

Abstract
While combination antiretroviral therapy (cART) durably suppresses HIV replication, virus persists in CD4+ T-cells that harbor latent but spontaneously inducible and replication-competent provirus. One strategy to inactivate these viral reservoirs involves the use of agents that continue to reinforce HIV latency even after their withdrawal. To identify new chemical leads with such properties, we investigated a series of naturally-occurring flavones (chrysin, apigenin, luteolin, and luteolin-7-glucoside (L7G)) and functionally-related cyclin dependent kinase 9 (CDK9) inhibitors (flavopiridol and atuveciclib) which are reported or presumed to suppress HIV replication in vitro. We found that, while all compounds inhibit provirus expression induced by latency-reversing agents in vitro, only aglycone flavonoids (chrysin, apigenin, luteolin, flavopiridol) and atuveciclib, but not the glycosylated flavonoid L7G, inhibit spontaneous latency reversal. Aglycone flavonoids and atuveciclib, but not L7G, also inhibit CDK9 and the HIV Tat protein. Aglycone flavonoids do not reinforce HIV latency following their in vitro withdrawal, which corresponds with their ability to also inhibit class I/II histone deacetylases (HDAC), a well-established mechanism of latency reversal. In contrast, atuveciclib and flavopiridol, which exhibit little or no HDAC inhibition, continue to reinforce latency for 9 to 14+ days, respectively, following their withdrawal in vitro. Finally, we show that flavopiridol also inhibits spontaneous ex vivo viral RNA production in CD4+ T cells from donors with HIV. These results implicate CDK9 inhibition (in the absence of HDAC inhibition) as a potentially favorable property in the search for compounds that durably reinforce HIV latency.
Keywords: antiviral agent, cyclin-dependent kinase (CDK), flavonoid, histone deacetylase (HDAC), human immunodeficiency virus (HIV), viral latency
Graphical Abstract
1. Introduction
While combination antiretroviral therapy (cART) has reduced worldwide morbidity and mortality due to HIV/AIDS, it does not target resting CD4+ T-cells that contain integrated and replication-competent proviruses. In addition, even in virally suppressed individuals on cART, long-term HIV infection associates with increased risks of chronic inflammation and comorbidities like cancers and cardiovascular disease [1, 2]. Inhibiting provirus expression from HIV reservoirs could suppress HIV replication and reduce the risks of chronic inflammation by lowering viral antigen production. Toward this goal, several chemical compounds have been described to inhibit provirus expression in the co-presence of proviral stimuli [3 - 5]. Moreover, a subset of these agents continue to reinforce HIV latency even following their withdrawal from cell culture or discontinuation in animal models [6 - 8]. These first-generation compounds, colloquially referred to as “block-and-lock” agents [6 - 8], are particularly important for emerging efforts to achieve durable, cART-free HIV remission in humans.
The HIV Tat protein is a major driver of provirus transcription and HIV latency reversal [9]. In the absence of Tat, transcription initiates at the viral long-terminal repeat (LTR) but readily pauses or aborts. The activation domain of Tat binds the host positive transcription elongation factor B (P-TEFb) complex, consisting of cyclin-dependent kinase 9 (CDK9) and cyclin T1, and recruits it to the LTR [10]. Phosphorylation of RNA polymerase II by CDK9 then promotes productive HIV transcription and latency reversal [11 - 12]. Among preclinical agents that block this process, didehydro cortistatin A (dCA) directly interferes with the Tat basic domain and the ability of Tat to interact with the transactivation-responsive element, an RNA stem loop structure formed at the 5’ end of viral RNA, thereby preventing P-TEFb recruitment to the LTR [10]. Following prolonged in vitro treatment of primary cells from people living with HIV on suppressive cART, and subsequent withdrawal of the compound, HIV latency continued to be reinforced, even upon strong activation stimuli. dCA also delayed viral rebound in a mouse model of HIV following its discontinuation [6, 13], thereby demonstrating that reinforced latency is a viable pharmacological strategy in vivo. dCA appears to reinforce latency by favoring a repressive epigenetic environment at the HIV LTR that results in stable and long-term histone deacetylation and recruitment of transcription-repressing complexes [14]. Additional compounds including sudemycin D6 and ZL-0580 also continue to reinforce latency following withdrawal by targeting upstream host factors of the Tat/P-TEFb interaction (SF3B1 and BRD4, respectively) [15 - 16]. However, despite these recent advances, the extent to which HIV latency can be durably reinforced by targeting additional cellular factors is not known. As such, there exists an opportunity for the identification of additional “block-and-lock”-class agents.
One group of compounds that may reinforce HIV latency following withdrawal are the flavones, a subset of plant-based flavonoid compounds characterized by two 6-carbon phenyl rings, the A and B rings, attached by a third 3-carbon linker, the C ring (Figure 1A). Some naturally-occurring flavones, including chrysin, apigenin, and luteolin (Figure 1B), are known to inhibit HIV replication as well as provirus expression concurrently induced by latency-reversing agents (LRAs) in vitro [17 - 19]. These and additional flavones including wogonin, tricin, and flavopiridol have also been shown to act on CDK9 [20 - 22], which is consistent with the potential to reinforce latency following withdrawal. However, some of these flavones have also been reported to inhibit cellular histone deacetylase (HDAC) activity [23 - 25]. As HDAC inhibition is a well-established pathway for proviral expression and HIV latency reversal [26], it is unclear whether this activity may conflict with ability of these agents to reinforce latency following withdrawal.
Figure 1.

Structures of flavonoids and CDK9 inhibitors. (A) Flavone backbone with numbered carbons. (B) Chemical structures of flavonoids chrysin, apigenin, luteolin, L7G, and naringenin. (C) Chemical structures of CDK9 inhibitors flavopiridol and atuveciclib.
Here we investigate the ability of flavones and functionally-related agents to promote HIV latency and show that compounds which inhibit CDK9, without concomitant inhibition of HDACs, also continue to promote latency that persists even following their withdrawal.
2. Materials and Methods
2.1. Cells and reagents
Jurkat T cells (Clone E6-1) were obtained from the American Tissue Culture Collection. CEM-derived GXR25 GFP-reporter T cells (CEM-GXR) have been previously described [27]. J-Lat T cells (clones 9.2 and 10.6) were obtained from the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH (contributed by Dr. Eric Verdin [28]). OM-10.1 macrophages were also obtained from the NIH AIDS Reagent Program (contributed by Dr. Salvatore Butera [29]). Jurkat-Tat-Tat-Dendra+HIV LTR-mCherry (“JurTat”) cells were described previously [58] and obtained as a gift from Dr. Leor Weinberger. HEK-293T cells were purchased commercially (Lenti-X 293T; Clontech Laboratories). All cell lines were maintained for no more than 20 passages.
Peripheral blood mononuclear cells (PBMCs) were obtained from three uninfected donors following written informed consent. Study protocols were approved by the Institutional Review Boards of Simon Fraser University and the University of British Columbia – Providence Health Care Research Institute. CD4+ T cells were additionally obtained from five HIV-1 infected donors on stably suppressive combination antiretroviral therapy (< 50 copies/mL of plasma viral load) for at least 3 years following written informed consent. These participants were recruited in accordance with human subject research guidelines of the United States Department of Health and Human Services under the supervision of the Wistar and Philadelphia FIGHT institutional review boards.
PBMCs and cell lines were cultured in R10+ media [RPMI 1640 with HEPES and L-Glutamine, 10% Fetal Bovine Serum (FBS), 100 U of penicillin/mL, and 100 μg of streptomycin/mL (Sigma-Aldrich (Oakville, ON, Canada)]. HEK-293T cells (Clontech) were cultured in Dulbecco’s Modified Eagle Medium with 4.5 g/L Glucose and L-Glutamine (Lonza), supplemented with FBS and antibiotics as above. Primary CD4+ T cells were cultured in R20+ media, which was identical to R10+ media except for 20% FBS.
Apigenin, chrysin, luteolin, L7G, naringenin, and flavopiridol were purchased as powder from Sigma-Aldrich, then dissolved in DMSO and stored at −20°. TNFα, panobinostat, prostratin, HMBA, and human IL-2 were obtained from Sigma-Aldrich. JQ1 and atuveciclib were purchased from Cayman Chemical Co. (Ann Arbor, MI, USA) and AdooQ Bioscience (Irvine, CA, USA), respectively. All compounds were reported ≥ 95% pure. ADP-Glo Kinase Assays and HDAC-Glo I/II Assays were obtained from Promega (Madison, WI, USA), and purified CDK9/Cyclin T1 was obtained from Sigma-Aldrich. pNL4.3, expressing the NL4.3 strain of HIV-1, was obtained from the NIH AIDS Reagent Program (contributed by Dr. Malcolm Martin [30]). pCMV-Tat, an HIV-1 Tat expression vector, and pCMV-ΔTat, its corresponding control lacking Tat, were previously described by Malim et al. [31] and provided as a gift from Dr. Brendan Bell.
2.2. Virus production
HEK-293T cells were transfected with 15 μg pNL4.3 via Lipofectamine LTX (Invitrogen; Burlington, ON, Canada) according to manufacturer’s instructions. Cell free supernatants containing HIV-1NL4.3 virions were collected after 48 hours and stored at −80°C. To estimate viral titers, CEM-GXR cells were infected with known volumes of HIV-1NL4.3 stock for 48 hours. The percentage of GFP-expressing cells was quantified and used to estimate MOIs [27].
2.3. HIV replication assays in PBMCs
Infectivity studies involving PBMCs were performed as described by Mwimanzi et al. [32]. Briefly, PBMCs from 3 donors without HIV were activated for 3 days with 5 μg/mL phytohemagglutinin in R10+ media, infected with HIV-1NL4.3 for 6 hours (MOI = 0.003), then pelleted and resuspended in R10+ media supplemented with 100 U/mL IL-2 (Sigma-Aldrich), seeded into 96 well U-bottom plates at 150,000 cells/well, and treated with test agents or 0.1% DMSO. On day 3 post-infection, media, drug, and IL-2 were replenished. Supernatant p24Gag was quantified on day 6 post-infection by ELISA (Xpress Bio, Frederick, MD, USA) as per manufacturer’s instructions. 0.1% DMSO vehicle and uninfected controls served as positive and negative controls for p24Gag detection, as we previously observed that 0.1% DMSO has no effect on HIV replication [33].
2.4. Viability detection assays
To quantify cytotoxicity of test agents on PBMCs, the above procedure was replicated with uninfected PBMCs from the same donors. On day 6 post-treatment, culture viability was assessed using Guava Viacount dye (Luminex; Toronto, ON, Canada) and flow cytometry, as per manufacturer’s instructions. The proportion of live cells in treated conditions were normalized to that of 0.1% DMSO vehicle control, which we previously reported has no effect on cell viability [33].
To measure cytotoxicity in Jurkat cells in the presence of test agents, cells were seeded in 96 well plates at 200,000 cells/well and treated for 24 hours. Following incubation, culture viability was analyzed using Guava Viacount dye as per manufacturer’s instructions.
2.5. Provirus expression assays
J-Lat 9.2 or 10.6 cells were seeded in 96 well plates at 200,000 cells/well and co-treated with a pro-inflammatory cytokine (10 ng/mL TNFα) or an LRA (10 μM prostratin, 0.1 μM panobinostat, 10 μM JQ1, or 10 mM HMBA) in the presence of test agents at defined concentrations or 0.1% DMSO vehicle control for 24 hours. Cells were then examined for GFP expression by flow cytometry. In a subset of experiments, cells were fixed and permeabilized using the Cytofix/Cytoperm fixation kit (BD Biosciences; Mississauga, ON, Canada), stained for intracellular p24Gag (KC57-RD1 antibody, Beckman Coulter; Mississauga, ON, Canada), and examined for p24Gag expression by flow cytometry.
OM-10.1 cells were seeded as above and cotreated with 1 ng/mL TNFα and test agents or 0.1% DMSO control for 24 hours. Cells were fixed, permeabilized, stained for p24Gag as above, and examined for p24Gag expression by flow cytometry.
2.6. Latency reinforcement assays
For studies involving inhibition of spontaneous provirus expression or reinforced HIV latency in vitro, J-Lat 10.6 cells were seeded in 96 well plates at 50,000 cells/well in 200 μL and treated with compounds or 0.1% DMSO vehicle control. Every 3 – 4 days, cells were examined for GFP expression by flow cytometry and/or stained for p24Gag expression using the cytofix/cytoperm protocol described above. Remaining cells were re-suspended in fresh media, and drug was replenished to the same final concentration. Incubation was continued for 21 days, at which point cells were pelleted, washed extensively with PBS, and re-suspended in fresh R10+ media. Washed cells were then analyzed for GFP expression every 24 to 72 hours for up to 14 additional days.
2.7. HIV Tat-induced expression assays
5 x 106 CEM-GXR cells were resuspended in 200 μL of Opti-MEM media (Thermo Fisher; Mississauga, ON, Canada) containing 1.6 μg of pCMV-Tat or pCMV-ΔTat, then transfected in 0.4 cm cuvettes using a GenePulser MXCell electroporation system (Bio-Rad; Mississauga, ON, Canada) (single 250 V square-wave pulse for 25 milliseconds). Transfected cells were rested for 10 minutes at room temperature, diluted in R10+, then seeded into 96 well plates at 100,000 cells/well and treated with test agents or 0.1% DMSO vehicle control. After 24 hours, Tat-driven GFP expression was detected by flow cytometry.
JurTat cells were stimulated with 500 ng/mL doxycycline (Dox, Sigma-Aldrich) and seeded at 200,000 cells/well in 96 well plates in the presence of compounds of 0.1% DMSO control. Tat-Dendra and mCherry reporter expression were then analyzed by flow cytometry after 24 hours.
2.8. Kinase activity assays
The ADP-Glo kinase assay kit (Promega; Madison, WI, USA) was used to measure CDK9 kinase activity in the presence of test agents by quantifying the levels of ATP converted to ADP. Kinase reactions were performed in white 384 well plates, 10 μL final volumes per well, and following manufacturer’s instructions. Briefly, reagents and inhibitors were diluted in the supplied kinase reaction buffer. 30 ng of CDK9/cyclin T1 was incubated with 10 μM ATP, 0.2 μg/mL PDKTide peptide substrate, and test agents. No-enzyme control conditions were included to represent signal background, while no-inhibitor conditions represented maximum kinase activity. After 40 minutes, kinase reactions were stopped by addition of 10 μl ADP-Glo reagent per well, and mixtures were incubated at room temperature for 40 minutes. 20 μl of Kinase Detection Reagent was added, and luminescence intensity was measured after 30 minutes using an Infinity M200 multimode plate reader (Tecan Life Sciences; Morrisville, NC, USA). Data were then normalized to no-inhibitor controls following background subtraction.
2.9. HDAC activity assays
HDAC activity was examined using the HDAC-Glo I/II Assay Kit (Promega; Madison, WI, USA) as per manufacturer’s instructions. Briefly, HDAC reactions were performed in white 384 well plates, 20 μL final volume/well. Compounds were diluted to desired concentrations in the provided buffer and added to wells. Jurkat cells were then resuspended in Phenol-Red- and FBS-free RPMI 1640 and seeded into wells at 3,000 cells/well. Cells treated with 0.1% DMSO served as a baseline HDAC activity control, while wells containing only media were included to control for signal background. Following incubation at 37 °C for 90 minutes, 20 μL of HDAC-Glo I/II Reagent plus 1% Triton-X100 (prepared as per manufacturer’s instructions) was added to each well. Plates were mixed for 30 seconds, then incubated at room temperature for 30 minutes. Luminescence was detected by an Infinity M200 multimode plate reader (Tecan Life Sciences; Morrisville, NC, USA). Resulting data were then normalized to the no-inhibitor control following background subtraction.
2.10. Measures of viral RNA production in primary CD4+ T cell cultures
CD4+ T cells from donors with HIV were isolated from PBMCs by negative selection using the EasySepTM Human CD4+ T cell Enrichment Kit (STEM-CELL, Vancouver, BC, Canada) and cultured as described previously [56], 106 cells were then treated with 0.1% DMSO or 30 nM flavopiridol for 24 hours in 1 mL cultures. Alternatively, 106 cells (in 1 mL) were treated with DMSO or flavopiridol for 72 hours, washed extensively and resuspended in fresh R20+ plus IL-2 and 0.1% DMSO or 30 nM flavopiridol, incubated for an additional 72 – 96 hours, washed extensively and resuspended in fresh R20+ plus IL-2 without compounds, and incubated for a further 24 hours. Following incubation, cells were counted visually by trypan blue staining.
Viral RNA was extracted from culture supernatants using a QIAamp Viral RNA Mini kit (Qiagen, Germantown, MD, USA) and subjected to quantitative RT-PCR (qRT-PCR) as described previously using a C1000 Thermal Cycler and CFX96 Real-Time system (Bio-Rad, Hercules, CA, USA) and as described previously [59]. The limit of detection of supernatant viral RNA was 200 copies/mL as determined through repeating end point detection from serial dilution of the AcroMetrix HIV-1 Panel (Thermo Fisher, Waltham, MA, USA).
2.11. Data and statistical analysis
Flow cytometry data were analyzed using FlowJo v. 10.5.3 (FlowJo LLC; Ashland, OR, USA). Background GFP in uninfected CEM-GXR cultures or unstimulated J-Lat 9.2 cultures was set to 0.05%, while background GFP in unstimulated J-Lat 10.6 varied between 5-15% relative to background GFP expression observed in unstimulated J-Lat 9.2 cells. Half-maximal effective and cytotoxic concentrations (EC50s and CC50s, respectively) were calculated from at least 3 independent experiments, using the linear regression function in GraphPad Prism v. 8.2.1 (GraphPad; San Diego, CA, USA). For assay assessing inhibition of provirus expression, compound EC50s were calculated after normalizing results from treated cells to those of LRA-only controls. For J-Lat 10.6 cells, background GFP from unstimulated cells was subtracted from all samples prior to normalizing. All data are presented as mean ± SD from at least 3 independent experiments. Statistically significant differences in provirus expression were determined by one sample t test, with a p value < 0.05 considered significant.
3. Results
3.1. Selection of flavones and CDK9 inhibitors for latency reinforcement studies
To identify novel agents that can reinforce HIV latency, we first focused on a series of naturally-occurring flavones including chrysin, apigenin, and luteolin, as these were previously reported to suppress HIV replication and/or concurrently inhibit LRA-induced provirus expression in cell line models (Figure 1) [17 - 19]. On the A ring, all three share hydroxyls at the C5 and C7 positions. On the B ring, chrysin features no hydroxylation, apigenin exhibits hydroxylation at C4’ and luteolin exhibits hydroxylation at C3’ + C4’ (Figure 1A-B). We also examined the glycoside derivative of luteolin, luteolin-7-glucoside (L7G), as this moiety’s effect on HIV inhibition is unknown (Figure 1B). Also included was the flavanone naringenin, which structurally resembles apigenin but lacks the C2-3 double bond characteristic of flavones. As this difference substantially reduces the ability to inhibit HIV [17 - 18], naringenin was included as a negative control.
Flavones are reported to target host factors in addition to CDK9. For example, chrysin, apigenin, and luteolin also inhibit HDACs [23 - 25], which could potentially counteract the compounds' pro-latency effects. To investigate this, we also assessed the semi-synthetic flavone flavopiridol and the non-flavonoid atuveciclib (also called BAY 1143572; Figure 1C). Flavopiridol is a pan-CDK inhibitor reported to act on CDK9 and HIV transcription at low nanomolar concentrations [22]; however, it also inhibits other cellular CDKs [34]. In contrast, atuveciclib, while less potent than flavopiridol, inhibits CDK9 with > 100-fold selectivity over other CDKs [35]. To date, the ability of these compounds to continue to reinforce HIV latency following withdrawal has previously not been reported.
3.2. Flavones and atuveciclib suppress HIV replication in PBMCs
While most compounds in Figure 1 suppress HIV replication in cell lines [17 - 18, 22], only flavopiridol has been investigated in primary cells with regard to dose-dependence [36]. To confirm the antiviral activities of these compounds using a standardized method, we assessed their suppression of HIV replication in PBMCs from 3 uninfected donors (Figure 2). PBMCs were infected in vitro with HIV-1NL4.3 for 6 hours (multiplicity of infection (MOI) = 0.003), washed, and treated with compounds at multiple concentrations or with 0.1% DMSO vehicle control. Cells were washed and replaced with fresh compound at Day 3 post-infection, and viral production was evaluated at Day 6 post-infection by measuring supernatant p24Gag by ELISA. Using this system, we observed suppression of HIV replication for all flavones and atuveciclib (Figure 2A). For example, HIV replication was suppressed 26.3 ± 24.8% by 1 μM L7G and 77.3 ± 32.7% by 10 μM L7G. Results across all assessed L7G concentrations corresponded to a half-maximal effective concentration (EC50) of 2.6 ± 0.8 μM (Table 1). Corresponding EC50s for other flavonoids included 9.2 ± 2.9 μM for chrysin, 5.0 ± 1.6 μM for apigenin, and 12.7 ± 3.2 μM for luteolin, 0.020 ± 0.003 μM for flavopiridol, and 3.4 ± 0.7 μM for atuveciclib (Figure 2A; Table 1). In contrast, no antiviral activity was observed with naringenin at up to 30 μM.
Figure 2.

Flavones and CDK9 inhibitors suppress HIV replication in PBMCs. (A) Effects of compounds after 6 days’ incubation on HIV-1NL4.3 replication in infected PBMC cultures, as measured by supernatant p24Gag levels. (B) Effects of compounds after 6 days’ incubation on cell viability in uninfected PBMCs, as measured using Viacount dye. For A and B, results are presented relative to PBMCs treated with 0.1% DMSO and generated from 3 independent donors. Horizontal dotted lines denote activities at 100 and 50% of cells treated with 0.1% DMSO.
Table 1.
Effects of flavonoids and CDK9 inhibitors on active HIV replication and cell viability in PBMCs.
| Compound | EC50 (μM) | CC50 (μM) | Selectivity Index (CC50/EC50) |
|---|---|---|---|
| Chrysin | 9.2 ± 2.9 | > 30 | > 3.3 |
| Apigenin | 5.0 ± 1.6 | 19.8 ± 6.5 | 4.0 |
| Luteolin | 12.7 ± 3.2 | 13.9 ± 5.9 | 1.1 |
| L7G | 2.6 ± 0.8 | 18.3 ± 2.5 | 7.0 |
| Naringenin | > 30 | > 30 | n.d. |
| Flavopiridol | 0.020 ± 0.003 | 0.22 ± 0.04 | 11.0 |
| Atuveciclib | 3.4 ± 0.67 | 24.4 ± 1.5 | 7.2 |
To assess compound toxicity in this cell model, we treated uninfected PBMCs from the same donors for 6 days as described above and measured viability using Viacount dye (Figure 2B) [32]. In these studies, no toxicity was observed with up to 30 μM chrysin, suggesting that suppression of HIV replication is not due to primary cell toxicity. While toxicity was observed at high concentrations of apigenin, L7G, flavopiridol, and atuveciclib (half-maximal cytotoxic concentrations (CC50s) = 19.8 ± 6.4, 18.3 ± 2.5, 0.25 ± 0.06 and 24.7 ± 4.9 μM, respectively), these compounds also suppressed HIV replication at concentrations without obvious toxicity (Selectivity Index (SI), or CC50/EC50 = 4.0, 7.0, 11.0 and 7.2, respectively; Table 1). Alternatively, luteolin was toxic to PBMCs at concentrations that suppressed HIV replication, with a CC50 of 13.9 ± 5.9, which yielded a SI of 1.1. No toxicity was observed with naringenin up to 30 μM. These results confirm that most flavones and atuveciclib, but not luteolin, suppress HIV replication in PBMCs at concentrations that do not affect cell viability.
3.3. Flavones and atuveciclib inhibit LRA-induced provirus expression
We next examined the ability of these compounds to inhibit LRA-induced provirus expression in the J-Lat cell line model of HIV latency [28]. J-Lat cells are derived from Jurkat T cells and contain a latent provirus featuring a frameshift mutation in Env and a GFP reporter in place of Nef. Provirus expression can be induced via treatment with LRAs and measured by analysis of GFP production in live-gated cells.
We first assessed the activities of compounds in J-Lat 9.2 cells, which do not express GFP in the absence of stimulation (Figure 3A). Cells were treated for 24 hours with LRAs including the NF-κB stimulator and pro-inflammatory cytokine TNFα, the HDAC inhibitor panobinostat, or the protein kinase C activator prostratin, after which GFP expression was measured in live cells by flow cytometry (Figure 3B-D). On average, treatment of J-Lat 9.2 cells with 10 ng/mL TNFα, 0.1 μM panobinostat, or 10 μM prostratin induced GFP expression in 10.4 ± 1.7%, 10.8 ± 1.4%, or 10.7 ± 2.7% of cells, respectively (Figure 3B-D, dotted lines), consistent with previous observations [37]. However, co-administration of chrysin, apigenin, luteolin, or L7G inhibited induction of GFP by all LRAs in live-gated J-Lat 9.2 cells (Figure 3B-D, Table 2). For example, in the presence of 10 ng/mL TNFα, provirus expression was inhibited 22.5 ± 24.9% by 3 μM chrysin and 88.3 ± 8.8% by 10 μM chrysin (Figure 3B). Together, the data from the chrysin concentration series corresponded to an EC50 of 4.0 ± 0.4 μM for this compound (Figure 3B; Table 2). Corresponding EC50s for other flavonoids were 5.6 ± 0.6 μM for apigenin, 7.6 ± 0.6 μM for luteolin, and 4.5 ± 0.6 μM for L7G (Figure 3B; Table 2). Similar results were observed by these flavonoids when provirus expression was induced with 0.1 μM panobinostat (respective EC50s = 2.8 ± 0.4, 4.1 ± 0.4, 6.8 ± 0.8, and 4.3 ± 0.4 μM; Figure 3C), while the activities of luteolin and L7G were weaker in J-Lat 9.2 cells stimulated with 10 μM prostratin (respective EC50s = 4.3 ± 1.2, 7.3 ± 1.1, 20.6 ± 2.6, and 17.3 ± 1.1 μM; Figure 3D; Table 2). In contrast, naringenin exhibited little to no activity in these experiments (EC50s >30 μM), while flavopiridol was notably more potent than the natural flavones, with EC50S of 0.008 ± 0.0004, 0.028 ± 0.003, and 0.009 ± 0.001 μM against induction with TNFα, panobinostat, and prostratin, respectively. Finally, atuveciclib inhibited LRA-induced provirus expression with generally more efficacy than the naturally occurring flavones but less than flavopiridol (EC50s of 1.7 ± 0.2, 3.6 ± 0.8, and 1.8 ± 0.1 μM, vs. induction by TNFα, panobinostat, and prostratin, respectively).
Figure 3.


Flavones, flavopiridol, and atuveciclib inhibit provirus expression in the simultaneous presence of LRAs. A, Representative examples of GFP expression in J-Lat 9.2 cells treated with 0.1% DMSO vehicle control, 10 μM prostratin, or prostratin plus 10 μM apigenin. Values indicate percent GFP-positive cells. (B-D) Effect of compounds after 24 hours’ incubation on LRA-induced provirus expression in J-Lat 9.2 cells. Cells were treated with 10 ng/mL TNFα (B), 0.1 μM panobinostat (C), or 10 μM prostratin (D) in the concurrent presence of test compounds at defined concentrations. Horizontal dotted lines denote average % GFP expression in cells treated with LRA only. E, Representative examples of GFP expression in J-Lat 10.6 cells treated with 0.1% DMSO, 10 μM JQ1, or JQ1 plus 10 μM apigenin. Values indicate percent GFP-positive cells. (F-H) Effect of compounds after 24 hours’ incubation on LRA-induced provirus expression in J-Lat 10.6 cells. Cells were treated with 10 ng/mL TNFα (F), 10 μM JQ1 (G), or 10 mM HMBA (H) in the concurrent presence of test compounds at defined concentrations. Red dotted lines denote percent spontaneous GFP expression in J-Lat 10.6 cells treated with 0.1% DMSO, and black dotted lines denote percent GFP expression in the presence of LRA only. I, Representative examples of viral p24Gag protein expression in OM-10.1 cells treated with 0.1% DMSO, 1 ng/mL TNFα, or TNFα plus 10 μM apigenin. Values indicate percent p24Gag-positive cells. (J) Effect of compounds after 24 hours’ incubation on concurrent 1 ng/mL TNFα-induced provirus expression in OM-10.1 cells. Black dotted line denotes p24Gag production in the presence of TNFα only. (K-L) Effects of compounds after 24 hours’ incubation on Jurkat (K) and OM10.1 (L) cell viability, as measured using Viacount dye. In K-L, data are presented relative to viability of 0.1% DMSO vehicle control-treated cells (black dotted lines).
Table 2.
Effects of flavonoids and CDK9 inhibitors on inhibiting LRA-induced provirus expression in J-Lat 9.2, J-Lat 10.6, and OM10.1 cells.
| J-Lat 9.2 | |||
|---|---|---|---|
| Compound | TNFα EC50 (μM) |
Panobinostat EC50 (μM) |
Prostratin EC50 (μM) |
| Chrysin | 4.0 ± 0.4 | 2.8 ± 0.4 | 4.3 ± 1.2 |
| Apigenin | 5.6 ± 0.6 | 4.1 ± 0.4 | 7.3 ± 1.1 |
| Luteolin | 7.6 ± 0.6 | 6.8 ± 0.8 | 20.6 ± 2.6 |
| L7G | 4.5 ± 0.6 | 4.3 ± 0.4 | 17.3 ± 2.2 |
| Naringenin | >30 | >30 | >30 |
| Flavopiridol | 0.008 ± 0.0004 | 0.028 ± 0.003 | 0.009 ± 0.001 |
| Atuveciclib | 1.7 ± 0.2 | 3.6 ± 0.8 | 1.8 ± 0.1 |
| J-Lat 10.6 | OM-10.1 | |||
|---|---|---|---|---|
| Compound | TNFα EC50 (μM) |
JQ1 EC50 (μM) |
HMBA EC50 (μM) |
TNFα EC50 (μM) |
| Chrysin | 6.3 ± 0.7 | 7.7 ± 1.5 | 6.7 ± 1.4 | 4.7 ± 0.3 |
| Apigenin | 6.2 ± 0.3 | 7.2 ± 1.1 | 5.2 ± 0.8 | 4.1 ± 0.8 |
| Luteolin | 13.6 ± 0.8 | 15.4 ± 1.5 | 26.5 ± 5.8 | 10.3 ± 0.6 |
| L7G | 11.9 ± 0.5 | 16.4 ± 3.3 | 10.1 ± 3.5 | 10.3 ± 1.2 |
| Naringenin | >30 | >30 | >30 | >30 |
| Flavopiridol | 0.019 ± 0.001 | 0.014 ± 0.002 | 0.099 ± 0.002 | 0.013 ± 0.0008 |
| Atuveciclib | 4.3 ± 0.2 | 2.4 ± 0.2 | 1.8 ± 0.1 | 1.8 ± 0.09 |
To assess whether these activities extended to proviruses integrated at a different host genomic site, we evaluated the ability of compounds to inhibit LRA-induced provirus expression in J-Lat 10.6 cells. Unlike J-Lat 9.2 cells, untreated J-Lat 10.6 cells feature a basal level of 5-15% GFP-positive cells at rest (Figure 3E; Figure F-H, red dotted lines) [37] and are more responsive to latency reversal, with 80.3 ± 2.6% GFP-positive cells induced by 10 ng/mL TNFα after 24 hours (Figure 3F, black dotted line). Furthermore, when treated with 10 μM of the BRD4 inhibitor JQ1 or 10 mM of the PI3K/Akt activator HMBA [26, 38 - 41], we observed GFP production in 35.9 ± 2.0% and 34.6 ± 1.4% of cells, respectively (Figure 3G-H, black dotted lines). In contrast, JQ1 and HMBA did not stimulate GFP expression in J-Lat 9.2 cells (data not shown), consistent with previous studies indicating that latency reversal in J-Lat 10.6 cells is more responsive to these activators [28, 37]. However, as in J-Lat 9.2 cells, compounds consistently inhibited provirus expression in live-gated J-Lat 10.6 cells induced by LRAs (Figures 3F-H; Table 2). For example, in cells stimulated with 10 ng/mL TNFα, chrysin, apigenin, luteolin, and L7G inhibited provirus expression with EC50s of 6.3 ± 0.7, 6.2 ± 0.3, 13.6 ± 0.8, and 11.9 ± 0.5 μM, respectively (Figure 3F). In cells stimulated with 10 μM JQ1, chrysin, apigenin, luteolin, and L7G inhibited provirus expression with EC50s of 7.7 ± 1.5, 7.2 ± 1.1, 15.4 ± 1.5, and 16.4 ± 3.3 μM, respectively (Figure 3G). In cells stimulated with 10 mM HMBA, chrysin, apigenin, luteolin, and L7G inhibited provirus expression with EC50s of 6.7 ± 1.4, 5.2 ± 0.8, 26.5 ± 5.8, and 10.1 ± 3.5 μM, respectively (Figure 3H). In contrast, naringenin did not inhibit any LRA-induced provirus expression. Also as observed in J-Lat 9.2 cells, flavopiridol was substantially more potent than the natural flavones, with EC50S of 0.019 ± 0.001, 0.014 ± 0.002, and 0.099 ± 0.002 μM upon stimulation of cells with TNFα, JQ1, and HMBA, respectively. Atuveciclib also inhibited provirus expression induced by these LRAs, also with more efficacy than natural flavones but less than flavopiridol (EC50s of 4.3 ± 0.2, 2.4 ± 0.2, and 1.8 ± 0.1 μM upon induction with TNFα, JQ1, and HMBA, respectively).
To verify that these results were not restricted to Jurkat-based cells, we treated the OM-10.1 macrophage cell line, which is latently infected with a full-length, infectious provirus [29], with 1 ng/mL TNFα in the presence or absence of test compounds and measured intracellular p24Gag expression in live-gated cells by flow cytometry (Figure 3I-J). While p24Gag production was minimal in untreated cells, 1 ng/mL TNFα induced p24Gag production in an average of 90.6 ± 4.0% of live-gated cells (Figure 3J, dotted line). Chrysin, apigenin, luteolin, and L7G inhibited TNFα-induced provirus expression with EC50s of 4.7 ± 0.3, 4.1 ± 0.8, 10.3 ± 0.6, and 10.3 ±1.2 μM, respectively; Figure 3J; Table 2), while no inhibition was observed with naringenin. Flavopiridol and atuveciclib also continued to be more potent (EC50s = 0.013 ± 0.0008 and 1.8 ± 0.09 μM, respectively).
To confirm that inhibition of LRA-induced provirus expression was not due to cellular toxicity, we treated parental Jurkat and OM10.1 cells with compounds for 24 hours and assessed viability using Viacount dye as described above (Figure 3K-L). We observed minimal toxicity when both cell lines were treated with up to 30 μM of the natural flavones, 0.1 μM flavopiridol, or 10 μM atuveciclib; that is, at concentrations where provirus expression was inhibited. At higher concentrations some toxicity was observed: 0.3 μM flavopiridol and 30 μM atuveciclib reduced viability by 38.8 ± 25% and 61.3 ± 2.6%, respectively, in Jurkat cells compared to 0.1% DMSO vehicle-treated controls.
Taken together, these results indicate that the four naturally occurring flavones, in addition to flavopiridol and atuveciclib, inhibit provirus expression induced by 5 LRAs with different functional mechanisms and without concomitant cellular toxicity. They also support the targeting of HIV at a post-integration stage across three in vitro models with different thresholds of latency reversal.
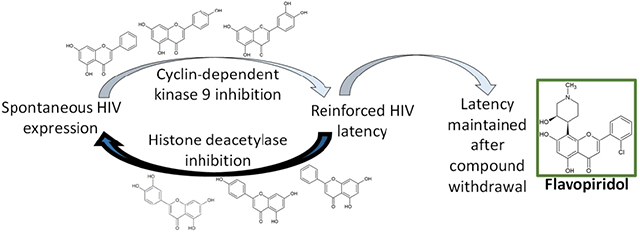
3.4. Aglycone flavones and atuveciclib, but not L7G, inhibit spontaneous latency reversal over time
Established “block-and-lock” agents, like dCA, which continue to reinforce HIV latency after their withdrawal, can also inhibit spontaneous latency reversal in cell lines after long-term culture [13]. We therefore asked whether compounds assessed here could also inhibit spontaneous latency reversal in this manner. To explore this, we used the J-Lat 10.6 cell line which, as described above and previously [37], features a proportion of cells that spontaneously produce GFP and provirus expression detectible by flow cytometry (Figure 4A). J-Lat 10.6 cells were treated with test compounds or 0.1% DMSO vehicle control for 21 days, during which time GFP and intracellular p24 production were assessed every 3 to 4 days (Figure 4A).
Figure 4.

Aglycone flavones, flavopiridol, and atuveciclib, but not L7G, inhibit spontaneous provirus expression. (A) Representative flow cytometry plots showing GFP and viral p24Gag protein expression in J-Lat 10.6 cells following 15 days’ incubation in the absence or presence of 10 μM chrysin or L7G. (B) Effects of compounds on spontaneous provirus expression in J-Lat 10.6 cells over 21 days. (C) Effects of compounds on Jurkat cell viability, as measured using Viacount dye, over 21 days. For (B-C), results are presented relative to cells treated with 0.1% DMSO over the same time course (black dotted lines).
Notably, continuous treatment with 10 μM chrysin, 10 μM apigenin, and 15 μM luteolin steadily reduced the percentage of GFP-positive cells (i.e., inhibited spontaneous latency reversal) over 21 days by 87.8 ± 9.1%, 86.7 ± 11.7%, and 55.4 ± 1.4%, respectively, when compared to cells treated concurrently with 0.1% DMSO vehicle control (Figure 4B). Conversely, and despite its ability to inhibit LRA-induced provirus expression above, spontaneous latency reversal was highly variable, but not inhibited, over 21 days’ treatment with 10 μM L7G (Figures 4A-B). These results suggest that chrysin, apigenin, and luteolin (but not L7G) act via distinct mechanisms that inhibit spontaneous latency reversal in vitro. As expected, 10 μM naringenin had only marginal effects on spontaneous latency reversal after 21 days (i.e., no more than 31.5 ± 2.0% inhibition at Day 21; Figure 4B). Notably, long term treatment with either 10 nM flavopiridol or 10 μM atuveciclib also inhibited spontaneous latency reversal in this cell model (Figure 4B): 21 days of flavopiridol treatment reduced spontaneous GFP expression by 73.8 ± 9.6%, while atuveciclib abolished > 95% of spontaneous GFP production after 9 days, with a 96.5 ± 0.7% decrease after 21 days (Figure 4B). No major impact on cell viability was observed for any compound over 21 days (Figure 4C), indicating that the observed reduction in spontaneous latency reversal is not attributable to cell toxicity.
In summary, these results indicate that most flavones and the CDK9 inhibitor atuveciclib, but not L7G, inhibit spontaneous latency reversal in long-term culture.
3.5. Inhibition of spontaneous latency reversal correlates with inhibiting CDK9 and Tat-dependent transactivation
The observation that both aglycone flavones and atuveciclib can inhibit spontaneous HIV latency reversal in vitro, but L7G cannot, led us to ask whether these observations corresponded to inhibition of CDK9 and CDK9-dependent, Tat-induced provirus expression. We therefore assessed the ability of these compounds to inhibit CDK9 activity using a kinase enzymatic assay that uses a luminescence reporter to quantify the levels of ATP converted to ADP by cell-free, purified CDK9-Cyclin T1 complexes. As shown in Figure 5A, the control CDK9 inhibitors flavopiridol and atuveciclib inhibited CDK9-dependent luminescence with half-maximal inhibitory concentrations (IC50S) of 0.0014 ± 0.0008 and 0.035 ± 0.020 μM (Table 3), consistent with published values for CDK9 inhibition [22, 35]. Chrysin, apigenin, and luteolin also inhibited CDK9 activity in this assay with IC50s of 0.021 ± 0.010, 0.21 ± 0.10, and 0.072 ± 0.030 μM, respectively. In contrast, L7G inhibited CDK9 with an IC50 of 6.9 ± 3.9 μM, or 96-fold higher than its aglycone analogue, luteolin. As expected, naringenin had minimal activity at these concentrations (IC50 > 30 μM).
Figure 5.

Compounds that inhibit spontaneous latency reversal also inhibit CDK9 and Tat-dependent transactivation. (A) CDK9 kinase activity in the presence of compounds, as measured using the ADP-Glo kinase assay. Data are presented relative to CDK9 activity in the presence of 0.1% DMSO. (B) Representative flow cytometry data of GFP expression in CEM-GXR cells transfected with an empty plasmid vector (pCMV-ΔTat) or constitutive Tat expression vector (pCMV-Tat) in the absence or presence of 10 μM apigenin or L7G. (C) Effects of compounds after 24 hours’ incubation on Tat-induced GFP expression in CEM-GXR cells following 24 hours’ incubation. Data are presented relative to Tat-transfected cells treated with 0.1% DMSO. (D) Representative flow cytometry data of mCherry expression and Dendra fluorescence in JurTat cells during no treatment, Dox treatment, or Dox plus 15 μM apigenin. (E) Effect of select compounds (as denoted by the same colors and shapes used in C) on Tat-transactivated mCherry expression in JurTat cells. Data are presented as the ratio of mCherry:Dendra fluorescence and shown relative to the mCherry:Dendra fluorescence in cells treated with 0.1% DMSO. Horizontal dotted lines in A, B, and E denote activities at 100 and 50% of cells treated with 0.1% DMSO.
Table 3.
Effects of flavonoids and CDK9 inhibitors on CDK9 activity, Tat-dependent transactivation, and cellular HDAC activity.
| Compound | CDK9 inhibition IC50 (μM) |
Tat-dependent transactivation EC50 (μM) |
HDAC Inhibition IC50 (μM) |
|---|---|---|---|
| Chrysin | 0.021 ± 0.010 | 7.1 ± 1.0 | 24.0 ± 4.8 |
| Apigenin | 0.21 ± 0.10 | 8.5 ± 0.9 | 47.2 ± 6.7 |
| Luteolin | 0.072 ± 0.030 | 22.2 ± 5.0 | > 90 |
| L7G | 6.9 ± 3.9 | > 30 | > 90 |
| Naringenin | > 30 | > 30 | 88.7 ± 32.1 |
| Flavopiridol | 0.0014 ± 0.0008 | 0.050 ± 0.003 | > 1 |
| Atuveciclib | 0.035 ± 0.020 | 5.8 ± 0.4 | > 90 |
To determine whether the ability of compounds to inhibit CDK9 was relevant to provirus expression in vitro, we transfected CEM-GXR cells, which contain an integrated GFP reporter driven by the HIV LTR [27], with a constitutively-active Tat expression vector (pCMV-Tat). Following 24 hours’ incubation, Tat-induced GFP expression was measured by flow cytometry in live-gated cells. In these assays, an average of 15.7 ± 7.9% of cells were GFP-positive following pCMV-Tat transfection, while no GFP was observed in cells transfected with a control plasmid lacking Tat (pCMV-ΔTat; Figure 5B). Notably, when pCMV-Tat-transfected CEM-GXR cells were treated with chrysin, apigenin, or luteolin, GFP expression was inhibited with EC50s of 7.1 ± 1.0, 8.5 ± 0.9, and 22.2 ± 5.0 μM respectively (Figure 5B-C; Table 3). In contrast, L7G was unable to inhibit GFP expression at up to 30 μM with only marginally improved activity over the negative control naringenin. Additionally, and as expected for CDK9 inhibitors, flavopiridol and atuveciclib inhibited Tat-driven GFP in this model with EC50s of 0.050 ± 0.003 and 5.8 ± 0.4 μM.
To verify that these observations occur independent of cell line, we next tested the activities of a subset of compounds using Jurkat Tet-Tat-Dendra+HIV LTR-mCherry (“JurTat”) cells [58]. In this cell line, an inducible Tet-on promoter drives expression of a Tat-Dendra fusion protein. Tat-Dendra, in turn, drives expression of an LTR-regulated mCherry reporter. As a result, doxycycline (Dox)-treated JurTat cells should induce both Dendra fluorescence and mCherry expression, while inhibitors of Tat-driven transcription should selectively inhibit mCherry over Dendra fluorescence.
To test this hypothesis, JurTat cells were stimulated with 500 ng/mL Dox in the presence or absence of flavonoids for 24 hours, and mCherry and Dendra were monitored by flow cytometry (Figure 5D). In these cells, addition of Dox induced mCherry expression in an average of 13.1 ± 5.3% of cells, while 15 μM apigenin reduced mCherry expression to an average of 1.8 ± 0.7% of cells, (i.e., 13.7% of the fluorescence observed in cells treated with only Dox). In contrast, while Dox induced Dendra expression in an average of 43.6 ± 9.3% of cells, 15 μM apigenin reduced Dendra expression to an average of 17.4 ± 6.5% of cells (i.e., 39.9% of the fluorescence observed in cells treated with only Dox). Thus, residual mCherry fluorescence in the presence of 15 μM apigenin is only 34.3% of residual Dendra fluorescence (i.e. 13.7% / 39.9%), supporting disproportional inhibition of mCherry by apigenin in this assay. Corresponding ratios were then calculated for a subset of compounds (apigenin, luteolin, L7G, and naringenin) from dose-response data, revealing similar inhibition profiles to observed in CEM-GXR cells (Figure 5E). While the selective quenching of mCherry fluorescence is likely to be influenced in part by the secondary, partial block of Tat-Dendra production [58], results in the presence of these compounds largely agreed with those of obtained in CEM-GXR cells, with EC50s of 14.5 ± 1.5 and 9.5 ± 1.5 μM for apigenin and luteolin, respectively. In contrast, neither naringenin nor L7G substantially affected the ratio of mCherry:Dendra fluorescence at up to 30 μM.
Taken together, these results indicate that the aglycone flavones and atuveciclib, but not L7G, inhibit both CDK9 and Tat-dependent transactivation, which further correlates with ability to inhibit spontaneous latency reversal in vitro.
3.6. The extent to which compounds continue to reinforce HIV latency following withdrawal inversely correlates with HDAC inhibition
“Block-and-lock” compounds like dCA reinforce a HIV latent state that persists following their withdrawal from cell culture, in part by favoring a repressive epigenetic, histone deacetylated environment at the HIV LTR [13]. In contrast, some flavonoids including apigenin, chrysin, and luteolin are reported to cause global hyperacetylation through anti-HDAC or pro-histone acetyltransferase activities [42 - 43]. As HDAC inhibition is a well-established pathway for enhancing provirus expression and HIV latency reversal [26], this activity could counteract the ability of these compounds to reinforce a latent state that persists following withdrawal. In contrast, CDK9 inhibitors that do not possess HDAC inhibitory activity may reinforce latency in a more durable manner.
To test this possibility, we examined global intracellular HDAC activity in the presence of these flavones and atuveciclib using an assay that quantifies class I and II HDAC activity in Jurkat cells via a cell-permeable, acetylated, luminogenic peptide substrate. Increased deacetylation of the substrate by cellular HDACs results in increased luminescence, while co-incubation with HDAC inhibitors decreases luminescence. Using this assay, 0.1 μM of the control pan-HDAC inhibitor panobinostat reduced cellular HDAC activity by 88.0 ± 10.9% (Figure 6A), consistent with our previous observations [44]. In contrast, chrysin and apigenin exhibited weaker but detectable HDAC inhibition, with IC50s of 24.0 ± 4.8 and 47.2 ± 6.7 μM, respectively (Figure 6B; Table 3). At 10 μM, both chrysin and apigenin inhibited HDAC activity by 37.9 ± 7.7% and 19.4 ± 5.4%, respectively, indicating detectable HDAC inhibition at concentrations that also inhibit provirus expression. Weaker but detectable activity was also observed with luteolin, which inhibited 10.1 ± 1.6% and 42.6 ± 9.5% of HDAC activity at 10 and 100 μM, respectively (Figure 6B). Intriguingly, atuveciclib also exhibited weak HDAC inhibition at high concentrations, with 11.3 ± 12.9% and 29.7 ± 14.3% block of cellular HDAC activity at 10 and 90 μM. In contrast, no HDAC inhibition was observed with up to 1 μM flavopiridol.
Figure 6.

Compound inhibition of HDACs inversely correlates with reinforced HIV latency following culture withdrawal. (A) Examples of cellular HDAC activity in the absence or presence of 0.1 μM panobinostat, as measured using the HDAC-Glo I/II assay. (B) Effects of compounds after 90 minutes’ incubation on Jurkat cell HDAC activity. (C-D) GFP expression in J-Lat 10.6 cells treated with compounds or 0.1% DMSO for 21 days (pre-wash), then washed extensively and incubated for an additional 24 hours (C) or up to 14 days (D) in the absence of compounds (post-wash). Data are presented relative to cells treated with 0.1% DMSO. Horizontal dotted lines in B-D denote activities at 100 and 50% of cells treated with 0.1% DMSO.
To determine whether these compounds continued to reinforce HIV latency following withdrawal, J-Lat 10.6 cells were treated with compounds for 21 days, extensively washed, and subsequently incubated in drug-free media. After 24 hours, spontaneous GFP production in live-gated cells was quantified by flow cytometry. In control cultures treated with 0.1% DMSO, an average 11.1 ± 2.0% cells expressed GFP after 21 days’ incubation. After being washed and left untreated for 24 hours, an average 10.5 ± 2.2% cells expressed GFP, suggesting that the washing process does not affect spontaneous latency reversal. In contrast, 24 hours after washing, GFP expression in cells previously treated with either 10 μM chrysin, apigenin, or luteolin rebounded to an average of 70.9 ± 65.6%, 80.6 ± 80.5%, or 79.9 ± 26.6% of vehicle-treated control cells (Figure 6C), values that were not significantly different from control levels (p > 0.05; one sample t test). The large variance in provirus rebound in these cells suggests that reinforcement of HIV latency by these natural flavones is unstable and readily reversible following their withdrawal from culture.
In contrast, spontaneous GFP expression in cells previously treated with 10 nM flavopiridol or 10 μM atuveciclib did not rebound to control levels: after 24 hours, cells previously treated with these compounds rebounded to only 32.4 ± 3.6% and 53.0 ± 0.8% of control GFP levels, respectively, values that were significantly lower than control levels for cultures treated with flavopiridol or atuveciclib (p < 0.0001; one sample t test), but not with chrysin, apigenin, or luteolin. This suggests that flavopiridol and atuveciclib are capable of establishing a latent state that persists for at least short durations following their removal. The marked differences in GFP expression following removal of these two compounds was nevertheless notable: while minimal GFP rebound was observed following wash-out of flavopiridol (i.e., 26.2 ± 9.6% GFP expression of untreated cells before wash-out vs. 32.4 ± 3.6% after wash-out, for a 1.2-fold increase in GFP), the rebound following wash-out of atuveciclib was more substantial (3.4 ± 0.7% GFP expression of untreated cells before wash-out vs. 53.0 ± 0.8% after wash-out, for a 15.6-fold increase; Figure 6C).
Intriguingly, GFP expression in flavopiridol and atuveciclib-treated cultures also remained below control levels for several days following their withdrawal (Figure 6D). For example, 14 days following washout, GFP levels in cells previously treated with 10 nM flavopiridol expressed GFP at only 59.7 ± 15.2% of control levels (representing a 2.3-fold increase from GFP levels during treatment). In contrast, 11 days following wash-out of atuveciclib, GFP expression levels had returned to those of untreated controls (Figure 6D).
Overall, these results suggest that naturally occurring flavones that possess HDAC inhibitory activity do not reinforce latency following their removal. Atuveciclib, which possesses weak HDAC inhibitory activity, induces a latent state that reverses slowly upon compound removal. In contrast, flavopiridol, which lacks HDAC inhibitory activity, establishes a latent state that persists for at least 14 days after wash-out.
3.6. Flavopiridol inhibits spontaneous viral RNA production following withdrawal in primary CD4+ T cells from donors with HIV
We next asked whether the ability of flavopiridol to reinforce HIV latency following its removal extends to primary cells. We therefore investigated flavopiridol’s effect on HIV RNA levels in culture supernatants from CD4+ T cells isolated from five donors with HIV using qRT-PCR (Figure 7). When cultured for 24 hours in the presence of 0.1% DMSO, HIV RNA levels in CD4+ T cell culture supernatants were 2.4*108 ± 4.8*108 viral copies per mL on average, compared to 1.7*109 ± 3.1*109 viral copies/mL in cell cultures treated with 30 nM flavopiridol (Figure 7A). This slight but consistent increase in viral RNA (average 7.1-fold) did not correspond to changes in cell viability, as measured by direct live cell counting (average 1.1*106 ± 6.2*105 cells/mL in 0.1% DMSO-treated cultures vs. 1.2*106 ± 5.6*105 cells/mL in flavopiridol-treated cultures; Figure 7B), suggesting that effects are not likely attributable to toxicity.
Figure 7.

Effects of flavopiridol on spontaneous viral RNA production in cultures of primary CD4+ T cells from donors with HIV. (A) Absolute viral RNA copies per mL of supernatant in primary CD4+ cultures following 24 hours’ treatment with 0.1% DMSO or 30 nM flavopiridol. (B) Effects of culture treatments in (A) on cell viability. (C) Absolute viral RNA copies per mL supernatant in primary CD4+ cultures following 6 – 7 days’ treatment with 0.1% DMSO or 30 nM flavopiridol, extensive cell washing, and further incubation in the absence of compounds for 24 hours. (D) Effects of culture treatments in (C) on cell viability. For each panel, shapes and colors denote individual donors. In A and C, dotted lines indicate limit of detection, and empty shapes indicate no viral RNA detected.
To determine flavopiridol’s effect on HIV RNA levels in culture supernatant following lengthier treatment and washout, CD4+ cells from the same five donors were cultured in the presence of 0.1% DMSO or 30 nM flavopiridol for 6 – 7 days. Cells were then extensively washed and incubated in the absence of compounds for an additional 24 hours, and supernatant viral RNA was monitored again by qRT-PCR (Figure 7C). In these studies, cultures treated with 0.1% DMSO contained an average of 3.7*105 ± 1.2*104 viral copies/mL, indicating a ~3 log reduction in spontaneous viral RNA production at 24 hours post-washout. In contrast, viral RNA production was reduced in 4 of 5 flavopiridol-treated cultures (average 1.2*104 ± 1.6*104 viral copies/mL across 5 cultures), with no viral RNA detected in two cultures and an average 96.8% reduction in total viral RNA across all cultures (Figure 7C). Similar to results following 24 hours’ culture, no major effects on viability were observed in cells treated with flavopiridol after 6 – 7 days (average 1.3*106 ± 2.4*105 cells/mL in 0.1% DMSO-treated cultures vs. 1.3*106 ± 3.9*105 cells/mL in flavopiridol-treated cultures; Figure 7D). Taken together, these results broadly recapitulate the cell line data described above and further support that flavopiridol inhibits spontaneous viral production in infected primary CD4+ cells.
4. Discussion
Compounds that establish a latent HIV state that persists following their withdrawal [13 - 16] hold particular therapeutic promise, as these could eventually lead to the development of cART-free remission-based therapies or a “functional” HIV cure. Toward this goal, we explored a series of naturally occurring flavones, in addition to the semi-synthetic flavone flavopiridol and its functional analogue atuveciclib, as CDK9 inhibitors with the potential to reinforce HIV latency following their discontinuation in vitro. We chose this set of compounds based on reports that the naturally-occurring flavones chrysin, apigenin, and luteolin suppress active HIV replication and inhibit provirus expression at low micromolar concentrations with no obvious cell toxicities at antiviral concentrations [8, 17 - 19]. Flavopiridol has also been reported to inhibit active replication in PBMCs [36]. Our studies using in vitro HIV-infected PBMCs agree with these results derived largely from HIV-infected cell lines.
However, we also show for the first time that all compounds assessed here, except the negative control naringenin, inhibit provirus expression induced by multiple LRAs, indicating that their mechanism overrides the effects of several of these agents. We also show the extent of atuveciclib’s anti-HIV activity and formally demonstrate that highly selective inhibition of CDK9 (i.e. > 100-fold selectivity over other CDKs [35]) is sufficient to suppress active HIV replication in PBMCs. We further show that long-term culture with aglycone flavones or atuveciclib, but not the glycosylated L7G, inhibits spontaneous HIV latency reversal over time, and this outcome correlates with inhibition of CDK9 and Tat transactivation. Intriguingly, we also found that prolonged incubation with flavopiridol (which unlike chrysin, apigenin, and luteolin does not target class I/II HDACs) induces a latent state in vitro that persists for at least 14 days following its withdrawal from cell culture. Over time, flavopiridol also inhibits spontaneous viral RNA production in isolated CD4+ T cells from HIV-infected donors, although future studies are needed to investigate the extent of reinforced latency by flavopiridol and other agents ex vivo. Finally, we demonstrate that atuveciclib also reinforces latency for short periods in vitro following withdrawal; however, this effect is lost by 11 days following withdrawal and correlates with weak but detectible HDAC inhibition. Figure 8A summarizes the results from these studies.
Figure 8.

Model for reinforced HIV latency following compound withdrawal. A, Summary of bioactivities of compounds described here. B, CDK9 inhibition inhibits spontaneous provirus expression and reinforces HIV latency even following compound withdrawal. However, ability to reinforce latency is sensitive to and prevented by simultaneous HDAC inhibition.
Taken together, our results indicate that inhibition of CDK9 corresponds with HIV transcriptional quiescence and reinforced latency. This in turn identifies CDK9 as a potential target for therapies designed to permanently silence HIV reservoirs in humans. However, our results also indicate that concomitant ability to inhibit histone deacetylases, even weakly, may counteract the latency-reinforcing activities induced by CDK9 inhibition (Figure 8B). Future therapeutic leads that target CDK9 with the goal of long-term, cART-free HIV remission may therefore require exceptional selectivity for CDK9. Future studies to assess the ability of CDK9 inhibitors to reinforce latency ex vivo, either alone or in combination with other candidate agents like dCA, and/or using CD4+ T-cells obtained from persons living with HIV on suppressive cART, are therefore warranted.
Flavonoids are a diverse chemical group of plant metabolites with a variety of functions including pigmentation, cell signalling, and protection from ultraviolet radiation, among others [45]. Thousands of flavonoids have been identified from plants or synthesized, and many feature diverse biological activities including antioxidative, antibacterial, antiviral, or anticancer properties [46 - 47]. Many flavonoids, including the naturally occurring ones studied here, are reported to have multiple cellular targets and presumably act on several cellular pathways. However, the natural flavones assessed here appear to have selective anti-HIV functions, as they suppress HIV replication and inhibit HIV latency reversal at lower concentrations than those that cause significant cytotoxicity. This is consistent with similar observations for other host factor-based HIV inhibitors such as sudemycin D6 and digoxin, where low concentrations block virus replication but do not obviously affect host cell physiology [15, 48]. These results suggest that the overall in vitro effects of flavones at low concentrations seem to be antiviral, and whereas cells may compensate in this context for the partial loss-of-function of host factors such as CDK9, HIV cannot. We note that these observations contrast with those of several LRAs, where concentrations that induce HIV latency reversal often overlap with those causing cytotoxicity [37, 56, 57].
The ability of these flavones to inhibit provirus expression induced by multiple classes of LRAs further suggests that they are acting on pathways required for general latency reversal as opposed to pathways distinct to each LRA. In particular, we note that all active compounds assessed here can inhibit provirus expression induced by JQ1, which is an LRA that acts directly on CDK9 activity and recruitment of transcriptional complexes to the HIV promoter [49 - 50]. This observation is in line with the ability of flavopiridol to inhibit proviral expression due to a chalcone derivative LRA that promotes CDK9 phosphorylation and enhances p-TEFb activity [51]. These results are also consistent with a model of CDK9 as a primary regulator of HIV transcription, as shown by the ability of both flavopiridol and atuveciclib to inhibit provirus expression by multiple LRAs. However, as L7G also inhibits provirus expression by all LRAs but does not target CDK9 at the same concentrations, additional antiviral mechanisms at the level of viral transcription must also be at play.
Notably, we observed that aglycone flavones and atuveciclib can suppress spontaneous latency reversal in vitro following 21 days in culture, while L7G cannot. These results correspond with the ability of flavones to inhibit both CDK9/Tat-dependent reporter expression, where L7G was less effective (and in the case of Tat inhibition, ineffective). Intriguingly, an in silico docking analysis of the aglycone flavone wogonin binding to CDK9 by Polier et al. (2011) predicted that these molecular interactions involved docking of wogonin’s A ring into the ATP-binding pocket of CDK9 [20]. Wogonin structurally resembles chrysin, differing only by the addition of a methoxy group on C8 of the A ring (Figure 1A). It is therefore reasonable to hypothesize that chrysin, and by extension apigenin and luteolin, may bind in a similar orientation. In contrast, the glycosyl moiety of L7G, which is present on C7 of the A ring, may force L7G to bind to CDK9 in a different orientation and impact its inhibitory potential. The inability of L7G to suppress spontaneous latency reversal in vitro, strongly block CDK9 enzymatic activity, and inhibit Tat-dependent reporter expression in our studies suggest that the glycosyl group of L7G may be responsible for its distinct activity profile.
A therapeutic strategy to achieve cART-free HIV remission may require compounds that establish a latent state that persists even after their discontinuation. For example, dCA treatment creates a state that is resistant to spontaneous latency reversal for days to weeks after withdrawal [13]. In contrast, the reinforced latency observed during long-term in vitro treatment with apigenin, chrysin, or luteolin did not persist following their withdrawal. However, these compounds were also confirmed as HDAC inhibitors at concentrations where spontaneous latency reversal was inhibited [42]. As HDAC inhibitors confer de-repressed epigenetic features at the HIV promoter [26], concurrent HDAC inhibition may counter the ability of these flavonoids to promote a repressive epigenetic environment upon CDK9 inhibition (Figure 8B). These mechanistic details require further study.
Consistent with this hypothesis, long-term treatment with the CDK9 inhibitor flavopiridol (which suppresses active HIV replication, inhibits provirus expression, and inhibits spontaneous latency reversal, but does not inhibit cellular HDAC activity), continued to establish a latent state that persisted for at least 14 days following withdrawal in vitro. In contrast, atuveciclib, which in our hands was functionally equivalent to flavopiridol with the exception of weak ability to inhibit HDACs, was only temporarily following its withdrawal: within 11 days, spontaneous latency reversal rebounded to baseline levels, consistent with the expectation that HDAC inhibition counteracts latency-reinforcing effects. Notably, both flavopiridol and atuveciclib have undergone several phase I/II clinical trials for cancers associated with CDK dysregulation, although neither is currently approved for clinical use [52 - 54]. Interestingly, both inhibitors target HIV replication and provirus expression in this study at concentrations lower than those causing cytotoxicity [22, 35, 55], suggesting that they may have more efficacy against HIV than tumor cells. Efficacy studies involving these agents and potentially other CDK9 inhibitors to reinforce latency in primary cells from HIV-infected donors and/or animal models of HIV are thus warranted.
There are currently no licensed antiretrovirals that target HIV transcription or expression from latently infected cells. Inhibitors of provirus expression could potentially supplement existing HIV therapies, for example by blocking residual transcription from proviruses to reduce chronic inflammation during long-term cART. Additionally, durable reinforcement of HIV latency may lead to cART-free HIV remission-based therapies. Flavonoids and other agents that selectively target CDK9 may be useful in achieving these objectives.
Acknowledgements:
We thank Silven Read and Khumoekae Richard for superb technical assistance, Bruce Ganase for assistance with participant recruitment, Brendan Bell for pCMV-Tat and pCMV-ΔTat plasmids, Leor Weinberger and Brandon Razooky for JurTat cells, and Jocelyn Rivera-Ortiz, Nicolas Chomont, and Amélie Pagliuzza for additional experimental and technical guidance. Funding was provided by the Canadian Institutes for Health Research (CIHR PJT-153057) (I.T., M.A.B., Z.L.B.), the Canadian Foundation for AIDS Research (CANFAR) (I.T., M.A.B., Z.L.B.) and the New Frontiers in Research Fund – Explorations (NFRFE-2018-01386) (I.T.). This work was also supported through the Sub-Saharan African Network for TB/HIV Research Excellence (SANTHE) (I.T., K.A.M.), a DELTAS African Initiative [grant # DEL-15-006]. The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)’s Alliance for Accelerating Excellence in Science in Africa (AESA) and supported by the New Partnership for Africa’s Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust [grant # 107752/Z/15/Z] and the UK government. This work was also supported by the following grants to L.J.M.: Beyond Antiretroviral Treatment (BEAT)-HIV Delaney Collaboratory Grant UM1AI126620, co-funded by NIAID, NIMH, NINDS, and NIDA, National Institutes of Health. It was also supported by Merck, Inc; the Philadelphia Field Initiating Group for HIV-1 Trials (Philadelphia FIGHT); the CLAWS Foundation, the Robert I. Jacobs Fund of The Philadelphia Foundation; Estate of Rusty Miller and Martha Stengel and the Summerhill Charitable Trust; Penn Center for AIDS Research Grant P30 AI 045008; and the Herbert Kean, M.D., Family Professorship. C.S. was supported by a CIHR Frederick Banting and Charles Best MSc Award. M.A.B. holds a Tier 2 Canada Research Chair in viral pathogenesis and immunity. Z.L.B. is supported by a Scholar Award from the Michael Smith Foundation for Health Research.
Abbreviations:
- cART
combination antiretroviral therapy
- CC50
half-maximal cytotoxic concentration
- CDK9
cyclin-dependent kinase 9
- dCA
didehydro cortistatin A
- Dox
doxycycline
- EC50
half-maximal effective concentration
- FBS
fetal bovine serum
- HDAC
histone deacetylase
- HIV
human immunodeficiency virus
- IC50
half-maximal inhibitory concentration
- L7G
luteolin-7-glucoside
- LRA
latency-reversing agent
- LTR
long-terminal repeat
- MOI
multiplicity of infection
- PBMC
peripheral blood mononuclear cell
- P-TEFb
positive transcription elongation factor B
- qRT-PCR
quantitative reverse transcription polymerase chain reaction
- SI
selectivity index
Footnotes
Conflict of Interest: The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chawla A, Wang C, Patton C, Murray M, Punekar Y, de Ruiter A, and Steinhart C (2018) A review of long-term toxicity of antiretroviral treatment regimens and implications for an aging population. Infect. Dis. Ther 7, 183–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zicari S, Sessa L, Cotugno N, Ruggiero A, Morrocchi E, Concato C, Rocca S, Zangari P, Manno EC, and Palma P (2019) Immune activation, inflammation, and non-AIDS co-morbidities in HIV-infected patients under long-term ART. Viruses. 11, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Besnard E, Hakre S, Kampmann M, Lim HW, Hosmane NN, Martin A, Bassik MC, Verschueren E, Battivelli E, Chan J, Svensson JP, Gramatica A, Conrad RJ, Ott M, Greene WC, Krogan NJ, Siliciano RF, Weissman JS, and Verdin E (2016) The mTOR complex controls HIV latency. Cell Host Microbe. 14, 785–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hayashi T, Jean M, Huang H, Simpson S, Santoso NG, and Zhu J (2017) Screening of an FDA-approved compound library identifies levosimendan as a novel anti-HIV-1 agent that inhibits viral transcription. Antiviral Res. 146, 76–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vargas B, Giacobbi NS, Sanyal A, Venkatachari NJ, Han F, Gupta P, and Sluis-Cremer N (2019) Inhibitors of signaling pathways that block reversal of HIV-1 latency. Antimicrob. Agents Chemother. 63, e01744–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kessing CF, Nixon CC, Li C, Tsai P, Takata H, Mousseau G, Ho PT, Honeycutt JB, Fallahi M, Trautmann L, Garcia JV, and Valente ST (2017) In vivo suppression of HIV rebound by didehydro-cortistatin A, a “block-and-lock” strategy for HIV-1 treatment. Cell Rep 21:600–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marsden MD and Zack JA (2019) HIV cure strategies: A complex approach for a complicated viral reservoir? Future Virol. 14, 5–8 [Google Scholar]
- 8.Darcis G, Van Driessche B, and Van Lint C (2017) HIV latency: Should we shock or lock? Trends Immunol. 38, 217–228 [DOI] [PubMed] [Google Scholar]
- 9.Delannoy A, Poirier M, Bell B (2019) Cat and mouse: HIV transcription in latency, immune evasion and cure/remission strategies. Viruses. 11, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mancebo HSY, Lee G, Flygare J, Tomassini J, Luu P, Zhu Y, Peng J, Blau C, Hazuda D, Price D, and Flores O (1997) P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev. 11, 2633–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ping YH and Rana TM (2001) DSIF and NELF interact with RNA polymerase II elongation complex and HIV-1 tat stimulates P-TEFb-mediated phosphorylation of RNA polymerase II and DSIF during transcription elongation. J. Biol. Chem 276, 12951–12958 [DOI] [PubMed] [Google Scholar]
- 12.Asamitsu K, Fujinaga K, and Okamoto T (2018) HIV Tat/P-TEFb interaction: A potential target for novel anti-HIV therapies. Molecules. 23, 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mousseau G, Kessing CF, Fromentin R, Trautmann L, Chomont N, and Valente ST (2015) The tat inhibitor didehydro-cortistatin a prevents HIV-1 reactivation from latency. mBio. 6, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li C, Mousseau G, and Valente ST (2019) Tat inhibition by didehydro-Cortistatin A promotes heterochromatin formation at the HIV-1 long terminal repeat. Epigenetics Chromatin. 12, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kyei GB, Meng S, Ramani R, Niu A, Lagisetti C, Webb TR, and Ratner L (2018) Splicing factor 3B subunit 1 interacts with HIV tat and plays a role in viral transcription and reactivation from latency. mBio. 9, e01423–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niu Q, Liu Z, Alamer E, Fan X, Chen H, Endsley J, Gelman BB, Tian B, Kim JH, Michael NL, Robb ML, Ananworanich J, Zhou J, and Hu H (2019) Structure-guided drug design identifies a BRD4-selective small molecule that suppresses HIV. J. Clin. Invest 129, 3361–3373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu CQ, Chen K, Shi Q, Kilkuskie RE, Cheng YC, and Lee KH (1994) Anti-aids agents, 10. Acacetin-7-O-β-d-galactopyranoside, an anti-HIV principle from chrysanthemum morifolium and a structure-activity correlation with some related flavonoids. J. Nat. Prod 57, 42–51 [DOI] [PubMed] [Google Scholar]
- 18.Critchfield JW, Butera ST, and Folks TM (1996) Inhibition of HIV activation in latently infected cells by flavonoid compounds. AIDS Res. Hum. Retroviruses 12, 39–46 [DOI] [PubMed] [Google Scholar]
- 19.Mehla R, Bivalkar-Mehla S, and Chauhan A (2011) A flavonoid, luteolin, cripples HIV-1 by abrogation of Tat function. PLoS One. 6, e27915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Polier G, Ding J, Konkimalla BV, Eick D, Ribeiro N, Köhler R, Giaisi M, Efferth T, Desaubry L, Krammer PH, and Li-Weber M (2011) Wogonin and related natural flavones are inhibitors of CDK9 that induce apoptosis in cancer cells by transcriptional suppression of Mcl-1. Cell Death Dis. 2, e182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sadanari H, Fujimoto KJ, Sugihara Y, Ishida T, Takemoto M, Daikoku T, and Murayama T (2018) The anti-human cytomegalovirus drug tricin inhibits cyclin-dependent kinase 9. FEBS Open Bio. 8, 646–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chao SH, Fujinaga K, Marion JE, Taube R, Sausville EA, Senderowicz AM, Peterlin BM, and Price DH (2000) Flavopiridol inhibits P-TEFb and blocks HIV-1 replication. J. Biol. Chem 275, 28345–28348 [DOI] [PubMed] [Google Scholar]
- 23.Pandey M, Kaur P, Shukla S, Abbas A, Fu P, and Gupta S (2012) Plant flavone apigenin inhibits HDAC and remodels chromatin to induce growth arrest and apoptosis in human prostate cancer cells: In vitro and in vivo study. Mol. Carcinog 51, 952–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pal-Bhadra M, Ramaiah MJ, Reddy TL, Krishnan A, Pushpavalli SNCVL, Babu KS, Tiwari AK, Rao JM, Yadav JS, and Bhadra U (2012) Plant HDAC inhibitor chrysin arrest cell growth and induce p21WAF1 by altering chromatin of STAT response element in A375 cells. BMC Cancer. 12, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Attoub S, Hassan AH, Vanhoecke B, Iratni R, Takahashi T, Gaben AM, Bracke M, Awad S, John A, Kamalboor HA, Al Sultan MA, Arafat K, Gespach C, and Petroianu G (2011) Inhibition of cell survival, invasion, tumor growth and histone deacetylase activity by the dietary flavonoid luteolin in human epithelioid cancer cells. Eur. J. Pharmacol 651, 18–25 [DOI] [PubMed] [Google Scholar]
- 26.Shirakawa K, Chavez L, Hakre S, Calvanese V, and Verdin E (2013) Reactivation of latent HIV by histone deacetylase inhibitors. Trends Microbiol. 21, 277–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brockman MA, Tanzi GO, Walker BD, and Allen TM (2006) Use of a novel GFP reporter cell line to examine replication capacity of CXCR4- and CCR5-tropic HIV-1 by flow cytometry. J. Virol. Methods 131, 134–142 [DOI] [PubMed] [Google Scholar]
- 28.Jordan A, Bisgrove D, and Verdin E (2003) HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 22, 1868–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Butera ST, Perez VL, Wu BY, Nabel GJ, and Folks TM (1991) Oscillation of the human immunodeficiency virus surface receptor is regulated by the state of viral activation in a CD4+ cell model of chronic infection. J. Virol. 65, 4645–4653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, and Martin MA (1986) Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol 59, 284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malim MH, Hauber J, Fenrick R, and Cullen BR (1988) Immunodeficiency virus rev trans-activator modulates the expression of the viral regulatory genes. Nature. 335, 181–183 [DOI] [PubMed] [Google Scholar]
- 32.Mwimanzi P, Tietjen I, Miller SC, Shahid A, Cobarrubias K, Kinloch NN, Baraki B, Richard J, Finzi A, Fedida D, Brumme ZL, and Brockman MA (2016) Novel acylguanidine-based inhibitor of HIV-1. J. Virol 90, 9495–9508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tietjen I, Ntie-Kang F, Mwimanzi P, Onguéné PA, Scull MA, Idowu TO, Ogundaini AO, Meva'a LM, Abegaz BM, Rice CM, Andrae-Marobela K, Brockman MA, Brumme ZL, and Fedida D (2015) Screening of the pan-African natural product library identifies ixoratannin A-2 and boldine as novel HIV-1 inhibitors. PLoS One. 10, e0121099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Senderowicz AM (1999) Flavopiridol: The first cyclin-dependent kinase inhibitor in human clinical trials. Invest. New Drugs 17, 313–320 [DOI] [PubMed] [Google Scholar]
- 35.Lücking U, Scholz A, Lienau P, Siemeister G, Kosemund D, Bohlmann R, Briem H, Terebesi I, Meyer K, Prelle K, Denner K, Bömer U, Schäfer M, Eis K, Valencia R, Ince S, von Nussbaum F, Mumberg D, Ziegelbauer K, Klebl B, Choidas A, Nussbaumer P, Baumann M, Schultz-Fademrecht C, Rühter G, Eickhoff J, and Brands M (2017) Identification of atuveciclib (BAY 1143572), the first highly selective, clinical PTEFb/CDK9 inhibitor for the treatment of cancer. ChemMedChem. 12, 1776–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Biglione S, Byers SA, Price JP, Nguyen VT, Bensaude O, Price DH, and Maury W (2007) Inhibition of HIV-1 replication by P-TEFb inhibitors DRB, seliciclib and flavopiridol correlates with release of free P-TEFb from the large, inactive form of the complex. Retrovirology. 4, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richard K, Williams DE, Dilip de Silva E, Brockman MA, Brumme ZL, Andersen RJ, and Tietjen I (2018) Identification of novel HIV-1 latency-reversing agents from a library of marine natural products. Viruses. 10, 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Osborn L, Kunkel S, and Nabel GJ (1989) Tumor necrosis factor α and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor κB. Proc. Natl. Acad. Sci. USA 86, 2336–2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams SA, Chen LF, Kwon H, Fenard D, Bisgrove D, Verdin E, and Greene WC (2004) Prostratin antagonizes HIV latency by activating NF-κB. J. Biol. Chem 279, 42008–42017 [DOI] [PubMed] [Google Scholar]
- 40.Zhu J, Gaiha GD, John SP, Pertel T, Chin CR, Gao G, Qu H, Walker BD, Elledge SJ, and Brass AL (2012) Reactivation of Latent HIV-1 by Inhibition of BRD4. Cell Rep. 2, 807–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Contreras X, Barboric M, Lenasi T, and Peterlin BM (2007) HMBA releases P-TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLoS Pathog. 3, 1459–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh P, Tomar RS, and Rath SK (2015) Anticancer potential of the histone deacetylase inhibitor-like effects of flavones, a subclass of polyphenolic compounds: a review. Mol. Biol. Rep 42, 1515–1531 [DOI] [PubMed] [Google Scholar]
- 43.Godoy LD, Lucas JE, Bender AJ, Romanick SS, and Ferguson BS (2017) Targeting the epigenome: Screening bioactive compounds that regulate histone deacetylase activity. Mol. Nutr. Food Res 61, 1600744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Divsalar DN, Simoben CV, Schonhofer C, Richard K, Sippl W, Ntie-Kang F, and Tietjen I (2020) Novel histone deacetylase inhibitors and HIV-1 latency-reversing agents identified by large-scale virtual screening. Front. Pharmacol 11, 905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samanta A, Das G, and Das S (2011) Roles of flavonoids in plants. Int J Pharm Sci Tech 6:12–35. [Google Scholar]
- 46.Chahar MK, Sharma N, Dobhal MP, and Joshi YC (2011) Flavonoids: A versatile source of anticancer drugs. Pharmacogn. Rev 5, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang T, Li Q, and Bi K (2018) Bioactive flavonoids in medicinal plants: Structure, activity and biological fate. Asian J. Pharm. Sci 13, 12–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wong RW, Balachandran A, Ostrowski MA, and Cochrane A (2013) Digoxin suppresses HIV-1 replication by altering viral RNA processing. PLoS Pathog. 9, e1003241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Z, Guo J, Wu Y, and Zhou Q (2013) The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing BRD4 inhibition of Tat-transactivation. Nucleic Acids Res. 41, 277–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boehm D, Calvanese V, Dar RD, Xing S, Schroeder S, Martins L, Aull K, Li PC, Planelles V, Bradner JE, Zhou MM, Siliciano RF, Weinberger L, Verdin E, and Ott M (2013) BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell cycle. 12, 452–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu J, Ao MT, Shao R, Wang HR, Yu D, Fang MJ, Gao X, Wu Z, Zhou Q, and Xue YH (2017) A chalcone derivative reactivates latent HIV-1 transcription through activating P-TEFb and promoting Tat-SEC interaction on viral promoter. Sci. Rep 7, 10657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shapiro GI (2004) Preclinical and clinical development of the cyclin-dependent kinase inhibitor flavopiridol. Clin. Cancer Res 10, 4270s–4275s [DOI] [PubMed] [Google Scholar]
- 53.Wiernik PH (2016) Alvocidib (flavopiridol) for the treatment of chronic lymphocytic leukemia. Expert Opin. Investig. Drugs. 25, 729–734 [DOI] [PubMed] [Google Scholar]
- 54.ClinicalTrials.gov. Bethesda (MD): National Library of Medicine (US). Identifier NCT02345382, Phase I dose escalation of BAY1143572 in subjets with acute leukemia; 2018. June 25 [cited 9 September 2020]; Available from: https://clinicaltrials.gov/ct2/show/NCT02345382 [Google Scholar]
- 55.Ali A, Ghosh A, Nathans RS, Sharova N, O’Brien S, Cao H, Stevenson M, and Rana TM (2009) Identification of flavopiridol analogues that selectively inhibit positive transcription elongation factor (P-TEFb) and block HIV-1 replication. ChemBioChem. 10, 2072–2080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Richard K, Schonhofer C, Giron LB, Rivera-Ortiz J, Read S, Kannan T, Kinloch NN, Shahid A, Felicke R, Wappler S, Imming P, Harris M, Brumme ZL, Brockman MA, Mounzer K, Kossenkov AV, Abdel-Mohsen M, Andrae-Marobela K, Montaner LJ, and Tietjen I (2020) The African natural product knipholone anthrone and its analogue anthralin (dithranol) enhance HIV-1 latency reversal. J. Biol. Chem 295, 14084–14099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abner E and Jordan A (2019) HIV “shock and kill” therapy: In need of revision. Antiviral Res. 166, 19–34. [DOI] [PubMed] [Google Scholar]
- 58.Razooky BS, Pai A, Aull K, Rouzine IM, and Weinberger LS (2015) A hardwired HIV latency program. Cell. 160, 990–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yuan Z, Wang N, Niu W, Li Q, and Guo J (2017) Controlling multi-cycle replication of live-attenuated HIV-1 using an unnatural genetic switch. ACS Synth. Biol 6, 721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]