetting. Globally, rapid transcriptional and architectural resetting associated with mitotic retention of H3K27 acetylation, supporting a bookmarking function. Indeed, mitotic depletion of H3K27ac impaired the early reactivation of bookmarked, stem cell-associated genes. However, 3D chromatin reorganization remained largely unaffected, suggesting these processes are driven by distinct forces upon mitotic exit. This study uncovers principles and mediators of PSC molecular resetting during self-renewal.
Keywords: Mitosis, pluripotent stem cells, transcription reactivation, 3D chromatin organization, bookmarking, H3K27ac, enhancer-promoter interaction, cell identity, PRO-seq, Hi-C
eTOC Blurb
Pelham-Webb et al. characterize the kinetics of transcriptional reactivation and 3D chromatin reorganization in pluripotent stem cells after cell division and examine the degree of coordination between these processes. Furthermore, they interrogate the role of mitotic bookmarking factors in the rapid molecular resetting of stem cell identity.
Graphical Abstract
INTRODUCTION
Mitosis (MIT) is accompanied by global transcriptional silencing (Gottesfeld and Forbes, 1997; Taylor, 1960), dissociation of transcription factors and cofactors from target genes (Martínez-Balbás et al., 1995) and profound reorganization of 3D chromatin architecture (Earnshaw and Laemmli, 1983; Marsden and Laemmli, 1979). Faithful propagation of cell identity relies on the proper reestablishment of cell-type defining molecular features during mitotic exit and entry to the next Gap 1 (G1) phase. Therefore, MIT-to-G1 transition is of critical importance for cell fate decisions (Boward et al., 2016; Soufi and Dalton, 2016) and a unique time window to study the principles of transcriptional and architectural resetting (Pelham-Webb et al., 2020).
Work in cycling human and mouse cell lines has captured the distinct folding states of mitotic and interphase chromosomes (Gibcus et al., 2018; Liang et al., 2015b; Naumova et al., 2013; Ou et al., 2017) and uncovered key events for their dynamic reorganization during mitotic exit (Abramo et al., 2019; Dileep et al., 2015; Nagano et al., 2017; Pelham-Webb et al., 2020; Zhang et al., 2019). Different studies have reported distinct waves of transcriptional reactivation during mitotic release and a global, transient spike in transcription during G1 entry (Hsiung et al., 2016; Palozola et al., 2017). However, the interplay between architectural resetting and transcriptional reactivation during mitotic exit, the underlying mechanisms, and the potential significance for cell fate inheritance control remain unknown.
One proposed mechanism for faithful heritability of cell identity is mitotic bookmarking (Michelotti et al., 1997), which refers to the partial retention of features such as DNA and histone modifications (Margueron and Reinberg, 2010; Zaidi et al., 2010), epigenetic modulators (Blobel et al., 2009; Dey et al., 2009), and DNA-binding transcription factors (TFs) (Caravaca et al., 2013; Deluz et al., 2016; Festuccia et al., 2016; Kadauke et al., 2012; Liu et al., 2017b; Teves et al., 2018; Teves et al., 2016). Few of these features have been tested for their functional role in the resetting and/or maintenance of cell identity. Recent work using advanced genomics, proteomics, and imaging technologies has further expanded the repertoire of mitotically retained features, including acetylation of histone tail residues such as H3K27ac (Behera et al., 2019; Javasky et al., 2018; Kang et al., 2020; Liu et al., 2017a; Liu et al., 2017b); chromatin accessibility at promoters (Hsiung et al., 2015; Teves et al., 2016); and low-level transcription (Liang et al., 2015a; Liu et al., 2017a; Palozola et al., 2017). To what extent mitotic retention of any of these features promotes reestablishment of cell type-specific gene expression and 3D chromatin landscape upon G1 entry remains largely unexplored (Palozola et al., 2019; Pelham-Webb et al., 2020).
MIT-to-G1 transition is shown to be the critical time window for pluripotent stem cells (PSC) to decide either to self-renew in culture or differentiate towards different lineages upon proper stimulation (Coronado et al., 2013; Pauklin et al., 2016; Pauklin and Vallier, 2013; Sela et al., 2012; Singh et al., 2015). Intriguingly, G1 phase is exceptionally short in PSCs (~3hr) compared to somatic cells (Savatier et al., 2002), suggesting the presence of efficient mechanisms of molecular resetting. Understanding how the PSC-defining features and functions resume after mitosis might allow us to better harness their potential for biomedical applications (Evans, 2011; Tabar and Studer, 2014).
Here, we used PRO-seq, in situ Hi-C, and high-resolution 4C-seq to characterize and integrate transcriptional and architectural changes during M-to-G1 in mouse PSCs. This approach revealed distinct patterns of transcriptional and topological resetting, their temporal interconnections, and potential underlying mechanisms. We also provide experimental evidence for the role of H3K27ac mitotic bookmarking in promoting faster transcriptional, but not architectural, resetting, suggesting that the two processes are partially uncoupled and/or driven by distinct forces during mitotic exit.
RESULTS
Mitotic arrest and release of pluripotent stem cells into G1
We characterized the molecular resetting of mouse PSCs after cell division in a mitotic release time course (Figures 1A and S1A). PSCs arrested in mitosis (MIT) by nocodazole treatment were isolated by mitotic shake-off (Liu et al., 2017b). Mitotic purity was verified by H3Ser10p staining (Hendzel et al., 1997) followed by Fluorescence Activated Cell Sorting (FACS) (Figures 1B) and immunofluorescence (Figure 1C); cell preparations with <95% purity were discarded. Mitotic cells were released in full medium and collected at different time intervals for evaluation of DNA content (Figure 1D) and cell cycle markers using the FUCCI2a system (Figure 1E) (Mort et al., 2014). Consistent with prior studies (Coronado et al., 2013), 1 hour after release the majority of PSCs entered G1 (55%-70% 2N population), while 2-3 hours later S phase began (Figures S1B and S1C). Therefore, we chose 1 hour and 3 hours post-release to enrich for early G1 (EG1) and late G1 (LG1), respectively. Asynchronous (ASYN) PSCs (>70% in S/G2 phase) were analyzed to represent a later stage of resetting (Figure 1E).
Figure 1.
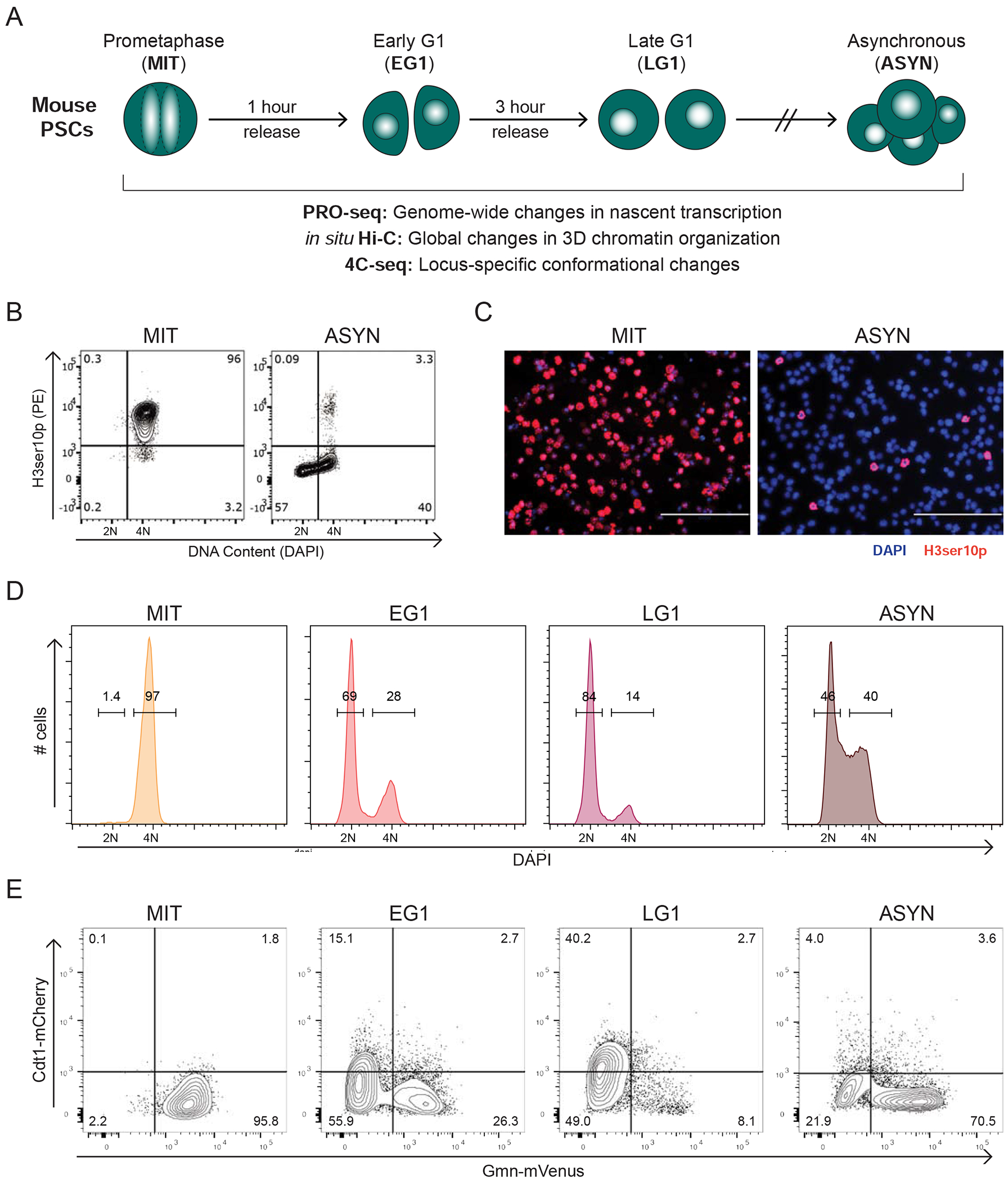
Mitotic arrest and release of pluripotent stem cells into G1 (A) Strategy for collecting and profiling mouse pluripotent stem cells (PSCs) in Mitosis (MIT), Early G1 (EG1), Late G1 (LG1), and untreated, asynchronous cells (ASYN). (B-C) FACS plots (B) and representative immunofluorescence images (C) showing H3ser10 phosphorylation (H3ser10p), a mitosis-specific histone mark, and DAPI in ASYN and MIT populations. (D) FACS histograms showing the percentage of cells with DNA content of 2N (G1) and 4N (G2/M) during a representative mitotic release time course. (E) FACS plots from a representative time course with FUCCI2a cells indicating the percent of cells in EG1 (Cdt1−, Gmn−), LG1 (Cdt1+, Gmn−), and S/G2/M (Gmn+). See also Figure S1
Distinct waves of gene and enhancer reactivation during mitotic exit
To capture transcriptional resetting, we applied a slightly modified version of Precision nuclear run-on sequencing (PRO-seq) more suitable for mitotic cells (Figures S1D and S1E), including spike-in controls to account for global transcriptional changes (Table S1) (see Methods). We found a high correlation between biological replicates and a clear separation of mitotic samples from the rest (Figure S2A). As expected (Gottesfeld and Forbes, 1997; Konrad, 1963; Taylor, 1960), global transcription was dramatically decreased during mitosis, but some genes (n=4008) showed residual expression (normalized RPKM >1) (Figure 2A), in agreement with recent studies (Liang et al., 2015a; Liu et al., 2017a; Palozola et al., 2017).
Figure 2.
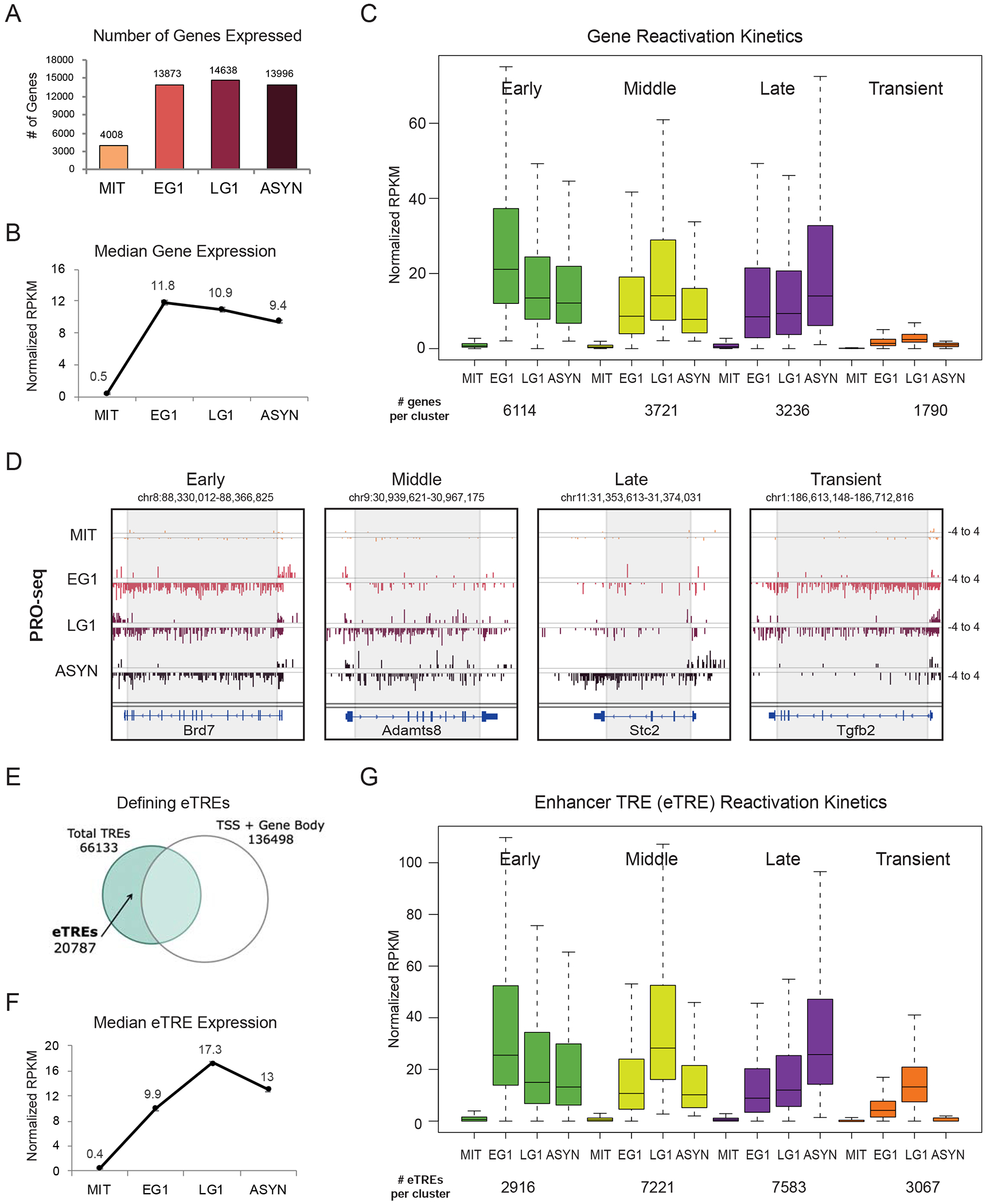
Distinct waves of gene and enhancer reactivation during mitotic exit (A) Number of genes expressed at each time point (RPKM>1 after normalization to Drosophila spike-in control and number of cells). (B) Median expression at each time point of all expressed genes. Error bars show +/− SEM of two biological replicates. (C) Genes were assigned to a group based on when they reached their maximal expression level (EG1 = early, LG1 = middle, ASYN = late). Transient genes were defined as normalized RPKM <2 in ASYN but >2 in EG1 and/or LG1. Box plots depict the median transcriptional activity across the time course; number of genes per cluster listed below. (D) Genome browser tracks of PRO-seq data (plus and minus strands) for one gene from each reactivation group. Grey box approximates the region used to quantify genic PRO-seq signal. (E) Strategy for identifying enhancer transcriptional regulatory elements (eTREs) using PRO-seq and published TSS data. Total TREs were defined using dREG. (F) Median expression at each time point of all 20787 identified eTREs. Error bars show +/−SEM of two biological replicates. (G) Patterns of eTRE transcriptional reactivation kinetics, defined as in (C). Box plots depict the median transcriptional activity across the time course. See also Figures S1, S2 and Tables S1, S6
Globally, transcription was rapidly reset by EG1 (Figure 2A) when median expression levels were highest (Figure 2B), but reactivation kinetics was variable among genes. We therefore clustered genes into different groups based on when they reached their maximal expression level: early G1 (Early), late G1 (Middle), or asynchronous (Late) (Figures 2C and 2D; Table S1). We also identified a group of genes (n=1790) that were transiently expressed in EG1 and/or LG1 but decreased to low levels (normalized RPKM <2) in ASYN (Transient). Reactivation patterns of randomly selected genes were validated by RT-qPCR analysis of pre-mRNA transcripts in an independent release time course and in sorted cells (Figure S2B and S2C). With the exception of Transient genes, which showed much lower overall expression in ASYN cells (Figure 2C and S2D), median transcriptional levels were similar across gene clusters. By plotting median gene lengths per cluster, we ruled out that faster or slower reactivation kinetics reflect a bias for shorter or longer genes (Figure S2E). Thus, gene reactivation during mitotic exit in PSCs is an asynchronous process independent of absolute transcriptional levels or rate of RNA polymerase elongation.
To track the transcriptional reactivation of enhancers, we generated a genomewide atlas of transcriptional regulatory elements (TREs) (Danko et al., 2015), excluding TREs within 1kb of TSS or gene body of protein-coding and non-coding genes (Figure 2E). The resulting 20,787 high-confidence enhancer TREs (eTREs) showed overall a slower reactivation (Figure 2F) compared to genes (Figure 2B), suggesting potentially different requirements or mechanisms of resetting. Nevertheless, we could still identify eTREs that followed Early, Middle, Late, or Transient reactivation kinetics similar to those observed in genes (Figures 2G and S2F; Table S1).
Stem cell genes and enhancers are rapidly reactivated upon mitotic exit
To gain insight into the biological relevance of the observed reactivation kinetics, we performed Gene Ontology (GO) analysis (McLean et al., 2010). Metabolic and signal transduction processes were enriched in the Middle gene cluster, while the Late gene group preferentially included genes involved in chromosome segregation and cell division, such as Cdca2 and Aurka (Figures 3A and 3B). Middle and Late eTREs associated with similar functions (Figure S3A). General “housekeeping” processes like transcription, RNA splicing, and protein transport were mostly associated with the Early gene group (Figure 3A). Notably, only the Early gene group enriched for terms associated with stem cell maintenance (Figure 3A) and showed significant overrepresentation (pvalue = 2.79E-15, Fisher’s exact test) within a published ESC/iPSC gene expression signature (Papadimitriou et al., 2016) (Figure 3C). Early reactivated eTREs also enriched for stem cell regulation categories (Figure S3A) and showed overrepresentation (p-value = 2.33E-5, Fisher’s exact test) of previously-defined PSC super-enhancers (SEs) (Whyte et al., 2013) (Figure S3B), which were reactivated at significantly faster rate and higher levels compared to all other eTREs (Figure 3D).
Figure 3.
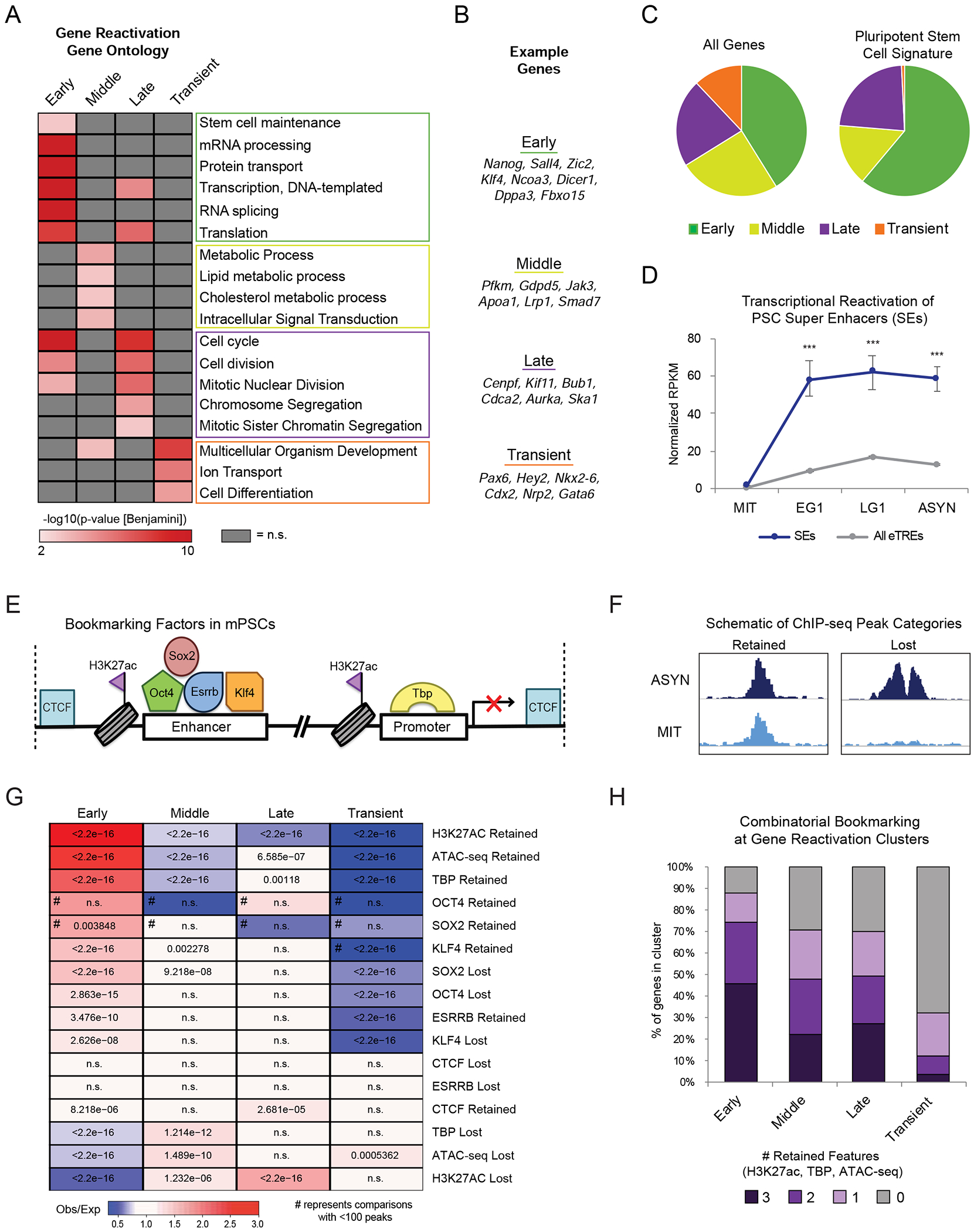
Bookmarked, stem cell genes and enhancers are rapidly reactivated upon mitotic exit (A) Heat map indicates the enrichment (-log10(pvalue)) of selected top Gene Ontology (GO) terms in each gene reactivation cluster. Adjusted (Benjamini) p-value <0.01was used as cut-off. Not significant terms are shown in grey. (B) Example genes in each reactivation group corresponding to top GO terms. (C) Pie charts show the distribution of all 14861 expressed genes (“All Genes”) versus 478 genes defined in an ESC/iPSC signature (Papadimitriou et al., 2016) (“Pluripotent Stem Cell Signature”) within the reactivation clusters. (D) Transcriptional reactivation of eTREs that overlap with defined PSC SEs (n=255 eTREs) (Whyte et al., 2013) compared to all other eTREs (n=20532 eTREs). Error bars show upper and lower limit of the 95% confidence interval. Asterisks indicate significance (*** p<0.0001) for SEs vs. Other eTREs, two-sided Wilcoxon’s rank sum test. (E) Schematic depicting the various proteins and histone modifications that have been identified as putative mitotic bookmarks in PSCs and have available ChIP-seq both in mitotic and asynchronous cells. (F) Schematic showing how we categorized published ChIP-seq peaks as Retained or Lost during mitosis. (G) Relative enrichment or depletion of the Retained/Lost peaks for each bookmarking factor at the promoters (+/−2.5kb from TSS) of each gene reactivation cluster. Color indicates ratio of observed (Obs) versus expected (Exp) frequency and p-value (two-sided Fisher’s exact test) is indicated if significant (p<0.01). Comparisons using <100 overlapping peaks are denoted with a hash mark (#). (H) Stacked bar plot showing the percent of genes in each transcriptional reactivation cluster that retained all, some, or none of the three bookmarks (H3K27ac, TBP, ATAC-seq). See also Figure S3 and Tables S2, S6
Intriguingly, transiently activated genes and eTREs enriched for developmental and differentiation processes, and included lineage-specific regulators such as Gata6, Cdx2 and Pax6 (Figures 3A, 3B, and S3A). In contrast with the other three gene clusters, promoters of Transient genes were not occupied by pluripotency TFs or transcriptional coactivators, but instead bound by Polycomb Repressive Complex 1 and 2 (PRC1/2) components (EZH2, SUZ12, JARID2, RING1B) (Figure S3C). About 20% of Transient genes were previously identified as high-confidence bivalent genes (Asenjo et al., 2020), supporting the notion that they are in a repressed or poised transcriptional state.
Enhancer reactivation patterns mirror the kinetics of target genes
In order to examine the relative reactivation kinetics of enhancers and their target genes, we assigned eTREs to their most proximal gene based on linear distance (<20kb) (Figure S3D), and to one or more distal target genes (>20kb) (Figure S3E) based on long-range chromatin contacts previously detected by H3K27ac HiChIP (Di Giammartino et al., 2019). Both approaches showed matched kinetics of eTREs and target genes at frequencies significantly higher than expected (Figures S3D and S3E), suggesting a coordinated process. However, the fast activation of many Early genes could not be explained by any proximal or looped Early eTRE, raising the possibility that resetting of these genes may be promoter-driven and enhancer-independent.
Mitotic bookmarking predicts rapid transcriptional reactivation
Since chromatin state and TF binding in asynchronous cells could not adequately explain the distinct transcriptional reactivation kinetics (see Figure S3C), we turned our attention to the mitotic state. We performed ATAC-seq in asynchronous and mitotic cells and utilized published ChIP-seq datasets for putative mitotic bookmarks in PSCs (Figure 3E). For each feature, we defined peaks as “Retained” (present in both mitotic and asynchronous cells) or “Lost” (present only in asynchronous cells) (Figure 3F, see also methods) and then examined their enrichment around gene promoters and eTREs (+/−2.5kb). This analysis showed that promoters of Early-reactivated genes had a significantly higher likelihood to retain H3K27ac, chromatin accessibility, and TBP binding compared to all other gene groups (Figure 3G; Table S2). In agreement, more than 85% of Early gene promoters overlapped with at least one Retained peak from H3K27ac, ATAC-seq, or TBP, and many (45%) retained all three features (Figure 3H), suggesting an additive effect of bookmarking. Faster reactivation of eTREs also associated with mitotic retention of features such as H3K27ac and chromatin accessibility (Figure S3F; Table S2). These findings establish a strong association between rapid transcriptional reactivation and mitotic retention of only few of the previously reported bookmarks.
Chromosomal compartments and domain boundaries are established in EG1 in coordination with transcriptional reactivation
In parallel with the transcriptional changes during mitotic exit, we also characterized the dynamics of 3D chromatin reorganization with the goal to understand their temporal interconnections upon resetting of PSC identity. Principle Component Analysis (PCA) confirmed consistency between in situ Hi-C replicates and indicated gradual changes in the course of mitotic release (Figure S4A). Consistent with previous studies (Abramo et al., 2019; Gibcus et al., 2018; Nagano et al., 2017; Naumova et al., 2013; Zhang et al., 2019), chromatin architecture was dramatically altered in mitosis (Figure S4B), with an almost complete loss of compartments, topologically-associating domains (TADs), and long-range interactions (Figure 4).
Figure 4.
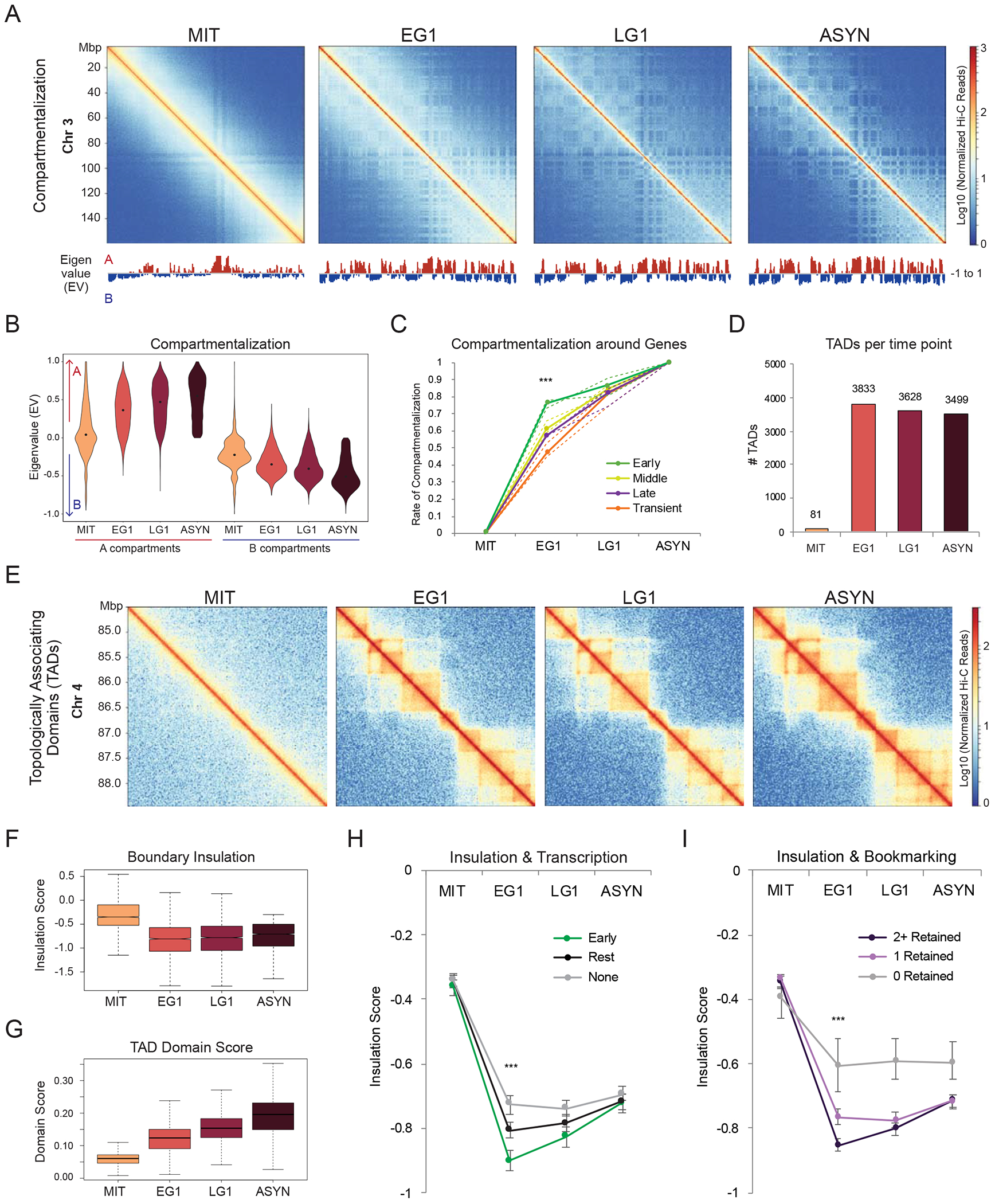
Chromosomal compartments and domain boundaries are established in EG1 in coordination with transcriptional reactivation (A) Hi-C interaction heatmaps (log10 Normalized Hi-C reads) of chromosome 3 for each time point to illustrate compartment reformation. The eigenvalues (EV) of each matrix at 100kb resolution are shown below. (B) Violin plot depicting the compartmentalization (eigenvalue, EV) for all 100kb bins across the genome at each time point. Bins with EV>0 in asynchronous cells are called A, while bins with EV<0 are called B. (C) Line plot showing rate of compartmentalization during mitotic exit for compartments (100kb bins) containing genes from the four gene reactivation clusters, prioritized as Early (n=4208) > Middle (n=1861) > Late (n=921) > Transient (n=903). Lines indicate the median compartmentalization rate for each Hi-C replicate (dashed) or for the averaged replicates (solid). Asterisks indicate significance (*** p<0.0001) for Early versus any other cluster at EG1, two-sided Wilcoxon’s rank sum test. (D) Number of TADs identified at each time point. (E) Hi-C interaction maps (log10 Normalized Hi-C reads) for each time point of a region on chromosome 4 (chr4:84,500,000-88,500,000), illustrating TAD dynamics. (F-G) Box plots showing the (F) insulation scores of TAD boundaries (n=3519 in asynchronous cells) and (G) domain scores of TADs (n=3499) across the time course. (H-I) Median insulation scores at each time point for (H) TAD boundaries containing: at least one early gene or eTRE (Early, n=985), only later activated genes or eTREs (Middle, Late, and/or Transient) (Rest, n=1182), or no active genes or eTREs (None, n=1352) or (I) TAD boundaries that retain none (n=176), one (n=1414) or two or more (n=1929) bookmarking features (CTCF, H3K27ac, ATAC-seq, TBP). For both (H-I) Asterisks indicate significance (*** p<0.0001) for all pairwise comparisons at EG1, two-sided Wilcoxon’s rank sum test. See also Figure S4 and Tables S3, S6
The majority (~85%) of A and B compartments (Lieberman-Aiden et al., 2009) (Figure S4C) were reestablished by EG1, though they continued to segregate and gain strength at later time points (Figures 4A and 4B) as previously reported (Abramo et al., 2019; Kang et al., 2020; Nagano et al., 2017; Zhang et al., 2019). Almost all expressed genes were located within A compartments (defined as open, gene-rich and active regions), with the exception of 462 Transient genes within B compartments (Figure S4D). Although absolute compartmentalization scores were similar between early, middle, and late reactivated genes in asynchronous cells (Figure S4E), compartments that harbored early reactivated genes (and eTREs) reached their final compartmentalization strengths at a significantly faster rate (Figures 4C and S4F). These findings show that the recovery rate of chromosomal compartments correlates with the kinetics of transcriptional resetting.
TADs were also disrupted in mitosis and largely reset by EG1 (Figures 4D and 4E), in agreement with previous reports (Abramo et al., 2019; Nagano et al., 2017; Zhang et al., 2019). Boundary insulation was rapidly recovered (Figure 4F), while TAD domain score, which measures the ratio of intra-TAD versus overall connectivity (Dixon et al., 2012; Stadhouders et al., 2018), increased more gradually during G1 entry (Figure 4G). A substantial fraction (~20%) of asynchronous TADs were formed by merging of smaller TADs in EG1 and/or LG1 (data not shown), supporting a recently proposed bottom-up model of TAD formation (Zhang et al., 2019). Although resetting of domain scores did not correlate with transcriptional reactivation kinetics, boundaries that overlapped with at least one Early gene/eTRE showed significantly faster and stronger insulation compared to boundaries with later reactivated genes/eTREs or with no transcriptional activity (Figure 4H). Moreover, we observed an association between spikes in transcription and boundary insulation during EG1 (Figure S4G). Together, these results suggest that the timing and degree of insulation upon mitotic exit associates with local transcriptional activity.
We next tested whether mitotic retention of histone marks and/or chromatin-bound proteins is linked to faster architectural resetting. Indeed, boundaries that retained H3K27ac, CTCF, or other bookmarking features (TBP and ATAC-seq) during mitosis established significantly stronger insulation by EG1 compared to boundaries that lost these features (Figure S4H-K). Mitotic retention of multiple factors (Figure 4I) showed even stronger early insulation. Intriguingly, 95% of TAD boundaries (3343/3519) in PSCs were bookmarked by at least one factor (H3K27ac, TBP, CTCF, ATAC-seq). Given that the degree of mitotic retention is variable among cell types, as shown for CTCF (Oomen et al., 2019; Owens et al., 2019; Zhang et al., 2019), the relevance of boundary bookmarking remains to be investigated.
Active regulatory elements and bookmarked regions engage rapidly in chromatin looping, while structural loops reform at a slower rate
Next, we assessed the kinetics of chromatin looping during mitotic exit. Significant contacts were called with Fit-Hi-C (Ay et al., 2014) at 20kb resolution (qval<10−3 for both replicates). To further increase confidence, we intersected our contacts with recent high-resolution Micro-C data in mESC (Hsieh et al., 2020), resulting in 14,091 common loops (Figure S5A; Table S3; see methods). Similar to the progressive increase in TAD domain score (Figure 4G), loops re-formed in a gradual manner (Figure S5B). K-means clustering across all time points identified three groups of interactions with different kinetics of resetting: Fast, Gradual, and Slow (Figures 5A and 5B; Table S3). All groups enriched for binding of the classical architectural factors CTCF and cohesin (Figure 5C) and showed a progressive increase in size (Figure S5C). This suggests that more distal contacts are formed slower than closer ones, in agreement with the loop extrusion model (Alipour and Marko, 2012; Davidson et al., 2019; Fudenberg et al., 2016; Golfier et al., 2020; Kim et al., 2019; Sanborn et al., 2015). Examples of differential loop kinetics are presented by Hi-C contact maps (Figures 5D and S5D). Association analysis revealed that Fast established contacts were strongly enriched for active histone marks, binding of pluripotency TFs, and transcriptional machinery (Figure 5C), while Gradual and Slow showed only weak or no association with transcriptional regulatory features. Moreover, chromatin contacts with H3K27ac in both anchors were overall stronger and faster than loops with only CTCF and/or cohesin (Figure S5E). This supports the notion that active regulatory loops are reformed prior to structural loops upon mitotic exit (Zhang et al., 2019). Fast established loops were also preferentially enriched for mitotic retention of H3K27ac and TBP, suggesting that mitotic bookmarking by these factors might also promote rapid loop reformation (Figure 5E). The lack of correlation between CTCF bookmarking and loop reformation kinetics is in agreement with the recent finding that cohesin recruitment, and not CTCF binding, is the rate limiting step for establishment of structural loops upon mitotic exit (Zhang et al., 2019).
Figure 5.
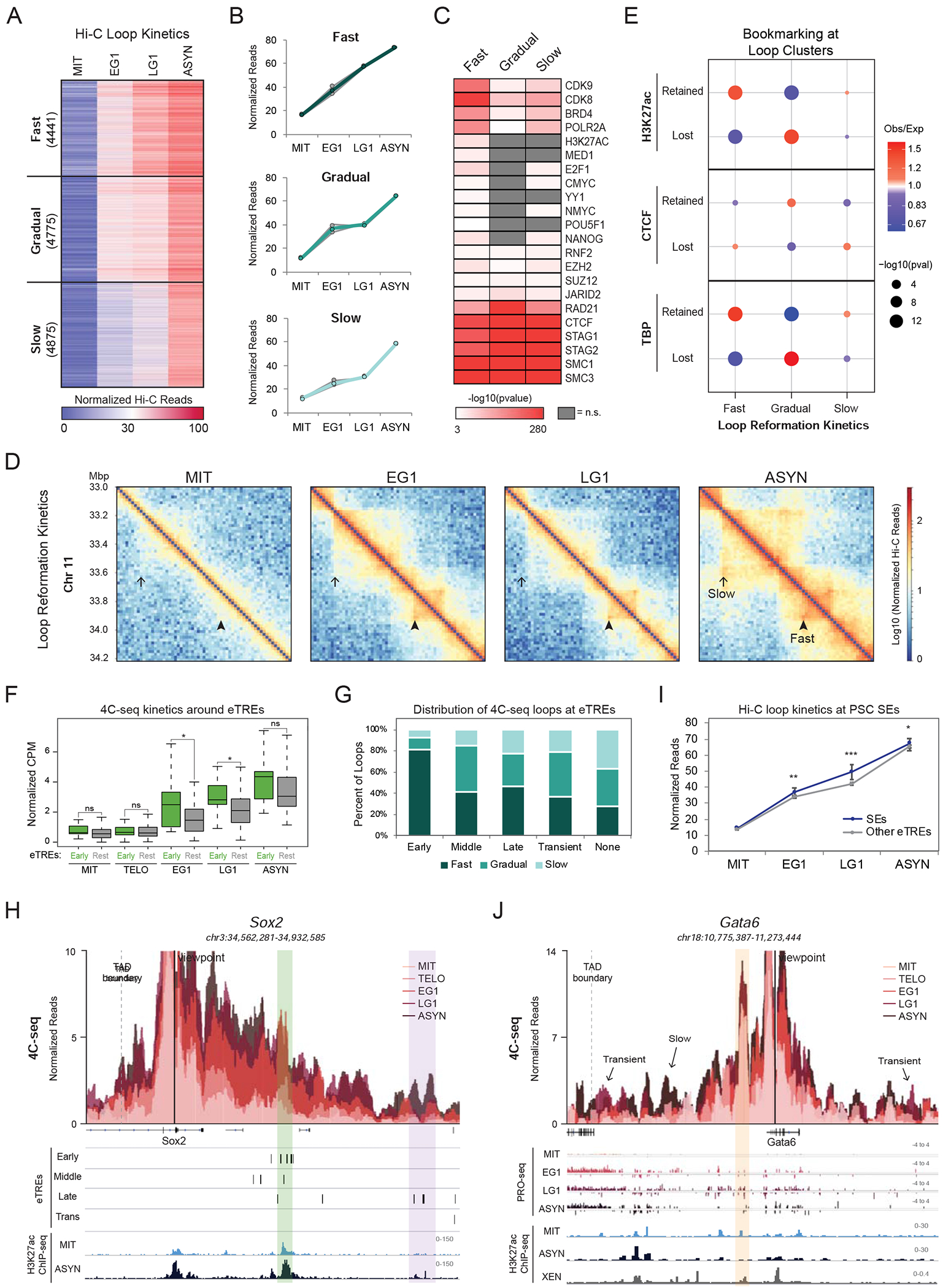
Enhancer-promoter contacts are rapidly reformed with different patterns at early or transiently activated genes (A-B) K-means clustering of 14091 Hi-C loops (see methods) identified three clusters with different reformation kinetics (Fast, Gradual, Slow). (A) Heat map illustrating the normalized Hi-C reads for each contact over the time course. Number of loops per cluster is shown. (B) Line plots depicting the median of normalized read counts for each cluster over the time course either for each replicate (grey lines) or for the average of the replicates (colored lines). (C) Heat map showing select factors and marks that are enriched at the loop anchors of each Hi-C cluster (shown in A) as calculated from LOLA analysis (Sheffield and Bock, 2016) (see Methods). Color bar indicates significance of enrichment (-log10(pvalue)), while not significant (q>0.001) terms are shown in grey. (D) Hi-C interaction maps (log10 Normalized Hi-C reads) at 10kb resolution of a region on chromosome 11 (chr11:33,000,000-34,200,000) at each time point. Examples of a Fast and Slow reestablished contacts are indicated. (E) Relative enrichment of Hi-C loop clusters for the presence of Retained or Lost H3K27ac, CTCF, or TBP ChIP-seq peaks (see Figures 3E-G). Size of dots indicates p-value (two-sided Fisher’s exact test) and color indicates ratio of observed (Obs) versus expected (Exp) frequency. (F) Box plot showing the 4C-seq contact frequency over the time course for loop anchors that overlap with or are nearby (<5kb) to Early eTREs (Early, n=17 loops) versus anchors that contain only Middle, Late, or Transient eTREs (Rest, n=48 loops). Asterisks indicate significance (*p<0.05) for Early vs. Rest, two-sided Wilcoxon’s rank sum test. Not significant (ns) indicates p>0.05. (G) Stacked bar plot showing the percent of Fast, Gradual, and Slow reformed 4C-seq loops overlapping or near (<5kb) to Early (n=27), Middle (n=46), Late (n=49), or Transient (n=19) eTREs. Any loop anchor >5kb from all eTREs was shown as “None” (n=167). (H) 4C-seq data is represented as average CPM around the viewpoint (Sox2 promoter) with each time point shown as an overlapping bar plot. Genome browser tracks underneath show eTRE reactivation clusters (Early, Middle, Late, and Transient) and H3K27ac ChIP-seq in mitotic and asynchronous cells. The Sox2 super-enhancer (Early eTREs) is highlighted in green while another enhancer region (Late eTREs) is highlighted in purple. (I) Line plot showing Hi-C contact strength for loops with an anchor containing a PSC SE (Whyte et al., 2013) (n=253) versus loops containing any other eTREs (n=6073). Error bars show upper and lower limit of the 95% confidence interval. Asterisks indicate significance (* p<0.01, ** p<0.001, *** p<0.0001) for SEs vs. Other eTREs, two-sided Wilcoxon’s rank sum test. (J) 4C-seq data representation (as shown in (H)) around the Gata6 promoter. Genome browser tracks underneath show raw PRO-seq reads for each time point, H3K27ac ChIP-seq in mitotic and asynchronous cells, and H3K27ac ChIP-seq in asynchronous eXtraembryonic ENdoderm (XEN) stem cells. A retained contact between the Gata6 promoter and a proximal enhancer is highlighted in orange. Arrows indicate visually-detected transient or slow formed contacts. See also Figure S5 and Tables S3, S5, S6
Different patterns of chromatin loops characterize early or transiently activated genes and enhancers that promote self-renewal or differentiation.
The coordinated reactivation of enhancers and their target genes during mitotic exit (see Figures S3D and S3E) suggests a functional link between chromatin looping and transcriptional reactivation. On the other hand, the overall rapid recovery of transcriptional activity during G1 (see Figures 2B and 2F) compared to the slower reestablishment of long-range chromatin contacts (see Figures 5A and S5B) argues for partial uncoupling. To address this, we examined the temporal relationship between transcriptional reactivation and fine-scale architectural resetting using the Hi-C loop clusters. Although the levels of transcriptional activity were significantly higher for genes within the Fast cluster (Figure S5F), the transcriptional recovery rates were indistinguishable across loop clusters (and vice versa (Figure S5G)). This suggests a weak or no association between transcriptional reactivation and loop reformation kinetics, as recently proposed for a different cell type (Kang et al., 2020).
We considered this may be due to technical limitations of our Hi-C analysis, which misses short-range interactions (<60kb) (see methods), underrepresents regulatory loops (Bonev et al., 2017; Di Giammartino et al., 2019; Mumbach et al., 2016; Mumbach et al., 2017), and does not cover a large fraction (>50%) of expressed genes and eTREs. We therefore revisited the relationship between transcriptional reactivation and loop formation by performing high-resolution 4C-seq around promoters of 11 example genes with distinct reactivation patterns and biological relevance (Tables S3 and S5). In addition to the previous mitotic release time course, we also included an earlier time point (30 minutes release, see Figure S1B) to capture chromatin contacts during anaphase/telophase (TELO). For each viewpoint, we called significant contacts based on differential normalized 4C-seq signal between any two time points (Fold change >3, p<0.05) and additional filtering (see methods). The 232 high-confidence contacts (ranging from 9 to 590kb distance) showed gradual and nonsynchronous reformation kinetics (Figure S5H; Table S3), consistent with our Hi-C results. While proximal loops were often reformed first, many loci showed clear exceptions, with more distal loops reaching their maximal strength prior to proximal contacts (Figure S5I). Interestingly, loops connecting genes to Early eTREs were significantly faster reformed than those contacting later (Middle, Late, Transient) eTREs or no eTREs (Figures 5F and 5G). For example, at the Sox2 locus (a critical regulator of pluripotency and reprogramming) we observed rapid contact reformation between the promoter and super enhancer (Whyte et al., 2013), which contains multiple Early eTREs, and much slower interaction with nearby Late eTREs (Figure 5H). This was true for all rapidly reactivated genes tested, which formed at least one fast loop with an Early eTRE (Figure S5J). This suggests that early enhancer-promoter communication might be sufficient for initial gene activation, while later loops may stabilize gene expression or provide additional layers of regulation. Re-analysis of our Hi-C data confirmed the faster reformation of contacts around pluripotent stem cell super-enhancers (Whyte et al., 2013) (Figure 5I). These results demonstrate a degree of coordination between architectural reorganization and transcriptional reactivation during mitotic exit, particularly around stem cell-related genes and enhancers.
Although the vast majority of observed 4C-seq contacts were abrogated in mitosis, we observed several noteworthy exceptions. The transiently expressed Gata6 locus, encoding a master regulator of the endodermal fate (Schrode et al., 2014), had a strong, proximal contact that was maintained in mitosis and G1 phase (Figure 5J). This anchor corresponded to a putative enhancer in eXtra-embryonic ENdoderm (XEN) cells (unpublished H3K27ac ChIP-seq from our group), implying that this persistent contact may pre-program activation of Gata6 during G1 and/or upon endoderm differentiation. We also discovered transiently formed loops during G1 (Figure 5J), which occurred within the same TAD and coincided with the transient activation of genes or eTREs at their anchors. These loops differ from recently reported “aberrant” G1 inter-TAD chromatin contacts (Zhang et al., 2019), but their role during self-renewal and differentiation in stem cells remains to be determined.
Taken together, long-range chromatin contacts are re-established upon mitotic exit in complex and asynchronous patterns that only partly associate with the transcriptional reactivation of involved elements.
Loss of mitotic H3K27ac perturbs transcriptional reactivation during G1 entry with no effects on 3D chromatin reorganization
Prompted by the strong association of mitotic H3K27ac with rapid resetting of transcription and looping/insulation (Figures 3G, S3F, S4H, and 5E), we decided to experimentally test the bookmarking function of this mark. We depleted H3K27ac during mitosis by treating PSCs with the selective p300/CBP inhibitor A485 (p300i) (Lasko et al., 2017) for three hours prior to mitotic shake-off (MIT). Then, we released in full medium without p300i or Nocodazole for 1hr (EG1) to detect early, direct effects on transcriptional reactivation and topological resetting. To distinguish between specific effects of p300i on M-to-G1 resetting versus “generic” effects of global H3K27ac loss, we also collected Asynchronous cells (ASYN) treated and released (ASYN+1) for the same duration (Figure 6A). FACS analysis of DNA content at each time point showed no effects of the treatment on mitotic purity or cell cycle release (data not shown). Western blot analysis and H3K27ac ChIP-seq revealed a strong reduction of H3K27ac upon p300i treatment and a quick recovery upon wash off (Figures 6B, S6A, and S7).
Figure 6.
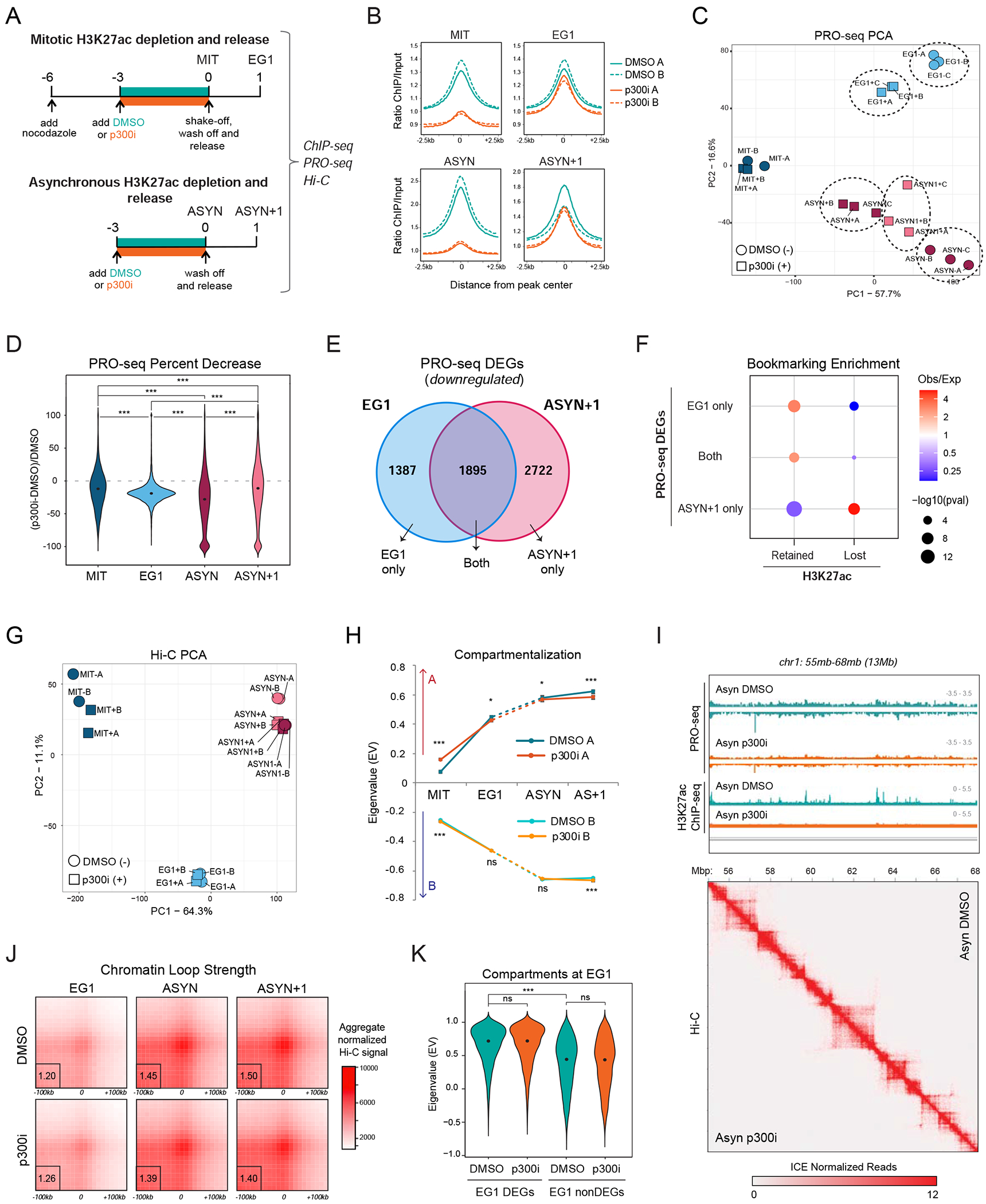
Loss of mitotic H3K27ac perturbs transcriptional reactivation during G1 entry with no effects in 3D chromatin reorganization (A) Schematic illustration of our strategy for selectively depleting H3K27ac during mitosis and in asynchronous cells by treating for 3 hrs with the p300/CBP inhibitor A485 (p300i) or DMSO control prior to releasing into fresh media without the inhibitor for 1hr (see methods). (B) Average line plot showing the H3K27ac signal (ratio ChIP/input) for DMSO- (teal) and p300i- (orange) treated samples at each time point. Solid and dashed lines indicate individual replicates. Line plots are centered (+/−2.5kb) around the 29,965 peaks called in MIT, EG1, and/or ASYN DMSO samples. (C) Principal Component Analysis (PCA) analysis of MIT, EG1, ASYN, and ASYN+1 PRO-seq samples after DMSO (−) or p300i (+) treatment and recovery. A, B and C indicate independent replicates. (D) Violin plot shows the percent decrease in PRO-seq signal in p300i- vs. DMSO-treated samples for all genes expressed at each time point (RPKM>1). Asterisks indicate significance (***p<0.0001) for all pairwise comparisons, two-sided Wilcoxon’s rank sum test. (E) Venn diagram of the overlap between downregulated, differentially-expressed genes (DEGs) after p300i treatment in EG1 and ASYN+1 (see also Figure S6B). (F) Relative enrichment or depletion of Retained or Lost H3K27ac ChIP-seq peaks at the promoter (+/−2.5kb from TSS, at least 1bp overlap) of downregulated DEGs (EG1 only, ASYN+1 only, or Both, see Figure 6E). Size of dots indicates p-value (two-sided Fisher’s exact test) and color indicates ratio of observed (Obs) versus expected (Exp) frequency. Genes containing multiple peak types were prioritized by Retained>Lost. (G) Principal Component Analysis (PCA) analysis of Hi-C data (based on compartment eigenvalues for each 100kb bin) for MIT, EG1, ASYN, and ASYN+1 samples after DMSO (−) or p300i (+) treatment and recovery. (H) Line plot shows median compartmentalization (eigenvalue) of A compartments (top) and B compartments (bottom) in DMSO- and p300i-treated Hi-C samples at each time point. Error bars show upper and lower limit of the 95% confidence interval. Asterisks indicate significance (* p<0.01, ** p<0.001, *** p<0.0001) for DMSO versus p300i, two-sided Wilcoxon’s rank sum test. Not significant (ns) indicates p>0.01. (I) Genome browser tracks for chr1:55,000,000-68,000,000 (13Mb) showing the PRO-seq data (merge of triplicates) and H3K27ac ChIP-seq data (one replicate) for ASYN DMSO and ASYN p300i samples, indicating the global decrease in transcription and H3K27ac levels after p300 inhibition. Below, the Hi-C heatmap for the same region shows no effect on 3D chromatin architecture (ASYN DMSO on upper right; ASYN p300i on lower left). Hi-C data was visualized using Juicebox (Robinson et al., 2018) at 20kb resolution. (J) Aggregated peak analysis (APA) showing the aggregate Hi-C signal of DMSO- and p300i-treated Hi-C samples at each time point centered around our high-confidence Hi-C loops as identified in Figure 5A (see methods). Bin size, 10kb. The APA score, calculated as the ratio of the number of contacts in the central bin to the mean number of contacts in the lower-left corner, is shown at the lower left (outlined by a black box). (K) Violin plot shows compartmentalization (eigenvalue) during EG1 after DMSO or p300i treatment/recovery, focusing specifically around compartments containing EG1 DEGs and nonDEGs. Asterisks indicate significance (*** p<0.0001) for all relevant pairwise comparisons, two-sided Wilcoxon’s rank sum test. Not significant (ns) indicates p>0.01. See also Figures S6, S7 and Tables S4, S6
PRO-seq analysis showed good correlation among replicates and a clear separation between p300i treated/released (+) samples compared to DMSO controls (−) (Figure 6C). Overall, p300i treatment caused a global and significant decrease in transcriptional activity at every timepoint (Figure 6D and S6B). The most pronounced effects were in the ASYN treated cells with 8198 differentially transcribed genes (DEGs: 7797 down, 401 up, compared to respective DMSO samples) (>1.2FC, pval<0.05) (Figure 6D and S6B; Table S4), supporting a functional role of p300 activity and H3K27ac levels in the maintenance of transcription (Kang et al., 2020; Martire et al., 2020; Raisner et al., 2018). Despite the global recovery of H3K27ac at EG1 and ASYN+1 (Figure 6B), transcriptional activity was significantly impaired at both timepoints (Figure 6D) with common but also stage-specific downregulated DEGs (Figure 6E). EG1 DEGs enriched for genes of the Early reactivated cluster, while ASYN+1 DEGs showed overrepresentation of Late reactivated genes (Figure S6C). Moreover, EG1 DEGs were strongly enriched for H3K27ac Retained peaks (bookmarking), while ASYN+1 DEGs associated with Lost H3K27ac peaks during mitosis (Figure 6F). Notably, genes important for stem cell identity, including Esrrb, Nanog, Pou5f1, Nodal, and Zfp42, were preferentially enriched in EG1 DEG when compared to unaffected genes (EG1 nonDEG) (Figure S6D). These results provide experimental support for the functional role of mitotic H3K27ac in rapid post-mitotic transcriptional reactivation of bookmarked and PSC-associated genes.
In contrast with the significant and broad transcriptional defects upon p300i treatment, our Hi-C analysis did not detect any notable effects on the 3D chromatin (re)organization. Principal Component Analysis clustered all samples based on cell cycle stage (MIT, EG1 or ASYN) and did not clearly distinguish between p300i- or DMSO-treatment (Figure 6G). Indeed, A and B compartmentalization across all time points was almost identical between DMSO and p300i-treated samples (Figures 6H and S6E) with <1.5% of the genome showing significant differences in compartment scores (eigenvalues) around weakly compartmentalized regions (Figure S6F; Table S4). Similarly, TAD boundary insulation, domain score, and loop strength showed minimal global alterations (Figures S6G, S6H, 6I, and 6J) and very few or no significant changes at any time point (Table S4). Specifically, we failed to detect any significant topological changes in EG1, even when comparing chromatin architecture around differential (EG1 DEG) and non-differential (EG1 nonDEG) genes (Figures 6K, S6I-K). Although we cannot exclude that fine-scale and locus-specific reorganization might still occur, these results demonstrate that neither mitotic nor interphase depletion of H3K27ac by p300i disrupt global 3D chromatin architecture.
Together, these experiments document a functional role of mitotic H3K27ac bookmarking for transcriptional reactivation but not for architectural resetting during G1 entry. Moreover, these results support that 3D chromatin (re)organization is (i) largely independent of p300 activity and H3K27ac levels when these are perturbed temporarily and (ii) potentially uncoupled from transcriptional activity.
DISCUSSION
The MIT-to-G1 transition poses substantial challenges for the maintenance of cell identity in self-renewing cells. Our study integrates transcriptional and architectural changes during mitotic exit in mouse PSCs and revealed distinct — and partly coordinated — waves of molecular resetting, with a particularly rapid resetting around PSC-associated genes and enhancers. Moreover, we provided experimental support for the bookmarking function of H3K27ac for rapid post-mitotic transcriptional activation (model in Figure 7).
Figure 7.
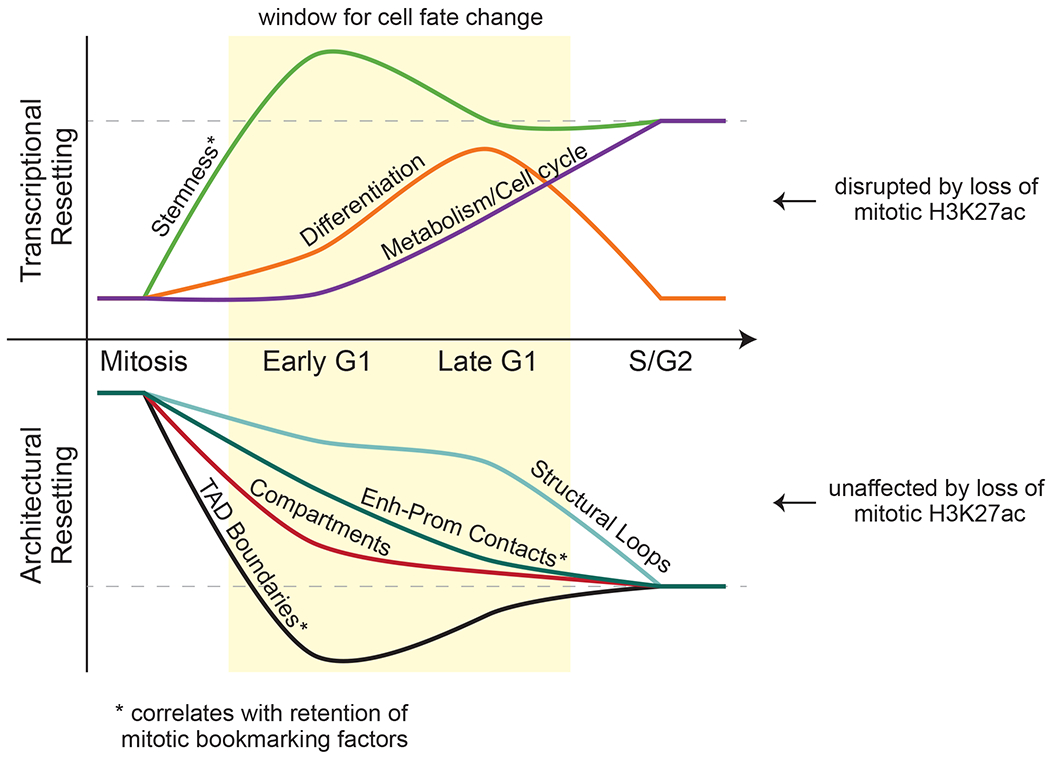
Resetting of PSC identity during G1 entry Model summarizing the transcriptional and topological resetting of pluripotent stem cell identity during G1 entry. The top half describes the waves of transcriptional (re)activation while the bottom shows the kinetics of 3D chromatin architecture reorganization. In both cases, the dashed line indicates full resetting of the interphase state. Transcriptional reactivation is disrupted by loss of H3K27ac bookmarking while architectural resetting is unaffected.
Transcriptional shutdown during mitosis and a global transcriptional spike during G1 entry have also been reported in different cell types (Hsiung et al., 2016; Palozola et al., 2017; Teves et al., 2018). However, in contrast with somatic cells (Palozola et al., 2017), we observed preferential rapid reactivation of genes associated with PSC identity compared to the slower turn on of housekeeping genes, suggesting cell type-specific differences in transcriptional resetting upon mitotic exit. This may reflect the constant need of fast-cycling PSCs to balance between self-renewal and differentiation (Evans, 2011) and the particularly critical role of G1 phase in this decision (Boward et al., 2016). As pluripotency regulators such as NANOG degrade during mitosis (Liu et al., 2017b), this rapid reactivation of the stemness program may be critical for self-renewal. On the other hand, the observed transient activation of developmental genes in G1 may constitute a temporal “priming” step that can tip the balance from self-renewal to lineage specification (Coronado et al., 2013; Pauklin et al., 2016; Pauklin and Vallier, 2013; Singh et al., 2013; Singh et al., 2015). This is in agreement with previous reports of “noisy” de-repression of lineage regulators during G1 in self-renewing human (Singh et al., 2013) and mouse (Asenjo et al., 2020) PSCs. Future single-cell analyses will be required to determine the degree of co-expression of different developmental regulators in PSCs exiting mitosis and the underlying mechanisms.
Our study also revealed distinct and nonsynchronous patterns of topological reorganization in PSC during mitotic exit and provided insights into their underlying mechanisms. We confirmed that compartmentalization and TAD boundary insulation were rapidly reestablished in early G1 (Abramo et al., 2019; Dileep et al., 2015; Nagano et al., 2017; Naumova et al., 2013; Zhang et al., 2019), while long-range contacts were formed overall at a slower rate. It is important to acknowledge that nocodazole-mediated mitotic arrest might have affected the normal kinetics of 3D chromatin reorganization upon mitotic exit, and that the presence of not-yet released mitotic cells within our bulk EG1 and LG1 populations might have limited our ability to detect some of the early established contacts. In agreement with previous studies (Abramo et al., 2019; Zhang et al., 2019), most of our Hi-C detected chromatin contacts were bound by CTCF and cohesin and their size was inversely correlated with the rate of recovery, further supporting that CTCF/cohesin-mediated loop extrusion (Alipour and Marko, 2012; Davidson et al., 2019; Fudenberg et al., 2016; Golfier et al., 2020; Kim et al., 2019; Sanborn et al., 2015) is a major mechanism of chromatin reorganization upon mitotic exit. Importantly, both our Hi-C and 4C-seq analyses revealed that active regulatory loops marked by H3K27ac and binding of TFs/cofactors are reset at a faster rate than purely structural CTCF/cohesin loops, as also shown in erythroblasts (Zhang et al., 2019). This asynchrony could reflect a faster recruitment of factors and complexes that stall Cohesin and stabilize looping around active regulatory regions upon mitotic exit. Alternatively, it could suggest the presence of extrusion-independent forces for contact formation through affinity/segregation of chromatin marks and factors that are enriched in actively transcribed regions (Di Giammartino et al., 2020; Kim and Shendure, 2019; Nuebler et al., 2018; Rada-Iglesias et al., 2018; Stadhouders et al., 2019).
Mitotic bookmarking by protein factors or histone marks has been previously proposed to promote rapid transcriptional reactivation of cell identity genes during G1 entry (Kadauke and Blobel, 2013; Zaidi et al., 2010). However, direct links between mitotic retention and reactivation kinetics are rather sparse and contradictory (Behera et al., 2019; Blobel et al., 2009; Caravaca et al., 2013; Deluz et al., 2017; Dey et al., 2009; Festuccia et al., 2016; Hsiung et al., 2016; Kadauke et al., 2012; Oh et al., 2020; Owens et al., 2019; Teves et al., 2018) and are usually based on limited numbers of genes or global RNA levels. By tracking genome-wide transcriptional activity at single base-pair resolution (using PRO-seq) and integrating ChIP-seq data of previously reported bookmarking factors in mouse PSCs, we found that only mitotic H3K27ac and selected DNA-bound factors, such as TBP and KLF4, were strongly associated with rapid transcriptional resetting. Mitotic loss of these marks/factors was associated with a slower transcriptional reactivation, suggesting that “de novo” re-assembly of a favorable chromatin state and/or transcriptional complex during G1 entry are rate-limiting factors.
We validated the bookmarking function of H3K27ac by showing that mitotic depletion of this mark by p300 inhibition impaired the early (EG1) transcriptional reactivation of thousands of bookmarked genes, including key pluripotency regulators. Importantly, H3K27ac is also detected on the mitotic chromatin of other cells types, and was recently shown to be important for transcriptional memory in a human osteosarcoma line (Kang et al., 2020). This suggests a more universal bookmarking function of this mark, which might promote the rapid recruitment of important transcriptional regulators and cofactors, such as BRD4 (Behera et al., 2019). Intriguingly, genes that were affected by p300i in EG1 were also characterized by a relatively high mitotic transcriptional activity (not shown), raising the possibility that mitotic transcription per se, might also function as bookmark (Palozola et al., 2017; Palozola et al., 2019). Moreover, we cannot exclude that additional p300/CBP targets (Weinert et al., 2018), such as other histone acetylated residues (H3K18ac, H3K122ac) that can be maintained on mitotic chromatin (Behera et al., 2019), might also contribute to rapid transcriptional activation.
Our computational analyses showed that H3K27ac bookmarking was also associated with rapid topological resetting upon mitotic exit. However, temporal p300 inhibition either during mitosis or in asynchronous cells showed no significant effects on global 3D chromatin (re)organization, despite drastic depletion of H3K27ac and global transcriptional defects. This discrepancy underlines the need for functional validation of computational predictions, and highlights the complex interconnections between transcription, epigenome, and 3D topology (Di Giammartino et al., 2020; Hildebrand and Dekker, 2020; Rada-Iglesias et al., 2018; Rowley and Corces, 2018; van Steensel and Furlong, 2019). Our findings support — at least a partial — functional uncoupling between genomic architecture and chromatin and/or transcriptional state, as proposed in other recent studies (Crump et al., 2019; Jiang et al., 2020; Ray et al., 2019). However, since our experimental perturbations did not fully eliminate transcription, it is plausible that the remaining transcriptional activity promotes proper chromatin organization, in agreement with its previously instructive role in genome folding (Busslinger et al., 2017; Heinz et al., 2018; Hsieh et al., 2020; Zhang et al., 2020a; Zhang et al., 2020b). Systematic and acute perturbations of all candidate regulators of post-mitotic resetting coupled with high resolution 3D genomics and super-resolution imaging technologies will help elucidate these cause-and-effect relationships.
In conclusion, our findings provide insights into the order of molecular events during G1 entry and constitute a stepping stone towards understanding the mechanisms that ensure faithful transcriptional and architectural resetting and ultimately the maintenance of stem cell identity.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Effie Apostolou (efa2001@med.cornell.edu).
Materials Availability
FUCCI2a iPSC lines are available upon request.
Data and Code Availability
All genomics data (Hi-C, ATAC-seq, RNA-seq, 4C-seq, ChIP-seq, and PRO-seq) generated during this study are available at GEO under accession code GSE138965. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138965
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell line generation
Mouse embryonic fibroblasts (MEFs) were isolated from E13.5 embryos from R26Fucci2aR (Mort et al., 2014) mice in a mixed C57BL/6, DBA/2, and CD1 background (gift from Hadjantonakis lab (MSKCC)). Male MEFs were co-infected with a FUGW-rtTA (Maherali et al., 2008) and TRE-OKSM (STEMCCA) cassette (Sommer et al., 2009) according to standard protocols (Di Giammartino et al., 2019) and were then reprogrammed in the presence of 1μ/ml of doxycycline and 50μg/ml of ascorbic acid for 12 days prior to dox withdrawal for establishment of transgene-free induced PSCs (iPSCs). Individual iPSC clones were picked and screened for successful reprogramming based on expression of multiple pluripotency markers.
Cell culture conditions
Two independent, male iPSC clones (described above) were used as biological replicates for all analyses in this study. iPSCs were cultured at 37°C on irradiated feeder cells in FBS/LIF+2i conditions. Specifically, KO-DMEM media (Invitrogen) supplemented with 15% heat-inactivated fetal bovine serum, GlutaMAX, penicillin-streptomycin, non-essential amino acids, beta-mercaptoethanol, and 1000 U/ml LIF, plus 2i (1uM MEK inhibitor (Stemgent 04-0006) and 3uM GSK3 inhibitor (Stemgent 04-0004-10)) was used.
METHOD DETAILS
Mitotic arrest, release, and cell cycle analysis
To arrest cells in mitosis, 80-90% confluent mouse iPSCs were passaged 1:3 on gelatinized plates the day before synchronization. Nocodazole (200ng/ml) (Sigma, M1404) was added to the medium for 6hr prior to collection by mitotic shake-off. 1/3 of cells were processed immediately (for mitotic time point), while the remaining cells were washed 2x with PBS and released into pre-warmed (37°C) media and plated on gelatin. Cells were then collected at 1 hr and 3hrs after release. For each time point, a fraction of cells were used for estimation of synchronization and release efficiency by FACS (BD FACS Canto II analyzer and BD FACS Diva v8.0.3 software) after ethanol fixation and staining with 300nM DAPI (Biolegend, 422801). Aliquots from each time point were also tested for their cell cycle stage by using a BD FACS Aria II to assess the FUCCI2a markers (Cdt1-mCherry, Gmn-mVenus). Any mitotic shake-off experiment with <95% 4N cells was discarded. Asynchronous cells were processed identically but without Nocodazole treatment. FlowJo (v10.6.1) software was utilized for analysis and representation of FACS data.
H3K27ac depletion/recovery using p300 inhibitor
Cells were prepared for synchronization as above. After 3 hours of Nocodazole treatment, either 20uM p300/CBP catalytic inhibitor (A485, Tocris #6387) or the same volume DMSO was added to the media for the remaining 3 hours of synchronization. After mitotic shake-off, half of cells were processed immediately (mitotic time point, MIT) while the remainder were washed and released into fresh media, without nocodazole or inhibitors, and then collected after 1hr (Early G1, EG1). For asynchronous treatment and release, the cells were treated and collected as above, but without Nocodazole. After the 3hr treatment with A485 or DMSO, cells were either collected by trypsinization (ASYN) or washed and released into fresh media for 1 hr (ASYN+1) before collection. For each time point, cells were processed for Western Blot, cell cycle analysis by FACS, H3K27ac ChIP-seq, PRO-seq, and Hi-C. All downstream processing steps were performed as described in the respective sections of the methods. Hi-C was performed using the Arima-HiC kit (Arima, cat # A510008) and the KAPA Hyper Prep Kit (KAPA, cat # KK8502) according to manufacturer’s instructions.
Validation of mitotic purity
The efficiency of synchronization and mitotic shake-off was estimated by FACS after ethanol fixation and staining with H3Ser10p antibody (Abcam, ab47297) and DAPI. Mitotic purity was further validated after the 1% formaldehyde fixation step of Hi-C (see below). Aliquots of mitotic and asynchronous cells were removed after fixation and 60K cells were cytospun at 1000rpm for 5mins onto a slide, following by immunofluorescence staining with H3Ser10p and DAPI. For validation of mitotic purity after PRO-seq permeabilization, aliquots of mitotic and asynchronous cells were resuspended in PBS + 300nM DAPI immediately after permeabilization and analyzed by FACS and microscopy. Image analysis utilized FIJI (Schindelin et al., 2012).
FUCCI2a FACS sorting
Prior to sorting, a partial synchronization and release was performed to enrich for cells in G1. Initially, the cells were treated as for mitotic arrest (above). After 5hrs Nocodazole treatment, cells were kept in the dish (no shake-off), gently washed 2x with PBS, and then released into pre-warmed (37°C) media for 2hrs. Cells were then collected in bulk and a BD FACS Aria II was utilized to sort cells in early G1 (mCherry- Gmn-), late G1 (mCherry+ Gmn−) and S/G2 (Gmn+). An aliquot of each sorted population was rechecked for purity and cell cycle analysis while remaining cells were processed for pre-mRNA RT-qPCR.
pre-mRNA RT-qPCR
RNA was isolated using RNeasy Mini Kit (Qiagen, 74106), and DNase treated (Qiagen, 79254). cDNA was prepared using iScript Reverse Transcription Supermix (Biorad, 1708841). Real Time PCR was performed using PowerUp SYBR Green Master Mix (Applied Biosystems, A25742) and pre-mRNA primer sequences are listed in Table S5.
Western Blot
Cells were resuspended in 1X Laemmli Buffer. Samples were sonicated for 5 cycles using the Bioruptor Pico (Diagenode) and then boiled for 5 mins. Samples were then used for Western Blot analysis using the following antibodies: H3K27ac (ab4729), H3ser10p (ab47297), and H3 (ab1791).
ATAC-seq
ATAC-seq was performed as previously described (Buenrostro et al., 2013). In brief, a total of 50,000 cells were washed once with 50μL of cold PBS and resuspended in 50μL lysis buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.2% (v/v) IGEPAL CA-630). They were then centrifuged for 10min at 800xg at 4°C, followed by the addition of 50μL transposition reaction mix (25μL TD buffer, 2.5¼L Tn5 transposase and 22.5μL ddH2O) using reagents from the Nextera DNA library Preparation Kit (Illumina #FC-121-103). Samples were then incubated at 37°C for 30min. DNA was isolated using a ZYMO Kit (D4014). ATAC-seq libraries were prepared using NEBNext High-Fidelity 2X PCR Master Mix (NEB, #M0541), a uniquely barcoded primer per sample, and a universal primer. Samples were first subjected to 5 cycles of initial amplification. To determine the suitable number of cycles required for the second round of PCR (to minimize PCR bias) the library was assessed by quantitative PCR (Buenrostro et al., 2015). Briefly, a 5μL aliquot of the initial amplification sample was used for 20 cycles of qPCR. Linear Rn versus cycle was plotted to determine cycle number corresponding to 1/3 of maximum fluorescent intensity. For each sample, the remaining 45μL of initial PCR product was therefore further amplified for 5 more cycles using Nextera primers. Samples were subject to a dual size selection (0.55x–1.5x) using SPRIselect beads (Beckman Coulter, B23317). Finally, the ATAC libraries were sequenced on an Illumina Hi-Seq (2500) platform for 50bp paired-end reads.
PRO-seq
PRO-seq was performed as described previously (Kwak et al., 2013), with a few adjustments. All steps were performed on ice. For each replicate per time point, 2 million cells were collected, washed briefly in PBS, and resuspended in Buffer P (10mM Tris-HCl pH 8.0, 10% glycerol, 10mM KCl, 5mM MgCl2, 250mM sucrose, 0.5mM DTT, 0.1% IGEPAL) at the cell density of 2×106 cells/ml. After 1min of incubation, 5× volume of Buffer W (10mM Tris-HCl pH 8.0, 10% glycerol, 10mM KCl, 5mM MgCl2, 250mM sucrose, 0.5mM DTT) was added to dilute the IGEPAL before centrifugation at 1000xg for 5min. Supernatant was discarded and the permeabilized cells were resuspended in Buffer F (5mM Tris-HCl pH 8.5, 40% glycerol, 5mM MgCl2, 0.5mM DTT, 5 mM DTT, 1μL/ml RNase inhibitor (SUPERaseIN, Ambion)) at a density of 2×106 cells/100μl and immediately frozen in liquid nitrogen. Samples were stored at −80°C until library preparation. PRO-seq libraries were prepared using biotin-NTP-supplemented run-ons from 0.2-0.5 × 106 permeabilized cells with Drosophila nuclei spike-in. NRO-RNA was purified using Norgen RNA isolation columns (Norgen Biotek cat. # 17200), followed by base hydrolysis on ice for 10min. After the first bead enrichment and 3’-ligation, the NRO-RNA was again bound to beads. The decapping, end repair, and 5’-ligation reactions were all performed on the beads in 20μl reactions with rotation. Samples in the original, untreated mitotic-release time course were then reverse transcribed while bound to beads in 20μl reactions and cDNA was eluted twice with 25μl ddH2O by heating for 1min at 80°C, returning tubes to magnet, and collecting supernatant. Samples in the DMSO/p300i time course were eluted from beads with TRIZOL following the 5’ ligation, precipitated, and then reverse transcribed as in (Mahat et al., 2016). Test amplifications were performed on 5% of each library and fully amplified libraries were concentrated using Monarch PCR clean-up columns (NEB, T1030S). Libraries were sequenced on an Illumina NextSeq500 for 75bp single end reads.
RNA-seq
Total RNA from 300,000 asynchronous cells per replicate was prepared with TRIZOL (Life technologies #15596018) following manufacturer’s instructions. Libraries were generated by the Weill Cornell Genomics core facility using the Illumina TruSeq stranded mRNA library preparation kit (#20020594) and sequenced on an Illumina HiSeq4000 platform on SE50 mode.
Hi-C
In situ Hi-C was performed as described previously (Rao et al., 2014). In brief, 2 million cells (per replicate per time point) were crosslinked in 1% formaldehyde at RT for 10 minutes and quenched with 125mM glycine for 5 mins at RT. Cells were resuspended in lysis buffer (10mM Tris-HCl (pH 8.0), 10mM NaCl, 0.2% lgepal CA630 (Sigma)) and chromatin was digested overnight with 100U MboI (NEB, R0147M). Fragmented ends were labeled with biotin-14-dATP (Life Tech, 19524-016). Overnight ligation was performed using 2000U T4 DNA Ligase (NEB, M0202), and then samples were reverse cross-linked with Proteinase K (NEB, P8102). DNA was ethanol precipitated overnight, washed, and then sonicated in a Diagenode Bioruptor 300 (8 cycles, 30sec on/off, medium setting). SPRIselect beads (Beckman Coulter, B23317) were used for a 300-500 bp size selection. Aliquots were removed after digestion, ligation, sonication, and size selection and run on an agarose gel for quality control. Dynabeads MyOne Streptavidin T1 beads (Invitrogen, 65602) were used for biotin pulldown. Repair of fragmented ends was performed followed by dA-tailing. 1X NEB Quick Ligation Reaction Buffer (NEB, B2200S) was used for ligation of SeqCap Adapter Kit A (Roche, 7141530001) indexes. Library amplification was performed directly off of the T1 beads using KAPA HiFi HotStart ReadyMix and primers (KAPA, KK2620) and 8 PCR cycles. Finally, a 1X cleanup was performed using SPRIselect beads. The Hi-C libraries were sequenced on an Illumina Hi-Seq 4000 platform for 50bp paired-end reads. For the p300i and DMSO treatment experiments shown in Figures 6 and S6, Hi-C was performed using the Arima-HiC kit (Arima, cat # A510008) and the KAPA Hyper Prep Kit (KAPA, cat # KK8502) according to manufacturer’s instructions.
4C-seq
For each sample, 2 million cells were crosslinked in 1% formaldehyde at RT for 10 min and quenched with 125 mM glycine for 5 min. The cell pellets were washed twice in PBS and resuspended in 300μl lysis buffer (10mM Tris-HCl (pH 8.0), 10mM NaCl, 0.2% lgepal CA630 (Sigma I8896)), then incubated on ice for 20 min. Following centrifugation at 2500xg for 5 min at 4°C, the pellet was resuspended in 50uL of 0.5% SDS and incubated for 10 min at 65°C. SDS was quenched with 145uL ddH2O and 25uL of 10% TritonX-100 for 15 mins at 37°C. 25ul of CutSmart buffer (NEB B7204S) was added with 10ul DpnII enzyme (NEB R0543M) and the samples were incubated overnight at 37°C with 700rpm rotation. After confirming the digestion efficiency, the enzyme was inactivated by incubating at 65°C for 20 mins. The samples were then diluted with 669μl ddH2O, 120μl T4 ligation buffer (NEB B0202), 60μl 10mM ATP (NEB P0756S), 120μl 10% Triton X-100, 6μl 20mg/ml BSA and 5μl 400U/μl T4 DNA Ligase (NEB M0202) for 3h on a rotor at RT. The samples were then treated with proteinase K and reverse crosslinked overnight. Following RNAse treatment, phenol/chloroform extraction and DNA precipitation, the pellets were dissolved in 100μl of 10mM Tris pH 8. The second digestion was performed by adding 20μl 10x buffer B (Fermentas), 10μl Csp6I (Fermentas, ER0211), 80μl ddH2O, and incubating overnight at 37°C with 700rpm rotation. After confirming the second digestion efficiency, the enzyme was again inactivated. Another ligation was performed by adding 300μl T4 ligation buffer, 150μl 10mM ATP, 5μl T4 DNA Ligase, and ddH2O to 3mL and incubating overnight at 16°C. The DNA was purified by phenol/chloroform extraction and ethanol precipitation, resulting in the 4C-template. For library preparation, primers were designed around the TSS were for each example gene according to criteria previously described (Krijger et al., 2020). Library preparation was then performed using the inverse PCR strategy described in (Krijger et al., 2020). Briefly, 200 ng of 4C-template DNA was used to PCR amplify the libraries using the Roche Expand long template PCR system (Roche, 11681842001). Primers were removed using SPRIselect beads (Beckman Coulter, B23317). A second round of PCR was performed using the initial PCR library as a template, with overlapping primers to add the P5/P7 sequencing primers and indexes.
The libraries were sequenced on a HiSeq4000 in SE150 mode. All of the primer sequences can be found in Table S5.
H3K27ac ChIP-seq
ChIP-seq was performed as previously described (Liu et al., 2017b), with a few alterations. 5 million cells were used per replicate for each time point. Cells were crosslinked in 1% formaldehyde at RT for 10 minutes and quenched with 125mM glycine for 5 mins at RT. As a normalization control (Orlando et al., 2014), 5 million formaldehyde-fixed Drosophila nuclei were added to each sample. Cell pellets were washed twice in PBS and resuspended in 300ul lysis buffer (10mM Tris pH8, 1mM EDTA, 0.5% SDS). Cells were sonicated in a Pico bioruptor device (10 cycles 30sec on/off,) and spun down 10 minutes at 4°C at maximum speed. 5% of each sample was aliquoted and frozen to later use as input. Supernatants were diluted 5 times with dilution buffer (0.01% SDS, 1.1% triton,1.2mM EDTA,16.7mM Tris pH8, 167mM NaCl) and incubated with H3K27ac antibody (3ug/10M total cells) (ab4729) O/N with rotation at 4°C. Next day, protein G Dynabeads (ThermoScientific), preblocked with 1mg/ml BSA protein, were added (30ul Dynabeads per sample) and incubated for 3.5 hours at 4°C. Beads were immobilized on a magnet and washed twice in low salt buffer (0.1% SDS,1% triton, 2mM EDTA, 150mM NaCl, 20mM Tris pH8), twice in high salt buffer (0.1% SDS,1% triton, 2mM EDTA, 500mM NaCl, 20mM Tris pH8), twice in LiCl buffer (0.25M LiCl, 1% NP40, 1% deoxycholic acid (sodium salt), 1mM EDTA, 10mM Tris pH8) and once in TE buffer. DNA was then eluted from the beads by incubating with 150ul elution buffer (1% SDS, 100mM NaHCO3) for 30 minutes at 65°C (vortexing every 10min). Supernatants were collected and reverse-crosslinked by incubation at 65°C O/N in presence of proteinase K. After RNase A treatment for 1hr at 37°C, DNA was purified using a ZYMO Kit (D4014). 25ng of immunoprecipitated material and input were used for ChIP-seq library preparation using the KAPA Hyper prep kit (KK8502) according to manufacturer’s instructions. Libraries were sequenced on an Illumina NextSeq2000 platform on SR100 mode.
QUANTIFICATION AND STATISTICAL ANALYSIS
ATAC-seq analysis
Bowtie 2 aligner (v2.2.6) (Langmead and Salzberg, 2012) with the following parameters –very-sensitive -X 2000 was used to map raw sequencing data on mm10 genome while filtering of duplicate reads and low quality reads (Q<20) was performed with the use of picard tools (MarkDuplicates command, v2.12.2) (Picard) and samtools (v1.8) (Li et al., 2009). All reads in Blacklisted genomic regions and chrM were removed from downstream analysis. Filtered paired-end reads were corrected for tn5 insertion position at each read end by shifting +4 / −5 bp from the positive and negative strand respectively. Peak calling was performed in the merged aligned reads from all time points in order to build an accessibility genome atlas (ATAC ATLAS) for the PSC cell cycle with the use of MACS2 peak calling algorithm (v2.1.1) (Zhang et al., 2008). Visualization of sequencing data on the IGV browser (Robinson et al., 2011) was perfomed with the use of bedGraph and BigWig files generated with the use of bedtools (genomeCoverageBed) (Quinlan and Hall, 2010) and bedGraphToBigWig command. All files displayed were normalized to sequencing depth and RPM (reads per million) values were generated for each BigWig file. Identification of “retained” and “lost” accessible sites was performed with the use of intersectBed for all accessible sites in mitotic and asynchronous after peak calling with MACS2.
PRO-seq analysis
Sequencing of PRO-seq in single-end 75bp fragments was performed for each time point. Cutadapt (v1.4.2) (Martin, 2011) was used in order to trim adaptors from sequenced reads and keep only the first 25bp of each read. Trimmed sequencing data were aligned with the use of bowtie 2 aligner in both drosophila (Dm6 version) and mouse genome (mm10 version). All reads that mapped to ribosomal RNA were excluded from the adaptor-filtered files and the remaining reads were first mapped to the Drosophila genome and the unmapped reads were isolated and aligned to the mm10 mouse genome. All aligned files were filtered for low quality reads, and reads that were mapped in blacklisted regions and chrM. Only uniquely aligned reads on the mappable genome (Umap 24 bp) were used to calculate the percentage of aligned reads in each replicate. For downstream analysis we used all protein coding genes from ensemble GRCm38.95 version while gene expression was calculated based on the number of reads within the gene body after discarding the first and last 500bp [TSS + 500bp, TES -500bp] in order to avoid getting signal from the promoter and transcription end site which are usually rich in PRO-seq signal. Based on the number of cells used for each experiment and the mapped reads in Drosophila genome (in millions) we normalized gene expression levels after scaling all reads to the sample with the minimum sequencing depth (Table S1) and then normalized to the size of the gene (Kb). Normalized RPKM values were used for downstream analysis for more than 14,000 protein coding and long non-coding genes that were found to be expressed (normalized RPKM >=1) at least in one of the 4 time points.
The same analysis steps were applied for the PRO-seq experiments with p300i or DMSO (presented in Figures 6 and S6). An additional scaling factor was also applied to normalize data across different runs and account for batch effects. In order to calculate the transcriptional changes per time-point we generated percentage of transcriptional decrease based on the following formula: 100*(p300i(t) - DMSO(t))/DMSO(t), where (t) stands for each time-point. Differential analysis of genes per time-point was conducted with the use of DESeq algorithm (Anders and Huber, 2010), comparing each p300i-treated sample to the respective DMSO. Only genes with more than 20% of transcriptional changes and p-adjusted lower than 0.05 were considered as transcriptionally affected by p300i (DEGs).
TRE and eTRE identification
Detection of regulatory elements (TRE) was performed in the merged PRO-seq aligned reads from each timepoint with the use of dREG algorithm (Danko et al., 2015; Wang et al., 2019). All identified TREs were extended +/−100bp from both ends and those that were included in a region +/− 1kb from any transcript were discarded from the downstream analysis. The remaining TREs were identified as enhancer TREs (eTRE) and their expression levels were calculated as in PRO-seq analysis with the only difference of the size normalization which covered the whole eTRE region.
RNA-seq analysis
TopHat2 (Kim et al., 2013) with default setting for paired end data was used to align the data to the mouse genome (mm10) and samtools for transforming the file formats, filtering low quality reads and sorting the paired end reads. HTseq-count (Anders et al., 2015) on mapped reads was used to calculate the raw counts per transcript and additional RPKM normalization was performed in R.
Hi-C analysis
HiCexplorer (v1.8) (Ramirez et al., 2018) was used to analyze the 50b paired-end reads generated for Hi-C experiments in asynchronous, mitotic cells and cells in Early and Late G1 phase. Bowtie2 aligner was used to align each pair independently and HiCexlorer tools hicBuildMatrix, hicNormalize and hicCorrectMatrix were utilized in order to construct the 20kb HiC matrices , normalized for sequencing depth and perform iterative correction method (ICE) normalization (Imakaev et al., 2012). The same algorithms were used for analyzing p300i treated and untreated asynchronous, asynchronous +1h, mitotic and Early G1 phase cells. Both .h5 files and .hic files for visualizing Compartments, TADs and loops were generated with the use of HiCExplorer and juicer_tools (version 1.19.02) (Durand et al., 2016) pre-command in multiple resolutions. Custom scripts, hicConvertFormat, and juicer tools were used for the conversion of .h5 to .hic files.
Compartment analysis
In addition to 20kb normalized matrices, 100kb matrices were generated with the use of hicMergeMatrixBins in order to visualize larger chromatin structures. Compartment identification was performed with the use of Cscore algorithm (v1.1) (Zheng and Zheng, 2018) for each replicate and chromosome separately with the use of the following parameter minDis = 1000000. All compartments were assigned to active (A) and inactive (B) compartments based on gene density in each 100kb region.
TAD analysis
Matrices in 20kb resolution were used to identify topologically associated domains (TADs) with hicFindTADs and the use of the following parameters, “--minDepth 120000 --maxDepth 420000 --step 40000 --delta 0.01--thresholdComparisons 0.01 --correctForMultipleTesting FDR”. Boundaries calculated in each replicate were merged and only boundaries with an insulation score <-0.3 were considered as TAD boundaries for that time point. TAD domain score was calculated as described in (Stadhouders et al., 2018).
Chromatin contact analysis
Cis-chromatin interactions were calculated for each replicate with the use of Fit-Hi-C (v1.1.0) (Ay et al., 2014) with the use of the following parameters “-r 20000 -L 40000 -U 10000000”. Only loops that were common in both replicates with a q-value <0.01 and more than 5 contacts were scored as valid loops, which resulted in 52,489 called contacts. In order to increase stringency of our called loops we focused on the common loops between our dataset and deep-sequenced Micro-C from mouse ESC cells (GSE130275) (Hsieh et al., 2020). HiCCUPS (juicer_tool version 1.19.02) was applied on the 2.6B billion .hic matrix provided by GEO and more than 23,000 contacts were called in the mESC Micro-C. Overlap of the two datasets after extending newly called Micro-C loops by one 10kb bin led to a group of 14,091 highly-confidence contacts which we used for all downstream analyses. Contact strength per time point was calculated based on the normalized Hi-C reads, while different kinetics of loop reestablishment was estimated after k-means clustering of the z-transformed normalized Hi-C reads across all timepoints. Enrichment analysis of previously published data with LOLA (Sheffield and Bock, 2016) was performed on the accessible sites that overlapped with the anchors of each loop cluster. APA plots and APA metrics at 10kb resolution were generated with Juicer with the following parameters “apa -w 10 -r 10000”. Aggregate signal from +/−10 bins around both anchors for each loop was plotted as a heatmap in R.
Virtual 4C
We used filtered Hi-C read pairs as described above and in (Di Giammartino et al., 2019) before binning and normalizing each replicate. We extracted read pairs from the 10kb bin that the read mate maps around the virtual viewpoint. We defined successive overlapping windows for each chromosome at a 10kb resolution, overlapping by 90% of their length. We then counted the second mapped read mate in all overlapping bins. Read counts were normalized to the total sequencing depth of the respective replicate. Visualization was done using normalized read counts per condition.
4C-seq analysis
All sequenced reads were demultiplexed with the use of fastx-toolkit while VP primer was trimmed with the use of fastq_trimmer. Only reads containing the RE site next to the VP were considered for analysis. Bowtie2 with very-sensitive option was used for aligning the trimmed reads (size > 40bp) to the mouse genome (mm10) while samtools was used to filter for high quality uniquely mapped reads. Smoothening of the 4C signal was performed by calculating the number of raw reads within the 10kb binned genome with a sliding window of 500bp followed by sequencing depth normalization of the cis interaction identified around the viewpoint (+/− 1MB). R and DESeq were used to depict the differential enrichment between the sliding windows and bins with an enrichment of >3 CPM, pvalue <0.05 and fold >2 between any two time points were scored as differential interacting regions. Significant bins per time point were merged and only interactions with more than 3 bins were scored as interaction regions. Interacting regions that were less than 1kb were discarded.
ChIP-seq enrichment analysis
Enrichment analysis of previously published data in ESC cells was performed with LOLA (Sheffield and Bock, 2016). For gene and loop clusters the enrichment analysis was performed on the accessible sites that overlapped with the promoter (TSS+/−2.5kb) or with either of the anchors. All the accessible sites (ATAC ATLAS) identified in the time course were used as a background set and transcription factors or histone modifications with a ‘-log10(p-value)> 3’ were considered significant and presented in the heat maps.
ChIP-seq analysis of H3K27ac DMSO/p300i dataset
All reads were first aligned to Drosophila genome (DM6) with bowtie2 aligner, followed by filtering of low quality reads (q>20), duplicates, chrM and blacklisted regions with the use of samtools, picard tools and bedtools. All unaligned reads were exported and mapped to the mouse genome (mm10 genome version) followed by the same analysis steps. MACS2 with default settings was used to call H3K27ac peaks. The new mitotic and asynchronous peaks were then overlapped with previous published peaks (GSE92846) and the common “Retained” and “Lost” H3K27ac peaks were used for downstream analysis. All mouse reads were normalized to drosophila reads and bigWig files were generated with the use of deepTools (version 3.5.0) (Ramírez et al., 2016) and bigWigCompare tool. For all replicates per time-point, ChIP was normalized to the corresponding input and deeptools were utilized to calculate enrichment of ChIP signal around bookmarked regions.
ChIP-seq analysis of published data
We used the following published ChIP-seq datasets from asynchronous/interphase and mitotic mouse ESCs GSE75066 (ESRRB), GSE122589 (OCT4, SOX2), GSE131356 (CTCF), GSE109962 (TBP), GSE92846 (KLF4, H3K27ac). When possible, ChIP-seq data sets that had utilized DSG fixation were used (OCT4, SOX2 (Festuccia et al., 2019)), as this has been shown to capture mitotic TF binding better than traditional formaldehyde fixation (Festuccia et al., 2019; Teves et al., 2016). After merging individual replicates and respective inputs, all data were mapped to mm10 with bowtie2 and --local -very-sensitive-local option. Filtering of low-quality mapped reads and duplicate removal was performed with samtools and picard tools while additional filtering of mm10 black-regions was done with bedtools. MACS2 with default settings was used to call peaks in mitotic and in asynchronous/interphase cells. Intersection of called peaks in these 2 cell states led to the classification as Retained or Lost peaks for each transcription factor and histone modification.
Gene ontology and pathway analysis
David knowledgebase (Dennis et al., 2003) was used for annotating transcripts to biological process and signaling pathways. ENSEMBL transcript ids were used as an input. Only processes and pathways with a corrected (Benjamini) p-value <0.01 were considered significant. For assigning biological processes and pathways to genomic regions (for eTRE clusters) we used GREAT analysis software (v3.0.0) (McLean et al., 2010) with the use of the ‘Basal plus extension’ option and with 2.5kb proximal upstream, 1kb proximal downstream and distal 250kb parameters. Only processes and pathways with Hyper FDR Q-value <0.05 were considered significant.
Assigning gene-eTRE pairs for enrichment analysis
We paired genes and eTREs with two different approaches.
1. Distance: eTREs were assigned to genes in a distance +/− 20kb from the TSS of gene. If multiple eTREs were within +/−20kb, the closest eTRE was assigned to the gene. A total of 4779 gene-eTRE pairs were used for this analysis.
2. Chromosomal interactions: All HiChIP loops (10kb resolution) that were common in two different ESC cell lines (Di Giammartino et al., 2019) were used to pair genes and eTREs. Loops with a unique eTRE in only one of the two anchors and at least one TSS in the looped anchor were scored as eTRE-gene loops. A total of 12411 gene-eTRE pairs were used for this analysis.
Statistical Methods
All median comparisons were performed with the use of two-sided Wilcoxon’s rank sum test. P-values and summary statistics for all variables tested are provided in Table S6. K-means with default settings (“Hartigan-Wong” algorithm) was performed in R (3.4.4 version) for Hi-C and 4C-seq using the average normalized values from the two replicate experiments for each timepoint. Two-sided Fisher’s exact test was used to calculate significance of enrichment between various groups of eTREs, genes, loops, ATAC-seq peaks, H3K27ac ChIP-seq peaks, TBP ChIP-seq peaks, and CTCF ChIP-seq peaks. In the enrichment plots, size of dots indicates significance of the enrichment while color indicates Observed over Expected ratio. Comparison of the distribution of different gene clusters around boundaries was performed in R with the use of Kolmogorov-Smirov test. Median and their corresponding 95% confidence interval was calculated in R with the use of “DescTools” library. Identification of differential compartments, TAD domains, and TAD boundaries was performed in R with the use of Welch’s t-test between DMSO and p300i treated replicates at each time-point. Only regions with a difference of at least 0.2 (compartments), 0.1 (domains), and 0.05 (boundaries) and p-value <0.01 were considered significantly affected after p300 inhibition.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal Histone H3 (phospho S10) | Abcam | Cat# ab47297 |
| Rabbit polyclonal to Histone H3 (acetyl K27) | Abcam | Cat# ab4729 |
| Rabbit polyclonal to Histone H3 | Abcam | Cat# ab1791 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Nocodazole | Sigma | Cat# M1404 |
| MEK inhibitor (PD0325901) | Stemgent | Cat# 04-0006 |
| GSK inhibitor (CHIR99021) | Stemgent | Cat# 04-0004-10 |
| A 485 (p300/CBP inhibitor) | Tocris | Cat# 6387 |
| Critical Commercial Assays | ||
| Nextera DNA library Preparation Kit | Illumina | Cat# FC-121-103 |
| SeqCap Adapter Kit A | Roche | Cat# 7141530001 |
| KAPA HiFi HotStart ReadyMix and primers | KAPA | Cat# KK2620 |
| KAPA Hyper Prep Kit | KAPA | Cat# KK8502 |
| Arima-HiC Kit | Arima | Cat# A510008 |
| Deposited Data | ||
| Raw and processed data of ATAC-seq libraries in mitotic and asynchronous mouse PSCs |
This study | GEO: GSE138965 |
| Raw and processed data of Hi-C libraries for mitotic release time course in mouse PSCs |
This study | GEO: GSE138965 |
| Raw and processed data of PRO-seq libraries for mitotic release time course in mouse PSCs |
This study | GEO: GSE138965 |
| Raw and processed data of 4C-seq libraries at 11 loci for mitotic release time course in mouse PSCs |
This study | GEO: GSE138965 |
| Raw and processed data of RNA-seq libraries in asynchronous mouse PSCs |
This study | GEO: GSE138965 |
| Raw and processed data of H3K27ac ChIP-seq libraries after DMSO or p300i (A485) treatment in mitosis, early G1, and asynchronous mouse PSCs |
This study | GEO: GSE138965 |
| Raw and processed data of PRO-seq libraries after DMSO or p300i (A485) treatment in mitosis, early G1, and asynchronous mouse PSCs |
This study | GEO: GSE138965 |
| Raw and processed data of Hi-C libraries after DMSO or p300i (A485) treatment in mitosis, early G1, and asynchronous mouse PSCs |
This study | GEO: GSE138965 |
| H3K27ac and KLF4 ChIP-seq in mitotic and asynchronous mouse embryonic stem cells |
Liu et al., 2017b | GEO: GSE92846 |
| TBP ChIP-seq in mitotic and asynchronous mouse embryonic stem cells |
Teves et al., 2018 | GEO: GSE109962 |
| ESRRB ChIP-seq in mitotic and asynchronous mouse embryonic stem cells |
Festuccia et al., 2016 | GEO: GSE75066 |
| CTCF ChIP-seq in mitotic and asynchronous mouse embryonic stem cells |
Owens et al., 2019 | GEO: GSE131356 |
| OCT4 and NANOG ChIP-seq in mitotic and asynchronous mouse embryonic stem cells |
Festuccia et al., 2019 | GEO: GSE122589 |
| H3K27ac Hi-ChIP in asynchronous mouse embryonic stem cells |
Di Giammartino et al., 2019 | GEO: GSE113431 |
| Micro-C in mouse embryonic stem cells: valid pairs and .hic matrix files |
Hsieh et al., 2020 | GEO: GSE130275 |
| Drosophila reference genome (Dm6) | Berkeley Drosophila Genome Project |
https://www.ncbi.nlm.nih.gov/assembly/GCF_000001215.4/ |
| Mouse reference genome GRCm38 (mm10) | Genome Reference Consortium |
https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.20/ |
| Experimental Models: Cell Lines | ||
| FUCCI2a iPSC clone A | This study | N/A |
| FUCCI2a iPSC clone B | This study | N/A |
| Oligonucleotides | ||
| See Table S5 for all oligonucleotide sequences | This study Krijger et al., 2020 |
N/A |
| Software and Algorithms | ||
| FlowJo (v10.6.1) | FlowJo | http://docs.flowjo.com/d2/ |
| FIJI (ImageJ v2.0.0) | Schindelin et al., 2012 | https://imagej.net/Fiji |
| Bowtie 2 aligner (v2.2.6) | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Tophat2 (v2.1.0) | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Picard (MarkDuplicates command, v2.12.2) | Broad Institute | https://broadinstitute.github.io/picard/ |
| Samtools (v1.7) | Li et al., 2009 | http://samtools.sourceforge.net/ |
| MACS2 (v2.1.1) | Zhang et al., 2008 | https://pypi.org/project/MACS2/#description |
| IGV browser | Robinson et al., 2011 | https://software.broadinstitute.org/software/igv/ |
| Bedtools (v2.25.0) | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| Htseq-count (0.5.4.p3) | Anders et al., 2015 | https://htseq.readthedocs.io/en/release_0.11.1/history.html#version-0-5-4 |
| DeepTools (version 3.0.2) | Ramirez et al, 2016 | https://deeptools.readthedocs.io/en/develop/index.html# |
| Fastx-toolkit (0.0.13) | FastX-Toolkit | http://hannonlab.cshl.edu/fastx_toolkit/ |
| Cutadapt (v1.4.2) | Martin, 2011 | https://doi.org/10.14806/ej.17.1.200 |
| dREG | Danko et al., 2015; Wang et al., 2019 | https://github.com/Danko-Lab/dREG |
| HiCexplorer (v1.8) | Ramirez et al., 2018 | https://hicexplorer.readthedocs.io/en/latest/ |
| FitHiC (1.1.3) | Ay et al., 2014 | https://noble.gs.washington.edu/proj/fit-hi-c/ |
| R (3.4.4) | R Core Team, 2020 | https://www.R-project.org/ |
| LOLA (v1.0.0) | Sheffield and Bock, 2016 | http://code.databio.org/LOLA/ |
| DESeq | Anders and Huber, 2010 | https://bioconductor.org/packages/release/bioc/html/DESeq.html |
| Cscore algorithm (v1.1) | Zheng and Zheng, 2018 | https://github.com/scoutzxb/CscoreTool |
| David knowledgebase (v6.8) | Dennis et al., 2003 | https://david.ncifcrf.gov/ |
| GREAT analysis (v3.0.0) | McLean et al., 2010 | http://great.stanford.edu/great/public-3.0.0/html/ |
| Juicebox | Robinson et al., 2018 | https://www.aidenlab.org/juicebox/ |
| Juicer tools (v1.19.02) | Durand et al., 2016 | https://github.com/aidenlab/juicer |
HIGHLIGHTS.
Rapid resetting of the stem cell program and transient activation of lineage genes
Chromatin contacts around stem cells enhancers reform faster than structural loops
Chromatin reorganization partially associates with transcriptional levels and kinetics
Loss of mitotic H3K27ac perturbs transcriptional, but not architectural, resetting
ACKNOWLEDGEMENTS
We are grateful to Matthias Stadtfeld, Ari Melnick, Dan Landau, Chris Barbieri, Danwei Huangfu, Steve Josefowicz, Kat Hadjantonakis, Todd Evans, and members of the Apostolou, Tsirigos, Danko and Stadtfeld laboratories for critical feedback on this work. We also thank Gerd Blobel for continuous brainstorming, Charles Danko and Alexandra Chivu for advice on dREG analysis and eTRE interpretation, Boaz Aronson and Pedro Rocha for 4C-seq primer design, and Laurianne Scourzic for FACS analysis and sorting. The R26Fucci2aR mice were a generous gift from the Hadjantonakis lab. This work was supported by the NIH Director’s New Innovator Award (DP2DA043813), the National Institute of General Medical Sciences ( NIGMS ) (grants 1R01GM138635 to E.A. and 1R35GM128857 to L.C. ) and the Tri-Institutional Stem Cell Initiative by the Starr Foundation. BPW is supported by the NICHD (T32HD060600 and F30HD097926), as well as by a Medical Scientist Training Program grant from NIGMS (T32GM007739) to the Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD-PhD Program. A.T. is supported by the American Cancer Society (RSG-15-189-01-RMC), the Leukemia and Lymphoma Society, and the St. Baldrick’s Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL EXCEL TABLES
Table S1. PRO-seq stats and gene and eTRE clusters, Related to Figure 2
Table S2. Bookmarking Enrichment Plots, Related to Figure 3
Table S3. Hi-C and 4C-seq stats and loop clusters, Related to Figures 4 and 5
Table S4. PRO-seq, H3K27ac ChIP-seq and Hi-C stats for p300i and DMSO treated samples, Related to Figure 6
Table S5. Primers used in this study, Related to Figures S2 and 5
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abramo K, Valton A-L, Venev SV, Ozadam H, Fox AN, and Dekker J (2019). A chromosome folding intermediate at the condensin-to-cohesin transition during telophase. Nature cell biology 21, 1393–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alipour E, and Marko JF (2012). Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Res 40, 11202–11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biology 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asenjo HG, Gallardo A, López-Onieva L, Tejada I, Martorell-Marugán J, Carmona-Sáez P, and Landeira D (2020). Polycomb regulation is coupled to cell cycle transition in pluripotent stem cells. Science Advances 6, eaay4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ay F, Bailey TL, and Noble WS (2014). Statistical confidence estimation for Hi-C data reveals regulatory chromatin contacts. Genome Res 24, 999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behera V, Stonestrom AJ, Hamagami N, Hsiung CC, Keller CA, Giardine B, Sidoli S, Yuan Z-F, Bhanu NV, Werner MT, et al. (2019). Interrogating Histone Acetylation and BRD4 as Mitotic Bookmarks of Transcription. Cell Reports 27, 400–415.e405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobel GA, Kadauke S, Wang E, Lau AW, Zuber J, Chou MM, and Vakoc CR (2009). A Reconfigured Pattern of MLL Occupancy within Mitotic Chromatin Promotes Rapid Transcriptional Reactivation Following Mitotic Exit. Molecular Cell 36, 970–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonev B, Cohen NM, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot J-P, Tanay A, et al. (2017). Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 171, 557–572.e524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boward B, Wu T, and Dalton S (2016). Concise Review: Control of Cell Fate Through Cell Cycle and Pluripotency Networks. Stem Cells 34, 1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busslinger GA, Stocsits RR, van der Lelij P, Axelsson E, Tedeschi A, Galjart N, and Peters J-M (2017). Cohesin is positioned in mammalian genomes by transcription, CTCF and Wapl. Nature 544, 503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caravaca JM, Donahue G, Becker JS, He X, Vinson C, and Zaret KS (2013). Bookmarking by specific and nonspecific binding of FoxA1 pioneer factor to mitotic chromosomes. Genes & Development 27, 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronado D, Godet M, Bourillot P-Y, Tapponnier Y, Bernat A, Petit M, Afanassieff M, Markossian S, Malashicheva A, Iacone R, et al. (2013). A short G1 phase is an intrinsic determinant of naïve embryonic stem cell pluripotency. Stem Cell Res 10, 118–131. [DOI] [PubMed] [Google Scholar]
- Crump NT, Ballabio E, Godfrey L, Thorne R, Repapi E, Kerry J, Tapia M, Hua P, Filippakopoulos P, Davies JOJ, et al. (2019). BET inhibition disrupts transcription but retains enhancer-promoter contact. bioRxiv, 848325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danko CG, Hyland SL, Core LJ, Martins AL, Waters CT, Lee HW, Cheung VG, Kraus WL, Lis JT, and Siepel A (2015). Identification of active transcriptional regulatory elements from GRO-seq data. Nature Methods 12, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson IF, Bauer B, Goetz D, Tang W, Wutz G, and Peters J-M (2019). DNA loop extrusion by human cohesin. Science 366, 1338. [DOI] [PubMed] [Google Scholar]
- Deluz C, Friman ET, Strebinger D, Benke A, Raccaud M, Callegari A, Leleu M, Manley S, and Suter DM (2016). A role for mitotic bookmarking of SOX2 in pluripotency and differentiation. Genes & Development 30, 2538–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, and Lempicki RA (2003). DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4, P3. [PubMed] [Google Scholar]
- Dey A, Nishiyama A, Karpova T, McNally J, and Ozato K (2009). Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Molecular Biology of the Cell 20, 4899–4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giammartino DC, Kloetgen A, Polyzos A, Liu Y, Kim D, Murphy D, Abuhashem A, Cavaliere P, Aronson B, Shah V, et al. (2019). KLF4 is involved in the organization and regulation of pluripotency-associated three-dimensional enhancer networks. Nature Cell Biology 21, 1179–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giammartino DC, Polyzos A, and Apostolou E (2020). Transcription factors: building hubs in the 3D space. Cell Cycle 19, 2395–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dileep V, Ay F, Sima J, Vera DL, Noble WS, and Gilbert DM (2015). Topologically associating domains and their long-range contacts are established during early G1 coincident with the establishment of the replication-timing program. Genome Res 25, 1104–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, and Aiden EL (2016). Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Systems 3, 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnshaw WC, and Laemmli UK (1983). Architecture of metaphase chromosomes and chromosome scaffolds. J Cell Biol 96, 84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans M (2011). Discovering pluripotency: 30 years of mouse embryonic stem cells. Nat Rev Mol Cell Biol 12, 680–686. [DOI] [PubMed] [Google Scholar]
- Festuccia N, Dubois A, Vandormael-Pournin S, Tejeda EG, Mouren A, Bessonnard S, Mueller F, Proux C, Cohen-Tannoudji M, and Navarro P (2016). Mitotic binding of Esrrb marks key regulatory regions of the pluripotency network. Nature Cell Biology 18, 1139–1148. [DOI] [PubMed] [Google Scholar]
- Festuccia N, Owens N, Papadopoulou T, Gonzalez I, Tachtsidi A, Vandoermel-Pournin S, Gallego E, Gutierrez N, Dubois A, Cohen-Tannoudji M, et al. (2019). Transcription factor activity and nucleosome organization in mitosis. Genome Res 29, 250–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, and Mirny LA (2016). Formation of Chromosomal Domains by Loop Extrusion. Cell Rep 15, 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibcus JH, Samejima K, Goloborodko A, Samejima I, Naumova N, Nuebler J, Kanemaki MT, Xie L, Paulson JR, Earnshaw WC, et al. (2018). A pathway for mitotic chromosome formation. Science 359, eaao6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golfier S, Quail T, Kimura H, and Brugués J (2020). Cohesin and condensin extrude DNA loops in a cell cycle-dependent manner. eLife 9, e53885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesfeld JM, and Forbes DJ (1997). Mitotic repression of the transcriptional machinery. Trends Biochem Sci 22, 197–202. [DOI] [PubMed] [Google Scholar]
- Heinz S, Texari L, Hayes MGB, Urbanowski M, Chang MW, Givarkes N, Rialdi A, White KM, Albrecht RA, Pache L, et al. (2018). Transcription Elongation Can Affect Genome 3D Structure. Cell 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, Bazett-Jones DP, and Allis CD (1997). Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma 106, 348–360. [DOI] [PubMed] [Google Scholar]
- Hildebrand EM, and Dekker J (2020). Mechanisms and Functions of Chromosome Compartmentalization. Trends in Biochemical Sciences 45, 385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh T-HS, Cattoglio C, Slobodyanyuk E, Hansen AS, Rando OJ, Tjian R, and Darzacq X (2020). Resolving the 3D Landscape of Transcription-Linked Mammalian Chromatin Folding. Molecular Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiung CCS, Bartman CR, Huang P, Ginart P, Stonestrom AJ, Keller CA, Face C, Jahn KS, Evans P, Sankaranarayanan L, et al. (2016). A hyperactive transcriptional state marks genome reactivation at the mitosis-G1 transition. Genes & Development 30, 1423–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiung CCS, Morrissey CS, Udugama M, Frank CL, Keller CA, Baek S, Giardine B, Crawford GE, Sung M-H, Hardison RC, et al. (2015). Genome accessibility is widely preserved and locally modulated during mitosis. Genome Research 25, 213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J, and Mirny LA (2012). Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Methods 9, 999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javasky E, Shamir I, Gandhi S, Egri S, Sandler O, Rothbart SB, Kaplan N, Jaffe JD, Goren A, and Simon I (2018). Study of mitotic chromatin supports a model of bookmarking by histone modifications and reveals nucleosome deposition patterns. Genome Res 28, 1455–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Huang J, Lun K, Li B, Zheng H, Li Y, Zhou R, Duan W, Wang C, Feng Y, et al. (2020). Genome-wide analyses of chromatin interactions after the loss of Pol I, Pol II, and Pol III. Genome Biology 21, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadauke S, and Blobel GA (2013). Mitotic bookmarking by transcription factors. Epigenetics & Chromatin 6, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadauke S, Udugama MI, Pawlicki JM, Achtman JC, Jain DP, Cheng Y, Hardison RC, and Blobel GA (2012). Tissue-specific mitotic bookmarking by hematopoietic transcription factor GATA1. Cell 150, 725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Shokhirev MN, Xu Z, Chandran S, Dixon JR, and Hetzer MW (2020). Dynamic regulation of histone modifications and long-range chromosomal interactions during postmitotic transcriptional reactivation. Genes Dev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, and Shendure J (2019). Mechanisms of Interplay between Transcription Factors and the 3D Genome. Mol Cell 76, 306–319. [DOI] [PubMed] [Google Scholar]
- Kim Y, Shi Z, Zhang H, Finkelstein IJ, and Yu H (2019). Human cohesin compacts DNA by loop extrusion. Science 366, 1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konrad CG (1963). PROTEIN SYNTHESIS AND RNA SYNTHESIS DURING MITOSIS IN ANIMAL CELLS. J Cell Biol 19, 267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijger PHL, Geeven G, Bianchi V, Hilvering CRE, and de Laat W (2020). 4C-seq from beginning to end: A detailed protocol for sample preparation and data analysis. Methods 170, 17–32. [DOI] [PubMed] [Google Scholar]
- Kwak H, Fuda NJ, Core LJ, and Lis JT (2013). Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339, 950–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino EL, Hansen TM, Risi RM, Frey R, Manaves V, et al. (2017). Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 550, 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang K, Woodfin AR, Slaughter BD, Unruh JR, Box AC, Rickels RA, Gao X, Haug JS, Jaspersen SL, and Shilatifard A (2015a). Mitotic Transcriptional Activation: Clearance of Actively Engaged Pol II via Transcriptional Elongation Control in Mitosis. Molecular Cell 60, 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z, Zickler D, Prentiss M, Chang FS, Witz G, Maeshima K, and Kleckner N (2015b). Chromosomes Progress to Metaphase in Multiple Discrete Steps via Global Compaction/Expansion Cycles. Cell 161, 1124–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Chen S, Wang S, Soares F, Fischer M, Meng F, Du Z, Lin C, Meyer C, DeCaprio JA, et al. (2017a). Transcriptional landscape of the human cell cycle. Proceedings of the National Academy of Sciences 114, 3473–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Pelham-Webb B, Di Giammartino DC, Li J, Kim D, Kita K, Saiz N, Garg V, Doane A, Giannakakou P, et al. (2017b). Widespread Mitotic Bookmarking by Histone Marks and Transcription Factors in Pluripotent Stem Cells. Cell Rep 19, 1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahat DB, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, Waters CT, Munson K, Core LJ, and Lis JT (2016). Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat Protoc 11, 1455–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maherali N, Ahfeldt T, Rigamonti A, Utikal J, Cowan C, and Hochedlinger K (2008). A high-efficiency system for the generation and study of human induced pluripotent stem cells. Cell Stem Cell 3, 340–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, and Reinberg D (2010). Chromatin structure and the inheritance of epigenetic information. Nat Rev Genet 11, 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden MPF, and Laemmli UK (1979). Metaphase chromosome structure: Evidence for a radial loop model. Cell 17, 849–858. [DOI] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal; Vol 17, No 1: Next Generation Sequencing Data Analysis. [Google Scholar]
- Martónez-Balbás MA, Dey A, Rabindran SK, Ozato K, and Wu C (1995). Displacement of sequence-specific transcription factors from mitotic chromatin. Cell 83, 29–38. [DOI] [PubMed] [Google Scholar]
- Martire S, Nguyen J, Sundaresan A, and Banaszynski LA (2020). Differential contribution of p300 and CBP to regulatory element acetylation in mESCs. BMC Mol Cell Biol 21, 55–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelotti EF, Sanford S, and Levens D (1997). Marking of active genes on mitotic chromosomes. Nature 388, 895–899. [DOI] [PubMed] [Google Scholar]
- Mort RL, Ford MJ, Sakaue-Sawano A, Lindstrom NO, Casadio A, Douglas AT, Keighren MA, Hohenstein P, Miyawaki A, and Jackson IJ (2014). Fucci2a: a bicistronic cell cycle reporter that allows Cre mediated tissue specific expression in mice. Cell Cycle 13, 2681–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, and Chang HY (2016). HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat Methods 13, 919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumbach MR, Satpathy AT, Boyle EA, Dai C, Gowen BG, Cho SW, Nguyen ML, Rubin AJ, Granja JM, Kazane KR, et al. (2017). Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat Genet 49, 1602–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Lubling Y, Várnai C, Dudley C, Leung W, Baran Y, Cohen NM, Wingett S, Fraser P, and Tanay A (2017). Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 547, 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumova N, Imakaev M, Fudenberg G, Zhan Y, Lajoie BR, Mirny LA, and Dekker J (2013). Organization of the mitotic chromosome. Science 342, 948–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuebler J, Fudenberg G, Imakaev M, Abdennur N, and Mirny LA (2018). Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proceedings of the National Academy of Sciences 115, E6697–E6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oomen ME, Hansen AS, Liu Y, Darzacq X, and Dekker J (2019). CTCF sites display cell cycle-dependent dynamics in factor binding and nucleosome positioning. Genome Res 29, 236–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, Fritz CC, Bradner JE, and Guenther MG (2014). Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep 9, 1163–1170. [DOI] [PubMed] [Google Scholar]
- Ou HD, Phan S, Deerinck TJ, Thor A, Ellisman MH, and O’Shea CC (2017). ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science 357, eaag0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens N, Papadopoulou T, Festuccia N, Tachtsidi A, Gonzalez I, Dubois A, Vandormael-Pournin S, Nora EP, Bruneau B, Cohen-Tannoudji M, et al. (2019). CTCF confers local nucleosome resiliency after DNA replication and during mitosis. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palozola KC, Donahue G, Liu H, Grant GR, Becker JS, Cote A, Yu H, Raj A, and Zaret KS (2017). Mitotic transcription and waves of gene reactivation during mitotic exit. Science 358, 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palozola KC, Lerner J, and Zaret KS (2019). A changing paradigm of transcriptional memory propagation through mitosis. Nature reviews. Molecular cell biology 20, 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadimitriou E, Pantazaka E, Castana P, Tsalios T, Polyzos A, and Beis D (2016). Pleiotrophin and its receptor protein tyrosine phosphatase beta/zeta as regulators of angiogenesis and cancer. Biochim Biophys Acta 1866, 252–265. [DOI] [PubMed] [Google Scholar]
- Pauklin S, Madrigal P, Bertero A, and Vallier L (2016). Initiation of stem cell differentiation involves cell cycle-dependent regulation of developmental genes by Cyclin D. Genes & Development 30, 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauklin S, and Vallier L (2013). The cell-cycle state of stem cells determines cell fate propensity. Cell 155, 135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelham-Webb B, Murphy D, and Apostolou E (2020). Dynamic 3D Chromatin Reorganization during Establishment and Maintenance of Pluripotency. Stem Cell Reports 15, 1176–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard Picard Toolkit. B. Institute, ed. (GitHub Repository). [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Grosveld FG, and Papantonis A (2018). Forces driving the three-dimensional folding of eukaryotic genomes. In Mol Syst Biol, p. e8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raisner R, Kharbanda S, Jin L, Jeng E, Chan E, Merchant M, Haverty PM, Bainer R, Cheung T, Arnott D, et al. (2018). Enhancer Activity Requires CBP/P300 Bromodomain-Dependent Histone H3K27 Acetylation. Cell Rep 24, 1722–1729. [DOI] [PubMed] [Google Scholar]
- Ramirez F, Bhardwaj V, Arrigoni L, Lam KC, Gruning BA, Villaveces J, Habermann B, Akhtar A, and Manke T (2018). High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat Commun 9, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep sequencing data analysis. Nucleic Acids Res 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. (2014). A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray J, Munn PR, Vihervaara A, Lewis JJ, Ozer A, Danko CG, and Lis JT (2019). Chromatin conformation remains stable upon extensive transcriptional changes driven by heat shock. Proceedings of the National Academy of Sciences of the United States of America 116, 19431–19439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Turner D, Durand NC, Thorvaldsdóttir H, Mesirov JP, and Aiden EL (2018). Juicebox.js Provides a Cloud-Based Visualization System for Hi-C Data. Cell Syst 6, 256–258.e251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley MJ, and Corces VG (2018). Organizational principles of 3D genome architecture. Nat Rev Genet 19, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn AL, Rao SS, Huang SC, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, et al. (2015). Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci U S A 112, E6456–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savatier P, Lapillonne H, Jirmanova L, Vitelli L, and Samarut J (2002). Analysis of the cell cycle in mouse embryonic stem cells. Methods Mol Biol 185, 27–33. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrode N, Saiz N, Di Talia S, and Hadjantonakis A-K (2014). GATA6 levels modulate primitive endoderm cell fate choice and timing in the mouse blastocyst. Developmental cell 29, 454–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sela Y, Molotski N, Golan S, Itskovitz-Eldor J, and Soen Y (2012). Human embryonic stem cells exhibit increased propensity to differentiate during the G1 phase prior to phosphorylation of retinoblastoma protein. Stem Cells 30, 1097–1108. [DOI] [PubMed] [Google Scholar]
- Sheffield NC, and Bock C (2016). LOLA: enrichment analysis for genomic region sets and regulatory elements in R and Bioconductor. Bioinformatics 32, 587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AM, Chappell J, Trost R, Lin L, Wang T, Tang J, Matlock BK, Weller KP, Wu H, Zhao S, et al. (2013). Cell-cycle control of developmentally regulated transcription factors accounts for heterogeneity in human pluripotent cells. Stem Cell Reports 1, 532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AM, Sun Y, Li L, Zhang W, Wu T, Zhao S, Qin Z, and Dalton S (2015). Cell-Cycle Control of Bivalent Epigenetic Domains Regulates the Exit from Pluripotency. Stem Cell Reports 5, 323–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer CA, Stadtfeld M, Murphy GJ, Hochedlinger K, Kotton DN, and Mostoslavsky G (2009). Induced pluripotent stem cell generation using a single lentiviral stem cell cassette. Stem Cells 27, 543–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A, and Dalton S (2016). Cycling through developmental decisions: how cell cycle dynamics control pluripotency, differentiation and reprogramming. Development 143, 4301–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadhouders R, Filion GJ, and Graf T (2019). Transcription factors and 3D genome conformation in cell-fate decisions. Nature 569, 345–354. [DOI] [PubMed] [Google Scholar]
- Stadhouders R, Vidal E, Serra F, Di Stefano B, Le Dily F, Quilez J, Gomez A, Collombet S, Berenguer C, Cuartero Y, et al. (2018). Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat Genet 50, 238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabar V, and Studer L (2014). Pluripotent stem cells in regenerative medicine: challenges and recent progress. Nat Rev Genet 15, 82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JH (1960). Nucleic acid synthesis in relation to the cell division cycle. Ann N Y Acad Sci 90, 409–421. [DOI] [PubMed] [Google Scholar]
- Teves SS, An L, Bhargava-Shah A, Xie L, Darzacq X, and Tjian R (2018). A stable mode of bookmarking by TBP recruits RNA polymerase II to mitotic chromosomes. eLife 7, e35621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves SS, An L, Hansen AS, Xie L, Darzacq X, and Tjian R (2016). A dynamic mode of mitotic bookmarking by transcription factors. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, and Furlong EEM (2019). The role of transcription in shaping the spatial organization of the genome. Nature Reviews Molecular Cell Biology 20, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Chu T, Choate LA, and Danko CG (2019). Identification of regulatory elements from nascent transcription using dREG. Genome Res 29, 293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert BT, Narita T, Satpathy S, Srinivasan B, Hansen BK, Schölz C, Hamilton WB, Zucconi BE, Wang WW, Liu WR, et al. (2018). Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/p300 Acetylome. Cell 174, 231–244.e212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi SK, Young DW, Montecino MA, Lian JB, van Wijnen AJ, Stein JL, and Stein GS (2010). Mitotic bookmarking of genes: a novel dimension to epigenetic control. Nat Rev Genet 11, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Huang P, Sharma M, Keller CA, Giardine B, Zhang H, Gilgenast TG, Phillips-Cremins JE, Hardison RC, and Blobel GA (2020a). Alteration of genome folding via contact domain boundary insertion. Nature Genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Emerson DJ, Gilgenast TG, Titus KR, Lan Y, Huang P, Zhang D, Wang H, Keller CA, Giardine B, et al. (2019). Chromatin structure dynamics during the mitosis-to-G1 phase transition. Nature 576, 158–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Übelmesser N, Josipovic N, Forte G, Slotman JA, Chiang M, Gothe H, Gusmao EG, Becker C, Altmüller J, et al. (2020b). RNA polymerase II is required for spatial chromatin reorganization following exit from mitosis. bioRxiv, 2020.2010.2027.356915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, and Zheng Y (2018). CscoreTool: fast Hi-C compartment analysis at high resolution. Bioinformatics 34, 1568–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All genomics data (Hi-C, ATAC-seq, RNA-seq, 4C-seq, ChIP-seq, and PRO-seq) generated during this study are available at GEO under accession code GSE138965. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138965