

 This work is licensed under a
This work is licensed under a Abstract
Cellular senescence is now recognized as one of the hallmarks of aging. Herein, we examine current findings on senescence of the vascular endothelium and its impacts on age-related vascular diseases. Endothelial senescence can result in systemic metabolic changes, implicating senescence in chronic diseases such as diabetes, obesity and atherosclerosis. Senolytics, drugs that eliminate senescent cells, afford new therapeutic strategies for control of these chronic diseases.
Keywords: endothelium, aging, age-related disease, senescence, vascular dysfunction
Introduction
The endothelial cell (EC) monolayer forms the inner cellular lining of all blood vessels forming a critical interface between blood and tissue (1). Vascular endothelium is involved in physiological functions, which include regulation of blood fluidity, hemostasis and clotting, vascular tone, immune responses, inflammation, angiogenesis, and metabolism (2).
Dysfunction of the endothelium is a major contributor to cardiovascular diseases (CVD) such as stroke, atherosclerosis, hypertension and diabetes (see review by Hadi et al. (3)) and more recently has been shown to play a role in the severe response to COVID-19 (4). Chronological aging is the dominant risk factor for CVD, cancer and neurodegenerative diseases (5) and indeed endothelial dysfunctions including arterial stiffening (6), impaired neovascularization (7) and loss of tissue-barrier function are evident in age-related diseases (8). This review will focus on cellular aging or senescence of the vascular endothelium.
Cellular senescence
Aging is defined as a gradual decline in organism function and is underlined by cellular aging. Current biological hallmarks of cellular aging include increased cellular/oxidative stress, DNA damage, telomere shortening, stem cell depletion, mitochondria dysfunction, epigenetics and ncRNA dysregulation, loss of proteostasis and cellular senescence (9). Cellular senescence is characterized by permanent cell cycle arrest and distinct changes in cell morphology, metabolism, chromatin reorganization, gene profiles and activation of a proinflammatory secretome, termed the senescence associated secretory phenotype, SASP (10, 11, 12). The SASP can include cytokines (e.g. IL-1α/β, IL-6), chemokines (e.g. CXCLs, CCLs), growth factors (e.g. VEGF, FGF), proteases (e.g. MMPs, TIMPs) and lipids (13). Different aging pathways can induce the senescence state but the SASP phenotype will be cell type and stimulant specific (14, 15). The SASP can maintain the senescence phenotype in the cells, induce senescence in neighboring cells and influence the inflammatory state of the microenvironment (13). The SASP is also essential for activating a specific inflammatory profile that is responsible for the removal of the senescence cells (16, 17).
Senescence was originally described as a potent mechanism, together with apoptosis, for controlling cell proliferation and malignant transformation. Now it is known to contribute to development (18). During embryonic development, cellular senescence is induced by cell fusion to form the outer layer of the placenta and contributes to the normal functioning placenta (19). Further, senescence contributes to pathogenic conditions such as liver fibrosis, (17) yet is essential for tissue homeostasis (20) and wound healing (21). In adults, senescence is a response to stress, triggered to halt the increase in potentially dysfunctional cells. However, the accumulation of senescent cells with age contributes to age-related-pathologies as is seen for example in renal dysfunction (22) and CVD (23). The clearance of senescent cells is immune-mediated and the increase in senescent cells with age maybe partially attributed to the aging immune system or immunosenescence (24, 25). In addition, senescent cells have the ability to evade the immune system by altering the expression of major histocompatibility complex (MHC) HLA-E (26), a key recognition signal required by natural killer cells and differentiated T-cells to clear senescent cells.
Pathways of senescence
Broadly speaking, senescence can be divided into telomere-dependent replicative senescence (RS) or Hayflick’s limit (27) and stress-induced premature senescence (SIPS) (28). In vitro, replicative senescence can be induced through continual passaging of cells, as some cells (e.g. fibroblasts, ECs, immune cells) have a division lifespan. When the division capacity of these cells has been exhausted, based on their telomere shortening, they enter growth arrest and become senescent. There is substantial evidence that RS plays an important physiological role in tumor suppression (29).
Unlike RS, SIPS is considered to be a telomere shortening-independent process and is a rapid response characterized by random DNA damage in the genome followed by activation of the DNA damage response (DDR). SIPS can be induced by for example, oncogenic stress (30), metabolic stress (31), inflammation (32), and oxidative stress (33).
Most senescence is mediated through activation of the p21/p53 and p16/retinoblastoma (RB) protein tumor suppressor pathways (Fig. 1) (34). Activation of the DDR pathway and telomere dysfunction commonly induces p21/p53 dependent senescence while other stresses/stimuli are more often associated with the p16/RB pathway (34). The preference toward one pathway vs another appears to be cell type-specific (34, 35), with variation across species (36) and also stimulant dependent. For example, telomere dysfunction can lead to activation of the p53 or p16/RB pathway in human cells, but will only trigger the p53 pathway in rodent cells (36).
Figure 1.
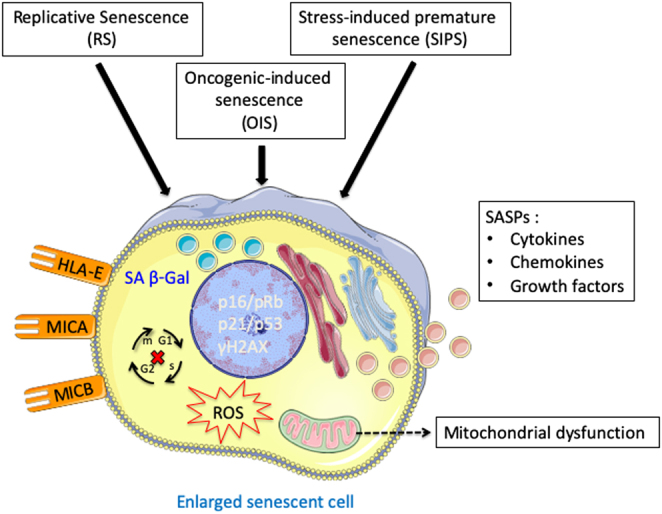
A typical senescent phenotype that is shared between different cell types and activation senescent pathways.
Senescence markers
There is no single marker that defines a senescent cell (37) and this has hindered the field. A panel of markers in combination with either p21, p16 or p53 is used to denote senescence (Table 1).
Table 1.
The different markers of senescence that are commonly used.
| Senescence marker | Description |
|---|---|
| SA β–Gal | Senescence-associated beta-galactosidase is an enzymatic stain that can be used on cells and tissues (Fig. 1) (38), with its activity associated with an increase in lysosomes biogenesis found in senescent cells (39). |
| γH2AX | Histone H2A variant is DDR marker that is phosphorylated at serine 139 upon DNA double-strand breaks (40). |
| γH2AX can also be used to detect nuclear membrane bleb structures that contain cytosolic chromatin fragments (CCFs) in the cytoplasmic compartment of senescent cells (41, 42). | |
| CCFs are known to be involved in the secretion of proinflammatory SASP through the activation of the cGAS-STING-NFκB pathway (42). | |
| Recently, it has been shown that impairment of autophagy is linked to CCFs formation, and metformin-induced activation of autophagy can reduce CCFs levels and suppress SASP secretion (43). | |
| CCFs and SASP profile in senescent primary human fibroblasts have also been associated with mitochondrial dysfunction via a retrograde ROS-JNK signaling pathway (44). | |
| Lamin B1 | Is a protein of the nuclear envelope and its expression is reduced during SIPS-associated nuclear membrane blebbing (45, 46). |
| Ki67, BrdU, PCNA | Are typical proliferation markers used to detect growth arrest (47, 48). High expression of these markers correlates with a high proliferation rate, and their decrease in expression is used in conjunction with other markers to confirm cellular senescence. |
Pathways that regulate senescence/SASP
The SASP is driven largely by proinflammatory pathways involving NFκB, mTOR and p38/MAPK (38, 39, 40) often activated through the metabolic state of the cell or through paracrine effects by surrounding senescent cells.
At least four interdependent nutrient-sensing pathways act in the induction of senescence. First, NAD+/NADH pathway that involves NAD regulated AMPK (5’ AMP-activated protein kinase) is upstream of the p38/MAPK-NFκB axis and also impacts on the proinflammatory SASP in an independent senescence growth arrest pathway (41). In contrast, pharmacological inhibition of cluster of differentiation 38 (CD38), a nicotinamide nucleotidase (NADase), was shown to reverse the age‐related decline in NAD+ levels in muscle and liver, and reduce telomere‐associated DNA damage in mice. Interestingly, the SASP secretome was shown to induce CD38 expression and increase CD38‐NADase activity in non‐senescent cells, leading to the suggestion that during aging the SASP may contribute to NAD+ decline by upregulation of CD38 (42). Although these studies had conflicting results on the role of NAD+ on inflammation, it shows that NAD+ is critical for SASP regulation and the differences may be due to the differences in cell type and/or the type of senescence induction.
Secondly, the dysfunction of mitochondria as a major energy and reactive oxygen producers, has recently been linked to the regulation of cellular senescence and SASP, mediated through AMPK (43). However, such mitochondrial dysfunction can lead to both a proinflammatory SASP as well as a low inflammatory senescent phenotype (termed mitochondrial dysfunction-associated senescence, MiDAS) driven through AMPK-mediated p53 activation (43) and with a lowered NAD+/NADH ratio.
Thirdly, mTOR another known energy sensor, regulates mitochondrial homeostasis and negatively regulates autophagy. Inhibition of mTOR with rapamycin has been shown to suppress SASP through the MAPKAPK2 pathway (44).
Fourthly, the energy sensor SIRT1, belongs to a family of histone and protein deacetylases, and is positively associated with longevity through calorie restriction (45). In the heart, the deficiency of SIRT1 has been linked to oxidative stress and inflammation, with senescence of ECs and VSMCs mediated through the autophagy pathway. Overexpression of SIRT1 protected against ischemia-reperfusion injury in the heart through increased autophagy, NOS, FOXO3 activation, and deacetylation of NFκB (46).
Lastly, miRNA are upstream regulators of senescence. MiRs such as miR-217 and miR-34a increases EC senescence, through SIRT1 downregulation. Indeed, there are endothelial miRs that can also regulate SASP such as miR-155 and miR-21 (see review by Yamakuchi and Hashiguchi (47)). As most of these endothelial miRs were identified using in vitro studies, more in vivo studies are required to delineate their roles in age-related diseases.
Endothelial cell senescence in age-related diseases
CVD and cerebrovascular diseases are the leading causes of death in the elderly population (48). EC dysfunction is a well-accepted hallmark of age-related vascular dysfunction, with the initiation of abnormal inflammatory and thrombotic circuits, arterial stiffening and oxidative stress being central to its biology. Importantly, for our understanding of vascular aging, senescent EC accumulate in aging tissues and contribute to tissue dysfunction (49, 50, 51). Structural and functional changes in senescent ECs are summarized in Figs 2 and 3.
Figure 2.

General scheme depicting the structural and functional changes in senescent ECs. Black lines represent tight or adhesion junction interactions between the ECs.
Figure 3.
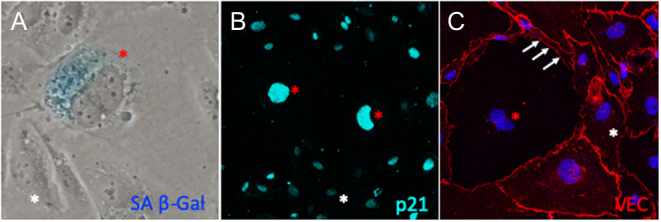
Examples of H2O2-induced senescent phenotype in human umbilical endothelial cells (HUVECs) from our lab. Senescent HUVECs depicted by red asterisk had upregulated (A) SA b-Gal, (B) nuclear p21 expression and reduced (C) VE-cadherin compared to non-senescent ECs (white asterisk).
In vitro studies defining senescent ECs have demonstrated that senescence can be induced by many of the stimuli associated with aging, such as hypoxia, disturbed flow and oxidative stress (52), high glucose conditions (53), β-amyloid peptides (54) and chronic inflammatory conditions (55). These senescent ECs generate a proinflammatory SASP similar to all other cell types. The senescence is induced through the classic senescence signaling pathways, p21/p53 and p16/RB. However, ECs can also express a non-activated, potentially anti-inflammatory senescent population, as we have previously described (56), which is also induced by age-related stress (52) and by overexpression of the vascular protective gene ARHGAP18 (a.k.a. SENEX) (56). This anti-inflammatory senescent phenotype is mediated through caveolae and inhibition of NFκB (57). The presence of the proinflammatory (defined as E1) and anti-inflammatory (defined as E2) senescent ECs suggests a duality of senescence in the vasculature-to both promote and limit inflammation. Confirmation of the existence in vivo of these cell types awaits identification of selective markers, which will allow investigations into their role in initiation and progression of disease.
Senescent ECs have been identified in many age-associated diseases. The following have been most widely studied.
Atherosclerosis
Atherosclerosis is considered a chronic inflammatory disease, characterized by enhanced leucocyte recruitment into the lesions. Further, endothelial dysfunction is a well-known initiating event in atherogenesis (58). Plaque accumulation in the vascular system is seen from the aorta to coronary arteries (59). These plaque formations usually begin with the deposition of small cholesterol crystals in the intima and the underlying smooth muscle layer. At late stages, these plaques in the arteries can lead to clot formation and thrombosis, which will result in obstruction of blood flow and even plaque rupture.
Senescent foamy macrophages have damaging roles during the onset of atherosclerosis in mice (60). Almost two decades ago, senescent ECs were described in atherosclerotic lesions from autopsied patients (23). SA-β-gal positive ECs found at the atherosclerotic lesions were enlarged and flattened in comparison to those at non-lesional areas. Telomere lengths in aortic ECs decrease as a function of donor age and have been observed in patients with atherosclerosis (61, 62). Aortic ECs isolated from aged mice were reported to have a flat and enlarged morphology (63) and human aortic ECs induced into senescence showed EC dysfunction with enhanced proinflammatory ICAM-1 expression and reduced eNOS activity (23). Further, a higher fraction of ECs from patients with abdominal aortic aneurysm (AAA), another clinical manifestation of atherosclerosis, were positive for Ki67 demonstrating a higher proliferation rate compared to control patients (64), a likely harbinger of telomere-dependent replicative senescence.
In the context of atherosclerosis, there are a number of likely stimuli for senescence induction of the endothelium. The areas of hemodynamic stress or disturbed blood flow such as arterial bifurcations and curved areas such as the iliac and thoracic artery, and the aortic arch, are areas of atherosclerosis development. ECs in these areas are senescent, induced via the p53/p21 pathway (65). Our lab has demonstrated that disturbed flow mimicking these atherosclerotic regions, induces EC senescence (52). It is proposed that the proinflammatory SASP produced by the senescent ECs will lead to a chronic sterile inflammatory environment with vascular remodeling. Indeed, SASP cytokines such as MCP-1, PDGFB, TNFα and IL-6 are atherogenic (66) and can increase monocyte infiltration, EC turnover and smooth muscle cell movement into the sub-intimal space (67).
Hyperlipidemia and lipid oxidation are high-risk factors for atherosclerosis (68). Hypercholesterolemia is associated with increased blood levels of oxidized low-density lipoprotein (ox-LDL), well known to be involved in EC dysfunction and which induces EC senescence (69, 70). Interestingly, the protective high-density lipoproteins (HDL), which are also anti-inflammatory (71) can inhibit senescence induction (Powter EE, Rye K-A and Gamble JR, unpublished results). Some risk factors that can increase ox-LDL levels include high-fat diet, smoking, diabetes and stress (72). Many of these risk factors also correspond with an increase in free radicals, which can affect NO bioavailability, damage the endothelium and induce EC permeability. Such changes cause accumulation of LDL in the sub-endothelial space resulting in enhanced EC expression of adhesion molecules such as vascular cell adhesion molecule 1 (VCAM1), E- and P-Selectins to promote leukocyte recruitment (73). CD9, a novel senescence-related marker was reported to be increased in aged human arteries and mice (74). In mouse models of atherosclerosis, CD9 expression was co-expressed with SA-β-gal staining and was found the be upregulated in ECs and macrophages within lesions. Genetic deletion and antibody treatment against CD9 further showed CD9 is critical for inducing EC senescence and exacerbating atherosclerotic lesions. Another study reported angiopoietin-like 2 is increased in senescent aortic endothelium found in atherosclerosis mouse model and knockdown of this protein in vivo reduced plaque progression (75).
Beyond regulating blood pressure, angiotensin II (Ang II), a principal effector of the renin-angiotensin system (RAS), is an important signaling molecule involved in atherogenic stimuli, such as induction of oxidative stress, secretion of proinflammatory cytokines and adhesion molecules within the vessel wall (76). Ang II can induce EC senescence and dysfunction, which in turns impairs NO vasoactive function and hemodynamic stress response (77). Concurrently, the levels of angiotensin-converting enzyme (ACE) and ET-1 levels are increased (78). Clinical and experimental animal studies of ACE inhibitors have shown these prevent the progression of atherosclerosis (79, 80). Valsartan, a well-known AngII receptor blocker, has been shown to attenuate AngII-induced EC senescence and inflammation (77).
Multiple studies have shown that disturbed blood flow (81), LDL (82) and AngII (83) increase the pathogenesis of atherosclerosis by affecting Notch signaling. The Notch pathway is important in vascular development and vascular modeling (84). Ligand activation of this pathway results in the cleavage of transmembrane Notch receptor protein into intracellular and extracellular parts, whereby the intracellular domain translocates into the nucleus for transcriptional activity (84). One study showed basic-helix-loop-helix transcriptional repressors Hey1 and Hes1, which are targets of Notch activation, were increased in senescent luminal ECs within atherosclerotic plaques in ApoE−/− mouse model and patients with coronary artery disease (85). The authors found that enhanced Notch1 activation resulted in telomere-dependent senescence. However, enhanced Notch1 decreased senescence in HUVECs via telomere-independent pathway (86). Further, constitutive activation of Notch pathway via increased expression of Notch1 intracellular domain was shown to induce premature senescence through p16 signaling. Apart from the usual Notch ligands (e.g. Delta, Serrate, LAG-2 family), other atherogenic proteins such as thrombospondins 1/2 and periostin, to name a few, have been shown to modulate Notch signaling (see review by Wang (87)). Both proteins are extracellular matrix proteins, and affect vascular processes like angiogenesis. Thrombospondin-1 is associated with endothelial senescence (88), but evidence of periostin and EC senescence is lacking.
Obesity
Studies in both human and mice demonstrate physiological aging can drive senescence in adipocytes and/or the EC population but importantly, that endothelial senescence alone can drive systemic metabolic dysfunction.
Age has been associated with significant changes in metabolism, leading to age-dependent increases in body weight, reduced insulin sensitivity and changes in lipid metabolism (89). Indeed, CVD is the leading cause of morbidity and mortality in obese individuals. White adipose tissue (WAT) is an organ responsible for regulating systemic energy homeostasis and is composed of visceral and s.c. WAT. Disruption of adipose tissue function has been linked to a chronic inflammatory state that can deregulate vascular homeostasis (90). Precursor cells required for maintaining adipocyte turnover and normal WAT function have been found to be senescent in obese human individuals and mice (91, 92). Further, studies on ECs isolated from adipose tissue biopsies from obese subjects showed that the visceral region had more inflammation and significantly higher senescent ECs compared to leaner subjects (93, 94). The ECs can be induced into senescence by conditioned medium from visceral adipose tissue from obese but not normal individuals, showing the potential paracrine effects of SASP (93). Mouse models have been used to dissect the role of obesity on the adipocyte and EC compartments. In endothelial-specific progeroid mice, the ECs were induced into early senescence. These mice showed metabolic impairment through adipose tissue dysfunction, specifically in s.c. inguinal WAT (95). The senescent endothelial-derived SASP (mainly IL-1a), induced senescent-like features in mature adipocytes but not on pre-adipocytes, indicating heighten the risk of senescent adipocytes with age (95). In normal mice on a high-calorie diet, p53/p21-driven EC senescence was observed in the aorta, skeletal muscle and lung. In endothelial-specific p53-deficient mice on a high-calorie diet, there was improved insulin sensitivity and reduced fat deposition (96). In contrast, overexpression of p53 in endothelium caused metabolic abnormalities in mice. Thus, endothelial senescence alone can drive systemic metabolic dysfunction thus providing a functional link between aging, the vasculature and metabolic disease.
Diabetes
Type II diabetes mellitus (T2DM), also known as adult-onset diabetes, is a progressive condition in which the body becomes resistant to normal insulin function and/or gradual loss of insulin production in the pancreas. The reduction of insulin sensitivity is associated with a decrease in mass and function of insulin-producing pancreatic β–cells, leading to gradual high blood glucose or hyperglycemia. Many studies have shown that obesity and T2DM are closely linked (97), although diabetes can also occur in patients that are not obese and obese patients may not become diabetic. Aging is a known risk factor for both of these metabolic diseases, thus, suggesting that cellular senescence may provide a nexus between aging and metabolic disorders. Indeed β–cell senescence has been identified in pancreatic islets isolated from aged male mice, human donors with T2DM or with high BMI (98).
Vascular dysfunction has been widely reported in diabetic patients (99) and animal models (100). Of these diabetic retinopathy (DR) is a well-known microvascular complication of T2DM. Vision impairment in patients with DR is caused by macular edema and neovascularization. In vitro studies have shown that hyperglycemia can induce EC senescence (53, 101). Further, senescent ECs are found in aortic and retinal regions in rodent models of diabetes (102, 103). P53 activation is seen in the vasculature in both diabetic human and animal studies (101, 102, 103) and is associated with activation by hyperglycemic-induced production of advanced glycation end products (AGEs) and oxidative stress pathways (104). AGEs have been shown to induce EC dysfunction through p38/MAPK and ERK1/2 signaling pathways (105), which are associated with senescence and cell cycle regulation, respectively. One of the common secondary symptoms of T2DM is chronic non-healing wound. Diabetic patients are at risk of developing diabetic foot ulcers, a type of chronic non-healing wound, which can lead to lower limb amputation and death. Vascular dysfunction in diabetic patients has been associated with local tissue hypoxia and lower limb neuropathy resulting in poor healing of wounds (see review by Chao and Cheing (106)). Campisi et al. have demonstrated the need for fibroblasts and EC senescence in normal wound healing at least in animal models (43). Normal would healing is divided into four stages: (a) hemostasis, (b) inflammation, (c) proliferation and (d) remodeling. Fibroblasts and ECs are known to proliferate during physiological wound healing and senescence of these cells is required at the remodeling stages. However, in diabetic wounds, EC proliferation or neo-angiogenesis is compromised (107), resulting in impairment of vascular circulation to the wound. Consequently, the wound becomes hypoxic, leading to upregulation of p53, the upstream regulator of p21-dependent senescence. In diabetic murine wounds the senescence is observed mostly in macrophages (108) and the SASP from the accumulated senescent macrophages promotes fibroblast senescence and fibrosis. Topical silencing of p53 can increase EC numbers and improve diabetic wound healing (109). Thus, the difference between normal wound healing and diabetic wound healing would suggest that cell-type-specific senescence is important likely through the release of cell-type-specific SASP.
The EC compartment in diabetes is also compromised. Senescent ECs have been associated with inflammation and impaired angiogenesis in diabetic rats (110) and the endothelial progenitor cells (EPCs) derived from bone marrow are reduced and dysfunctional in diabetic patients (111). EPCs are known for maintaining vascular homeostasis and compensatory angiogenesis, which are also critical for wound healing. Thus, although senescent ECs exert a beneficial effect on normal wound healing, in pathological wound repair they appear to be detrimental.
Cancer
Age is a risk factor for cancer development. The vasculature in solid tumors is both functionally and structurally abnormal, with chronic uncontrolled angiogenesis. In line with this proliferative phenotype, senescent ECs have been identified in human glioma likely activated to curb the proliferative response (112). However, this is also at the expense of promoting a proinflammatory milieu with increased Notch seen in human carcinomas, melanoma and human colorectal carcinomas (113). Endothelial Notch1 activity is linked to metastasis by promoting senescent, proinflammatory endothelium (114). In contrast, endothelial Notch3 activity was shown to limit tumor growth through apoptosis (115). In mice, ECs had a high expression of p16 in the primary tumor and metastatic sites in the lungs with constitutive active Notch 1. The senescent ECs were inflammatory, expressing high levels of the adhesion molecule VCAM1 that is associated with increased neutrophil infiltration into the tumor, a cell type associated with poor prognosis (114). The senescent cancer cells may also either induce angiogenesis or induce the ECs to undergo senescence through SASP. Thus, the EC senescence is both beneficial (inhibition of angiogenesis) but also detrimental (proinflammatory, facilitating neutrophil infiltration), which is consistent with the understanding of senescence in tumors, to act as a double-edged sword (29).
Dementia
Clinical-pathology studies have suggested an overlap in cognitive impairment found in clinical Alzheimer’s disease (AD), with vascular pathologies (e.g. microinfarcts and microbleeds) (116). Comorbidity studies have found that patients with vascular dementia have higher prevalence of CVDs such as diabetes, atherosclerosis, coronary artery diseases and cardiac arrhythmia (117).
The major hallmarks of AD, the most common form of dementia, include the accumulation of aggregated extracellular amyloid and the intracellular-accumulation of neurofibrillary tangles, which can lead to neurodegeneration. AD can be characterized as familial or hereditary but the majority of AD cases are age-related.
The blood-brain barrier (BBB), which is part of the neurovascular unit, consists of ECs, pericytes, and astrocytes and regulates solute passage between the blood and the brain. BBB breakdown occurs early, even before deposition of amyloid-beta and cognitive decline (118, 119) suggesting an intact BBB is essential. The concept that endothelial dysfunction is a critical link in the development of AD, is gaining strength. Reduction in the cerebral blood flow, impaired hemodynamic response and vascular reactivity have been detected in patients in the early stages of AD and across people with normal aging-to-mild cognitive impairment (120). The increased deposition of amyloid plaque in brain microvessels, termed cerebral amyloid angiopathy is associated with microbleeds and cerebral hemorrhages (121). However, drug trials involving the use of anti-amyloid have shown that although it is effective in reducing plaque burden in AD patients, it did not reverse neurovascular dysfunction or improve cognitive function (122).
Senescence of microglia, astrocytes, pericytes, oligodendrocytes, oligodendrocyte progenitor cells, and neurons (123, 124, 125, 126, 127, 128) have been found in the brain and this senescence has been attributed to the development of AD and Parkinson’s disease. Although senescent ECs have not been identified in the brain of patients with these neurodegenerative diseases, many in vitro studies have shown that oligomeric β-amyloid can induce SIPS in ECs (54, 129).
Although a definitive understanding of EC senescence and AD development is still lacking, evidence is accumulating of its potential importance. A number of senescence-associated markers are seen in the endothelium in the brain. For example, a recent population-based study has shown positive correlation between DNA damage and p53 in the endothelium in human brain samples of AD (130). Further, the authors concluded that endothelial DNA damage and senescence is associated with aging and it may occur independently of AD pathology. A recent single-cell RNASeq study by Kiss et al. has detected a higher prevalence of senescent brain ECs in aged mice (51). These senescent brain ECs expressed a unique SASP profile that was different from senescent microglia, astrocytes and oligodendrocytes. Senescent ECs expressed increased levels of Kitl, Plat, Igfbp7, Cxcl12, and Ctnnb1. Cxcl12 and Ctnnb1 are related to leukocyte transendothelial migration, suggesting a mechanism for increased inflammation. Tissue plasminogen activator (TPA or Plat), is essential for clot lysis on the endothelial cell surface (131) and Ctnnb1 (β-catenin) is involved in cell–cell adhesion (132). Studies from progeroid mice have shown that senescent EC have poor tight junction organization (127) and tight junction proteins claudin-5 and occludin are found to decrease in expression in AD mice (133). A further example is Notch1 where studies have suggested it maybe a potential biomarker for AD (134, 135). Interestingly, the gamma secretase enzyme, involved in the cleavage of Notch receptor, is one of two enzymes primarily involved in the processing of the amyloid precursor protein into neurotoxic β-amyloid peptides. In vitro Notch1 expression was shown to be increased by β-amyloid treatment in brain ECs (134) and Notch1 activation induces EC senescence (85). Further, activated Notch1 is associated with vascular pathology in AD. Chronic hippocampal expression of cleaved Notch intracellular domain NICD was found to promote vascular thickening and amyloid deposition in a rat model of early AD and chronic activation of Notch1 led to a decreased cerebral blood flow in early AD in a transgenic rat model (136). Thus, identification of senescent ECs in AD, and their role in the BBB awaits further investigation.
Senotherapeutics
The damaging role of senescent cells in disease development has really been cemented through the use of transgenic mice (e.g. INK-ATTAC, p16-TMR) that can selectively ablate senescent cells (137). With loss of senescent cells, these mice showed a reduced incidence of age-related pathologies in kidney, heart and brain.
The field of senolytics, drugs that selectively eliminate senescent cell, is gaining momentum. Dasatinib and Quercetin (D+Q) were some of the first senolytics, which remove senescent cells in vitro and in progeroid mice through targeting of the anti-apoptotic pathways in senescence (138). Long term oral treatment of D+Q have been shown to improve vascular function in aged or atherosclerotic mice at a level similar to genetic clearance of senescent cells in aged INK-ATTAC mice (139). The D+Q combination has been shown to efficiently reduce senescence cell burden in phase I trials for several senescence-related diseases such as diabetic kidney disease (NCT02848131) (140). Further, the D+Q combination have also been shown to alleviate physical dysfunction seen in senescence-associated disease such as idiopathic pulmonary fibrosis (NCT02874989) (141), however, it remains unclear whether senescent cells burden was reduced.
Recently, two further senolytics have been trialed. Navitoclax, which targets the Bcl-2 family of anti-apoptotic factors (142) and Venclexta (a small molecule BH3 mimetic) which blocks diabetes by targeting senescence (143). Further studies have shown that D+Q exhibit cell and pathway selectivity (140), thus demonstrating the need for expansion of our senolytic repertoire to provide cell- and disease-specific drugs for future use. A recent study has shown that sustained ablation of p16 senescent liver sinusoidal ECs promotes liver and perivascular fibrosis (144) which can be detrimental to healthspan. Thus, selective and time-dependent removal of the senescent population of cells is likely to influence the success of senolytics in the clinic. In contrast to senolytics that target the anti-apoptotic pathway, a novel study has demonstrated a drug called SSK1, which specifically eliminates senescent cells through the activity of lysosomal β-galactosidase with more superior efficacy than common senolytics (145). Other types of senescence targeting drugs with different specificities are being developed. These include senomorphics, drugs that modulate the inflammatory SASP profile without killing senescent cells. Studies using senolytics and senomorphics on disease/senescent endothelium have been well-reviewed by Kim and Kim (146). Finally, senescence immunotherapy provides another therapeutic strategy. At present, it is being used for treatment of cancers but it is emerging as a promising alternative to senolytics to clear senescent cells under the notion that different immune cells are capable of selectively identifying and removing unique senescent cells. Current senescent immunotherapy findings can be found in reviews by Kim and Kim (146) and Song et al. (147).
Summary and future directions
ECs play a critical role in vascular homeostasis, regulation of inflammation and thrombosis and maintenance of organ function. This review outlined our existing understanding of senescent ECs and their contribution to cardiovascular disease, metabolic disease and dementia. Although senescence was initially considered as an all-encompassing phenotypic change, it is now apparent that each cell type exhibits an unique and distinguishing senescence phenotype, one that may also be tissue specific. Hence our understanding of endothelial senescence is still in its infancy. Current findings have indicated that specific depletion of senescent cells reverses age-related changes and prolongs life span. However, caution should be urged as cellular senescence also plays important physiological roles such as in tissue development, wound healing and tumor inhibition. To achieve optimal success in targeting senescence it will be imperative to have a thorough knowledge of the senescent cell type at play in disease, and their spatiotemporal expression in order to deliver the most appropriate senolytic, senomorphic or drug combination.
Declaration of interest
Jennifer Gamble is a Senior Editor of Vascular Biology. Jennifer Gamble was not involved in the review or editorial process for this paper, on which she is listed as an author.
Funding
The authors are supported by the National Health and Medical Research Council of Australia Ideas Grant (#GNT1183057).
Author contribution statement
K K T, P C, Y Z, M A V and J R G contributed to writing this review.
References
- 1.Atkins GB, Jain MK, Hamik A.Endothelial differentiation: molecular mechanisms of specification and heterogeneity. Arteriosclerosis, Thrombosis, and Vascular Biology 2011. 31 1476–14. ( 10.1161/ATVBAHA.111.228999) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feletou M.The Endothelium: Part 1: Multiple Functions of the Endothelial Cells-Focus on Endothelium-Derived Vasoactive Mediators. Morgan & Claypool Life Sciences: San Rafael (CA), 2011. Available from: https://www.ncbi.nlm.nih.gov/books/NBK57149/ [PubMed] [Google Scholar]
- 3.Hadi HA, Carr CS, Suwaidi JAl.Endothelial dysfunction: cardiovascular risk factors, therapy, and outcome. Vascular Health and Risk Management 2005. 1 183–1. [PMC free article] [PubMed] [Google Scholar]
- 4.Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P, Baluha A, Bar N, Bona RD, Burns AJet al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet: Haematology 2020. 7 e575–e582. ( 10.1016/S2352-3026(2030216-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Niccoli T, Partridge L.Ageing as a risk factor for disease. Current Biology 2012. 22 R741–R7. ( 10.1016/j.cub.2012.07.024) [DOI] [PubMed] [Google Scholar]
- 6.Donato AJ, Machin DR, Lesniewski LA.Mechanisms of dysfunction in the aging vasculature and role in age-related disease. Circulation Research 2018. 123 825–848. ( 10.1161/CIRCRESAHA.118.312563) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonham CA, Kuehlmann B, Gurtner GC.Impaired neovascularization in aging. Advances in Wound Care 2020. 9 111–126. ( 10.1089/wound.2018.0912) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blau CW, Cowley TR, O’Sullivan J, Grehan B, Browne TC, Kelly L, Birch A, Murphy N, Kelly AM, Kerskens CMet al. The age-related deficit in LTP is associated with changes in perfusion and blood-brain barrier permeability. Neurobiology of Aging 2012. 33 1005.e23–1005.e35. ( 10.1016/j.neurobiolaging.2011.09.035) [DOI] [PubMed] [Google Scholar]
- 9.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G.The hallmarks of aging. Cell 2013. 153 1194–1. ( 10.1016/j.cell.2013.05.039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS.Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008. 133 1019–10. ( 10.1016/j.cell.2008.03.039) [DOI] [PubMed] [Google Scholar]
- 11.Campisi J, d’Adda di Fagagna F.Cellular senescence: when bad things happen to good cells. Nature Reviews: Molecular Cell Biology 2007. 8 729–7. ( 10.1038/nrm2233) [DOI] [PubMed] [Google Scholar]
- 12.Hernandez-Segura A, Nehme J, Demaria M.Hallmarks of cellular senescence. Trends in Cell Biology 2018. 28 436–453. ( 10.1016/j.tcb.2018.02.001) [DOI] [PubMed] [Google Scholar]
- 13.Coppe JP, Desprez PY, Krtolica A, Campisi J.The senescence-associated secretory phenotype: the dark side of tumor suppression. Annual Review of Pathology 2010. 5 99–118. ( 10.1146/annurev-pathol-121808-102144) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McHugh D, Gil J.Senescence and aging: causes, consequences, and therapeutic avenues. Journal of Cell Biology 2018. 217 65–77. ( 10.1083/jcb.201708092) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katsuumi G, Shimizu I, Yoshida Y, Minamino T.Vascular senescence in cardiovascular and metabolic diseases. Frontiers in Cardiovascular Medicine 2018. 5 18. ( 10.3389/fcvm.2018.00018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sagiv A, Burton DG, Moshayev Z, Vadai E, Wensveen F, Ben-Dor S, Golani O, Polic B, Krizhanovsky V.NKG2D ligands mediate immunosurveillance of senescent cells. Aging 2016. 8 328–3. ( 10.18632/aging.100897) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW.Senescence of activated stellate cells limits liver fibrosis. Cell 2008. 134 657–6. ( 10.1016/j.cell.2008.06.049) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe Jet al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013. 155 1119–11. ( 10.1016/j.cell.2013.10.041) [DOI] [PubMed] [Google Scholar]
- 19.Chuprin A, Gal H, Biron-Shental T, Biran A, Amiel A, Rozenblatt S, Krizhanovsky V.Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes and Development 2013. 27 2356–23. ( 10.1101/gad.227512.113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Childs BG, Baker DJ, Kirkland JL, Campisi J, van Deursen JM.Senescence and apoptosis: dueling or complementary cell fates? EMBO Reports 2014. 15 1139–11. ( 10.15252/embr.201439245) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adams PD.Healing and hurting: molecular mechanisms, functions, and pathologies of cellular senescence. Molecular Cell 2009. 36 2–14. ( 10.1016/j.molcel.2009.09.021) [DOI] [PubMed] [Google Scholar]
- 22.Docherty MH, O’Sullivan ED, Bonventre JV, Ferenbach DA.Cellular senescence in the kidney. Journal of the American Society of Nephrology 2019. 30 726–736. ( 10.1681/ASN.2018121251) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I.Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation 2002. 105 1541–154. ( 10.1161/01.cir.0000013836.85741.17) [DOI] [PubMed] [Google Scholar]
- 24.Hazeldine J, Lord JM.The impact of ageing on natural killer cell function and potential consequences for health in older adults. Ageing Research Reviews 2013. 12 1069–10. ( 10.1016/j.arr.2013.04.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akbar AN, Henson SM, Lanna A.Senescence of T lymphocytes: implications for enhancing human immunity. Trends in Immunology 2016. 37 866–876. ( 10.1016/j.it.2016.09.002) [DOI] [PubMed] [Google Scholar]
- 26.Pereira BI, Devine OP, Vukmanovic-Stejic M, Chambers ES, Subramanian P, Patel N, Virasami A, Sebire NJ, Kinsler V, Valdovinos Aet al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8(+) T cell inhibition. Nature Communications 2019. 10 2387. ( 10.1038/s41467-019-10335-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hayflick L.The limited in vitro lifetime of human diploid cell strains. Experimental Cell Research 1965. 37 614–6. ( 10.1016/0014-4827(6590211-9) [DOI] [PubMed] [Google Scholar]
- 28.Brack C, Lithgow G, Osiewacz H, Toussaint O.EMBO WORKSHOP REPORT: Molecular and cellular gerontology Serpiano, Switzerland, September 18–22, 1999. EMBO Journal 2000. 19 1929–19. ( 10.1093/emboj/19.9.1929) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Campisi J.Aging and cancer: the double-edged sword of replicative senescence. Journal of the American Geriatrics Society 1997. 45 482–48. ( 10.1111/j.1532-5415.1997.tb05175.x) [DOI] [PubMed] [Google Scholar]
- 30.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW.Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997. 88 593–602. ( 10.1016/s0092-8674(0081902-9) [DOI] [PubMed] [Google Scholar]
- 31.Wang C, Maddick M, Miwa S, Jurk D, Czapiewski R, Saretzki G, Langie SA, Godschalk RW, Cameron K, von Zglinicki T.Adult-onset, short-term dietary restriction reduces cell senescence in mice. Aging 2010. 2 555–5. ( 10.18632/aging.100196) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dumont P, Balbeur L, Remacle J, Toussaint O.Appearance of biomarkers of in vitro ageing after successive stimulation of WI-38 fibroblasts with IL-1alpha and TNF-alpha: senescence associated beta-galactosidase activity and morphotype transition. Journal of Anatomy 2000. 197 529–5. ( 10.1046/j.1469-7580.2000.19740529.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Q, Ames BN.Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. PNAS 1994. 91 4130–4. ( 10.1073/pnas.91.10.4130) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Campisi J.Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 2005. 120 513–5. ( 10.1016/j.cell.2005.02.003) [DOI] [PubMed] [Google Scholar]
- 35.Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J.Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO Journal 2003. 22 4212–4222. ( 10.1093/emboj/cdg417) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacobs JJ, de Lange T.Significant role for p16INK4a in p53-independent telomere-directed senescence. Current Biology 2004. 14 2302–230. ( 10.1016/j.cub.2004.12.025) [DOI] [PubMed] [Google Scholar]
- 37.van Deursen JM.The role of senescent cells in ageing. Nature 2014. 509 439–4. ( 10.1038/nature13193) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodier F, Muñoz DP, Teachenor R, Chu V, Le O, Bhaumik D, Coppé JP, Campeau E, Beauséjour CM, Kim SHet al. DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. Journal of Cell Science 2011. 124 68–81. ( 10.1242/jcs.071340) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu Set al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nature Cell Biology 2015. 17 1049–10. ( 10.1038/ncb3195) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Freund A, Patil CK, Campisi J.P38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO Journal 2011. 30 1536–15. ( 10.1038/emboj.2011.69) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nacarelli T, Lau L, Fukumoto T, Zundell J, Fatkhutdinov N, Wu S, Aird KM, Iwasaki O, Kossenkov AV, Schultz Det al. NAD(+) metabolism governs the proinflammatory senescence-associated secretome. Nature Cell Biology 2019. 21 397–407. ( 10.1038/s41556-019-0287-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chini C, Hogan KA, Warner GM, Tarragó MG, Peclat TR, Tchkonia T, Kirkland JL, Chini E.The NADase CD38 is induced by factors secreted from senescent cells providing a potential link between senescence and age-related cellular NAD(+) decline. Biochemical and Biophysical Research Communications 2019. 513 486–493. ( 10.1016/j.bbrc.2019.03.199) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan Aet al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metabolism 2016. 23 303–3. ( 10.1016/j.cmet.2015.11.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, Raguz S, Acosta JC, Innes AJ, Banito Aet al. Erratum: mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nature Cell Biology 2015. 17 1370. ( 10.1038/ncb3243) [DOI] [PubMed] [Google Scholar]
- 45.Chen D, Bruno J, Easlon E, Lin SJ, Cheng HL, Alt FW, Guarente L.Tissue-specific regulation of SIRT1 by calorie restriction. Genes and Development 2008. 22 1753–175. ( 10.1101/gad.1650608) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nadtochiy SM, Yao H, McBurney MW, Gu W, Guarente L, Rahman I, Brookes PS.SIRT1-mediated acute cardioprotection. American Journal of Physiology: Heart and Circulatory Physiology 2011. 301 H1506–H15. ( 10.1152/ajpheart.00587.2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamakuchi M, Hashiguchi T.Endothelial cell aging: how miRNAs contribute? Journal of Clinical Medicine 2018. 7 170. ( 10.3390/jcm7070170) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yazdanyar A, Newman AB.The burden of cardiovascular disease in the elderly: morbidity, mortality, and costs. Clinics in Geriatric Medicine 2009. 25 563–57, vii. ( 10.1016/j.cger.2009.07.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yousefzadeh MJ, Zhao J, Bukata C, Wade EA, McGowan SJ, Angelini LA, Bank MP, Gurkar AU, McGuckian CA, Calubag MFet al. Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell 2020. 19 e13094. ( 10.1111/acel.13094) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rossman MJ, Kaplon RE, Hill SD, McNamara MN, Santos-Parker JR, Pierce GL, Seals DR, Donato AJ.Endothelial cell senescence with aging in healthy humans: prevention by habitual exercise and relation to vascular endothelial function. American Journal of Physiology: Heart and Circulatory Physiology 2017. 313 H890–H895. ( 10.1152/ajpheart.00416.2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kiss T, Nyúl-Tóth Á, Balasubramanian P, Tarantini S, Ahire C, DelFavero J, Yabluchanskiy A, Csipo T, Farkas E, Wiley Get al. Single-cell RNA sequencing identifies senescent cerebromicrovascular endothelial cells in the aged mouse brain. GeroScience 2020. 42 429–444. ( 10.1007/s11357-020-00177-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coleman PR, Chang G, Hutas G, Grimshaw M, Vadas MA, Gamble JR.Age-associated stresses induce an anti-inflammatory senescent phenotype in endothelial cells. Aging 2013. 5 913–9. ( 10.18632/aging.100622) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yokoi T, Fukuo K, Yasuda O, Hotta M, Miyazaki J, Takemura Y, Kawamoto H, Ichijo H, Ogihara T.Apoptosis signal-regulating kinase 1 mediates cellular senescence induced by high glucose in endothelial cells. Diabetes 2006. 55 1660–166. ( 10.2337/db05-1607) [DOI] [PubMed] [Google Scholar]
- 54.Donnini S, Solito R, Cetti E, Corti F, Giachetti A, Carra S, Beltrame M, Cotelli F, Ziche M.Abeta peptides accelerate the senescence of endothelial cells in vitro and in vivo, impairing angiogenesis. FASEB Journal 2010. 24 2385–23. ( 10.1096/fj.09-146456) [DOI] [PubMed] [Google Scholar]
- 55.Khan SY, Awad EM, Oszwald A, Mayr M, Yin X, Waltenberger B, Stuppner H, Lipovac M, Uhrin P, Breuss JM.Premature senescence of endothelial cells upon chronic exposure to TNFalpha can be prevented by N-acetyl cysteine and plumericin. Scientific Reports 2017. 7 39501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Coleman PR, Hahn CN, Grimshaw M, Lu Y, Li X, Brautigan PJ, Beck K, Stocker R, Vadas MA, Gamble JR.Stress-induced premature senescence mediated by a novel gene, SENEX, results in an anti-inflammatory phenotype in endothelial cells. Blood 2010. 116 4016–40. ( 10.1182/blood-2009-11-252700) [DOI] [PubMed] [Google Scholar]
- 57.Powter EE, Coleman PR, Tran MH, Lay AJ, Bertolino P, Parton RG, Vadas MA, Gamble JR.Caveolae control the anti-inflammatory phenotype of senescent endothelial cells. Aging Cell 2015. 14 102–11. ( 10.1111/acel.12270) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ross R.Atherosclerosis – an inflammatory disease. New England Journal of Medicine 1999. 340 115–1. ( 10.1056/NEJM199901143400207) [DOI] [PubMed] [Google Scholar]
- 59.Heusch G, Libby P, Gersh B, Yellon D, Böhm M, Lopaschuk G, Opie L.Cardiovascular remodelling in coronary artery disease and heart failure. Lancet 2014. 383 1933–19. ( 10.1016/S0140-6736(1460107-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM.Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016. 354 472–477. ( 10.1126/science.aaf6659) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Okuda K, Khan MY, Skurnick J, Kimura M, Aviv H, Aviv A.Telomere attrition of the human abdominal aorta: relationships with age and atherosclerosis. Atherosclerosis 2000. 152 391–39. ( 10.1016/s0021-9150(9900482-7) [DOI] [PubMed] [Google Scholar]
- 62.Chang E, Harley CB.Telomere length and replicative aging in human vascular tissues. PNAS 1995. 92 11190–1119. ( 10.1073/pnas.92.24.11190) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Uraoka M, Ikeda K, Kurimoto-Nakano R, Nakagawa Y, Koide M, Akakabe Y, Kitamura Y, Ueyama T, Matoba S, Yamada Het al. Loss of bcl-2 during the senescence exacerbates the impaired angiogenic functions in endothelial cells by deteriorating the mitochondrial redox state. Hypertension 2011. 58 254–2. ( 10.1161/HYPERTENSIONAHA.111.176701) [DOI] [PubMed] [Google Scholar]
- 64.Cafueri G, Parodi F, Pistorio A, Bertolotto M, Ventura F, Gambini C, Bianco P, Dallegri F, Pistoia V, Pezzolo Aet al. Endothelial and smooth muscle cells from abdominal aortic aneurysm have increased oxidative stress and telomere attrition. PLoS ONE 2012. 7 e35312. ( 10.1371/journal.pone.0035312) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Warboys CM, de Luca A, Amini N, Luong L, Duckles H, Hsiao S, White A, Biswas S, Khamis R, Chong CKet al. Disturbed flow promotes endothelial senescence via a p53-dependent pathway. Arteriosclerosis, Thrombosis, and Vascular Biology 2014. 34 985–9. ( 10.1161/ATVBAHA.114.303415) [DOI] [PubMed] [Google Scholar]
- 66.Sprague AH, Khalil RA.Inflammatory cytokines in vascular dysfunction and vascular disease. Biochemical Pharmacology 2009. 78 539–5. ( 10.1016/j.bcp.2009.04.029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gimbrone Jr MA, Garcia-Cardena G.Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circulation Research 2016. 118 620–6. ( 10.1161/CIRCRESAHA.115.306301) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nilsson J, Regnström J, Frostegård J, Stiko A.Lipid oxidation and atherosclerosis. Herz 1992. 17 263–26 [PubMed] [Google Scholar]
- 69.Raman KG, Gandley RE, Rohland J, Zenati MS, Tzeng E.Early hypercholesterolemia contributes to vasomotor dysfunction and injury associated atherogenesis that can be inhibited by nitric oxide. Journal of Vascular Surgery 2011. 53 754–7. ( 10.1016/j.jvs.2010.09.038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang TC, Chen YJ, Chang SF, Chen CH, Chang PY, Lu SC.Malondialdehyde mediates oxidized LDL-induced coronary toxicity through the Akt-FGF2 pathway via DNA methylation. Journal of Biomedical Science 2014. 21 11. ( 10.1186/1423-0127-21-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cockerill GW, Rye KA, Gamble JR, Vadas MA, Barter PJ.High-density lipoproteins inhibit cytokine-induced expression of endothelial cell adhesion molecules. Arteriosclerosis, Thrombosis, and Vascular Biology 1995. 15 1987–19. ( 10.1161/01.atv.15.11.1987) [DOI] [PubMed] [Google Scholar]
- 72.Rajendran P, Rengarajan T, Thangavel J, Nishigaki Y, Sakthisekaran D, Sethi G, Nishigaki I.The vascular endothelium and human diseases. International Journal of Biological Sciences 2013. 9 1057–10. ( 10.7150/ijbs.7502) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kattoor AJ, Pothineni NVK, Palagiri D, Mehta JL.Oxidative stress in atherosclerosis. Current Atherosclerosis Reports 2017. 19 42. ( 10.1007/s11883-017-0678-6) [DOI] [PubMed] [Google Scholar]
- 74.Cho JH, Kim EC, Son Y, Lee DW, Park YS, Choi JH, Cho KH, Kwon KS, Kim JR.CD9 induces cellular senescence and aggravates atherosclerotic plaque formation. Cell Death and Differentiation 2020. 27 2681–2696. ( 10.1038/s41418-020-0537-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Caland L, Labbé P, Mamarbachi M, Villeneuve L, Ferbeyre G, Noly PE, Carrier M, Thorin-Trescases N, Thorin É.Knockdown of angiopoietin-like 2 induces clearance of vascular endothelial senescent cells by apoptosis, promotes endothelial repair and slows atherogenesis in mice. Aging 2019. 11 3832–3850. ( 10.18632/aging.102020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schmidt-Ott KM, Kagiyama S, Phillips MI.The multiple actions of angiotensin II in atherosclerosis. Regulatory Peptides 2000. 93 65–77. ( 10.1016/s0167-0115(0000178-6) [DOI] [PubMed] [Google Scholar]
- 77.Li R, Mi X, Yang S, Yang Y, Zhang S, Hui R, Chen Y, Zhang W.Long-term stimulation of angiotensin II induced endothelial senescence and dysfunction. Experimental Gerontology 2019. 119 212–220. ( 10.1016/j.exger.2019.02.012) [DOI] [PubMed] [Google Scholar]
- 78.Schiffrin EL.Canadian Institutes of Health Research Multidisciplinary Research Group on Hypertension. Beyond blood pressure: the endothelium and atherosclerosis progression. American Journal of Hypertension 2002. 15 115S–122S. ( 10.1016/s0895-7061(0203006-6) [DOI] [PubMed] [Google Scholar]
- 79.Candido R, Jandeleit-Dahm KA, Cao Z, Nesteroff SP, Burns WC, Twigg SM, Dilley RJ, Cooper ME, Allen TJ.Prevention of accelerated atherosclerosis by angiotensin-converting enzyme inhibition in diabetic apolipoprotein E-deficient mice. Circulation 2002. 106 246–2. ( 10.1161/01.cir.0000021122.63813.32) [DOI] [PubMed] [Google Scholar]
- 80.Dagenais GR, Pogue J, Fox K, Simoons ML, Yusuf S.Angiotensin-converting-enzyme inhibitors in stable vascular disease without left ventricular systolic dysfunction or heart failure: a combined analysis of three trials. Lancet 2006. 368 581–58. ( 10.1016/S0140-6736(0669201-5) [DOI] [PubMed] [Google Scholar]
- 81.Rizzo P, Mele D, Caliceti C, Pannella M, Fortini C, Clementz AG, Morelli MB, Aquila G, Ameri P, Ferrari R.The role of notch in the cardiovascular system: potential adverse effects of investigational notch inhibitors. Frontiers in Oncology 2014. 4 384. ( 10.3389/fonc.2014.00384) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zeng Q, Song R, Ao L, Xu D, Venardos N, Fullerton DA, Meng X.Augmented osteogenic responses in human aortic valve cells exposed to oxLDL and TLR4 agonist: a mechanistic role of Notch1 and NF-kappaB interaction. PLoS ONE 2014. 9 e95400. ( 10.1371/journal.pone.0095400) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ozasa Y, Akazawa H, Qin Y, Tateno K, Ito K, Kudo-Sakamoto Y, Yano M, Yabumoto C, Naito AT, Oka Tet al. Notch activation mediates angiotensin II-induced vascular remodeling by promoting the proliferation and migration of vascular smooth muscle cells. Hypertension Research 2013. 36 859–8. ( 10.1038/hr.2013.52) [DOI] [PubMed] [Google Scholar]
- 84.Gridley T.Notch signaling in the vasculature. Current Topics in Developmental Biology 2010. 92 277–309. ( 10.1016/S0070-2153(1092009-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu ZJ, Tan Y, Beecham GW, Seo DM, Tian R, Li Y, Vazquez-Padron RI, Pericak-Vance M, Vance JM, Goldschmidt-Clermont PJet al. Notch activation induces endothelial cell senescence and pro-inflammatory response: implication of Notch signaling in atherosclerosis. Atherosclerosis 2012. 225 296–303. ( 10.1016/j.atherosclerosis.2012.04.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yoshida Y, Hayashi Y, Suda M, Tateno K, Okada S, Moriya J, Yokoyama M, Nojima A, Yamashita M, Kobayashi Yet al. Notch signaling regulates the lifespan of vascular endothelial cells via a p16-dependent pathway. PLoS ONE 2014. 9 e100359. ( 10.1371/journal.pone.0100359) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang MM.Notch signaling and Notch signaling modifiers. International Journal of Biochemistry and Cell Biology 2011. 43 1550–15. ( 10.1016/j.biocel.2011.08.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gao Q, Chen K, Gao L, Zheng Y, Yang YG.Thrombospondin-1 signaling through CD47 inhibits cell cycle progression and induces senescence in endothelial cells. Cell Death and Disease 2016. 7 e2368. ( 10.1038/cddis.2016.155) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Barton M.Obesity and aging: determinants of endothelial cell dysfunction and atherosclerosis. Pflugers Archiv 2010. 460 825–8. ( 10.1007/s00424-010-0860-y) [DOI] [PubMed] [Google Scholar]
- 90.Kwaifa IK, Bahari H, Yong YK, Noor SM.Endothelial dysfunction in obesity-induced inflammation: molecular mechanisms and clinical implications. Biomolecules 2020. 10 291. ( 10.3390/biom10020291) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gustafson B, Nerstedt A, Smith U.Reduced subcutaneous adipogenesis in human hypertrophic obesity is linked to senescent precursor cells. Nature Communications 2019. 10 2757. ( 10.1038/s41467-019-10688-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H, Khosla S, Jensen MD, Kirkland JL.Fat tissue, aging, and cellular senescence. Aging Cell 2010. 9 667–6. ( 10.1111/j.1474-9726.2010.00608.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Villaret A, Galitzky J, Decaunes P, Estève D, Marques MA, Sengenès C, Chiotasso P, Tchkonia T, Lafontan M, Kirkland JLet al. Adipose tissue endothelial cells from obese human subjects: differences among depots in angiogenic, metabolic, and inflammatory gene expression and cellular senescence. Diabetes 2010. 59 2755–27. ( 10.2337/db10-0398) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pellegrinelli V, Rouault C, Veyrie N, Clément K, Lacasa D.Endothelial cells from visceral adipose tissue disrupt adipocyte functions in a three-dimensional setting: partial rescue by angiopoietin-1. Diabetes 2014. 63 535–5. ( 10.2337/db13-0537) [DOI] [PubMed] [Google Scholar]
- 95.Barinda AJ, Ikeda K, Nugroho DB, Wardhana DA, Sasaki N, Honda S, Urata R, Matoba S, Hirata KI, Emoto N.Endothelial progeria induces adipose tissue senescence and impairs insulin sensitivity through senescence associated secretory phenotype. Nature Communications 2020. 11 481. ( 10.1038/s41467-020-14387-w) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yokoyama M, Okada S, Nakagomi A, Moriya J, Shimizu I, Nojima A, Yoshida Y, Ichimiya H, Kamimura N, Kobayashi Yet al. Inhibition of endothelial p53 improves metabolic abnormalities related to dietary obesity. Cell Reports 2014. 7 1691–1703. ( 10.1016/j.celrep.2014.04.046) [DOI] [PubMed] [Google Scholar]
- 97.Eckel RH, Kahn SE, Ferrannini E, Goldfine AB, Nathan DM, Schwartz MW, Smith RJ, Smith SR.Obesity and type 2 diabetes: what can be unified and what needs to be individualized? Journal of Clinical Endocrinology and Metabolism 2011. 96 1654–16. ( 10.1210/jc.2011-0585) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Aguayo-Mazzucato C, Andle J, Lee TB, Midha A, Talemal L, Chipashvili V, Hollister-Lock J, van Deursen J, Weir G, Bonner-Weir S.Acceleration of beta cell aging determines diabetes and senolysis improves disease outcomes. Cell Metabolism 2019. 30 129.e4–142.e4. ( 10.1016/j.cmet.2019.05.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Torimoto K, Okada Y, Mori H, Tanaka Y.Relationship between fluctuations in glucose levels measured by continuous glucose monitoring and vascular endothelial dysfunction in type 2 diabetes mellitus. Cardiovascular Diabetology 2013. 12 1. ( 10.1186/1475-2840-12-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Algenstaedt P, Schaefer C, Biermann T, Hamann A, Schwarzloh B, Greten H, Rüther W, Hansen-Algenstaedt N.Microvascular alterations in diabetic mice correlate with level of hyperglycemia. Diabetes 2003. 52 542–54. ( 10.2337/diabetes.52.2.542) [DOI] [PubMed] [Google Scholar]
- 101.Orimo M, Minamino T, Miyauchi H, Tateno K, Okada S, Moriya J, Komuro I.Protective role of SIRT1 in diabetic vascular dysfunction. Arteriosclerosis, Thrombosis, and Vascular Biology 2009. 29 889–8. ( 10.1161/ATVBAHA.109.185694) [DOI] [PubMed] [Google Scholar]
- 102.Brodsky SV, Gealekman O, Chen J, Zhang F, Togashi N, Crabtree M, Gross SS, Nasjletti A, Goligorsky MS.Prevention and reversal of premature endothelial cell senescence and vasculopathy in obesity-induced diabetes by ebselen. Circulation Research 2004. 94 377–3. ( 10.1161/01.RES.0000111802.09964.EF) [DOI] [PubMed] [Google Scholar]
- 103.Shosha E, Xu Z, Narayanan SP, Lemtalsi T, Fouda AY, Rojas M, Xing J, Fulton D, Caldwell RW, Caldwell RB.Mechanisms of diabetes-induced endothelial cell senescence: role of arginase 1. International Journal of Molecular Sciences 2018. 19 1215. ( 10.3390/ijms19041215) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Strycharz J, Drzewoski J, Szemraj J, Sliwinska A.Erratum to ‘Is p53 involved in tissue-specific insulin resistance formation?’ Oxidative Medicine and Cellular Longevity 2017. 2017 8036902. ( 10.1155/2017/8036902) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ren X, Ren L, Wei Q, Shao H, Chen L, Liu N.Advanced glycation end-products decreases expression of endothelial nitric oxide synthase through oxidative stress in human coronary artery endothelial cells. Cardiovascular Diabetology 2017. 16 52. ( 10.1186/s12933-017-0531-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chao CY, Cheing GL.Microvascular dysfunction in diabetic foot disease and ulceration. Diabetes/Metabolism Research and Reviews 2009. 25 604–6. ( 10.1002/dmrr.1004) [DOI] [PubMed] [Google Scholar]
- 107.Okonkwo UA, Chen L, Ma D, Haywood VA, Barakat M, Urao N, DiPietro LA.Compromised angiogenesis and vascular Integrity in impaired diabetic wound healing. PLoS ONE 2020. 15 e0231962. ( 10.1371/journal.pone.0231962) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wilkinson HN, Clowes C, Banyard KL, Matteuci P, Mace KA, Hardman MJ.Elevated local senescence in diabetic wound healing is linked to pathological repair via CXCR2. Journal of Investigative Dermatology 2019. 139 1171, .e6–1181.e6. ( 10.1016/j.jid.2019.01.005) [DOI] [PubMed] [Google Scholar]
- 109.Nguyen PD, Tutela JP, Thanik VD, Knobel D, Allen RJ, Chang CC, Levine JP, Warren SM, Saadeh PB.Improved diabetic wound healing through topical silencing of p53 is associated with augmented vasculogenic mediators. Wound Repair and Regeneration 2010. 18 553–55. ( 10.1111/j.1524-475X.2010.00638.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bitar MS.Diabetes impairs angiogenesis and induces endothelial cell senescence by up-regulating thrombospondin-CD47-dependent signaling. International Journal of Molecular Sciences 2019. 20 673. ( 10.3390/ijms20030673) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rosso A, Balsamo A, Gambino R, Dentelli P, Falcioni R, Cassader M, Pegoraro L, Pagano G, Brizzi MF.p53 mediates the accelerated onset of senescence of endothelial progenitor cells in diabetes. Journal of Biological Chemistry 2006. 281 4339–4. ( 10.1074/jbc.M509293200) [DOI] [PubMed] [Google Scholar]
- 112.Charalambous C, Virrey J, Kardosh A, Jabbour MN, Qazi-Abdullah L, Pen L, Zidovetzki R, Schönthal AH, Chen TC, Hofman FM.Glioma-associated endothelial cells show evidence of replicative senescence. Experimental Cell Research 2007. 313 1192–1. ( 10.1016/j.yexcr.2006.12.027) [DOI] [PubMed] [Google Scholar]
- 113.Colombo M, Mirandola L, Chiriva-Internati M, Basile A, Locati M, Lesma E, Chiaramonte R, Platonova N.Cancer cells exploit notch signaling to redefine a supportive cytokine milieu. Frontiers in Immunology 2018. 9 1823. ( 10.3389/fimmu.2018.01823) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wieland E, Rodriguez-Vita J, Liebler SS, Mogler C, Moll I, Herberich SE, Espinet E, Herpel E, Menuchin A, Chang-Claude Jet al. Endothelial Notch1 activity facilitates metastasis. Cancer Cell 2017. 31 355–367. ( 10.1016/j.ccell.2017.01.007) [DOI] [PubMed] [Google Scholar]
- 115.Lin S, Negulescu A, Bulusu S, Gibert B, Delcros JG, Ducarouge B, Rama N, Gadot N, Treilleux I, Saintigny Pet al. Non-canonical NOTCH3 signalling limits tumour angiogenesis. Nature Communications 2017. 8 16074. ( 10.1038/ncomms16074) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schneider JA, Bennett DA.Where vascular meets neurodegenerative disease. Stroke 2010. 41 (Supplement) S144–S14. ( 10.1161/STROKEAHA.110.598326) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vogelgsang J, Wolff-Menzler C, Kis B, Abdel-Hamid M, Wiltfang J, Hessmann P.Cardiovascular and metabolic comorbidities in patients with Alzheimer’s disease and vascular dementia compared to a psychiatric control cohort. Psychogeriatrics 2018. 18 393–401. ( 10.1111/psyg.12338) [DOI] [PubMed] [Google Scholar]
- 118.Ujiie M, Dickstein DL, Carlow DA, Jefferies WA.Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation 2003. 10 463–4. ( 10.1038/sj.mn.7800212) [DOI] [PubMed] [Google Scholar]
- 119.Webster SJ, Bachstetter AD, Nelson PT, Schmitt FA, Van Eldik LJ.Using mice to model Alzheimer’s dementia: an overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Frontiers in Genetics 2014. 5 88. ( 10.3389/fgene.2014.00088) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sweeney MD, Kisler K, Montagne A, Toga AW, Zlokovic BV.The role of brain vasculature in neurodegenerative disorders. Nature Neuroscience 2018. 21 1318–1331. ( 10.1038/s41593-018-0234-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Samuraki M, Matsunari I, Yoshita M, Shima K, Noguchi-Shinohara M, Hamaguchi T, Ono K, Yamada M.Cerebral amyloid angiopathy-related microbleeds correlate with glucose metabolism and brain volume in Alzheimer’s disease. Journal of Alzheimer’s Disease 2015. 48 517–5. ( 10.3233/JAD-150274) [DOI] [PubMed] [Google Scholar]
- 122.Selkoe DJ, Hardy J.The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Molecular Medicine 2016. 8 595–608. ( 10.15252/emmm.201606210) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bhat R, Crowe EP, Bitto A, Moh M, Katsetos CD, Garcia FU, Johnson FB, Trojanowski JQ, Sell C, Torres C.Astrocyte senescence as a component of Alzheimer’s disease. PLoS ONE 2012. 7 e45069. ( 10.1371/journal.pone.0045069) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Al-Mashhadi S, Simpson JE, Heath PR, Dickman M, Forster G, Matthews FE, Brayne C, Ince PG, Wharton SB.Medical Research Council Cognitive Function and Ageing Study. Oxidative glial cell damage associated with white matter lesions in the aging human brain. Brain Pathology 2015. 25 565–5. ( 10.1111/bpa.12216) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Flanary BE, Sammons NW, Nguyen C, Walker D, Streit WJ.Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Research 2007. 10 61–74. ( 10.1089/rej.2006.9096) [DOI] [PubMed] [Google Scholar]
- 126.Jurk D, Wang C, Miwa S, Maddick M, Korolchuk V, Tsolou A, Gonos ES, Thrasivoulou C, Saffrey MJ, Cameron Ket al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012. 11 996–1004. ( 10.1111/j.1474-9726.2012.00870.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yamazaki Y, Baker DJ, Tachibana M, Liu CC, van Deursen JM, Brott TG, Bu G, Kanekiyo T.Vascular cell senescence contributes to blood-brain barrier breakdown. Stroke 2016. 47 1068–10. ( 10.1161/STROKEAHA.115.010835) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang P, Kishimoto Y, Grammatikakis I, Gottimukkala K, Cutler RG, Zhang S, Abdelmohsen K, Bohr VA, Misra Sen J, Gorospe Met al. Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nature Neuroscience 2019. 22 719–728. ( 10.1038/s41593-019-0372-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Singh Angom R, Wang Y, Wang E, Pal K, Bhattacharya S, Watzlawik JO, Rosenberry TL, Das P, Mukhopadhyay D.VEGF receptor-1 modulates amyloid beta 1–42 oligomer-induced senescence in brain endothelial cells. FASEB Journal 2019. 33 4626–4637. ( 10.1096/fj.201802003R) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Garwood CJ, Simpson JE, Al Mashhadi S, Axe C, Wilson S, Heath PR, Shaw PJ, Matthews FE, Brayne C, Ince PGet al. DNA damage response and senescence in endothelial cells of human cerebral cortex and relation to Alzheimer’s neuropathology progression: a population-based study in the Medical Research Council Cognitive Function and Ageing Study (MRC-CFAS) cohort. Neuropathology and Applied Neurobiology 2014. 40 802–8. ( 10.1111/nan.12156) [DOI] [PubMed] [Google Scholar]
- 131.Suzuki Y, Yasui H, Brzoska T, Mogami H, Urano T.Surface-retained tPA is essential for effective fibrinolysis on vascular endothelial cells. Blood 2011. 118 3182–318. ( 10.1182/blood-2011-05-353912) [DOI] [PubMed] [Google Scholar]
- 132.Mehta D, Malik AB.Signaling mechanisms regulating endothelial permeability. Physiological Reviews 2006. 86 279–367. ( 10.1152/physrev.00012.2005) [DOI] [PubMed] [Google Scholar]
- 133.Kook SY, Hong HS, Moon M, Ha CM, Chang S, Mook-Jung I.Abeta(1)(-)(4)(2)-RAGE interaction disrupts tight junctions of the blood-brain barrier via Ca(2)(+)-calcineurin signaling. Journal of Neuroscience 2012. 32 8845–88. ( 10.1523/JNEUROSCI.6102-11.2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cho SJ, Yun SM, Jo C, Jeong J, Park MH, Han C, Koh YH.Altered expression of Notch1 in Alzheimer’s disease. PLoS ONE 2019. 14 e0224941. ( 10.1371/journal.pone.0224941) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Brai E, Raio NA, Alberi L.Erratum to: Notch1 hallmarks fibrillary depositions in sporadic Alzheimer’s disease. Acta Neuropathologica Communications 2016. 4 90. ( 10.1186/s40478-016-0360-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cuello AC, Hall H, Do Carmo S.Experimental pharmacology in transgenic rodent models of Alzheimer’s disease. Frontiers in Pharmacology 2019. 10 189. ( 10.3389/fphar.2019.00189) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki Aet al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016. 530 184–18. ( 10.1038/nature16932) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg Met al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 2015. 14 644–6. ( 10.1111/acel.12344) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, Hagler M, Jurk D, Smith LA, Casaclang-Verzosa Get al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016. 15 973–97. ( 10.1111/acel.12458) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, Herrmann SM, Jensen MD, Jia Q, Jordan KLet al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. EBiomedicine 2019. 47 446–456. ( 10.1016/j.ebiom.2019.08.069) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, Prata L, Masternak MM, Kritchevsky SB, Musi Net al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBiomedicine 2019. 40 554–563. ( 10.1016/j.ebiom.2018.12.052) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wilson WH, O’Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, Tulpule A, Dunleavy K, Xiong H, Chiu YLet al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet: Oncology 2010. 11 1149–11. ( 10.1016/S1470-2045(1070261-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Thompson PJ, Shah A, Ntranos V, Van Gool F, Atkinson M, Bhushan A.Targeted elimination of senescent beta cells prevents Type 1 diabetes. Cell Metabolism 2019. 29 1045, .e10–1060.e10. ( 10.1016/j.cmet.2019.01.021) [DOI] [PubMed] [Google Scholar]
- 144.Grosse L, Wagner N, Emelyanov A, Molina C, Lacas-Gervais S, Wagner KD, Bulavin DV.Defined p16(high) senescent cell types are indispensable for mouse Healthspan. Cell Metabolism 2020. 32 87.e6–99.e6. ( 10.1016/j.cmet.2020.05.002) [DOI] [PubMed] [Google Scholar]
- 145.Cai Y, Zhou H, Zhu Y, Sun Q, Ji Y, Xue A, Wang Y, Chen W, Yu X, Wang Let al. Elimination of senescent cells by beta-galactosidase-targeted prodrug attenuates inflammation and restores physical function in aged mice. Cell Research 2020. 30 574–589. ( 10.1038/s41422-020-0314-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kim EC, Kim JR.Senotherapeutics: emerging strategy for healthy aging and age-related disease. BMB Reports 2019. 52 47–55. ( 10.5483/BMBRep.2019.52.1.293) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Song P, An J, Zou MH.Immune clearance of senescent cells to combat ageing and chronic diseases. Cells 2020. 9 671. ( 10.3390/cells9030671) [DOI] [PMC free article] [PubMed] [Google Scholar]