

Abstract
βγ-crystallins are the primary structural and refractive proteins found in the vertebrate eye lens. Because crystallins are not replaced after early eye development, their solubility and stability must be maintained for a lifetime, which is even more remarkable given the high protein concentration in the lens. Aggregation of crystallins caused by mutations or post-translational modifications can reduce crystallin protein stability and alter intermolecular interactions. Common post-translational modifications that can cause age-related cataract include deamidation, oxidation, and tryptophan derivatization. Metal ion binding can also trigger reduced crystallin solubility via a variety of mechanisms. Interprotein interactions are critical to maintaining lens transparency: crystallins can undergo domain swapping, disulfide bonding, and liquid-liquid phase separation, all of which can cause opacity depending on the context. Important experimental techniques for assessing crystallin conformation in the absence of a high-resolution structure include dye-binding assays, circular dichroism, fluorescence, light scattering, and transition metal FRET.
Keywords: βγ-crystallin, cataract, long-lived proteins, protein aggregation, protein solubility
1. Introduction
The focusing power of the vertebrate eye is determined in part by the concentration and distribution of the extremely long-lived crystallin proteins making up the lens. In terrestrial organisms, the air/water interface at the cornea also plays a major role[1], whereas in fish, this functionality is entirely determined by the shape and composition of the lens.[2,3,4] Crystallins have evolved from diverse small, soluble proteins[5,6] and comprise the majority of the lens tissue.[7,8] The majority of crystallins in the vertebrate lens belong to two superfamilies, the chaperone α-crystallins and the structural and refractive βγ-crystallins. Unlike most proteins, which are subject to continuous turnover in the cell, crystallins are generally not replaced after their expression during early development. Crystallin aggregation leads to cataract, a leading cause of blindness and a WHO priority eye disease.[9] In addition to their biomedical relevance, the long-term solubility and stability of crystallin proteins also represent a fascinating physical chemistry problem. How do these small, globular proteins maintain their solubility and transparency for prolonged periods, even at concentrations upwards of 400 mg/mL? In terms of understanding the underlying phenomena, crystallin longevity is thus the flip side of protein deposition diseases. Understanding the factors that enable these proteins to resist aggregation in the crowded lens environment for many years is critical to forming a complete picture of protein solubility and aggregation.
In this review, we summarize the major types of structural crystallins found in vertebrate lenses, their evolutionary origins, and important structural features. We also consider the many routes to cataract formation from a physical chemistry perspective. We describe some of the experimental techniques that are most commonly used for studies of crystallins. Hereditary cataract can be caused by mutations to any of the major crystallin proteins that change their folding stability or surface properties. Other mutations can create conditions under which crystallin solubility is compromised. Reduced crystallin solubility can also result from post-translational modifications (PTMs), which can take the form of deamidation, oxidation, interactions with metal cations, proteolysis, or derivatization with sugars. UV light irradiation, a common factor in age-related cataract, is one pathway to several types of crystallin damage. We discuss the role of protein-protein interactions in mediating aggregation. Finally, we describe how liquid-liquid phase separation (LLPS) can occur in lens proteins, even under conditions where they are fully folded.
2. βγ-crystallins evolved from a common origin and fulfill structural and refractive functions
βγ-crystallins are the primary structural and optical proteins of the lens; they maintain transparency, increase the refractive index, and protect the retina from UV light damage. In the lens fiber cells, these proteins exist as an amorphous, dense liquid where transparency is governed by the short-range spatial order, similar to a glass.[10] The key regulators of βγ-crystallin expression are the transcription factors c-MAF and Pax6.[11] Pax6 gene regulation is conserved amongst all crystallins, including vertebrate α-crystallins and unrelated taxon-specific crystallins (e.g., jellyfish J2and J3-crystallins and scallop Ω-crystallins). Recent evidence suggests that Pax6 regulation of these proteins may have arisen from pre-existing cis-sites that are affiliated with small heat shock proteins.[12]
The sequence and structural similarity of lens β and γ crystallins indicated that the two shared a common ancestor.[13,14] The βγ-crystallins share a common fold characterized by two double Greek key motifs (Figure 1) and a per-monomer molecular mass of approximately 21 kDa. The overall symmetry of the βγ-crystallins suggests that the crystallins were formed from two gene duplication events, probably after their recruitment to serve a refractive function.[15] The first duplication event transitioned the single Greek key motif into a wedge-like domain containing two Greek keys, and the second event formed the dimer.[16,8] The sequence similarity between motifs one and three and motifs two and four (Figure 1), but not in any other combination, are further evidence supporting these duplication events.
Figure 1:

HγS displayed to highlight the conserved structure among γ-crystallins. Protein structures are rendered using UCSF Chimera.[17] The N-terminus is circled and the C-terminus is boxed. Greek-key motifs are numbered from the N-terminus to the C-terminus. Conserved tryptophan side-chains are shown as sticks. (A) HγS (PDB ID: 2M3T) with individual Greek key motifs highlighted in red and blue. The left and right side are two wedge-like β-sandwich domains, where one Greek key (red) binds to the other (blue). The hydrophobic interdomain interface holds the two blue Greek key motifs together. (B) HγS rotated 90°. (C) Schematic of the Greek key motif. Each Greek key is formed by antiperiplanar β-sheets, which are continuously connected via linker sequences.
In γ-crystallins, each domain is encoded by a single exon, whereas in β-crystallins each domain is encoded by two exons separated by an intron. These alternative splicing patterns present the question of when the divergence of the β- and γ-crystallin families occurred: were γ-crystallins formed from the β-type gene encoding pattern through intron loss, or were β- and γ-crystallins formed from distinct duplication events? The presence of the βB2-crystallin outside of the lens is evidence for the former evolutionary progression.[18] The later evolutionary progression is supported by the existence of a two-domain crystallin-like protein from the sponge Geodia that lacks introns.[19] The relative likelihoods of these evolutionary progressions are considered in a review by Alessio[19], who concludes that the progression from the single Greek key motif to two-domain proteins were separate events for the β- and γ-crystallins. In this hypothesis, the initial fusion event formed dimers both with and without introns between the motif sequences. The dimers with introns then further combined to create the β-crystallins, whereas the latter formed γ-crystallins.
The structures of several βγ-crystallins are well characterized, providing a basis for comparison among paralogs (related proteins in the same organism) and orthologs (homologous proteins in different organisms). The structural building block of all of the βγ-crystallins can be seen in a relative of the vertebrate crystallins, the tunicate βγ-crystallin (PDB ID: 2BV2) (Figure 2A).[20] The structure of tunicate βγ-crystallin consists of a single domain with the familiar double Greek key motif (Figure 1). Along the path to modern vertebrate βγ-crystallins, a second double Greek key domain was added (Figure 2B), and the ability to bind divalent cations was lost, possibly as a result of selection for increased refractive index.[21] The single domain βγ-crystallin of the tunicate Ciona intestinalis is expressed in the light- and depth-sensing organs, but also in the palps.[20] This protein binds Ca2+[20] and is greatly stabilized by doing so.[24] It also has a higher refractive index than would be predicted by amino acid sequence alone[25], giving it a unique position as a single-domain intermediate form with both Ca2+ binding and refractive function. Like the tunicate crystallin, other crystallins from phylum Chordata[26] and the more distantly related microbial orthologs[27,28,29] have provided a wealth of background information about the evolution of βγ-crystallins.
Figure 2: Types of βγ-crystallins.

(A) Tunicate βγ-crystallin (PDB ID: 2BV2)[20] is monomeric and has a single domain. This protein can bind two calcium ions (shown in red). (B) HγS (PDB ID:2M3T)[22] in monomeric but has two double Greek key domains. (C) β-crystallins have the same domain organization, but are typically dimeric. Human βB2-crystallin can form domain-swapped dimers (PDB ID: 1YTQ).[23]
Characterization of βγ-crystallins’ biophysical properties provides insight into the adaptation of the double-Greek key motif to its function in the eye lens. The advent of polydispersity in the eye lens from both a diverse set of βγ-crystallins and oligomeric domain-swapped β-crystallins appears to have increased the overall protein content that can be accommodated in the lens.[6] The linker sequence, the N- and C-terminal extensions, and the hydrophobic residues at interfaces between Greek key motifs, are all key structural features that promote domain association in βγ-crystallins. The extensive and sometimes contradictory data revealing the extent to which these structural features drive domain association in βγ-crystallins were reviewed and were used to generate a likely sequence for the evolutionary progression.[19,8] Experiments with chimeric protein sequences show that the exchange of a β-type linker sequence or extensions for γ-type features in a β-crystallin can promote stabilization of a typically dimeric protein as a monomer. This suggests that amino acid substitutions in the interdomain linker sequence could be the key to divergence of β- and γ-crystallins. Similarly, hydrophobic residues at the β-crystallin interdomain interfaces tend to promote dimerization more than in γ-crystallins, suggesting that substitutions in this region were critical for the divergence of β- and γ-crystallins.
3. The βγ-crystallins share a very stable fold
The structures of wild-type human γ-crystallins have been solved, as have many cataract-related variants and structural crystallins from other vertebrates, including mouse[30], chicken[31], and zebrafish.[32] NMR structures of HγB[33], HγC (PDB ID: 2NBR)[34], HγS (PDB ID: 2M3T)[22], and γD (PDB ID: 2KLJ)[35] have been reported, as well as a crystal structure of HγD (PDB ID: 1HK0).[36] β-crystallins are typically dimeric, sometimes with multiple possible configurations. For example, human βB2-crystallin has been observed in different states in solution, including a domain-swapped dimer in a crystal structure (PDB ID: 1YTQ)[23] (Figure 2C) and a face-en-face dimer, without domain swapping.[37] Studies of these molecules have mostly focused on two aspects: the molecular basis of their extraordinary stability and solubility, and how changes due to mutation or post-translational modification alter their biophysical properties. The γ-crystallins have shown to be more stable than β-crystallins in solution, suggesting that the inherent stability of the Greek key motif is not the only contributor to the stability of these proteins; the interdomain interface also contributes to the overall stabilization.[38,39] In general, the N-terminal domain is less stable than the C-terminal domain. The C-terminal domain is more conserved across the human lens paralogs, suggesting that it was strongly selected for via evolution, as it was necessary for stabilization. The N-terminal domain may therefore be more modular and able to adapt to new functions. There are several aromatic residues that increase stabilization through pi-stacking, notably in conserved tyrosine corners[40], and conserved tryptophans in the hydrophobic core, which will be discussed in Section 4.[41] A thorough account of these and other known factors that contribute to the stability of βγ-crystallins were reviewed by Serebryany and King.[42]
HγS and HγD have different solubility thresholds, and different variants of these proteins exhibit a wide range of aggregation propensities.[43] The recent NMR structure of wild-type HγC shows that this protein is a typical wellfolded, highly soluble γ-crystallin.[34] An NMR structure of zebrafish γM-crystallin (Danio rerio) (PDB ID: 2M3C) reveals different organization of hydrophobic packing in the N-terminal domain relative to human γ-crystallins, probably due to the absence of one of the generally strongly conserved Trp residues in the hydrophobic core.[32] Further experiments using solution NMR and femtosecond fluorescence spectroscopy of strategically placed Trp probes have shown that water molecules in the hydration shell of this protein exhibit slow dynamics[44], in contrast to the fast surface water dynamics shown for HγS.[45] This discrepancy may reflect the different environments occupied by these proteins, as the protein concentration in the fish lens exceeds 1000 mg/mL, more than double the concentration for the already crowded human lens.
4. Biophysical techniques for studying crystallins
Three-dimensional structures have been solved for many βγ-crystallin proteins using both X-ray crystallography and solution-state NMR, showing the similarities conferred by their characteristic fold as well as important differences in the details. X-ray crystallography is the traditional method of choice for solving biomolecular structures, as excellent tools are available for rapid data collection and structure determination once suitable crystals are formed. However, because of their high solubility, crystallins are often difficult to crystallize, and their solution structures and dynamics are of particular interest due their being in solution in the biologically relevant state. Therefore, many structural studies have been performed using solution-state NMR, as reviewed in[46] and discussed for several cataract-related variants in Section 5. In addition to providing ensembles of structures in solution, this method enables the investigation of dynamics over a wide range of timescales and to detect rare conformational states.[47,48,49] For example, the solution NMR dynamics of HγD-W42R showed a partially unfolded state in exchange with the native-like structure, yielding insight into its increased susceptibility to proteolysis.[50] NMR can also be used to probe protein-ligand and protein-protein interactions, as has been demonstrated for γS-crystallin and its aggregation-prone variants binding to a fluorescent dye[51,52] and to the molecular chaperone αB-crystallin.[22,53]
Building on the strong foundation of information about the monomers, the most urgent set of problems facing this field is the detailed investigation of the insoluble aggregates found in cataract. HPLC and GPC analysis of cataract samples have shown that a cataract is made mostly of noncrosslinked crystallin proteins.[54] Amorphous-looking aggregates have been observed for HγD in the presence of copper and zinc ions[55] and for the HγD-P23T variant[56], whereas HγS and its G18V variant display a mixture of amorphous aggregates and amyloid fibrils, with the relative populations varying based on the aggregation conditions.[57,58] Amyloid fibrils have also been observed in UV-induced cataracts in both porcine[59] and human lenses[60], raising the question of how prevalent each type of aggregate is, and under what conditions. A characteristic signature has been identified in the 2DIR spectrum that can detect amyloid secondary structure, even if the individual domains contain only 4–5 β-strands.[61] It has been reported that aggregates in cataract lenses can be disrupted by small molecules such as rosmarinic acid[62] or lanosterol and other cholesterol derivatives,[63,64,65] preventing and even reversing cataract formation. However, other researchers report that full transparency is not achieved by these compounds[66], and that lanosterol works in canine but not human lenses[67], underscoring the need for continued research on the mechanism of crystallin aggregation and how it can be prevented or reversed.
For large complexes or insoluble aggregates, such as the amorphous or amyloid aggregates formed in cataract, solid-state NMR with magic angle spinning (MAS) is a useful structural method.[68,69,70,71] This technique can be used either on its own or synergistically with other methods including small-angle X-ray scattering (SAXS), or for larger complexes, electron microscopy. Solid-state NMR has been used to show native-like structure in aggregates of the P23T variant of HγD-crystallin[56], and to solve the structures of αB-crystallin complexes.[72] Solid-state NMR is a promising technique for future detailed studies of the aggregates found in cataract because it has provided some of the earliest detailed information available about the structural arrangement of amyloid fibrils[73], and it continues to be a workhorse technique for solving new fibril structures.[74] Solid-state NMR structures have been solved for fibrils formed for Aβ(1−42)[75,76,77], finding multiple polymorphs, including one that displays the same antibody reactivity as amyloid plaques from Alzheimer’s brain samples.[78] Structural models generated from solid-state NMR and electron microscopy data have revealed structures of different polymorphs of the Aβ(1–40) peptide.[79,80,81] Although parallel, in-register β-sheets appear in both polymorphs, many other structural features differ, including the overall symmetry of the complex, the conformation of the inter-strand regions, and some of the intermolecular contacts. We anticipate that this methodology will prove similarly useful for elucidating the structures of crystallin aggregates. Cryo-electron microscopy can be used for aggregates or complexes involving α-crystallin oligomers and their client proteins, although it is not currently applicable to individual crystallin molecules due to the lower size limit of approximately 38 kDa for single particles.[82]
Fortunately, in many cases relevant biophysical questions can be answered even in the absence of a detailed structural model. Along the way to high-resolution structure determination, several relatively simple experiments can be used to test hypotheses about stability, solubility and protein-protein interactions. Circular dichroism (CD) spectroscopy provides a rapid means of assessing the protein’s secondary structure. In CD, the absorbance of a beam composed of equal intensities of left and right circularly polarized light is measured after passing through the sample. Because proteins are highly chiral molecules, the protein backbone absorbs different amounts of left-rotating or right-rotating light, depending on its secondary structure features.[83] The effective rotation of the plane of the beam is then recorded as a function of the frequency, providing an estimate of the relative amounts of α-helix, β-sheet, and random coil. Different variants of the same protein can be compared to each other, enabling detection of partial unfolding due to mutation or PTMs. The CD spectrum can also be monitored with increasing temperature or denaturant concentration to measure protein unfolding. If the secondary structure is already known or not needed, unfolding can alternatively be monitored using fluorescence spectroscopy experiments.
Built right into the βγ-crystallins is a set of very sensitive fluorescent probes that allow for characterization of disturbances to the protein structure, in the form of four highly conserved tryptophan residues. A common feature of vertebrate lens βγ-crystallins is an arrangement of two tryptophans in the hydrophobic core of each domain, which naturally acts as a FRET pair to protect the retina from UV light.[84] The fluorescence of these tryptophans is strongly affected by the surrounding chemical environment, making them excellent reporters of the overall fold of the protein. This method is most useful in comparative mode, investigating differences between variants or measuring unfolding during thermal or chemical denaturation. Gathering a large body of data about how crystallin stability is affected by temperature, denaturants, mutation, or modification is essential for understanding the specific mechanisms of protein folding and aggregation.
Light scattering is an accessible method for studying protein-protein interactions and, importantly, aggregation of βγ-crystallins. Scattering experiments are complementary to spectroscopic measurements: the latter elucidate different aspects of the internal structure of the molecule, based on electronic structure, vibrational modes, or the arrangements of nuclear or electron spins, while the former provide information about properties of the proteins as physical particles. Light scattering techniques for investigating protein solution properties can be split into two categories, Static Light Scattering (SLS) and Dynamic Light Scattering (DLS). SLS measures the intensity of light scattered from particles in a solution, which depends on the hydrodynamic radius and mass.[85] Because scattering also depends on the molecular orientation, the intensity of scattering is measured at a variety of angles in what is known as Multi-Angle Light Scattering (MALS). Particularly when combined with size exclusion chromatography to isolate individual species[86], MALS enables determination of the second virial coefficient or A2[87,88], which effectively reports on the favorability of protein-protein interactions. A2 is a measurement of the mean potential force between two particles in a solution.[89] In contrast, DLS measures fluctuations in scattering intensity at a single angle over time, which can be used to estimate particle size as well as the diffusion coefficient.[90] This makes DLS an excellent tool for investigating aggregation of particles over time, or as a function of changing conditions. DLS is often used to characterize the aggregation of crystallins as a function of temperature or pH, or upon addition of salts or other solution components.
Small-molecule fluorophores, such as Thioflavin T (ThT) and 8-anilino-1-naphthalenesulfonic acid (ANS), are useful probes for investigating crystallin structure and interactions. These fluorophores bind to the protein of interest, producing observable changes in their fluorescence spectra such as frequency shifts and changes in intensity (Figure 3). ThT fluoresces strongly when bound to amyloid fibrils[91,92,93], enabling detection of fibrils and monitoring the kinetics of their formation.[94,95] ANS and bis-ANS are commonly used as a probe of exposed hydrophobic surface area, although the binding mode also involves electrostatic interactions between the negatively charged sulfate groups on the dye and positive charges on the target protein.[96,97] For example, ANS fluorescence has been used to characterize the placement of hydrophobic patches on HγS to better understand the α-crystallin-client protein binding interface.[52] These small molecule probes can be used to quickly and inexpensively probe the aggregation state of a protein and detect partially unfolded intermediates.
Figure 3:

ANS binding can be used to probe hydrophobic surface area. (A) ANS is a mostly hydrophobic small molecule with a negatively charged sulfate group. This allows the molecule to bind to exposed hydrophobic pockets on a protein, as shown in this docking simulation of a predicted structure for the G18A variant of γS-crystallin binding to ANS. The protein structure is color coded from blue (hydrophobic) to orange (hydrophilic). (B) When ANS binds to a protein, it will fluoresce more strongly; hydrophobic surface exposure correlates with intensity, as shown in this simulation of a γS-G18A (green) compared to γS-WT (blue) suggesting that the hydrophobic core of the protein is more exposed in the variant. (C) This florescence in the bound state is caused by the absorption of a photon, causing electronic excitation; when the electron relaxes back to the ground state, energy is released as a lower-energy photon.
The absorption spectra of transition metals overlap with the fluorescence emission spectra of protein functional groups, thereby providing a donor (fluorophore)/ acceptor (metal) pair. Non-fluorescent transition metals quench donor emission over a distance range between 10 and 20 Å, on account of a small Förster distance (Ro) that is on the scale of the biomarcomolecule’s length. The short Ro coupled with an inherent inverse-sixth-power distance dependence makes tmFRET a powerful tool for studying intramolecular interactions.[98] A schematic illustrating tmFRET is shown in (Figure 4). The wide range of absorption spectra between transition metals and transition metal chelator complexes allows for donor/acceptor-pair tuning.[98] Furthermore, transition metals are advantageous acceptors, as they can be used in either native or synthetic metal-binding sites, can be reversibly bound through chelation, and are less restricted by orientation as they have several absorption dipoles.[98] For proteins without native metal binding sites, di-histidine binding sites can be introduced through mutagenesis, or chelators such as EDTA can be attached with short linkers.[99]
Figure 4: Transition metal Förster resonance energy transfer (tmFRET) and an example of a crystallin protein where it is useful.

(A) A model of one of the Cu2+-binding sites of HγS, showing the Cys residues that coordinate the metal ion FRET donor, and the nearby Trp 47, which acts as a FRET acceptor. (B) Schematic depiction of the interactions in (A), with key distances labeled. Both (A) and (B) are based on data from.[58] (C) Drawing showing the approximate positions of relevant spectral peaks. Trp in a protein has a strong absorption peak centered at around 280 nm, and fluorescence at around 350 nm (although this can vary considerably depending on the local environment.) Transition metals such as Cu2+ often have a relatively broad absorbance peak at around 400 nm, allowing for substantial overlap with the Trp fluorescence, and hence FRET. (D) Jablonski diagram indicating the various energy transfer mechanisms, including excitation, fluorescence, FRET, and vibrational relaxation (small red arrows.)
5. Cataract-related mutations in structural crystallins often cause increased aggregation propensity
So far, there are over 30 reported mutations associated with hereditary cataract, many of which are summarized in a review by Vendra et al.[100] To give a few examples, structures have been solved for cataract-related variants of HγD-crystallin, including γD-R58H[36], γD-P23T[101,102], γD-R76S[103], γD-V75D[104], and a variety of deamidation variants.[105] For HγS, structures have been solved for γS-G18V[22], γS-G57W[106,107], and the deamidation variants N14D and N76D.[108] Themes have emerged from biophysical and structural characterization of βγ-crystallin variants, including relatively subtle structural differences from wild-type, increased hydrophobic surface exposure, higher susceptibility of the N-terminal domain to unfolding, and the formation of multiple unfolding intermediates.
The solution-state NMR structures of the G18V[22] and G57W[107] variants of HγS (HγS-G18V and HγS-G57W, respectively) are both similar to that of the wild-type protein (HγS-WT) without major structural rearrangement. However, for both proteins, biophysical experiments had previously showed that these variants are less stable and more aggregation prone.[109,110,106] Solid-state NMR spectra of HγD-P23T aggregated under physiological conditions are consistent with the local structure of the protein being nearly identical to HγS-WT.[56] These are described as amorphous-looking; however, their solid state NMR peak dispersion and linewidths are consistent with a high degree of structural homogeneity.[56]
Although they are not misfolded, HγS-G18V[52], HγS-G57W[107], and γD-P23T[56] all have increased hydrophobic surface area compared to γS-WT, as well as subtle but widespread perturbations to the N-terminal domain (NTD), where each mutation is located, whereas the C-terminal domain (CTD) remains unchanged. Despite their minimal structural perturbations, cataract-related variants of γS-crystallin behave differently in the crowded solution of the eye lens, as evidenced by differing interactions with surface water[45] and with small molecules in the lens.[111]
The isolated domains of HγS and HγD are each very stable, although in both cases the NTD is less stable than the CTD.[38] The many observations of crystallin aggregation mediated by partial unfolding of the NTD, while largely leaving the CTD unchanged, furthers the hypothesis that the NTD is stabilized by the interdomain interface.[39,112,113,38] A particularly dramatic example of this effect occurs in HγD-V75D, where denaturation experiments in urea revealed that it is possible to unfold the entire NTD while the CTD retained a native structure.[104] The residues of the NTD occupy more conformations than those found in the CTD[114], and the NTD shows a higher degree of flexibility in the HγD-G57W variant over WT.[115] In a cataract-related variant of mouse γS-crystallin, a single mutation, F9S in the first β-strand destabilizes the first Greek key motif, promoting unfolding of the NTD under even mild heat stress.[116] The HγS-Y67N, was recently discovered in a Chinese family with autosomal dominant nuclear congenital cataracts.[117] In this protein, structural destabilization leading to reduced thermal and chemical stability relative to HγS-WT[118] takes the form of disrupting one of the tyrosine corners that are essential for stabilizing Greek key motifs.[107]
Another example of this unfolding mechanism was found in the zebrafish γM7 crystallin, which has a CTD that is highly similar to that of HγS and, in contrast, an N-terminus with key sequence differences, especially between residues 67–72. Specifically, this protein lacks one of the strongly conserved tryptophan residues found in other vertebrate γ-crystallins, leading to a restructuring of the loop between β8 and β7. This amplifies hydrophobic packing, presumably to compensate for the loss of the bulky Trp in the hydrophobic core while maintaining the Greek key motif. These structural differences likely reflect the evolutionary constraints of the fish crystallins to perform in a more densely packed eye lens that is under less UV stress compared to its land counterparts.[32]
Destabilization is strongly associated with aggregation, although the correlation is not always straightforward.[110,42] Complete unfolding is often prevented by intramolecular hydrophobic contacts, salt bridges, disulfide bonds, or by the chaperone activity of α-crystallin. This can give rise to an accumulation of partially unfolded intermediates, which may be more physiological relevant than completely denatured proteins, given the preserving nature of the eye lens[119], though there is some evidence of fully denatured proteins in cataractous lenses as well.[120]
Similar aggregation-prone variants are also observed in β-crystallins. Compared to their WT counterparts, the mutants HβB2-W59C[121], HβB2-W151C[121], HβB2-S175G/H181Q[122], and HβB2-R188H[123] all have increased hydrophobic exposure, decreased stability, decreased solubility and an increased propensity to aggregate. The dependence of the NTD stability on that of the CTD is highlighted in the comparison of stability and solubility between the HβB2-W59C and HβB2-W151C variants.[121] Both of these tryptophan residues are conserved in all human lens βγ-crystallins, although these studies indicate that mutating them is not equivalent in terms of structural disruption. Every form of characterization used indicates that the W151C mutant is far more perturbed relative to WT than W59C. Both mutations of HβB2-S175G/H181Q are in the fourth Greek key motif. Hydrogen/deuterium exchange mass spectrometry experiments revealed significant conformational changes in the loop region that transverses the CTD and connects the two Greek keys[122], suggesting that this loop region is critical for stabilization. In HβB2, R188 forms an ion pair with E75 in the intradomain interface between the Greek keys.[23] HβB2-R188H disrupts this pairing and, therefore, the equilibrium among different oligomeric states: this variant exists mostly as a tetramer, rather than the native dimer.[123] The reduced solubility and stability can therefore be associated with the loss of stabilization from homodimer formation. HβB2V187E aggregated rapidly upon expression in bacteria, and was found almost solely in inclusion bodies.[123] Molecular dynamics simulations suggested the mutation completely disrupted folding of the domain. Across all crystallins, a hydrophobic residue is conserved in this position, supporting the idea that this position is a critical part of the hydrophobic core. In contrast, the CTD was only slightly loosened by the HβB2-V187M mutation, a conversion to another hydrophobic residue.[123]
At neutral pH, HβB1-R233H does not greatly alter the degree of hydrophobic exposure or solubility compared to HβB1-WT HβB1. The mutation slightly alters the structure and increased aggregation propensity at high temperatures. However, this aggregation did not correlate to decreased stability; in fact, HβB1-R233H had an increased stability compared to WT. Rather, the increased aggregation propensity may be due to the increased hydrophobic character of the C-terminal extension.[124] Increased hydrophobicity of the C-terminal extension was also considered as the cause of aggregation for HβB1-X253R, a cataract-associated variant with a 26-amino acid C-terminal extension. The mutation had only minor effects on stability and structure, but a large increase in the aggregation propensity.[125]
Heterooligomerization has been shown to increase the solubility of β-crystallins[126,127,128] and coexpression has been used as a tool to stabilize and solubilize these proteins.[129] In general, acidic β-crystallins are less stable than basic β-crystallins.[130,131] Although HβB1-R233H increased the stability of the protein in isolation, the βA3/βB1 heterodimer was destabilized. The formation of the heterodimer was unaffected, suggesting that R233 plays a role in heterodimer stabilization.[124] Xi et. al. argue that these trends in stability imply that R233 destabilizes βB1 and plays a functional role in beneficial heterodimer formation rather than contributing to the inherent stability. This could be extended to the C-terminal extension, generally, as the X253R mutation also weakened the dimerization of βA3/βB1. Therefore, mutations in this region could cause cataract by disturbing heteroligomerization in vivo.[125] The HβA4-G64W variant is destabilized and improperly folded. This mutation caused an increase in aggregation that was not ameliorated by coexpression with HβB1. Therefore, HβA4-G64W is likely unable to bind to HβB1, suggesting a role for G64 in heterodimerization.[132] HβB2-R188H was unable to stabilize HβA3 like WT, suggesting this mutation also affects the heterodimer formation. In contrast to the monomeric γ-crystallins, β-crystallin functionality is not a simple function of individual protein solubility and stability: these proteins must be considered in the context of the chemical milieu of the lens.
6. Age-related cataract often results from post-translational modification
Although studying the aggregation mechanism of cataract-associated variants of the γ-crystallins is very instructive from the standpoint of protein biophysics and sequence-structure-function relationships, most instances of cataract disease are not congenital but result from post-translational modifications (PTMs) accumulated during aging.[133] These PTMs include deamidation, glycation, acetylation, and products of oxidative damage[134], all of which can dramatically effect protein stability and intermolecular interactions. Here we discuss the chemical mechanisms underpinning the formation of these PTMs, as well as their implications for protein solubility.
6.1. Deamidation
Deamidation is a spontaneous process where the amide group on an asparagine or glutamine side chain is replaced with a carboxylic acid (Figure 5). The mechanism proceeds via a cyclic intermediate, which leads to a mixture of the D and L versions of both aspartate and isoaspartate.[135] These deamidation events play a functional role in some tissues[136,137], but in structural lens proteins they are primarily a deleterious effect of aging and exposure to UV radiation.[138,139] In irradiated rat lenses, proteomic analysis revealed many different PTMs; however deamidation of Asp and Glu residues were among the most common.[140] The degree to which deamidation at any particular site contributes to the formation of cataract is an open question, as the solubility impact appears to depends on both which isomer is formed and where in the protein it is located.[141,142,143] Many detailed investigations of specific deamidation-mimicking variants have been performed, some examples of which are discussed below.
Figure 5: Deamidation Mechanisms.
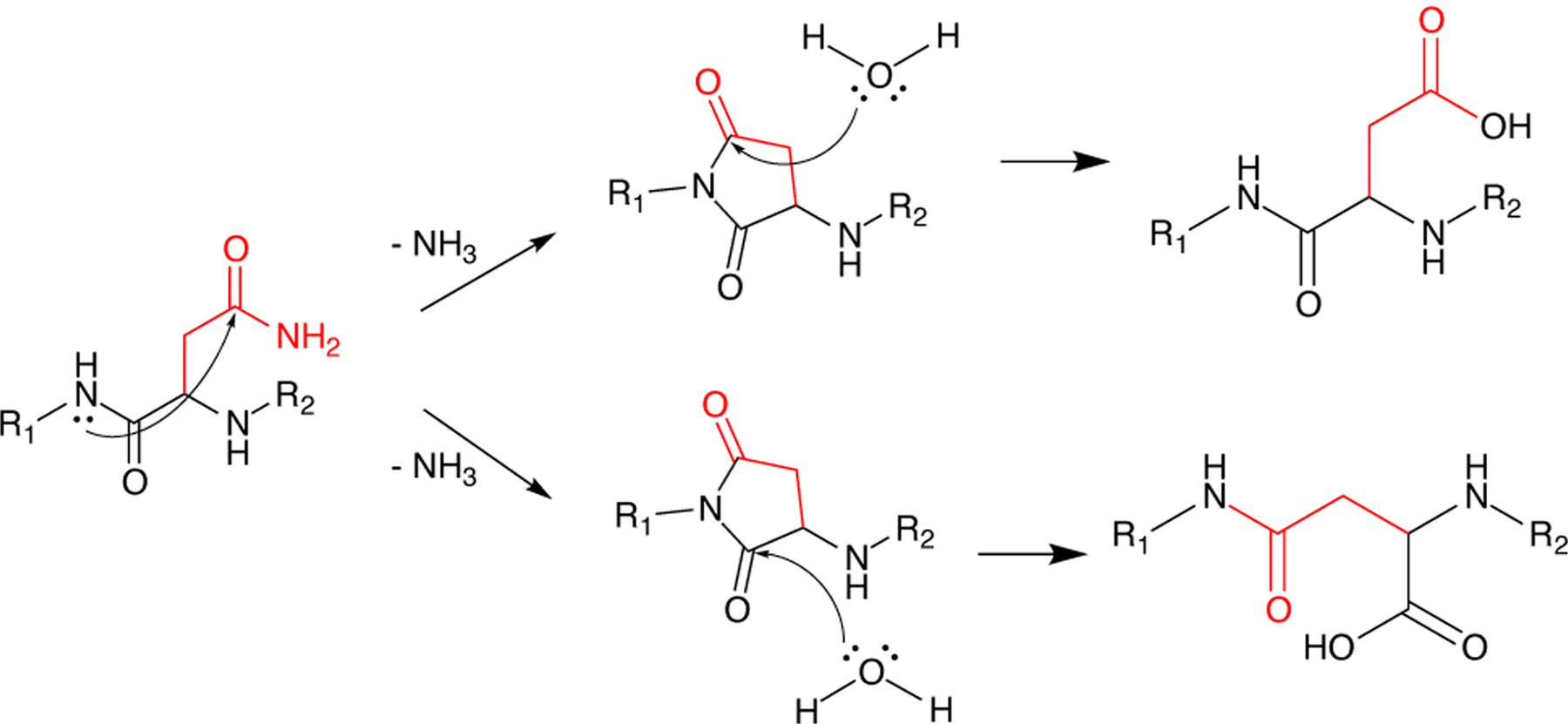
Deamidation of asparagine and glutamine residues yield their acidic counterparts, altering the biophysical properties of the protein. Backbone amide attack of asparagine or glutamine residues result in succinimide or glutarimide intermediates, respectively. Hydrolysis results in either conversion of the starting amide to an acidic residue (top) or breaking of backbone amide bonds to form iso-aspartic acid (γ or iso-glutamic acid (bottom), respectively.
In human βB1, deamidation leads to destabilization and a less compact dimer structure, whereas truncation of either the N- or C-terminal extensions does not impact stability with respect to urea denaturation.[144,145] βB2-[146] and βA3-crystallins[147] are similarly destabilized by single or double deamidation, although their overall structures remain largely native-like. Deamidations also impact the dynamics of β-crystallins in the context of homo- and heterodimers: enhanced flexibility, especially for βB2, may promote the formation of larger oligomers that could lead to cataract.[148]
Deamidation also causes destabilization and altered intermolecular interactions in γ-crystallins. Although the structures of the N76D and N143D variants of HγS are similar to HγS-WT, these variants exhibit increased attractive forces to other proteins.23456789[149] The deamidation-mimicking N76D and N14D variants of HγS exhibit increased aggregation propensity and are more susceptible to oxidative damage.[108] This investigation also showed that both the N- and CTDs of these variants are more flexible and expose more of the hydrophobic core, leading to two possible drivers of aggregation. One is that the increase in flexibility drives aggregation directly via the presence of aggregation-prone unfolding intermediates, and the other is that deamidation events make the protein more susceptible to other PTMs, further impacting solubility. HγS-N76D has an increased propensity for dimer formation and decreased resistance to denaturants relative to HγS-WT, although it is resistant to thermal aggregation.[150] A triple mutant mimicking the effects of multiple deamidations (HγSN14D/N76D/N143D) is more destabilized and aggregation prone than any of the single mutants, and resists solubilization by αA-crystallin.[151] On the other hand, a thorough study of HγD where all 7 of its Asn residues were individually mutated to Asp revealed no major changes in stability, solubility, or aggregation propensity. The structures of all these variants, as measured by both x-ray crystallography and solution NMR were also largely unchanged. This provides evidence that deamidation of individual residues in HγD does not initiate aggregation leading to cataract, although the imapct of multiple deamidations is not addressed.[105] An interesting topic for future investigation is whether deamidation variants of HγD are more susceptible to oxidation and other deleterious modification, as observed for HγS by Forsythe et al.[108], which would provide a mechanism for their involvement in cataractogenesis even if the impact of the deamidations themselves on protein structure is minimal.
6.2. Oxidation
Another important set of modifications that are relevant to crystallin aggregation are those brought about by oxidative damage. Although the lens contains a reservoir of glutathione[152], it is depleted with age[153,154], and thus oxidative damage is one of the most abundant types of post-translational modifications in the lens nucleus.[155] Oxidative damage can truncate the protein through backbone cleavage[156] or sidechain modification. In particular, the sidechains of Met, Trp, and Cys are prone to oxidationdriven modifications that accumulate over time and are found in high rates in cataractous lenses.[157,158] Cysteine oxidation and the formation of disulfide bonds can perturb native structure or form intermolecular disulfide bridges that decrease solubility and lead to larger aggregates.[159] Tryptophan oxidation has been shown to destabilize the protein[160], and would also disrupt the UV-filtering capability of the two highly conserved tryptophan pairs[161,84], which effectively absorb potentially damaging radiation and redistributes the energy via thermal motion.[162]
Despite the various protective features in the lens, frequent exposure to UV radiation can cause free radical formation that leads to oxidation products. Cations of redoxactive metals such as iron[163] and copper are also implicated in crystallin oxidation. It is difficult to predict and isolate all the potential products; however, oxidative damage has been localized to specific side chains such as methionine, cysteine, and tryptophan in proteins of mouse lenses.[158] The chemical and biophysical properties of bovine γ-crystallins are altered after treatment with a strong oxidizing agent, with increased hydrophobic exposure and more disulfide-based protein cross-linking.[164] A more detailed discussion of disulfide cross-linking in the crystallins can be found in section 7.2. In addition to their baseline increase in aggregation propensity, many cataract-related crystallin variants also have increased susceptibility to oxidative damage. Six cataract-related variants of HγD- and HγC were found to be more aggregation-prone than WT after treatment with UV light or hydrogen peroxide.[165] Aggregates with different morphologies have been reported upon oxidation. UV-B irradiated mixtures of α, β, and γ-crystallins result in amyloid fibril based aggregates[166], while amorphous-looking aggregates have been formed from UV-irradiated HγS.[57] Small-molecule antioxidants that can prevent or mitigate oxidative damage are of great interest, as they may have some prophylactic value for cataract prevention. Compounds such as (−)-epigallocatenchingallate[167,168,169], herperetin[170] and green tea flavonols[171] have been shown to prevent oxidative damage to crystallins in vitro. These compounds could serve as an important starting point for finding effective preventative measures for age related cataract.
6.3. Proteolysis
Truncation of crystallins is another form of age-related damage observed in aged lenses; however, proteolysis has a complex role in the development and maintenance of the healthy lens. Enzymatic digestion by proteases in the eye lens has a beneficial function during lens development and even into adulthood.[172] The ubiquitin-proteasome pathway is functional in lens fiber cells[173], although activity of both the ubiquitination machinery and and proteasomes are reduced with age, probably due to depletion of the necessary enzymes.[174] While it is active, this cellular disposal system can remove crystallins that are damaged by oxidation or repeated exposure to UV light, but as the lens ages and the diffusion barrier between the nucleus and the outer part of the lens becomes more pronounced[175], damaged proteins are accumulated rather than removed by proteolysis.[176] The ubiquitin-dependent proteolytic pathway is critical for the degradation of oxidatively modified proteins[172], which otherwise aggregate and bind to membranes[177,178], contributing to the hindrance of diffusion in aged lenses. At least one congenital cataract mutation where the lack of solubility is due to increased sensitivity to these natural housekeeping proteases. The S129R variant of human βB1-crystallin has enhanced sensitivity to proteolysis, and is associated with an autosomal dominant congenital cataract-microcornea syndrome.[179]
Non-enzymatic cleavage of crystallins is also an important process in the lens, as the long-lived crystallin proteins are susceptible to various forms of damage that render them aggregation-prone, especially in the absence of a functional ubiquitin-proteasome system, as in the aging lens. This type of modification is often concomitant with both oxidative damage and deamidation. At neutral to mildly basic pH, spontaneous deamidation of Asp and Gln residues often occurs via formation of a succinimide intermediate[180,181], but if formation of this intermediate is prevented by the local tertiary structure, the amide nitrogen of the backbone may act as the nucleophile instead of the sidechain NH2, leading to backbone cleavage either as a competing pathway, or after deamidation has occurred (Figure 5).[182,183] As expected from the dependence on three-dimensional structure, these modifications are not randomly distributed but instead depend on the neighboring residues.[184,185] The side chains of nearby residues may also be involved in the chemistry, with serine being particularly likely to participate in truncation[186] or racemization, albeit via distinct chemical mechanisms.[187] In some cases, backbone cleavage can also lead to interprotein cross-link, further compromising solubility.[188,189]
6.4. Tryptophan derivatives in the lens
Tryptophan metabolites produced from the kynurenine pathway in the anterior cortical epithelial cells (Figure 6) are capable of mitigating the damage of frequent ultraviolet radiation exposure in the eye lens. These small molecules absorb some of the radiation that passes through the cornea, particularly in the 300–400nm wavelength range, and dissipate the energy as vibrational motion.[190,133] These UV-filtering metabolites are depleted in aged lenses[191], making the lens proteins more susceptible to UV-induced damage.[192] Although kynurenine and its derivatives play a photoprotective role on their own, their chemical reactivity can also promote oxidative damage and aggregation and are under investigation as an accelerant of brunescent cataracts. Oxidized tryptophan metabolites can form covalent linkages to crystallins or GSH, a major route of 3OHKyn depletion.[192]
Figure 6:

The formation of tryptophan metabolites through the kynurenine pathway. Enzymes are indicated adjacent to each arrow. At the beginning and the end of the pathway are drawings illustrating the healthy lens and the development of brunescent cataract as a result of tryptophan metabolite production.[193,194]
The covalent linkage of tryptophan metabolites to nucleophilic residues, especially lysine and histidine, destabilizes the protein, leading to cataractogenisis.[195,196] A possible source of oxidation could be through hydrogen peroxide produced from the interactions 3-hydroxykynurenine (3OHKyn) and 3-hydroxyanthranilic acid (3OHAA) with Cu2+.[197] The 3OHKyn and 3OHAA precursors (Figure 6) lack the o-aminophenol group and do not promote metal ion-mediated hydrogen peroxide, emphasizing the necessity of the hydroxyl group for redox activity, possibly due to resonance stabilization of the radical species.[197]
Cross-linked crystallins are found extensively in cataractous lenses.[198,191] Tryptophan metabolites are known to form covalent linkages on crystallins and are readily oxidized into reactive species. Indeed, incubations of α-crystallin with 3OHKyn or 3OHAA produce crosslinks that are greatly amplified in the presence of Cu2+, even without UV exposure. Similarly, incubations of 3OHKyn and 3OHAA with bovine lens protein (BLP) in the dark produced no appreciable amount of higher molecular weight species until Cu2+ or, to a lesser extent, Fe2+ was co-incubated.[190] Cu2+ was rapidly converted to Cu1+ with accompanying oxidation of the o-aminophenol group. One proposed mechanism of cross-linking requires two equivalents of metal ion per 3OHAA or 3OHKyn to perform one-electron transfers that form quinonimine species that are susceptible to Michael additions.[190]These investigations highlight the complexity of crystallin aggregation in the eye lens and support the assertion that elevated levels of metal ions found in the eye are causative agents of cataract.
7. Metal ion-induced interactions
The ubiquitous βγ-crystallins of the vertebrate lens are believed to have evolved from ancestral Ca2+-binding proteins, and orthologs that bind divalent cations are found in a variety of organisms, including many that lack eyes. The pathway for crystallin evolution is generally assumed to be selection for stability and solubility, often followed by loss of the original activity, in this case metal binding, along the way to gaining its new optical function.[6,21] This process is illustrated by the Neurospora crassa abundant perithecial protein (APP), an early crystallin ortholog that is highly aggregation-resistant at high protein concentrations and lacks functional calcium ion binding sites.[199] Unlike related proteins, human βγ-crystallins lack the ability to bind Ca2+.[200]
Increased divalent cation concentration in the eye lens, which can be caused by aging[163], smoking[201,202], or diabetes[203,204], is associated with lens opacification and cataract formation. Each crystallin protein has idiosyncratic interactions with each metal ion, even among crystallins in the same family and for ions of the same charge and similar size. Here we discuss several mechanisms by which metal ions can induce aggregation. Though these mechanisms are considered separately, even in vitro synergistic interactions between different mechanisms is likely to be important, and in vivo additional factors such as cellular density[205], protein diversity, point mutations, and post translational modifications will also affect metal ion-crystallin interactions.
Metal ions in the eye lens can destabilize crystallins through coordination of residues that are forced to adopt distorted conformations to make contact with the ion. As with hereditary point mutations, this structural perturbation can lead to aggregation. For example, tryptophan fluorescence and CD experiments show that human HγD is significantly destabilized and often aggregates in the presence of Cu2+ and Zn2+.[55] Separately from or in addition to their destabilizing effects, coordination of metal ions can also lead to intermolecular metal ion-bridged aggregates: these are often detected by testing whether metal chelators such as EDTA or DTT diminish or reverse aggregation.
Copper and zinc ions, which have distinct bioinorganic properties, interact very differently with human γ-crystallins. Zinc has been found in higher concentrations[206] in cataractous eye lenses, whereas copper is less abundant but has a higher propensity for inducing aggregation. HγD does not appear to interact with Mn2+, Ca2+, Fe2+, or Ni2+.[55] Copper (II)-induced aggregation of HγD exhibits biphasic behavior with respect to concentration: partial unfolding dominated at low Cu2+ concentration, whereas ion-mediated disulfide bonding becomes more important at high (> 4 equivalents) Cu2+ concentration. A deeper investigation into copper mediated disulfide bond formation indicated Cys111 as a highly likely position for cross-linking formation[159] In contrast to Cu2+-induced aggregation, no biphasic behavior was observed for Zn2+-induced aggregation of HγD. No disulfide bonds were formed, and there was little evidence of HγD destabilization in the presence of Zn2+, consistent with this ion’s lack of redox activity.[207] These observations suggest the formation of intermolecular cation-bridged species. Further corroboration of this mechanism came when the elimination of His22 of HγD eliminated aggregation. Conversely, the introduction of a histidine at the homologous positions in HγC and HγS promoted Zn2+-induced aggregation of these proteins that did not occur with either wild-type protein.[207]
Cu2+ and Zn2+ also play contrasting roles in the aggregation of HγS. As with HγD, both Zn2+ and Cu2+ strongly promote aggregation, whereas a variety of other metal ions induce it weakly or not at all.[208] Zn2+-induced aggregation was markedly reduced when all solvent-exposed Cys residues were replaced by Ser, and addition of EDTA abolished all aggregation in both the variant and WT protein, consistent with Cys-mediated bridging. Surprisingly, removal of the solvent-exposed cysteine residues accelerated and increased the extent of Cu2+-induced aggregation.[208] Two distinct binding events were observed using tmFRET and ITC experiments: one at the site of the cysteine loop and the other at a different location, possibly involving normally buried residues that become accessible due to partial unfolding.[58] The Cu2+-induced aggregates could be partially resolubilized using EDTA, and further resolubilized by DTT, which alone produced stable monomers, indicating different aggregation pathways.[58] The presence of oxidized cysteine and methionine residues found through mass spectrometric analysis of trypsin digests suggested that Cu2+ was being reduced to Cu+, which would allow for the possibility that Cu+ is forming metal ion-bridged aggregates that can only be broken by DTT and not EDTA.[208,58] As was the case for HγD, HγS aggregation studies suggest that zinc ion-induced aggregation is primarily promoted by bridging, whereas copper ion-induced aggregation proceeds though a complex mixture of oxidation, destabilization, and metal bridging mechanisms.
HγS and HγC are also susceptible to mercury ion-induced aggregation through destabilization and intermolecular disulfide bonding. Increasing equivalents of Hg2+ ions steadily decreased thermal stability and increased aggregation and unfolding.[209] Investigations of the N- and C-terminal domains separately found that only the NTD was susceptible to intermolecular disulfide bonds. HγC aggregation in the presence of mercury ions exhibits biphasic behavior. At Hg2+ < 3 equivalents, aggregation increased and thermal stability decreased with increasing Hg2+ to protein ratios. After 3 equivalents, thermal stability began to increase and aggregation decreased, although there was still an increase relative to the protein in the absence of mercury. Studies of the individual N- and C-terminal domains found that each domain followed the pattern seen at low equivalents, suggesting that the biphasic behavior requires both domains to be present. There is evidence of intermolecular disulfide bonds, suggesting that both destabilization and oxidative mechanisms are necessary for aggregation.[209] For this process and for Cu2+-induced aggregation of HγS, more detailed investigation, for example NMR chemical shift perturbation, would be necessary to determine the origin of the biphasic behavior.
8. Protein-protein interactions
Altered protein-protein interactions are an important driver of crystallin aggregation, leading to varied aggregation pathways. MD simulations of the R85D variant of HγD suggest that the biggest difference between R85D and wild-type is an increase in hydrogen bonds, leading to a more rigid protein that is better able to form stable intermolecular contacts, leading to aggregation.[210] MD simulations of the W42R variant of HγS suggest that the critical structural change is a loss of hydrophobic interactions holding the two domains together.[211] Computational models of a variety of HγD variants suggested that increases in attractive forces between proteins leads to an increase in aggregation propensity.[212] Simulating HγD under acidic conditions shows that the CTD forms a novel β-sheet.[213] The increased aggregation propensity of HγS-G75V could be related to an increase in the exposure of the hydrophobic core.[214] To provide a more accurate model of charge states influencing the potential energy landscape, a screened electrostatic model was used to define the charge state of HγD, accounting for the effects of neighboring residues on local pKa values of potentially charged side-chains.[215] All of this is confounded by the fact that aggregates are formed by many different mechanisms, often producing a mixed population of aggregates. For example, UV-irradiated samples of HγS-G18V formed amorphous-looking aggregates, while those aggregated under low pH conditions were more fibrillar in nature, although both types are often present in the same sample.[57] HγD also forms both native-like and amyloid aggregates[216,56], with both having been observed in samples from cataractous lenses.[60,217,122]
8.1. Condensation/ domain swapping
Domain swapping occurs when two or more identical protein chains or monomers swap elements of their structure to form dimers or open-ended chains.[218] Although this type of exchange or swapping is often found in disease-related aggregates, it can also be used to control and modulate protein function.[219] For example, domain swapping is observed in the native structure of β-crystallins, which comprise highly similar Greek key domains connected by a short inter-domain linker region.[37] Although γ-crystallins are monomeric, β-crystallins are dimers or multimers and exist as a wide range of homo- and hetero-oligomers.[37] βB2-crystallin exists as a domain-swapped dimer in the crystal structure.[37]
In γ-crystallins, domain swapping does not occur in the native state. When it does occur, it can make either closed dimers, which are soluble, or long polymeric chains, which can lead to aggregation. This process often involves partially unfolded states that swap portions of β-sheets to form extended oligomers. For example, HγD can partially unfold in either the NTD[220] or the CTD[221], allowing mixed β-sheets containing strands from both monomers. In oxidation-mimicking point variants where tryptophan is replaced by glutamic acid (HγD-W42E, W68E, W130E and W156E), aggregation occurs via domain-swapped polymerization.[222] In HγD-W42E and W42Q, an intramolecular disulfide bond traps the protein in a partially unfolded conformation that exposes β-strand edges, facilitating this kind of aggregation.[223] Surprisingly, mixing this variant with the wild-type protein exacerbates its aggregation[160], which has important implications for how oxidation damage can promote cataract, as oxidized proteins in vivo are in contact with a large reservoir of undamaged ones.
8.2. Disulfide bonds
Intra- and intermolecular disulfide bonds are common PTMs, particularly in aged and cataractous lenses where protective antioxidants are depleted.[155] Intramolecular disulfide bonds may perform an oxidoreductase function, serving as a buffering system through a disulfide exchange mechanism, especially as a last resort mechanism after eye lens antioxidants have been depleted.[224] Key to this theory was the observation that aggregation of the W42Q variant was initiated by WT HγD in a prion-like model.[160] The proximal cause of aggregation is the transfer of a disulfide bond from an oxidized WT HγD to the variant. The transferred disulfide bond acts as a thermodynamic sink, trapping the protein in a conformation that favors aggregation.[224] Using this exchange model as a buffering system would require the formation of thermodynamically equivalent disulfide bonds. Furthermore, oxidized WT HγD, which contains only one disulfide bond between Cys108 and Cys110, is capable of oxidizing the Cys32-Cys41 disulfide bond (Figure 7), leading to aggregation.[224] Other investigators have found independently that Cu2+ can oxidize the intramolecular Cys108-Cys110 disulfide bond on HγD (Figure 7).[159] In protein disulfide isomerases (PDIs), a family of enzymes that catalyze the oxidation, reduction, and isomerization of disulfides, intra- and intermolecular disulfide exchange is rapid and reversible.[225] PDIs have a CXXC motif; however, the functional sequence can be further minimized, as viable PDI mimics were prepared with a CXC motif.[226] Other examples of redox-functionalized disulfide bonds in CXC motifs are found in thiol oxidase Erv2p[227] and Hsp33.[228] This is relevant to crystallin disulfide exchange because HγD contains a similar CSC motif, which is believed to form a strained disulfide bond that is susceptible to reductive cleavage.
Figure 7: Disulfide bond schematic view.

(A) Cu2+ oxidization of the intramolecular Cys108-Cys110 disulfide bond on HγD (PDB: 1HK0). (B) Oxidization of the Cys32-Cys41 disulfide bond leading to aggregation of HγD (PDB: 1HK0).
In summary, βγ-crystallin aggregation can result from mutations or a number of environmental factors. A schematic illustrating the currently known types of βγ-crystallin aggregation is shown in Figure 8.
Figure 8:

Schematic representation of several possible aggregation pathways for γ-crystallins. Rectangular shapes indicate stable states, whereas rounded shapes denote transient species. Species shown on blue backgrounds are in solution; pink backgrounds indicate insoluble aggregates. Red lines indicate modifications to the protein structure. This schematic is not a comprehensive representation of all potential HγS- aggregation pathways, but instead illustrates currently known mechanisms.
8.3. LLPS
Liquid-liquid phase separation (LLPS) occurs when a protein solution spontaneously separates into dense liquid droplets dispersed in a dilute solution.[229,230] LLPS can be triggered by changes in temperature, pH, salt concentration, or interactions with other macromolecules.[229] This type of phase separation is a functional mechanism for cells to organize and maintain different cellular compartments[231], producing membraneless organelles to segregate particular chemical components without lipid bilayers.[231,232] The dark side of LLPS is protein aggregation: aggregation of misfolded proteins in neurodegenerative diseases often starts with the formation of dense liquid droplets.[231]
Some structural crystallins and their mixtures are susceptible to formation of LLPS, often in response to lowered temperature, a phenomenon called cold cataract, which is schematically depicted in Figure 9. This phenomenon has been known in the eye lens since at least the early 1980s, when light scattering experiments were first used to characterize the size of the droplets in different vertebrate lenses.[233,234,235] Bovine γB-crystallin undergoes LLPS near 0 °C, but this transition can be suppressed by the addition of an appropriate concentration of α-crystallin.[236] Cold cataract resistance in fish is of particular interest, because deep-sea fish are adapted to withstand low temperature and high pressure, a challenging environment for the lens proteins. The Antarctic toothfish, Dissostichus mawsoni, has at least 13 expressed γ-crystallin paralogs.[237] Although the whole lens of D. mawsoni resists cold cataract down to at least −12 °C, below which it freezes solid[238], the individual γ-crystallins vary greatly in cold-cataract resistance despite their high sequence identity.[239] Wild-type toothfish γM8b phase separates at −4.8 °C, but mutating only three Arg residues on the surface to Lys makes it more resistant to cold cataract than any of the wild-type proteins tested, with an onset temperature of −13.7 °C, illustrating the importance of specific protein-protein interactions. Conversely, mutating three Lys residues to Arg raises the onset temperature of LLPS by an equivalent amount, to 3.5 °C[239], demonstrating that LLPS can be controlled by making subtle changes to protein surface properties, even in well-folded proteins. The importance of specific surface interactions were hinted at in early studies, for example the finding that phase separation induces major changes to the Raman spectrum of tyrosine in lens proteins.[240] Recently, high pressure was found to drive phase-separated solutions of human and rat γD-crystallin back into homogenous solution, hinting at a mechanism for deep sea fish to avoid cold cataract. Addition of trimethylamine-N-oxide (TMAO), which is found in the tissues of organisms living at high pressure[241], induced LLPS under the same conditions[242], suggesting that this piezolyte may assist in the formation of functional membraneless organelles in deep ocean environments.
Figure 9: Schematic view of liquid-liquid phase separation (LLPS).

(A) Drawing of the human eye, showing the position of the lens. (B) A γ-crystallin protein in a homogeneous phase. (C) In LLPS, the solution partitions into protein-rich and protein-poor phases, forming droplets and producing reversible opacification.
9. Conclusion and outlook
All of the βγ-crystallins share very similar three-dimensional structures, but they differ in details such as surface properties and the lengths of the extensions at the termini. Even in cataract-related variants, the basic structural unit comprising two double Greek-key domains is maintained, and unfolding is often minor and transient or happens only under stress conditions. However, because of the unforgiving constraints on lens protein solubility, even subtle structural changes can have a large impact on their solution properties. Small differences in hydrophobic exposure and salt-bridging interactions, which can be observed using CD, intrinsic fluorescence, dye binding, and light scattering experiments, appear to be main drivers of βγ-crystallin aggregation, along with alterations in the surface hydration layer. These observations are in contrast to the large-scale misfolding and structural rearrangement often observed in other protein deposition diseases. The altered protein dynamics resulting from these small changes can be observed in NMR experiments[243], informing our understanding of solvent accessibility and expansion of the βγ-crystallin interactome to include new disulfide bonds, metal-protein interactions, and protein-small molecule interactions, potentially allowing for future design of therapeutics that target vulnerable conformational states.
For the next phase of crystallin research, it is critical to move beyond the solution-phase behavior of monomers and dimers toward high-resolution structures of crystallin aggregates and a detailed understanding of their formation kinetics. One potential complication for studying βγ-crystallin aggregation is the difficulty in distinguishing native protein, amorphous aggregates, and amyloid fibrils, all of which have primarily β-sheet secondary structure. Sorting out and classifying the different species involved will require a mixture of physical chemistry and structural biology techniques. Fortunately, the intense interest in amyloid fibrils over the last two decades has yielded a useful suite of experimental modalities for distinguishing different β-sheet structures. In particular, solid-state NMR continues to provide many of the high-resolution structures available for amyloid fibrils.[74] Optical spectroscopy provides another route to distinguishing aggregates and oligomers with similar secondary structures, as variations in β-sheet structure correlate with infrared (IR) spectroscopic observables.[244] 2D IR spectroscopy has been used to observe the divergence of different fibril polymorphs in human islet amyloid polypeptide (hIAPP) from a common intermediate in solution[245], and to monitor the transformation of Aβ oligomers to fibrils.[246] A recently developed topological classification for amyloid fibril structures[247,248] could also be extended to provide a more systematic description of β-sheet oligomers and non-amyloid aggregates.
For proteins that are supposed to be chemically inert, crystallins undergo rich and varied chemistry that always seems to surprise the investigator. In this review we have discussed PTMs that arise from interaction with products of the kynurenine pathway, metal ion interactions, oxidations and deamidations that have been uncovered in lenses with age-related cataract. Recent evidence suggests that disulfide bond formation between solvent exposed cysteines in γ-crystallins may serve as a buffering system, through disulfide exchange, to prevent harmful oxidations.[58,224,249] Formation of these dimers appears to be a last resort buffer, since high concentrations of these dimers eventually promote the formation of larger aggregates. The mechanisms by which aggregates are formed in the eye lens, whether they be amorphous-looking aggregates, amyloid fibrils, or a combination of the two, is still largely unknown. Understanding these phenomena requires details of protein-protein interactions that happen during normal homeostasis and after perturbation. Here we reviewed protein-protein interactions via domain-swapping and condensation, disulfide bond formation, and LLPS. Identification of site-specific interactions via NMR and X-ray crystallography in known cataract-causing variants and PTMs represent an important basis for developing an overall understanding. The vertebrate lens proteins serve as a counterpoint to the emphasis on amyloid fibril formation as the mechanism underlying protein deposition diseases: understanding protein solubility also requires studying highly soluble proteins, the flip side of aggregation. In addition to the direct impact on cataract, understanding how these extraordinary proteins maintain their solubility will enable the design of highly soluble proteins and provide more general insight into hydration of complex molecules with a variety of functional groups.
Acknowledgements
Our crystallin work is supported by NIH grants 2R01EY021514 to R.W.M and 1R01EY025328 to R.W.M. and D.J. Tobias and NSF grant DMR-2002837 to R.W.M. and D.J. Tobias. M.A.S.P. is supported by the HHMI Gilliam Fellowship for Advanced Study. K.W.R was supported by the NSF Training Grant DGE-1633631. This work builds on discussions at the 2018 Crystallin Satellite Biophysical Society Meeting in San Francisco, and we are grateful to the participants for ideas and suggestions.
Biographies

Megan Rocha is a graduate student in Prof. Rachel Martin’s lab at the University of California, Irvine. She received her BA in chemistry at Whitman College in 2017. Her undergraduate research focused on the synthesis of a noncovalent macrocyclic proteasome inhibitors. Currently, she is working toprobe the biophysical properties that determine the solubility and refractivity of crystallins.

Marc Sprague-Piercy first found a passion for science studying the development of the visual system of Xenopus laevis as a community college student. He soon found a deeper interest in understanding how the structure of a protein, at the level of atoms and bonds, determines the function of that protein. He is currently studying the molecular mechanisms that underpin protein aggregation and chaperone activity in the human eye lens.

Ashley Kwok obtained her B.S. in Chemistry from the University of California, Irvine in 2020. She has worked as an Undergraduate Researcher under the guidance of Professor Rachel Martin since 2019. Her undergraduate research focuses on aggregation-prone variants of human lens γS-crystallin and their relationship to cataract disease.

Kyle W. Roskamp is a research scientist at Second Genome. He received his Bachelor of Arts from Pomona College, double majoring in Chemistry and Mathematics, and his Ph.D. in Chemistry from UC-Irvine. At UCI he worked with Prof. Rachel Martin to investigate the protein aggregation pathways of the γ-crystallins. He also worked to characterize the evolutionary adaptations of γ-crystallins for functionality in the lens.

Rachel W. Martin is a Professor of Chemistry and Molecular Biology & Biochemistry at the University of California, Irvine. She obtained her B.S. from Arizona State University in 1997 and her Ph.D. from Yale University in 2002. Her postdoctoral work at UC Berkeley focused on low-field, mobile NMR instrumentation and the NMR of oriented biological samples. In 2005, she started her own laboratory at UC Irvine. Current research directions include investigating the stability and solubility of long-lived proteins, developing instrumentation and methodology for NMR spectroscopy, and the discovery of enzymes from genomic data.
Footnotes
Conflict of interest
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
References
- [1].Patel S, Marshall J, Fitzke FWI 1995. J. Cataract Refractive Surg 11, 100–105. [DOI] [PubMed] [Google Scholar]
- [2].Jagger W 1992. Vis. Res 32, 1271–1284. [DOI] [PubMed] [Google Scholar]
- [3].Kröger RHH, Campbell MCW, Munger R, Fernald RD 1994. Vis. Res 34, 1815–1822. [DOI] [PubMed] [Google Scholar]
- [4].Garner LF, Smith GT, Yao S, Augusteyn RC 2001. Vis. Res 41, 973–979. [DOI] [PubMed] [Google Scholar]
- [5].Wistow GJ, Piatigorsky J 1988. Annu. Rev. Biochem 57, 479–504. [DOI] [PubMed] [Google Scholar]
- [6].Slingsby C, Wistow GJ, Clark AR 2013. Protein Sci. 22, 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Horwitz J, Bova MP, Ding L-L, Haley DA, Stewart PL 1999. Eye 13, 403–408. [DOI] [PubMed] [Google Scholar]
- [8].Bloemendal H, de Jong W, Jaenicke R, Lubsen NH, Slingsby C, Tardieu A 2004. Prog. Biophys. Mol. Biol 86, 407–485. [DOI] [PubMed] [Google Scholar]
- [9].World Health Organization ”Priority Eye Diseases”, can be found under https://www.who.int/blindness/causes/priority/en/index1.html, 2020.
- [10].Delaye M, Tardieu A 1983. Nature 302, 415–417. [DOI] [PubMed] [Google Scholar]
- [11].Cvekl A, McGreal R, Liu Wei Prog. Mol. Biol. Transl. Sci, vol. 134 2015. 129–167. [DOI] [PubMed] [Google Scholar]
- [12].Cvekl A, Zhao Y, McGreal R, Xie Q, Gu X, Zheng D 2017. Genome Biol. Evol 9, 2075–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Piatigorsky J 1984. Cell 38, 620. [DOI] [PubMed] [Google Scholar]
- [14].Jaenicke R 1994. Naturwissenschaften 81, 423–429. [DOI] [PubMed] [Google Scholar]
- [15].Piatigorsky J, Wistow G 1991. Science 252, 1078–1079. [DOI] [PubMed] [Google Scholar]
- [16].Blundell T, Lindley P, Miller L, Moss D, Slingsby C, Tickle I, Turnell B, Wistow G 1981. Nature 289, 771–777. [DOI] [PubMed] [Google Scholar]
- [17].Pettersen E, Goddard T, Huang C, Couch G, Greenblatt D, Meng E, F. T.E. 2004. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- [18].Wistow G 1993. Trends Biochem. Sci 18, 301–306. [DOI] [PubMed] [Google Scholar]
- [19].D’Alessio G 2002. Eur. J. Biochem 269, 3122–3130. [DOI] [PubMed] [Google Scholar]
- [20].Shimeld SM, Purkiss AG, Dirks RP, Bateman OA, Slingsby C, Lubsen NH 2005. Curr. Biol 15, 1684–1689. [DOI] [PubMed] [Google Scholar]
- [21].Suman SK, Mishra A, Yeramala L, Rastogi ID, Sharma Y 2013. Biochemistry 52, 9047–9058. [DOI] [PubMed] [Google Scholar]
- [22].Kingsley CN, Brubaker WD, Markovic S, Diehl A, Brindley AJ, Oschkinat H, Martin RW 2013. Structure 21, 2221–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Smith MA, Bateman OA, Jaenicke R, Slingsby C 2007. Protein Sci. 16, 615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kozlyuk N, Sengupta S, Bierma JC, Martin RW 2016. Biochemistry 55, 6961–6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Khago D, Bierma JC, Roskamp KW, Kozlyuk N, Martin RW 2018. J. Phys.: Condens. Matter 30, 435101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Riyahi K, Shimeld SM 2007. Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol 147, 347–357. [DOI] [PubMed] [Google Scholar]
- [27].Mishra A, Krishnan B, Srivastava SS, Sharma Y 2014. Prog. Biophys. Mol. Biol 115, 42–51. [DOI] [PubMed] [Google Scholar]
- [28].Mishra A, Krishnan B, Raman R, Sharma Y 2016. Biochim. Biophys. Acta, Gen. Subj 1860, 299–303. [DOI] [PubMed] [Google Scholar]
- [29].Jaenicke R, Slingsby C 2001. Crit. Rev. Biochem. Mol. Biol 36, 435–499. [DOI] [PubMed] [Google Scholar]
- [30].Wu Z, Delaglio F, Wyatt K, Wistow G, Bax A 2005. Protein Sci. 14, 3101–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sagar V, Chaturvedi SK, Schuck P, Wistow G 2017. Structure 25, 1068–1078.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mahler B, Chen Y, Ford J, Thiel C, Wistow G, Wu Z 2013. Biochemistry 52, 3579–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ebersbach H, Fiedler E, Scheuermann T, Fiedler M, Stubbs MT, Reimann C, Proetzel G, Rudolph R, Fiedler U 2007. J. Mol. Biol 372, 172–185. [DOI] [PubMed] [Google Scholar]
- [34].Dixit K, Pande A, Pande J, Sarma SP 2016. Biochemistry 55, 3136–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang J, Zuo X, Yu P, Byeon I-JL, Jung J, Wang X, Dyba M, Seifert S, Schwieters CD, Qin J, Gronenborn AM, Wang Y-X 2009. J. Am. Chem. Soc 131, 10507–10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Basak A, Bateman O, Slingsby C, Pande A, Asherie N, Ogun O, Benedek GB, Pande J 2003. J. Mol. Biol 328, 1137–1147. [DOI] [PubMed] [Google Scholar]
- [37].Xi Z, Whitley MJ, Gronenborn AM 2017. Structure 25, 496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mills-Henry IA, Thol SL, Kosinski-Collins MS, Serebryany E, King JA 2019. Biophys. J 117, 269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Flaugh SL, Kosinski-Collins MS, King J 2005. Protein Sci. 14, 569–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hemmingsen JM, Gernert KM, Richardson JS, Richardson DC 1994. Protein Sci. 3, 1927–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kong F, King J 2011. Protein Sci. 20, 513–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Serebryany E, King JA 2014. Prog. Biophys. Mol. Biol 115, 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhang K, Zhao W-J, Yao K, Yan Y-B 2019. Biochemistry 58, 2499–2508. [DOI] [PubMed] [Google Scholar]
- [44].Houston P, Macro N, Kang M, Chen L, Yang J, Wang L, Wu Z, Zhong D 2020. J. Am. Chem. Soc 142, 3997–4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Huang K-Y, Kingsley CN, Sheil R, Cheng C-Y, Bierma JC, Roskamp KW, Khago D, Martin RW, Han S 2016. J. Am. Chem. Soc 138, 5392–5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Martin RW 2014. eMagRes 3, 139–152. [Google Scholar]
- [47].Grutsch S, Brüschweiler S, Tollinger M 2016. PLoS Comput. Biol 12, e1004620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sekhar A, Kay LE 2019. Annu. Rev. Biophys 48, 297–319. [DOI] [PubMed] [Google Scholar]
- [49].Arthanari H, Takeuchi K, Dubey A, Wagner G 2019. Curr. Opin. Struct. Biol 58, 294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ji F, Jung J, Koharudin LMI, Gronenborn AM 2013. J. Biol. Chem 288, 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bharat SV, Shekhtman A, Pande J 2014. Biochem. Biophys. Res. Commun 443, 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Khago D, Wong EK, Kingsley CN, Freites JA, Tobias DJ, Martin RW 2016. Biochim. Biophys. Acta, Gen. Subj 1860, 325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Sprague-Piercy MA, Wong E, Roskamp KW, Fakhoury JN, Freites JA, Tobias DJ, Martin RW 2020. Biochim. Biophys. Acta, Gen. Subj 1864, 129502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sher M, Hameed A, Noreen S, Fayyaz-ur-Rehman M, Hussain MA, Bukhari SNA 2015. Analyst 140, 6392–6397. [DOI] [PubMed] [Google Scholar]
- [55].Quintanar L, Domínguez-Calva JA, Serebryany E, Rivillas-Acevedo L, Haase-Pettingell C, Amero C, King JA 2016. ACS Chem. Biol 11, 263–272. [DOI] [PubMed] [Google Scholar]
- [56].Boatz JC, Whitley MJ, Li M, Gronenborn AM, van der Wel PC 2017. Nat. Commun 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Roskamp KW, Montelongo DM, Anorma CD, Bandak DN, Chua JA, Malecha KT, Martin RW 2017. Invest. Ophthalmol. Visual Sci 58, 2397–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Roskamp KW, Azim S, Kassier G, Norton-Baker B, Sprague-Piercy MA, Miller RD, Martin RW 2020. Biochemistry 59, 2371–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhang TO, Alperstein AM, Zanni MT 2017. J. Mol. Biol 429, 1705–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Alperstein AM, Ostrander JS, Zhang TO, Zanni MT 2019. Proc. Natl. Acad. Sci. U. S. A 116, 6602–6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lam AR, Moran S, Preketes NK, Zhang T, Zanni MT, Mukamel S 2013. J. Phys. Chem. B 117, 15436–15443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chemerovski-Glikman M, Mimouni M, Dagan Y, Haj E, Vainer I, Allon R, Blumenthal EZ, Adler-Abramovich L, Segal D, Gazit E, ZayitSoudry S 2018. Sci. Rep 8, 9341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zhao L, Chen X-J, Zhu J, Xi Y-B, Yang X, Hu L-D, Ouyang H, Patel SH, Jin X, Lin D, Wu F, Flagg K, Cai H, Li G, Cao G, Lin Y, Chen D, Wen C, Chung C, Wang Y, Qiu A, Yeh E, Wang W, Hu X, Grob S, Abagyan R, Su Z, Tjondro HC, Zhao X-J, Luo H, Hou R, Jefferson J, Perry P, Gao W, Kozak I, Granet D, Li Y, Sun X, Wang J, Zhang L, Liu Y, Yan Y-B, Zhang K 2015. Nature 523, 607–611. [DOI] [PubMed] [Google Scholar]
- [64].Makley LN, McMenimen KA, DeVree BT, Goldman JW, McGlasson BN, Rajagopal P, Dunyak BM, McQuade TJ, Thompson AD, Sunahara R, Klevit RE, Andley UP, Gestwicki JE 2015. Science 350, 674–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Chen X-J, Hu L-D, Yao K, Yan Y-B 2018. Biochem. Biophys. Res. Commun 506, 868–873. [DOI] [PubMed] [Google Scholar]
- [66].Daszynski DM, Santhoshkumar P, Phadte AS, Sharma KK, Zhong HA, Lou MF, Kador PF 2019. Sci. Rep 9, 8459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Shanmugam PM, Barigali A, Kadaskar J, Borgohain S, Mishra DKC, Ramanjulu R, Minija C 2015. Indian J. Ophthalmol 63, 888–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Meier BH, Riek R, Böckmann A 2017. Trends Biochem. Sci 42, 777–787. [DOI] [PubMed] [Google Scholar]
- [69].van der Wel PC 2017. Solid State Nucl. Magn. Reson 88, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Jaroniec CP 2019. J. Magn. Reson 306, 42–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Martin RW, Kelly JE, Kelz JI 2019. J. Struct. Biol 206, 73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Jehle S, Rajagopal P, Bardiaux B, Markovic S, Kühne R, Stout JR, Higman VA, Klevit RE, van Rossum BJ, Oschkinat H 2010. Nat. Struct. Mol. Biol 17, 1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Benzinger TLS, Gregory DM, Burkoth TS, Miller-Auer H, Lynn DG, Botto RE, Meredith SC 1998. Proc. Natl. Acad. Sci. U. S. A 95, 13407–13412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Meier B, Riek R, Böckmann A 2017. Trends Biochem. Sci 42, 777–787. [DOI] [PubMed] [Google Scholar]
- [75].Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Döbeli H, Schubert D, Riek R 2005. Proc. Natl. Acad. Sci. U. S. A 102, 17342–17347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Xiao Y, Ma B, McElheny D, Parthasarathy S, Long F, Hoshi M, Nussinov R, Ishii Y 2015. Nat. Struct. Mol. Biol 22, 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev I, Rosay M, Donovan KJ, Michael B, Wall J, Linse S, et al. 2016. J. Am. Chem. Soc 138, 9663–9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Vandersteen A, Hubin E, Sarroukh R, De Baets G, Schymkowitz J, Rousseau F, Subramaniam V, Raussens V, Wenschuh H, Wildemann D, Broersen K 2012. FEBS Lett. 586, 4088–4093. [DOI] [PubMed] [Google Scholar]
- [79].Petkova AT, Yau W-M, Tycko R 2006. Biochemistry 45, 498–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Paravastu AK, Leapman RD, Yau W-M, Tycko R 2008. Proc. Natl. Acad. Sci. U. S. A 105, 18349–18354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Sgourakis NG, Yau W-M, Qiang W 2015. Structure 23, 216–227. [DOI] [PubMed] [Google Scholar]
- [82].Henderson R 1995. Q. Rev. Biophys 28, 171–193. [DOI] [PubMed] [Google Scholar]
- [83].Berova N, Nakanishi K, Woody RW. John Wiley & Sons, Hoboken, 2000. [Google Scholar]
- [84].Chen J, Callis PR, King J 2009. Biochemistry 48, 3708–3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Some D 2013. Biophys. Rev 5, 147–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Sahin E, Roberts Ther CJ. Proteins 2012. Springer; 403–423. [Google Scholar]
- [87].Ma Y, Acosta DM, Whitney JR, Podgornik R, Steinmetz NF, French RH, Parsegian VA 2015. J. Biol. Phys 41, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Wu D, Zhou H-X 2016. Biophys. J 110, 43a. [Google Scholar]
- [89].Neal B, Asthagiri D, Lenhoff A 1998. Biophys. J 75, 2469–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Berne BJ, Pecora R Dynamic Light Scattering: With Applications to Chemistry, Biology, and Physics Courier Corporation, Chelmsford: 2000. [Google Scholar]
- [91].Naiki H, Higuchi K, Hosokawa M, Takeda T 1989. Anal. Biochem 177, 244–249. [DOI] [PubMed] [Google Scholar]
- [92].Khurana R, Coleman C, Ionescu-Zanetti C, Carter SA, Krishna V, Grover RK, Roy R, Singh S 2005. J. Struct. Biol 151, 229–238. [DOI] [PubMed] [Google Scholar]
- [93].Xue C, Lin TY, Chang D, Guo Z 2017. R. Soc. Open Sci 4, 160696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Sebastiao M, Quittot N, Bourgault S 2017. Anal. Biochem 532, 83–86. [DOI] [PubMed] [Google Scholar]
- [95].Qin Z, Sun Y, Jia B, Wang D, Ma Y, Ma G 2017. Langmuir 33, 5398–5405. [DOI] [PubMed] [Google Scholar]
- [96].Matulis D, Lovrien R 1998. Biophys. J 74, 422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Bothra A, Bhattacharyya A, Mukhopadhyay C, Bhattacharyya K, Roy S 1998. J. Biomol. Struct. Dyn 15, 959–966. [DOI] [PubMed] [Google Scholar]
- [98].Zagotta WN, Gordon MT, Senning EN, Munari MA, Gordon SE 2016. J. Gen. Physiol 147, 201–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Taraska JW, Zagotta WN 2010. Neuron 66, 170–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Vendra VPR, Khan I, Chandani S, Muniyandi A, Balasubramanian D 2016. Biochim. Biophys. Acta, Gen. Subj 1860, 333–343. [DOI] [PubMed] [Google Scholar]
- [101].Jung J, Byeon I-JL, Wang Y, King J, Gronenborn AM 2009. Biochemistry 48, 2597–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Ji F, Koharudin LM, Jung J, Gronenborn AM 2013. Proteins: Struct., Funct., Bioinf 81, 1493–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Ji F, Jung J, Gronenborn AM 2012. Biochemistry 51, 2588–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Whitley MJ, Xi Z, Bartko JC, Jensen MR, Blackledge M, Gronenborn AM 2017. Biophys. J 112, 1135–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Guseman AJ, Whitley MJ, González JJ, Rathi N, Ambarian M, Gronenborn AM 2020. Structure , DOI: 10.1016/j.str.2020.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Khan I, Chandani S, Balasubramanian D 2016. Mol. Vision 22, 771–782. [PMC free article] [PubMed] [Google Scholar]
- [107].Bari KJ, Sharma S, Chary KVR 2019. J. Struct. Biol 205, 72–78. [DOI] [PubMed] [Google Scholar]
- [108].Forsythe HM, Vetter CJ, Jara KA, Reardon PN, David LL, Barbar EJ, Lampi KJ 2019. Biochemistry 58, 4112–4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Ma Z, Piszczek G, Wingfield PT, Sergeev YV, Hejtmancik JF 2009. Biochemistry 48, 7334–7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Brubaker WD, Freites JA, Golchert KJ, Shapiro RA, Morikis V, Tobias DJ, Martin RW 2011. Biophys. J 100, 498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].He Y, Kang J, Song J 2020. Biochem. Biophys. Res. Commun 533, 913–918. [DOI] [PubMed] [Google Scholar]
- [112].Mills IA, Flaugh SL, Kosinski-Collins MS, King JA 2007. Protein Sci. 16, 2427–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Bari KJ, Sharma S, Chary KVR 2019. Biochem. Biophys. Res. Commun 517, 499–506. [DOI] [PubMed] [Google Scholar]
- [114].Bari KJ, Sharma S, Chary KVR 2019. Biochem. Biophys. Res. Commun 514, 946–952. [DOI] [PubMed] [Google Scholar]
- [115].Bari KJ, Sharma S, Chary KVR 2019. Biochem. Biophys. Res. Commun 511, 679–684. [DOI] [PubMed] [Google Scholar]
- [116].Lee S, Mahler B, Toward J, Jones B, Wyatt K, Dong L, Wistow G, Wu Z 2010. J. Mol. Biol 399, 320–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Zhang T, Yan L, Leng Y, Chen C, Ma L, Wang Q, Zhang J, Cao L 2018. Gene 675, 9–14. [DOI] [PubMed] [Google Scholar]
- [118].Bari KJ, Sharma S, Chary KV 2018. Biochem. Biophys. Res. Commun 506, 862–867. [DOI] [PubMed] [Google Scholar]
- [119].Acosta-Sampson L, King J 2010. J. Mol. Biol 401, 134–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Moreau KL, King JA 2012. Trends Mol. Med 18, 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Zhao W-J, Xu J, Chen X-J, Liu H-H, Yao K, Yan Y-B 2017. Int. J. Biol. Macromol 103, 764–770. [DOI] [PubMed] [Google Scholar]
- [122].Song I-K, Na S, Paek E, Lee K-J 2020. bioRxiv , DOI: 10.1101/2020.05.26.116228. [DOI] [Google Scholar]
- [123].Zhang K, Zhao W-J, Leng X-Y, Wang S, Yao K, Yan Y-B 2014. Biochim. Biophys. Acta, Mol. Basis Dis 1842, 44–55. [DOI] [PubMed] [Google Scholar]
- [124].Xi Y-B, Zhao W-J, Zuo X-T, Tjondro HC, Li J, Dai A-B, Wang S, Yan Y-B 2014. Biochim. Biophys. Acta, Mol. Basis Dis 1842, 2216–2229. [DOI] [PubMed] [Google Scholar]
- [125].Leng X-Y, Li H-Y, Wang J, Qi L-B, Xi Y-B, Yan Y-B 2016. Biochem. J 473, 2087–2096. [DOI] [PubMed] [Google Scholar]
- [126].Chan MP, Dolinska M, Sergeev YV, Wingfield PT, Hejtmancik JF 2008. Biochemistry 47, 11062–11069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Sergeev Y, Hejtmancik J, Wingfield P 2004. Biochemistry 43, 415–424. [DOI] [PubMed] [Google Scholar]
- [128].Slingsby C, Bateman O 1990. Biochemistry 29, 6592–6599. [DOI] [PubMed] [Google Scholar]
- [129].Marín-Vinader L, Onnekink C, van Genesen ST, Slingsby C, Lubsen NH 2006. FEBS J. 273, 3172–3182. [DOI] [PubMed] [Google Scholar]
- [130].Srivastava K, Gupta R, Chaves JM, Srivastava OP 2009. Biochemistry 48, 7179–7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Bateman O, Sarra R, Van Genesen S, Kappe G, Lubsen N, Slingsby C 2003. Exp. Eye Res 77, 409–422. [DOI] [PubMed] [Google Scholar]
- [132].Li W, Ji Q, Wei Z, Chen Y.-l., Zhang Z, Yin X, Aghmiuni SK, Liu M, Chen W, Shi L, et al. 2019. Exp. Eye Res 186, 107712. [DOI] [PubMed] [Google Scholar]
- [133].Truscott RJ, Friedrich MG 2016. Biochim. Biophys. Acta, Gen. Subj 1860, 192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Srivastava OP, Srivastava K, Chaves JM, Gill AK 2017. Biochem. Biophys. Rep 10, 94–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Geiger T, Clarke S 1987. J. Biol. Chem 262, 785–794. [PubMed] [Google Scholar]
- [136].Reissner KJ, Aswad DW 2003. Cell. Mol. Life Sci 60, 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Müller MM 2018. Biochemistry 57, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Lampi KJ, Ma Z, Hanson SR, Azuma M, Shih M, Shearer TR, Smith DL, Smith JB, David LL 1998. Exp. Eye Res 67, 31–43. [DOI] [PubMed] [Google Scholar]
- [139].Lampi KJ, Wilmarth PA, Murray MR, David LL 2014. Prog. Biophys. Mol. Biol 115, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Kim I, Saito T, Fujii N, Kanamoto T, Fujii N 2016. Amino Acids 48, 2855–2866. [DOI] [PubMed] [Google Scholar]
- [141].Warmack R, Shawa H, Liu K, Lopez K, Loo J, Horwitz J, Clarke S 2019. J. Biol. Chem 294, 12203–12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Lyon YA, Collier MP, Riggs DL, Degiacomi MT, Benesch JLP, Julian RR 2019. J. Biol. Chem 294, 7546–7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Guseman AJ, Gronenborn AM 2019. J. Biol. Chem 294, 7556–7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Lampi KJ, Oxford JT, Bachinger HP, Shearer TR, David LL, Kapfer DM 2001. Exp. Eye Res 72, 279–288. [DOI] [PubMed] [Google Scholar]
- [145].Kim YH, Kapfer DM, Boekhorst J, Lubsen NH, Bächinger HP, Shearer TR, David LL, Feix JB, Lampi KJ 2002. Biochemistry 41, 14076–14084. [DOI] [PubMed] [Google Scholar]
- [146].Lampi KJ, Amyx KK, Ahmann P, Steel EA 2006. Biochemistry 45, 3146–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Takata T, Oxford JT, Demeler B, Lampi KJ 2008. Protein Sci. 17, 1565–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Lampi KJ, Murray MR, Peterson MP, Eng BS, Yue E, Clark AR, Barbar E, David LL 2016. Biochim. Biophys. Acta, Gen. Subj 1860, 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Pande A, Mokhor N, Pande J 2015. Biochemistry 54, 4890–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Ray NJ, Hall D, Carver JA 2016. Biochimica et Biophysica Acta 1860, 315–324. [DOI] [PubMed] [Google Scholar]
- [151].Vetter CJ, Thorn DC, Wheeler SG, Mundorff C, Halverson K, Carver JA, David LL, Lampi KJ 2020. bioRxiv , doi: 10.1101/2020.02.21.960237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Giblin FJ 2000. J. Ocul. Pharmacol. Ther 16, 121–135. [DOI] [PubMed] [Google Scholar]
- [153].Fecondo JV, Augusteyn RC 1983. Exp. Eye Res 36, 15–23. [DOI] [PubMed] [Google Scholar]
- [154].Wei M, Xing K-Y, Fan Y-C, Libondi T, Lou MF 2015. Invest. Ophthalmol. Visual Sci 56, 598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Hains PG, Truscott RJ 2007. J. Proteome Res 6, 3935–3943. [DOI] [PubMed] [Google Scholar]
- [156].Stadtman ER 1992. Science 257, 1220–1224. [DOI] [PubMed] [Google Scholar]
- [157].Truscott RJ 2005. Exp. Eye Res 80, 709–725. [DOI] [PubMed] [Google Scholar]
- [158].Kim I, Saito T, Fujii N, Kanamoto T, Chatake T, Fujii N 2015. Biochem. Biophys. Res. Commun 466, 622–628. [DOI] [PubMed] [Google Scholar]
- [159].Ramkumar S, Fan X, Wang B, Yang S, Monnier VM 2018. Biochim. Biophys. Acta, Mol. Basis Dis 1864, 3595–3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Serebryany E, King JA 2015. J. Biol. Chem 290, 11491–11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Chen J, Flaugh SL, Callis PR, King J 2006. Biochemistry 45, 11552–11563. [DOI] [PubMed] [Google Scholar]
- [162].Schafheimer N, King J 2013. Photochem. Photobiol 89, 1106–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Garner B, Roberg K, Qian M, Eaton JW, Truscott RJ 2000. Exp. Eye Res 71, 599–607. [DOI] [PubMed] [Google Scholar]
- [164].Hajjari S, Masoudi R, Javadi S, Hemmateenejad B, Yousefi R 2016. Protein Pept. Lett 23, 78–86. [DOI] [PubMed] [Google Scholar]
- [165].Zhao W-J, Yan Y-B 2018. Int. J. Biol. Macromol 108, 665–673. [DOI] [PubMed] [Google Scholar]
- [166].Cetinel S, Semenchenko V, Cho J-Y, Sharaf MG, Damji KF, Unsworth LD, Montemagno C 2017. PloS One 12, e0177991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Chaudhury S, Ghosh I, Saha G, Dasgupta S 2015. Int. J. Biol. Macromol 77, 287–292. [DOI] [PubMed] [Google Scholar]
- [168].Chaudhury S, Bag S, Bose M, Das AK, Ghosh AK, Dasgupta S 2016. Mol. BioSyst 12, 2901–2909. [DOI] [PubMed] [Google Scholar]
- [169].Chaudhury S, Dutta A, Bag S, Biswas P, Das AK, Dasgupta S 2018. Spectrochim. Acta, Part A 192, 318–327. [DOI] [PubMed] [Google Scholar]
- [170].Rana S, Ghosh KS 2020. Arch. Biochem. Biophys 679, 108204. [DOI] [PubMed] [Google Scholar]
- [171].Chaudhury S, Roy P, Dasgupta S 2017. Biochimie 137, 46–55. [DOI] [PubMed] [Google Scholar]
- [172].Sharma KK, Santhoshkumar P 2009. Biochim. Biophys. Acta, Gen. Subj 1790, 1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Pereira P, Shang F, Hobbs M, Girão H, Taylor A 2003. Exp. Eye Res 76, 623–631. [DOI] [PubMed] [Google Scholar]
- [174].Whitcomb EA, Shang F, Taylor A 2013. Invest. Ophthalmol. Visual Sci 54, ORSF31–ORSF36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Sweeney MHJ, Truscott RJW 1998. Exp. Eye Res 67, 587–595. [DOI] [PubMed] [Google Scholar]
- [176].Taylor A, Jahngen-Hodge J, Huang LL, Jacques P 1991. Age 14, 65–71. [Google Scholar]
- [177].Cobb BA, Petrash JM 2002. Biochemistry 41, 483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Mainali L, O’Brien WJ, Timsina R 2021. Curr. Eye Res , 185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Wang S, Zhao W-J, Liu H, Gong H, Yan Y-B 2013. Biochim. Biophys. Acta, Mol. Basis Dis 1832, 302–311. [DOI] [PubMed] [Google Scholar]
- [180].Reissner KJ, Aswad DW 2003. CMLS Cell. Mol. Life Sci 60, 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Klaene J, Ni W, Alfaro J, Zhou Z 2014. J. Pharm. Sci 103, 3033–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Catak S, Monard G, Aviyente V, Ruiz-López MF 2008. J. Phys. Chem. A 112, 8752–8761. [DOI] [PubMed] [Google Scholar]
- [183].Aki K, Okamura E 2017. J. Pept. Sci 23, 28–37. [DOI] [PubMed] [Google Scholar]
- [184].Aswad DW, Paranandi MV, Schurter BT 2000. J. Pharm. Biomed. Anal 21, 1129–1136. [DOI] [PubMed] [Google Scholar]
- [185].Radkiewicz JL, Zipse H, Clarke S, Houk KN 2001. J. Am. Chem. Soc 123, 3499–3506. [DOI] [PubMed] [Google Scholar]
- [186].Su S-P, Lyons B, Friedrich M, McArthur JD, Song X, Xavier D, Truscott RJW, Aquilina JA 2012. Aging Cell 11, 1125–1127. [DOI] [PubMed] [Google Scholar]
- [187].Lyons B, Jamie J, R.J.W. T 2014. Amino Acids 46, 199–207. [DOI] [PubMed] [Google Scholar]
- [188].Friedrich MG, Wang Z, Schey KL, Truscott RJW. 2019. Biochem. J 476, 3817–3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Wang Z, Friedrich MG, Truscott RJW, Schey KL 2019. Biochim. Biophys. Acta, Proteins Proteomics 1867, 831–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Tweeddale HJ, Hawkins CL, Janmie JF, Truscott RJ, Davies MJ 2016. Free Radical Res. 50, 1116–1130. [DOI] [PubMed] [Google Scholar]
- [191].Wood AM, Truscott RJ 1993. Exp. Eye Res 56, 317–325. [DOI] [PubMed] [Google Scholar]
- [192].Bova LM, Sweeney MH, Jamie JF, Truscott RJ 2001. Invest. Ophthalmol. Visual Sci 42, 200–205. [PubMed] [Google Scholar]
- [193].Malina HZ, Martin XD 1996. Graefe’s Arch. Clin. Exp. Ophthalmol 234, 457–462. [DOI] [PubMed] [Google Scholar]
- [194].Chiarugi A, Rapizzi E, Moroni F, Moroni F 1999. FEBS Lett. 453, 197–200. [DOI] [PubMed] [Google Scholar]
- [195].Parker NR, Jamie JF, Davies MJ, Truscott RJ 2004. Free Radic. Biol. Med 37, 1479–1489. [DOI] [PubMed] [Google Scholar]
- [196].Mizdrak J, Hains PG, Truscott RJ, Jamie JF, Davies MJ 2008. Free Radic. Biol. Med 44, 1108–1119. [DOI] [PubMed] [Google Scholar]
- [197].Goldstein LE, Leopold MC, Huang X, Atwood CS, Saunders AJ, Hartshorn M, Lim JT, Faget KY, Muffat JA, Scarpa RC, et al. 2000. Biochemistry 39, 7266–7275. [DOI] [PubMed] [Google Scholar]
- [198].Chen Y, Reid G, Simpson R, Truscott R 1997. Exp. Eye Res 65, 835–840. [DOI] [PubMed] [Google Scholar]
- [199].Pawar AD, Kiran U, Raman R, Chandani S, Sharma Y 2019. Biochem. Biophys. Res. Commun 516, 796e800. [DOI] [PubMed] [Google Scholar]
- [200].Srivastava SS, Raman R, Kiran U, Garg R, Chadalawada S, Pawar AD, Sankaranarayanan R, Sharma Y 2018. Mol. Microbiol 110, 955–972. [DOI] [PubMed] [Google Scholar]
- [201].Ramakrishnan S, Sulochana K, Selvaraj T, Rahim AA, Lakshmi M, Arunagiri K 1995. Br. J. Ophthalmol 79, 202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Cekic O 1998. Br. J. Ophthalmol 82, 186–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Aydin E, Cumurcu T, Özuĝurlu F, Özyurt H, Sahinoglu S, Mendil D, Hasdemir E 2005. Biol. Trace Elem. Res 108, 33–41. [DOI] [PubMed] [Google Scholar]
- [204].Dawczynski J, Blum M, Winnefeld K, Strobel J 2002. Biol. Trace Elem. Res 90, 15–23. [DOI] [PubMed] [Google Scholar]
- [205].Fagerholm PP, Philipson BT, Lindström B 1981. Exp. Eye Res 33, 615–620. [DOI] [PubMed] [Google Scholar]
- [206].Langford-Smith A, Tilakaratna V, Lythgoe PR, Clark SJ, Bishop PN, Day AJ 2016. PLoS One 11, e0147576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Dominguez-Calva JA, Haase-Pettingell C, Serebryany E, King JA, Quintanar L 2018. Biochemistry 57, 4959–4962. [DOI] [PubMed] [Google Scholar]
- [208].Roskamp KW, Kozlyuk N, Sengupta S, Bierma JC, Martin RW 2019. Biochemistry 58, 4505–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Dominguez-Calva J, Perez-Vazquez M, Serebryany E, King J, Quintanar L 2018. JBIC, J. Biol. Inorg. Chem 23, 1105–1118. [DOI] [PubMed] [Google Scholar]
- [210].Karunakaran R, Srikumar PS 2018. Mol. Cell. Biochem 449, 55–62. [DOI] [PubMed] [Google Scholar]
- [211].Wong EK, Prytkova V, Freites JA, Butts CT, Tobias DJ 2019. Biochemistry 58, 3691–3699. [DOI] [PubMed] [Google Scholar]
- [212].O’Brien CJ, Blanco MA, Costanzo JA, Enterline M, Fernandez EJ, Robinson AS, Roberts CJ 2016. Protein Eng., Des. Sel 29, 231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Chang C-K, Wang SS-S, Lo C-H, Hsiao H-C, Wu JW 2017. J. Biomol. Struct. Dyn 35, 1042–1054. [DOI] [PubMed] [Google Scholar]
- [214].Zhu S, Xi X-B, Duan T-L, Zhai Y, Li J, Yan Y-B, Yao K 2018. Int. J. Biol. Macromol 117, 807–814. [DOI] [PubMed] [Google Scholar]
- [215].Wahle CW, Martini KM, Hollenbeck DM, Langner A, Ross DS, Hamilton JF, Thurston GM 2017. Phys. Rev. E 96, 032415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Moran SD, Zhang TO, Zanni MT 2014. Protein Sci. 23, 321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Paviani V, de Melo PJ, Avakin A, Mascio PD, Ronsein GE, Augusto O 2020. Free Radic. Biol. Med 160, 356–367. [DOI] [PubMed] [Google Scholar]
- [218].Bennett MJ, Schlunegger MP, Eisenberg D 1995. Protein Sci. 4, 2455–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Mascarenhas NM, Gosavi S 2017. Prog. Biophys. Mol. Biol 128, 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Garcia-Manyes S, Giganti D, Badilla CL, Lezamiz A, Perales-Calvo J, Beedle AE, Fernández JM 2016. J. Biol. Chem 291, 4226–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Das P, King JA, Zhou R 2011. Proc. Natl. Acad. Sci. U. S. A 108, 10514–10519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Serebryany E, Takata T, Erickson E, Schafheimer N, Wang Y, King JA 2016. Protein Sci. 25, 1115–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Serebryany E, Woodard JC, Adkar BV, Shabab M, King JA, Shakhnovich EI 2016. J. Biol. Chem 291, 19172–19183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Serebryany E, Yu S, Trauger SA, Budnik B, Shakhnovich EI 2018. J. Biol. Chem 293, 17997–18009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Oka OB, Yeoh HY, Bulleid NJ 2015. Biochem. J 469, 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Woycechowsky KJ, Raines RT 2003. Biochemistry 42, 5387–5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Gross E, Sevier CS, Vala A, Kaiser CA, Fass D 2002. Nat. Struct. Biol 9, 61–67. [DOI] [PubMed] [Google Scholar]
- [228].Jakob U, Muse W, Eser M, Bardwell JC 1999. Cell 96, 341–352. [DOI] [PubMed] [Google Scholar]
- [229].Hyman AA, Weber CA, Jülicher F 2014. Annu. Rev. Cell Dev. Biol 30, 39–58. [DOI] [PubMed] [Google Scholar]
- [230].Alberti S, Gladfelter A, Mittag T 2019. Cell 176, 419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Feng Z, Chen X, Wu X, Zhang M 2019. J. Biol. Chem 294, 14823–14835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Dignon GL, Zheng W, Kim YC, Mittal J 2019. ACS Cent. Sci 5, 821–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Benedek GB, Clark JI, Serrallach EN, Young CY, Mengel L, Sauke T, Bagg A, Benedek K 1979. Philos. Trans. R. Soc., A 293, 329–340. [Google Scholar]
- [234].Delaye M, Clark JI, Benedek GB 1982. Biophys. J 37, 647–656. [PMC free article] [PubMed] [Google Scholar]
- [235].Siezen RJ, Fisch MR, Slingsby C, Benedek GB 1985. Proc. Natl. Acad. Sci. U. S. A 82, 1701–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Thurston GM 2006. J. Chem. Phys 124, 134909. [DOI] [PubMed] [Google Scholar]
- [237].Kiss AJ, Cheng C-HC 2008. Comp. Biochem. Physiol., Part D: Genomics Proteomics 3, 155–171. [DOI] [PubMed] [Google Scholar]
- [238].Kiss AJ, Mirarefi AY, Ramakrishnan S, Zukoski CF, DeVries AL, Cheng C-HC 2004. J. Exp. Biol 207, 4633–4649. [DOI] [PubMed] [Google Scholar]
- [239].Bierma JC, Roskamp KW, Ledray AP, Kiss AJ, Cheng C-HC, Martin RW 2018. J. Mol. Biol 430, 5151–5168. [DOI] [PubMed] [Google Scholar]
- [240].Mizuno A, Ozaki Y, Itoh K, Matsushima S, Iriyama K 1984. Biochem. Biophys. Res. Commun 119, 989–994. [DOI] [PubMed] [Google Scholar]
- [241].Yancey PH 2005. J. Exp. Biol 208, 2819–2830. [DOI] [PubMed] [Google Scholar]
- [242].Cinar S, Cinar H, Chan HS, Winter R 2019. J. Am. Chem. Soc 141, 7347–7354. [DOI] [PubMed] [Google Scholar]
- [243].Kleckner IR, Foster MP 2011. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics 1814, 942–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Lomont JP, Ostrander JS, Ho J-J, Petti MK, Zanni MT 2017. J. Phys. Chem. B 121, 8935–8945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Buchanan LE, Maj M, Dunkelberger EB, Cheng P-N, Nowick JS, Zanni MT 2018. Biochemistry 57, 6470–6478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Lomont JP, Rich KL, Maj M, Ho J-J, Ostrander JS, Zanni MT 2018. J. Phys. Chem. B 122, 144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Grazioli G, Yu Y, Unhelkar MH, Martin RW, Butts CT 2019. The J. Phys. Chem. B 123, 5452–5462. [DOI] [PubMed] [Google Scholar]
- [248].Yu Y, Grazioli G, Unhelkar MH, Martin R, Butts CT 2020. Sci. Rep 10, Article number 15668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Thorn DC, Grosas AB, Mabbitt PD, Ray NJ, Jackson CJ, Carver JA 2019. J. Mol. Biol 431, 483–497. [DOI] [PubMed] [Google Scholar]