

 This work is licensed under a
This work is licensed under a Abstract
The terms ‘idiopathic short stature’ (ISS) and ‘small for gestational age’ (SGA) were first used in the 1970s and 1980s. ISS described non-syndromic short children with undefined aetiology who did not have growth hormone (GH) deficiency, chromosomal defects, chronic illness, dysmorphic features or low birth weight. Despite originating in the pre-molecular era, ISS is still used as a diagnostic label today. The term ‘SGA’ was adopted by paediatric endocrinologists to describe children born with low birth weight and/or length, some of whom may experience lack of catch-up growth and present with short stature. GH treatment was approved by the FDA for short children born SGA in 2001, and by the EMA in 2003, and for the treatment of ISS in the US, but not Europe, in 2003. These approvals strengthened the terms ‘SGA’ and ‘ISS’ as clinical entities. While clinical and hormonal diagnostic techniques remain important, it is the emergence of genetic investigations that have led to numerous molecular discoveries in both ISS and SGA subjects. The primary message of this article is that the labels ISS and SGA are not definitive diagnoses. We propose that the three disciplines of clinical evaluation, hormonal investigation and genetic sequencing should have equal status in the hierarchy of short stature assessments and should complement each other to identify the true pathogenesis in poorly growing patients.
Keywords: linear growth, short stature, small for gestational age, idiopathic short stature, genotyping, growth hormone therapy
Introduction
Normal human linear growth results from an evolutionary process expressing the sum effect of multiple genes linked to survival (1). Throughout the growing phase of life, increase in height is both an indicator of good health and a sensitive marker of illness (2). Understanding the physiology of linear growth initially focused on the growth hormone (GH)-insulin-like growth factor (IGF)-1 axis as the key regulatory process (3). However, the development of genetic studies in growth physiology, notably next generation sequencing technology, is identifying new molecular mechanisms, particularly in genes involved in growth plate physiology and chondrogenesis, that modulate growth and when disturbed may lead to short stature (4, 5).
The factors influencing linear growth can be categorised in various ways, but we favour a category of factors playing a role within the growth plate vs factors acting on the growth plate from elsewhere in the body. This categorisation leads to two groups, that is, primary and secondary growth disorders (4, 5, 6). The majority of primary growth disorders consists of clinically defined syndromes and skeletal dysplasias caused by variants of genes associated with growth plate physiology such as rare variants with a large effect, for example, achondroplasia caused by a heterozygous variant in FGFR3. It is noteworthy that also less rare variants with a moderate effect (7) or common variants with a small effect (as demonstrated in genome-wide association studies (GWAS)) (8) have been associated with body stature. Population GWAS studies recently identified 3290 near-independent SNPs associated with height, including 1185 height-associated SNPs located within loci not previously identified. The near-independent genome-wide significant SNPs explained ∼24.6% of the variance of height in a population of 700,000 European individuals (9). In the international classification of paediatric endocrine diagnoses (www.icped.org) (6), short children born small for gestational age (SGA) is classified under primary growth disorders according to the absence of a known factor outside of the growth plate, and thus the expectation that the cause may reside in the growth plate.
The second set of factors includes hormones, nutrition, inflammatory cytokines and extracellular fluid, and can be congenital or acquired. The best known example of an endocrine growth disorder is a defect of the GH–IGF-1 axis, but other hormonal factors include thyroxine, glucocorticoids and sex steroids, sometimes in interaction with each other (10). Nutritional deficits are known to cause growth faltering (2), as well as increased concentrations of inflammatory cytokines (e.g. in juvenile arthritis or Crohn disease) (2) or disorders of extracellular fluid (e.g. renal tubular acidosis).
From the clinical perspective, the child referred with short stature statistically has a height more than 1.6 standard deviations (s.d.) below the mean of the population or of the family, using the Hermanussen and Cole definition of target height (11). In practical terms, the perception by the child or parents that height is below average is sufficient to warrant a specialist referral. Similarly, children with unexplained shifts in growth percentiles may need evaluation. Therefore children referred to a paediatric endocrinologist can have highly variable degrees of ‘short stature’ or growth failure and equally wide ranges of potential aetiologies from complete normality to a growth pattern which will lead to adult height below the target height range, that is, less than 1.6 s.d. below the mid-parental height.
Early investigation and diagnosis of short stature is important (12). The investigation protocol combines clinical skills of history-taking, and observation with laboratory tests aimed to identify non-endocrine causes or hormonal abnormalities which can allow the clinician to commit to a diagnosis and thereby explain the child’s growth pattern (13). Paediatric endocrinologists need to be trained in the clinical skills of history-taking and phenotyping. The inclusion of genotyping in the investigation process in certain situations has led to important discoveries, which have not only identified new causes of short stature, but also clarified the physiology of human growth regulation (14, 15, 16, 17) and in some cases indicated the correct therapeutic approach (18). Clinicians need to understand the genetic tools available and their interpretation. A strategy that combines the three disciplines of clinical assessment, endocrine evaluation and genetic sequencing, each having the same degree of importance, is required to probe and identify the pathogenesis in cases with uncertain aetiologies.
Idiopathic short stature
The term ‘idiopathic short stature’ (ISS) was applied to short children without a known aetiology over 35 years ago, before the era of molecular investigation. A current definition of ISS will be discussed subsequently. However, ISS is not a definitive diagnosis; Its original use as a description of children with short stature, who did not have GH deficiency, served a purpose in its day, but now clinicians need to take the investigation of such children further, with new opportunities presenting a realistic chance of identifying causative pathogeneses. As precision medicine attempts to personalise diagnosis and therapy, new genetic discoveries in the GH–IGF-1 axis and growth plate chondrogenesis need representation in the diagnostic protocol. The practice of labelling children as having ISS, after initial clinical, endocrine and radiological assessment, without further observation, evaluation and delineation of pathogenesis, is failing to optimise short stature management.
The origin of the ISS designation
The diagnosis of GH deficiency in children entered clinical practice in the late 1960s with the demonstration of GH release following stimulation by insulin-induced hypoglycaemia (19) or acute administration of glucagon and other GH secretagogues (20). GH stimulation tests permitted diagnosis of GH deficiency, and thereby separated GH deficient patients from those with similar appearance but normal GH secretion. Towards the end of the era of administration of pituitary hGH, which terminated in 1985 due to the Creutzfeldt–Jakob epidemic, the anticipation of the availability of recombinant hGH (rhGH), first synthesised in 1979 (21), led to short- and long-term studies of hGH therapy in subjects with so-called ‘normal variant short stature’ (22) or labelled as ‘short normal’ children (23). Interestingly, the heterogeneity of ‘short normal’ children was clear at that time as judged by their wide variability of growth responses to hGH therapy (24, 25). A conference, convened at the NIH in November 1983 to discuss the future use of rhGH in short children without GH deficiency, concluded that in a society that ‘values tallness’, controlled research studies of rhGH in such patients were authorised (26). At that meeting, ISS as a diagnostic group acquired scientific respectability.
ISS as an indication for rhGH therapy
When rhGH was approved by the FDA in October 1985 for use in children with ‘inadequate GH’, this was contingent on the establishment of post-marketing surveillance. Genentech, the makers of the approved rhGH, Protropin, set up the National Cooperative Growth Study (NCGS), within which ISS was a designated diagnostic group (27). ‘Idiopathic short stature’, thus became an established label for short children with normal GH secretion, normal birth weight and absence of chromosomal defects or chronic illness and was soon adopted throughout the paediatric endocrinology community. Alternative terms were proposed such as ‘idiopathic growth failure’ (IGF) or ‘growth failure of unknown aetiology’ (GFUE) to change the emphasis from ‘stature’ to ‘growth’ or growth rate (2). However, ISS prevailed and 35 years later, remains a popular designation for short children with no defined aetiology. Of note, patients with ‘normal variant short stature’ specifically those with familial short stature and constitutional growth delay were not excluded from the ‘ISS’ designation.
Randomised clinical trials with rhGH were led by industry and produced positive growth-promoting results (28, 29), and ISS was soon referred to as ‘a condition termed idiopathic short stature’ (28) or a ‘diagnostic category’ (30). Positive data confirmed the effects of rhGH therapy, even compared with placebo-treated controls (29). Predictably, these results lead to approval of ISS by the FDA in 2003 as an indication for rhGH therapy, under the criteria of height <−2.25 s.d. without evidence of underlying disease or GH deficiency and short expected adult height. This decision had major implications on clinical practice as suddenly 400,000 children in the USA were eligible for rhGH therapy (31). Similar applications to the European Medicines Agency were unsuccessful, largely due to the absence of data showing a positive effect on quality of life (32).
The FDA approval for GH therapy of ISS consolidated this category of patients in the minds of paediatricians with data on efficacy and safety accumulating in international databases such as the Kabi international growth study (KIGS) and NCGS (33). Results of cohorts of ISS subjects were, and still are, being regularly analysed and published (34, 35), and are used as the basis for management guidelines (5, 36). ISS is also used as a diagnostic category in the International Classification of Pediatric Endocrine Disorders (www.icped.org) (6). Notably, the ISS patients treated with rhGH responded inconsistently and in particular, growth during year 1 of therapy did not predict the response in year 2, which emphasised the marked heterogeneity of patients carrying the ISS label (37).
Current definition of ISS and its sub-classification
The definition of ISS is clinically important because inclusion of a patient within this designation may, in certain societies where ISS is approved for rhGH therapy, provide an indication for this treatment. ISS is currently defined as short stature with height <−2 SDS, normal birth size (birth weight and length >−2 SDS), absence of abnormal physical features and normal general screening investigations, normal body proportions and absence of major dysmorphic features (38).
The components of ISS were critically appraised in two reviews by Wit et al. in 2008 (39, 56). A Consensus Statement on ISS management was also published in 2008 (40). ISS is subdivided into familial short stature (FSS) with normal or delayed bone age and non-familial short stature (NFSS) with normal or delayed bone age (38, 39, 40, 41). The definitions proposed for FSS and NFSS are based on the calculation of ‘conditional’ target height (cTH), which is adjusted for the correlation between maternal and paternal heights, so-called assortative mating, and for the correlation between children’s height SDS and mid-parental height SDS (11). The definition of FSS is height SDS = cTH SDS ± 1.6 and of NFSS, height SDS < cTH SDS −1.6 based on the fact that 95% of healthy children have height SDS = cTH SDS ± 1.6 (the TH range).
It is likely that in most subjects with FSS their short stature is related to inheritance of polygenic variants from both parents with multiple small negative effects on height. However, a copy number variant (CNV) or monogenic defect is also possible, particularly if there is a pattern of dominant inheritance. The inheritance of multiple variants in the same or different growth-related pathways may occur (17). In children with NFSS and a slow tempo of constitutional delay of growth and puberty is statistically the most likely diagnosis, particularly if bone age is delayed and the family history is positive for delayed puberty. However, also a recessive or de novo pathogenic gene variant or CNV should be considered. It is in the NFSS group that defects associated with an adult height below parental target height are most likely to occur.
In the time when GH was considered as the major factor in growth regulation, ‘ISS’ was used to describe children who fall between GH deficiency and GH insensitivity in the GH–IGF-1 axis continuum model. According to this model, ISS subjects should have a normal physiological equilibrium between GH sensitivity and GH deficiency, which is the case with those with FSS, where no endocrine defect in the child or parents has been identified. However, since the discovery that most genes associated with growth have no direct relationship with the GH–IGF-I axis (9), it appears more useful to think in terms of another conceptual framework for understanding short and tall stature that is centred not on the GH–IGF-1 axis, but rather on the growth plate (4). In the 21st century, 35 years after its inception, ISS describes a highly heterogeneous group of short patients and no longer qualifies as a single final diagnosis.
Short stature related to birth size small for gestational age (SGA)
The majority of infants born SGA, defined as birth weight and/or length <−2 SDS for gestational age (42) show spontaneous catch-up growth into the normal range (i.e. height >−2 SDS) by the age of 2 to 3 years (43). By the age of 8 years, approximately 90% of SGA children have reached a normal height (44). However, the 10% who remain short have a high risk of adult short stature with height below their target height range, particularly if their birth length was <−2 SDS (45). These subjects require referral to a paediatric endocrinologist (42) and will comprise approximately 20% of all short stature referrals (46). In paediatric endocrine publications, the term ‘SGA’ has gradually transformed from the description of low birth size (birth weight and/or length) into a diagnostic label indicating a ‘short individual born SGA’ (42).
Although originally clinicians may have thought that the low birth size can be considered the ‘cause’ of short stature in childhood and adulthood, it now appears more likely that a short child born SGA should be viewed as a child with an unspecified growth disorder of prenatal onset. SGA is therefore similarly inherent ‘idiopathic’ as ISS. Indeed, a continuum of ‘non-syndromic short stature’ has been demonstrated in a study of children born SGA (n = 45) or with appropriate gestational age (n = 137) (47), so the distinction between short SGA and non-SGA children is artificial. An umbrella term ‘non-syndromic short stature’ has been used for short SGA and non-SGA children without dysmorphic features (14). We suggest that in a future revision of the International Classification of Pediatric Endocrine Diagnoses, short children born SGA should be classified in the same group as ISS.
Returning to our primary message, the designation of SGA is not a diagnosis and further investigations are needed to identify the precise aetiology where possible leading to optimal clinical management (48). These further investigations are essentially genetic studies.
Endocrine abnormalities in patients initially considered ISS or SGA
In the 1980s and 1990s, the study of childhood linear growth focused on the function of different components of the GH–IGF-1 axis, and enormous progress in the understanding of this axis was made (10). The original somatomedin hypothesis, published in 1957 (49), was up-dated 50 years later (3) showing that the IGF system played a key role in growth regulation with both circulating and peripherally produced IGF-1 having individual roles (50). IGF-1 deficiency was reported to occur in a proportion of short patients with normal GH secretion (51), which placed some ISS patients in an intermediate position between GH deficiency and GH resistance, although some overlap existed.
Evidence was accumulating that some ISS patients had a degree of functional GH insensitivity (52) with a broad range of generation of IGF-1 in response to GH. The important study by Cohen et al. in 2007 reported that high doses of rhGH were needed to reach a serum IGF-1 concentration of +2 s.d. (53). Evidence of subnormal generation of IGF-1 was also demonstrated in the elegant studies by Buckway and Selva of responses in the IGF-1 generation test (IGFGT). Compared to normal control subjects, ISS patients had basal IGF-1 levels in the lower half of the normal range and after GH stimulation on days 5 and 8 of the IGFGT, IGF-1 levels were significantly lower than normal, regardless of GH dose (54, 55). These hormonal findings challenge the definition of ISS which states that there is no endocrine abnormality. The molecular basis of these results was not apparent at that time.
Variants in genes regulating GH action with phenotypes consistent with ISS or SGA
ISS patients may have variable GH sensitivity and IGF-I concentrations (39) and consistent with this, a proportion have a diminished response to rhGH therapy (56, 57). Therefore, it has been suggested that less deleterious GHR gene defects may cause ISS associated with features of GH insensitivity (58). Numerous studies of ISS cohorts have reported heterozygous GHR variants occurring with a frequency ranging from 5 to 15.5% (59). It has also been noted that GHR sequence changes are common in children with ISS, with many also identified in control subjects and normal stature family members (60).
Since the start of genetic investigations of short stature phenotypes in the late 1980s, a number of pathogenic variants have been discovered in children labelled as having ISS or SGA. In 2019, Storr et al. published an extensive review of mild or ‘non-classical’ abnormalities of GH action (60). Mild forms of GH insensitivity can be broadly divided into three categories; (i) aberrations of GH signalling caused by homozygous or heterozygous variants of genes encoding the GH receptor (GHR) or signal transducer and activator of transcription 5B (STAT5B) (59, 60, 61, 62, 63, 64); ii) defects of IGF-I secretion (IGF1), transport (IGFALS) and bioavailability (PAPPA2) (65, 66, 67, 68, 69, 70, 71, 72) and iii) IGF-I insensitivity (IGF1R) (73). Most patients with GH1, GHR and STAT5B defects are born with a normal birth size, most patients with IGF1 and IGF1R variants with a low birth size, while mean birth weight of patients with IGFALS defects is approximately −2 SDS (73). A paternally transmitted heterozygous variant of IGF2 is one of the genetic causes of Silver–Russell syndrome (74). A summary of phenotypic and endocrine features of genetic defects consistent with ISS is shown in Table 1.
Table 1.
Summary of phenotypic and biochemical features of defects causing GH insensitivity originally labelled as ISS or SGAa.
| Genetic defect | Gene defect | |||||||
|---|---|---|---|---|---|---|---|---|
| GHR heterozygous | GHR pseudo-exon, homozygous | STAT5B heterozygous | IGFI heterozygous | IGF2 heterozygous (paternal) | IGFALS homozygous or heterozygous | PAPPA2 homozygous | IGF1R heterozygous | |
| Facial dysmorphism | − | Mid-face hypoplasia +/− | Mid-face hypoplasia +/− | Micro-gnathia | Relative macrocephaly | − | Long thin nose, small chin | − |
| Microcephaly | − | − | − | +/− | +/− | +/− | +/− | +/− |
| Feeding problems | − | − | − | + | + | − | − | + |
| Intellectual delay | − | − | − | − | +/− | − | − | +/− |
| Pubertal delay | − | − | +/− | − | +/− | + | − | − |
| Immune deficiency | − | − | + | − | − | − | − | − |
| Hypoglycaemia | − | + | −/+ | − | n/r | − | − | − |
| Hyper-insulinaemia | − | − | − | +/− | n/r | + | + | − |
| IGF-1 | ↓ | n/↓ | ↓ | n/↓ | n/↑ | ↓ | ↑ | n/↑ |
| IGFBP-3 | ↓ | n/↓ | ↓ | n | n/↑ | ↓ | ↑ | n/r |
| ALS | n/↓ | n/↓ | +/− | n | n/r | ↓ | ↑ | n/r |
| GH | ↑ | n/↑ | ↑ | n/↑ | n/↑ | ↑ | ↑ | n/↑ |
| GHBP deficiency | +/− | − | − | − | − | − | − | n/r |
aThis table presents genetic variants presenting with mild or moderate short stature; it does not contain characteristics of homozygous defects of GHR, STAT5B and IGF-I;
+, positive; −, negative; +/−, predominantly positive; −/+, predominantly negative; ↑, increased; ↓, decreased.
ALS, acid labile subunit; GHBP, growth hormone binding protein; IGFBP-3, IGF binding protein-3; n, normal; n/r, not reported.
Gene variants affecting growth plate chondrogenesis with phenotypes consistent with ISS or SGA
Genetic defects which impair chondrogenesis are likely to cause some degree of body disproportion; however, this may be mild and not noticed by the clinician, resulting in the child being labelled as ISS or SGA (48). Out of the multiple reported genes (14), we discuss four examples.
SHOX haploinsufficiency
Pathogenic variants of the gene encoding short stature homeobox (SHOX), located at the tip of the X and Y chromosome, and deletions or duplications of the SHOX enhancer regions, impair chondrocyte differentiation in the growth plate. A gene dose effect is apparent as homozygous or compound heterozygous inactivating SHOX variants cause Langer mesomelic dysplasia, while heterozygous variants cause a milder skeletal dysplasia, Leri–Weill dyschondrosteosis, with the classical Madelung deformity of the wrist, or present clinically as ISS. SHOX haploinsufficiency is caused more frequently by CNVs than by single nucleotide variants (75), and is reported to account for 2–15% of children presenting with ISS (14, 76). In a series of 521 patients with short stature due to SHOX haploinsufficiency, treated under the licence for rhGH, 44% were documented to have non-syndromic short stature (77). The extent of the SHOX haploinsufficiency phenotype is not yet well defined (77, 78).
Fibroblast growth factor (FGF) signalling
Several FGFs and their receptors play a role in the growth plate. The FGF receptor 3, encoded by FGFR3, acts as a negative regulator of chondrogenesis, so heterozygous activating variants impair bone elongation resulting in short-limbed skeletal dysplasia (14). There is a range of phenotypes, from classical achondroplasia to hypochondroplasia presenting as a relatively mild skeletal dysplasia, even with normal body proportions (79) and showing some response to rhGH therapy (80).
CNP-NPR2 pathway
C-natriuretic peptide (CNP) encoded by NPPC is a local, positive regulator of growth plate function (14). Homozygous inactivating variants of NPR2, which encodes the main CNP receptor, cause severe skeletal dysplasia, but relatives who are heterozygous for these mutations have a relatively mild growth defect with phenotypes similar to SHOX deficiency (81). Heterozygous NPR2 variants are thought to account for 2–6% of cases of ISS (82, 83).
ACAN mutations
The growth plate is situated between the epiphysis and metaphysis of the long bones, and chondrogenesis proceeds with osteoblasts, osteoclasts and blood vessels transforming the newly formed cartilage into bone. Aggrecan is the most abundant proteoglycan in hyaline cartilage and crucial to the structure and function of the growth plate. Variants in ACAN which encodes for aggrecan are associated with a range of growth defects which may be severe or mild, that is presenting as ISS or SGA (14). Patients carrying heterozygous variants of ACAN can reach an adult height of 150–152 cm without further dysmorphic features (84). Hauer et al. performed sequence analyses in 428 families with short stature, and the results showed that heterozygous nonsense variants of ACAN were identified in six families, that is 1.4%. The mean height SDS value of the affected subjects was −3.2 s.d., that is consistent with a label of ISS (85). In a study of WES of 200 short stature patients, Hauer et al. also identified heterozygous carriers of recessive skeletal dysplasia alleles in 3.5% of cases. These were notably ACAN and NPR2 defects, with ACAN being the most commonly mutated known short stature-associated gene with a frequency of 2.5% (86).
Diagnostic approach and investigations of patients with ISS or SGA
Clinical assessment
Schemes for the clinical assessment of ISS or SGA patients have been published in a number of reviews (13, 39) and in the 2008 Consensus Statements on management of ISS (40) and SGA (42). A general scheme for short stature assessment in patients labelled as ISS or SGA is shown in Fig. 1. Clinical skills are crucial in eliciting a detailed family history, including consanguinity, heights of parents, grandparents and siblings. Birth weight and length, gestation, and developmental milestones need to be documented, and potential symptoms from the major body systems need to be elicited by direct questioning. SGA is a highly heterogeneous state, with potential influences from geographical, maternal, paternal, placental, environmental and foetal factors (48). The diagnostic approach to the SGA child should similarly start with a search for clues for a primary or secondary growth disorder. Key primary growth disorders associated with SGA are clinically defined syndromes associated with growth failure such as Silver–Russell, Noonan, Prader–Willi, Bloom, 3M and neurofibromatosis type 1 syndromes and skeletal dysplasias (growth plate chondrogenesis disorders) such as ACAN, FGFR3, NPR2 and SHOX mutations (14). IGF-1 receptor (IGF1R) defects have been categorised under secondary growth disorders (6). A careful medical history, including details of maternal life-style, such as smoking and alcohol, environmental conditions during pregnancy are essential to elicit. Post-natal developmental delay, feeding difficulties and behaviour problems are also important to probe for by direct questioning.
Figure 1.
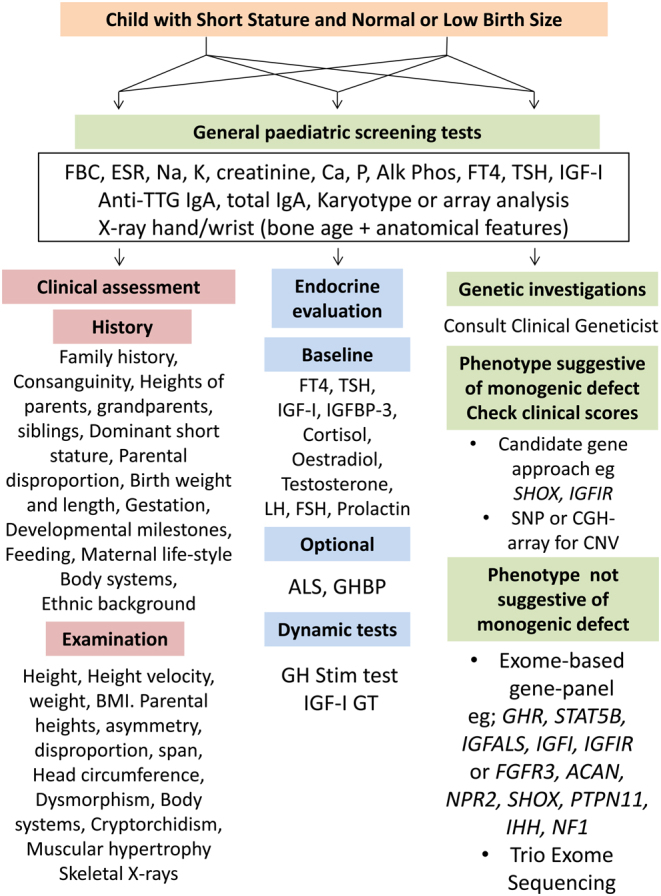
Scheme for investigation of ISS and ‘idiopathic’ SGA giving equal status to clinical assessment, endocrine evaluation and genetic investigations (12, 91). CVS, cardiovascular system; Anti-TTG, anti-tissue transglutaminase; IGF-I-GT, IGF-I generation test; CGH-array, comparative genomic hybridisation; CNV, copy number variation.
Physical examination should include measurement of height, sitting height, sitting height/height ratio SDS, arm span and head circumference (13). Documentation of dysmorphic features which may be subtle, as in 3M syndrome and IGF1R variants, is important. Body asymmetry and disproportion, microcephaly or relative macrocephaly, heart murmur, cryptorchidism and muscular hypertrophy should be sought. Accurate height measurement with documentation of severe short stature (height <–3 SDS) and height SDS similar to one parent are also relevant clues (13).
General paediatric screening tests
The suggested scheme for laboratory screening tests for exclusion of general paediatric pathology, including thyroid function, IGF-1, IGFBP-3 and karyotype in girls proposed in the 2008 ISS Consensus Statement (40) was challenged in 2013 by Sisley et al. who reported that in 232 short patients who were healthy and had normal physical appearance, 98.7% had normal screening tests (87). The message was that in healthy short children screening tests were not cost-effective. To some extent this view was perpetuated in the recent GRS ‘Guidelines’ article (5) which stated that ‘Laboratory tests should be guided by clinical features rather than routinely applied to all patients with short stature ... Clinical discretion should be applied to the scope of testing for non-endocrine disease’.
In the context of ISS and unexplained SGA, it is essential that routine haematological and biochemical screening tests are performed (Fig. 1). Chronic illnesses such as coeliac and Crohn’s disease need to be excluded as do sex chromosome defects such as mosaic Turner syndrome (2). The ISS group with NFSS are most likely to have occult illness, being shorter than their parental target height, so pathological causes need to be excluded before endocrine assessment. Exclusion of underlying pathology is essential before therapy with GH is considered (13).
In short children born SGA, rhGH therapy may be prescribed in many of these children and normal renal, liver and haematological status are important to document before this therapy is initiated. Laboratory screening should consist of full blood count, ESR, IGF-1, FT4, TSH, test for coeliac disease, electrolyte and renal and liver function tests, calcium, phosphate and alkaline phosphatase (13, 40). A hand/wrist bone age X-ray with examination for features of skeletal dysplasia should be performed in all referred short children. A karyotype or SNP array analysis (for detecting Turner syndrome, and with the latter tool also for copy number variants and uniparental disomy) in all girls with height <–2 SDS or height SDS <TH SDS –1.6 should also be performed (13). If serum IGF-1 is low, a GH stimulation test or an IGFGT is further options for identification of an endocrine aetiology.
Investigation of the GH-IGF-1 axis
Hormonal assessment should follow the guidelines, published previously (40) and reviewed in the recent GRS Consensus Statement (5). Important messages are highlighted in this publication such as an IGF-1 assay with reliable reference data with ranges for paediatric ages should be used, and serum IGF-1 is influenced by many factors such as assay methodology, age, nutrition and chronic illness (88). An IGF-1 level >0 SDS at any age makes GHD less likely but not impossible (5, 88). The GH stimulation test (GHST) has been criticised over the years for not a physiological stimulus and for lack of reproducibility. Examination of the timing of peak GH in certain tests such as the arginine and levodopa test will increase their sensitivity (89) (Fig. 1).
The GHST remains of key importance in the assessment of short children potentially labelled as ISS or SGA. An abnormal GHST, with a peak GH level of <7 or <10 μg/L in the US (5) and auxological features consistent with GHD, immediately removes a patient from an ‘idiopathic’ category. A low IGF-1 value without knowledge of GH status cannot distinguish GHD from GH insensitivity (51). Longitudinal observation and monitoring may lead to re-evaluation of the GH-IGF-I axis in some inadequately growing children that may involve repeat GHST. Once the distinction between GHD and non-GHD short stature has been made, further investigations to define aetiology such as measurements of IGFBP-3, ALS and an IGFGT, with its recognised disadvantages (90), are options. However, the designations of FSS or NFSS still apply, now with GHD or non-GHD added, but genetic investigations are indicated to identify a causative genetic pathogenesis and thereby remove the patient from the undefined category of ISS or SGA.
Genetic studies for the further evaluation of aetiology
Genetic testing was briefly mentioned in the 2008 ISS Consensus Statement (39), but advances in genetic techniques and particularly next generation sequencing have resulted in increasing yields of positive genetic diagnoses in short children labelled ISS with and without SGA (14, 15). A range of genetic techniques are available, but those should be prioritised which can provide positive causative answers. Targeted evaluation of a single gene (candidate gene sequencing) is recommended for a child who shows characteristic clinical or laboratory features of a well-known genetic syndrome. However, in most cases there is not a strong suspicion for a certain syndrome (e.g. children presenting with mild GHI or minor dysmorphic features), so that the candidate gene approach has largely been overtaken by exome sequencing (ES), using targeted gene panels, supplemented by chromosomal microarray to detect both sequence variants and CNVs (91, 98). ES is likely to identify many novel genetic variants of uncertain causative significance (91), and functional studies will be required to assess pathogenicity (92). When the genetic variant is rare or novel, incorporating phenotype/genotype correlation and familial segregation is critical in the interpretation of causation. Support from the bio-informatic and genetic experts may be required (5).
Using a combination of targeted genetic testing and exome sequencing (ES), Hauer et al. reported a diagnostic yield of 33% in 565 cases of unexplained short stature (85, 86). A change in the paradigm of growth assessment has been proposed, prioritising genetic testing in favour of the relatively low yield of endocrine assessment (17). However, in our view, genetic testing is an integral part of the assessment of short stature and should be given equal status to clinical and endocrine assessment (Fig. 1). Schemes and indications have been proposed in a number of recent reports (14, 15, 17, 91, 92, 93).
Which short patients should have genetic investigations?
Short children with normal or low birth size are clinically heterogeneous and may harbour a range of molecular aetiologies for their short stature in up to 40% of cases (91, 93, 94, 95, 96). In general, genetic investigations are indicated if there are positive clinical diagnostic clues for a monogenic disorder. Examples of such clues include severe short stature, microcephaly or relative macrocephaly, dysmorphic features, disproportion, a positive family history, and a low birth size (SGA) (13). In general, patients with a clinical suspicion of monogenic disorders in whom a clear diagnosis will enhance clinical management in terms of explanation, prognosis, choice of therapy or genetic counselling, are good candidates for genetic testing. Examples include soft dysmorphic features suggestive of Noonan syndrome, patients with possible mild GH insensitivity (60) and patients with borderline body disproportion suggestive of SHOX, ACAN or NPR2 genetic variants (4, 97).
Regarding the implications for the choice of therapy, the principal aim of genetic investigations is to identify monogenic disorders having a significant effect on growth (17). In children with ISS, one would expect that these defects would be found mainly in the NFSS subjects, where either parents with normal stature may be carriers of a recessive gene or the patient carries a de novo pathogenic gene variant. An example of apparent NFSS of genetic origin is the pseudoexon GHR variant which can present with mild short stature and normal IGF-1 levels (63).
Many of the FSS patients are likely to have polygenic short stature having inherited common gene variants of small effect size (91). However, if dominant inheritance of short stature is traceable in one or more generations, monogenic defects as in NPR2 (91) and SHOX mutations (14), dominant negative GHR mutations (61) and Noonan syndrome defects (94) are likely.
In short children born SGA the frequency of genetic causes appears to be higher than in ISS, although direct comparative studies are awaited. Extreme examples include genetic defects in fundamental cellular processes, which can produce severe global growth deficiencies known as primordial dwarfism where pre- and post-natal growth is severely affected. A more extensive overview of gene variants associated with SGA was reviewed by Finken et al. (48). As with ISS, the primary aim of genetic studies in SGA subjects is to identify monogenic defects. These may be predicted by the use of clinical scoring systems, such as the Netchine–Harbison system for Silver–Russell syndrome (99), the recently published scoring system for IGFR mutations (73) and concordance with characteristics of ACAN variants (97, 100, 101) or Madelung deformity which is pathognomonic for SHOX haploinsufficiency. Positive scores can be followed up by candidate gene sequencing or, preferably using a hypothesis-free approach, with a genome-based targeted gene panel, including a check for copy number variations and uniparental disomy.
In a study of 55 unexplained cases of short SGA subjects, sequencing using a targeted gene panel or ES gave a positive diagnostic yield of 15% (16). Heterozygous pathogenic or likely pathogenic genetic variants in 8 of the 55 patients, were identified in genes already associated with growth disorders. Four of the genes were associated with growth plate development, IHH (n = 2), NPR2 (n = 2), SHOX (n = 1), and ACAN (n = 1), and two with the RAS/MAPK pathway, PTPN11 (n = 1) and NF1 (n = 1) (16). In the case of a dysmorphic SGA child where the diagnosis is uncertain, ES was particularly helpful in identifying the mutation in the BLM gene confirming the diagnosis of Bloom syndrome (102). In special cases where a novel monogenic disorder is suspected, ES in a ‘trio’, that is patient and both parents, can be performed. It is likely that future bio-informatic analyses of next generation sequencing technology will show that in many cases SGA is caused by a combination of multiple (epi)genetic variants (103).
We believe that a case can be made for genetic assessment of short children born SGA, because the pre-test likelihood of detecting a genetic condition is high (in the order of 30–40%), particularly if a child presents with additional features as summarised previously. Another reason is that it is important to be sure that the child does not has a syndrome for which rhGH treatment is contraindicated (e.g. Bloom syndrome) (102) or debatable, such as NF-I (105). A third reason is that the diagnosis of IGF1R haploinsufficiency or genetic syndromes affecting growth plate function (e.g. SHOX haploinsufficiency) implies that a higher dosage of rhGH is indicated to generate an appropriate growth response.
Which short patients should not have genetic investigations?
It is reasonable to assume that when the label of ISS or SGA is given to a child with short stature, a detailed auxological examination would have been performed to exclude body disproportion and major dysmorphic features. Similarly, endocrine investigations to exclude GH deficiency and GH insensitivity (GHI) should have been performed. So, the decision about genetic testing must be made by the clinician responsible for the child’s care by subjectively weighing the diagnostic clues for a primary growth disorder and their severity (13). In general, the greater the severity of short stature, the more likely is it that there is an identifiable genetic defect (5, 91), but the number and severity of any additional congenital anomalies or dysmorphic features, evidence of a skeletal dysplasia, associated intellectual disability, microcephaly or relative macrocephaly should be added to the interpretation (13, 17, 91). This agrees with the recommendation that not all short children should have genetic investigations (5). Specifically, children with mild familial short stature without any additional abnormal clinical features should not be tested. Similarly, children with delayed bone age with expected adult heights within the normal range with a positive family history for delayed puberty for the family should in general not be tested as they may have constitutional delay of growth and puberty.
Labelling a child as ISS or SGA without further attempts to clarify aetiology can be damaging to clinical outcome
In clinical medicine, ‘diagnosis’ generally means that the aetiology of a condition has been identified. As we stated previously, ISS and SGA are not final diagnoses and the designation ‘idiopathic’ (explicit in ISS, implicit in SGA) means that no aetiology has been identified. We accept that ISS and SGA are terms that will continue to be used. The FDA licences for rhGH therapy in ISS and SGA increases the temptation for clinicians to use these labels in order to prescribe rhGH either as short-term or long-term therapy.
This approach can be both psychologically damaging, when an over-optimistic height prognosis is attached to such therapy and also counterproductive when a pathogenesis supporting alternative therapy such as rhIGF-1 would be more effective. A case in point is the molecular disorder of PAPPA2 mutations. Affected children have mild short stature with subtle dysmorphic features. If labelled as ISS or SGA, hGH therapy might well have been prescribed. In fact, identifying and understanding the pathogenesis will lead to therapy with rhIGF-1, as the mutation results in deficiency of free IGF-1, and positive responses to rhIGF-1 therapy have now been reported (18, 104). The same argument applies to mild GHR mutations, where responses to rhIGF-1 have also been documented (63), in contrast to lack of evidence of responses to rhGH. Genetic identification of IGF1R defects can be compared to published experience of rhGH therapy in such patients (73) rather than to non-specific responses to hGH in idiopathic SGA subjects.
The new development of C-type natriuretic peptide analogues, such as Vosoritide, and the long-acting form of TransCon CNP, which are currently being assessed for treatment of growth plate defects would, if shown to be effective, be a further example of the inappropriate use of rhGH in patients (106, 107). It should be mentioned that patients receiving new research preparations such as the CNP analogues should be followed in specialist centres with experience of paediatric bone disorders.
Conclusions
The challenge of identifying the accurate aetiology in a child with short stature focuses on three key disciplines; (i) clinical skills, (ii) evidence-based endocrine assessment and (iii) genetic testing and data interpretation. The clinician plays a vital role in eliciting an accurate history, carefully documenting the phenotype and observing subtle clinical characteristics. The endocrine assessment is arguably less precise with decisions made on the basis of somewhat empirical deviations from normative data and unphysiological dynamic tests. Genetic investigations provide the hope of accurate causative findings which may clarify physiological mechanisms. These three disciplines should be mutually complementary. We accept that labels such as ISS and SGA are useful and will continue to be used. However, they should not be the end-points of evaluation of the patient. The use of the previous three disciplines is available and indicated in children with unexplained short stature with the aim of accurately categorising their pathogeneses, and thereby gradually eroding the large groups of patients currently carrying the labels ‘idiopathic short stature’ or ‘SGA’.
Declaration of interest
Prof. Robert Rapaport has had a Consultancy contract with OPKO. The other authors have nothing to disclose.
Funding
This work did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
References
- 1.Rosenfeld RG.Insulin-like growth factors and the basis of growth. New England Journal of Medicine 2003. 349 2184–2186. ( 10.1056/NEJMp038156) [DOI] [PubMed] [Google Scholar]
- 2.Graber E, Rapaport R.Growth and growth disorders in children and adolescents. Pediatric Annals 2012. 41 e1–e9. ( 10.3928/00904481-20120307-07) [DOI] [PubMed] [Google Scholar]
- 3.Kaplan SA, Cohen P.The somatomedin hypothesis 2007: 50 years later. Journal of Clinical Endocrinology and Metabolism 2007. 92 4529–4535. ( 10.1210/jc.2007-0526) [DOI] [PubMed] [Google Scholar]
- 4.Baron J, Sävendahl L, De Luca F, Dauber A, Phillip M, Wit JM, Nilsson O.Short and tall stature: a new paradigm emerges. Nature Reviews: Endocrinology 2015. 11 735–746. ( 10.1038/nrendo.2015.165) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collett-Solberg PF, Ambler G, Backeljauw PF, Bidlingmaier M, Biller BMK, Boguszewski MCS, Cheung PT, Choong CSY, Cohen LE, Cohen P.et al. Diagnosis, genetics, and therapy of short stature in children: a Growth Hormone Research Society International Perspective. Hormone Research in Paediatrics 2019. 92 1–14. ( 10.1159/000502231) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ranke MB, Quigley C.Eds). International Classification of Pediatric Endocrine Diagnoses 2016. (available at: www.icped.org) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marouli E, Graff M, Medina-Gomez C, Lo KS, Wood AR, Kjaer TR, Fine RS, Lu Y, Schurmann C, Highland HM.et al. Rare and low-frequency coding variants alter human adult height. Nature 2017. 542 186–190. ( 10.1038/nature21039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo MH, Hirschhorn JN, Dauber A.Insights and implications of genome-wide association studies of height. Journal of Clinical Endocrinology and Metabolism 2018. 103 3155–3168. ( 10.1210/jc.2018-01126) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yengo L, Sidorenko J, Kemper KE, Zheng Z, Wood AR, Weedon MN, Frayling TM, Hirschhorn J, Yang J, Visscher PM.et al. GIANT Consortium meta-analysis of genome-wide association studies for height and body mass index in ~700000 individuals of European ancestry. Human Molecular Genetics 2018. 27 3641–3649. ( 10.1093/hmg/ddy271) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blum WF, Alherbish A, Alsagheir A, El Awwa A, Kaplan W, Koledova E, Savage MO.The growth hormone-insulin-like growth factor-I axis in the diagnosis and treatment of growth disorders. Endocrine Connections 2018. 7 R212–R222. ( 10.1530/EC-18-0099) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hermanussen M, Cole J.The calculation of target height reconsidered. Hormone Research 2003. 59 180–183. ( 10.1159/000069321) [DOI] [PubMed] [Google Scholar]
- 12.Savage MO, Backeljauw PF, Calzada R, Cianfarani S, Dunkel L, Koledova E, Wit JM, Yoo HW.Early detection, referral, investigation, and diagnosis of children with growth disorders. Hormone Research in Paediatrics 2016. 85 325–332. ( 10.1159/000444525) [DOI] [PubMed] [Google Scholar]
- 13.Wit JM, Kamp GA, Oostdijk W.on behalf of the Dutch Working Group on Triage and Diagnosis of Growth Disorders in Children. Towards a rational and efficient diagnostic approach in children referred for growth failure to the general paediatrician. Hormone Research in Paediatrics 2019. 91 223–240. ( 10.1159/000499915) [DOI] [PubMed] [Google Scholar]
- 14.Wit JM, Oostdijk W, Losekoot M, van Duyvenvoorde HA, Ruivenkamp CA, Kant SG.MECHANISMS IN ENDOCRINOLOGY: Novel genetic causes of short stature. European Journal of Endocrinology 2016. 174 R145–R. ( 10.1530/EJE-15-0937) [DOI] [PubMed] [Google Scholar]
- 15.Argente J, Pérez-Jurado LA.Genetic causes of proportionate short stature. Best Practice and Research: Clinical Endocrinology and Metabolism 2018. 32 499–522. ( 10.1016/j.beem.2018.05.012) [DOI] [PubMed] [Google Scholar]
- 16.Freire BL, Homma TK, Funari MFA, Lerario AM, Vasques GA, Malaquias AC, Arnhold IJP, Jorge AAL.Multigene sequencing analysis of children born small for gestational age with isolated short stature. Journal of Clinical Endocrinology and Metabolism 2019. 104 2023–2030. ( 10.1210/jc.2018-01971) [DOI] [PubMed] [Google Scholar]
- 17.Dauber A.Genetic testing for the child with short stature-has the time come to change our diagnostic paradigm? Journal of Clinical Endocrinology and Metabolism 2019. 104 2766–2769. ( 10.1210/jc.2019-00019) [DOI] [PubMed] [Google Scholar]
- 18.Cabrera-Salcedo C, Mizuno T, Tyzinski L, Andrew M, Vinks AA, Frystyk J, Wasserman H, Gordon CM, Hwa V, Backeljauw P.et al. Pharmacokinetics of IGF-1 in PAPP-A2-deficient patients, growth response, and effects on glucose and bone density. Journal of Clinical Endocrinology and Metabolism 2017. 102 4568–4577. ( 10.1210/jc.2017-01411) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frasier SD.The serum growth-hormone response to hypoglycemia in dwarfism. Journal of Pediatrics 1967. 71 625–6. ( 10.1016/s0022-3476(67)80197-5) [DOI] [PubMed] [Google Scholar]
- 20.Vanderschueren-Lodeweyckx M, Wolter R, Malvaux P, Eggermont E, Eeckels R.The glucagon stimulation test: effect of plasma growth hormone and on immunoreactive insulin, cortisol, and glucose in children. Journal of Pediatrics 1974. 85 182–187. ( 10.1016/s0022-3476(74)80389-6) [DOI] [PubMed] [Google Scholar]
- 21.Goeddel DV, Heyneker HL, Hozumi T, Arentzen R, Itakura K, Yansura DG, Ross MJ, Miozzari G, Crea R, Seeburg PH.Direct expression in Escherichia coli of a DNA sequence coding for human growth hormone. Nature 1979. 281 544–548. ( 10.1038/281544a0) [DOI] [PubMed] [Google Scholar]
- 22.Rudman D, Kutner MH, Blackston RD, Cushman RA, Bain RP, Patterson JH.Children with normal-variant short stature: treatment with human growth hormone for six months. New England Journal of Medicine 1981. 305 123–131. ( 10.1056/NEJM198107163050302) [DOI] [PubMed] [Google Scholar]
- 23.Van Vliet G, Styne DM, Kaplan SL, Grumbach MM.Growth hormone treatment for short stature. New England Journal of Medicine 1983. 309 1016–1022. ( 10.1056/NEJM198310273091703) [DOI] [PubMed] [Google Scholar]
- 24.Plotnick LP, Van Meter QL, Kowarski AA.Human growth hormone treatment of children with growth failure and normal growth hormone levels by immunoassay: lack of correlation with somatomedin generation. Pediatrics 1983. 71 324–327. [PubMed] [Google Scholar]
- 25.Gertner JM, Genel M, Gianfredi SP, Hintz RL, Rosenfeld RG, Tamborlane WV, Wilson DM.Prospective clinical trial of human growth hormone in short children without growth hormone deficiency. Journal of Pediatrics 1984. 104 172–176. ( 10.1016/s0022-3476(84)80987-7) [DOI] [PubMed] [Google Scholar]
- 26.Underwood LE.Report of the conference on uses and possible abuses of biosynthetic human growth hormone. New England Journal of Medicine 1984. 311 606–608. ( 10.1056/NEJM198408303110925) [DOI] [PubMed] [Google Scholar]
- 27.Hintz RL.The importance of the National Cooperative Growth Study (NCGS). In Deciphering Growth, pp. 131–141. Ed Carel J-C.Berlin Heidelberg: Springer-Verlag, 2005. [Google Scholar]
- 28.Hintz RL, Attie KM, Baptista J, Roche A.Effect of growth hormone treatment on adult height of children with idiopathic short stature. Genentech Collaborative Group. New England Journal of Medicine 1999. 340 502–507. ( 10.1056/NEJM199902183400702) [DOI] [PubMed] [Google Scholar]
- 29.Leschek EW, Rose SR, Yanovski JA, Troendle JF, Quigley CA, Chipman JJ, Crowe BJ, Ross JL, Cassorla FG, Blum WF.et al. National Institute of Child Health and Human Development-Eli Lilly & Co. Growth Hormone Collaborative Group. Journal of Clinical Endocrinology and Metabolism 2004. 89 3140–3148. ( 10.1210/jc.2003-031457) [DOI] [PubMed] [Google Scholar]
- 30.Cohen P.Controversy in clinical endocrinology: problems with reclassification of insulin-like growth factor I production and action disorders. Journal of Clinical Endocrinology and Metabolism 2006. 91 4235–4236. ( 10.1210/jc.2006-1641) [DOI] [PubMed] [Google Scholar]
- 31.Swatz Topor L, Feldman HA, Bauchner H, Cohen L.Variation in methods of predicting adult height for children with idiopathic short stature. Pediatrics 2010. 126 937–944. ( 10.1542/peds.2009-3649) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ranke MB, Wit JM.Growth hormone – past, present and future. Nature Reviews: Endocrinology 2018. 14 285–300. ( 10.1038/nrendo.2018.22) [DOI] [PubMed] [Google Scholar]
- 33.Ranke MB, Lindberg A, Price DA, Darendeliler F, Albertsson-Wikland K, Wilton P, Reiter EO.KIGS International Board. Age at growth hormone therapy start and first-year responsiveness to growth hormone are major determinants of height outcome in idiopathic short stature. Hormone Research 2007. 68 53–62. ( 10.1159/000098707) [DOI] [PubMed] [Google Scholar]
- 34.Kaplowitz PB, Shulman DI, Frane JW, Jacobs J, Lippe B.Characteristics of children with best and poorest first- and second-year growth during rhGH therapy: data from 25years of the Genentech National Cooperative Growth Study (NCGS). International Journal of Pediatric Endocrinology 2013. 2013 9. ( 10.1186/1687-9856-2013-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sävendahl L, Polak M, Backeljauw P, Blair J, Miller BS, Rohrer TR, Pietropoli A, Ostrow V, Ross J.Treatment of children with GH in the United States and Europe: long-term follow-up from NordiNet® IOS and ANSWER Program. Journal of Clinical Endocrinology and Metabolism 2019. 104 4730–4742. ( 10.1210/jc.2019-00775) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grimberg A, Allen DB.Growth hormone treatment for growth hormone deficiency and idiopathic short stature: new guidelines shaped by the presence and absence of evidence. Current Opinion in Pediatrics 2017. 29 466–471. ( 10.1097/MOP.0000000000000505) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deodati A, Cianfarani S.Impact of growth hormone therapy on adult height of children with idiopathic short stature: systematic review. BMJ 2011. 342 c7157. ( 10.1136/bmj.c7157) [DOI] [PubMed] [Google Scholar]
- 38.Ranke MB.Towards a consensus on the definition of idiopathic short stature. Hormone Research 1996. 45 (Supplement 2) 64–66. ( 10.1159/000184851) [DOI] [PubMed] [Google Scholar]
- 39.Wit JM, Clayton PE, Rogol AD, Savage MO, Saenger PH, Cohen P.Idiopathic short stature: definition, epidemiology, and diagnostic evaluation. Growth Hormone and IGF Research 2008. 18 89–110. ( 10.1016/j.ghir.2007.11.004) [DOI] [PubMed] [Google Scholar]
- 40.Cohen P, Rogol AD, Deal CL, Saenger P, Reiter EO, Ross JL, Chernausek SD, Savage MO, Wit JM.2007 ISS Consensus Workshop participants. Consensus statement on the diagnosis and treatment of children with idiopathic short stature: a summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology Workshop. Journal of Clinical Endocrinology and Metabolism 2008. 93 4210–4217. ( 10.1210/jc.2008-0509) [DOI] [PubMed] [Google Scholar]
- 41.Inzaghi E, Reiter E, Cianfarani S.The challenge of defining and investigating the causes of idiopathic short stature and finding an effective therapy. Hormone Research in Paediatrics 2019. 92 71–83. ( 10.1159/000502901) [DOI] [PubMed] [Google Scholar]
- 42.Clayton PE, Cianfarani S, Czernichow P, Johannsson G, Rapaport R, Rogol A.Management of the child born small for gestational age through to adulthood: a consensus statement of the International Societies of Pediatric Endocrinology and the Growth Hormone Research Society. Journal of Clinical Endocrinology and Metabolism 2007. 92 804–810. ( 10.1210/jc.2006-2017) [DOI] [PubMed] [Google Scholar]
- 43.Hokken-Koelega AC, De Ridder MA, Lemmen RJ, Den Hartog H, De Muinck Keizer-Schrama SM, Drop SL.Children born small for gestational age: do they catch up? Pediatric Research 1995. 38 267–271. ( 10.1203/00006450-199508000-00022) [DOI] [PubMed] [Google Scholar]
- 44.de Ridder MA, Engels MA, Stijnen T, Hokken-Koelega AC.Small for gestational age children without early catch-up growth: spontaneous growth and prediction of height at 8 years. Hormone Research 2008. 70 203–20. ( 10.1159/000137660) [DOI] [PubMed] [Google Scholar]
- 45.Chaussain JL, Colle M, Ducret JP.Adult height in children with prepubertal short stature secondary to intrauterine growth retardation. Acta Paediatrica 1994. 399 72–7. ( 10.1111/j.1651-2227.1994.tb13294.x) [DOI] [PubMed] [Google Scholar]
- 46.Grote FK, Oostdijk W, De Muinck Keizer-Schrama SM, van Dommelen P, van Buuren S, Dekker FW, Ketel AG, Moll HA, Wit JM.The diagnostic work up of growth failure in secondary health care; an evaluation of consensus guidelines. BMC Pediatrics 2008. 8 21. ( 10.1186/1471-2431-8-21) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caliebe J, Martin DD, Ranke MB, Wit JM.The auxological and biochemical continuum of short children born small for gestational age (SGA) or with normal birth size (idiopathic short stature). International Journal of Pediatric Endocrinology 2010. 2010 852967. ( 10.1155/2010/852967) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finken MJJ, van der Steen M, Smeets CCJ, Walenkamp MJE, de Bruin C, Hokken-Koelega ACS, Wit JM.Children born small for gestational age: differential diagnosis, molecular genetic evaluation, and implications. Endocrine Reviews 2018. 39 851–894. ( 10.1210/er.2018-00083) [DOI] [PubMed] [Google Scholar]
- 49.Salmon WD, Daughaday WH.A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. Journal of Laboratory and Clinical Medicine 1957. 49 825–836. [PubMed] [Google Scholar]
- 50.LeRoith D.Clinical relevance of systemic and local IGF-I: lessons from animal models. Pediatric Endocrinology Reviews 2008. 5 (Supplement 2) 739–7. [PubMed] [Google Scholar]
- 51.Ranke MB.Defining insulin-like growth factor-I deficiency. Hormone Research 2006. 65 (Supplement 1) 9–14. ( 10.1159/000090641) [DOI] [PubMed] [Google Scholar]
- 52.Park P, Cohen P.Insulin-like growth factor (IGF-I) measurements in growth hormone (GH) therapy of idiopathic short stature (ISS). Growth Hormone and IGF Research 2005. 15 (Supplement A) S13–S20. ( 10.1016/j.ghir.2005.06.011) [DOI] [PubMed] [Google Scholar]
- 53.Cohen P, Rogol AD, Howard CP, Bright GM, Kappelgaard AM, Rosenfeld RG.American Norditropin Study Group. Insulin growth factor-based dosing of growth hormone therapy in children: a randomized, controlled study. Journal of Clinical Endocrinology and Metabolism 2007. 92 2480–2486. ( 10.1210/jc.2007-0204) [DOI] [PubMed] [Google Scholar]
- 54.Buckway CK, Guevara-Aguirre J, Pratt KL, Burren CP, Rosenfeld RG.The IGF-I generation test revisited: a marker of GH sensitivity. Journal of Clinical Endocrinology and Metabolism 2001. 86 5176–51. ( 10.1210/jcem.86.11.8019) [DOI] [PubMed] [Google Scholar]
- 55.Selva KA, Buckway CK, Sexton G, Pratt KL, Tjoeng E, Guevara-Aguirre J, Rosenfeld RG.Reproducibility in patterns of IGF generation with special reference to idiopathic short stature. Hormone Research 2003. 60 237–246. ( 10.1159/000074038) [DOI] [PubMed] [Google Scholar]
- 56.Wit JM, Reiter EO, Ross JL, Saenger PH, Savage MO, Rogol AD, Cohen P.Idiopathic short stature: management and growth hormone treatment. Growth Hormone and IGF Research 2008. 18 111–135. ( 10.1016/j.ghir.2007.11.003) [DOI] [PubMed] [Google Scholar]
- 57.Savage MO, Burren CP, Rosenfeld RG.The continuum of growth hormone-IGF-I axis defects causing short stature: diagnostic and therapeutic challenges. Clinical Endocrinology 2010. 72 721–728. ( 10.1111/j.1365-2265.2009.03775.x) [DOI] [PubMed] [Google Scholar]
- 58.Pedicelli S, Peschiaroli E, Violi E, Cianfarani S.Controversies in the definition and treatment of idiopathic short stature (ISS). Journal of Clinical Research in Pediatric Endocrinology 2009. 1 105–115. ( 10.4008/jcrpe.v1i3.53) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sjoberg M, Salazar T, Espinosa C, Dagnino A, Avila A, Eggers M, Cassorla F, Carvallo P, Mericq MV.Study of GH sensitivity in Chilean patients with idiopathic short stature. Journal of Clinical Endocrinology and Metabolism 2001. 86 4375–4381. ( 10.1210/jcem.86.9.7850) [DOI] [PubMed] [Google Scholar]
- 60.Storr HL, Chatterjee S, Metherell LA, Foley C, Rosenfeld RG, Backeljauw PF, Dauber A, Savage MO, Hwa V.Nonclassical GH insensitivity: characterization of mild abnormalities of GH action. Endocrine Reviews 2019. 40 476–505. ( 10.1210/er.2018-00146) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vairamani K, Merjaneh L, Casano-Sancho P, Sanli ME, David A, Metherell LA, Savage MO, Del Pozo JS, Backeljauw PF, Rosenfeld RG.et al. Novel dominant-negative GH receptor mutations expands the spectrum of GHI and IGF-I deficiency. Journal of the Endocrine Society 2017. 1 345–358. ( 10.1210/js.2016-1119) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Metherell LA, Akker SA, Munroe PB, Rose SJ, Caulfield M, Savage MO, Chew SL, Clark AJ.Pseudoexon activation as a novel mechanism for disease resulting in atypical growth-hormone insensitivity. American Journal of Human Genetics 2001. 69 641–646. ( 10.1086/323266) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chatterjee S, Shapiro L, Rose SJ, Mushtaq T, Clayton PE, Ten SB, Banghoo A, Kumbattae U, Dias R, Savage MO.et al. Phenotypic spectrum and responses to recombinant human IGF-1 (rhIGF-1) therapy in patients with homozygous intronic pseudoexon growth hormone receptor mutations. European Journal of Endocrinology 2018. 178 481–489. ( 10.1530/EJE-18-0042) [DOI] [PubMed] [Google Scholar]
- 64.Klammt J, Neumann D, Gevers EF, Andrew SF, Schwartz ID, Rockstroh D, Colombo R, Sanchez MA, Vokurkova D, Kowalczyk J.et al. Dominant-negative STAT5B mutations cause growth hormone insensitivity with short stature and mild immune dysregulation. Nature Communications 2018. 9 2105. ( 10.1038/s41467-018-04521-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heath KE, Argente J, Barrios V, Pozo J, Díaz-González F, Martos-Moreno GA, Caimari M, Gracia R, Campos-Barros A.Primary acid-labile subunit deficiency due to recessive IGFALS mutations results in postnatal growth deficit associated with low circulating insulin growth factor (IGF)-I, IGF binding protein-3 levels, and hyperinsulinemia. Journal of Clinical Endocrinology and Metabolism 2008. 93 1616–1624. ( 10.1210/jc.2007-2678) [DOI] [PubMed] [Google Scholar]
- 66.Dauber A, Muñoz-Calvo MT, Barrios V, Domené HM, Kloverpris S, Serra-Juhé C, Desikan V, Pozo J, Muzumdar R, Martos-Moreno GÁ.et al. Mutations in pregnancy-associated plasma protein A2 cause short stature due to low IGF-I availability. EMBO Molecular Medicine 2016. 8 363–374. ( 10.15252/emmm.201506106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van Duyvenvoorde HA, van Setten PA, Walenkamp MJ, van Doorn J, Koenig J, Gauguin L, Oostdijk W, Ruivenkamp CA, Losekoot M, Wade JD.et al. Short stature associated with a novel heterozygous mutation in the insulin-like growth factor 1 gene. Journal of Clinical Endocrinology and Metabolism 2010. 95 E363–E36. ( 10.1210/jc.2010-0511) [DOI] [PubMed] [Google Scholar]
- 68.Fuqua JS, Derr M, Rosenfeld RG, Hwa V.Identification of a novel heterozygous IGF1 splicing mutation in a large kindred with familial short stature. Hormone Research in Paediatrics 2012. 78 59–66. ( 10.1159/000337249) [DOI] [PubMed] [Google Scholar]
- 69.Batey L, Moon JE, Yu Y, Wu B, Hirschhorn JN, Shen Y, Dauber A.A novel deletion of IGF1 in a patient with idiopathic short stature provides insight Into IGF1 haploinsufficiency. Journal of Clinical Endocrinology and Metabolism 2014. 99 E153–E159. ( 10.1210/jc.2013-3106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fofanova-Gambetti OV, Hwa V, Wit JM, Domené HM, Argente J, Bang P, Högler W, Kirsch S, Pihoker C, Chiu HK.et al. Impact of heterozygosity for acid-labile subunit (IGFALS) gene mutations on stature: results from the international acid-labile subunit consortium. Journal of Clinical Endocrinology and Metabolism 2010. 95 4184–4191. ( 10.1210/jc.2010-0489) [DOI] [PubMed] [Google Scholar]
- 71.Domené HM, Scaglia PA, Martínez AS, Keselman AC, Karabatas LM, Pipman VR, Bengolea SV, Guida MC, Ropelato MG, Ballerini MG.et al. Heterozygous IGFALS gene variants in idiopathic short stature and normal children: impact on height and the IGF system. Hormone Research in Paediatrics 2013. 80 413–423. ( 10.1159/000355412) [DOI] [PubMed] [Google Scholar]
- 72.Işık E, Haliloglu B, van Doorn J, Demirbilek H, Scheltinga SA, Losekoot M, Wit JM.Clinical and biochemical characteristics and bone mineral density of homozygous, compound heterozygous and heterozygous carriers of three novel IGFALS mutations. European Journal of Endocrinology 2017. 176 657–667. ( 10.1530/EJE-16-0999) [DOI] [PubMed] [Google Scholar]
- 73.Walenkamp MJE, Robers JML, Wit JM, Zandwijken GRJ, van Duyvenvoorde HA, Oostdijk W, Hokken-Koelega ACS, Kant SG, Losekoot M.Phenotypic features and response to GH treatment of patients with a molecular defect of the IGF-1 receptor. Journal of Clinical Endocrinology and Metabolism 2019. 104 3157–3171. ( 10.1210/jc.2018-02065) [DOI] [PubMed] [Google Scholar]
- 74.Begemann M, Zim B, Santen G, Wirthgen E, Soellner L, Büttel HM, Schweizer R, van Workum W, Binder G, Eggermann T.Paternally inherited IGF2 mutation and growth restriction. New England Journal of Medicine 2015. 373 349–356. ( 10.1056/NEJMoa1415227) [DOI] [PubMed] [Google Scholar]
- 75.Fukami M, Seki A, Ogata T.SHOX haploinsufficiency as a cause of syndromic and non syndromic short stature. Molecular Syndromology 2016. 7 3–11. ( 10.1159/000444596) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Binder G.Short stature due to SHOX deficiency: genotype, phenotype, and therapy. Hormone Research in Paediatrics 2011. 75 81–89. ( 10.1159/000324105) [DOI] [PubMed] [Google Scholar]
- 77.Benabbad I, Rosilio M, Child CJ, Carel JC, Ross JL, Deal CL, Drop SL, Zimmermann AG, Jia N, Quigley CA.et al. Safety outcomes and near-adult height gain of growth hormone-treated children with SHOX deficiency: data from an observational study and a clinical trial. Hormone Research in Paediatrics 2017. 87 42–50. ( 10.1159/000452973) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shapiro S, Klein GW, Klein ML, Wallach EJ, Fen Y, Godbold JH, Rapaport R.SHOX gene variants: growth hormone/insulin-like growth factor-1 status and response to growth hormone treatment. Hormone Research in Paediatrics 2015. 83 26–35. ( 10.1159/000365507) [DOI] [PubMed] [Google Scholar]
- 79.Kant SG, Cervenkova I, Balek L, Trantirek L, Santen GWE, de Vries MC, Duyvenvoorde HA, van der Wielen MJR, Verkerk AJ, Uitterlinden AG.et al. A novel variant of FGFR3 causes proportionate short stature. European Journal of Endocrinology 2015. 172 763–7. ( 10.1530/EJE-14-0945) [DOI] [PubMed] [Google Scholar]
- 80.Pinto G, Cormier-Daire V, Le Merrer M, Samara-Boustani D, Baujat G, Fresneau L, Viaud M, Souberbielle JC, Pineau JC, Polak M.Efficacy and safety of growth hormone treatment in children with hypochondroplasia: comparison with an historical cohort. Hormone Research in Paediatrics 2014. 82 355–363. ( 10.1159/000364807) [DOI] [PubMed] [Google Scholar]
- 81.Hisado-Oliva A, Garre-Vázquez AI, Santaolalla-Caballero F, Alberta Belinchón A, Barreda-Bonis AC, Vasques GA, Ramirez J, Luzuriaga C, Carlone G, González-Casado I.et al. Heterozygous NPR2 mutations cause disproportionate short stature, similar to Léri-Weill dyschondrosteosis. Journal of Clinical Endocrinology and Metabolism 2015. 100 E1133–E. ( 10.1210/jc.2015-1612) [DOI] [PubMed] [Google Scholar]
- 82.Vasques GA, Amano N, Docko AJ, Funari MFA, Quedas EPS, Nishi MY, Arnhold IJ, Hasegawa T, Jorge AA.Heterozygous mutations in natriuretic peptide receptor-B (NPR2) gene as a cause of short stature in patients initially classified as idiopathic short stature. Journal of Clinical Endocrinology and Metabolism 2013. 98 E1636–E1644. ( 10.1210/jc.2013-2142) [DOI] [PubMed] [Google Scholar]
- 83.Wang SR, Jacobsen CM, Carmichael H, Edmund AB, Robinson JW, Olney RC, Miller TC, Moon JE, Mericq V, Potter LR.et al. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) gene as a cause of short stature. Human Mutation 2015. 36 474–481. ( 10.1002/humu.22773) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nilsson O, Guo MH, Dunbar N, Popovic J, Flynn D, Jacobsen C, Lui JC, Hirschhorn JN, Baron J, Dauber A.Short stature, accelerated bone maturation, and early growth cessation due to heterozygous aggrecan mutations. Journal of Clinical Endocrinology and Metabolism 2014. 99 E1510–E. ( 10.1210/jc.2014-1332) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hauer NN, Sticht H, Boppudi S, Büttner C, Kraus C, Trautmann U, Zenker M, Zweier C, Wiesener A, Jamra RA.et al. Genetic screening confirms heterozygous mutations in ACAN as a major cause of idiopathic short stature. Scientific Reports 2017. 7 12225. ( 10.1038/s41598-017-12465-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hauer NN, Popp B, Schoeller E, Schuhmann S, Heath KE, Hisado-Oliva A, Klinger P, Kraus C, Trautmann U, Zenker M.et al. Clinical relevance of systematic phenotyping and exome sequencing in patients with short stature. Genetics in Medicine 2018. 20 630–638. ( 10.1038/gim.2017.159) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sisley S, Trujillo MV, Khoury J, Backeljauw P.Low incidence of pathology detection and high cost of screening in the evaluation of asymptomatic short children. Journal of Pediatrics 2013. 163 1045–1051. ( 10.1016/j.jpeds.2013.04.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wit JM, Bidlingmaier M, de Bruin C, Oostdijk W.A proposal for the interpretation of serum IGF-I concentration as part of Laboratory Screening in children with growth failure. Journal of Clinical Research in Pediatric Endocrinology 2020. 12 130–139. ( 10.4274/jcrpe.galenos.2019.2019.0176) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yau M, Chacko E, Regelmann MO, Annunziato R, Wallach EJ, Chia D, Rapaport R.Peak growth hormone response to combined stimulation test in 315 children and correlations with metabolic parameters. Hormone Research in Paediatrics 2019. 92 36–44. ( 10.1159/000502308) [DOI] [PubMed] [Google Scholar]
- 90.Coutant R, Dörr HG, Gleeson H, Argente J.Diagnosis of endocrine disease: limitations of the IGF1 generation test in children with short stature. European Journal of Endocrinology 2012. 166 351–357. ( 10.1530/EJE-11-0618) [DOI] [PubMed] [Google Scholar]
- 91.Dauber A, Rosenfeld RG, Hirschhorn JN.Genetic evaluation of short stature. Journal of Clinical Endocrinology and Metabolism 2014. 99 3080–30. ( 10.1210/jc.2014-1506) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Argente J, Tatton-Brown K, Lehwalder D, Pfäffle R.Genetics of growth disorders-which patients require genetic testing? Frontiers in Endocrinology 2019. 10 602. ( 10.3389/fendo.2019.00602) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Murray PG, Clayton PE, Chernausek SD.A genetic approach to evaluation of short stature of undetermined cause. Lancet: Diabetes and Endocrinology 2018. 6 564–574. ( 10.1016/S2213-8587(18)30034-2) [DOI] [PubMed] [Google Scholar]
- 94.Plachy L, Dusatkova P, Maratova K, Petruzelkova L, Zemkova D, Elblova L, Kucerova P, Toni L, Kolouskova S, Snajderova M.et al. NPR2 variants are frequent among children with familiar short stature and respond well to growth hormone therapy. Journal of Clinical Endocrinology and Metabolism 2020. 105 dgaa037. ( 10.1210/clinem/dgaa037) [DOI] [PubMed] [Google Scholar]
- 95.Wang SR, Carmichael H, Andrew SF, Miller TC, Moon JE, Derr MA, Hwa V, Hirschhorn JN, Dauber A.Large-scale pooled next-generation sequencing of 1077 genes to identify genetic causes of short stature. Journal of Clinical Endocrinology and Metabolism 2013. 98 E1428–E. ( 10.1210/jc.2013-1534) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wit JM, van Duyvenvoorde HA, Scheltinga SA, de Bruin S, Hafkenscheid L, Kant SG, Ruivenkamp CA, Gijsbers AC, van Doorn J, Feigerlova E.et al. Genetic analysis of short children with apparent growth hormone insensitivity. Hormone Research in Paediatrics 2012. 77 320–333. ( 10.1159/000338462) [DOI] [PubMed] [Google Scholar]
- 97.Nilsson O.Aggrecanopathies highlight the need for genetic evaluation of ISS children. European Journal of Endocrinology 2020. 183 C9–C10. ( 10.1530/EJE-20-0420) [DOI] [PubMed] [Google Scholar]
- 98.Zahnleiter D, Uebe S, Ekici AB, Hoyer J, Wiesener A, Wieczorek D, Kunstmann E, Reis A, Doerr HG, Rauch A.et al. Rare copy number variants are a common cause of short stature. PLoS Genetics 2013. 9 e1003365. ( 10.1371/journal.pgen.1003365) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wakeling EL, Brioude F, Lokulo-Sodipe O, O’Connell SM, Salem J, Bliek J, Canton AP, Chrzanowska KH, Davies JH, Dias RP.et al. Diagnosis and management of Silver-Russell syndrome: first international consensus statement. Nature Reviews: Endocrinology 2017. 13 105–124. ( 10.1038/nrendo.2016.138) [DOI] [PubMed] [Google Scholar]
- 100.Van der Steen M, Pfundt R, Maas SJWH, Bakker-van Waarde WM, Odink RJ, Hokken-Koelega ACS.ACAN gene mutations in short children born SGA and response to growth hormone treatment. Journal of Clinical Endocrinology and Metabolism 2017. 102 1458–1467. ( 10.1210/jc.2016-2941) [DOI] [PubMed] [Google Scholar]
- 101.Gkourogianni A, Andrew M, Tyzinski L, Crocker M, Douglas J, Dunbar N, Fairchild J, Funari MFA, Heath KE, Jorge AAL.et al. Clinical characterization of patients with autosomal dominant short stature due to aggrecan mutations. Journal of Clinical Endocrinology and Metabolism 2017. 102 460–469. ( 10.1210/jc.2016-3313) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cottrell E, Ladha T, Borysewicz-Sańczyk H, Sawicka B, Savage MO, Bossowski AT, Storr HL.The value of whole exome sequencing for genetic diagnosis in a patient with Bloom syndrome. Journal of Endocrinological Investigation 2020. [epub]. ( 10.1007/s40618-020-01433-z) [DOI] [PubMed] [Google Scholar]
- 103.Stalman SE, Solanky N, Ishida M, Alemán-Charlet C, Abu-Amero S, Alders M, Alvizi L. Baird W, Demetriou C, Henneman P.et al. Genetic analyses in small-for-gestational-age newborns. Journal of Clinical Endocrinology and Metabolism 2018. 103 917–925. ( 10.1210/jc.2017-01843) [DOI] [PubMed] [Google Scholar]
- 104.Muñoz-Calvo MT, Barrios V, Pozo J, Chowen JA, Martos-Moreno GÁ, Hawkins F, Dauber A, Domené HM, Yakar S, Rosenfeld RG.et al. Treatment with recombinant human insulin-like growth factor-1 improves growth in patients with PAPP-A2 deficiency. Journal of Clinical Endocrinology and Metabolism 2016. 101 3879–3883. ( 10.1210/jc.2016-2751) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Howell SJ, Wilton P, Lindberg A, Shalet SM.Growth hormone replacement and the risk of malignancy in children with neurofibromatosis. Journal of Pediatrics 1998. 133 201–205. ( 10.1016/s0022-3476(98)70245-8) [DOI] [PubMed] [Google Scholar]
- 106.Breinholt VM, Rasmussen CE, Mygind PH, Kjelgaard-Hansen M, Faltinger F, Bernhard A, Zettler J, Hersel U.TransCon CNP, a sustained-release C-type natriuretic peptide prodrug, a potentially safe and efficacious new therapeutic modality for the treatment of comorbidities associated with fibroblast growth factor receptor 3-related skeletal dysplasias. Journal of Pharmacology and Experimental Therapeutics 2019. 370 459–471. ( 10.1124/jpet.119.258251) [DOI] [PubMed] [Google Scholar]
- 107.Savarirayan R, Tofts L, Irving M, Wilcox W, Bacino CA, Hoover-Fong J, Ullot Font R, Harmatz P, Rutsch F, Bober MB.et al. Once-daily, subcutaneous vosoritide therapy in children with achondroplasia: a randomised, double-blind, phase 3, placebo-controlled, multicentre trial. Lancet 2020. 396 684–692. ( 10.1016/S0140-6736(20)31541-5) [DOI] [PubMed] [Google Scholar]