

Abstract
NO sensitive soluble guanylyl cyclase (sGC) plays a key role in mediating physiological functions of NO. Genetic alterations of the GUCY1A3, coding for the α1 subunit of sGC, are associated with several cardiovascular dysfunctions. A rare sGC variant with Cys517→Tyr substitution in the α1subunit, has been associated with moyamoya disease and achalasia. In this report we characterize the properties of this rare sGC variant. Purified α1C517Yβ1 sGC preserved only ~25% of its cGMP-forming activity and showed an elevated Km for GTP substrate. However, the mutant enzyme retained a high affinity for and robust activation by NO, similar to wild type sGC. Purified α1C517Yβ1 enzyme was more sensitive to specific sGC heme oxidizers and less responsive to heme reducing agents. When expressed in COS7 cells, α1C517Yβ1 sGC showed a much stronger response to cinaciguat or gemfibrozil, which targets apo-sGC or sGC with ferric heme, as compared to its NO response or the relative response of the wild type sGC. A stronger response to cinaciguat was also observed for purified α1C517Yβ1 in the absence of reducing agents. In COS7 cells, αCys517β sGC was less stable than the wild type enzyme under normal conditions and exhibited accelerated degradation upon induction of cellular oxidative stress. We conclude that diminished cGMP-forming activity of this sGC variant is aggravated by its high susceptibility to oxidative stress and diminished protein stability. The combination of these deficiencies contribute to the severity of observed moyamoya and achalasia symptoms in human carriers of this rare α1C517Yβ1 sGC variant.
Keywords: Nitric oxide, cGMP, soluble guanylyl cyclase, oxidative stress, protein stability
1. INTRODUCTION
Nitric Oxide signaling is essential for many physiologic processes in mammals. NO-sensitive guanylyl cyclase (NO-GC), usually referred to as soluble guanylyl cyclase (sGC), plays an important role in NO signaling. NO-sensitive guanylyl cyclase is a heterodimeric protein composed of one α and one β subunit. Both subunits are needed for a functional catalytic site, which uses GTP to synthesize secondary cellular messenger cGMP. Humans and mice have two functional isoforms of the α subunit (α1 and α2) and one functional β1 isoform. The heterodimer α1β1 (GC-1) is ubiquitously expressed and has a higher level of expression than the α2β1 (GC-2) counterpart, which has tissue-specific expression, and is more prevalent in brain, kidney and placenta. The N-terminal portion of the β1 subunit harbors the heme moiety. Under physiologic conditions, sGC heme contains ferrous iron which binds NO with subnanomolar affinity [1, 2], triggering conformational changes that activate several hundred fold the cGMP-forming activity of the catalytic site.
This enzyme is a key mediator of most physiological functions of NO and is often referred to as NO receptor. E.g., NO-stimulated sGC relaxes vascular smooth muscle cells (VSMC), facilitates repair of injured endothelium [3, 4], prevents adhesion and migration of leukocytes [5], reduces platelet aggregation [6], decreases neointimal growth of injured blood vessels by inhibiting proliferation and migration of VSMC [7], improves oxygenation [8],promotes motility along the gastrointestinal tract [9, 10], and participates in neurotransmission [11]
Diminished sGC function contributes to cardiovascular disorders, such as coronary artery disease, atherosclerosis and hypertension [12, 13]. Mice lacking β1 subunit, develop hypertension [14], and have significant problems with gastrointestinal motility [10]. Similarly, mice carrying heme-deficient and NO-unresponsive β1 sGC mutant, also have impaired smooth muscle relaxation, develop hypertension, gastroparesis and erectile dysfunction [9, 15, 16]. Genome-wide association [17, 18] and familial genetic studies [19–21] also confirm the crucial function of NO receptor in human cardiovascular physiology.
Two independent studies identified a number of rare mutation variants in the human GUCY1A3 gene coding for the α1 subunit that are associated with early onset hypertension, stenosis of the terminal part of internal carotid arteries, development of moyamoya-like angiopathy, as well as achalasia - dysfunction of esophageal motility and swallowing [20, 21]. All mutations identified by Herve and colleagues lead to premature stop codons, predicting catalytically inactive sGC variants, and result in undetectable level of sGC protein [20]. The study by Wallace and colleagues also identified a frameshift mutations coding for truncated α1 subunit and dysfunctional sGC [21]. Besides these mutations causing truncated dysfunctional sGC, a single-nucleotide missense mutation coding for the Cys517→Tyr substitution in the α1 sGC subunit was associated with the development of moyamoya disease and achalasia in the carriers of this rare variant of α1 sGC [21]. Preliminary biochemical characterization of recombinant α1C517Yβ1 heterodimer demonstrated the same sensitivity for NO donors as the wild type sGC [21], but a diminished catalytic activity and a fractionally increased Km for GTP. Previous studies of mice lacking α1 subunit demonstrated a robust redundancy of the NO-sGC signaling. It has been shown that even a small amount of α2 subunit of sGC may be sufficient to compensate for the lack of functional α1 [22]. Therefore, it seems incongruous that individuals carrying the α1C517Yβ1 mutant with moderately attenuated of sGC function exhibit symptoms of moyamoya disease of same severity as individuals lacking functional α1β1 sGC. To solve this conundrum, in this report we further evaluated biochemical properties of the α1C517Yβ1 mutant and its stability in different cellular conditions.
2. MATERIAL AND METHODS
2.1. Reagents:
Triethanolamine, dithiotreitol, imidazole, aminolevulinic acid, actinomycin D, rotenone, antimycin, protease inhibitor cocktail for His-tagged proteins, salts were from Sigma-Aldrich (St. Louis, MO). Fetal bovine serum, DMEM/F12 media, penicillin-streptomycin, Lipofectamine with Plus reagent were form Thermo Fischer (Waltham, MA). DEAE-FF Sepharose media, Superdex 200 and Hitrap desalting columns were from GE Lifesciences (Chicago, IL). His-Bind agarose was from EMDMillipore (Billerica, MA). α-[32P]GTP was from Perkin Elmer (Waltham, MA).
2.2. Protein expression and purification.
Human sGC enzymes was expressed in Sf9 cells as detailed previously [23] with minor modifications. In brief, 6 liters of Sf9 cells were harvested 72 hours after being infected with viruses expressing α1 and β1 sGC subunits. For the expression of α1C517Yβ1 sGC, baculovirus expressing α1C517Y variant was used. Cell pellets were lyzed by sonication and 100,000 × g supernatant was applied onto a 50 ml DEAE-FF Sepharose column. sGC-containing fractions were eluted with 350 mM NaCl and loaded on a Ni-agarose column (30 ml bed volume). Following a wash with 40 mM imidazole, sGC was eluted with 200 mM imidazole. sGC-containing fractions were pooled and concentrated on a 10 kDa centrifugal filter concentrator (Millipore, Bedford, MA) and subjected to gel-filtration on Superdex 200 equilibrated with 50 mM triethanolamine pH 7.4, 250 mM NaCl, 1 mM MgCl2 and 5 mM DTT. The sample was supplemented with 25% glycerol and stored at −80°C. To obtain enzyme preps free of exogenously added thiols, purified wild type or αCys517β sGC samples, were passed through a 5 ml Hitrap desalting column (GE Lifesciences) equilibrated with 50 mM triethanolamine pH 7.4, 250 mM NaCl, 1 mM MgCl2. To obtain wild type or mutant sGC enzyme containing ferric heme, the following strategy was applied: thiol-free sGC enzyme preps were treated with increasing amounts of sGC heme oxidizing agent ODQ (0.01–10 μM range) and 1 minute after each addition, the conversion from ferrous sGC (Soret peak at 432 nm) to ferric sGC heme (Soret peak at 393 nm) was recorded spectroscopically using an Agilent 8453 UV-Vis spectrophotometer. The oxidation was considered complete, when additional ODQ did not cause any further changes in recorded UV-vis spectrum. The excess of ODQ was removed by passing the preparations of sGC with oxidized heme through the Hitrap desalting column equilibrated with 50 mM triethanolamine pH 7.4, 250 mM NaCl, 1 mM MgCl2. The fractions containing sGC were confirmed by UV-Vis spectroscopy and used immediately for experiments.
2.3. sGC assay.
To probe the process of heme oxidation of wild type or α1C517Yβ1 sGC by recording the changes in cGMP-forming activity, 0.1 μg aliquots of purified wild type or mutant enzyme were incubated with different concentrations of ODQ for 1 minute at room temperature. After ODQ treatment, the enzyme samples were added to reaction buffer (50 mM triethanolamine pH 7.4, 100 μM EGTA, 2 mM MgCl2) and transferred to 37°C. cGMP-forming activity was initiated by adding 1 mM Mg2+-GTP/0.08 μCi of [α−32P]GTP mix containing water or 10 μM nitric oxide donor DEA-NO. To assay the effect of cinaciguat on sGC activity, 1 μM cinaciguat was added to the enzyme/reaction buffer mix 5 minutes before initiating the assay. To probe for the efficiency of heme reduction, 0.1 μg aliquots of purified ferric wild type or α1C517Yβ1 sGC were transferred to reaction buffer containing different amounts of DTT. After 5 minute incubation at 37°C in the presence of DTT, the activity assay was initiated by adding the Mg2+-GTP/[α−32P]GTP/NO mix. cGMP-forming activity was stopped after 10 minutes by adding zinc acetate and sodium carbonate and processed as described previously [24] to quantify the amount of generated cGMP.
2.4. UV-Vis studies of oxidation and reduction of sGC heme.
To compare the kinetics of heme reduction, ferric wild type or α1C517Yβ1 sGC samples were treated with 2 mM DTT and UV-Vis spectra of sGC were recorded at indicated times until no further changes in spectra were observed.
2.5. Transient transfection of COS-7 cells.
COS-7 cells were grown in DMEM/F12 media with 10% FBS and 100 U/ml penicillin, 100 μg/ml streptomycin in 6-well plates. When cells reached 80% confluency, they were transfected using Lipofectamine LTX with Plus reagent according to manufacturer’s protocol. 2.5 ug of each α1 and β1 plasmid were used per well. In some experiments, 48 hours after transfection, the cells were treated with 1 μM actinomycin D or the combination of 1 μM rotenone and 1 μM antimycin. Transfected cells were collected by trypsinization at indicated times. After collection, the cells were washed once in serum-free DMEM and re-suspended in 60 μl of lysis buffer (50 mM triethanolamine pH 7.4, with protease inhibitors). The cells were disrupted by sonication and the lysates were subjected to centrifugation at 20,000xg and then at 100,000xg. 100,000xg supernatant fractions were used for western blotting against α1 and β1 sGC and tubulin. 20,000xg pellet fractions were resuspended in 60 μl lysis buffer and used for western blotting against mitochondrial aconitase 2.
2.6. To measure sGC activity in COS-7 cell lysates
To measure sGC activity in COS-7 cell lysates the cells were harvested and washed as described above. The combined cell pellet collected from three wells was re-suspended in 250 μl of lysis buffer containing non-specific PDE inhibitors IBMX (1 mM), PDE5 inhibitor sildenafil (10 μM) and PDE9A inhibitor BAY73–6691 (10 μM). After sonication and 20,000xg centrifugation, 40 μl of supernatant was used in a 20 minute sGC assay as described above.
2.7. Western blotting
Western blotting was used to determine the level of α1 and β1 sGC subunits in transfected COS-7 cells. For detection of α1 sGC, β1 sGC and β-tubulin, 100,000xg supernatant fractions, 100 μg of total protein, were treated with Laemmli buffer and separated on 8% SDS-PAGE. For detection of mitochondrial aconitase, 20,000xg pellet fractions, were treated with Laemmli buffer and separated on 8% SDS-PAGE. The proteins were transferred on a PVDF membrane and blocked with 5% milk solution prepared in Tris-buffered saline containing 0.1% Tween 20. α1 sGC was detected by Western blotting using mouse monoclonal 1B3 antibodies raised against purified sGC (1:1000 dilution) [25]. Western blotting against β1 sGC was performed using polyclonal antibodies raised against terminal 15 aminoacids of the human β1 subunit (1:2000 dilution) [26]. Both antibodies were previously validated using recombinant sGC [25]-[26]. The level of mitochondrial aconitase in 20,000xg pellet fractions was evaluated using commercial antibodies (1:1000 dilution, GenTex). The results were visualized by enhanced chemiluminescence (GE Healthcare) using HRP-labeled secondary antibodies (1:8000 dilution) and captured on ChemiDoc MP imaging system (Bio-Rad). β-Tubulin was detected using rhodamine-conjugated anti-tubulin human Fab fragment (1:2000 dilution, BioRad). The amount of β-tubulin or the level of transiently expressed sGC subunits was quantified by densitometry using Image J software (NIH).
2.8. Molecular modeling of the human α1C517Yβ1 sGC.
The structure of mutant α1C517Yβ1 sGC was modeled based on the Cryo-EM structure of NO-stimulated wild type sGC (PDB ID: 6JT2) [27] using the SWISS-MODEL platform [28]. The model generated for α1C517Yβ1 sGC was visualized using the RasTop 2.2 Molecular Visualization Software.
2.9. Statistical Analysis.
Statistical analysis, nonlinear regression, calculation of IC50, EC50 and was performed using GraphPad Prism 5.1 (GraphPad, La Jolla, USA). Km values for GTP were determined from Lineweaver-Burk plots as reported previously [29]. All results are expressed as mean ± SD of at least three independent experiments. Comparisons between two groups were performed using the two tailed student t-test. For comparison of more than two groups, one-way ANOVA (analysis of variance) was performed followed by Tukey’s multi comparison test with Prism 5. Two-way ANOVA followed by Bonferroni’s multi comparison test was used for statistical evaluation of the dose-response curves. The p value <0.05 was considered statistically significant.
3. RESULTS.
3.1. Enzymatic characteristics of α1C517Yβ1 sGC.
Our first goal was to compare the enzymatic properties of recombinant wild type and α1C517Yβ1 sGC enzymes. Corroborating our previous findings [21], purified α1C517Yβ1 exhibited a ~3 fold lower basal specific activity than the wild type enzyme (Fig. 1A and Table 1). Diminished basal activity correlates with lower cGMP-forming activity following activation by NO donor DEA-NO. Although NO-stimulated activity of α1C517Yβ1 was lower, in a standard assay containing 1 mM DTT as thiol reducing agent, the fold stimulation of cGMP-forming activity by NO was statistically similar for α1C517Yβ1 and wild type sGC (Fig. 1A and Table 1). α1C517Yβ1 sGC has a higher Km for GTP substrate, which partially explains the attenuated cGMP-forming activity. The heme-independent sGC activator cinaciguat is known to preferentially target heme-deficient sGC or sGC with oxidized heme [30] and bind in the heme pocket of sGC [31]. The fibrate drug gemfibrozil was also shown to act as a heme-independent sGC activator [24]. Consistent with basal and NO-stimulated data, cinaciguat- and gemfibrozil-stimulated activities of recombinant α1C517Yβ1 sGC were also lower than for the wild type sGC (Fig. 1A and Table 1). However, the fold stimulation by these activators was the same for both enzymes (Fig. 1A).
Figure 1. Comparison of NO, cinaciguat and gemfibrozil response of purified wild type and α1Cys517Tyrβ1 and in COS7 cells.
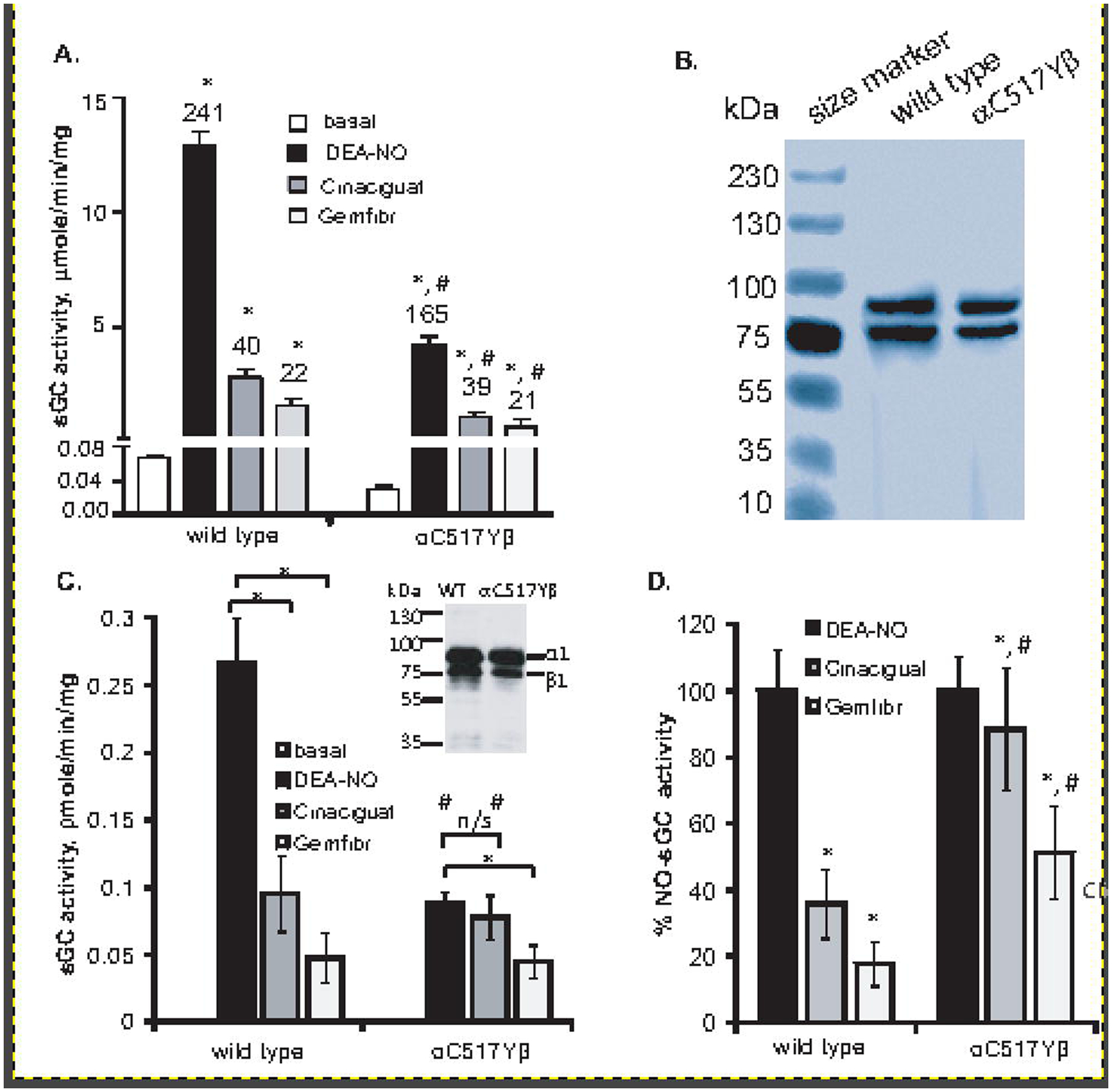
(A): Specific activity of purified wild type and α1Cys517Tyrβ1 in basal conditions and after stimulation with 10 μM DEA-NO, 1 μM Cinaciguat or 100 μM Gemfibrozil (Gemfibr). Numbers above bars indicate the fold stimulation over basal activity. The assay was performed with 1 mM DTT. Data are mean ± SD from three independent experiments performed in triplicate. * indicates p<0.05 vs basal activity (t-test); # indicates p< 0.05 vs. wild type values (one-way ANOVA followed by Tukey’s multi comparison test). (B) Coomassie staining of wild type and α1Cys517Tyrβ1 enzyme preparations. 5 μg purified protein was displayed on SDS-PAGE and stained with Coomassie blue dye. (C): cGMP-forming activity of COS7 cells transfected with plasmids expressing wild type sGC or the rare α1Cys517Tyrβ1 variant. The activity was measured in the presence of 10 μM DEA-NO, 1 μM Cinaciguat or 100 μM Gemfibrozil, without DTT. Data are mean ± SD from measurements performed in triplicates from three independent transfections. * indicates p<0.05 (indicated t-test); # indicates p< 0.05 vs. wild type values (one-way ANOVA followed by Tukey’s multi comparison test). n/s- difference not statistically significant. Inset: expression of α1 and β1 subunits in 100 μg of COS-7 cell protein lysate transfected by wild type or α1C517Yβ1 sGC. (D): cGMP-forming activity data from (C) presented as percent of NO-stimulated activity. * indicates p<0.05 vs NO-stimulated activity (t-test); # indicates p< 0.05 vs. wild type values (one-way ANOVA followed by Tukey’s multi comparison test).
Table 1.
Comparison of key enzymatic parameters of purified wild type and mutant sGC enzymes1.
| Wild type sGC α1β1 | Mutant α1Cys517Tyrβ1 | |||
|---|---|---|---|---|
| +DTT | No thiols assay | + DTT | No thiols assay | |
| Basal cGMP forming activity, μmole cGMP/min/mg | 0.065 ± 0.003 | 0.074 ± 0.005 | 0.026 ± 0.003 | 0.036 ± 0.009 |
| - fold activation | 253 ± 16 | 210 ± 19 | 163 ± 53 | 78.5 ± 18 * |
| - fold activation | 40 | 39 | 38.8 | 47 * |
| - NO-induced | 74.6 ± 6.1# | 68.9 ± 8.3# | 196 ± 9# | 176 ± 14# |
| DEA-NO EC50, nM: | 28 ± 6 | 32 ± 12 | 25 ± 12 | 28 ± 14 |
data are mean ± SD from at least three independent measurements performed in triplicates;
indicates p< 0.05 (Student t-test) vs assay with DTT;
indicates p< 0.05 (Student t-test) vs wild type values;
indicates p< 0.05 (Student t-test) vs basal values;
To compare the properties of the mutant and wild type sGC in cellular environment, COS7 cells were transiently transfected with plasmids expressing mutant or wild type α1 and β1 subunits. Both enzymes showed a comparable level of expression (Fig. 1C, inset). Similar to purified protein, cGMP-forming activity of cinaciguat- and gemfibrozil-stimulated lysates with wild type sGC was several times lower that NO-stimulated activity (Fig. 1A and B), constituting ~35% and 20% of NO-stimulated activity, respectively (Fig. 1D). Corroborating the data with purified protein, cellular α1C517Yβ1 sGC exhibited a lower NO-stimulated activity than cellular wild type sGC (Fig. 1C). However, cinaciguat- and gemfibrozil-stimulated activities of α1C517Yβ1 lysates were much higher relative to NO stimulation, constituting, respectively, 90% and 50% of NO-stimulation (Fig. 1C and D).
We hypothesized that the difference in redox environment of cellular milieu and in the standard sGC assay, which is performed in the presence of 1 mM DTT, may explain the observed different response to cinaciguat for α1C517Yβ1. Therefore, we compared the enzymatic activity, Km for GTP and the apparent NO EC50 for the wild type and mutant sGC, in the absence of DTT (Table 1). We found that in the absence of DTT, mutant sGC was substantially less responsive to stimulation by DEA-NO. In NO stimulated state, the Vmax for α1C517Yβ1 in the absence of DTT was lower (2.85 ± 0.15 μmole cGMP/min/mg) than in the presence of DTT (3.52 ± 0.3 μmole cGMP/min/mg). Moreover, in the absence of DTT, α1C517Yβ1 exhibited a statistically significant change in the fold stimulation by DEA-NO. In the presence of DTT and saturating concentrations of DEA-NO, α1C517Yβ1 exhibited a 163 ± 53 fold stimulation by NO, but in the absence of DTT this value decreased to 75.5 ± 18 fold. The observed difference in the fold stimulation by DEA-NO of the wild type enzyme (211 ±10 vs 146 ± 57 fold) was not statistically significant. Similarly, the response of purified wild type sGC to cinaciguat was not affected by the absence of DTT. In contrast to wild type sGC, α1C517Yβ1 mutant enzyme exhibited higher activity and stronger fold activation by cinaciguat in the absence of DTT. These data demonstrate that redox conditions strongly influence the properties of α1C517Yβ1.
3.2. α1C517Yβ1 is more sensitive to heme oxidation.
Decreased NO activation and increased cinaciguat activation of α1C517Yβ1 in the absence of DTT, lead us to explore the possibility that α1C517Yβ1 is more susceptible to heme oxidation. Therefore, we compared the sensitivity of the wild type and α1C517Y-β1 enzyme preparations to sGC heme oxidizing agent NS2028. Previous studies demonstrated that sGC with oxidized heme does not respond to stimulation by NO [32]. Therefore, we probed the change in the redox status of sGC heme by measuring NO-stimulated cGMP-forming activity in response to different doses of NS2028 in a DTT-free assay. As shown in Fig. 2, mutant α1C517Yβ1 sGC had a 0.037± 0.01 μM IC50 for NS2028, while the wild type sGC had a much higher IC50 for NS2028 (0.28 ±0.07 μM). These data support the notion that the heme of the α1C517Yβ1 mutant is much more sensitive to oxidation than the wild type sGC.
Figure 2. α1Cys517Tyrβ1 mutant is more susceptible to heme oxidation.
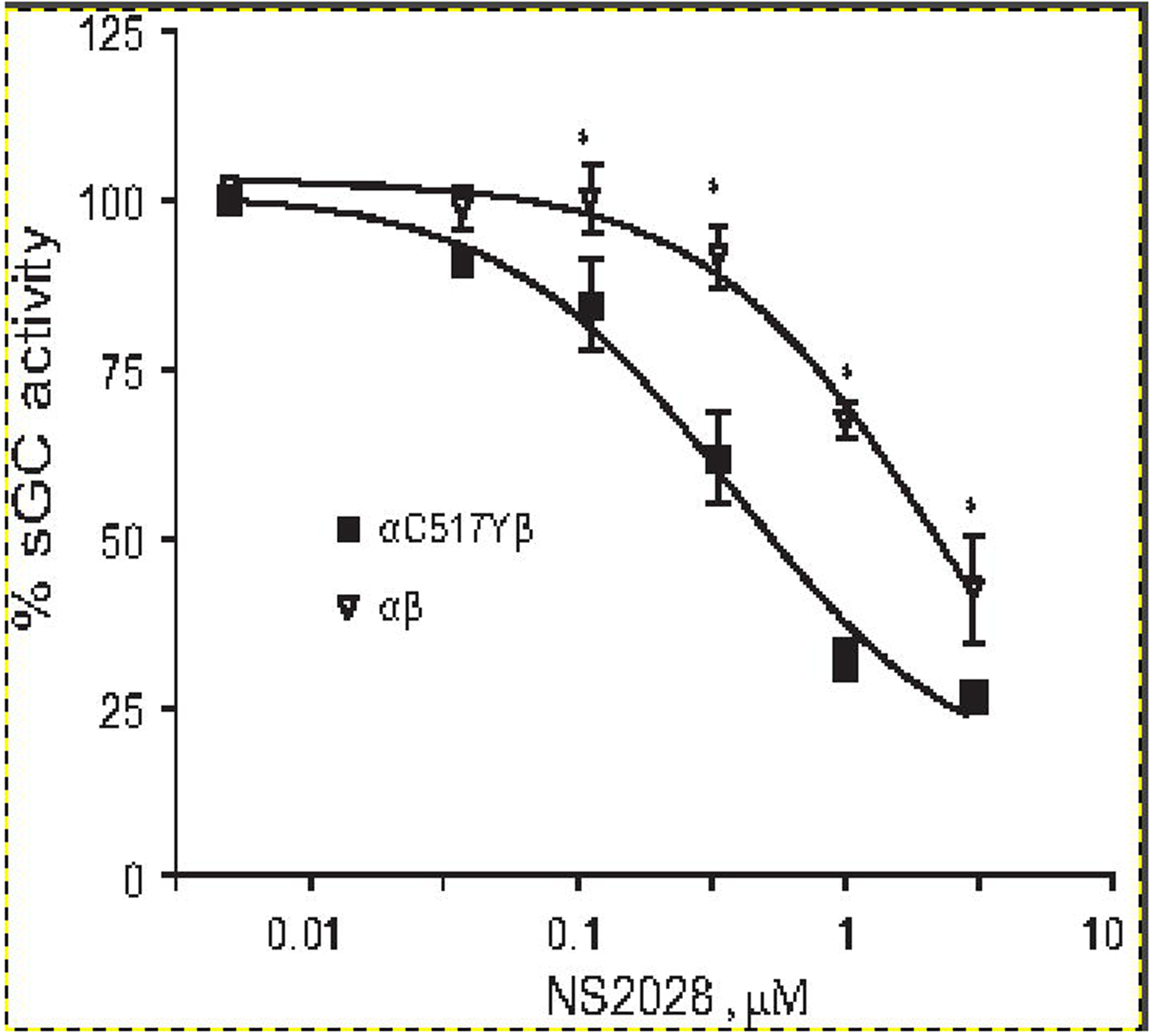
NO stimulated (10 μM DEA-NO) cGMP-forming activity of purified α1Cys517Tyrβ1 (filled squares) and wild type α1β1 sGC (open triangles) pre-treated for 5 minutes with indicated concentrations of heme-oxidizing NS2028 was determined in thiol-free conditions. Data are presented as % of maximal NO-stimulated activity determined in the absence of NS2028. Data are mean ± SD from three independent measurements performed in triplicates. Maximal activity was 16.02±0.32 and 3.2±0.12 μmole cGMP/min/mg for wild type and mutant sGC, respectively. EC50 for NS2028 was estimated at 2.18 ±0.15 μM for wild type sGC and 0.37 ± 0.13 μM for the mutant sGC. * - p<0.05 (two-way ANOVA followed by Bonferroni multi-comparison test).
3.3. α1C517Yβ1 is more resistant to heme reduction by DTT.
We have previously reported that DTT acts as an effective reducing agent for oxidized sGC heme and can restore the responsiveness of sGC with ferric heme to NO stimulation [33]. Similarly, hydrogen sulfide was shown to regulate the redox state of sGC heme [34]. Therefore, we investigated if Cys517→Tyr mutation affected the process of reduction of ferric sGC heme to a functional NO-competent ferrous state. We tested how NO-stimulated activities of the wild type and α1C517Yβ1 sGC are restored by different concentrations of DTT in the assay. As shown in Figure 3, >2 mM DTT almost completely restored NO-dependent activation of the wild type sGC containing ferric heme, corroborating our previous finding [33]. In contrast, NO-stimulated activity of the α1C517Yβ1 enzyme containing ferric heme was restored only by half, even in the presence of 5 mM DTT. These data point to a less efficient DTT-dependent reduction of α1C517Yβ1 heme.
Figure 3. DTT reactivation of ferric sGC.
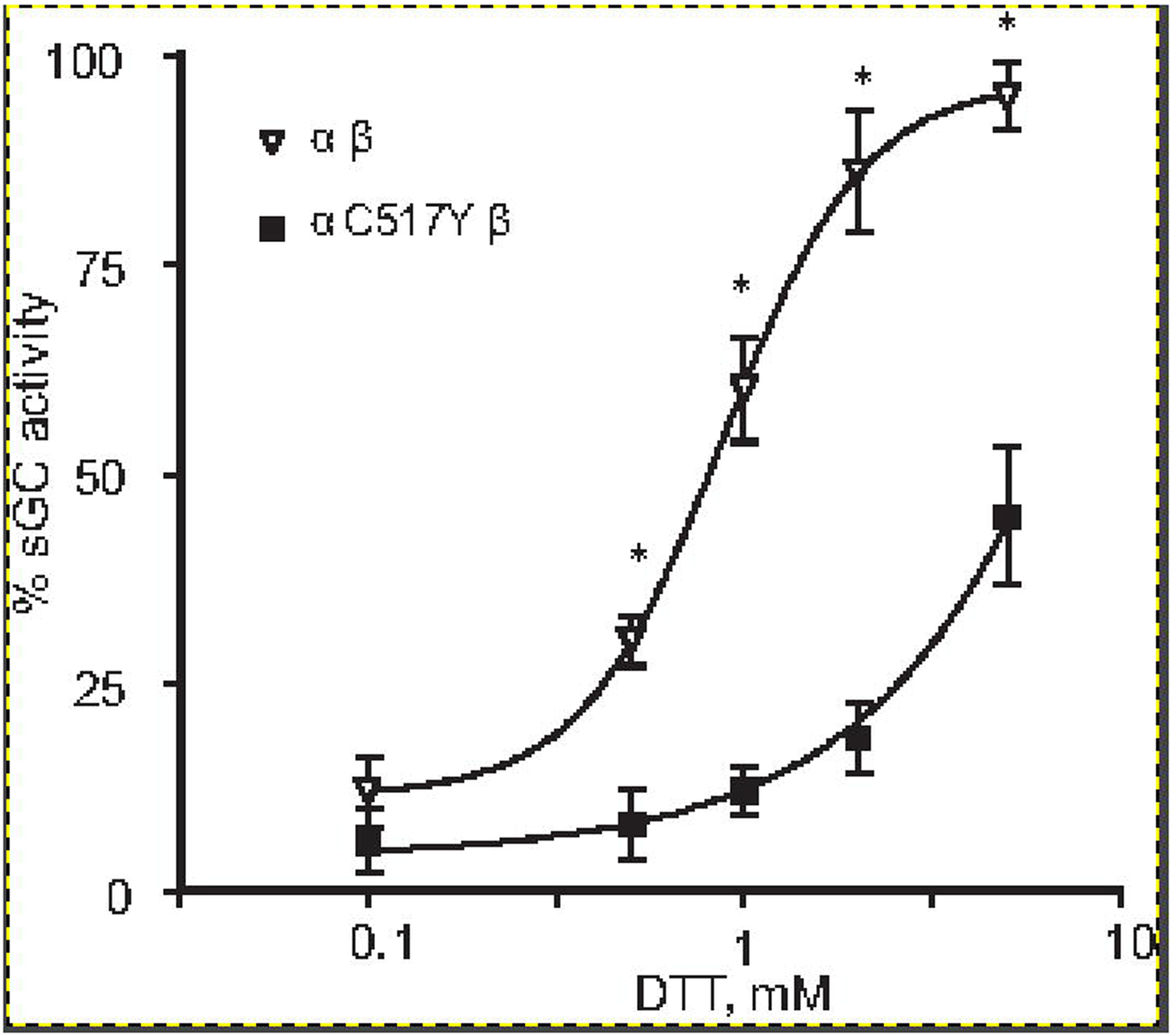
NO stimulated cGMP forming activity of purified α1Cys517Tyrβ1 (filled squares) and wild type α1β1 (open triangles) sGC fully oxidized by10 μM NS2028 was determined in the presence of indicated concentrations of DTT. Data are presented as % of maximal NO-stimulated activity determined with enzymes preps that were not exposed to NS2028. Data are mean ± SD from three independent measurements performed in triplicates. * - p<0.05 (two-way ANOVA followed by Bonferroni multi-comparison test).
To further characterize this feature of α1C517Yβ1 sGC, we compared the kinetics of DTT-dependent reduction of ferric heme for wild type and α1C517Yβ1 sGC. As demonstrated in Figure 4, in both cases 2 mM DTT converted the ferric sGC heme with a Soret peak at 393 nm into ferrous state with the maximum at 431 nm. However, the kinetics of this reduction process was markedly different. The reduction of the wild type sGC was almost entirely completed within first 100 seconds of incubation, exhibiting a half-life for ferric heme of ~22–28 seconds. At the same time, the calculated half-life of ferric α1C517Yβ1 in the presence of 2 mM DTT was almost 5 times longer (~ 112–127 sec). Even after 15 min of incubation, a substantial fraction of α1C517Yβ1 was still in the ferric state. Taken together, these data suggest that α1C517Yβ1 heme is more sensitive to oxidation, yet is more resistant to reduction by thiols.
Figure 4. Kinetics of heme reduction by DTT.
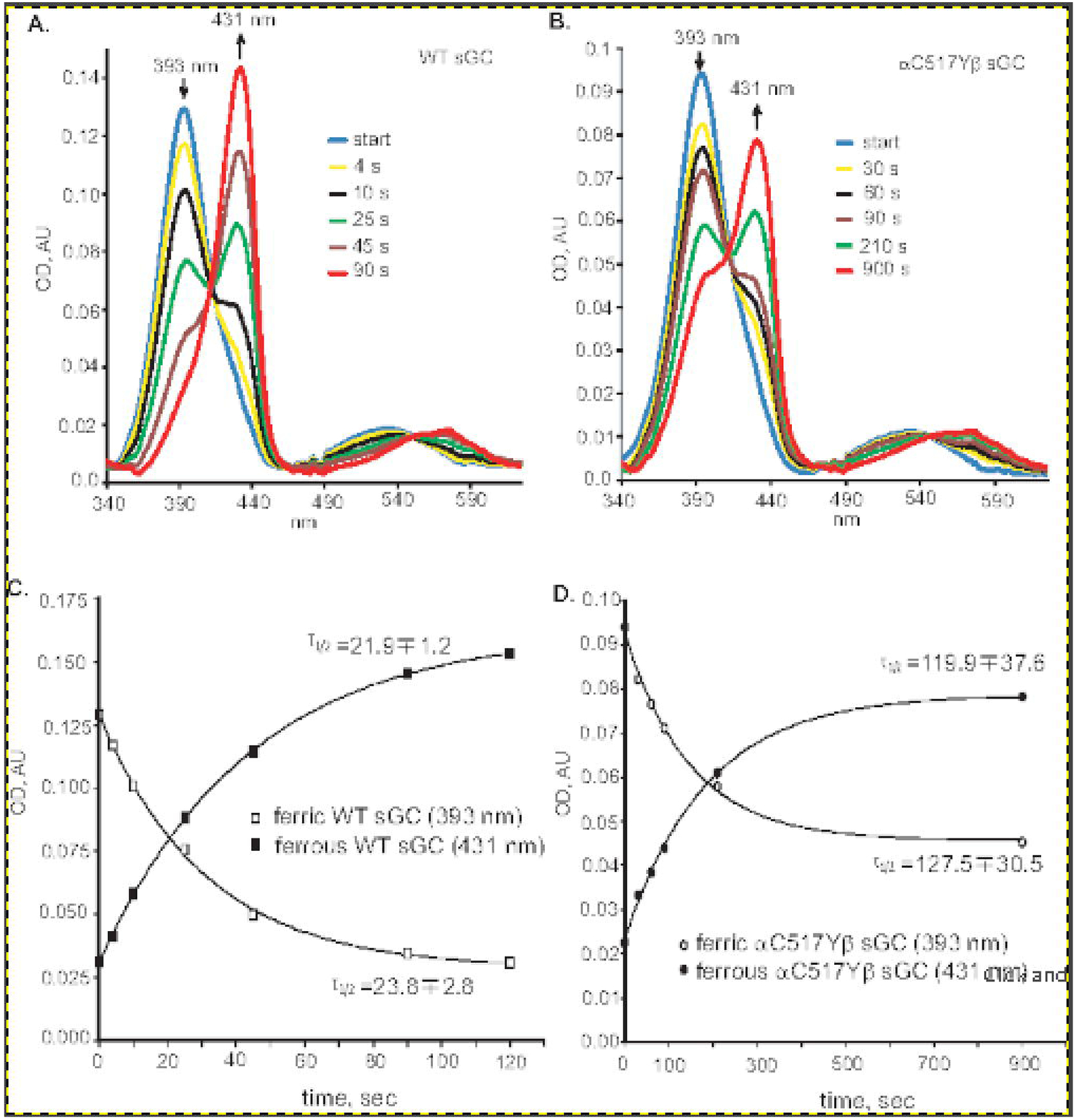
Wild type (A) or α1Cys517Tyrβ1 (B) sGC enzyme preparations containing ferric heme were supplemented with 2 mM DTT and the changes in the UV-Vis spectra were recorded at indicated time points at 25°C in 50 mM triethanolamine pH 7.4, 250 mM NaCl, 1 mM MgCl2. Arrows indicate the position and the direction of absorbance change for ferric and ferrous heme. (C and D): Kinetics of change in the absorbance of ferric (393 nm) and ferrous (430 nm) forms of wild type (C) or α1Cys517Tyrβ1 (D) sGC heme after the addition of DTT were derived from the data presented in (A) and (B). The calculated half-life of each species is indicated.
3.4. α1C517Yβ1 is unstable under cellular oxidative conditions.
Previous reports demonstrated that sGC with ferric heme has the propensity to lose the heme moiety [35]. Moreover, apo-sGC or sGC with ferric heme is more susceptible to degradation via proteasome-dependent mechanism [36]. Therefore, we investigated the stability of α1C517Yβ1 in COS-7 cells by determining the half-life of wild type and mutant α1 and β1 subunits when expressed in COS-7 cells. For this purpose, 48 hours after transfection with sGC expressing plasmids, the cells were treated with 1 μM actinomycin D, an inhibitor of RNA synthesis. By monitoring for 72 hours the changes in the level of α1 and β1 subunits in cells treated with actinomycin D we determine the half-life of α1 and β1 subunits for the wild type and mutant sGC. These data indicate that wild type sGC subunits have a half-life of > 96 hours in COS-7 cells (Fig. 5A and 5C). Under same conditions, the subunits of α1C517Yβ1 sGC were less stable, with the half-life for α1C517Y variant of ~58 hours and for the β1 subunit of ~63 hours (Fig. 5A and 5C). We also compared the stability of wild type and α1C517Yβ1 sGC in conditions of cellular oxidative stress induced by co-treatment with 1 μM rotenone and 1 μM antimycin A. Elevated cellular oxidative stress in response to this treatment was confirmed by the time-dependent decrease of mitochondrial aconitase Aco 2 (Fig. 5B). Aco2 is a widely used marker for oxidative stress, due to its high sensitivity to increased level of cellular superoxide radical and/or hydrogen peroxide [37, 38]. Under conditions of oxidative stress induced by rotenone and antimycin, the stability of wild type subunits slightly decreased, which was clearly observed at 72 hour time point (Fig. 5B and 5D). The estimated half-life for α1 and β1 subunits was ~90 and 80 hours, respectively. The α1C517Yβ1 variant was much less stable under oxidative stress. The estimated half-life for α1C517Y and β1 subunits was 28 and 42 hours, respectively. Moreover, after 72 hours of rotenone/antimycin treatment, only traces of α1C517Y or β1 subunits were observed. These data support that α1C517Yβ1 variant of sGC is unstable and prone to degradation, especially in conditions of cellular oxidative stress.
Figure 5. Diminished stability of α1Cys517Tyrβ1 sGC in COS7 cells.
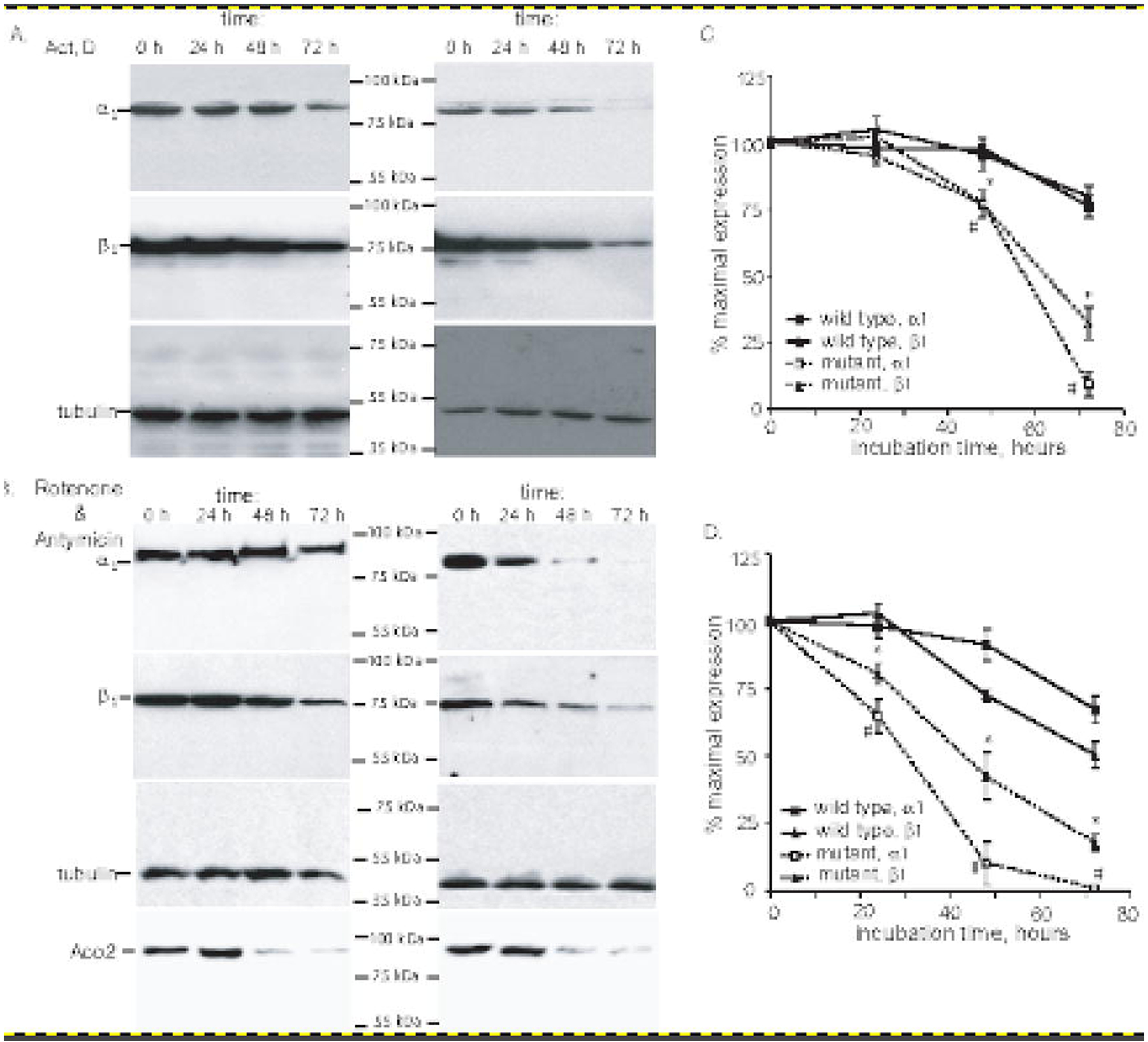
(A-B): COS-7 cells were transfected with plasmids expressing wild type or α1Cys517Tyrβ1 sGC. 48 hours post-transfection, the cells were treated with 1 μM actinomicyn D (A) or 1 μM rotenone and 1 μM antimycin (B). At indicated time points, the cells were lysed and 100,000xg supernatant were used for the detection of α1, β1 and β-tubulin by western blotting. Changes in the level of ROS sensitive mitochondrial aconitase (ACO2) was evaluated in 20,000xg pellets. Shown are representative blots of three independent transfections with similar results. (C-D): Densitometry results of western blots from (A-B) showing changes in the amount of α1 and β1 subunits in COS-7 cells expressing wild type or α1Cys517Tyrβ1 after actinomycin D (A) or rotenone+antimycin (B) treatments. Signals for α1 and β1 subunits are normalized to β-tubulin signal. Data are presented as % of signal obtained in control samples, before the exposure to actinomycin D (A) or rotenone+antimycin (B). Data are mean ± SD from three independent transfections.
To determine if accelerated degradation of α1C517Yβ1 variant is related to the increased heme sensitivity to oxidation, we monitored how sGC activity changes in conditions of increased cellular oxidative stress. We measured the cGMP-forming activity in lysates of COS7 cells exposed to 1 μM rotenone and1 μM antimycin for a different length of time. As expected, during the course of the treatment, total NO- and cinaciguat-stimulated cGMP-forming activity declined over time in both wild type and α1C517Yβ1-expressing cells (Fig. 6A and B). The decline in total sGC activity was consistent with observed decrease in sGC level (Fig. 5 C and D), exhibiting a more rapid decline in cells expressing α1C517Yβ1. To estimate the changes in specific activity of cellular wild type and mutant sGC variant during the treatment, total cGMP-forming activity was normalized to the level of β1 subunit detected by western blotting (Fig. 6 C and D). The dynamics of changes in apparent specific activity in NO-stimulated sample (Fig. 6C) exhibited the same tendency as the total activity (Fig. 5D). However, a marked difference in the apparent specific activity of the wild type α1β1 and the α1C517Yβ1 variant was observed in cinaciguat-treated samples. Wild type sGC exhibited only a modest increase in cinaciguat-responsive activity at 48- and 72-hour time points, respectively, 13 ± 8 % and 16 ± 13% (Fig. 6D). On the contrary, the α1C517Yβ1 variant exhibited a >30% increase in cinaciguat-stimulated activity at 24-hour point and a >250% increase at later time points (Fig. 6D). Similarly, 36 hours after treatment, normalized gemfibrozil-stimulated activity insignificantly increase for the wild type lysates, but was more than 60% higher for lysates containing α1C517Yβ1 variant. These data indicate that in rotenone/antimycin-treated cells the proportion of cellular α1C517Yβ1 with oxidized heme or without heme increases dramatically.
Figure 6. Time-dependent change in sGC activity in rotenone/antimycin-treated cells.
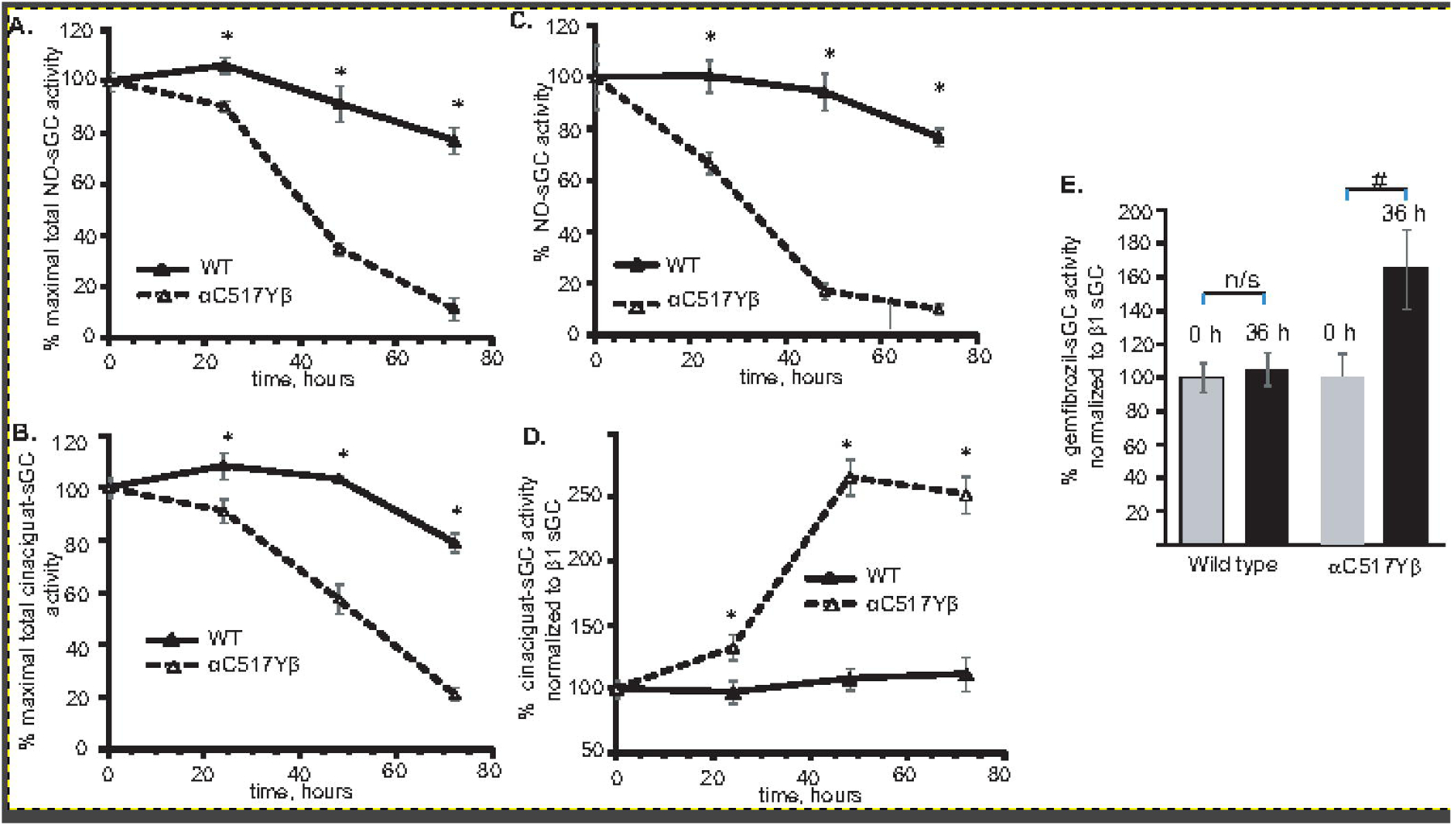
COS-7 cells transiently expressing wild type or α1Cys517Tyrβ1 sGC were treated with 1 μM rotenone and 1 μM antimycin. At indicated times, the cells were lysed and sGC activity in response to 10 μM DEA-NO (A) or 2 μM cinaciguat (B) was measured. Data normalized to total protein lysate are presented as % of cGMP-forming activity in non-treated cells. Data from three independent transfections are presented as mean ± SD. All activity measurements were performed in triplicates. (C-D): Apparent sGC specific activity calculated after normalization to the amount of β1 sGC determined by western blotting from (A-B). (E): Apparent specific activity of sGC stimulated by 100 μM gemfibrozil in control lysates and after 36 hour of rotenone/antymicin treatment. Data in (C-E) are presented as % of mean ± SD of controls. Data from three independent transfections are shown. All measurements were performed in triplicates. * indicates p<0.05, two-way ANOVA followed by Bonferroni multi-comparison test; # indicates p< 0.05 vs. control untreated sample (t-test); n/s – the difference is not statistically significant.
4. DISCUSSION.
Over the last half century, multiple pharmacological and in vivo studies demonstrated the importance of NO/cGMP signaling in cardiovascular physiology [39]. The development of high-throughput DNA sequencing opened the door for multicenter genome-wide association studies (GWAS) and studies of families with inherited cardiovascular problems. GWAS and familial genetic studies identified a number of mutations of GUCY1A3 gene, which codes for the α1 sGC subunit. Besides the common single nucleotide polymorphism locus in the non-coding region of GUCY1A3 gene [40], a number of rare point mutations have been identified in the GUCY1A3 gene. Most of the identified point missense mutations are associated with increased susceptibility to coronary artery disease (CAD) or myocardial infarction (MI) [19, 41]. These association stems from the partial depression of sGC expression levels and resulting decrease of cellular cGMP response to NO stimulation [42].
A number of identified rare variants of GUCY1A3 gene carries nonsense mutations or frameshift mutation resulting in early termination. These variants are predicted to result in nonfunctional α1 subunit and a sGC heterodimer incapable of synthesizing cGMP [19–21]. Most of these mutations have been identified in two familial studies of patients with moyamoya-like syndrome [20, 21]. These patients displayed a plethora of pathologic conditions, which included carotid artery stenosis, cerebral angiopathy, severe hypertension and problems with esophageal motility and swallowing. A recent report also described a patient with progressive moyamoya-like symptoms and achalasia, whose exome sequencing identified an undisclosed mutation in GUCY1A3 [43]. The mice deficient in sGC recapitulate many of the features exhibited by patients in these two studies, e.g. development of hypertension and affected motility of the smooth muscles in the GI tract [9, 10].
Interestingly, similar moyamoya-like syndrome, hypertension and achalasia have been described in homozygote carriers of Cys517→Tyr missense mutation of the α1 subunit [21]. Biochemical studies presented in this manuscript clearly indicate that α1C517Yβ1 sGC retained the ability to synthesize cGMP. However, under basal conditions or upon activation by NO or cinaciguat, the α1C517Yβ1 variant consistently exhibited a Vmax value that was 3–5 times lower than the wild type enzyme (Table 1). Another noticeable difference between wild type and α1C517Yβ1 sGC was the increased GTP-Km value for both basal and NO-stimulated conditions. Recently reported cryo-EM structure of NO-stimulated sGC [27] provides some structural cue to the changes caused by the Cys517→Tyr substitution. Modeling of the α1C517Yβ1 catalytic region based on structure of the wild type enzyme (Fig. 7) suggests that the much bulkier sidechain of the Tyr517 may come in close contact with the juxtaposed Tyr532 (Fig. 7C vs 7D). The close contact between Tyr517 and Tyr532 most likely create a strain and possible clash between the side chains of these two Tyr residues. As the model suggests, this results in some distancing of the alpha helixes containing them. As a result, the model predicts a shift in the position of Asp530 which directly interact with GTP substrate (Fig. 7A and B). Suboptimal positioning of the residue crucial for GTP-protein interaction is the probable structural cause for decreased GTP Km and diminished Vmax. Interestingly, a previous study reported that nitrosylation of rat α1 Cys 516 residue, which is homologous to Cys517 in humans, also impairs sGC catalytic properties [44]. Perhaps, the addition of the nitrosyl group has similar structural consequences, resulting in diminished cGMP catalysis. At the same time, under supporting redox conditions, the response of α1C517Yβ1 to NO stimulation does not seem to be significantly impaired, as attested by similar values of DEA-NO EC50 and comparable NO fold stimulation for wild type and α1C517Yβ1 sGC (Table 1). Considering that under normal conditions estimated intracellular concentration of GTP is ~100 μM [45, 46], and taking into consideration the GTP-Km and Vmax values of NO-stimulated α1C517Yβ1, the cells expressing α1C517Yβ1 should maintain at least a 25% rate of cGMP synthesis of cells expressing wild type sGC, provided that NO stimulation is similar.
Figure 7. Molecular modeling of changes in the α1 sGC catalytic domain induced by Cys517→Tyr substitution.
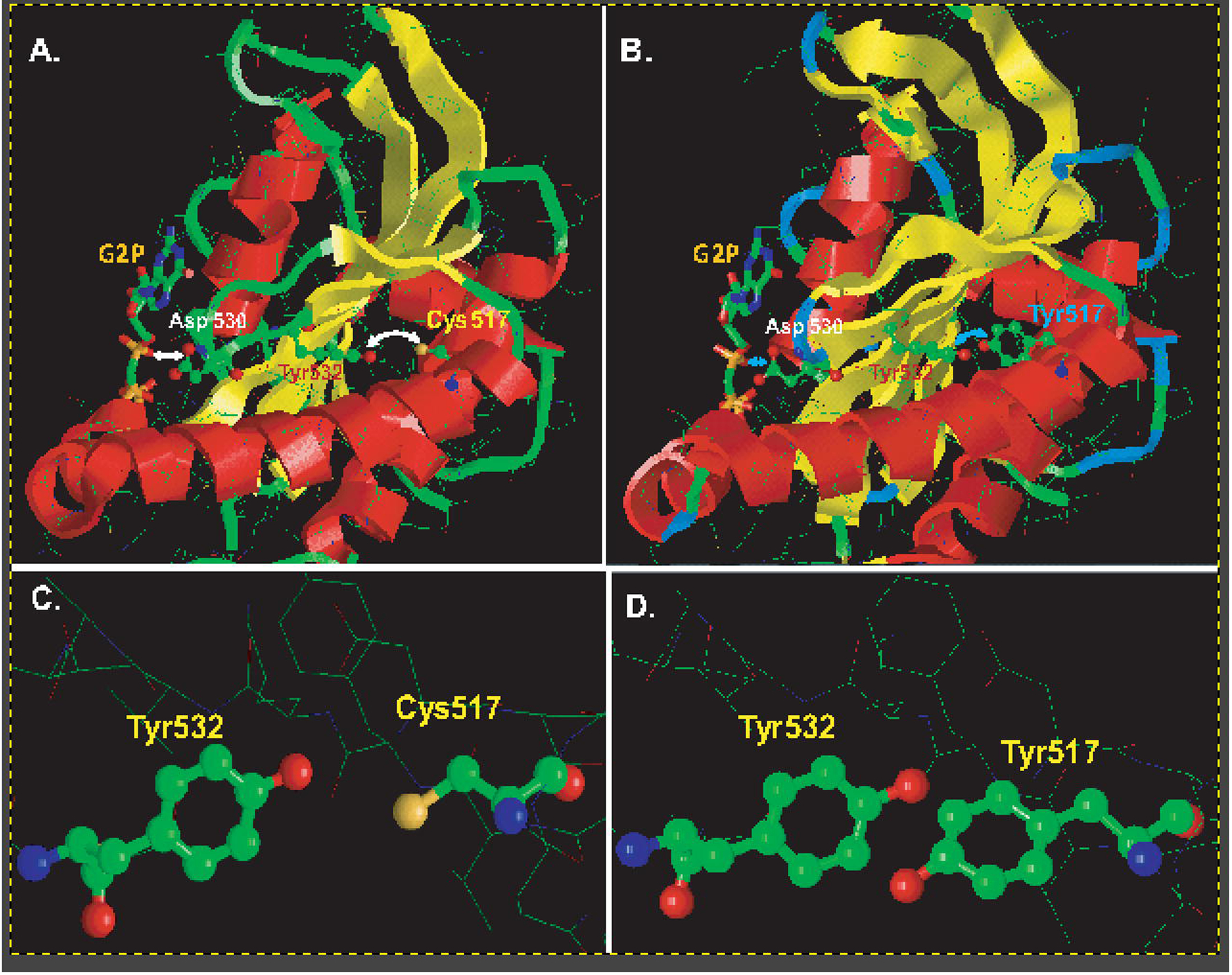
(A): Close up view of the α1 catalytic region of wild type sGC based on the cryo-EM structure of NO-stimulated human sGC (PDB 6JT2). The residues Cys517, Tyr532, Asp530 and the GTP analog are highlighted. (B): Close up view of the α1 catalytic region of the α1Cys517Tyrβ mutant. The model was generated using the SWISS-MODEL platform based on PDB 6JT2. The residues Tyr517, Tyr532, Asp530 and the GTP analog G2P (phosphomethylphosphonic acid guanylate ester) are highlighted. For simplicity of viewing, the catalytic region of the β1 subunit is omitted in both (A) and (B). The arrows highlight the cross-talk between residue 517 and Tyr532 and the associated distance between residues Asp530 and the GTP substrate analog. (C-D): Close up view of residues 517 and Tyr532 shows that in wild type sGC (C) Cys517 and Tyr 532 are more remote than the juxtaposed Tyr517 and Tyr532 in α1Cys517Tyrβ mutant (D).
The proper functioning of NO/sGC/cGMP signaling in vascular system is protected by its redundancy. In mouse aorta, 94% of sGC contains α1 subunit and only 6% of sGC contains α2 subunit [22]. It should be noted that recombinant GC-2 is less active than the predominant GC-1 sGC [47, 48]. Nevertheless, studies of transgenic mice completely lacking the α1 subunit demonstrated that the remaining amount of α2 sGC is sufficient to mediate proper vasoreactivity in response to NO [22]. This report clearly demonstrated that retaining even less that 6% of normal NO-sGC mediated synthesis of cGMP is sufficient to maintain proper vascular function in mice under normal conditions. Therefore, carriers of α1C517Yβ1, which retains much higher cGMP-forming activity, should exhibit a less severe phenotype. The expected phenotype should be closer to the mutant sGC variants with predisposition to MI or CAD, rather than the severe phenotype of carriers of catalytically inactive sGC.
In addition to depressed catalytic properties of the α1C517Yβ1 sGC mutant, current studies revealed that α1C517Yβ1 sGC function is strongly affected by redox conditions and redox agents, i.e. DTT. This conclusion is supported by findings that α1C517Yβ1 exhibits diminished NO-stimulated specific activity, while relative activation by cinaciguat is increased (Table 1). Ferrous heme proteins that bind NO, such as hemoglobin, are easily converted into a ferric state in the presence of oxygen [49]. The function of wild type sGC as high sensitivity and affinity receptor for NO is strongly dependent on its ability to maintain ferrous heme moiety, even when the NO-heme adduct is formed in aerobic conditions [50]. While the exact mechanism of this process is not entirely clear, it is most likely related to the unique ability of sGC heme to exclude oxygen as heme ligand [51]. The combination of enzymatic and spectroscopic measurements presented in current studies demonstrate that α1C517Yβ1 sGC heme is more susceptible to oxidation. That is clearly demonstrated by the lower IC50 for sGC heme-oxidizing agent NS2028 (Fig. 2). Moreover, the restoration of the functional ferrous status of α1C517Yβ1 heme from the NO-insensitive ferric state by reducing agent DTT is significantly impaired, as compared to the wild type sGC (Fig. 3 and 4). Although the exact mechanism of this impairment needs further investigation, it is reasonable to assume that structural changes in the catalytic domain caused by the Cys517→Tyr substitution also affected the function of the heme-binding HNOX domain of the β1 subunit of sGC. A substantial number of studies demonstrate that events in the catalytic domain of sGC may affect the properties of the heme domain. E.g., CO-sGC heme adduct, which is 6-coordinate, becomes a mixture of 5- and 6-coordinate in the presence of stimulators YC-1, but only when GTP binds to the catalytic domain, the heme-coordinating bond with His 105 residue is fully cleaved [52]. Similarly, the stability of NO-sGC heme complex is significantly affected by the presence of GTP substrate [53–55]. Structural information derived from single particle and cryo-EM studies of sGC structure [27, 56] provide strong evidence that in basal condition sGC’s catalytic domain and the heme-binding region are in close proximity to each other. Such proximity may provide the conduit needed for the cross-talk between the catalytic and heme-binding domains observed for α1C517Yβ1 sGC.
The function of sGC as highly sensitive NO receptor is highly susceptible to changes in cellular oxidative conditions due to the effects of various reactive oxygen species. E.g., earlier studies demonstrated that exposure of sGC from platelet lysates to superoxide anion [57] or of sGC from isolated aorta to peroxynitrite [58] significantly impairs sGC stimulation by NO. Multiple studies demonstrated that oxidation or loss of sGC heme caused by pharmacological agents, pathological conditions or reactive species [26, 59–61] undergoes proteosomal degradation [36, 61]. Therefore, higher sensitivity of α1C517Yβ1 to heme oxidizing agents and its impaired ability to efficiently restore the ferrous status of sGC heme strongly suggest that in conditions of oxidative stress, a substantial portion of this rare α1C517Yβ1 sGC variant may be NO-insensitive. Previous studies demonstrated that sGC with oxidized heme easily loses the heme moiety [35]. Therefore, we compared the stability of the wild type and α1C517Yβ1 sGC in COS7 cells under normal and oxidative stress conditions. Even under normal conditions, when de novo synthesis of sGC was inhibited, the stability of α1C517Yβ1 was diminished as compared with the wild type sGC (Fig. 4 A). The protein instability was even more evident, when the production of oxidative species in transfected cells was stimulated by inhibiting the mitochondrial electron transfer chain using a combination of rotenone and antimycin A (Fig. 4B). Mitochondrial aconitase is a good marker for cellular oxidative stress, since its activity is highly susceptible to various ROS and is quickly inactivated in response to elevated level of superoxide anion or hydrogen peroxide [38]. Once inactivated, mitochondrial aconitase is subject to ATP-dependent degradation by Lon protease [37]. Decreased level of aconitase 2 in the course of the treatment (Fig. 5B) clearly demonstrated that the combination of rotenone and antimycin A resulted in elevated cellular oxidative stress. Although in these set of experiments de novo protein synthesis was not inhibited, α1C517Yβ1 exhibited a significantly more rapid degradation. The dynamics of α1C517Yβ1 degradation coincided with significant increase in the apparent specific activity of α1C517Yβ1 in response to cinaciguat or gemfibrozil stimulation (Fig. 6D, E). The wild type enzyme had only a marginal increase in response to these agents. Since cinaciguat and gemfibrozil preferentially targets heme-less sGC or sGC with oxidized heme [62], our data prove elevated oxidation and loss of α1C517Yβ1 heme under cellular oxidative conditions, resulting in rapid degradation of α1C517Yβ1 sGC variant. Recent studies demonstrated that cytochrome B5 reductase 3 (cytB5R3) plays a role in maintaining sGC heme in a reduced state [63]. Additional studies are needed to determine if conformational changes caused by the Cys517→Tyr mutation interferes with protein interaction or makes the mutant less susceptible to heme-reductase function of cytB5R3.
As discussed above, the modeling of α1C517Yβ1 catalytic domain predicted a juxtaposition of Tyr 532 and the substituted Tyr517 (Fig. 7D). Multiple studies demonstrated that under oxidative conditions similar positioning in other proteins resulted in cross-linking of such tyrosines and the creation of dityrosine protein modification [64, 65]. Dityrosine modification has been shown to impair the function of modified proteins and to be a strong marker for selective degradation by proteasomes [66]. Although our efforts to detect dityrosine modification in recombinant α1C517Yβ1 exposed to different oxidative species were not successful (data not shown), it remains a possibility that such modification of α1C517Yβ1 may occur in conditions of elevated ROS or RNS. Nitrosylation of several cysteine residue of sGC, including α1Cys517, contribute to desensitization of sGC [44, 67–71] and may play a crucial role in thiol-based regulation of sGC function [72, 73]. It remains to be determined how these processes affect the stability of α1C517Yβ1.
In summary, we demonstrate that the deficiency of cGMP-forming activity of the rare α1C517Yβ1 variant is aggravated by a substantially enhanced rate of sGC heme oxidation and significant impairment of its reduction. This combination facilitates the degradation of α1C517Yβ1 protein during cellular oxidative stress, resulting in a considerably depressed NO-dependent cGMP synthesis. These data indicate that carriers of α1C517Yβ1 variant may rapidly loose sGC function following inflammatory response or unbalanced cellular redox homeostasis, which may contribute to the severity of vascular and gastrointestinal tract dysfunctions observed in these patients.
The function of sGC is currently modulated in clinic by NO-replacing nitrovasodilators, or heme-dependent sGC simulators, such as riociguat, vericiguat [74, 75]. These agents are being used to upregulate sGC function for the management of angina, pulmonary hypertension or heart failure [76]. However, these heme-dependent sGC activators may not be effective in supporting sGC function in patients carrying the α1C517Yβ1 sGC variant, due to an expected high proportion of heme-less or ferric sGC. However, these patients may benefit from NO-independent activators that stimulate sGC regardless of heme status. E.g., ataciguat and cinaciguat have been developed as sGC activators that primarily targets apo-sGC or sGC with ferric heme [30, 77]. Although these agents were not successful in clinical trials for the treatment of heart failure [78], it is highly relevant that they protect apo- and ferric sGC from degradation [36, 79] The anti-hyperlipidemic drug gemfibrozil, which is used in for the treatment of type IV hyperlipidemias, was also identified as a heme-independent sGC activator with vasoactive effect [24] and may have some potential off-label application. Biochemical characterization of the rare α1C517Yβ1 sGC presented in this report should help with devising some potential treatment of α1C517Yβ1 carriers that will offset the progression or alleviate the severity of developing symptoms and improve their quality of life.
ACKNOWLEDGMENT.
This work was supported by RO1 HL139838 (E.M. and I.S.) from the National Institutes of Health and Nazarbayev University travel grants (K.L. and Y.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES.
None.
CITED LITERATURE:
- 1.Roy B and Garthwaite J, Nitric oxide activation of guanylyl cyclase in cells revisited. Proc Natl Acad Sci U S A, 2006. 103(32): p. 12185–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Batchelor AM, et al. , Exquisite sensitivity to subsecond, picomolar nitric oxide transients conferred on cells by guanylyl cyclase-coupled receptors. Proc Natl Acad Sci U S A, 2010. 107(51): p. 22060–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noiri E, et al. , Permissive role of nitric oxide in endothelin-induced migration of endothelial cells. J Biol Chem, 1997. 272(3): p. 1747–52. [DOI] [PubMed] [Google Scholar]
- 4.Noiri E, et al. , Podokinesis in endothelial cell migration: role of nitric oxide. Am J Physiol, 1998. 274(1 Pt 1): p. C236–44. [DOI] [PubMed] [Google Scholar]
- 5.Kubes P, Suzuki M, and Granger DN, Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci U S A, 1991. 88(11): p. 4651–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Radomski MW, Palmer RM, and Moncada S, Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet, 1987. 2(8567): p. 1057–8. [DOI] [PubMed] [Google Scholar]
- 7.Seki J, et al. , FK409, a new nitric-oxide donor, suppresses smooth muscle proliferation in the rat model of balloon angioplasty. Atherosclerosis, 1995. 117(1): p. 97–106. [DOI] [PubMed] [Google Scholar]
- 8.Stasch JP, Pacher P, and Evgenov OV, Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation, 2011. 123(20): p. 2263–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cosyns SM, et al. , Heme deficiency of soluble guanylate cyclase induces gastroparesis. Neurogastroenterol Motil, 2013. 25(5): p. e339–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friebe A, et al. , Fatal gastrointestinal obstruction and hypertension in mice lacking nitric oxide-sensitive guanylyl cyclase. Proc Natl Acad Sci U S A, 2007. 104(18): p. 7699–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garthwaite J, Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci, 2008. 27(11): p. 2783–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vita JA, Endothelial function. Circulation, 2011. 124(25): p. e906–12. [DOI] [PubMed] [Google Scholar]
- 13.Xu J and Zou MH, Molecular insights and therapeutic targets for diabetic endothelial dysfunction. Circulation, 2009. 120(13): p. 1266–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Groneberg D, et al. , Smooth muscle-specific deletion of nitric oxide-sensitive guanylyl cyclase is sufficient to induce hypertension in mice. Circulation, 2010. 121(3): p. 401–9. [DOI] [PubMed] [Google Scholar]
- 15.Decaluwe K, et al. , Erectile Dysfunction in Heme-Deficient Nitric Oxide-Unresponsive Soluble Guanylate Cyclase Knock-In Mice. J Sex Med, 2017. 14(2): p. 196–204. [DOI] [PubMed] [Google Scholar]
- 16.Thoonen R, et al. , Cardiovascular and pharmacological implications of haem-deficient NO-unresponsive soluble guanylate cyclase knock-in mice. Nat Commun, 2015. 6: p. 8482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ehret GB, et al. , Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature, 2011. 478(7367): p. 103–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu X, et al. , Genome-wide association study in Han Chinese identifies four new susceptibility loci for coronary artery disease. Nat Genet, 2012. 44(8): p. 890–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Erdmann J, et al. , Dysfunctional nitric oxide signalling increases risk of myocardial infarction. Nature, 2013. 504(7480): p. 432–6. [DOI] [PubMed] [Google Scholar]
- 20.Herve D, et al. , Loss of alpha1beta1 soluble guanylate cyclase, the major nitric oxide receptor, leads to moyamoya and achalasia. Am J Hum Genet, 2014. 94(3): p. 385–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wallace S, et al. , Disrupted Nitric Oxide Signaling due to GUCY1A3 Mutations Increases Risk for Moyamoya Disease, Achalasia and Hypertension. Clinicla Genetics, 2016. 90(4):: p. 351–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mergia E, et al. , Spare guanylyl cyclase NO receptors ensure high NO sensitivity in the vascular system. J Clin Invest, 2006. 116(6): p. 1731–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharina I, et al. , Cobinamides are novel co-activators of NO receptor which target sGC catalytic domain. J Pharmacol Exp Ther, 2011. 340(3): p. 723–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharina IG, et al. , The fibrate gemfibrozil is an NO – and heme-independent activator of soluble guanylyl cyclase: in vitro studies. Br J Pharmacol, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin E, et al. , Alternative splicing impairs sGC function in aortic aneurysm. . Am J Physiol Heart Circ Physiol 2014. 307(11): p. H1565–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharina IG, et al. , Alpha1 soluble guanylyl cyclase (sGC) splice forms as potential regulators of human sGC activity. J Biol Chem, 2008. 283(22): p. 15104–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang Y, et al. , Structural insights into the mechanism of human soluble guanylate cyclase. Nature, 2019. 574(7777): p. 206–210. [DOI] [PubMed] [Google Scholar]
- 28.Waterhouse A, et al. , SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res, 2018. 46(W1): p. W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin E, et al. , A constitutively activated mutant of human soluble guanylyl cyclase (sGC): implication for the mechanism of sGC activation. Proc Natl Acad Sci U S A, 2003. 100(16): p. 9208–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmidt HH, Schmidt PM, and Stasch JP, NO- and haem-independent soluble guanylate cyclase activators. Handb Exp Pharmacol, 2009(191): p. 309–39. [DOI] [PubMed] [Google Scholar]
- 31.Martin F, et al. , Structure of cinaciguat (BAY 58–2667) bound to Nostoc H-NOX domain reveals insights into heme-mimetic activation of the soluble guanylyl cyclase. J Biol Chem, 2010. 285(29): p. 22651–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garthwaite J, et al. , Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol Pharmacol, 1995. 48(2): p. 184–8. [PubMed] [Google Scholar]
- 33.Martin E, Lee YC, and Murad F, YC-1 activation of human soluble guanylyl cyclase has both heme-dependent and heme-independent components. Proc Natl Acad Sci U S A, 2001. 98(23): p. 12938–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Z, et al. , Regulation of soluble guanylyl cyclase redox state by hydrogen sulfide. Pharmacol Res, 2016. 111: p. 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fritz BG, et al. , Oxidation and loss of heme in soluble guanylyl cyclase from Manduca sexta. Biochemistry, 2011. 50(26): p. 5813–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meurer S, et al. , Nitric oxide-independent vasodilator rescues heme-oxidized soluble guanylate cyclase from proteasomal degradation. Circ Res, 2009. 105(1): p. 33–41. [DOI] [PubMed] [Google Scholar]
- 37.Bota DA and Davies KJ, Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat Cell Biol, 2002. 4(9): p. 674–80. [DOI] [PubMed] [Google Scholar]
- 38.Tretter L and Ambrus A, Measurement of ROS homeostasis in isolated mitochondria. Methods Enzymol, 2014. 547: p. 199–223. [DOI] [PubMed] [Google Scholar]
- 39.Murad F, Shattuck Lecture. Nitric oxide and cyclic GMP in cell signaling and drug development. N Engl J Med, 2006. 355(19): p. 2003–11. [DOI] [PubMed] [Google Scholar]
- 40.Consortium CAD, et al. , Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet, 2013. 45(1): p. 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Do R, et al. , Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature, 2015. 518(7537): p. 102–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wobst J, et al. , Stimulators of the soluble guanylyl cyclase: promising functional insights from rare coding atherosclerosis-related GUCY1A3 variants. Basic Res Cardiol, 2016. 111(4): p. 51. [DOI] [PubMed] [Google Scholar]
- 43.Ramesh V and Sankar J, Moyamoya Disease 6 with Achalasia Due to GUCY1A3 Mutation in a Child. Neurol India, 2020. 68(5): p. 1253–1254. [DOI] [PubMed] [Google Scholar]
- 44.Crassous PA, et al. , Soluble guanylyl cyclase is a target of angiotensin II-induced nitrosative stress in a hypertensive rat model. Am J Physiol Heart Circ Physiol, 2012. 303(5): p. H597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hatakeyama K, Harada T, and Kagamiyama H, IMP dehydrogenase inhibitors reduce intracellular tetrahydrobiopterin levels through reduction of intracellular GTP levels. Indications of the regulation of GTP cyclohydrolase I activity by restriction of GTP availability in the cells. J Biol Chem, 1992. 267(29): p. 20734–9. [PubMed] [Google Scholar]
- 46.Otero AD, Transphosphorylation and G protein activation. Biochem Pharmacol, 1990. 39(9): p. 1399–404. [DOI] [PubMed] [Google Scholar]
- 47.Koglin M and Behrends S, Cloning and functional expression of the rat alpha(2) subunit of soluble guanylyl cyclase. Biochim Biophys Acta, 2000. 1494(3): p. 286–9. [DOI] [PubMed] [Google Scholar]
- 48.Sharina I, et al. , Cobinamides are novel coactivators of nitric oxide receptor that target soluble guanylyl cyclase catalytic domain. J Pharmacol Exp Ther, 2012. 340(3): p. 723–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Helms CC, Gladwin MT, and Kim-Shapiro DB, Erythrocytes and Vascular Function: Oxygen and Nitric Oxide. Front Physiol, 2018. 9: p. 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martin E, et al. , Soluble Guanylyl Cyclase: The Nitric Oxide Receptor. Methods Enzymol, 2005. 396PE: p. 478–492. [DOI] [PubMed] [Google Scholar]
- 51.Tsai AL, et al. , A “Sliding Scale Rule” for Selectivity among NO, CO, and O(2) by Heme Protein Sensors. Biochemistry, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Z, et al. , Resonance Raman Evidence for the Presence of Two Heme Pocket Conformations with Varied Activities in CO-Bound Bovine Soluble Guanylate Cyclase and Their Conversion. Biochemistry, 2005. 44(3): p. 939–46. [DOI] [PubMed] [Google Scholar]
- 53.Cary SP, Winger JA, and Marletta MA, Tonic and acute nitric oxide signaling through soluble guanylate cyclase is mediated by nonheme nitric oxide, ATP, and GTP. Proc Natl Acad Sci U S A, 2005. 102(37): p. 13064–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kharitonov VG, et al. , Dissociation of nitric oxide from soluble guanylate cyclase. Biochem Biophys Res Commun, 1997. 239(1): p. 284–6. [DOI] [PubMed] [Google Scholar]
- 55.Tsai AL, et al. , Dynamic Ligand Exchange in Soluble Guanylyl Cyclase (sGC): IMPLICATIONS FOR sGC REGULATION AND DESENSITIZATION. J Biol Chem, 2011. 286(50): p. 43182–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Campbell MG, et al. , Single-particle EM reveals the higher-order domain architecture of soluble guanylate cyclase. Proc Natl Acad Sci U S A, 2014. 111(8): p. 2960–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brune B, Schmidt KU, and Ullrich V, Activation of soluble guanylate cyclase by carbon monoxide and inhibition by superoxide anion. Eur J Biochem, 1990. 192(3): p. 683–8. [DOI] [PubMed] [Google Scholar]
- 58.Weber M, et al. , The effect of peroxynitrite on the catalytic activity of soluble guanylyl cyclase. Free Radic Biol Med, 2001. 31(11): p. 1360–7. [DOI] [PubMed] [Google Scholar]
- 59.Sharina IG and Martin E, The Role of Reactive Oxygen and Nitrogen Species in the Expression and Splicing of Nitric Oxide Receptor. Antioxid Redox Signal, 2017. 26(3): p. 122–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stasch JP, et al. , Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest, 2006. 116(9): p. 2552–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gerassimou C, et al. , Regulation of the expression of soluble guanylyl cyclase by reactive oxygen species. Br J Pharmacol, 2007. 150(8): p. 1084–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stasch JP, et al. , NO- and haem-independent activation of soluble guanylyl cyclase: molecular basis and cardiovascular implications of a new pharmacological principle. Br J Pharmacol, 2002. 136(5): p. 773–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rahaman MM, et al. , Cytochrome b5 Reductase 3 Modulates Soluble Guanylate Cyclase Redox State and cGMP Signaling. Circ Res, 2017. 121(2): p. 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Malencik DA and Anderson SR, Dityrosine as a product of oxidative stress and fluorescent probe. Amino Acids, 2003. 25(3–4): p. 233–47. [DOI] [PubMed] [Google Scholar]
- 65.Feeney MB and Schoneich C, Tyrosine modifications in aging. Antioxid Redox Signal, 2012. 17(11): p. 1571–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giulivi C and Davies KJ, Dityrosine and tyrosine oxidation products are endogenous markers for the selective proteolysis of oxidatively modified red blood cell hemoglobin by (the 19 S) proteasome. J Biol Chem, 1993. 268(12): p. 8752–9. [PubMed] [Google Scholar]
- 67.Fernhoff NB, et al. , Heme-assisted S-nitrosation desensitizes ferric soluble guanylate cyclase to nitric oxide. J Biol Chem, 2012. 287(51): p. 43053–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oppermann M, et al. , Regulation of vascular guanylyl cyclase by endothelial nitric oxide-dependent posttranslational modification. Basic Res Cardiol, 2011. 106(4): p. 539–49. [DOI] [PubMed] [Google Scholar]
- 69.Sayed N, et al. , Desensitization of soluble guanylyl cyclase, the NO receptor, by S-nitrosylation. Proc Natl Acad Sci U S A, 2007. 104(30): p. 12312–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sayed N, et al. , Nitroglycerin-induced S-nitrosylation and desensitization of soluble guanylyl cyclase contribute to nitrate tolerance. Circ Res, 2008. 103(6): p. 606–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Beuve A, et al. , Identification of novel S-nitrosation sites in soluble guanylyl cyclase, the nitric oxide receptor. J Proteomics, 2016. 138: p. 40–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Beuve A, Thiol-Based Redox Modulation of Soluble Guanylyl Cyclase, the Nitric Oxide Receptor. Antioxid Redox Signal, 2017. 26(3): p. 137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alapa M, et al. , Selective cysteines oxidation in soluble guanylyl cyclase catalytic domain is involved in NO activation. Free Radic Biol Med, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Armstrong PW, et al. , Vericiguat in Patients with Heart Failure and Reduced Ejection Fraction. N Engl J Med, 2020. 382(20): p. 1883–1893. [DOI] [PubMed] [Google Scholar]
- 75.Ghofrani HA, et al. , Riociguat: Mode of Action and Clinical Development in Pulmonary Hypertension. Chest, 2017. 151(2): p. 468–480. [DOI] [PubMed] [Google Scholar]
- 76.Sandner P, et al. , Soluble Guanylate Cyclase Stimulators and Activators. Handb Exp Pharmacol, 2019. [DOI] [PubMed] [Google Scholar]
- 77.Schindler U, et al. , Biochemistry and pharmacology of novel anthranilic acid derivatives activating heme-oxidized soluble guanylyl cyclase. Mol Pharmacol, 2006. 69(4): p. 1260–8. [DOI] [PubMed] [Google Scholar]
- 78.Erdmann E, et al. , Cinaciguat, a soluble guanylate cyclase activator, unloads the heart but also causes hypotension in acute decompensated heart failure. Eur Heart J, 2013. 34(1): p. 57–67. [DOI] [PubMed] [Google Scholar]
- 79.Evgenov OV, et al. , NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat Rev Drug Discov, 2006. 5(9): p. 755–68. [DOI] [PMC free article] [PubMed] [Google Scholar]